


Abstract
Several existing technologies enable short genomic alterations including generating indels and short nucleotide variants, however, engineering more significant genomic changes is more challenging due to reduced efficiency and precision. Here, we developed RecT Editor via Designer-Cas9-Initiated Targeting (REDIT), which leverages phage single-stranded DNA-annealing proteins (SSAP) RecT for mammalian genome engineering. Relative to Cas9-mediated homology-directed repair (HDR), REDIT yielded up to a 5-fold increase of efficiency to insert kilobase-scale exogenous sequences at defined genomic regions. We validated our REDIT approach using different formats and lengths of knock-in templates. We further demonstrated that REDIT tools using Cas9 nickase have efficient gene-editing activities and reduced off-target errors, measured using a combination of targeted sequencing, genome-wide indel, and insertion mapping assays. Our experiments inhibiting repair enzyme activities suggested that REDIT has the potential to overcome limitations of endogenous DNA repair steps. Finally, our REDIT method is applicable across cell types including human stem cells, and is generalizable to different Cas9 enzymes.
INTRODUCTION
Gene editing is a powerful technology for interrogating and engineering biological systems (1–10). Our ability to engineer the genome has been greatly accelerated since the development of various gene-editing tools such as ZFNs, TALENs, and most recently CRISPR technology. Derived from prokaryotic defense systems, CRISPR–Cas tools introduce site-specific DNA double-strand breaks (DSBs) which can then be repaired by endogenous DNA repair machineries to achieve genome editing. Many CRISPR-based tools are available for short-sequence modifications such as introducing random indels or install single-nucleotide variants (SNVs) (3–5). However, larger-scale editing via HDR, like the insertion of fluorescent/selectable markers or therapeutic transgene, is highly sought after in the engineering of model systems, therapeutic cell production, and gene therapy (8,9,11). However, HDR is significantly limited by the competing non-homologous end joining (NHEJ) and Microhomology-mediated end joining (MMEJ) mechanisms which are far more efficient and error prone. Prior studies have employed diverse methods to improve HDR efficiencies for altering kilobases of DNA sequence (12,13). They mainly fall into one of the three categories: (i) perturbation of cell state. In most cell types, HDR is most active in S/G2 phase, thus its efficiency can be increased by cell synchronization in S/G2 phases when performing gene-editing (14), timed delivery of the designer nucleases or restriction of nuclease activity to S/G2 phase (15–17); (ii) chemical or genetic perturbation of endogenous repair pathways. Exemplar strategies shown to improve precision gene-editing are: inhibition of NHEJ pathways by depleting key NHEJ factors such as DNA ligase IV (18–25), alternating the repair pathway choice in favor of HDR by supplementing HDR enhancers such as RS-1 or overexpressing DNA repair proteins such as RAD51 (26), recruiting HDR-related repair proteins to Cas9 such as dominant-negative p53-binding protein 1 (dn53BP1), CtIP, and RecA (27–31); 3) Modification and optimization of repair template. Rationally designed or chemically-modified donor DNAs could improve HDR efficiencies. For example, conjugation of the donor DNA to Cas enzymes through interactions between streptavidin and biotin, or chemically modified donor with mutant Cas9 containing non-natural amino acids (32–36). In addition, increasing the length of homology and using chemical modification to recruit the donor DNAs to Cas9 have both been shown to enhance HDR (37–41). While these methods improve HDR efficiencies, the issues of on- and off-target errors often remain. Previous works have shown that inhibition of NHEJ pathways could result in increased cell apoptosis, and follow-up studies have observed significant variability of performances, particularly for small-molecule enhancers (26,28,42). Moreover, most HDR-enhancing tools require DSBs, often resulting in deleterious cellular effects (43,44). Hence, there is a continued need for efficient, low-error methods, with less dependence on damage repair pathways and robustness across cell types and contexts (12,45).
In contrast to Cas9-mediated HDR in mammalian cells, phage single-stranded DNA-annealing proteins (SSAP), exemplified by the bacteriophage lambda bet (also called Redβ), achieve up to multi-kilobase genome recombineering in microbes (46–49). Evolutionarily, these phage SSAPs are a distinct family of recombination proteins, independent from the bacterial RecA or eukaryotic RAD51 (50–52). Biochemically, SSAPs have been shown to bind to either ssDNA or dsDNA substrate, and then promote the strand exchange between homologous DNA sequences through their ability to stimulate annealing of complementary single-stranded regions (51–53). The simplicity and efficiency of phage SSAP provide intriguing possibilities for gene-editing in higher eukaryotes. To date, many studies have leveraged SSAPs for in vitro and bacterial applications, they have not been used for mammalian gene editing (46,49). We hypothesized that SSAP may facilitate homology-directed genome editing in mammalian cells when coupled to programmable Cas9 for genomic targeting (Figure 1A). Additionally, we sought to use phage SSAP as prior biochemistry studies demonstrated that they may be ATP-independent (53). Thus, phage SSAPs could partially overcome the limitations of endogenous DNA repair pathways mediated by eukaryotic proteins such as RAD51, which are ATP-dependent (13).
Figure 1.

Testing microbial SSAPs for genome engineering in mammalian cells. (A) Schematic showing the model and assay to measure knock-in activity of phage SSAPs with CRISPR genome targeting in mammalian cells. Inset (lower-right) depicts the detailed vector designs. Note the sizes of elements are not drawn to scale. (B) Using the 2A-mKate genomic knock-in assay in HEK293T cells to measure editing efficiency of three major families of phage SSAPs. All results here and later are from replicate experiments (n = 3), with error bars representing standard error of the mean (SEM), unless otherwise noted. (C) Imaging verification of mKate knock-in efficiency at HSP90AA1 locus for the top candidate SSAP RecT protein. (D, E) Using protein-aptamer pairs, MCP-MS2 and PCP-PP7 (D), to verify the dependence of RecT’s gene-editing activity on target-recruitment, with negative, over-expression, and positive controls. All constructs were tested using the knock-in assay at two genomic loci (E). Donor DNAs are dsDNA with 200 + 400 bp (DYNLT1) or 200 + 200 bp (HSP90AA1) of homology arms (HAs). NLS, nuclear localization signal; BFP, blue fluorescent protein; NTC, non-targeting control; PCP, PP7 coat protein.
Here, we engineered and tested phage SSAPs for mammalian gene-editing, and identified a lead candidate based on the small phage protein RecT, which we termed RecT Editor via Designer-Cas9-Initiated Targeting (REDIT). We validated the ability of REDIT to promote knock-in gene-editing using different donor DNAs at various insertion sizes in mammalian cells, showing up to 5-fold higher efficiencies compared with Cas9 references. We demonstrated that REDIT stimulated precision HDR without elevating indel formations at on/off-target sites. We then confirmed it is applicable in various cell types, and compatible with different Cas9 proteins (SpCas9 and the more compact SaCas9). Most importantly, REDIT worked with the Cas9 nickase (Cas9n, or Cas9-D10A) to promote HDR in single- and double-nicking formats, with improved knock-in efficiencies over both wild-type and nicking Cas9 tools. REDIT double-nicking (REDITdn) tools significantly reduced unwanted editing events, as measured by genome-wide off-target assays, clonal knock-in junction Sanger sequencing, and genomic insertion site profiling. We further provided evidence that REDIT maintained its advantage over Cas9 methods under DNA repair enzyme inhibition, which is supported by prior work on the ATP-independent activity of phage SSAPs. Finally, we validated that REDIT tools have the ability to enhance gene-editing efficiencies in non-transformed human stem cells at endogenous targets. Taken together, these results indicate that phage SSAPs could enhance template-mediated HDR in mammalian cells at genomic loci when recruited by Cas9. In particular, the nicking REDITdn tools provide a method for precision knock-in in mammalian cells without requiring DSB formation. This method is relatively compact, efficient for knock-in and low-error. It has the potential to help study genomic variants in stem cell models for basic research, and serve as a gene therapy approach. When selection methods for precision knock-in is limited, or when the assay is sensitive to unwanted editing events, the REDITdn method is a useful and complementary addition to the increasingly powerful gene-editing toolbox.
MATERIALS AND METHODS
SSAP RecT search and evolutionary analysis
For the Pfam database (54), we retrieved the RecT family (PF03837) protein sequences from the online portal taking the full 1397 members of this family. For the PSI-BLAST mapping, RefSeq non-redundant protein database was downloaded from NCBI on 29 October 2019. We identified the three major families of SSAP enzymes from Bacteriophage lambda, Escherichia coli Rac prophage, and bacteriophage T7, and extracted the primary enzyme sequences as listed in supplementary sequences. For mapping RecT distribution, the database was searched with Rac prophage RecT (NP_415865.1) as query using Position-specific iterated (PSI)-BLAST (55) to retrieve protein homologs. Hits were clustered with CD-HIT (56) and representative sequences were selected from each cluster for multiple alignment with MUSCLE (57). Then, FastTree (58) was used for maximum likelihood tree reconstruction with default parameters.
Plasmids construction
For the sgRNA/Cas9 plasmids, the parent plasmid (Addgene Plasmid #64323, Ralf Kuehn Lab) was digested by restriction enzymes BbsI and XbaI (New England BioLabs) to remove the standard sgRNA scaffold, and the backbone was purified. The MS2 stem loop guide RNA scaffold was inserted by Gibson Assembly using NEBuilder HiFi DNA Assembly Master Mix. The insertion had two BbsI restriction sites before the scaffold sequence to allow downstream cloning of guides, and the sequence of the MS2 stem loop scaffold is shown in Supplementary sequence. The sgRNAs were then inserted using Golden Gate cloning. Primer sequences of all guides used are listed in Supplementary Table S1. For sgRNA/Cas9n (nickase) plasmids, the parent plasmid (Addgene Plasmid #74119, Steve Jackson Lab) was digested by restriction enzymes PciI and KpnI (New England BioLabs) to remove both of the two U6 promoters and original scaffolds, and the backbone was purified. A U6 promoter and a MS2 stem loop guide RNA scaffold was cloned into the backbone by Gibson Assembly using NEBuilder HiFi DNA Assembly Master Mix. Two BbsI restriction sites as above allow downstream cloning of guides. All guide sequences used in the study are listed in Supplementary Table S1.
For the SSAP plasmids, sequence-optimized DNA fragments corresponding to all SSAPs were designed based on human/mouse codon usage, then ordered from Genscript or Genewiz. The sequences of all SSAP tested are listed in Supplementary Table S3. For the MS2 aptamer designs, the vectors expressing SSAPs were derived from a published plasmid by Feng Zhang Lab (Addgene Plasmid #61423), where the P65-HSF1 insert portion was replaced with the fragments encoding the SSAP enzymes to generate MCP fusion with SSAP. For SunTag designs, we amplified the SunTag components from published plasmids by Ronald Vale Lab (Addgene plasmid #60903 for tandem GCN4 peptides, and plasmid #60907 for scFV), and then cloned these fragments in-frame with Cas9 and SSAPs. For the direct fusion designs, we cloned the SSAPs in-frame into in-house pCBH-Cas9 plasmid derived from the published PX330 plasmids (Addgene #42230). All plasmid constructions were performed using NEBuilder HiFi DNA Assembly Master Mix (New England BioLabs). All sequence verified REDIT plasmids with BbsI(SpCas9) and BsaI(SaCas9) cloning sites will be deposited to Addgene. A step-by-step protocol describing the cloning and usage of REDIT methods is provided in Supplementary Notes, along with a list of all REDIT plasmids.
Cell culture
Human Embryonic Kidney (HEK) 293T, A549, HeLa and HepG2 were maintained in Dulbecco's modified Eagle's medium (DMEM, Life Technologies), with 10% fetal bovine serum (FBS, HyClone), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies) at 37 °C with 5% CO2. Apart from HEK 293T cells (from ThermoFisher), other cell lines were obtained from American Type Culture Collection (ATCC). The identity of the cell lines are authenticated regularly by short tandem repeat (STR) assay and routinely tested for the presence of Mycoplasma using qPCR assay. hES-H9 cells were maintained in mTeSR1 medium (StemCell Technologies) at 37°C with 5% CO2. Culture plates were pre-coated with Matrigel (Corning) 12 h prior to use, and cells were supplemented with 10 μM Y27632 (Sigma) for the first 24 h after each passage. Culture media was changed every 24 h.
Transfection
HEK293T cells were seeded into 96-well plates (Corning) 12–24 h prior to transfection at a density of 30 000 cells/well, and 250 ng of total DNA was transfected per well. HeLa and HepG2 cells were seeded into 48-well plates (Corning) 1 day prior to transfection at a density of 50 000 and 30 000 cells/well respectively, and 400 ng of total DNA was transfected per well. Transfections were performed with Lipofectamine 3000 (Life Technologies) following the manufacturer's instructions.
Electroporation
For hES-H9 related transfection experiments, P3 Primary Cell 4D-NucleofectorTM X Kit S (Lonza) was used following the manufacturer's protocol. For each reaction, 300,000 cells were nucleofected with 4 μg total DNA using the DC100 Nucleofector Program.
Fluorescence-activated cell sorting (FACS)
mKate knockin efficiency was analyzed on a CytoFLEX flow cytometer (Beckman Coulter; Stanford Stem Cell FACS Core). 72 hours after transfection, cells were washed once with PBS and dissociated with TrypLE Express Enzyme (Thermo Fisher Scientific). Cell suspension was then transferred to a 96-well U-bottom plate (Thermo Fisher Scientific) and centrifuged at 300 × g for 5 min. After removing the supernatant, pelleted cells were resuspended with 50 μl 4% FBS in PBS, and cells were sorted within 30 min of preparation.
Sanger sequencing and NGS of knock-in junctions
HEK293T cells were transfected with plasmid DNA and DNA templates and harvested after 72 h for genomic DNA using the QuickExtract DNA Extraction Solution (Biosearch Technologies) following the manufacturer's protocol. The target genomic region was amplified using specific primers outside of the homology arms of the DNA template. PCR products were purified with Monarch PCR & DNA Cleanup Kit (New England BioLabs). 100 ng of purified product was sent for Sanger sequencing with target-specific primers (EtonBio or Genewiz).
Treatment with HR and cell cycle inhibitor
All inhibitors were ordered from Sigma-Aldrich. For different inhibitor assays, the cells were pretreated with Nocodazole (sigma, M1404, 50 ng/mL), or B02 (Sigma, SML0364, 10 uM), or RI-1 (Sigma, 553514-10MG-M, 1 uM) for 16 h. Then after washing and switching to fresh media, the cells were transfected with different plasmids using lipofectamine 3000 following the manufacturer's instruction. Three days later, the cells were analyzed on a CytoFLEX flow cytometer and genomic DNA were also harvested for sequencing validation as above.
Cloning and colony sequencing experiment and analysis
To validate the precision of individual knock-in editing events at both junctions, we performed cloning and colony sequencing of edited cells to measure the full knock-in insertion precision at both 5′ and 3′ junctions using Sanger sequencing. Briefly, 72 h after transfection, cells were replated by limiting dilution to isolate single clones of edited cells, and genomic DNA was extracted using QuickExtract DNA Extraction Solution (Biosearch Technologies). 250 ng total genomic DNA was used for the TOPO TA cloning experiments. The knock-in target genomic regions were amplified using TA colony primers (completely outside of the homology arms of the donor DNA to avoid template amplification) for DYNLT1 or HSP90AA1 locus (Supplementary Table S2) using Phusion Flash High-Fidelity PCR Master Mix (ThermoScientific, F-548L). We then purified the targeted PCR products using Gel extraction kit (New England BioLabs,T1020L) following the manufacturer's instructions. Add A-tailing to the PCR products using Taq polymerase(New England BioLabs, M0273S) through incubate at 72°C for 30 min. TOPO TA cloning reactions were then set up following the manufacturer's instructions (Thermo Scientific, K457501, Topo TA kit for sequencing) and transformed into Top10 competent cells. We then send the colonies for RCA Sanger sequencing using M13F (5′-GTAAAACGACGGCCAG-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′) primers. We requested at least 48 colonies to be sequenced per genomic locus per test condition (Genewiz and QuintaraBio). The sequencing trace results from the Sanger sequencing were analyzed using SnapGene.
Next-generation sequencing library preparation
Seventy-two hours after transfection, genomic DNA was extracted using QuickExtract DNA Extraction Solution. 200 ng total DNA was used for NGS library preparation. Genes of interest were amplified using specific primers (Supplementary Table 2) for the first round PCR reaction. Illumina adapters and index barcodes were added to the fragments with a second round PCR using the primers listed in Supplementary Table 2. Round 2 PCR products were purified by gel electrophoresis on a 2% agarose gel using the Monarch DNA Gel Extraction Kit (New England BioLab). The purified product was quantified with Qubit dsDNA HS Assay Kit (Thermo Fisher) and sequenced on an Illumina MiSeq system using paired-end PE300 kits. All sequencing data has been deposited to NCBI archive with accession code as listed in Data Availability.
High-throughput sequencing data analysis
Processed (demultiplexed, trimmed, and merged) sequencing reads were analyzed to determine editing outcomes using CRISPPResso2 (59) by aligning sequenced amplicons to reference and expected HDR amplicons. The quantification window was increased to 10 bp surrounding the expected cut site to better capture diverse editing outcomes, but substitutions were ignored to avoid inclusion of sequencing errors. Only reads containing no mismatches to the expected amplicon were considered for HDR quantification; reads containing indels that partially matched the expected amplicons were included in the overall reported indel frequency. The computation work was supported by the SCG cluster hosted by the Genetics Bioinformatics Service Center (GBSC) at the Department of Genetics of Stanford. All customized scripts for data analysis will be deposited to Github under Cong Lab and made available for download.
Statistical analysis
Unless otherwise stated, all statistical analysis and comparison were performed using t-test, with 1% false-discovery-rate (FDR) using a two-stage step-up method of Benjamini, Krieger and Yekutieli. All experiments were performed in triplicates unless otherwise noted to ensure sufficient statistical power in the analysis.
Determination of editing at predicted Cas9 off-target sites
To evaluate off-target editing activity at known Cas9 off-target sites, same genomic DNA extracts for knock-in analysis were used as template for PCR amplification of top predicted off-targets sites (high scored as predicted by http://crispor.org) for the EMX1, VEGFA guides that have been published (60,61). All primer sequences are listed in Supplementary Table S2.
iGUIDE off-target analysis
Genome-wide, unbiased off-target analysis was performed following the iGUIDE pipeline based on Guide-seq invented previously (62,63). HEK293T cells were transfected in 20 uL Lonza SF Cell Line Nucleofector Solution on a Lonza Nucleofector 4D with program DS-150 according to the manufacturer's instructions. 300 ng of gRNA–Cas9 plasmids (or 150 ng of each gRNA–Cas9n plasmid for the double nickase), 150 ng of the effector plasmids, and 5 pmol of double stranded oligonucleotides (dsODN) were transfected. Cells were harvested after 72 h for genomic DNA using Agencourt DNAdvance reagent kit. 400 ng of purified gDNA which was then fragmented to an average of 500 bp and ligated with adaptors using NEBNext Ultra II FS DNA Library Prep kit following manufacturer's instructions. Two rounds of nested anchored PCR from the oligo tag to the ligated adaptor sequence were performed to amplify targeted DNA, and the amplified library was purified, size-selected and sequenced using Illumina Miseq V2 PE300 or V3 PE600. Sequencing data was analyzed using the published iGUIDE pipeline, with the addition of a downsampling step which ensures an unbiased comparison across samples.
GIS-seq and analysis
Genome-wide, unbiased off-target analysis of mKate knock-in was performed using GIS-seq, a method that is developed through integrating features from several genomic insertion analysis methods as detailed below. To obtain small-size genomic DNA suitable for downstream amplification comparable to the digestion step in LAM-PCR (64), we leveraged the NEB Fragmentase to enzymatically fragment genomic DNA and perform end-repair. To remove donor DNA thus avoid template amplification, we extracted the high-molecular-weight (HMW) genomic DNA with the DNAdvance genomic DNA kit (A48705, Beckman Coulter) as in the TLA method (65), and to amplify the knock-in sites, we used Nextera adaptor ligation followed by PCR using primers that bind to the Nextera adaptor on one end, and the knock-in fragment on the other end (with plus and minus for 5′ and 3′ junctions respectively), similar to the process described in iGUIDE/GUIDE-seq (62,63), based on simplified version of GUIDE-seq protocol (https://www.protocols.io/view/guide-seq-simplified-library-preparation-protocol-wikfccw). For all samples, HEK293T cells were transfected with plasmids and donor DNA as described above in the Transfection section. The genomic DNA was size-selected to make sure no template remained via the DNAdvance kit and 400ng of purified gDNA which was then fragmented to an average of 500bp, ligated with adaptors, and size-selected using NEBNext Ultra II FS DNA Library Prep kit following manufacturer's instructions. Two rounds of nested anchored PCR from the end of the knock-in sequence to the ligated adaptor sequence were performed to amplify targeted DNA, and the amplified library was purified, size-selected as described in the NEBNext Ultra II FS DNA library Prep kit protocol. Final libraries were sequenced using Illumina Miseq V2 PE300 or V3 PE600 kits. Sequencing data was analyzed to determine any off target insertion events with all the analysis code deposited to Github (detailed in the Availability section).
RESULTS
Identification of RecT as optimal candidate SSAP for mammalian gene-editing
While it is potentially difficult to engineer microbial SSAPs to function in mammalian contexts, they have been shown to harbor significant metagenomic diversity with distinctive activities depending on host organisms (49,50,53,66,67). Hence, we began our search for gene-editing phage proteins by examining different families of phage SSAPs (50,68). To harness them for eukaryotic genome editing, we engineered and evaluated three major types of phage SSAPs, Escherichia phage lambda Bet (LBet), Escherichia coli Rac prophage RecT (RecT) and Bacteriophage T7 gp2.5 (gp2.5), for their ability to facilitate HDR in mammalian cells. First, we cloned these SSAPs into mammalian expression vectors to functionally connect them to the CRISPR–Cas9 system via an RNA aptamer. Here we took advantage of the modularity of aptamer strategy: Cas9s and gRNAs bearing MS2 stem-loops remain constant, and we could conveniently fuse different candidate SSAPs to the MS2 coat protein (MCP) for the screening (Figure 1A). Further, we used the Streptococcus pyogenes Cas9 (69) given SpCas9’s robustness and popularity (hereafter referred to as ‘Cas9’). To measure HDR-mediated editing, we employed a genomic knock-in assay (70) where we constructed a T2A-mKate cassette (∼1-kb in length), flanked by ∼200-bp homology arms (HA) on both sides, which match the last exon (in-frame before the stop codon) of target genes, e.g. DYNLT1 and HSP90AA1. Upon precise knock-in, the percentage of mKate-positive cells allows the quantification of HDR efficiency (Figure 1A). Our screen revealed that phage SSAPs could indeed stimulate HDR to variable extents in human cells relative to Cas9 reference (Figure 1B). The SSAP RecT was most efficient and compact: canonical RecT is ∼270 amino acids (AA) long and induced the greatest increase in HDR, about 3-fold versus Cas9 (Figure 1B). This trend was consistent across genomic loci and was verified using fluorescence microscopy and Sanger sequencing of positive clones (Figure 1C; Supplementary Figure S1 and S2). We noticed that there is a small but noticeable level of fluorescence in the control group using Cas9 bearing non-targeting but valid guide RNA, termed ‘NTC’ control (Figure 1B). The NTC signals are comparable to the ‘no donor’ control, where we delivered Cas9 and guide RNA but without donor DNA. Hence, the majority of these background signals are likely from non-specific flow cytometry noise and cellular autofluorescence, alongside signals from off-target insertions of the template, as previously observed in knock-in experiments (25,71,72). Nonetheless, because of this observation, we included NTC controls in later experiments to assess this background.
Based on these results, while the SSAP RecT is promising as a potential gene-editing tool, we note the importance of checking if background template insertion/expression or promiscuous activity of RecT could lead to false positive signals. Hence, we performed validation tests with the same donor DNA alongside a set of controls: the NTC control; the NTC control with over-expression of RecT, and Cas9 with on-target guideRNA. We confirmed that the background in our assay is low, and that simply over-expressing RecT yielded only background-level editing (Figure 1E). Another caveat of the results is whether the increased HDR efficiency via SSAP is truly specific to target locus, i.e. the observed knock-in is dependent on recruiting SSAP to genomic targets via Cas9-guideRNA. This is a critical point as it is usually sufficient to just over-express RecT-like SSAPs for bacteria recombineering (46,49). To this end, we generated non-recruiting fusions between RecT and the PP7 coat protein (PCP), which recognizes PP7 but not MS2 aptamers (Figure 1D). Our results indicated that RecT functioned in a recruitment-dependent manner (Figure 1E). Given this initial success, we chose to focus on RecT as our candidate SSAP for further development.
Apart from the canonical Rac prophage RecT from E. coli, to measure if other RecT-like SSAPs could be utilized for mammalian genome engineering, we performed a screen of RecT-like SSAPs for their ability to improve knock-in efficiencies in human cells. Taking advantage of pioneering work on mining SSAPs from metagenomic sequences (49,50,68), we searched the Pfam and the NCBI non-redundant protein database via PSI-BLAST (54,55) for RecT-like SSAPs, and examined their diversity (Supplementary Figure S3A). Then, we prioritized compact candidates, sampling across evolutionary subtrees with a cut-off at 300AA based on the length distribution of RecT homologs (Supplementary Figure S3B). We codon-optimized a total of 16 SSAPs (including the E. coli Rac prophage RecT as EcRecT) for mammalian expression, and performed the screen using the genome knock-in assay. This screen revealed that several homologs could significantly stimulate genomic knock-in in human cells, demonstrating the generalizability of using SSAP for gene-editing, with EcRecT as the most active candidate (Supplementary Figure S3C and D). Thus, we centered on EcRecT (simplified as RecT hereafter) for downstream optimization and investigation.
Optimization and validation of RecT-based genome engineering method
To develop a useful RecT gene-editing tool, we bear in mind the following considerations: (i) What is the optimal design to use this SSAP for mammalian gene-editing? (ii) What are the efficiencies using this new tool compared with Cas9 benchmarks and existing HDR-enhancing methods? (iii) What are the design guidelines and capacities when using RecT SSAP for gene-editing? (iv) What are the occurrences of editing errors/indels at on/off-target sites, and can we mitigate these unwanted events? (v) What is the tool's dependence on endogenous repair pathways and would it perform reliably across contexts?
First, we compared three parallel strategies to recruit RecT SSAP to Cas9 genome-targeting machinery: direct fusion, SunTag-based recruitment (73), and, as previously used in the screen, MS2-aptamer fusion (Figure 1A, 2A; Supplementary Figure S4A). When we directly fused RecT to the Cas9 at its N- or C-term via peptide linkers, we did not observe significant stimulation of HDR efficiencies above Cas9 (Supplementary Figure S4B). In parallel, to employ the protein-based SunTag design to recruit multiple copies of the SSAP as in the MS2-aptamer design, we fused RecT to the anti-GCN4 single-chain variable fragment (scFv) that allows binding to a Cas9 bearing tandem GCN4 tag (Figure 2A). The SunTag design significantly increased HDR efficiencies to ∼2-fold better than Cas9 (Figure 2B). Nonetheless, across the strategies we tested, the best-performing design is the MS2-aptamer. Hence, to further engineer RecT for gene-editing, we zoomed in on this MS2-aptamer chassis.
Figure 2.

Development of REDIT tools via characterizing and optimizing RecT designs. (A) Schematic showing the SunTag-based recruitment of SSAP RecT to Cas9-guideRNA complex for gene-editing, with inset showing construct maps (not drawn to scale). (B) Quantify and compare genome editing efficiencies of SunTag and the original MS2-based strategies for recruiting phage SSAPs. Assays performed at two endogenous loci, DYNLT1 and HSP90AA1. (C) NLS and linker design to engineer and improve gene-editing efficiencies using SSAP RecT. Different NLS sequences were placed internally or fused to the N- or C-terminus of RecT via different linkers. (D) Measure the gene-editing efficiencies of different designs from (C) and identify the optimal design as REDIT. Assays performed at HSP90AA1 and DYNLT1 loci, shown as relative efficiencies to Cas9 reference. Donor DNAs have 200+400bp (DYNLT1) or 200 + 200 bp (HSP90AA1) of HAs. Statistical analysis here and later were performed using t-test, with 1% false-discovery-rate (FDR) using a two-stage step-up method of Benjamini, Krieger and Yekutieli, unless otherwise noted (*P < 0.05, **P < 0.01, ***P < 0.001).
During initial tests, we noticed incomplete nuclear-targeting of RecT in mammalian cells when fused to the originally selected nuclear localization signal (NLS) (Supplementary Figure S5). Therefore, we tested different monopartite or bipartite NLSs with different linkers (Figure 2C). Our screen identified the C-terminal SV40-NLS with a modified E-XTEN linker as the optimal choice (Figure 2D). This new construct more than doubled the gene-editing efficiencies relative to the original RecT design, consistently achieving up to 5-fold improvement compared with Cas9 (Figure 2D). We termed this design RecT Editor via Designer-Cas9-Initiated Targeting (REDIT).
Next, to benchmark this optimized RecT design, we compared it with three categories of existing HDR-enhancing tools (Figure 3A and B): DNA repair enzyme CtIP fusion with the Cas9 (Cas9-HE) (29), a fusion of the functional domain (amino acid 1 to 110) of human Geminin protein with the Cas9 (Cas9-Gem) (16), and a small-molecule enhancers of HDR via cell cycle control (14), Nocodazole (15,74). Across endogenous targets tested, the optimized RecT design had favorable performance compared with three alternative strategies (Figure 3C). Furthermore, the RecT-based REDIT design, which putatively acted through the SSAP activity independently from the other approaches, may synergize with existing methods. To test this hypothesis, we combined the SSAP with three different approaches (conveniently through the MS2-aptamer) (Figure 3A, right). Our results validated that REDIT method could indeed further enhance the HDR-promoting activities of the tested tools (Figure 3C). Together, these results verified the potential to use phage SSAP for mammalian genome engineering, and that an optimized design as in the REDIT tool significantly improved knock-in efficiencies compared with Cas9 references. REDIT also has the potential to synergistically work with existing HDR enhancement tools.
Figure 3.
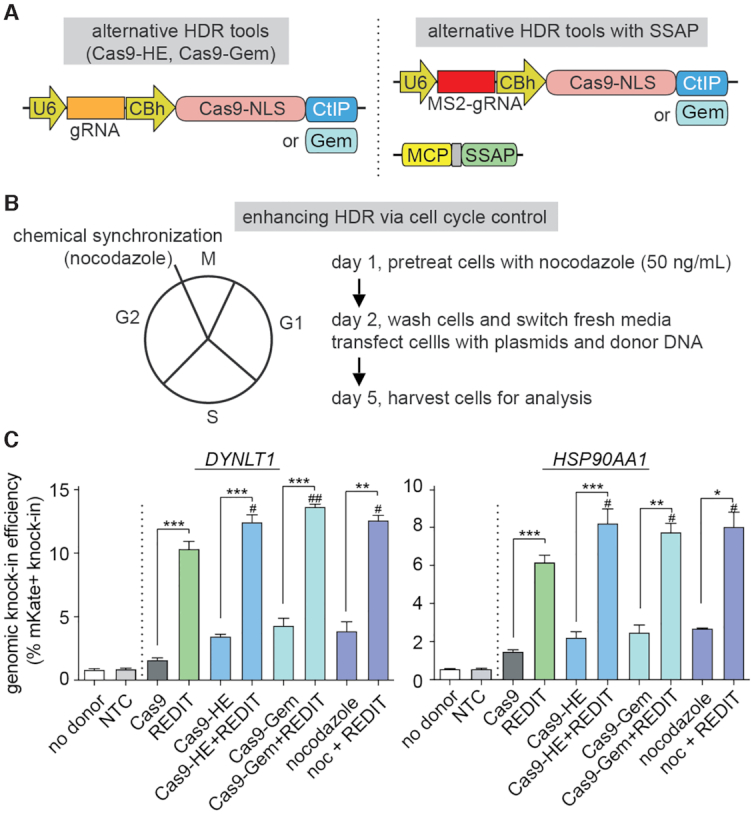
Comparing and combining the REDIT method with alternative HDR-enhancing gene-editing approaches. (A) Schematics showing alternative HDR-enhancing approaches via fusing functional domains, CtIP or Geminin (Gem), to Cas9 protein. (Left) Plasmids of individual benchmarks used in panel C; (Right) Plasmids combining the SSAP activity of REDIT with the alternative tools via MS2-aptamer recruitment. (B) Alternative small-molecule HDR-enhancing approach through cell cycle control. Nocodazole was used to synchronize cells at the G2/M boundary (Left), showing experiment procedures and timeline (Right). (C) Comparison of gene-editing efficiencies using REDIT and alternative HDR-enhancing tools, Cas9-HE (CtIP fusion), Cas9-Gem (Geminin fusion), and Nocodazole (noc), along with combination of REDIT with these methods (Cas9-HE/Cas9-Gem/noc+REDIT). Donor DNAs have 200 + 400 bp (DYNLT1) or 200 + 200bp (HSP90AA1) of HAs. All assays performed with no donor, NTC and Cas9 (no enhancement) controls. #P < 0.05, compared to REDIT; ##P < 0.01, compared to REDIT.
Characterization of design guidelines, capacity and specificity of the REDIT method
To consider REDIT as a knock-in editing method for future applications, we investigated the template design guideline, editing capacity, and specificity of REDIT. We performed three sets of experiments to measure: (i) activity of the REDIT method with different template designs, (ii) efficiencies of short- and long-sequence editing using this tool and (iii) specificity of this method through examining indels at on/off-target sites. Firstly, we quantified the effect of template HA lengths on the editing efficiency of REDIT when using the canonical HDR donor bearing HAs of at least 100 bp on each side (Figure 4A, left). We observed higher HDR rates for both Cas9 and RecT groups with increasing HA lengths, and REDIT effectively stimulated HDR over Cas9 using HA lengths as short as ∼100bp each side (Figure 4A; Supplementary sequences). When supplied with a longer template bearing 600–800 bp total HA, RecT achieved over 10% HDR efficiencies for kb-scale knock-in without selection, significantly higher than the 2–3% efficiency when only using Cas9 (Figure 4A, left). Recent reports identified that using donor DNAs with shorter HAs (usually between 10 and 50 bp) could significantly stimulate knock-in efficiencies thanks to the high repair activities from the Microhomology-mediated end joining (MMEJ) pathway (75–80). We thus tested the knock-in efficiencies of SSAP-based method compared with Cas9, using donor DNA with 0bp (NHEJ-based), 10bp or 50bp (MMEJ-based) HAs. Our results demonstrated that short-HA donors leveraging MMEJ mechanisms yielded higher editing efficiencies compared with HDR donors (Figure 4A, right). At the same time, REDIT tool was able to enhance the knock-in efficiencies as long as there is HA present (no effect for the 0bp NHEJ donor) (Figure 4A, right). This effect is particularly significant with the 10 bp donors, which we chose for further characterization and comparison with the HDR donors.
Figure 4.

Test template design guideline, junction precision, and capacity of REDIT gene-editing methods. (A) Homology arm (HA) length test comparing different template designs of HDR donors (longer HAs) or NHEJ/MMEJ donors (zero/shorter HAs) using REDIT and Cas9 references. Top and bottom are two genomic loci tested using mKate knock-in assay. (B) Design of junction profiling assay through isolation of knock-in clones, followed by genomic PCR using primers (fwd, rev) binding outside donor to avoid template amplification. Paired Sanger sequencing of the PCR products reveal homologous and non-homologous edits at the 5′- and 3′- junctions. (C) Percentage of colonies with indicated junction profiles from the Sanger sequencing of knock-in clones as in panel B. Indels are insertions or deletions detected from sequencing of ∼48 colonies per condition per locus. Editing methods and donor DNA listed at the bottom (HA lengths indicated in bracket). (D) Knock-in efficiencies using a 2-kb cassette to insert dual-GFP/mKate tags to validate REDIT methods with Cas9 references. HA lengths of donor DNAs indicated at the bottom.
Next, we sought to understand if the enhanced editing efficiencies using the two top template designs (HDR and MMEJ donors) could lead to better yield for precision knock-in, as defined by indel-free insertion events (Figure 4B). To this end, we clonally isolated the knock-in cells, then amplified the target genomic region using primers binding completely outside of the donor DNAs for colony Sanger sequencing. Our junction sequencing analysis (∼48 colonies per gene per condition) revealed varying degrees of indels at the 5′- and 3′- knock-in junctions, including at single or both junctions (Figure 4C). Overall, HDR donors have better precision than MMEJ donors, and REDIT could modestly improve the knock-in yield compared with Cas9 references, though junction indels are still observed (Figure 4C). Thus, for scenarios where correct knock-in product yield is critical, REDIT with PCR-based templates are beneficial, whereas the use of MMEJ donor DNA is better if maximizing knock-in efficiencies is a major consideration.
Furthermore, we sought to compare the efficiencies of REDIT and Cas9 when making different lengths of editing to gauge their editing capacity. For longer edits, we used 2-kb knock-in cassettes (Figure 4D), and for shorter edits with single-stranded oligo donors (ssODN) (Supplementary Figure S6A). When we increased the knock-in sequence length to ∼2-kb using a dual-mKate/GFP template, REDIT maintained its HDR-promoting activity compared with Cas9 across endogenous targets tested (Figure 4D). For ssODN tests, at two well-established loci EMX1 and VEGFA, we used REDIT and Cas9 to introduce 12–16-bp exogenous sequences (Supplementary sequences). As ssODN templates are short (<100 bp HAs on each side), we could leverage next-generation sequencing (NGS) to quantify the editing events. We observed comparable levels of indels between Cas9 and REDIT groups, and improved HDR efficiencies using REDIT (Supplementary Figure S6A), consistent with results using dsDNA donors.
The yield of knock-in experiments not only depends on on-target modification, but also will be influenced by off-target effects. Hence, to quantitatively understand off-target effects of REDIT versus the native Cas9, we used the EMX1 and VEGFA targets above, given these loci have well-established off-target sites (OTSs) from prior work (60–62). We profiled editing events at potential OTSs using two complementary approaches. We initially measured indels at known OTSs for EMX1 and VEGFA gRNAs (60,61), demonstrating that REDIT generated similar indel levels as Cas9 (Supplementary Figure S6B). Seeking a more thorough investigation of genome-wide off-target effects, we applied the unbiased GUIDE-seq analyses (62). Across both EMX1 and VEGFA gRNAs, we identified comparable numbers and intensities of OTSs between REDIT and Cas9 groups (Supplementary Figure S7). Thus, SSAP-based REDIT method could boost precision knock-in editing with comparable specificity and off-target profiles as the native Cas9.
Developing nicking-based REDIT method with high efficiency and improved genome-wide specificity
The precision and specificity data above pointed to a remaining challenge for this method due to on/off-target errors. Hence, we sought to mitigate this issue. As DSB-induced indels from NHEJ/MMEJ repair are the major source of such unwanted events (81,82), we looked into nicking Cas9 enzymes that do not generate DSBs (69,83,84). Based on prior biochemical and biophysical studies of RecT-like SSAPs (53,85), we reasoned that RecT may not rely on DSB thanks to its unusual ATP-independent SSAP activity: as one of the DNA strands is released via nicking activity, RecT’s high affinity for ss/dsDNA could allow its attachment to genomic and donor DNAs and then promote their exchange (86–88). Thus, to alleviate unwanted DNA damages and editing errors, we designed REDIT with Cas9 nickases (Cas9n-D10A). Cas9 nickases have been shown to mitigate off-target effects but reduced HDR efficiencies (88). Surprisingly, REDIT using Cas9n-D10A consistently enabled higher editing rates than Cas9 nickases, and also compared favorably against wild-type Cas9 (Figure 5A and B). For both single-nicking (Cas9n) and double-nick (Cas9dn) designs, REDIT tools raised HDR up to 5-fold versus the respective Cas9n and Cas9dn references, achieving up to 10% efficiency for kb-scale knock-in (Figure 5B). We termed the two nicking variants as REDITn and REDITdn, respectively, and focused later characterization on the more efficient REDITdn variant.
Figure 5.

Developing nicking variants of REDIT method as an efficient nickase-mediated gene-editing tool. (A, B) Left side showing schematics of the respective gene-editing approaches using wild-type Cas9 (wtCas9) or nickase Cas9 (Cas9n-D10A). The SSAP-based REDIT methods are separated into REDIT (wtCas9), REDITn (single-nicking Cas9n), and REDITdn (double-nicking Cas9dn). Right side showing the knock-in efficiencies of each tool at three genomic targets, compared with corresponding Cas9 references. Donor DNAs have 200 + 400 bp (DYNLT1) or 200+200bp (HSP90AA1, ACTB) of HAs. All assays performed with non-targeting controls (NTC).
Similar to the previous tests for REDIT, we used different donor DNAs to understand the template design guidelines for REDITdn. Consistent with REDIT, the REDITdn tool maintained its ability to enhance knock-in efficiencies when using HDR donors (≥100 bp HAs), up to 6-fold compared to Cas9dn (Supplementary Figure S8A). And as expected for a nickase-based tool, MMEJ donors were not able to yield higher efficiencies than HDR donors for REDITdn, but retained the general trend that longer HAs supported better performance using REDITdn. We next used ssODN as previously to characterize the precision of REDITdn with deep sequencing. Targeted NGS verified that REDITdn could promote HDR and reduced indel formation (Supplementary Figure S8B-C). As a more definitive test, we also examined individual knock-in events via Sanger sequencing of clonal knock-in cells, as previously done for the wild-type Cas9 and REDIT. The results of junction profiling indicated that REDITdn could improve the yield of knock-in, with more intact editing products compared with Cas9dn references (Figure 6A).
Figure 6.

Characterizing the specificity of nickase-based REDITdn gene-editing tool. (A) Percentage of colonies with indicated junction profiles from the Sanger sequencing of knock-in clones. Indels are insertions or deletions detected from sequencing of ∼48 colonies per condition per locus (as in Figure 4B). (B) Genomic-wide off-target site (OTS) counts via GUIDE-seq in HEK293T cells comparing REDIT and Cas9, REDITdn with Cas9dn, showing reduced off-target effects when using REDITdn. (C) Schematic showing the design, procedures, and analysis steps for GIS-seq to measure genome-wide insertion sites of the knock-in cassettes. High-molecular-weight (HMW) genomic DNA purification was needed to remove potential contamination from donor DNAs. Donor DNAs had 200 bp HAs each side. (D) Representative GIS-seq results showing plus/minus reads at on-target locus DYNLT1. The expected 2A-mKate knock-in site before the stop codon of the last exon are the center of the trimmed reads (reads clipped to remove 2A-mKate cassette). The template mutations help to avoid gRNA targeting and distinguish genomic and edited reads are labeled. (E) Summary of top GIS-seq insertion sites comparing Cas9dn and REDITdn groups, showing the expected on-target insertion site (highlighted) and reduced number of identified off-target insertion sites when using REDITdn. (Left) DYNLT1 and (Right) ACTB loci with MLE calculated from the distribution of filtered and trimmed GIS-seq reads.
Given single-strand nicks may still lead to off-target effects (81,82,89), we further performed GUIDE-seq to measure the genome-wide OTSs of REDIT and REDITdn, benchmarked against Cas9 and Cas9dn. With DYNLT1 gRNAs, REDITdn had only one detected off-target locus, a noticeable reduction of OTS counts from the 11 sites for Cas9 and 5 for Cas9dn (Figure 6B, Supplementary Figure S9). Specifically, a predicted OTS located at KIF6 was highly enriched in the Cas9 and REDIT groups, but not detected using REDITdn (Supplementary Figure S9).
As we observed significant reductions of indel formation with REDITdn, we wondered whether this method may improve editing precision by reducing random donor insertion, another DSB-related error during gene-editing. This is particularly important for genomic knock-in experiments, where imperfect knock-in could lead to significant concerns when the downstream applications are sensitive to confounding editing events, such as mechanistic investigation and therapeutic gene insertion. To this end, we profiled knock-in accuracy via mapping genome-wide insertion sites when using REDITdn compared with Cas9dn references. We adapted concepts from GUIDE-seq (62,63), LAM-PCR (64) and TLA (65) (detailed in the Methods section) to develop a NGS-based assay to identify genome-wide insertion sites (GIS), or GIS-seq (Figure 6C). Using GIS-seq, we obtained NGS read clusters/peaks representing knock-in insertion sites (Figure 6D, showing representative reads from the on-target site). We then applied GIS-seq to DYNLT1 and ACTB loci to measure the knock-in accuracy of Cas9dn and REDITdn. Sequencing results indicated that, when considering sites with high confidence based on maximum likelihood estimation, REDITdn had less off-target insertion sites identified compared with Cas9dn (Figure 6E). Together, our clonal Sanger sequencing of knock-in junctions (Figure 6A), GUIDE-seq analysis (Figure 6B), and GIS-seq results (Figure 6C–E) indicated that REDITdn is an efficient method with the ability to insert kilobase-length sequences with less unwanted editing events.
Understanding the performance of REDIT method under repair pathway inhibition and in non-transformed human stem cells
Most long-sequence editing methods depend on endogenous repair pathways following DNA cleavage and DSB formation, whereas REDITdn facilitated HDR via Cas9 nicking. This suggests REDIT may be less dependent on the HDR branch of mammalian DNA repair pathways. To investigate this, we leveraged specific inhibitors to block RAD51, an ATP-dependent homology-search and strand-invasion enzyme and the RecA-like recombination protein important for homology-directed repair (90,91). We measured the sensitivity of REDIT’s ability to promote HDR in the presence or absence of two distinctive pharmacological inhibitors of RAD51, B02 and RI1 (Figure 7A) (90,91). As expected, for Cas9-based editing, RAD51 inhibition significantly lowered HDR efficiencies (Figure 7B, C, Cas9 and Cas9dn groups). Intriguingly, RAD51 inhibition decreased REDIT and REDITdn efficiencies only moderately, as both REDIT/REDITdn methods maintained significantly higher knock-in efficiencies compared with Cas9/Cas9dn under RAD51 inhibition (Figure 7B-C). These observations led us to a hypothetical REDIT model where REDIT acts alongside the endogenous HDR: activated by Cas9 cutting or nicking (12,92), REDIT increases overall knock-in efficiencies, with RecT acts independently from RAD51 to promote HDR via its SSAP activity (53). Additional mechanistic studies using genetic/biochemical assays are required to understand and validate this model, and could provide insights for further optimizing the REDIT methods.
Figure 7.

Probing the dependence of REDIT gene-editing on endogenous DNA repair and applying REDIT methods for human stem cell engineering. (A) model showing the editing process and major repair pathways involved when using REDIT or Cas9 for gene-editing, the HDR pathway are highlighted for chemical perturbation (inhibition of RAD51). Donor DNAs with 200 + 200 bp HAs are used for all inhibitor experiments. (B, C) Relative knock-in efficiency of REDIT tools compared with Cas9 reference treated with RAD51 inhibitor B02 and RI-1, or vehicle-treated. Panel B is for the wtCas9-based REDIT and Cas9, and panel C is for Cas9 nickase-based REDITdn and Cas9dn. All conditions were measured with 1-kb knock-in assay at two genomic loci (DYNLT1 and HSP90AA1). (D) Knock-in efficiencies in hESCs (H9) using REDIT and REDITdn tested across three genomic loci, compared with corresponding Cas9 and Cas9dn references. (E, F) Original flow cytometry plots of mKate knock-in results in hESCs using REDIT, REDITdn with Cas9, Cas9dn, and NTC controls. Donor DNAs in the hESC experiments have 200 + 200 bp HAs across all loci tested.
Finally, to validate REDIT in different contexts, we applied REDIT tools in human embryonic stem cells (hESCs) to test their ability to engineer long sequences in non-transformed human cells (44,72,88). We observed robust stimulation of HDR across all three genomic sites (HSP90AA1, ACTB, OCT4/POU5F1) using REDIT and REDITdn (Figure 7D-E). Of note, REDIT and REDITdn editing used donor DNAs with 200-bp HAs on each side, and achieved up to over 5% efficiency for kb-scale gene-editing without selection (Figure 7D, Supplementary Figure S10), compared with ∼1% efficiency using non-REDIT methods (88). Additionally, we also validated that REDIT methods could improve knock-in efficiencies in A549 (lung-derived), HepG2 (liver-derived), and HeLa (cervix-derived) cells, demonstrating up to ∼15% kb-scale genomic knock-in without selection (Supplementary Figure S11). This improvement was up to 4-fold higher than the Cas9 groups, supporting the potential of using REDIT methods in different cell types.
DISCUSSION
REDIT methods harmonize the RNA-guided programmability of CRISPR genome-targeting with the SSAP activity of phage RecT (46,47,49,50,53,66,68). The nicking REDITdn tool enables long-sequence knock-in with significantly lower unwanted indels or random template insertions, as it will not induce significant levels of DSB formation and the ensuing DNA damage responses (10,12). This feature may provide advantages in research and therapeutic applications. For example, REDIT would be suitable for making large knock-in when efficiency is limited, or when minimizing undesirable errors is a prime concern, or for genome engineering in vivo where selection methods are not readily available to remove unwanted editing events (93). For a preliminary survey of its potential, we validated that REDIT is compatible with Staphylococcus aureus Cas9 (SaCas9) (94), a compact CRISPR system suitable for viral delivery (Supplementary Figure S12). This offered further support to the applicability of RecT-like SSAP for gene-editing. As recent work has provided novel Cas9 enzymes with enhanced activity and specificity (8,82,89,95), we expect that REDIT could be combined with them conveniently. And, as shown in the combination with Cas9-HE, Cas9-Gem, and cell cycle control methods (Figure 3), additional methods such as template recruitment, long single-stranded donor, and enhanced delivery may allow REDIT to achieve better absolute efficiencies beyond this work.
At the same time, REDITdn will be particularly relevant for improving the yield and interpretability of gene-editing experiments given the improved on-target precision and reduced off-target effects (Figure 6) (81,82). This SSAP-inspired gene-editing tool thus offers a new and complementary approach to existing CRISPR toolkits (1,3–7,9). Finally, while we provided some initial evidence that REDIT tools are at least partially in parallel with the endogenous HDR repair pathway, further studies on RecT-like SSAP enzyme in mammalian cell contexts are needed to fully understand the mechanisms of this process. It will be exciting to use chemical or genetic approaches to probe the nicking-mediated gene-editing facilitated by SSAP. Additionally, we expect SSAP HDR-enhancing activity may also facilitate other gene-editing tools, not limited to Cas9-based but also for ZFN and TALEN tools, awaiting future confirmation. This could open broader opportunities when Cas9 methods are limited.
DATA AVAILABILITY
All plasmids have been deposited to Addgene (with ID numbers 164802–164807) for open, free distribution and will be released upon publication. All next-generation sequencing data, including data from the targeted endogenous genome loci profiling and on/off-target analysis, are deposited to the NCBI Sequence Read Archive database with accession code PRJNA633116, PRJNA666273, PRJNA666282. Customized scripts for data analysis are deposited and available at Github under Cong Lab (https://github.com/cong-lab).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the entire Le Cong and Michael Cleary laboratory for their support, Dr Ravi Dinesh and Dr Mengdi Wang for comments on the manuscript, Feng Pan, Xin Yang, Li Zhu for generous help. We are grateful to the following scientists: pU6-(BbsI)_CBh-Cas9-T2A-BFP was a gift from Dr Ralf Kühn (Addgene # 64323). Cas9-nickase was a gift from Dr Keith Joung (Addgene # 53372). pCas9-CtIP and pCas9-Geminin was a gift from Jean-Paul Concordet (Addgene plasmid, #109402, #109403).
Contributor Information
Chengkun Wang, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA; Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA.
Jason K W Cheng, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA; Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA.
Qianhe Zhang, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA; Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA.
Nicholas W Hughes, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA; Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA.
Qiong Xia, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA; Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA.
Monte M Winslow, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA; Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA.
Le Cong, Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA; Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA; Wu Tsai Neuroscience Institute, Stanford University, Stanford, CA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [1R35HG011316 to L.C.]; Donald and Delia Baxter Foundation Faculty Scholar award; the computational work is supported by NIH [1S10OD023452 to Stanford Genomics Cluster (SCG)]. Funding for open access charge: Institutional funds from Stanford University.
Conflict of interest statement. Stanford University has filed a patent application based on this work. The authors will make the reagents openly available to the academic community through Addgene.
REFERENCES
- 1. Gaj T., Gersbach C.A., Barbas C.F.. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013; 31:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carroll D. Genome engineering with targetable nucleases. Annu. Rev. Biochem. 2014; 83:409–439. [DOI] [PubMed] [Google Scholar]
- 3. Doudna J.A., Charpentier E.. The new frontier of genome engineering with CRISPR–Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 4. Hsu P.D., Lander E.S., Zhang F.. Development and applications of CRISPR–Cas9 for genome engineering. Cell. 2014; 157:1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim H., Kim J.-S.. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014; 15:321–334. [DOI] [PubMed] [Google Scholar]
- 6. Sander J.D., Joung J.K.. CRISPR–Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014; 32:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang W., Marraffini L.A.. CRISPR–Cas: new tools for genetic manipulations from bacterial immunity systems. Annu. Rev. Microbiol. 2015; 69:209–228. [DOI] [PubMed] [Google Scholar]
- 8. Pickar-Oliver A., Gersbach C.A.. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019; 20:490–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anzalone A.V., Koblan L.W., Liu D.R.. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020; 38:824–844. [DOI] [PubMed] [Google Scholar]
- 10. Urnov F.D., Rebar E.J., Holmes M.C., Zhang H.S., Gregory P.D.. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010; 11:636–646. [DOI] [PubMed] [Google Scholar]
- 11. Wang D., Zhang F., Gao G.. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 2020; 181:136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yeh C.D., Richardson C.D., Corn J.E.. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019; 21:1468–1478. [DOI] [PubMed] [Google Scholar]
- 13. Pawelczak K.S., Gavande N.S., VanderVere-Carozza P.S., Turchi J.J.. Modulating DNA repair pathways to improve precision genome Engineering. ACS Chem. Biol. 2018; 13:389–396. [DOI] [PubMed] [Google Scholar]
- 14. Urnov F.D., Miller J.C., Lee Y.-L., Beausejour C.M., Rock J.M., Augustus S., Jamieson A.C., Porteus M.H., Gregory P.D., Holmes M.C.. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005; 435:646–651. [DOI] [PubMed] [Google Scholar]
- 15. Lin S., Staahl B.T., Alla R.K., Doudna J.A.. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014; 3:e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gutschner T., Haemmerle M., Genovese G., Draetta G.F., Chin L.. Post-translational regulation of Cas9 during G1 enhances Homology-Directed repair. Cell Rep. 2016; 14:1555–1566. [DOI] [PubMed] [Google Scholar]
- 17. Howden S.E., McColl B., Glaser A., Vadolas J., Petrou S., Little M.H., Elefanty A.G., Stanley E.G.. A Cas9 variant for efficient generation of Indel-Free knockin or gene-corrected human pluripotent stem cells. Stem Cell Rep. 2016; 7:508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bozas A., Beumer K.J., Trautman J.K., Carroll D.. Genetic analysis of zinc-finger nuclease-induced gene targeting in Drosophila. Genetics. 2009; 182:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beumer K.J., Trautman J.K., Bozas A., Liu J.-L., Rutter J., Gall J.G., Carroll D.. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:19821–19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morton J., Davis M.W., Jorgensen E.M., Carroll D.. Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:16370–16375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R.. Increasing the efficiency of homology-directed repair for CRISPR–Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015; 33:543–548. [DOI] [PubMed] [Google Scholar]
- 22. Maruyama T., Dougan S.K., Truttmann M.C., Bilate A.M., Ingram J.R., Ploegh H.L.. Increasing the efficiency of precise genome editing with CRISPR–Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015; 33:538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Canny M.D., Moatti N., Wan L.C.K., Fradet-Turcotte A., Krasner D., Mateos-Gomez P.A., Zimmermann M., Orthwein A., Juang Y.-C., Zhang W.et al.. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR–Cas9 genome-editing efficiency. Nat. Biotechnol. 2018; 36:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robert F., Barbeau M., Éthier S., Dostie J., Pelletier J.. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015; 7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nambiar T.S., Billon P., Diedenhofen G., Hayward S.B., Taglialatela A., Cai K., Huang J.-W., Leuzzi G., Cuella-Martin R., Palacios A.et al.. Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant. Nat. Commun. 2019; 10:3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song J., Yang D., Xu J., Zhu T., Chen Y.E., Zhang J.. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016; 7:10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paulsen B.S., Mandal P.K., Frock R.L., Boyraz B., Yadav R., Upadhyayula S., Gutierrez-Martinez P., Ebina W., Fasth A., Kirchhausen T.et al.. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR–Cas9 genome editing. Nat. Biomed. Eng. 2017; 1:878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P.. CRISPR–Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019; 10:2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Charpentier M., Khedher A.H.Y., Menoret S., Brion A., Lamribet K., Dardillac E., Boix C., Perrouault L., Tesson L., Geny S.et al.. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018; 9:1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tran N.-T., Bashir S., Li X., Rossius J., Chu V.T., Rajewsky K., Kühn R.. Enhancement of precise gene editing by the association of Cas9 with homologous recombination factors. Front. Genet. 2019; 10:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rees H.A., Yeh W.-H., Liu D.R.. Development of hRad51–Cas9 nickase fusions that mediate HDR without double-stranded breaks. Nat. Commun. 2019; 10:2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carlson-Stevermer J., Abdeen A.A., Kohlenberg L., Goedland M., Molugu K., Lou M., Saha K.. Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Nat. Commun. 2017; 8:1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gu B., Posfai E., Rossant J.. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018; 36:632–637. [DOI] [PubMed] [Google Scholar]
- 34. Savic N., Ringnalda F.C., Lindsay H., Berk C., Bargsten K., Li Y., Neri D., Robinson M.D., Ciaudo C., Hall J.et al.. Covalent linkage of the DNA repair template to the CRISPR–Cas9 nuclease enhances homology-directed repair. eLife. 2018; 7:e33761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Z., Wang Y., Wang S., Gorzalski A.J., McSwiggin H., Yu T., Castaneda-Garcia K., Prince B., Wang H., Zheng H.et al.. Efficient genome editing by CRISPR-Mb3Cas12a in mice. J. Cell Sci. 2020; 133:jcs240705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ling X., Xie B., Gao X., Chang L., Zheng W., Chen H., Huang Y., Tan L., Li M., Liu T.. Improving the efficiency of precise genome editing with site-specific Cas9-oligonucleotide conjugates. Sci. Adv. 2020; 6:eaaz0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song F., Stieger K.. Optimizing the DNA donor template for homology-directed repair of double-strand breaks. Mol. Ther. - Nucleic Acids. 2017; 7:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Renaud J.-B., Boix C., Charpentier M., De Cian A., Cochennec J., Duvernois-Berthet E., Perrouault L., Tesson L., Edouard J., Thinard R.et al.. Improved genome editing efficiency and flexibility using modified oligonucleotides with TALEN and CRISPR–Cas9 nucleases. Cell Rep. 2016; 14:2263–2272. [DOI] [PubMed] [Google Scholar]
- 39. Roth T.L., Li P.J., Blaeschke F., Nies J.F., Apathy R., Mowery C., Yu R., Nguyen M.L.T., Lee Y., Truong A.et al.. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell. 2020; 181:728–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B.et al.. Chemically modified guide RNAs enhance CRISPR–Cas genome editing in human primary cells. Nat. Biotechnol. 2015; 33:985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DiNapoli S.E., Martinez-McFaline R., Gribbin C.K., Wrighton P.J., Balgobin C.A., Nelson I., Leonard A., Maskin C.R., Shwartz A., Quenzer E.D.et al.. Synthetic CRISPR/Cas9 reagents facilitate genome editing and homology directed repair. Nucleic Acids Res. 2020; 48:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Srivastava M., Nambiar M., Sharma S., Karki S.S., Goldsmith G., Hegde M., Kumar S., Pandey M., Singh R.K., Ray P.et al.. An inhibitor of nonhomologous end-joining abrogates double-Strand break repair and impedes cancer progression. Cell. 2012; 151:1474–1487. [DOI] [PubMed] [Google Scholar]
- 43. Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J.. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018; 24:927–930. [DOI] [PubMed] [Google Scholar]
- 44. Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C.et al.. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018; 24:939–946. [DOI] [PubMed] [Google Scholar]
- 45. Sander J.D., Joung J.K.. CRISPR–Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014; 32:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pines G., Freed E.F., Winkler J.D., Gill R.T.. Bacterial recombineering: genome engineering via phage-based homologous recombination. ACS Synth. Biol. 2015; 4:1176–1185. [DOI] [PubMed] [Google Scholar]
- 47. Court D.L., Sawitzke J.A., Thomason L.C.. Genetic engineering using homologous recombination. Annu. Rev. Genet. 2002; 36:361–388. [DOI] [PubMed] [Google Scholar]
- 48. Esvelt K.M., Wang H.H.. Genome-scale engineering for systems and synthetic biology. Mol. Syst. Biol. 2013; 9:641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wannier T.M., Nyerges A., Kuchwara H.M., Czikkely M., Balogh D., Filsinger G.T., Borders N.C., Gregg C.J., Lajoie M.J., Rios X.et al.. Improved bacterial recombineering by parallelized protein discovery. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:13689–13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Iyer L.M., Koonin E.V., Aravind L.. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52. BMC Genomics. 2002; 3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kmiec E., Holloman W.K.. Beta protein of bacteriophage lambda promotes renaturation of DNA. J. Biol. Chem. 1981; 256:12636–12639. [PubMed] [Google Scholar]
- 52. Muyrers J.P., Zhang Y., Buchholz F., Stewart A.F.. RecE/RecT and Redalpha/Redbeta initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev. 2000; 14:1971–1982. [PMC free article] [PubMed] [Google Scholar]
- 53. Noirot P., Kolodner R.D.. DNA strand invasion promoted by Escherichia coli RecT protein. J. Biol. Chem. 1998; 273:12274–12280. [DOI] [PubMed] [Google Scholar]
- 54. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A.et al.. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Altschul S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinforma. Oxf. Engl. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Price M.N., Dehal P.S., Arkin A.P.. FastTree 2 – Approximately Maximum-Likelihood trees for large alignments. PLoS One. 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E.et al.. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019; 37:224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D.. High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat. Biotechnol. 2013; 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P.et al.. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR–Cas nucleases. Nat. Biotechnol. 2015; 33:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nobles C.L., Reddy S., Salas-McKee J., Liu X., June C.H., Melenhorst J.J., Davis M.M., Zhao Y., Bushman F.D.. iGUIDE: an improved pipeline for analyzing CRISPR cleavage specificity. Genome Biol. 2019; 20:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schmidt M., Schwarzwaelder K., Bartholomae C., Zaoui K., Ball C., Pilz I., Braun S., Glimm H., von Kalle C.. High-resolution insertion-site analysis by linear amplification–mediated PCR (LAM-PCR). Nat. Methods. 2007; 4:1051–1057. [DOI] [PubMed] [Google Scholar]
- 65. Cain-Hom C., Splinter E., van Min M., Simonis M., van de Heijning M., Martinez M., Asghari V., Cox J.C., Warming S.. Efficient mapping of transgene integration sites and local structural changes in Cre transgenic mice using targeted locus amplification. Nucleic Acids Res. 2017; 45:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Abbasi M.N., Fu J., Bian X., Wang H., Zhang Y., Li A.. Recombineering for genetic engineering of natural product biosynthetic pathways. Trends Biotechnol. 2020; 38:715–728. [DOI] [PubMed] [Google Scholar]
- 67. Mosberg J.A., Lajoie M.J., Church G.M.. Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics. 2010; 186:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lopes A., Amarir-Bouhram J., Faure G., Petit M.-A., Guerois R.. Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51 and Gp2.5 remote homologs. Nucleic Acids Res. 2010; 38:3952–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pinder J., Salsman J., Dellaire G.. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015; 43:9379–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mosiman V.L., Patterson B.K., Canterero L., Goolsby C.L.. Reducing cellular autofluorescence in flow cytometry: an in situ method. Cytometry. 1997; 30:151–156. [PubMed] [Google Scholar]
- 72. Zhu Z., Verma N., González F., Shi Z.-D., Huangfu D.. A CRISPR/Cas-mediated selection-free knockin strategy in human embryonic stem cells. Stem Cell Rep. 2015; 4:1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tanenbaum M.E., Gilbert L.A., Qi L.S., Weissman J.S., Vale R.D.. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014; 159:635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang J.-P., Li X.-L., Li G.-H., Chen W., Arakaki C., Botimer G.D., Baylink D., Zhang L., Wen W., Fu Y.-W.et al.. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017; 18:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nakade S., Tsubota T., Sakane Y., Kume S., Sakamoto N., Obara M., Daimon T., Sezutsu H., Yamamoto T., Sakuma T.et al.. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014; 5:5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Paix A., Folkmann A., Goldman D.H., Kulaga H., Grzelak M.J., Rasoloson D., Paidemarry S., Green R., Reed R.R., Seydoux G.. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E10745–E10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hisano Y., Sakuma T., Nakade S., Ohga R., Ota S., Okamoto H., Yamamoto T., Kawahara A.. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 2015; 5:8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sakuma T., Nakade S., Sakane Y., Suzuki K.-I.T., Yamamoto T.. MMEJ-assisted gene knock-in using TALENs and CRISPR–Cas9 with the PITCh systems. Nat. Protoc. 2016; 11:118–133. [DOI] [PubMed] [Google Scholar]
- 79. Kanca O., Zirin J., Garcia-Marques J., Knight S.M., Yang-Zhou D., Amador G., Chung H., Zuo Z., Ma L., He Y.et al.. An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. eLife. 2019; 8:e51539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tatiossian K.J., Clark R.D.E., Huang C., Thornton M.E., Grubbs B.H., Cannon P.M.. Rational selection of CRISPR–Cas9 guide RNAs for homology-directed genome editing. Mol. Ther. J. Am. Soc. Gene Ther. 2020; 29: 10.1016/j.ymthe.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. O’Geen H., Yu A.S., Segal D.J.. How specific is CRISPR/Cas9 really. Curr. Opin. Chem. Biol. 2015; 29:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tsai S.Q., Joung J.K.. Defining and improving the genome-wide specificities of CRISPR–Cas9 nucleases. Nat. Rev. Genet. 2016; 17:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A.et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thomason L.C., Costantino N., Court D.L.. Examining a DNA replication requirement for bacteriophage λ Red- and Rac Prophage RecET-Promoted recombination in Escherichia coli. mBio. 2016; 7:e01443-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A.. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014; 507:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Richardson C.D., Ray G.J., DeWitt M.A., Curie G.L., Corn J.E.. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR–Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016; 34:339–344. [DOI] [PubMed] [Google Scholar]
- 88. Ran F.A., Hsu P.D., Lin C.-Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y.et al.. Double nicking by RNA-Guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 154:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lin Y., Cradick T.J., Brown M.T., Deshmukh H., Ranjan P., Sarode N., Wile B.M., Vertino P.M., Stewart F.J., Bao G.. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014; 42:7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Huang F., Motlekar N.A., Burgwin C.M., Napper A.D., Diamond S.L., Mazin A.V.. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011; 6:628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Budke B., Logan H.L., Kalin J.H., Zelivianskaia A.S., Cameron McGuire W., Miller L.L., Stark J.M., Kozikowski A.P., Bishop D.K., Connell P.P.. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012; 40:7347–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jasin M., Haber J.E.. The democratization of gene editing: insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst.). 2016; 44:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M.. Gene therapy comes of age. Science. 2018; 359:eaan4672. [DOI] [PubMed] [Google Scholar]
- 94. Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S.et al.. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kim D., Luk K., Wolfe S.A., Kim J.-S.. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu. Rev. Biochem. 2019; 88:191–220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All plasmids have been deposited to Addgene (with ID numbers 164802–164807) for open, free distribution and will be released upon publication. All next-generation sequencing data, including data from the targeted endogenous genome loci profiling and on/off-target analysis, are deposited to the NCBI Sequence Read Archive database with accession code PRJNA633116, PRJNA666273, PRJNA666282. Customized scripts for data analysis are deposited and available at Github under Cong Lab (https://github.com/cong-lab).