


Abstract
Post-transcriptional control is essential to safeguard structural and metabolic changes in enucleated reticulocytes during their terminal maturation to functional erythrocytes. The timely synthesis of arachidonate 15-lipoxygenase (ALOX15), which initiates mitochondria degradation at the final stage of reticulocyte maturation is regulated by the multifunctional protein HNRNPK. It constitutes a silencing complex at the ALOX15 mRNA 3′ untranslated region that inhibits translation initiation at the AUG by impeding the joining of ribosomal 60S subunits to 40S subunits. To elucidate how HNRNPK interferes with 80S ribosome assembly, three independent screens were applied. They consistently demonstrated a differential interaction of HNRNPK with RPS19, which is localized at the head of the 40S subunit and extends into its functional center. During induced erythroid maturation of K562 cells, decreasing arginine dimethylation of HNRNPK is linked to a reduced interaction with RPS19 in vitro and in vivo. Dimethylation of residues R256, R258 and R268 in HNRNPK affects its interaction with RPS19. In noninduced K562 cells, RPS19 depletion results in the induction of ALOX15 synthesis and mitochondria degradation. Interestingly, residue W52 in RPS19, which is frequently mutated in Diamond-Blackfan Anemia (DBA), participates in specific HNRNPK binding and is an integral part of a putative aromatic cage.
INTRODUCTION
The control of protein synthesis involves the interaction of RNA-binding proteins (RBPs) with specific sequence elements, which are often located in the untranslated regions (UTRs) of mRNAs. Thereby regulatory messenger ribonucleoprotein (mRNP) complexes are formed, which facilitate cell-specific temporal and spatial mRNA translation (1,2). In particular, for differentiating erythroid cells, which undergo enucleation in the course of erythroblast to reticulocyte transition (3–6) and lack mRNA synthesis capacity during the maturation of reticulocytes to erythrocytes, post-transcriptional control is indispensable (7–12).
Heterogeneous ribonucleoprotein K (HNRNPK) is a multifunctional protein that interacts with DNA, RNA and proteins, and modulates the activity of genes and their products on different levels and in various cell types (13–15). Its function in mRNA translation regulation in erythroid cells has been studied in detail. HNRNPK interacts with CU-rich elements in the 3’UTR of specific mRNAs (10,16–18). Notably, it regulates the translation of MYH9 (18), SRC (16) and ALOX15 (10,19) mRNAs, which contribute to the proper erythropoiesis progression. MYH9 encoded NMHC IIA is involved in enucleation (18). The tyrosine kinase C-SRC phosphorylates HNRNPK in late reticulocytes (20,21) (see below). ALOX15 (arachidonate 15-lipoxygenase), previously referred to as reticulocyte 15-lipoxygenase, catalyzes mitochondrial membrane phospholipid dioxygenation at the onset of the mitochondrial breakdown in mature reticulocytes (22–24).
ALOX15 mRNA translation control restricts enzyme synthesis and activity to terminal erythroid maturation. This is critical as ALOX15 activity in premature erythroid cells would disturb their energy metabolism (25,26). Silencing of ALOX15 mRNA translation is mediated by HNRNPK that constitutes an inhibitory complex at the 3’UTR differentiation control element (DICE), in concert with HNRNPE1 and DDX6 (10,16,27,28). HNRNPK inhibits the joining of the ribosomal 60S subunit to the 40S subunit at the initiator AUG (19). A similar mechanism has been described for IGF2BP1, which binds to the ACTB mRNA 3’UTR and interferes with 60S ribosomal subunit joining in neuronal cells (29).
ALOX15 synthesis activation in mature reticulocytes is regulated in a complex manner (11). C-SRC catalyzed phosphorylation of Y458 in KH domain 3 (KH3) of HNRNPK abrogates its DICE-binding activity (20,21,30) and cleavage of KH3 by CASP3 safeguards the release from the inhibitory condition (31). Notably, the timing of C-SRC mediated HNRNPK phosphorylation is dependent on its arginine methylation state. In premature erythroid cells, protein arginine methyltransferase 1 (PRMT1) asymmetrically dimethylates the protein on five specific arginine residues (HNRNPKR5met) (16,32). Declining PRMT1 expression during erythroid maturation results in HNRNPKR5met replacement by nonmethylated HNRNPK (16).
However, until now it was unclear how the 3’UTR inhibitory complex connects to the translation initiation machinery and interferes with 60S subunit joining. Here, we characterize ribosomal protein 19 (RPS19) as the functional link between DICE-bound HNRNPK and the ribosomal 40S subunit, which enables ALOX15 mRNA translation inhibition. Through the use of three independent purification strategies, we identified RPS19 that is localized at the 40S subunit head and extends into its functional center (33–35). We demonstrate that the HNRNPK methylation state determines RPS19 binding in vitro and in vivo. Interestingly, dimethylated arginines are required for the HNRNPK–RPS19 interaction, which declines during induced erythroid maturation. Noteworthy, a W52R variant of RPS19 adversely affects HNRNPKR5met binding. Among others, the related mutation is linked to Diamond-Blackfan Anemia (DBA), a congenital red-cell aplasia (36). A direct relation between RPS19- and HNRNPK-mediated ALOX15 mRNA silencing was demonstrated by RPS19 depletion from K562 cells, which triggers ALOX15 expression and loss of mitochondria, independent of erythroid maturation.
MATERIALS AND METHODS
Plasmids
The tri-cistronic His-HNRNPKR5met expression plasmid has been described in (17) and pET16b-HNRNPK-5RG in (32). To generate RPS19 wt expression vectors, the RPS19 coding sequence was amplified from K562 cDNA and cloned in the pET16b BamHI site (Merck, Darmstadt, Germany) or between BamHI/EcoRI of pGEX(4T-1) (GE Healthcare). Plasmids encoding RPS19 variants RPS19-W52R and -F21A were created by site-directed mutagenesis. pSUPERIOR-Scr, -RPS19 #1 and -RPS19 #2 were produced by cloning of synthetic oligonucleotides encoding shRNA sequences (see Supplementary Material) with flanking BglII and HindIII sites into pSUPER-DUPER (kind gift of J. Lüscher-Firzlaff). pSUPER-DUPER was EcoRI/HindIII digested and the respective fragments cloned into pSUPERIOR.neo+gfp (Oligoengine, Seattle, WA, USA). pDNA6-tetR was a kind gift of J. Vervoorts-Weber.
Cell culture and induction of erythroid maturation
K562 cells (DSMZ ACC 10) were cultured in RPMI-1640 (10% FBS, 1× penicillin/streptomycin) (16). Cells were routinely authenticated and tested for mycoplasma contamination. Erythroid maturation was induced with 1.5 mM sodium butyrate (Merck, Darmstadt, Germany, B5887) (16). Cytoplasmic and total extracts were prepared as in (16).
ES (PRMT1+/+) and (PRMT1−/−) cells (37) have been cultured as described (32).
Transfection experiments and fluorescence activated cell sorting (FACS)
K562 cells (1 × 106 cells, in RPMI without additives) were transfected with 500 pmol siRNAs (MWG) (Supplementary Material) by electroporation (0.36 kV, 100 μF) (Gene-Pulser-Xcell, Bio-Rad, Hercules, CA, USA) (16) and harvested 48 h post transfection (Figure 1).
Figure 1.

Simultaneous HNRNPE1 and PRMT1 depletion induces ALOX15 expression independent of erythroid maturation. K562 cells were transfected with PRMT1, HNRNPE1 and HNRNPK specific siRNAs individually or in combination or with a nonrelated control siRNA (ctrl.) and analyzed 48 h post-transfection. (A) Western blot analysis of siRNA transfected K562 cell extracts as depicted with antibodies specific for PRMT1, HNRNPE1, HNRNPK, nonmethylated HNRNPK (non-RmetK) (16) and GAPDH as loading control. (B) Immunofluorescence analysis with HNRNPE1, PRMT1 and ALOX15 specific antibodies, nuclei were stained with DAPI. (C) Immunofluorescence staining with antibodies against HNRNPE1, HNRNPK and ALOX15. Nuclei were detected with DAPI; scale bar: 20 μm. The average number of ALOX15 positive cells is indicated below panels (B and C).
To generate RPS19-depleted cells, K562 cells were transfected by electroporation (0.36 kV, 950 μF) with pSUPERIOR.neo+gfp vectors. Forty-eight hours post-transfection GFP-positive and GFP-negative cells were fractionated by FACS on a BD FACSAriaII instrument (BD Biosciences, Franklin Lakes, NJ, USA) for RT-qPCR and Western blot analysis.
RNA isolation and RT-qPCR
RNA was extracted using Trizol (Thermo Fisher Scientific, Carlsbad, CA, USA). For reverse transcription, 1 μg total RNA, random primer and the Maxima H minus First strand cDNA synthesis kit (Thermo Fisher Scientific, Carlsbad, CA, USA, K1682) were used. For polysome analysis, RNA was isolated from equal volumes of gradient fractions after addition of 0.1 ng blaR extraction control and equal volumes were subjected to reverse transcription. QPCR was performed with PowerSYBRGreen (Thermo Fisher Scientific, Carlsbad, CA, USA, 4368702) on a StepOnePlus PCR system (Thermo Fisher Scientific, Carlsbad, CA, USA), primer sequences are listed in the Supplementary Material. RNA levels were determined by the ΔΔCT method (38).
Western blot assays
Western blot assays were performed as described (28) with specific antibodies (Supplementary Material). Images were acquired with the LAS4000 system (GE Healthcare) and quantified using ImageQuant (GE Healthcare).
Immunofluorescence staining
Immunofluorescence staining was performed as in (16) with specific antibodies (Supplementary Material). Mitochondria were stained with MitoTracker Orange (Thermo Fisher Scientific, Carlsbad, CA, USA, M7511). Images were acquired with AxioVision on an Apotome 2 Microscope (Carl-Zeiss-Jena, Jena, Germany).
In vitro transcription
For in vitro transcription, the T7 MegaScript kit (Thermo Fisher Scientific, Carlsbad, CA, USA, AM1334) was used following manufacturer protocols. The blaR extraction control was amplified by PCR from pcDNA6-tetR using a forward primer (T7 promoter sequence, taatacgactcactatagatggccaagcctttgtctc) and the blaR qPCR rv primer (Supplementary Material).
Immunoprecipitation
For mass spectrometry analyses, HNRNPK was immunoprecipitated from 5 mg cytoplasmic extracts generated at day 0 and 8 of erythroid maturation using a commercial HNRNPK antibody (sc-28380, Santa Cruz, Dallas, TX, USA) as previously described (16). For Western blot assays, the immunoprecipitation (IPP) was performed with the HNRNPK antibody described in (31). HNRNPK IPP from ES (PRMT1+/+) and (PRMT1−/−) cells (Figure 2G and H) was performed with HNRNPK peptide-antibody #2 (31) coupled to 30 μl protein G-Sepharose, incubated 1.5 h at 4°C with 200 μg total extract in IPP buffer (16).
Figure 2.

HNRNPK arginine methylation affects RPS19 binding. (A) Purification strategies that identified HNRNPK interacting proteins of the translational silencing complex listed in Table 1. Left panel: DICE-BoxB-RNA linked to GSH-sepharose via GST-tagged λ peptide (45) was incubated with translation competent cytoplasmic extract (CX) of noninduced K562 cells (0 d) and eluted DICE-RNA-bound complexes were further purified by HNRNPK immunoprecipitation (IPP) (28). Middle panel: K562 CX (0 d) and CX of cells induced 8 days for erythroid maturation (8 d) were subjected to HNRNPK IPP to enrich proteins differentially associated with HNRNPK (16). Right panel: RNase A-treated K562 CX (0 d) was utilized for affinity purification employing either recombinant quantitatively asymmetrically dimethylated His-HNRNPKR5met (17,32) or nonmethylated His-HNRNPK, which represent the methylation state of endogenous HNRNPK at day 0 and 8 of erythroid maturation, respectively (16). (B) Specific detection of His-HNRNPKR5met and His-HNRNPK by monoclonal antibodies generated against asymmetrically dimethylated arginine residues (admR) (46) or nonmethylated HNRNPK (non-RmetK) (16), respectively. (C) Recombinant His-tagged methylated HNRNPKR5met (lanes 1–11) or nonmethylated HNRNPK (lanes 12–22) were immobilized on Ni-NTA agarose and incubated with RNase A treated CX of K562 cells (0 d). HNRNPK was eluted with imidazole and visualized by Coomassie staining, interacting RPS19 with a specific antibody. Co-eluted RPS19 was quantified below (n = 3). ACTB served as control. (D) Upper panel: HNRNPK and PRMT1 expression in K562 cells at 0 d and 8 d and non-RmetK level. GAPDH served as control. Lower panel: HNRNPK IPP indicating predominant HNRNPK methylation at 0 d detected with the admR antibody. (E) HNRNPK IPP (lanes 4, 9) and control IPP (lanes 5, 10) from K562 0 d CX (input, lanes 1–3) and 8 d CX (input, lanes 6–8). HNRNPK and RPS19 were analyzed by western blotting, levels of RPS19 shown beneath. Representative blots were quantified from three independent experiments. (F) Ribosomal association of ALOX15 (CDS 1986 nts) mRNA and HGB1 (CDS 444 nts) mRNA fractionated upon sucrose gradient centrifugation in three independent experiments (means ± s.d.; n = 3). (G) Upper panel: Levels of HNRNPK, PRMT1, non-RmetK and Vinculin in extracts of ES (PRMT1+/+) and (PRMT1−/−) cells. Lower panel: HNRNPK IPP verifying lack of HNRNPK methylation in ES (PRMT1−/−) with admR antibody. (H) HNRNPK IPP (lanes 2, 5) and control IPP (lanes 3,6) from ES (PRMT1+/+) (input, lane 1) and (PRMT1−/−) cell extracts (input, lane 4). HNRNPK and RPS19 were detected by Western blotting.
Recombinant protein expression
Expression and purification of His-HNRNPKR5met, His-HNRNPK (17) and His-HNRNPK-5RG (32) have been described. His-RPS19 wt, -W52R and -F21A were expressed as described (10,21), except that the proteins were dialyzed against 20 mM HEPES, pH 8.0, 150 mM KCl, 50 mM sodium citrate. GST fusion proteins were expressed and purified as detailed in (30) and dialyzed against 20 mM HEPES pH 8.0, 150 mM KCl and 5% glycerol.
His-HNRNPK pulldown
For mass spectrometric analyses, 10 mg cytoplasmic extract was digested with RNase A (Thermo Fisher Scientific, Carlsbad, CA, USA) and combined with either 1 mg His-HNRNPKR5met, His-HNRNPK or without protein in a total volume of 1 ml (adjusted with buffer A: 300 mM KCl, 20 mM Tris, pH 8.0, 10 mM imidazole, 10 mM sodium citrate, 10% sucrose). The mixture was loaded on a 100 μl Ni-NTA-micro column in a FPLC instrument (GE Healthcare, Chicago, IL, USA; 0.05 ml/min). The column was washed with 0.5 ml buffer A (0.05 ml/min) and complexes were eluted with a gradient from 0% to 50% buffer B (300 mM KCl, 20 mM Tris pH 8.0, 500 mM imidazole, 10 mM sodium citrate, 10% sucrose) in 1 ml (0.02 ml/min). Subsequently, buffer B was raised to 100% in 0.1 ml. The elution profile was monitored by Coomassie staining and fraction 13 chosen for mass spectrometry.
For Western blot analysis shown in Figure 2C, 1 mg cytoplasmic extract was digested with RNase A and incubated with 100 μg His-HNRNPKR5met or His-HNRNPK coupled to Ni-NTA in buffer A for 1 h at 4°C. Beads were washed twice with buffer A and His-HNRNPK-containing complexes were eluted stepwise with increasing concentration of buffer B. Elution steps in percent buffer B were: 1 = 10%, 2 = 20%, 3 = 30%, 4 = 40%, 5 = 50%, 6 = 50%, 7 = 100%. Beads were boiled in SDS sample buffer for Coomassie staining and Western blot analysis. In Figure 4A, 100 μg cytoplasmic extract was digested with RNase A and incubated with 10 μg His-HNRNPKR5met, His-HNRNPK or His-HNRNPK-5RG coupled to Ni-NTA in buffer A for 1 h at 4°C. Beads were washed twice with buffer A and His-HNRNPK-containing complexes were eluted by boiling in SDS sample buffer. In Figure 4I, 200 ng GST-RPS19 or the respective variants were incubated with 1 μg His-HNRNPKR5met coupled to Ni-NTA in buffer A, supplemented with 0.025% Triton X-100 for 1 h at 4°C. Beads were processed and eluted as in Figure 4A.
Figure 4.

RPS19 interacts predominantly with HNRNPKR5met derived peptide 1m. (A) His-tagged methylated HNRNPKR5met, nonmethylated HNRNPK and HNRNPK5RG were immobilized on Ni-NTA agarose and incubated with RNase A treated CX of noninduced K562 cells (0 d). Left panel: Input of HNRNPK variants visualized by Coomassie. Right panel: Interacting RPS19 detected with a specific antibody in a representative Western blot, quantification below (n = 3). (B) Cartoon representation of the Tudor domain of SPF30 bound to a symmetrically (left panel) or to an asymmetrically methylated arginine (middle panel), with aromatic side chains numbered by amino acid sequence. In both cases, all involved residues are placed with a distance between 4 and 7 Å from each other. An additional asparagine helps coordinating the Argmet residue (PDB-IDs: 4a4f, 4a4h). Right panel: Cartoon representation of RPS19 of the human 80S ribosome structure (PDB-ID: 6ek0; chain B of pdb-bundle 3). In addition to the three aromatic residues, which are positioned between 4 and 8,5 Å from each other and potentially form an aromatic cage for Argmet recognition, Asparagine 51 resides in close vicinity. Potentially the side chain of this residue could adopt a different orientation upon Argmet binding and thus help stabilizing their interaction. Furthermore, residues P31 and W33 are part of a loop region in the direct vicinity that could adopt a different conformation upon Argmet binding and thereby further stabilize its binding. Figures were made with the program PyMol 1.7. (C) Melting temperature (Tm) of His-RPS19 (wt) and variants as indicated, determined by the TSA (n = 3). (D) TSA analysis of the interaction of His-RPS19(wt) with HNRNPK derived peptides P1, P1m, P2 and P2m. The ΔTm relative to the reaction without peptide addition is shown (triplicates are shown in Supplementary Figure S3). (E) Interaction of His-RPS19, variant W52R and non-related control F21A with P1m examined by TSA, ΔTm relative to no peptide addition. (F) Input of recombinant His-RPS19 (upper panel) or GST-RPS19 variants (lower panel) visualized by Coomassie staining and a specific RPS19 antibody. (G) Biotinylated peptides P1m or P1 immobilized on Streptavidin beads were incubated with RNase A treated K562 CX (0d) (left panel) or His-RPS19 (right panel). Bound RPS19 in a representative western blot, quantified below (n = 3). (H) Immobilized biotinylated P1m was incubated with His-RPS19 variants, as indicated. Bound RPS19 in a representative Western blot, quantified below (n = 3). (I) Immobilized His-HNRNPKR5met was incubated with GST-RPS19 variants, as indicated. Representative Western blots of co-eluted HNRNPK and RPS19, quantified below (n = 3).
Proximity ligation assay (PLA)
The PLA (39) was performed with the Duolink II kit (Merck, Darmstadt, Germany, DUO92102). For each sample a conventional immunofluorescence sample and a PLA sample were processed in parallel. Shortly, K562 cells were spun on poly-L-Lysin coated coverslips, formaldehyde fixed and permeabilized with 0.2% Triton X-100 in PBS. After blocking with 2% BSA in PBS, coverslips were incubated with combinations of primary antibodies (Supplementary Material). After washing, cells were incubated with – and + Duolink probes, ligated and subjected to rolling circle amplification as described in the manufacturers protocol. Coverslips were mounted with Prolonged Gold Antifade reagent with DAPI (Thermo Fisher Scientific, Carlsbad, CA, USA, P36931). PLA signals per cell were counted on 10 images per conditions in three replicate experiments.
Thermal shift assay (TSA)
The TSA (40) was run with His-RPS19 wt and His-RPS19 variants (10 μM), which were assembled with 2.5× SYPRO Orange (Sigma-Aldrich) and indicated amounts of HNRNPK-derived peptides (PSL GmbH Heidelberg, Germany) in a total volume of 20 μl assay buffer (20 mM HEPES pH 8.0, 150 mM KCl, 50 mM sodium citrate). Reactions were performed on a StepOnePlus system (Thermo Fisher Scientific, Carlsbad, CA, USA) starting from 25°C rising in 1°C/min increments to 90°C. The increasing fluorescence signal caused by progressing SYPRO Orange binding to hydrophobic surfaces exposed upon denaturation was used to derive Tm values by Boltzmann sigmoidal fit (41). Peptides with asymmetrically dimethylated arginine residues underlined used in the thermal shift assay P1: FPMRGRGGFDRMPPGRGGR, P1m: FPMRGRGGFDRMPPGRGGR, P2: SPRRGPPPPPPGRGGRGGS, P2m: SPRRGPPPPPPGRGGRGGS.
Pulldown assay with biotinylated peptides
In Figure 4G, left panel, 0.4 μg biotinylated peptides P1m or P1 (PSL, Heidelberg, Germany) were pre-coupled to streptavidin-magnetic beads and incubated with 10 μg RNase A pre-treated K562 0d cell extract. In Figure 4G, right panel and 4H, 0.04 μg peptides P1m or P1 were pre-coupled to streptavidin-magnetic beads and incubated with 200 ng recombinant His-RPS19 or the respective variants for 1 h at room temperature in a total volume of 200 μl assay buffer (20 mM HEPES, pH 8.0, 150 mM KCl, 50 mM sodium citrate). Beads were washed three times for 10 min each with 0.5 ml binding buffer. Bound proteins were released by boiling in SDS sample buffer, separated by 15% SDS-PAGE and detected with specific antibodies.
Mass spectrometry analyses
Samples were separated on a NuPAGE 4%–12% Bis-Tris gel (Thermo Fisher Scientific, Carlsbad, CA, USA) and colloidal Coomassie stained. Lanes were divided in 23 fragments that were in gel digested with trypsin, extracted (42) and peptides were analyzed in a capillary HPLC coupled electrospray ionization quadrupole time of flight (ESI-Q-TOF, Ultima, Waters, Milford, MA, USA) mass spectrometer as described (28). Peptide assignments and data analysis were performed by Mascot, Matrix Science (London, UK) (28).
Surface sensing of translation assay (SUnSET)
In the SUnSET assay (43), K562 cells were incubated for 1 h with 5 μg/ml Puromycin (Merck, Darmstadt, Germany, P8833) before cell lysis. Incorporation of Puromycin into nascent protein chains was monitored by western blot detection with an anti-Puromycin antibody (Supplementary Material).
Polysome gradient fractionation
Sucrose gradient centrifugation was performed as described (44).
Statistical analysis
Statistical analyses were performed with GraphPad Prism. Assuming a Gaussian distribution, either Student’s t-test or ANOVA with multiple comparison test as indicated were applied to test for significant differences. Significance levels were defined as *P < 0.05, **P < 0.01, ***P < 0.001.
RESULTS
Reduced HNRNPK methylation results in induced ALOX15 expression in K562 cells
In order to ascertain whether erythroid maturation-dependent changes in asymmetric arginine methylation of HNRNPK are required for its function in the control of ALOX15 expression, we depleted PRMT1, HNRNPE1 and HNRNPK by RNAi from noninduced K562 cells (Figure 1A–C). A diminished PRMT1 level specifically enabled the expression of nonmethylated HNRNPK (Figure 1A, lanes 2 and 4). Notably, simultaneous PRMT1 and HNRNPE1 depletion activated ALOX15 expression in 4.5% of the cells (0.7% in control cells) (Figure 1B), consistent with ALOX15 induction by combined HNRNPK and HNRNPE1 knockdown in 10.3% depleted cells (0.7% under control conditions) (16) (Figure 1C). These results indicate that the HNRNPK methylation state is important for the regulation of ALOX15 mRNA translation.
HNRNPK arginine methylation affects RPS19 binding
Since disabled 80S ribosome formation causes the inhibition of ALOX15 mRNA translation initiation by HNRNPK bound to the 3’UTR DICE (10,19), three independent interaction screens were established to identify and characterize components of the HNRNPK-associated translational silencing complex (Figure 2A). DICE-RNA affinity chromatography with noninduced K562 cell extracts employing λ-GST-BoxB (45), combined with HNRNPK-immunoprecipitation (HNRNPK IPP) identified specific functional interactors (28) (Figure 2A, left panel). Furthermore, we enriched proteins, which bind differentially to HNRNPK from translation competent cytoplasmic extracts of noninduced K562 cells and cells induced for erythroid maturation (16) (Figure 2A, middle panel). Considering the changing HNRNPK methylation state during erythroid maturation (16), we employed recombinant quantitatively asymmetrically dimethylated His-HNRNPKR5met (17) and nonmethylated His-HNRNPK to isolate interacting proteins from RNase A-treated noninduced K562 cell extracts in a pulldown assay (Figure 2A, right panel). Methylated HNRNPK suitable for interaction studies was expressed in Escherichia coli from a tri-cistronic plasmid that encoded PRMT1 and HNRNPK in a 2:1 ratio (17). All five arginine residues (R256, 258, 268, 296 and 299) previously shown to be quantitatively asymmetrically dimethylated by PRMT1 in vivo by mass spectrometry and Edman degradation (32) were also quantitatively dimethylated in the recombinant protein expressed from the tri-cistronic plasmid in E. coli, as detected by mass spectrometry (17).
Fourteen proteins emerged in all screens, among them HNRNPK and DDX6 (10,28). Interestingly, under translational silencing conditions, the 40S ribosomal protein RPS19 was specifically enriched with HNRNPKR5met (Table 1).
Table 1.
Proteins consistently identified in all three purification strategies
| Gene ID | Ensembl | Gene name |
|---|---|---|
| CCAR2 | ENSG00000158941 | Cell cycle and apoptosis regulator 2 |
| DDX3X | ENSG00000215301 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 3, X-linked |
| DDX6 | ENSG00000110367 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 6 |
| ELAVL1 | ENSG00000066044 | ELAV (embryonic lethal, abnormal vision, Drosophila)-like 1 (Hu antigen R) |
| HNRNPK | ENSG00000165119 | Heterogeneous nuclear ribonucleoprotein K |
| HNRNPL | ENSG00000104824 | Heterogeneous nuclear ribonucleoprotein L |
| HNRNPU | ENSG00000153187 | Heterogeneous nuclear ribonucleoprotein U (scaffold attachment factor A) |
| MTREX | ENSG00000039123 | Mtr4 exosome RNA helicase |
| RPS19 | ENSG00000105372 | Ribosomal protein S19 |
| RPS3 | ENSG00000149273 | Ribosomal protein S3 |
| RPSA | ENSG00000168028 | Ribosomal protein SA |
| SERBP1 | ENSG00000142864 | SERPINE1 mRNA-binding protein 1 |
| WDR82 | ENSG00000164091 | WD repeat domain 82 |
| ZCCHC8 | ENSG00000033030 | Zinc finger, CCHC-domain containing 8 |
To verify the interaction with RPS19, we performed pulldown assays with either recombinant non-methylated HNRNPK or HNRNPKR5met as a bait. Their methylation status was validated with specific peptide antibodies against nonmethylated HNRNPK (non-RmetK) (16) and asymmetrically dimethylated arginine residues (admR) (46), respectively (Figure 2B). Both His-HNRNPKR5met and His-HNRNPK (17) were incubated with cytoplasmic extract of noninduced K562 cells. Remarkably, binding of RPS19 to nonmethylated HNRNPK was reduced to 69% compared to HNRNPKR5met (Figure 2C, lanes 5–11 and 16–22). No interaction with ACTB was detected (Figure 2C).
To investigate whether the interaction of endogenous HNRNPK and RPS19 changes during erythroid maturation, we performed HNRNPK immunoprecipitation experiments from translation competent cytoplasmic extracts of noninduced K562 cells (0 d) and after 8 days of erythroid maturation (8 d) (Figure 2D and E). In the latter stage a subset of K562 cells undergoes enucleation, expression of ALOX15 and mitochondria degradation (16). A comparison of both extracts revealed an overall decrease of HNRNPK, whereas the level of nonmethylated HNRNPK increased (Figure 2D, upper panel), as previously described (16). This was paralleled by diminished PRMT1 expression at day 8 (Figure 2D, upper panel) and a decline of HNRNPKR5met, detected with the specific admR-antibody (Figure 2D, lower panel). HNRNPK was specifically immunoprecipitated from both extracts and RPS19 co-precipitation at day 8 was strongly reduced (Figure 2E, lane 9) compared to day 0 (Figure 2E, lane 4), despite its much stronger expression at day 8 (Figure 2E, lanes 1–3 and 6–8).
To monitor the polysomal distribution of endogenous ALOX15 mRNA in noninduced K562 cells and at day 8 of induction, we performed sucrose gradient fractionation experiments employing equal amounts of translation competent cytosolic extracts. Cells were treated with cycloheximide prior to lysis and polysome fractionation (Figure 2F). At day 8 of erythroid maturation, when nonmethylated HNRNPK was elevated (Figure 2D) and RPS19 interaction declined (Figure 2E), ALOX15 mRNA (CDS 1986 nts) shifted from translational inactive mRNPs to monosomes and polysomes (Figure 2F, upper panel), while HBG1 (γ-globin mRNA, CDS 444 nts) did not (Figure 2F, lower panel). This is consistent with our earlier findings, which demonstrated that at day 8 of induction ALOX15 is expressed, enzymatically active and mitochondria are degraded (16).
To further study the methylation-dependent HNRNPK–RPS19 interaction, we made use of embryonic stem cells carrying homozygous Prmt1 gene null mutations ES (PRMT1−/−) (37) (Figure 2G and H). Methylation of HNRNPK can be detected in ES (PRMT1+/+), but not (PRMT1−/−) cells devoid of PRMT1 (32) (Figure 2G). HNRNPK IPP proved binding of methylated HNRNPK from ES (PRMT1+/+) cells to RPS19, which was strongly reduced in ES (PRMT1−/−) cells (Figure 2H, lanes 2 and 5), in line with the differential association in K562 cell extracts (Figure 2E). From these findings we conclude that arginine methylation of HNRNPK affects its interaction with RPS19.
To further validate this assumption, we analyzed the in vivo interaction of RPS19 and HNRNPK in K562 cells employing the proximity ligation assay (PLA) (39) (Figure 3A and B). To detect intracellular interactions, different proteins were stained with specific antibodies (Supplementary Figure S1A). Oligonucleotides coupled to the secondary antibody were amplified and hybridized with fluorescent PLA probes (Figure 3A and B). K562 cells exclusively incubated with PLA probes served as negative control (Figure 3A and B). The known interaction of DDX6 with the P-body marker DCP1A was observed at day 0 and 8 (Figure 3A and B). Importantly, when nonmethylated HNRNPK predominates at day 8 (Figure 2D), the number (Figure 3A and B) and intensity (Supplementary Figure S1B) of PLA signals, which indicate HNRNPK-RPS19 binding, were significantly diminished. In contrast, DCP1A and HNRNPL did not associate with RPS19, supporting the specificity of the assay (Figure 3A and B; Supplementary Figure S1B).
Figure 3.

Specific HNRNPK–RPS19 interaction diminishes during erythroid maturation. K562 cells were induced for erythroid maturation by addition of 1.5 mM sodium butyrate. Noninduced cells (0 d) and cells at day 8 of maturation (8 d) were employed for the PLA. (A) PLA staining with the indicated antibody combinations or PLA probes only (ctrl.). Staining of nuclei with DAPI. (B) Quantification of the PLA assay shown in (A). Number of PLA signals per cell was determined in 10 images of three independent experiments each (***P <0.001).
These data indicate that HNRNPKR5met binding to RPS19 in early erythroid maturation is abrogated when nonmethylated HNRNPK becomes abundant in mature K562 cells. This dynamic interaction points to a potential mechanism in the regulation of ALOX15 mRNA translation.
Asymmetric dimethylation of HNRNPK R256, R258 and R268 mediates the interaction with RPS19
In order to identify HNRNPK arginine residues that confer differential RPS19 binding, we incubated His-HNRNPKR5met, nonmethylated HNRNPK or the substitution variant His-HNRNPK5RG, which is not methylated by PRMT1 (32) (Figure 4A, left panel) with cytoplasmic extract of non-induced K562 cells (K562 CX 0 d). Co-precipitated RPS19 was analyzed by western blot (Figure 4A, right panel). HNRNPKR5met exhibited stronger RPS19 binding than HNRNPK and binding of HNRNPKR5G was further diminished (Figure 4A, right panel). From this, we conclude that the five quantitatively asymmetrically dimethylated arginine residues are critical for its binding to RPS19. Methylated arginine residues can be recognized by aromatic cages for instance of Tudor domains (47–52) (Figure 4B, left and middle panel). Based on the high-resolution cryo-EM structure of the human 80S ribosome (53), we identified an arrangement of the aromatic residues W52, F53 and Y54 in RPS19 that resembles aromatic cages of Tudor domains and thus might serve as potential interaction scaffold for Argmet (Figure 4B, right panel). These three aromatic residues are positioned between 4 and 8.5 Å from each other. N51 resides in close vicinity, potentially the side chain of that residue could adopt a different orientation upon Argmet binding and thus help stabilizing their interaction.
To shed light on HNRNPK and RPS19 residues that contribute to their interaction, we conducted thermal shift assays to follow temperature dependent RPS19 unfolding in the presence of ligands. Folding transitions were traced by SYPRO Orange dye (40,54). Temperature-dependent denaturation exposes hydrophobic residues, to which fluorescent dyes attach and increasing fluorescence signals were used to calculate Tm values (41) (Figure 4D and E). In the TSA we used two HNRNPK derived peptides, P1, which bears R256, R258, R268 and P2, containing R296, R299 either in their nonmethylated (P1, P2) or methylated state (P1m, P2m). All five arginine residues are specifically asymmetrically dimethylated by PRMT1 (32).
Furthermore, we expressed and purified RPS19 and variants exhibiting modifications of W52, F53 and Y54 residues adjacent to the putative Argmet interaction site (Figure 4B, right panel). Besides RPS19-W52R, equivalent F53 and Y54 versions either failed to be expressed or exhibited deviant melting curves (Supplementary Figure S2D and E). Therefore, we focused on RPS19W52R and used RPS19F21A as negative control. Interestingly, a RPS19 W52R mutation has been functionally linked to defective erythropoiesis in DBA (36) and an impaired RPS19 function has been reported (55). Specific amino acid exchanges affected RPS19 Tm (Figure 4C and Supplementary Figure S3), interactions where therefor analyzed as ΔTm values. A concentration dependent increase of RPS19 (wt) Tm in the presence of HNRNPK peptide P1m (Figure 4D) indicated that methylated R256, R258 and R268 are predominantly involved in RPS19 interaction. Therefore, we focused on peptide P1m to compare the interaction of RPS19W52R and RPS19F21A with the wild-type protein. Specifically, RPS19W52R exhibited an impaired P1m interaction (Figure 4E).
To verify this result, we performed in vitro interaction assays (Figure 4F–I) employing His- or GST-tagged RPS19 variants (Figure 4F upper and lower panel, respectively), biotinylated HNRNPK peptides P1 and P1m (Figure 4G and H) or recombinant His-HNRNPKR5met (Figure 4I). When cytoplasmic extract of non-induced K562 cells was incubated with P1m or P1 coupled to streptavidin beads, a stronger interaction of endogenous RPS19 with peptide P1m was detected (Figure 4G, left panel). Likewise, His-RPS19 was bound in the same way by P1m and P1 (Figure 4G, right panel). Furthermore, binding of His-RPS19W52R to P1m was strongly diminished compared to His-RPS19 or His-RPS19F21A (Figure 4H), consistent with the result of the TSA (Figure 4E). The interaction of GST-RPS19W52R with methylated HNRNPK5Rmet coupled to Ni-NTA was reduced compared to GST-RPS19 and GST-RPS19F21A (Figure 4I) confirming the differential binding observed for the methylated HNRNPK peptide P1m (Figure 4E and H). This strongly suggests that the binding motif of RPS19 containing W52 and methylated HNRNPK residues R256, R258 and R268 contribute to the interaction of the two proteins.
ALOX15 expression and mitochondria degradation are induced in RPS19 depleted cells independent of terminal erythroid differentiation
Having shown that RPS19 interacts with asymmetrically dimethylated HNRNPK in noninduced K562 cells, we examined the function of RPS19 in ALOX15 mRNA translational silencing (Figure 5). RPS19 specific shRNAs (#1 and #2) or control shRNA (ctrl.) were expressed from pSUPERIOR.neo+gfp plasmids in noninduced K562 cells. Forty-eight hours post transfection FACS separated GFP-positive (GFP pos) and negative (GFP neg) cells were analyzed by RT-qPCR and Western blot (Figure 5A and B). Expression of RPS19 shRNAs #1 and #2 in GFP positive cells specifically resulted in a significant decline of RPS19 mRNA (Figure 5A) and RPS19 protein was reduced to 0.65 for both shRNAs (Figure 5B). Importantly, immunofluorescence analysis of noninduced GFP-positive K562 cells revealed that RPS19 depletion resulted in the derepression of ALOX15 synthesis (Figure 5C, left panel) and in diminished mitochondria staining (Figure 5C, right panel). Therefore, we conclude that RPS19 contributes to ALOX15 mRNA silencing in noninduced K562 cells.
Figure 5.

RPS19 depletion results in ALOX15 expression and mitochondria degradation. (A) K562 cells were transfected with pSUPERIOR.neo+gfp shRNA plasmids encoding a control shRNA (ctrl.) or RPS19-specific shRNAs #1 and #2. GFP-positive and GFP-negative cells were separated 48 h post-transfection by FACS. RT-qPCR analysis with RPS19 mRNA primers, normalized to 18S rRNA (n = 4). (B) Upper panel: representative Western blot of extracts derived from shRNA-transfected K562 cells as in (A), detection of RPS19 and GAPDH. Lower panel: quantification of RPS19 protein in three independent experiments (ANOVA with Holm Sidak’s multiple comparison test,**P < 0.01). (C) Immunofluorescence analysis of K562 cells transfected as in (A), without cell sorting. Left panel: ALOX15 and RPS19 staining with specific antibodies, transfected cells show GFP fluorescence. Nuclei were detected with DAPI. Right panel: Staining of functional mitochondria with MitoTracker Orange, RPS19, GFP, nuclei as at the left; scale bar: 20 μm.
From these GFP positive cells (shRNA #1) five stable RPS19 knockdown clones, A–E were generated (Figure 6). All clones exhibited diminished expression of RPS19 mRNA (Figure 6A) and protein (Figure 6B). Considering the documented ubiquitination of RPS19 in erythroid cells (56,57), we assume that bands at higher molecular weight represent poly-ubiquitinated RPS19 (Figure 6B, left panel). Diminished RPS19 expression was associated with decreased cell proliferation compared to K562 wild-type cells (Figure 6C), which was consistently observed when erythroid maturation of K562 cells was induced (16) and in HNRNPK-depleted cells (18). Furthermore, a reduced level of RPS19 was linked to an increase in erythroid maturation-independent hemoglobin synthesis, mostly pronounced in clones A and E (Figure 6D). To investigate if RPS19 depletion has a systemic impact on mRNA translation, we performed a SUnSET analysis in K562 cells (43). Cytoplasmic extracts of equal cell numbers revealed that the incorporation of puromycin was diminished for all five shRNA clones compared to wild-type K562 cells (Figure 6E), demonstrating that RPS19 depletion results in globally impaired mRNA translation.
Figure 6.

Reduction of RPS19 expression in stable knockdown cells. Stable RPS19 shRNA-expressing cell lines A to E were established by serial dilution from GFP-positive cells (shRNA#1) (Figure 5). (A) RPS19 mRNA determined by RT-qPCR, normalized to 18S rRNA (n = 4). (B) Left panel: representative Western blot for RPS19, RPS3 and RPL19, loading control VINCULIN. Nonmodified RPS19 is indicated by an arrow. Right panel: quantification of RPS19, ubiquitinated (Ub-RPS19), RPS3 and RPL19 signals (n = 4). (C) Number of K562 wt and RPS19-depleted cells during induced maturation (n = 4). (D) Percentage of hemoglobin-positive cells detected by benzidine staining (n = 5). (E) SUnSET analysis of global mRNA translation by puromycin incorporation. K562 wt and RPS19 shRNA cell lines were either left untreated (lanes 1–6) or incubated for 1 h with 5 μg/ml puromycin (lanes 7–12). Left panel: representative Western blot with puromycin antibody, loading control VINCULIN. Right panel: quantification of Western blot following puromycin treatment of (n = 3). (F) 28S and 18S rRNA determined by RT-qPCR, normalized to blaR extraction control (n = 4). One-way ANOVA with Holm Sidak’s multiple comparison test, *P < 0.05, **P < 0.01, ***P < 0.001.
We further focused on RPS19-depleted K562 cell clone A, which showed a significant decline of RPS19 and RPS3 that was associated with a diminished RPL19 expression (Figure 6B). Likewise, in RPS19-deficient erythroblasts a decrease in the expression of 40S and 60S subunit proteins was observed (58). Consistently, 18S rRNA and 28S rRNA were also reduced (Figure 6F). Altogether, the analysis of equal cell numbers indicates a reduction of ribosomal subunits in RPS19 clone A.
RPS19 depletion results in derepression of ALOX15 mRNA translation
To dissect the influence of RPS19 depletion on endogenous ALOX15 mRNA translation, we analyzed the distribution of ribosomal complexes by 15–45% sucrose gradient centrifugation. For a direct comparison with the analysis in noninduced K562 cells and at day 8 of induction (Figure 2F), we used equal amounts of translation competent cytoplasmic extracts (Figure 7A–C), in contrast to Figure 6 where extracts of equal cell numbers were examined. Wild-type K562 cells and RPS19 depleted clone A (Figure 6) were treated with cycloheximide prior to lysis and polysome fractionation (Figure 7A). Noteworthy, the ALOX15 mRNA translocation to heavy polysome containing fractions (Figure 7B) demonstrated an enhanced ALOX15 mRNA translation in RPS19-depleted extract. In contrast, the distribution of HBG1 (γ-globin) mRNA remained unaffected (Figure 7C), proving the specific impact of RPS19 on ALOX15 mRNA translation. ALOX15 synthesis was specifically activated in clone A (10.6% ALOX15 positive cells), compared to wild-type K562 cells (1.08%) (Figure 7D, right panel). Furthermore, mitotracker staining was reduced in cells of clone A (Figure 7D, left panel). Nonspecific signals were excluded by immunofluorescence staining without primary antibodies (Supplementary Figure S4).
Figure 7.

RPS19 depletion results in derepression of ALOX15 mRNA translation. (A–C) K562 wt cells (left) or RPS19 shRNA clone A cells (right) were treated with cycloheximide, lysed and fractionated on 15–45% sucrose gradients. (A) Upper panels: representative λ = A260nm fractionation profiles. Lower panels: 18S and 28S rRNA distribution in gradient fractions analyzed by RT-qPCR, normalized to blaR extraction control and input RNA (n = 3, mean ± SEM). (B) Left panel: distribution of ALOX15 mRNA determined as in (A). Right panel: ALOX15 mRNA in heavy and light polysomes, monosomes and mRNP (including 40S) fractions, as indicated in the left panel (n = 3, mean ± SD, two-way ANOVA, **P < 0.01). (C) HBG1 mRNA assigned to fractions as in (B). (D) Left panel: Immunofluorescence analysis of noninduced K562 wt cells and RPS19 shRNA clone A with RPS19 and ALOX15 specific antibodies. Nuclei detected with DAPI, functional mitochondria with MitoTracker Orange; scale bar: 20 μm. Right panel: average number of ALOX15 positive cells.
In summary, these findings demonstrate that independent of induced erythroid maturation, RPS19 depletion results in derepression of ALOX15 mRNA translation.
DISCUSSION
Derepression of ALOX15 mRNA translation activates ALOX15 synthesis in mature reticulocytes (59). The timely expression of ALOX15, which induces mitochondria membrane degradation, is crucial for the controlled mitochondria clearance during terminal erythroid maturation (22). HNRNPK and HNRNPE1 mediate ALOX15 mRNA silencing by inhibiting translational initiation (10). While HNRNPE1 is present in the cytoplasm of noninduced K562 cells (16,28), HNRNPK, initially mainly nuclear, accumulates in the cytoplasm starting at day 4 of maturation (16). Cytoplasmic accumulation of HNRNPK has been shown to be mediated by ERK1/2-dependent phosphorylation of Ser284 and Ser353 (60). It is conceivable that association of nuclear HNRNPK with ALOX15 mRNA precedes HNRNPE1 binding, which strengthens the inhibition in the cytoplasm. As component of the α-complex, HNRNPE1 contributes also to α-globin mRNA stabilization in erythroid cells (8,9).
The activation of ALOX15 synthesis, which can be detected in PRMT1 and HNRNPE1 depleted noninduced K562 cells (Figure 1B), resembles ALOX15 mRNA translation activation when HNRNPK and HNRNPE1 expression is diminished (16) (Figure 1C). These findings motivated us to evaluate HNRNPK interacting proteins, which were conjointly identified in three independent screens (Figure 2A), comprising differential RNA-dependent protein interactions, changing HNRNPK arginine methylation during erythroid maturation (16,28) and protein binding by recombinant HNRNPK and HNRNPKR5met (Table 1). Besides DDX6, a component of the ALOX15 mRNA translational silencing complex that recruits ALOX15 mRNA to RNP granules, from which 60S ribosomal subunits are excluded (28), we identified HNRNPU, DDX3 and RPSA. These proteins were shown earlier to interact with HNRNPK in K562 cell extracts (61). An association of DDX3X with HNRNPK and the JUND mRNA was also found to be linked to translation regulation in pancreatic β cells (62).
Interestingly, we furthermore identified ribosomal protein RPS19 as a novel component of the ALOX15 mRNA translation silencing complex in pre-mature erythroid cells (Table 1). RPS19, which is restricted to eukaryotes and archaea (63), is essential for yeast viability and mice development (64,65). It is involved in 18S rRNA processing, 40S ribosomal subunit assembly and maturation (64,66,67). Localized at the head of the 40S subunit, RPS19 extends into the functional center (33,34,68,69). Biochemical evidence for an interaction of RPS19 with translation initiation factor EIF2 in conjunction with other ribosomal proteins (70) was confirmed in Cryo-EM structural studies of 80S ribosomes from Drosophila melanogaster and Homo sapiens (33) and an association of RPS19 with EIF3 was determined in 80S ribosomes of Saccharomyces cerevisiae and Triticum aestivum (69). Consistently, the human cap-dependent 48S translation pre-initiation complex possesses both interactions (71). The ALOX15 mRNA translation repressor complex specifically includes the stalled 40S subunit as detected by sucrose gradient fractionation of in vitro translation initiation reactions and in toe printing assays (10,19), but lacks components of the ribosomal 60S subunit, like RPL19, in K562 cells (28). This is concordant with the outcome of comparative interaction assays, where we detected 40S subunit components RPS3 and RPSA besides RPS19, but no 60S subunit protein (Table 1).
Notably, mutations in RPS19 are linked to defective ribosome function (66,72) and contribute to disturbed differentiation of pro-erythroblasts in DBA (55).
Based on the vital function of RPS19 in the 48S pre-initiation complex and in productive erythropoiesis, we focused on the role of RPS19 in ALOX15 mRNA translation regulation.
Binding studies employing recombinant HNRNPKR5met and nonmethylated HNRNPK (17) revealed a differential RPS19-HNRNPK interaction in cytoplasmic K562 cells extract (Figure 2B,C). This was observed as well in HNRNPK immunoprecipitations from extracts of noninduced K562 cells and at day 8 of erythroid maturation (Figure 2D,E) and furthermore in ES (PRMT1+/+) and (PRMT1−/−) cells (Figure 2G,H). These experiments provided evidence for an enhanced binding of HNRNPKR5met to RPS19. Interestingly, at day 8 of erythroid maturation and in ES (PRMT1−/−) cells when nonmethylated HNRNPK predominates, RPS19 binding was reduced (Figure 2). Consistently, in vivo the RPS19–HNRNPK interaction declined during erythroid maturation (Figure 3).
In vitro binding assays and thermal shift experiments uncovered that asymmetrically dimethylated arginines R256, R258 and R268 in HNRNPK affected the association with RPS19 (Figure 4). Moreover, they revealed that DBA-related W52 in RPS19 is critical for HNRNPK binding (Figure 4). Noteworthy, RPS19 W52 is a constituent of a potential aromatic cage for Argmet recognition (Figure 4B, right panel), which might stabilize the interaction with HNRNPK upon arginine methylation by PRMT1.
Under translational repression conditions, when HNRNPK interacts with the ALOX15 mRNA 3’UTR CU-rich DICE, we detected stalled 40S ribosomal subunits at the AUG in both, in vitro translation initiation and toe printing experiments (10,19). We hypothesize that the HNRNPK interplay with 40S subunit-bound RPS19 is augmented by arginine methylation causing structural alterations, which interfere with 80S ribosome formation. Previously, it has been shown in S. cerevisiae that 48S complex formation can be inhibited by Scd6, which binds eIF4G1 through its RGG-motifs (73). Furthermore, PRMT1 dependent Scd6 methylation augments its repressor activity (74).
It is worth pointing out that a transient as well as stable reduction of RPS19 expression results in the derepression of ALOX15 synthesis and hence leads to diminished mitochondria staining (Figures 5 and 7). RPS19 depleted K562 clone A exhibited significantly diminished proliferation capacity and a reduced translation efficiency (Figure 6). In contrast to that, the distribution of ALOX15 mRNA demonstrates an enrichment in translationally active heavy polysomes (Figure 7), indicating a specific derepression of ALOX15 mRNA translation. This can be explained by a decline in the formation of HNRNPK–RPS19–40S complexes, which suppress 60S subunit joining. Consequently, free HNRNPK replaces the inhibitory complexes and active 80S ribosomes can be assembled.
Our findings demonstrate the methylation-dependent HNRNPK–RPS19 interaction (Figures 2–4) and the substantial impact of RPS19 on ALOX15 mRNA translation (Figures 5 and 7). Therefore, we suggest that the enhanced interaction of methylated HNRNPK with RPS19 in the 40S subunit leads to the efficient suppression of ALOX15 mRNA translation initiation in premature erythroid cells. Specific association of HNRNPK-RPS19–40S complexes, which interfere with 80S ribosome formation is mediated by the 3’UTR CU-rich DICE (Figure 8, left panel). In terminal erythroid maturation, when nonmethylated HNRNPK predominates, release of RPS19 from HNRNPK enables 80S ribosome assembly and ALOX15 synthesis is facilitated (Figure 8, right panel).
Figure 8.
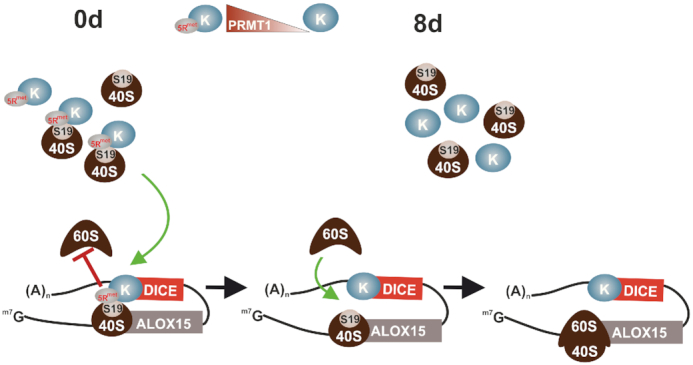
Methylation-dependent HNRNPK–RPS19 interaction regulates ALOX15 mRNA translation. ALOX15 is a key enzyme in erythroid cell maturation. The synthesis of ALOX15 is restricted to mature reticulocytes, where the enzyme initiates the degradation of mitochondria as a prerequisite for erythrocyte formation (22). Left panel: in erythroblasts the ALOX15 mRNA translation initiation is specifically silenced by the inhibition of 60S ribosomal subunit joining to the 40S subunit at the AUG (10,19). Specific inhibition is mediated by HNRNPK that constitutes an inhibitory complex at the CU-rich 3’UTR DICE (10,19). PRMT1-dependent asymmetric dimethylation of HNRNPK arginine residues (32) augments its interaction with RPS19 causing structural alterations that interfere with 80S ribosome formation. Right panel: during maturation the level of PRMT1 declines. This results in the replacement of methylated HNRNPK by the nonmethylated protein (16). In mature reticulocytes, when nonmethylated HNRNPK predominates, interaction of RPS19 with HNRNPK is abolished, enabling 80S ribosome assembly and ALOX15 mRNA translation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Carmen Tag (Department of Internal Medicine III, University Hospital RWTH Aachen) for FACS analysis and Master Students Julia Wette and Laura Gail for their initial contributions to experiments shown in Figure 5. The ALOX15 antibody was a kind gift of Hartmut Kühn (Charité Berlin).
Author Contributions: I.S.N.-d.V. performed experiments shown in Figures 1, 2, 4–6 and 7 and analyzed the data. R.S. implemented the PLA assay (Figure 3) and contributed initially to experiments shown in Figure 4C–E. B.M. established pulldown assays with recombinant nonmethylated and quantitatively asymmetrically dimethylated HNRNPK. G.M. supported the realization of the project. H.U. executed the mass spectrometry analysis, data processing and evaluation. D.N. essentially contributed to the identification of the RPS19 interaction scaffold (Figure 4B). D.H.O. and A.O.-L. designed the study, performed experiments shown in Figures 2B, D, G–H, 4A and 4F–I, analyzed and evaluated data and wrote the manuscript.
Notes
Present address: Isabel S. Naarmann-de Vries, Section of Bioinformatics and Systems Cardiology, Klaus Tschira Institute for Integrative Computational Cardiology, University Hospital Heidelberg, Germany.
Contributor Information
Isabel S Naarmann-de Vries, Department of Intensive Care Medicine, University Hospital, RWTH Aachen University, Aachen Germany.
Roberta Senatore, Department of Intensive Care Medicine, University Hospital, RWTH Aachen University, Aachen Germany.
Bodo Moritz, Institute of Pharmacy, Faculty of Natural Sciences, Martin-Luther-University Halle-Wittenberg, Halle (Saale), Germany.
Gernot Marx, Department of Intensive Care Medicine, University Hospital, RWTH Aachen University, Aachen Germany.
Henning Urlaub, Max-Planck-Institute for Biophysical Chemistry, Bioanalytical Mass Spectrometry Group, Göttingen, Germany; Department of Clinical Chemistry, University Medical Center, Göttingen, Germany.
Dierk Niessing, Institute of Pharmaceutical Biotechnology, Ulm University, Ulm, Germany; Institute of Structural Biology, Helmholtz Zentrum München, Neuherberg, Germany.
Dirk H Ostareck, Department of Intensive Care Medicine, University Hospital, RWTH Aachen University, Aachen Germany.
Antje Ostareck-Lederer, Department of Intensive Care Medicine, University Hospital, RWTH Aachen University, Aachen Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [OS 290/3-2 to A.O.-L., D.H.O.]. Funding for open access charge: Institutional funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Yamashita A., Takeuchi O.. Translational control of mRNAs by 3′-Untranslated region binding proteins. BMB Rep. 2017; 50:194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hentze M.W., Gebauer F., Preiss T.. Mathews M.B., Sonenberg N., Hershey J.W.B.. Cis-regulatory sequences and trans-acting factors in translational control. Translational Control in Biology and Medicine. 2007; 48:NY: Cold Spring Harbor Laboratory Press; 269–295. [Google Scholar]
- 3. Ji P., Murata-Hori M., Lodish H.F.. Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol. 2011; 21:409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keerthivasan G., Wickrema A., Crispino J.D.. Erythroblast enucleation. Stem Cells Int. 2011; 2011:139851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moras M., Lefevre S.D., Ostuni M.A.. From Erythroblasts to Mature Red Blood Cells: Organelle Clearance in Mammals. Front. Physiol. 2017; 8:1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palis J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014; 5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Waggoner S.A., Liebhaber S.A.. Regulation of alpha-globin mRNA stability. Exp. Biol. Med. (Maywood). 2003; 228:387–395. [DOI] [PubMed] [Google Scholar]
- 8. Kiledjian M., Wang X., Liebhaber S.A.. Identification of two KH domain proteins in the alpha-globin mRNP stability complex. EMBO J. 1995; 14:4357–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weiss I.M., Liebhaber S.A.. Erythroid cell-specific determinants of alpha-globin mRNA stability. Mol. Cell. Biol. 1994; 14:8123–8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ostareck D.H., Ostareck-Lederer A., Wilm M., Thiele B.J., Mann M., Hentze M.W.. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell. 1997; 89:597–606. [DOI] [PubMed] [Google Scholar]
- 11. Ostareck-Lederer A., Ostareck D.H.. Precision mechanics with multifunctional tools: how hnRNP K and hnRNPs E1/E2 contribute to post-transcriptional control of gene expression in hematopoiesis. Curr. Protein Pept. Sci. 2012; 13:391–400. [DOI] [PubMed] [Google Scholar]
- 12. Moore K.S., von Lindern M.. RNA binding proteins and regulation of mRNA translation in erythropoiesis. Front. Physiol. 2018; 9:910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Y., Wu W., Han Q., Wang Y., Li C., Zhang P., Xu H.. Post-translational modification control of RNA-binding protein hnRNPK function. Open Biol. 2019; 9:180239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ostareck D.H., Ostareck-Lederer A.. RNA-Binding proteins in the control of LPS-Induced macrophage response. Front. Genet. 2019; 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Z., Qiu H., He J., Liu L., Xue W., Fox A., Tickner J., Xu J.. The emerging roles of hnRNPK. J. Cell. Physiol. 2020; 235:1995–2008. [DOI] [PubMed] [Google Scholar]
- 16. Naarmann I.S., Harnisch C., Flach N., Kremmer E., Kuhn H., Ostareck D.H., Ostareck-Lederer A.. mRNA silencing in human erythroid cell maturation: heterogeneous nuclear ribonucleoprotein K controls the expression of its regulator c-Src. J. Biol. Chem. 2008; 283:18461–18472. [DOI] [PubMed] [Google Scholar]
- 17. Moritz B., Lilie H., Naarmann-de Vries I.S., Urlaub H., Wahle E., Ostareck-Lederer A., Ostareck D.H. Biophysical and biochemical analysis of hnRNP K: arginine methylation, reversible aggregation and combinatorial binding to nucleic acids. Biol. Chem. 2014; 395:837–853. [DOI] [PubMed] [Google Scholar]
- 18. Naarmann-de Vries I.S., Brendle A., Bahr-Ivacevic T., Benes V., Ostareck D.H., Ostareck-Lederer A.. Translational control mediated by hnRNP K links NMHC IIA to erythroid enucleation. J. Cell Sci. 2016; 129:1141–1154. [DOI] [PubMed] [Google Scholar]
- 19. Ostareck D.H., Ostareck-Lederer A., Shatsky I.N., Hentze M.W.. Lipoxygenase mRNA silencing in erythroid differentiation: The 3′UTR regulatory complex controls 60S ribosomal subunit joining. Cell. 2001; 104:281–290. [DOI] [PubMed] [Google Scholar]
- 20. Ostareck-Lederer A., Ostareck D.H., Cans C., Neubauer G., Bomsztyk K., Superti-Furga G., Hentze M.W.. c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol. Cell. Biol. 2002; 22:4535–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Messias A.C., Harnisch C., Ostareck-Lederer A., Sattler M., Ostareck D.H.. The DICE-binding activity of KH domain 3 of hnRNP K is affected by c-Src-mediated tyrosine phosphorylation. J. Mol. Biol. 2006; 361:470–481. [DOI] [PubMed] [Google Scholar]
- 22. Rapoport S.M., Schewe T.. The maturational breakdown of mitochondria in reticulocytes. Biochim. Biophys. Acta. 1986; 864:471–495. [DOI] [PubMed] [Google Scholar]
- 23. van Leyen K., Duvoisin R.M., Engelhardt H., Wiedmann M.. A function for lipoxygenase in programmed organelle degradation. Nature. 1998; 395:392–395. [DOI] [PubMed] [Google Scholar]
- 24. Grullich C., Duvoisin R.M., Wiedmann M., van Leyen K.. Inhibition of 15-lipoxygenase leads to delayed organelle degradation in the reticulocyte. FEBS Lett. 2001; 489:51–54. [DOI] [PubMed] [Google Scholar]
- 25. Hohne M., Thiele B.J., Prehn S., Giessmann E., Nack B., Rapoport S.M.. Activation of translationally inactive lipoxygenase mRNP particles from rabbit reticulocytes. Biomed. Biochim. Acta. 1988; 47:75–78. [PubMed] [Google Scholar]
- 26. Thiele B.J., Andree H., Hohne M., Rapoport S.M.. Regulation of the synthesis of lipoxygenase in erythroid cells. Acta Biol. Med. Ger. 1981; 40:597–602. [PubMed] [Google Scholar]
- 27. Ostareck-Lederer A., Ostareck D.H., Standart N., Thiele B.J.. Translation of 15-lipoxygenase mRNA is inhibited by a protein that binds to a repeated sequence in the 3′ untranslated region. EMBO J. 1994; 13:1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naarmann I.S., Harnisch C., Muller-Newen G., Urlaub H., Ostareck-Lederer A., Ostareck D.H.. DDX6 recruits translational silenced human reticulocyte 15-lipoxygenase mRNA to RNP granules. RNA. 2010; 16:2189–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huttelmaier S., Zenklusen D., Lederer M., Dictenberg J., Lorenz M., Meng X., Bassell G.J., Condeelis J., Singer R.H.. Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature. 2005; 438:512–515. [DOI] [PubMed] [Google Scholar]
- 30. Adolph D., Flach N., Mueller K., Ostareck D.H., Ostareck-Lederer A.. Deciphering the cross talk between hnRNP K and c-Src: the c-Src activation domain in hnRNP K is distinct from a second interaction site. Mol. Cell. Biol. 2007; 27:1758–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Naarmann-de Vries I.S., Urlaub H., Ostareck D.H., Ostareck-Lederer A.. Caspase-3 cleaves hnRNP K in erythroid differentiation. Cell Death Dis. 2013; 4:e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ostareck-Lederer A., Ostareck D.H., Rucknagel K.P., Schierhorn A., Moritz B., Huttelmaier S., Flach N., Handoko L., Wahle E.. Asymmetric arginine dimethylation of heterogeneous nuclear ribonucleoprotein K by protein-arginine methyltransferase 1 inhibits its interaction with c-Src. J. Biol. Chem. 2006; 281:11115–11125. [DOI] [PubMed] [Google Scholar]
- 33. Anger A.M., Armache J.P., Berninghausen O., Habeck M., Subklewe M., Wilson D.N., Beckmann R.. Structures of the human and Drosophila 80S ribosome. Nature. 2013; 497:80–85. [DOI] [PubMed] [Google Scholar]
- 34. Ben-Shem A., Garreau de Loubresse N., Melnikov S., Jenner L., Yusupova G., Yusupov M.. The structure of the eukaryotic ribosome at 3.0 A resolution. Science. 2011; 334:1524–1529. [DOI] [PubMed] [Google Scholar]
- 35. Jenner L., Melnikov S., Garreau de Loubresse N., Ben-Shem A., Iskakova M., Urzhumtsev A., Meskauskas A., Dinman J., Yusupova G., Yusupov M.. Crystal structure of the 80S yeast ribosome. Curr. Opin. Struct. Biol. 2012; 22:759–767. [DOI] [PubMed] [Google Scholar]
- 36. Draptchinskaia N., Gustavsson P., Andersson B., Pettersson M., Willig T.N., Dianzani I., Ball S., Tchernia G., Klar J., Matsson H.et al.. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999; 21:169–175. [DOI] [PubMed] [Google Scholar]
- 37. Pawlak M.R., Scherer C.A., Chen J., Roshon M.J., Ruley H.E.. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell. Biol. 2000; 20:4859–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 39. Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G.et al.. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006; 3:995–1000. [DOI] [PubMed] [Google Scholar]
- 40. Cimmperman P., Baranauskiene L., Jachimoviciute S., Jachno J., Torresan J., Michailoviene V., Matuliene J., Sereikaite J., Bumelis V., Matulis D.. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 2008; 95:3222–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huynh K., Partch C.L.. Analysis of protein stability and ligand interactions by thermal shift assay. Curr.Protoc. Protein Sci. 2015; 79:28.9.1–28.9.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shevchenko A., Wilm M., Vorm O., Mann M.. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996; 68:850–858. [DOI] [PubMed] [Google Scholar]
- 43. Schmidt E.K., Clavarino G., Ceppi M., Pierre P.. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 2009; 6:275–277. [DOI] [PubMed] [Google Scholar]
- 44. de Vries S., Naarmann-de Vries I.S., Urlaub H., Lue H., Bernhagen J., Ostareck D.H., Ostareck-Lederer A.. Identification of DEAD-box RNA helicase 6 (DDX6) as a cellular modulator of vascular endothelial growth factor expression under hypoxia. J. Biol. Chem. 2013; 288:5815–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Czaplinski K., Kocher T., Schelder M., Segref A., Wilm M., Mattaj I.W.. Identification of 40LoVe, a Xenopus hnRNP D family protein involved in localizing a TGF-beta-related mRNA during oogenesis. Dev. Cell. 2005; 8:505–515. [DOI] [PubMed] [Google Scholar]
- 46. Gross H., Hennard C., Masouris I., Cassel C., Barth S., Stober-Grasser U., Mamiani A., Moritz B., Ostareck D., Ostareck-Lederer A.et al.. Binding of the heterogeneous ribonucleoprotein K (hnRNP K) to the Epstein-Barr virus nuclear antigen 2 (EBNA2) enhances viral LMP2A expression. PLoS One. 2012; 7:e42106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sprangers R., Groves M.R., Sinning I., Sattler M.. High-resolution X-ray and NMR structures of the SMN Tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues. J. Mol. Biol. 2003; 327:507–520. [DOI] [PubMed] [Google Scholar]
- 48. Mathioudakis N., Palencia A., Kadlec J., Round A., Tripsianes K., Sattler M., Pillai R.S., Cusack S.. The multiple Tudor domain-containing protein TDRD1 is a molecular scaffold for mouse Piwi proteins and piRNA biogenesis factors. RNA. 2012; 18:2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen C., Jin J., James D.A., Adams-Cioaba M.A., Park J.G., Guo Y., Tenaglia E., Xu C., Gish G., Min J.et al.. Mouse Piwi interactome identifies binding mechanism of Tdrkh Tudor domain to arginine methylated Miwi. Proc. Natl. Acad. Sci. USA. 2009; 106:20336–20341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cote J., Richard S.. Tudor domains bind symmetrical dimethylated arginines. J. Biol. Chem. 2005; 280:28476–28483. [DOI] [PubMed] [Google Scholar]
- 51. Liu K., Guo Y., Liu H., Bian C., Lam R., Liu Y., Mackenzie F., Rojas L.A., Reinberg D., Bedford M.T.et al.. Crystal structure of TDRD3 and methyl-arginine binding characterization of TDRD3, SMN and SPF30. PLoS One. 2012; 7:e30375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tripsianes K., Madl T., Machyna M., Fessas D., Englbrecht C., Fischer U., Neugebauer K.M., Sattler M.. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat. Struct. Mol. Biol. 2011; 18:1414–1420. [DOI] [PubMed] [Google Scholar]
- 53. Natchiar S.K., Myasnikov A.G., Kratzat H., Hazemann I., Klaholz B.P.. Visualization of chemical modifications in the human 80S ribosome structure. Nature. 2017; 551:472–477. [DOI] [PubMed] [Google Scholar]
- 54. Pantoliano M.W., Petrella E.C., Kwasnoski J.D., Lobanov V.S., Myslik J., Graf E., Carver T., Asel E., Springer B.A., Lane P.et al.. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001; 6:429–440. [DOI] [PubMed] [Google Scholar]
- 55. Gregory L.A., Aguissa-Toure A.H., Pinaud N., Legrand P., Gleizes P.E., Fribourg S.. Molecular basis of Diamond-Blackfan anemia: structure and function analysis of RPS19. Nucleic Acids Res. 2007; 35:5913–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cretien A., Hurtaud C., Moniz H., Proust A., Marie I., Wagner-Ballon O., Choesmel V., Gleizes P.E., Leblanc T., Delaunay J.et al.. Study of the effects of proteasome inhibitors on ribosomal protein S19 (RPS19) mutants, identified in patients with Diamond-Blackfan anemia. Haematologica. 2008; 93:1627–1634. [DOI] [PubMed] [Google Scholar]
- 57. Nguyen A.T., Prado M.A., Schmidt P.J., Sendamarai A.K., Wilson-Grady J.T., Min M., Campagna D.R., Tian G., Shi Y., Dederer V.et al.. UBE2O remodels the proteome during terminal erythroid differentiation. Science. 2017; 357:eaan0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Horos R., Ijspeert H., Pospisilova D., Sendtner R., Andrieu-Soler C., Taskesen E., Nieradka A., Cmejla R., Sendtner M., Touw I.P.et al.. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood. 2012; 119:262–272. [DOI] [PubMed] [Google Scholar]
- 59. Thiele B.J., Andree H., Hohne M., Rapoport S.M.. Lipoxygenase mRNA in rabbit reticulocytes. Its isolation, characterization and translational repression. Eur. J. Biochem. 1982; 129:133–141. [DOI] [PubMed] [Google Scholar]
- 60. Habelhah H., Shah K., Huang L., Ostareck-Lederer A., Burlingame A.L., Shokat K.M., Hentze M.W., Ronai Z.. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat. Cell Biol. 2001; 3:325–330. [DOI] [PubMed] [Google Scholar]
- 61. Chen H.C., Lin W.C., Tsay Y.G., Lee S.C., Chang C.J.. An RNA helicase, DDX1, interacting with poly(A) RNA and heterogeneous nuclear ribonucleoprotein K. J. Biol. Chem. 2002; 277:40403–40409. [DOI] [PubMed] [Google Scholar]
- 62. Good A.L., Haemmerle M.W., Oguh A.U., Doliba N.M., Stoffers D.A.. Metabolic stress activates an ERK/hnRNPK/DDX3X pathway in pancreatic beta cells. Mol Metab. 2019; 26:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ban N., Beckmann R., Cate J.H., Dinman J.D., Dragon F., Ellis S.R., Lafontaine D.L., Lindahl L., Liljas A., Lipton J.M.et al.. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014; 24:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leger-Silvestre I., Caffrey J.M., Dawaliby R., Alvarez-Arias D.A., Gas N., Bertolone S.J., Gleizes P.E., Ellis S.R.. Specific Role for Yeast Homologs of the Diamond Blackfan Anemia-associated Rps19 Protein in Ribosome Synthesis. J. Biol. Chem. 2005; 280:38177–38185. [DOI] [PubMed] [Google Scholar]
- 65. Matsson H., Davey E.J., Draptchinskaia N., Hamaguchi I., Ooka A., Leveen P., Forsberg E., Karlsson S., Dahl N.. Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol. Cell. Biol. 2004; 24:4032–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Flygare J., Aspesi A., Bailey J.C., Miyake K., Caffrey J.M., Karlsson S., Ellis S.R.. Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood. 2007; 109:980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Juli G., Gismondi A., Monteleone V., Caldarola S., Iadevaia V., Aspesi A., Dianzani I., Proud C.G., Loreni F.. Depletion of ribosomal protein S19 causes a reduction of rRNA synthesis. Sci. Rep. 2016; 6:35026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lutsch G., Stahl J., Kargel H.J., Noll F., Bielka H.. Immunoelectron microscopic studies on the location of ribosomal proteins on the surface of the 40S ribosomal subunit from rat liver. Eur. J. Cell Biol. 1990; 51:140–150. [PubMed] [Google Scholar]
- 69. Armache J.P., Jarasch A., Anger A.M., Villa E., Becker T., Bhushan S., Jossinet F., Habeck M., Dindar G., Franckenberg S.et al.. Localization of eukaryote-specific ribosomal proteins in a 5.5-A cryo-EM map of the 80S eukaryotic ribosome. Proc. Natl. Acad. Sci. USA. 2010; 107:19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bommer U.A., Stahl J., Henske A., Lutsch G., Bielka H.. Identification of proteins of the 40 S ribosomal subunit involved in interaction with initiation factor eIF-2 in the quaternary initiation complex by means of monospecific antibodies. FEBS Lett. 1988; 233:114–118. [DOI] [PubMed] [Google Scholar]
- 71. Eliseev B., Yeramala L., Leitner A., Karuppasamy M., Raimondeau E., Huard K., Alkalaeva E., Aebersold R., Schaffitzel C.. Structure of a human cap-dependent 48S translation pre-initiation complex. Nucleic Acids Res. 2018; 46:2678–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Flygare J., Kiefer T., Miyake K., Utsugisawa T., Hamaguchi I., Da Costa L., Richter J., Davey E.J., Matsson H., Dahl N.et al.. Deficiency of ribosomal protein S19 in CD34+ cells generated by siRNA blocks erythroid development and mimics defects seen in Diamond-Blackfan anemia. Blood. 2005; 105:4627–4634. [DOI] [PubMed] [Google Scholar]
- 73. Rajyaguru P., She M., Parker R.. Scd6 targets eIF4G to repress translation: RGG motif proteins as a class of eIF4G-binding proteins. Mol. Cell. 2012; 45:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Poornima G., Shah S., Vignesh V., Parker R., Rajyaguru P.I.. Arginine methylation promotes translation repression activity of eIF4G-binding protein, Scd6. Nucleic Acids Res. 2016; 44:9358–9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.