


Abstract
The tumor suppressor BRCA1 is considered a master regulator of genome integrity. Although widely recognized for its DNA repair functions, BRCA1 has also been implicated in various mechanisms of chromatin remodeling and transcription regulation. However, the precise role that BRCA1 plays in these processes has been difficult to establish due to the widespread consequences of its cellular dysfunction. Here, we use nucleoplasmic extract derived from the eggs of Xenopus laevis to investigate the role of BRCA1 in a cell-free transcription system. We report that BRCA1-BARD1 suppresses transcription initiation independent of DNA damage signaling and its established role in histone H2A ubiquitination. BRCA1-BARD1 acts through a histone intermediate, altering acetylation of histone H4K8 and recruitment of the chromatin reader and oncogene regulator BRD4. Together, these results establish a functional relationship between an established (BRCA1) and emerging (BRD4) regulator of genome integrity.
Graphical Abstract
Graphical Abstract.
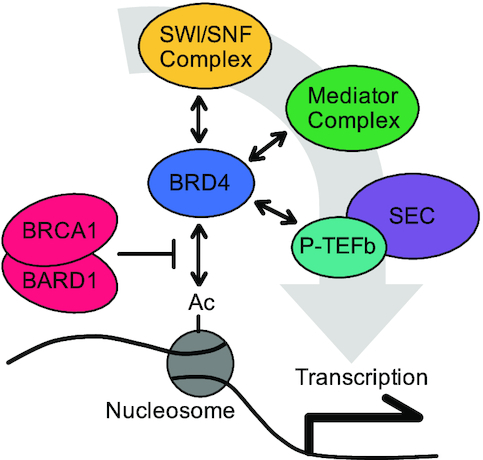
BRCA1-BARD1 regulates transcription initiation by limiting recruitment of the chromatin reader BRD4.
INTRODUCTION
BRCA1 (breast cancer susceptibility protein 1) is an established tumor suppressor that plays a critical role in the development of both sporadic and hereditary breast and ovarian cancer (1). Considered a ‘master regulator’ of genome integrity, BRCA1 has been linked to nearly all aspects of chromatin biology (2). Loss of BRCA1 leads to widespread defects in DNA repair, cellular stress signaling, cell cycle progression, apoptosis, chromatin condensation, and gene expression (3).
BRCA1’s different cellular activities are regulated by a variety of protein-protein interactions and post-translational modifications (2,4). BRCA1 forms a heterodimeric complex with BARD1 (BRCA1-associated RING domain protein 1). The two proteins interact through their respective N-terminal RING domains to form an E3 ubiquitin ligase (5). Binding to BARD1 also masks a nuclear export signal (NES) on BRCA1, thereby promoting nuclear retention and stability of the complex (6). BRCA1 contains a central coiled-coil domain that interacts with PALB2 (partner and localizer of BRCA2) and BRCA2 (breast cancer susceptibility protein 2) to stimulate RAD51 loading and homologous recombination (7). BRCA1 and BARD1 also contain tandem BRCT repeats in their C-termini that support interaction with phospho-proteins involved in damage signaling and protein localization (8).
In addition to its established DNA repair functions, BRCA1 has been linked to various mechanisms of transcription regulation (9). BRCA1 interacts with the RNA polymerase II holoenzyme (10) through RNA helicase A (11), and with the negative elongation factor (NELF) complex through COBRA1 (12). BRCA1 also binds to various transcription factors, including p53 (13), c-MYC (14), ERα (15), GATA3 (16) and others. BRCA1 has been shown to play a role in chromatin decondensation through interactions with SWI/SNF remodeling complexes (17–19) and the histone acetyltransferases P300 and CBP (20). Conversely, BRCA1 and BARD1 have been linked to chromatin condensation through interactions with the DNA methyltransferase DNMT3B and heterochromatin protein 1 (HP1) (21,22). BRCA1-BARD1 can also catalyze ubiqutination of histone H2A (23,24), which is generally associated with transcription repression (23,25).
Despite extensive study, the context and functional role that BRCA1 plays in transcription regulation remains poorly understood. We recently showed that nucleoplasmic extract (NPE) readily promotes transcription of plasmid-borne gene constructs (26), unlike traditional egg extracts where transcription is suppressed (27). Using the NPE system, we sought to investigate BRCA1’s role as a transcription regulator.
MATERIALS AND METHODS
Construction of plasmid substrates
pActin was generated as described previously (26). pCMV was purchased from Addgene (#11153). To create pBRCA1, the 5′ and 3′ regions of Xenopus laevis BRCA1 were amplified from sperm chromatin with the following primer pairs:
| 5′ region: | GCGCTGCCTAGGCCTGGGGCCAACATTTTTTTTTTAACTT and |
| TAGACCATGGTGGCTTGTCCTTTAGATAATACATTGGTTAAATGCAAT | |
| 3′ region: | ATAGCGGCCGCAATCACAGTGGAACTGGCCGGTTA and |
| CAGGAACTAGTCATCATTTAGAGAATCAACCTATCCAGCCTCAGA |
The resulting fragments were then cloned into pCMV using AvrII and NcoI (5′ region), or NotI and SpeI (3′ region). pCON was generated as described previously (28), and digested with BbsI to create pDSB. For pCarrier, the pFastBac1-BARD1 vector was used (29).
Incubations in nucleoplasmic extract
NPE was prepared as described previously (30). Briefly, sperm chromatin was incubated in interphase egg extract to form nuclei. Nuclei were then harvested and genomic material was removed by ultracentrifugation to create a soluble nuclear protein extract. For all reactions, NPE was supplemented with an ATP regeneration mix (6.5 mM phosphocreatine, 0.65 mM ATP, and 1.6 μg/ml creatine phosphokinase) and 1 mM DTT. All incubations were performed at 21°C. Extracts were placed at 21°C for 10 min prior to the addition of 2.5–10 ng/μl plasmid DNA, which represents the reaction start time. Where indicated, reactions were supplemented with 5 ng/uL pCON or pDSB, 5 mM caffeine (Fisher), 10–100 μM VE-821 (Abcam), 10–100 μM KU-5593 (Abcam), 20 μM ubiquitin vinyl sulfone (Fisher), 30 μM JQ1 (Sigma), or 40–400 nM recombinant BRCA1-BARD1. The endogenous level of BRCA1-BARD1 in NPE is estimated to be ∼40 nM. All reactions were performed at least two times with representative or averaged data shown. Where indicated, data were graphed with error bars representing ±1 standard deviation and P values determined using a two-tailed t-test: P < 0.05 (*), P < 0.005 (**), P < 0.0005 (***), P ≥ 0.05 (not significant, n.s.).
Reverse transcription quantitative PCR (RT-qPCR)
RNA was isolated from extract using the EZNA RNA Purification kit (Omega Bio-tek). cDNA was produced using QuantiTect Reverse Transcription kit (Qiagen) and then analyzed by quantitative PCR. cDNA generated by gene constructs was normalized to endogenous 18S rRNA to correct for differences in column recovery between time points and conditions. Unless indicated, data are graphed from amplification with the pActin (+10 to +153) primers. The following primer pairs were used:
| pActin: | (+10 to +153): CCCGCATAGAAAGGAGACA and GCCAGAACATAGACATTAAGAAGG; |
| (+174 to +289): TGAAATGGCCATGACTTGAG and GCAGTGCCCTGTAACAATGA; | |
| (+599 to +716): GCGCTTTACGTTAGCAATCC and AGGCTTTCAGTGAGCCAGTC | |
| pBRCA1: | CAACCTTTAGGTCTATTTCAACCCA and CCACCTTTAGGTCTATTTCAACCCA |
| pCMV: | AGCTGGACGGCGACGTAAAC and AGGTCAGGGTGGTCACGAGG |
| 18S rRNA: | GACCGGCGCAAGACGAACCA and TGCTCGGCGGGTCATGGGAA |
Antibodies and immunodepletion
RNA polymerase II antibodies were obtained from Bethyl Laboratories (A304-405A). TATA-binding protein (TBP) antibodies were obtained from Boster Biological Technology (PA1534). Histone H3 antibodies were obtained from Thermo Fisher (PA5-16183). Phospho-Chk1 (Ser345) and ubiquitin (P4D1) antibodies were obtained from Cell Signaling (#2341 and #3936, respectively). Ubiquityl-histone H2A antibodies were obtained from Millipore (05-678). Histone H4 acetyl-K5, K8 and K16 antibodies were obtained from Abclonal (A15233, A7258, and A5280, respectively). Histone H4 acetyl-K12 antibodies were gifted by Dr Hiroshi Kimura (Tokyo Institute of Technology, Tokyo, Japan) (31). Xenopus BRCA1 and BARD1 antibodies were generated by New England Peptide. BRD4 antibodies were provided by Dr Igor Dawid, NIH/NICHD (32). For depletion experiments, 10 μl of NPE was incubated with 4 μl of Protein-A Sepharose Fast Flow beads (GE Healthcare) that were pre-bound with either 16 μl of pre-immune (mock) or αBRCA1 serum for 1 h at 4°C. After three rounds of depletion, the resulting mock- and BRCA1-depleted extracts were immediately used for experiments.
Purification of recombinant BRCA1-BARD1
The full-length X. laevis BRCA1-BARD1 heterodimer was purified as previously described (29). Briefly, Sf9 cells were co-infected with recombinant baculoviruses containing FLAG-BRCA1 and HA-BARD1, or FLAG-BRCA1I26A and HA-BARD1. The resulting heterodimers were isolated by sequential affinity chromatography using anti-FLAG M2 agarose (Sigma, A2220) and anti-HA agarose (Sigma, A2095). We used site-directed mutagenesis (Agilent, 210518) to create the BRCA1I26A mutant from the parent FLAG-BRCA1 pFastBac vector using the following primers:
| Forward: | CTTCATCAGCTCTAAGCAGGCTGGGCACTCCAAATTCTTC |
| Reverse: | GAAGAATTTGGAGTGCCCAGCCTGCTTAGAGCTGATGAAG |
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously (33). Briefly, reaction samples were crosslinked in Egg Lysis Buffer (ELB: 10 mM HEPES–KOH pH 7.7, 50 mM KCl, 2.5 mM MgCl2 and 250 mM sucrose) containing 1% formaldehyde. Crosslinking was stopped with the addition of 125 mM glycine and then excess formaldehyde was removed with a Micro Bio-Spin 6 chromatography column (Bio-Rad). Samples were sonicated (Diagenode Bioruptor UCD-600 TS) and immunoprecipitated with the indicated antibodies. Crosslinks were then reversed, and the resulting DNA was isolated by phenol/chloroform extraction and ethanol precipitation. qPCR was then used to quantify recovered DNA as a fraction of total DNA (INPUT) and graphed. The following primers were used:
| pActin: | (−120 to +71): CCTCCTTCGTCCGCAGTTCC and GCTGGCGAACCGCTACTTGC |
Plasmid pull-down
Plasmids were isolated from extract as described previously (34). Briefly, reaction samples were added to LacI-coupled magnetic beads (Dynabeads M-280; Invitrogen) suspended in LacI pull-down buffer (10 mM HEPES pH 7.7, 2.5 mM MgCl2, 50 mM KCl, 250 mM sucrose, 0.25 mg/ml BSA and 0.02% Tween 20). Samples were then rotated at 4°C for 20 min, washed three times with LacI wash buffer (10 mM HEPES pH 7.7, 2.5 mM MgCl2, 50 mM KCl, 0.25 mg/ml BSA, and 0.02% Tween 20), dried, and suspended in 2× SDS sample buffer (100 mM Tris–HCl pH 6.8, 4% SDS, 0.2% bromophenol blue, 20% glycerol and 200 mM β-mercaptoethanol). The resulting DNA-bound proteins were then resolved by SDS-PAGE, and visualized by western blotting with the indicated antibodies. To visualize recovered DNA, pull-down samples were treated with 2 μg RNase at 37°C for 30 min, and then incubated with 20 μg proteinase K at 21°C for 16 h. DNA was resolved by agarose gel electrophoresis and visualized with SYBR Gold stain (Thermo Scientific).
Nuclease Digestion
Plasmid DNA was incubated in NPE at 20 ng/μl for 30 min. Reactions samples were then diluted 100-fold in Digestion Buffer (50 mM Tris–HCl pH 7.9 and 5 mM CaCl2) containing 1.5 U/ μl MNase (New England Biolabs). Digestion reactions were stopped at the indicated time points with 1/8th volume of Digestion Stop Solution (160 mM EDTA and 6.8% SDS). Samples were then incubated with 60 μg Proteinase K overnight at 21°C. DNA was isolated by phenol/chloroform extraction and ethanol precipitation. Resulting DNA intermediates were resolved by 1.5% agarose gel electrophoresis and visualized with SYBR Gold stain.
Chromatin decondensation assay
NPE was incubated with 1250 demembranated sperm chromatin per μl at 21°C. At the indicated time points, samples were withdrawn from the reaction and visualized by phase contrast light microscopy. Chromatin volume and density were determined by quantifying 2D area and average visual opacity of individual sperm chromatin, respectively.
In vitro ubiquitination assay
Reactions containing 2 mM ATP, 12 μM ubiquitin, 250 nM UBE1 (R&D Systems), 1.75 μM UBE2D1 (R&D Systems), and 17 nM of X. laevis BRCA1-BARD1 or BRCA1I26A-BARD1 were incubated for 30 min at 37°C. For control reactions, either ubiquitin or BRCA1-BARD1 was omitted. Reactions were stopped with 2x SDS sample buffer, resolved by SDS-PAGE, and visualized by western blotting with the indicated antibodies.
Mass spectrometry analysis
DNA-bound proteins were isolated by plasmid pull-down, resolved briefly by SDS-PAGE, stained with Coomassie Brilliant Blue, and then excised. The gel fragments were sent to the Taplin Mass Spectrometry Facility (Harvard Medical School, Boston, MA) for processing and analysis. The average sum intensity of proteins isolated above background (-DNA samples) was determined for each condition and quantified for enrichment over mock-depleted reactions (ΔMock). Proteins were ranked for enrichment in ΔBRCA1 reactions over +BRCA1-BARD1 reactions. Thus, a ‘high’ score represents proteins whose DNA binding was enriched in the absence of BRCA1 and reduced in the presence of elevated BRCA1-BARD1, and a ‘low’ score represents proteins whose DNA binding was lost in the absence of BRCA1 and increased in the presence of elevated BRCA1-BARD1. An extended graph showing distribution of ranked proteins is shown in Supplementary Figure S5.
RESULTS
BRCA1-BARD1 is necessary and sufficient to suppress transcription in NPE
A plasmid containing the 5′ and 3′ elements of the actb gene (pActin; Supplementary Figure S1A) was incubated in NPE that was immunodepleted with pre-immune (mock) or BRCA1 antibodies (Figure 1A) and formation of RNA was analyzed by reverse transcription quantitative PCR (RT-qPCR). In the absence of BRCA1, both the rate and duration of transcription activity increased compared to mock-depleted reactions (Figure 1B). Similar results were observed using different gene constructs (Supplementary Figure S1B–G), suggesting that BRCA1 suppressed transcription through a broad or sequence-independent mechanism. To confirm that the depletion effects were specifically due to removal of BRCA1-BARD1, we purified the full-length complex by tandem affinity pull-down (Figure 1C). When recombinant BRCA1-BARD1 was added to BRCA1-depleted extract, we saw that transcription activity was suppressed compared to both mock- and BRCA1-depleted reactions (Figure 1D). Recombinant BRCA1-BARD1 was also able to suppress transcription of undepleted extract in a dose-dependent manner (Figure 1E). Taken together, these results indicate that BRCA1-BARD1 is both necessary and sufficient to suppress a critical step during transcription in NPE.
Figure 1.

BRCA1-BARD1 is necessary and sufficient to suppress transcription in NPE. (A) Different amounts of mock-depleted (ΔMock) or BRCA1-depleted (ΔBRCA1) NPE was analyzed by western blot with the indicated antibodies. (B) pActin was incubated in mock- or BRCA1-depleted extract and RNA was quantified by RT-qPCR over time (n = 3). (C) Purified BRCA1-BARD1 (WT) and BRCA1I26A-BARD1 (I26A; used in Figure 4E) were resolved by SDS-PAGE and visualized by silver stain. (D) pActin was incubated in mock-depleted extract, BRCA1-depleted extract, or BRCA1-depleted extract supplemented with recombinant 200 nM BRCA1-BARD1. After 120 min, RNA was quantified by RT-qPCR (n = 2) (E) pActin was incubated in NPE supplemented with increasing amounts of BRCA1-BARD1. After 120 min, RNA was quantified by RT-qPCR (n = 3).
BRCA1 suppresses transcription independent of DNA damage signaling
BRCA1’s role in regulating gene expression has typically been described in the context of a DNA damage response (35–37). To test whether damage signaling is required for BRCA1-mediated transcription suppression, we supplemented mock- or BRCA1-depleted extract with caffeine, a broad kinase inhibitor that targets both ATM and ATR (38). In the presence of caffeine, phosphorylation of the checkpoint kinase Chk1 was completely blocked (Figure 2A), indicating general disruption of damage signaling. In both mock- and BRCA1-depleted reactions, caffeine reduced transcription activity ∼5-fold compared to a buffer control (Figure 2B). However, caffeine did not affect the relative increase in transcription caused by BRCA1 depletion (5.4- versus 4.1-fold), indicating that the extent of BRCA1-mediated suppression was unchanged. Similar effects were also seen in reactions supplemented with the ATR inhibitor VE-821 or the ATM inhibitor KU-5593. Both inhibitors were able to reduce Chk1 phosphorylation (Supplementary Figure S2C), but did not limit the increased transcription associated with BRCA1 loss (Supplementary Figure S2D).
Figure 2.

BRCA1 suppresses transcription independent of DNA damage signaling. (A, B) pActin was incubated in mock- or BRCA1-depleted extract supplemented with buffer or caffeine. Samples were withdrawn after 60 min and analyzed by western blot (A), or 120 min and RNA was quantified by RT-qPCR (B). (C, D) pActin was incubated in mock- or BRCA1-depleted extract for 60 min and then reactions were supplemented with 5 ng/μl pCON or pDSB. Samples were withdrawn after 120 min and analyzed by western blot (C), or 240 min and RNA was quantified by RT-qPCR (D). See Supplementary Figure S2A-B for experimental repeats. (E) pActin was incubated in mock- or BRCA1-depleted extract. After 30 min, samples were withdrawn, resolved by agarose gel electrophoresis, and visualized by SYBR Gold stain. Open circular (OC); supercoiled (SC); input (IN); molecular weight marker (MW).
We also tested whether exogenous DNA damage would affect BRCA1-mediated transcription suppression. Mock- or BRCA1-depleted reactions were supplemented with a second plasmid that was undamaged (pCON) or had been digested to create a double-strand break (pDSB). Chk1 phosphorylation was elevated in reactions supplemented with pDSB compared to pCON (Figure 2C), indicating induction of a DNA damage response. In both mock- and BRCA1-depleted reactions, pDSB increased transcription activity ∼2.7-fold compared to the pCON reactions (Figure 2D). However, the relative increase in transcription caused by BRCA1 depletion was again similar (2.0- versus 1.8-fold). Notably, BRCA1 depletion itself did not affect the topology or integrity of plasmid DNA incubated in extract (Figure 2E). Altogether, these results indicate that BRCA1 suppresses transcription independent of DNA damage signaling.
Transcription suppression involves a histone intermediate
In the embryos of rapidly developing species, transcription activity is regulated by the density of maternally-supplied histones (39,40). We previously showed that a similar mechanism also controls transcription in NPE (26), which supports nucleosome formation (Supplementary Figure S3A) (41). To test whether BRCA1’s ability to suppress transcription was linked to histone density, we compared transcription in mock- and BRCA1-depleted extracts incubated with 2.5 ng/ul pActin and increasing concentrations of a ‘carrier’ plasmid with no sequence homology. At lower DNA concentrations (when histone density is high), BRCA1 depletion increased transcription ∼9-fold (Figure 3A). At higher DNA concentrations (when histone density is low), transcription in BRCA1-depleted reactions was at or below the level found in mock-depleted reactions. Thus, BRCA1-mediated suppression is lost under conditions that support low histone density, arguing that BRCA1 functions through a histone intermediate.
Figure 3.

BRCA1 regulates transcription initiation through a histone intermediate. (A) 2.5 ng/μl pActin was incubated in mock- or BRCA1-depleted extract with increasing amounts of carrier plasmid. After 120 min, RNA was quantified by RT-qPCR (n = 2). For comparison, values were normalized to ΔMock reactions at each concentration. (B) pActin was incubated in mock- or BRCA1-depleted extract. Samples were withdrawn after 60 min and analyzed by ChIP with histone H3 antibodies (n = 3). (C) pActin was incubated in extract supplemented with buffer or 200 nM BRCA1-BARD1. Samples were withdrawn after 60 min and analyzed by ChIP with histone H3 antibodies (n = 3). (D, E) TBP- and RNAPII-ChIPs were performed as in (B) (n = 3). (F–H) Sperm chromatin was incubated in mock- or BRCA1-depleted extract. At the indicated time points, samples were visualized by phase contrast light microscopy (F). Chromatin volume (G) and density (H) were calculated at 150 min (n ≥ 12).
To investigate how BRCA1 suppresses transcription, DNA-bound proteins were analyzed by chromatin immunoprecipitation (ChIP) using primers that amplify the actb promoter region. We first tested whether histone binding was altered by analyzing DNA-bound histone H3 in reactions with mock- or BRCA1-depleted extract (Figure 3B), or reactions supplemented with buffer or recombinant BRCA1-BARD1 (Figure 3C). In each reaction, the level of DNA-bound histone H3 showed little or no change, ruling out the possibility that BRCA1 suppressed transcription by increasing histone density. We also analyzed recruitment of the transcription factor TATA-Binding Protein (TBP; Figure 3D) and RNAPII (Figure 3E). Compared to mock-depleted reactions, DNA-binding of both proteins increased in the absence of BRCA1. Although total transcription increased in the absence of BRCA1 (Supplementary Figure S3B), the relative efficiency of extension downstream from the promoter was unchanged (Supplementary Figure S3C). Together, these results support the idea that BRCA1 functions upstream of transcription initiation by restricting access of transcription factors to DNA. Consistent with this interpretation, we saw that BRCA1 depletion also increased the speed and extent of chromatin decondensation in NPE (Figure 3F–H).
Ubiquitin and E3 ligase activity are dispensable for transcription suppression
BRCA1 has been implicated in multiple mechanisms of chromatin remodeling that act upstream of transcription initiation (9). Of these, the best-characterized mechanism involves ubiquitination of histone H2A (H2A-Ub). H2A-Ub mediated by BRCA1-BARD1 has been linked to heterochromatin formation and transcription suppression (23). To test whether BRCA1-BARD1 regulates formation of H2A-Ub in extract, protein binding was analyzed by ChIP. We found that BRCA1 depletion led to a mild decrease in H2A-Ub relative to total histone levels (Figure 4A). Conversely, the addition of BRCA1-BARD1 to extract increased H2A-Ub ∼3-fold compared to buffer controls (Figure 4B). These results indicate that some ubiquitinated H2A is present in extract and that its formation is potentially regulated by BRCA1-BARD1.
Figure 4.

Ubiquitination is dispensable for BRCA1-mediated transcription suppression. (A) pActin was incubated in mock- or BRCA1-depleted extract. Samples were withdrawn after 60 min and analyzed by ChIP with histone H2A-Ub or H3 antibodies. Recovery of H2A-Ub over H3 is graphed (n = 2). (B) pActin was incubated in extract supplemented with buffer or 200 nM BRCA1-BARD1. Samples were analyzed by ChIP as in (A) (n = 2). (C, D) pActin was incubated in extract pre-treated with buffer or UbVS and then supplemented with buffer or 200 nM BRCA1-BARD1. Samples were withdrawn after 60 min and analyzed by western blot (C), or 120 min and RNA was quantified by RT-qPCR (D) (n = 2). (E) pActin was incubated in NPE supplemented with increasing amounts of BRCA1I26A-BARD1. After 120 min, RNA was quantified by RT-qPCR (n = 3). Results from Figure 1E are also shown for comparison.
To investigate the role of ubiquitination in transcription suppression, we pre-incubated extract with ubiquitin vinyl sulfone (UbVS), an irreversible de-ubiquitinase (DUB) inhibitor. In the presence of UbVS, ubiquitin turnover is blocked and free ubiquitin is rapidly depleted from extract (42), thereby preventing additional ubiquitination events (Figure 4C). Extracts treated with buffer or UbVS were then supplemented with buffer or BRCA1-BARD1 and transcription was analyzed by RT-qPCR. Despite the absence of free ubiquitin, BRCA1-BARD1 still suppressed transcription with the same efficiency as buffer-treated reactions (Figure 4D). We then tested whether BRCA1-BARD1’s ubiquitin ligase activity was required for transcription suppression. We generated a mutant BRCA1-BARD1 complex containing an I26A substitution in BRCA1 (Figure 1C), which retains interaction between BRCA1-BARD1, but disrupts E2 binding required for ubiquitin transfer (43) (Supplementary Figure S4). When the BRCA1I26A-BARD1 complex was added to extract, transcription was abolished in a dose-dependent manner, demonstrating nearly identical activity to the wild-type complex (Figure 4E). Taken together, these results indicate that ubiquitin and E3 ligase activity are dispensable for transcription suppression by BRCA1-BARD1.
BRCA1-BARD1 regulates DNA binding of the chromatin reader BRD4
To uncover alternative mechanisms of BRCA1-BARD1-mediated transcription suppression, we analyzed DNA binding in extract that was mock-depleted, BRCA1-depleted, or supplemented with BRCA1-BARD1. DNA-bound proteins were isolated by plasmid pull-down and identified by mass spectrometry (Supplementary Table S1). Results were ranked for enrichment in ΔBRCA1 over +BRCA1-BARD1 reactions to identify proteins whose DNA binding was increased in the absence of BRCA1 and also reduced in the presence of excess BRCA1-BARD1, or vice versa (Figure 5A). Factors involved in DNA repair and chromatin organization were highly represented throughout the data set (Supplementary Figure S5). As expected, members of the BRCA1-BARD1 complex scored ‘low’, indicating that their DNA binding was positively regulated by the presence of BRCA1-BARD1. We also found that four associated complexes involved in chromatin and transcription regulation (SWI/SNF, Mediator, P-TEFb, and the Super Elongation Complex) scored ‘high’, indicating that their DNA binding was negatively regulated by the presence of BRCA1-BARD1. Recruitment of these complexes to chromatin is regulated by the bromo- and extraterminal-domain (BET) protein BRD4 (Figure 5B), which also scored high. BRD4 is an epigenetic reader that binds to acetylated histones and is widely implicated in transcription activation (44). BRD4 is essential during embryogenesis (45) and plays an important role in cell differentiation later in development (46,47). BRD4 has also been shown to play a role in homologous recombination-mediated DNA repair (48), although a functional link to BRCA1 has not been established.
Figure 5.

BRCA1-BARD1 regulates DNA-binding of multiple transcription regulators. (A) pActin was incubated in extract that was mock-depleted, BRCA1-depleted, or supplemented with 200 nM BRCA1-BARD1. DNA-bound proteins were isolated by plasmid pull-down and analyzed by mass spectrometry. Results were normalized to ΔMock samples and ranked by enrichment of ΔBRCA1 over +BRCA1-BARD1 samples. Representative complex members are labeled. An extended graph showing distribution of ranked proteins is shown in Supplementary Figure S5. (B) Schematic showing the relationship between BRCA1, BRD4, and complexes identified in (A).
To confirm whether BRCA1 regulates the recruitment of BRD4 to DNA, pActin was incubated in mock- or BRCA1-depleted extract and DNA-bound proteins were analyzed by plasmid pull-down. We saw that BRD4 was present on DNA in mock-depleted reactions and increased in the absence of BRCA1 (Figure 6A). DNA binding of BRD4 was also analyzed in reactions supplemented with buffer or BRCA1-BARD1. In the presence of excess BRCA1-BARD1, BRD4 binding to DNA was completely blocked (Figure 6B). Thus, BRCA1 negatively regulates binding of BRD4 to DNA.
Figure 6.

BRCA1-BARD1 suppresses H4K8 acetylation and BRD4 binding. (A) pActin was incubated in mock- or BRCA1-depleted extract. At the indicated time points, DNA-bound proteins were isolated by plasmid pull-down and analyzed by western blot. Total recovery of plasmid DNA from mock- and BRCA1-depleted extracts is shown in Supplementary Figure S6. (B) pActin was incubated in extract supplemented with buffer or 200 nM BRCA1-BARD1 and DNA-bound proteins were analyzed as in (A). (C) pActin was incubated in extract supplemented with buffer or JQ1 (BETi). At the indicated time points, DNA-bound proteins were analyzed as in (A). (D) pActin was incubated in extract with increasing amounts of JQ1. After 120 min, RNA was quantified by RT-qPCR (n = 3). (E) pActin was incubated in NPE supplemented with buffer or JQ1. At the indicated time points, RNA was quantified by RT-qPCR. (F) pActin was incubated in mock- or BRCA1-depleted extract. Samples were withdrawn after 20 min and analyzed by ChIP with the indicated antibodies (n = 2). (G) pActin was incubated in extract supplemented with buffer or JQ1. Samples were withdrawn after 20 min and analyzed by ChIP with the indicated antibodies (n = 2).
BRD4 functions broadly in the genome, but is specifically linked to oncogenes like C-MYC, CCND1, KRAS, BCL2 and BRAF (44). To test whether BRD4 regulates transcription of our plasmid substrate, pActin was incubated in extract supplemented with buffer or an inhibitor of BET proteins called JQ1. JQ1 blocks interaction between BRD4 and acetyl-histones, leading to global reorganization of BRD4 in cells (49). When DNA-bound proteins were analyzed by plasmid pull-down, we saw that JQ1 severely reduced BRD4 accumulation compared to a buffer control (Figure 6C). Transcription of pActin was then quantified in reactions supplemented with buffer or JQ1. We found that transcription was reduced by JQ1 in a dose-dependent manner (Figure 6D) and remained inhibited for several hours (Figure 6E). These results argue that BRD4 is critical for transcription in our system. As such, negative regulation of BRD4 by BRCA1 represents a major mechanism of transcription suppression.
Association of BRD4 with chromatin is mediated primarily through its interaction with acetylated histones. BRD4 was shown to have the highest affinity for acetylation of histone H4 at lysines 5, 8, 12 and 16 (H4K5ac, H4K8ac, H4K12ac and H4K16ac) (50). To test whether BRCA1 regulates the formation of these chromatin marks, antibodies that specifically recognize each modification were used to ChIP plasmid-bound proteins in mock- or BRCA1-depleted reactions. We saw that BRCA1 depletion led to a significant increase in the level of H4K8ac, but not H4K5ac, H4K12ac or H4K16ac compared to mock-depleted reactions (Figure 6F). BRD4 contains a histone acetyl transferase (HAT) domain that has been linked to acetylation of multiple lysines on histones H3 and H4 (51). To test whether BRD4 also contributes to H4K8 acetylation in extract, ChIP was performed in reactions supplemented with buffer or JQ1. Although binding of BRD4 was severely reduced in the presence of JQ1, the level of H4K5ac, H4K8ac, H4K12ac and H4K16ac did not decrease (Figure 6G), indicating that their formation was not dependent on BRD4. Together, these results support a model where BRCA1 regulates BRD4 binding by suppressing acetylation of histone H4K8.
DISCUSSION
Here, we report that BRCA1-BARD1 suppresses transcription in nucleoplasmic extract (Figure 1). We show that transcription suppression was not dependent on damage signaling (Figure 2 and Supplementary Figure S2). However, DNA damage and other forms of cellular stress are likely to influence BRCA1-BARD1 activity by regulating expression or localization of the complex (52). We provide evidence that BRCA1-BARD1 acts through a histone intermediate (Figure 3A–C) and that it blocks transcription initiation (Figure 3D and E) by limiting access to chromatinized DNA (Figure 3F-H). Although limited histone H2A ubiquitination occurs in our system (Figure 4A and B), both ubiquitin and BRCA1-BARD1’s E3 ligase activity were dispensable for transcription suppression (Figure 4C-E), establishing the presence of an alternative mechanism. Depending on the cellular context, multiple mechanisms of suppression may provide redundant or finely tuned regulation of specific genes or genomic regions.
Using mass spectrometry, we identified proteins whose DNA-binding was regulated by BRCA1-BARD1 in extract (Figure 5A, Supplementary Figure S5, and Supplementary Table S1). We saw that BRD4, through interactions with SWI/SNF, Mediator, P-TEFb, and the Super Elongation Complex, was a potential candidate for regulation by BRCA1-BARD1 (Figure 5B). We then confirmed that BRCA1-BARD1 was a negative regulator of BRD4 binding by Western blot (Figure 6A and B) and showed that inhibition of BRD4 binding was sufficient to block transcription in our system (Figure 6C–E). BRD4 is recruited to active genes by acetyl-histones where it acts as a scaffold, stabilizing the transcription pre-initiation complex and stimulating elongation. When we looked at histone acetylations associated with BRD4 binding, we saw that BRCA1 specifically suppressed the level of H4K8ac (Figure 6F). BRD4’s recruitment to chromatin likely involves interaction with multiple histone acetylations (53). However, acetylation of H4K8 has been shown to greatly increase BRD4’s overall affinity for other acetyl-marks (50). Thus, regulation of H4K8 acetylation by BRCA1 may act as a regulatory switch for the stable association of BRD4 with chromatin.
BRD4 promotes expression of major oncogenes, establishing inhibition of BRD4 and other BET proteins as an emergent therapeutic strategy (54). Both BRCA1 loss and BET inhibition have been shown to cause synthetic lethality with poly-ADP ribose polymerase (PARP) inhibition (55,56). Our findings connect BRCA1 with BRD4, thereby identifying dysregulation of BRD4 as a common theme in both mechanisms of genome-induced cell death. Establishing the mechanisms that underlie these genetic interactions will continue to shed new light on the role they play in tumor suppression, cancer development, and therapeutic intervention.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Igor Dawid for BRD4 antibodies, Hiroshi Kimura for H4K12ac antibodies, and Shuo Qie for assistance with insect cell growth and expression. J.K.B. and D.T.L. designed and analyzed experiments; J.K.B. and G.F. performed experiments; J.K.B. and D.T.L. prepared the manuscript.
Contributor Information
John K Barrows, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC 29425, USA.
George Fullbright, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC 29425, USA.
David T Long, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC 29425, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35 GM119512 to D.T.L.]; South Carolina Clinical & Translational Research (SCTR) Institute [TL1 TR001451, UL1 TR001450 to J.K.B.]. Funding for open access charge: NIH, NIGMS [R35 GM119512].
Conflict of interest statement. None declared.
REFERENCES
- 1. Petrucelli N., Daly M.B., Pal T.. Adam M.P., Ardinger H.H., Pagon R.A.. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. GeneReviews® [Internet]. Seattle, WA: University of Washington. [Google Scholar]
- 2. Huen M.S., Sy S.M., Chen J.. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010; 11:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Venkitaraman A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002; 108:171–182. [DOI] [PubMed] [Google Scholar]
- 4. Savage K.I., Harkin D.P.. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015; 282:630–646. [DOI] [PubMed] [Google Scholar]
- 5. Hashizume R., Fukuda M., Maeda I., Nishikawa H., Oyake D., Yabuki Y., Ogata H., Ohta T.. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001; 276:14537–14540. [DOI] [PubMed] [Google Scholar]
- 6. Fabbro M., Rodriguez J.A., Baer R., Henderson B.R.. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J. Biol. Chem. 2002; 277:21315–21324. [DOI] [PubMed] [Google Scholar]
- 7. Zhang F., Ma J., Wu J., Ye L., Cai H., Xia B., Yu X.. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009; 19:524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leung C.C., Glover J.N.. BRCT domains: easy as one, two, three. Cell Cycle. 2011; 10:2461–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang X., Li R.. BRCA1-dependent transcriptional regulation: implication in tissue-specific tumor suppression. Cancers. 2018; 10:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scully R., Anderson S.F., Chao D.M., Wei W., Ye L., Young R.A., Livingston D.M., Parvin J.D.. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:5605–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson S.F., Schlegel B.P., Nakajima T., Wolpin E.S., Parvin J.D.. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998; 19:254–256. [DOI] [PubMed] [Google Scholar]
- 12. Ye Q., Hu Y.F., Zhong H., Nye A.C., Belmont A.S., Li R.. BRCA1-induced large-scale chromatin unfolding and allele-specific effects of cancer-predisposing mutations. J. Cell Biol. 2001; 155:911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang H., Somasundaram K., Peng Y., Tian H., Zhang H., Bi D., Weber B.L., El-Deiry W.S.. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene. 1998; 16:1713–1721. [DOI] [PubMed] [Google Scholar]
- 14. Wang Q., Zhang H., Kajino K., Greene M.I.. BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene. 1998; 17:1939–1948. [DOI] [PubMed] [Google Scholar]
- 15. Fan S., Ma Y.X., Wang C., Yuan R.Q., Meng Q., Wang J.A., Erdos M., Goldberg I.D., Webb P., Kushner P.J.et al.. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001; 20:77–87. [DOI] [PubMed] [Google Scholar]
- 16. Tkocz D., Crawford N.T., Buckley N.E., Berry F.B., Kennedy R.D., Gorski J.J., Harkin D.P., Mullan P.B.. BRCA1 and GATA3 corepress FOXC1 to inhibit the pathogenesis of basal-like breast cancers. Oncogene. 2012; 31:3667–3678. [DOI] [PubMed] [Google Scholar]
- 17. Hu Y.F., Hao Z.L., Li R.. Chromatin remodeling and activation of chromosomal DNA replication by an acidic transcriptional activation domain from BRCA1. Genes Dev. 1999; 13:637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miyake T., Hu Y.F., Yu D.S., Li R.. A functional comparison of BRCA1 C-terminal domains in transcription activation and chromatin remodeling. J. Biol. Chem. 2000; 275:40169–40173. [DOI] [PubMed] [Google Scholar]
- 19. Bochar D.A., Wang L., Beniya H., Kinev A., Xue Y., Lane W.S., Wang W., Kashanchi F., Shiekhattar R.. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000; 102:257–265. [DOI] [PubMed] [Google Scholar]
- 20. Pao G.M., Janknecht R., Ruffner H., Hunter T., Verma I.M.. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:1020–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Filipponi D., Muller J., Emelyanov A., Bulavin D.V.. Wip1 controls global heterochromatin silencing via ATM/BRCA1-dependent DNA methylation. Cancer Cell. 2013; 24:528–541. [DOI] [PubMed] [Google Scholar]
- 22. Lee Y.H., Kuo C.Y., Stark J.M., Shih H.M., Ann D.K.. HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Res. 2013; 41:5784–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu Q., Pao G.M., Huynh A.M., Suh H., Tonnu N., Nederlof P.M., Gage F.H., Verma I.M.. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011; 477:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalb R., Mallery D.L., Larkin C., Huang J.T., Hiom K.. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014; 8:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou W., Zhu P., Wang J., Pascual G., Ohgi K.A., Lozach J., Glass C.K., Rosenfeld M.G.. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell. 2008; 29:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barrows J.K., Long D.T.. Cell-free transcription in Xenopus egg extract. J. Biol. Chem. 2019; 294:19645–19654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang W.L., Shechter D.. Chromatin assembly and transcriptional cross-talk in Xenopus laevis oocyte and egg extracts. Int. J. Dev. Biol. 2016; 60:315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Räschle M., Knipscheer P., Enoiu M., Angelov T., Sun J., Griffith J.D., Ellenberger T.E., Schärer O.D., Walter J.C.. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008; 134:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joukov V., Groen A.C., Prokhorova T., Gerson R., White E., Rodriguez A., Walter J.C., Livingston D.M.. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell. 2006; 127:539–552. [DOI] [PubMed] [Google Scholar]
- 30. Walter J., Sun L., Newport J.. Regulated chromosomal DNA replication in the absence of a nucleus. Mol. Cell. 1998; 1:519–529. [DOI] [PubMed] [Google Scholar]
- 31. Zierhut C., Jenness C., Kimura H., Funabiki H.. Nucleosomal regulation of chromatin composition and nuclear assembly revealed by histone depletion. Nat. Struct. Mol. Biol. 2014; 21:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Toyama R., Rebbert M.L., Dey A., Ozato K., Dawid I.B.. Brd4 associates with mitotic chromosomes throughout early zebrafish embryogenesis. Dev. Dyn. 2008; 237:1636–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wolfe K.B., Long D.T.. Chromatin immunoprecipitation (ChIP) of plasmid-bound proteins in Xenopus egg extracts. Methods Mol. Biol. 2019; 1999:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fullbright G., Rycenga H.B., Gruber J.D., Long D.T.. p97 promotes a conserved mechanism of helicase unloading during DNA cross-link repair. Mol. Cell. Biol. 2016; 36:2983–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Siervi A., De Luca P., Byun J.S., Di L.J., Fufa T., Haggerty C.M., Vazquez E., Moiola C., Longo D.L., Gardner K.. Transcriptional autoregulation by BRCA1. Cancer Res. 2010; 70:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kleiman F.E., Wu-Baer F., Fonseca D., Kaneko S., Baer R., Manley J.L.. BRCA1/BARD1 inhibition of mRNA 3' processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005; 19:1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mullan P.B., Quinn J.E., Harkin D.P.. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene. 2006; 25:5854–5863. [DOI] [PubMed] [Google Scholar]
- 38. Sarkaria J.N., Busby E.C., Tibbetts R.S., Roos P., Taya Y., Karnitz L.M., Abraham R.T.. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999; 59:4375–4382. [PubMed] [Google Scholar]
- 39. Amodeo A.A., Jukam D., Straight A.F., Skotheim J.M.. Histone titration against the genome sets the DNA-to-cytoplasm threshold for the Xenopus midblastula transition. Proc. Natl. Acad. Sci. USA. 2015; 112:E1086–E1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prioleau M.N., Huet J., Sentenac A., Mechali M.. Competition between chromatin and transcription complex assembly regulates gene expression during early development. Cell. 1994; 77:439–449. [DOI] [PubMed] [Google Scholar]
- 41. Terui R., Nagao K., Kawasoe Y., Taki K., Higashi T.L., Tanaka S., Nakagawa T., Obuse C., Masukata H., Takahashi T.S.. Nucleosomes around a mismatched base pair are excluded via an Msh2-dependent reaction with the aid of SNF2 family ATPase Smarcad1. Genes Dev. 2018; 32:806–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Borodovsky A., Kessler B.M., Casagrande R., Overkleeft H.S., Wilkinson K.D., Ploegh H.L.. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001; 20:5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brzovic P.S., Keeffe J.R., Nishikawa H., Miyamoto K., Fox D. 3rd, Fukuda M., Ohta T., Klevit R.. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:5646–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donati B., Lorenzini E., Ciarrocchi A.. BRD4 and Cancer: going beyond transcriptional regulation. Mol. Cancer. 2018; 17:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chan S.H., Tang Y., Miao L., Darwich-Codore H., Vejnar C.E., Beaudoin J.D., Musaev D., Fernandez J.P., Benitez M.D.J., Bazzini A.A.et al.. Brd4 and P300 confer transcriptional competency during zygotic genome activation. Dev. Cell. 2019; 49:867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee J.E., Park Y.K., Park S., Jang Y., Waring N., Dey A., Ozato K., Lai B., Peng W., Ge K.. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017; 8:2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Najafova Z., Tirado-Magallanes R., Subramaniam M., Hossan T., Schmidt G., Nagarajan S., Baumgart S.J., Mishra V.K., Bedi U., Hesse E.et al.. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017; 45:127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mio C., Gerratana L., Bolis M., Caponnetto F., Zanello A., Barbina M., Di Loreto C., Garattini E., Damante G., Puglisi F.. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer. 2019; 144:755–766. [DOI] [PubMed] [Google Scholar]
- 49. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I.et al.. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jung M., Philpott M., Müller S., Schulze J., Badock V., Eberspächer U., Moosmayer D., Bader B., Schmees N., Fernández-Montalván A.et al.. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014; 289:9304–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Devaiah B.N., Case-Borden C., Gegonne A., Hsu C.H., Chen Q., Meerzaman D., Dey A., Ozato K., Singer D.S.. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016; 23:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rosen E.M. BRCA1 in the DNA damage response and at telomeres. Front. Genet. 2013; 4:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T.et al.. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stathis A., Bertoni F.. BET proteins as targets for anticancer treatment. Cancer Discov. 2018; 8:24–36. [DOI] [PubMed] [Google Scholar]
- 55. Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C.et al.. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921. [DOI] [PubMed] [Google Scholar]
- 56. Sun C., Yin J., Fang Y., Chen J., Jeong K.J., Chen X., Vellano C.P., Ju Z., Zhao W., Zhang D.et al.. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018; 33:401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.