

Abstract
Phages and other mobile genetic elements express anti-CRISPR proteins (Acrs) to protect their genomes from destruction by CRISPR–Cas systems. Acrs usually block the ability of CRISPR–Cas systems to bind or cleave their nucleic acid substrates. Here, we investigate an unusual Acr, AcrIF9, that induces a gain-of-function to a type I-F CRISPR–Cas (Csy) complex, causing it to bind strongly to DNA that lacks both a PAM sequence and sequence complementarity. We show that specific and non-specific dsDNA compete for the same site on the Csy:AcrIF9 complex with rapid exchange, but specific ssDNA appears to still bind through complementarity to the CRISPR RNA. Induction of non-specific DNA-binding is a shared property of diverse AcrIF9 homologues. Substitution of a conserved positively charged surface on AcrIF9 abrogated non-specific dsDNA-binding of the Csy:AcrIF9 complex, but specific dsDNA binding was maintained. AcrIF9 mutants with impaired non-specific dsDNA binding activity in vitro displayed a reduced ability to inhibit CRISPR–Cas activity in vivo. We conclude that misdirecting the CRISPR–Cas complex to bind non-specific DNA is a key component of the inhibitory mechanism of AcrIF9. This inhibitory mechanism is distinct from a previously characterized anti-CRISPR, AcrIF1, that sterically blocks DNA-binding, even though AcrIF1and AcrIF9 bind to the same site on the Csy complex.
INTRODUCTION
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins together represent an adaptive mechanism employed by many species of bacteria and archaea to destroy potentially harmful mobile genetic elements (MGEs), such as phages (1–3). CRISPR arrays are comprised of repeated DNA sequences interspersed with non-repeating DNA spacer sequences. Spacer DNA is often identical to MGE sequences. These arrays are transcribed and processed into CRISPR (cr)RNAs, comprised of one repeat and one spacer, that form complexes with Cas proteins. Using the spacer sequence as a guide, CRISPR–Cas complexes specifically bind DNA or RNA and mediate subsequent nucleolytic destruction of the targeted nucleic acid. CRISPR–Cas systems provide adaptive immunity against foreign DNA as segments of newly encountered MGEs can be incorporated into CRISPR arrays in the form of new spacers, providing defence against subsequent encounters with the same MGE.
CRISPR–Cas systems are tremendously diverse with 33 distinct subtypes distributed among 6 different types (2). Here, we focus on the type I-F system, which is found widely in Proteobacteria. The type I-F CRISPR–Cas complex, which is known as the Csy complex, comprises a 60 nucleotide (nt) crRNA with one molecule of Cas6f bound to the 3′-hairpin formed by the repeat sequence followed by 6 molecules of Cas7f bound to the 32 nt spacer (4,5). A complex of Cas5f and Cas8f are bound to another portion of the repeat sequence known as the handle, which lies at the 5′-end of the crRNA (6–8). Binding of dsDNA targets is initiated by Cas8f recognition of the protospacer adjacent motif (PAM) and subsequent separation of the DNA strands. Known as R-loop formation, this strand separation allows for hydrogen-bonding between the spacer region of the crRNA and the target strand of the DNA. Large conformation changes in the Csy complex occurring upon target DNA-binding lead to exposure of a Cas8f domain that recruits Cas3, the helicase-nuclease that mediates processive degradation of the targeted DNA (8).
The first five families of phage-encoded inhibitors of a CRISPR–Cas system, known as anti-CRISPR proteins (Acrs), were described in 2013. These Acrs blocked the activity of the type I-F system of Pseudomonas aeruginosa (Pae) (9). Since then, >90 families of Acrs have been described that act against many different types of CRISPR–Cas systems (10). Mechanistic and structural studies on Acrs have provided new insights into how CRISPR–Cas systems function and have illustrated the many fascinating ways by which small proteins can inhibit large protein-RNA complexes (11,12). In addition, a growing number of biotechnological applications for Acrs are being developed (13–15).
Structural and mechanistic investigations have been carried out on a number of Acrs specific to the I-F system (6–8,16–19). These studies have revealed four distinct mechanism of Acr action so far, raising the expectation that further investigation will reveal new modes of inhibition. To this end, we chose to investigate the AcrIF9 family, which is one of the largest and most diverse families of I-F Acrs (20). In a recent structural study on the Csy complex bound to AcrIF9 (Csy:F9), we and our collaborators found that this complex binds to dsDNA in a non-specific manner, requiring neither a PAM nor complementarity to the crRNA (21). Here, we have utilized a variety of biochemical approaches to characterize and understand this surprising property of the Csy:F9 complex. We have also used mutagenesis experiments to identify surface exposed positively charged residues in AcrIF9 and Cas8f that mediate the interaction with non-specific dsDNA. Our data highlight an important role for non-specific DNA-binding in the inhibitory mechanism of AcrIF9.
MATERIALS AND METHODS
In vivo assay for Acr activity
In vivo assays to detect Acr activity were carried out as originally described (9). pHERD30T (22) derived plasmids were used to express AcrIF9 homologues in P. aeruginosa strain UCBPP-PA14 (PA14), which possesses an active type I-F CRISPR–Cas system. Lysates of a CRISPR–Cas sensitive phage (DMS3m) or a CRISPR–Cas insensitive phage (DMS3) were spotted in ten-fold dilutions onto lawns of PA14 transformed with plasmids expressing Acrs of interest. A strain carrying pHERD30T was used as a negative control. Plates were incubated at 30°C overnight. Homologues to be tested were identified by PSI-BLAST (2 iterations) (23). The protein sequence alignment was constructed and analyzed using Jalview (24).
Expression and purification of Csy complex and Acrs
The P. aeruginosa Csy complex including crRNA was expressed from plasmids in Escherichia coli strain BL21(DE3) as previously described (5). Cas7f is tagged with 6xHis. To produce Csy:F9, the constructs expressing Csy complex and crRNA as stated above were co-expressed with pCDF-1b expressing untagged AcrIF9.
Cultures of E. coli BL21 (DE3) expressing the protein of interest were grown to an optical density (OD600) of 0.6 and then induced with 1 mM isopropyl-b-D-thiogalactoside (IPTG) for 16 h at 16 °C. Cells were collected by centrifugation at 7000 g for 15 min and resuspended in binding buffer (20 mM Tris pH 7.5, 200 mM NaCl, 5 mM imidazole, 1 mM tris (2-carboxyethyl)phosphine (TCEP)). The cells were lysed by sonication and the resulting lysates were centrifuged at 17 000 g for 25 min to remove cell debris. The supernatant was mixed with Ni-NTA beads and incubated for 1 hr at 4 °C. The lysates and the beads were then passed through a column, washed 5 times with wash buffer (20 mM Tris, pH 7.5, 200 mM NaCl, 30 mM imidazole, 5 mM β-mercaptoethanol) and then eluted in buffer containing 300 mM imidazole. Purified protein was dialysed into 20 mM Tris pH 7.5, 200 mM NaCl, 1 mM TCEP) overnight. Affinity-purified proteins were fractionated by size exclusion chromatography (SEC) using a GE Life Sciences Superdex 200 10/30 column. Fractions were collected in 1 ml volumes and monitored by optical density at 280 nm. Protein purity was assessed by visualization on Coomassie blue R250 stained SDS-PAGE gels.
Assessing Acr binding to the Csy complex
Purified 6xHis-tagged Csy complex (1000 nM) was bound to Ni-NTA beads and incubated with excess Acr (5000 nM) for 30 min at 4°C in binding buffer (20 mM Tris pH 7.5, 200 mM NaCl, 5 mM imidazole, 1 mM TCEP). Competitor Acr was added in equimolar concentration and incubated for 30 min at 4°C. Bound Csy complex and Acr were collected through centrifugation at 6000 × g for 2 min to remove unbound Acr. The reaction was then washed three times with wash buffer (20 mM Tris, pH 7.5, 200 mM NaCl, 30 mM imidazole, 5 mM β-mercaptoethanol) with a centrifugation step after each wash. The sample was then eluted in elution buffer (20 mM Tris, pH 7.5, 200 mM NaCl, 300 mM imidazole, 5 mM β-mercaptoethanol). The samples were visualized on Coomassie blue R250 stained SDS–PAGE gels. Each experiment was conducted at least three times and the same result was obtained each time. Single representation is shown in Supplementary Figure S2.
Site-directed mutagenesis
Complementary oligonucleotides comprising the codon to be mutated plus 20 nucleotides in both directions were synthesized by Eurofins Genomics. The entire plasmid template was then PCR amplified with the primers containing the mutations using Phusion DNA polymerase. Subsequently, the template was digested with DpnI and the PCR product was transformed into E. coli DH5α. Mutations were confirmed by DNA sequencing.
DNA binding assays
DNA molecules (sequences shown below) were synthesized (Eurofins Genomics) that contain 32 nucleotides that are either complementary (specific) to the crRNA in the Csy complex or scrambled (non-specific). The DNA was fluorescently labeled at the 5′ end with either 6-FAM or Cy5. To generate dsDNA, the labeled strand was mixed with an unlabeled complementary strand, heated to 100°C, and allowed to return slowly to room temperature. DNA binding reactions were conducted in a binding buffer (10 mM HEPES, pH 7.5, 1 mM MgCl2, 20 mM KCl, 1 mM TCEP, and 6% glycerol) at 37°C for 15 min. A DNA concentration of 100 nM was used in EMSA reactions with Csy or Csy:Acr complexes at 2000 nM. In competitive DNA binding experiments, the Csy complex, or Csy:F9 (1000 nM), were first incubated with 300 nM of DNA at 37°C for 15 min. Then the competitor DNA was added at increasing concentrations with the following ratios (1:1, 1:2, 1:4) and incubated at 37°C for another 15 min. For EMSA experiments with competing Acrs, the Csy complex was first incubated with ten-fold excess of one Acr for 1 hour at 4°C, and then equimolar amount of the competitor Acr was added and incubated under the same conditions. 100 nm DNA was then incubated with the resulting Acr-bound Csy complex at 37°C for 15 min. All EMSA reactions were resolved on native 4% or 6% polyacrylamide TBE gels. Gels were visualized with a Typhoon imager at 473 nm (6-FAM) and 635 nm (Cy5). Every EMSA experiment was carried out at least three times with reproducible results. Single representative gels are shown in figures.
For fluorescence polarization (FP) assays, Cy5 labeled DNA probes (4 nM) were incubated with purified Csy or Csy:F9 complexes at increasing concentrations (6.25,12.5, 25, 50,100, 200, 400, 800 nM) in a total volume of 50 ul in Greiner Bio-one 96 well black flat-bottom microplates. The samples were mixed with the assay buffer (20 mM HEPES pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.01% Triton X-100, 2 mM DTT, 0.1 mg/ml bovine gamma globulin) and incubated at 37°C for 30 min. The plate was then analyzed with a Tecan Microplate Reader Spark at 635 nM. The polarization signal was corrected to the reference (Cy5-DNA only) and the blank (assay buffer only). For competitive assays, the Csy or Csy:F9 complex (100 nM) was first incubated with Cy5-labeled DNA at 4 nM for 30 min at 37°C and then competed with increasing concentrations of unlabeled DNA (0, 0.5, 1, 2, 4, 8, 16, 32 nM) for 30 min at 37°C. All FP assays were performed at least three times. Average values are plotted with error bars representing standard deviation.
The sequences of the DNA used for DNA binding assays are shown below. Protospacer sequences complementary to the crRNA are in bold and underlined. PAMs are in red. In the 2× and 1× sequences we used a 24 nt protospacer instead of 32 nt in order to keep all dsDNA target sequences used at approximately the same length. We found that the Csy complex binds with similar affinity to a 24 nt protospacer.
| 50 bp DNASP target strand: |
| 5′:GAATGACCTACAGGTAGACGCGGACATCAAGCCCGCCGTGAAGGCATGTCAA |
| 50 bp DNANS: |
| 5′:GCGCACCTATTAACCGTTCGCAGAAACCAGTAGTAGTCCAAGCGACATGCAG |
| DNA2x target strand: |
| 5′:CGCGGACATCAAGCCCGCCGTGAAGGCATGTCCGCGGACATCAAGCCCGCCGTGAAGGCATGT |
| DNA1x target strand: |
| 5′:GTAGTAGTCCAACGGCATGTAATGACCTACGCGGACATCAAGCCCGCCGTGAAGGCATGT |
| 25 bp DNASP: |
| 5′:CAAGCCCGCCGTGAAGGCATGTCAA |
| 12 bp DNASP: |
| 5′:GCCGTGAAGGCA |
In vivo activity measurement for CsySba. The efficiency of transformation assay (EOT) was performed as described previously (25). CsySba was expressed in E. coli BL21-AI in the presence and absence of AcrIF9Vpa. A spacer targeting the ampicillin resistance cassette of pETDuet-1 was used to determine the EOT. EOT equals to the colony ratio between the colony count of the strain of interest and its corresponding Cas3 HD mutant strain, presented as percentages. Error bars represent the standard error of the mean, three replicates were quantified.
Bio-layer interferometry (BLI)
The conditions used in the BLI experiments were as described previously (26). AcrIF9Vpa and Csy complex were mixed at a molar ratio of 1:1 and incubated for 10 min at room temperature in BLI buffer (0.1 μM BSA and 0.01% Triton X-100). The Csy complex, Csy:F9, or AcrIF9Vpa alone, was tested against 100 nM of either dsDNASP or dsDNANS. Assays were performed in duplicate on the BLItz platform (FortéBio) using High Precision Streptavidin (SAX) Biosensors (FortéBio).
Phage plaquing assay
P. aeruginosa PA14 was transformed with a vector control or a construct expressing candidate Acr genes on pHERD30T. 150 μl of overnight culture and 10 μl of a lysate of phage DMS3m diluted 106-fold were mixed with soft LB agar and poured onto LB agar plates (MgSO4, 50 μg ml−1 gentamicin and 10 mM arabinose). The plates were grown overnight at 30°C. Individual plaques were counted.
Assay of phzM repression and growth curves
A crRNA was designed to target the Csy complex to the promoter region of the phzM gene to block the transcription of the gene and, thus, block pyocyanin production as described previously (16). DNA encoding the crRNA and the Acr of interest was cloned into P. aeruginosa expression vector pHERD30T and was used to transform WT PA14, or PA14 Δcas3, a strain lacking the Cas3 nuclease. The transformants were grown in LB overnight and subcultured into 5 ml King's A medium at a dilution of 1:100. The culture was grown at 37°C for 3 h to reach an OD of 0.6 then induced with 10 mM arabinose. The cells were then grown overnight at 37°C. Pyocyanin was extracted with an equal volume of chloroform then mixed with 2 ml of 0.2 M HCl. The resulting samples were quantitated by measuring absorbance at 520 nm.
Overnight cultures of WT PA14, or PA14 Δcas3 strains transformed with the crRNA-expression plasmids described above were diluted 1:100 in LB supplemented with 10 mM arabinose. 150 μl of each sample was then added to a 96-well plate and the optical density at 600 nm was measured every 30 min over the course of 6 hours.
RESULTS
AcrIF9 binds to Cas7f at a site overlapping with AcrIF1
Genes encoding four members of the AcrIF9 family found in strains of Vibrio parahaemolyticus (Vpa), Proteus penneri (Ppe), Aggregatibacter actinomycetemcomitans (Aac) and Xanthomonas fragariae (Xfr) were synthesized and expressed in Pae (Supplementary Figure S1A). All four homologs showed robust inhibition of the Pae type I-F CRISPR–Cas system (Supplementary Figure S1B). Expression of 6xHis-tagged versions of each homolog in E. coli followed by Ni-NTA purification showed that the homolog from Ppe was most suitable for further biochemical analysis due to it displaying the highest expression level and solubility. All subsequent studies were performed with this protein except where noted.
Two different recently solved cryo-EM structures of AcrIF9-bound Csy (Csy:F9) complex revealed a stoichiometry of two AcrIF9 monomers per complex (21,27). Remarkably, the binding sites of AcrIF9 overlap very closely with those of the previously characterized Acr, AcrIF1 (6,16). To confirm that AcrIF9 and AcrIF1 occupy overlapping sites on Cas7f, we conducted a competitive binding experiment. Untagged AcrIF9 was added to Csy complex containing 6xHis-tagged Cas7f that was pre-saturated with untagged AcrIF1. Purification of Acr-bound Csy complexes using Ni-NTA chromatography showed that the prior addition of AcrIF1 completely blocked the binding of AcrIF9 (Supplementary Figure S2). By contrast, prior addition of AcrIF9 or AcrIF1 to the Csy complex did not impede the binding of untagged AcrIF2 (Supplementary Figure S2), which binds to Cas8f (6,16). These results confirm that AcrIF9 and AcrIF1 bind to the same binding site on Cas7f.
AcrIF9 abolishes the DNA binding specificity of the Csy complex
Since AcrIF9 and AcrIF1 share binding sites on the Csy complex, it was expected that they would have the same effect on the Csy complex. AcrIF1 sterically blocks the hybridization of target DNA to the crRNA, thus strongly inhibiting the binding of both dsDNA and ssDNA (7). However, previous work indicated that AcrIF9 induced the Csy complex to bind DNA non-specifically, a property not observed for AcrIF1 (21). To investigate this surprising aspect of AcrIF9, Electrophoretic Mobility Shift Assays (EMSAs) were used to assess the effect of AcrIF9 on the DNA-binding activity of the Csy complex. As shown previously, binding of the Csy complex to a 50 bp dsDNA fragment containing a sequence matching the crRNA spacer and an appropriate Protospacer Adjacent Motif (PAM) causes a large change in the mobility of the fragment in a polyacrylamide gel ((16), Figure 1A). Addition of AcrIF9 to the Csy complex did not abrogate dsDNA binding, and the shifted band displayed a slower mobility and was smeared (Figure 1A). By contrast, a Csy complex bound to AcrIF1 (Csy:F1) displayed no dsDNA-binding ability, as was previously shown (16). When specific ssDNA was used as a binding substrate, the Csy:F9 complex bound as well as the Csy complex alone, but smearing and slower mobility was not observed. The Csy:F1 complex that displayed greatly reduced ssDNA binding (Figure 1A). Thus, AcrIF9 and AcrIF1 elicit very different effects on the DNA-binding activity of the Csy complex despite binding at overlapping sites on Cas7f.
Figure 1.

The Csy:F9 complex binds to DNA non-specifically. (A) The binding of Csy, Csy:F9 and Csy:F1 complexes to dsDNASP and ssDNASP was assessed on EMSA gels. (B) The same experiment shown in (A) was performed with dsDNANS and ssDNANS. (C) Increasing concentrations of Csy or Csy:F9 complex were added to constant concentrations of dsDNASP (left two panels) or dsDNANS (right panel). These experiments were performed using 50 bp DNA fragments 5′-labeled with Cyanine 5 (Cy5).
The smearing of the dsDNA band bound to Csy:F9 suggested that this complex may not recognize a specific site on the DNA. To test this idea, we performed a similar EMSA experiment using a 50 bp non-specific dsDNA sequence (dsDNANS) that possesses the same base composition as the specific target DNA, but the sequence is randomized. The PAM sequence is also absent. While the Csy complex on its own displayed no affinity for dsDNANS, Csy:F9 displayed robust binding activity (Figure 1B). Notably, binding of Csy:F9 to dsDNANS resulted in a similar supershifted and smeared band as was seen in the experiments with the specific target DNA (dsDNASP). Csy:F9 also bound to non-specific ssDNA (ssDNANS). AcrIF9 alone showed no binding to dsDNA (Supplementary Figure S3A). Titration experiments where concentrations of unbound Csy or Csy:F9 were incrementally increased indicated that the affinity of Csy:F9 for dsDNANS was similar to its affinity for dsDNASP and to the affinity of unbound Csy for dsDNASP. In each of these cases, the DNA was mostly bound at a complex concentration of 1000 nM (Figure 1C). In some gels the Csy:F9:DNA complexes appear to be stuck in the wells, which could result from aggregation of the Csy complex. To ensure that this was not the case, we ran EMSA experiments on lower percentage acrylamide gels and found that the Csy:F9:DNA complexes did fully migrate into the gels (Supplementary Figure S3B).
It should be noted that the EMSA results involving AcrIF9 presented here differ in appearance from those previously published (21). Here, we detected a much more pronounced supershift when Csy:F9 was bound to dsDNA, and we did not observe DNA binding inhibition at low concentrations of AcrIF9 as was previously observed. The previous assays used a constant concentration of both DNA and Csy complex, only increasing the concentration of AcrIF9. In the current experiments, the Csy:F9 complex was pre-formed through co-expression and purified as a complex. Increasing concentrations of the pre-formed complex were added to the reactions. The condition under which DNA-binding appeared to be inhibited previously utilized a lower Csy concentration and a substoichiometric concentration of AcrIF9. Despite these minor differences, the previous and current results are consistent in showing strong binding of dsDNANS by the Csy:F9 complex.
Multiple Csy:F9 complexes bind to non-specific DNA
To further investigate the DNA-binding properties of Csy:F9, we used a fluorescence polarization (FP) assay (28). The binding of fluorescently labeled DNA to the 350 kD Csy complex markedly reduces its tumbling rate causing an increased FP signal. Thus, the binding of DNA to Csy and Csy:F9 could be quantitated by monitoring the FP signal. Consistent with the EMSA results, the binding of Csy:F9 to both dsDNANS and dsDNASP was readily detected and occurred at Csy complex concentrations within the same range as required for binding of dsDNASP to the Csy complex on its own (Figure 2A). Dissociation constants (Kd values) calculated from these data showed the Csy complex binding dsDNASP with a Kd of 17 ± 6 nM while Csy:F9 bound dsDNASP and dsDNANS with apparent Kd values of 96 ± 33 nM and 73 ± 23 nM, respectively. These values were calculated assuming formation of a 1:1 Csy:DNA complex, which is likely not the case for Csy:F9 as is discussed below; thus, we use the term ‘apparent Kd’ and quote these values only to provide an estimate of the binding strength. Notably, the FP signal measured for the Csy:F9 complex binding to dsDNANS at saturation was nearly double that seen when dsDNASP was bound by Csy alone, suggesting that more than one molecule of Csy:F9 may be binding to each molecule of dsDNANS. Supporting this idea, the Csy:F9 EMSA titration experiments described above (Figure 1C) showed a gradual increase in size of the shifted band in the presence of pre-saturating concentrations of the Csy:F9 complex. This behavior is likely the result of additional molecules of Csy:F9 binding to the DNA as the concentration of the complex increases.
Figure 2.

Multiple Csy:F9 can bind to a single piece of dsDNA in a sequence-independent manner. (A) The binding of 50 bp Cy5-labeled dsDNASP and dsDNANS to Csy and Csy:F9 complexes was assessed by monitoring increase in fluorescence polarization (FP). Increasing concentration of complex were added to a constant concentration of dsDNA ligand. The type of DNA being bound by the indicated complex is shown in the legend. (B) Binding of increasing concentrations of Csy complex to a constant concentration of Cy5-labeled dsDNA2x was monitored on EMSA gels. (C) Binding of Cy5-labeled dsDNA2x to increasing concentrations of Csy and Csy:F9 complexes was monitored by FP. FP of Csy and Csy:F9 bound to the 50 bp dsDNASP from (A) are included for comparison. (D) Cy5 labeled dsDNA1x was pre-saturated with Csy complex, and then increasing concentrations of Csy:F9 complex were added. DNA-binding was monitored by EMSA. The error bars in (A) and (C) correspond to SD, n = 3.
To directly address the effect of multiple Csy complexes binding to a single DNA molecule, we designed a 60 bp dsDNA target sequence containing two 24 bp complementary binding sites for the Csy complex, each with their own PAMs and including the seed region (henceforth called dsDNA2X, Figure 2B). A 24 nt ssDNA molecule complementary to the 5′ end of the crRNA was previously shown to bind strongly to the Csy complex (16). EMSAs with dsDNA2X revealed the formation of two distinct bands when mixed with the Csy complex at high concentrations (Figure 2B). The more slowly moving band presumably resulted from the binding of two Csy complexes to one molecule of dsDNA. This band runs with a mobility similar to the band observed when the Csy:F9 complex is mixed with the dsDNASP or dsDNANS molecules tested above (Figure 1C). Binding of the Csy complex to dsDNA2X in FP experiments caused a doubling of the FP signal compared to binding of dsDNASP, resulting in a signal level similar to that seen when the Csy:F9 complex binds to dsDNASP, dsDNANS or dsDNA2X (Figure 2A, C). A final 60 bp molecule, called dsDNA1X, was synthesized that contained one 24 bp complementary binding site for the Csy complex at one end followed by a random sequence that could act as a non-specific binding site for Csy:F9 (Figure 2D). We first saturated dsDNA1X with the Csy complex, resulting in a single shifted band on the EMSA gel. Subsequent addition of Csy:F9 led to a stepwise slowing of the DNA mobility as the concentration of Csy:F9 was increased (Figure 2D). These data further demonstrate that the slowed mobility of the Csy:F9:DNA complex is the result of multiple complexes binding to a single molecule of DNA.
Having determined that multiple Csy:F9 complexes could bind a single dsDNA molecule, we used EMSA experiments to investigate the minimal DNA length required for the binding of multiple Csy:F9. These assays used shorter dsDNASP of 25 and 12 bp that maintained the PAM and complementarity to the seed sequence (5). It can be seen that binding of Csy:F9 to the 25 bp fragment still resulted in a supershift compared to binding by the Csy complex alone (Supplementary Figure S4). This supershift was similar to the supershift seen with the 50 bp fragment. However, the mobilities of Csy:F9 or Csy complex bound to the 12 bp dsDNASP fragment were very similar, indicating that this length of DNA molecule could accommodate only one Csy:F9 complex (Supplementary Figure S4). Binding of the Csy complex to this shortened DNA is much weaker than Csy:F9 likely because the Csy complex requires DNA complementarity to bind this fragment, which is limited due to the short fragment length. The non-specific binding mediated by Csy:F9 is not diminished when this small target is used, which further emphasizes the unique binding mechanism used by Csy:F9.
Csy:F9 binds dsDNANS and dsDNASP at an overlapping site, but ssDNASP is bound differently
The surprising ability of Csy:F9 to bind dsDNANS may involve a distinct surface on the Csy complex that is not normally engaged in DNA-binding. We performed competition EMSA experiments to determine whether dsDNANS and dsDNASP compete for the same binding site on Csy:F9. The Csy and Csy:F9 complexes were first pre-saturated with FAM-labeled dsDNASP and then increasing concentrations of Cy5-labeled dsDNANS were added. In reactions with Csy complex alone, addition of dsDNANS even at a 4-fold excess caused no reduction in binding to dsDNASP (Figure 3A, left panel). By contrast, addition of dsDNANS to Csy:F9 led to increased levels of free dsDNASP, even when the two types of DNA were present at equal concentrations (Figure 3A, left panel). Moreover, most of the dsDNASP was displaced at a dsDNANS:dsDNASP ratio of 4:1 (Figure 3A, left panel). Viewing the binding of dsDNANS by illuminating the same gel with light at 635 nm (absorption wavelength for Cy5) showed that no dsDNANS was bound to the Csy complex alone but Csy:F9 bound robustly to dsDNANS even at its lowest concentration (Figure 3A, right panel). Our observation that dsDNANS readily competes dsDNASP off of Csy:F9, implies that dsDNANS and dsDNASP bind to an overlapping site.
Figure 3.

DNA binding competition assays. (A) Csy or Csy:F9 complexes (1000 nM) were pre-saturated with 300 nM 6-FAM labeled dsDNASP, then increasing concentrations of Cy5-labeled dsDNANS were added as competitor. DNA-binding was monitored by EMSA. The ratio of dsDNASP to dsDNANS is shown over each lane. The same gel was irradiated at 473 nm to visualize the 6-FAM-labeled DNA (left panel) and at 673 nm to visualize the Cy5-labeled DNA (right panel). (B) Csy or Csy:F9 complexes were pre-mixed with Cy5-dsDNA at 4 nM then competed with increasing concentrations of unlabeled dsDNA as indicated on the x-axis. Relative fluorescence polarization (y-axis) is the FP value at each competitor concentration divided by the value observed in the absence of competitor DNA. (C) Competition experiments are similar to (B) except that experiments were performed with specific or non-specific ssDNA. (D) These competition EMSA experiments are similar to (A) except that complexes were pre-saturated with 6-FAM-labeled ssDNASP and the competitor was Cy5-labeled ssDNANS. The error bars in (B) and (D) correspond to SD, n = 3.
Binding competition experiments were also conducted using FP. Csy or Csy:F9 complexes were pre-saturated with labeled DNA and then challenged with increasing concentrations of non-labeled DNA. If the added non-labeled DNA competed for the same binding site as the labeled DNA, then a decrease in FP signal was expected as the labeled DNA would be competed off of the complex. In the case of the Csy:F9 complex, it can be seen that dsDNANS and dsDNASP competed with each other readily and to a similar degree regardless of whether the complex was pre-saturated with dsDNASP or dsDNANS (Figure 3B). By contrast, binding of the Csy complex to dsDNASP was competed negligibly by dsDNANS even at an 8-fold excess. Notably, the addition of excess levels of dsDNASP to the pre-formed Csy:dsDNASP complex resulted in little loss of FP signal (Figure 3B), implying that the off-rate of dsDNASP from Csy is considerably longer than the 30 min incubation period after which the competitor DNA was added.
To directly address the kinetics of dsDNA binding to Csy:F9, increasing concentrations of unlabeled dsDNANS were added to a complex pre-saturated with labeled dsDNASP and the dissociation of the labeled DNA was monitored over time using FP (Supplementary Figure S5A). Due to the experimental set-up, we were unable to measure time points shorter than 5 min. It can be seen that little change in signal was observed between the 5 and 30 min time points, indicating that the dsDNASP was completely dissociated within 5 min. The same dissociation kinetics were observed when the Csy:F9 complex was saturated with labeled dsDNANS and competed with unlabeled dsDNASP (Supplementary Figure S5B). By contrast, dsDNASP bound to the Csy complex alone was not competed off at all by dsDNANS after 30 min (Supplementary Figure S5C). Overall, these experiments show that bound dsDNA dissociates quickly from Csy:F9 complex, while dsDNA bound specifically to the Csy complex dissociates slowly.
Competition EMSA experiments conducted with ssDNA presented a different picture. While dsDNASP and dsDNANS readily competed for an overlapping site on Csy:F9, ssDNANS was unable to compete ssDNASP off of the Csy:F9 complex (Figure 3C). This result was similar to that obtained in testing the Csy complex where ssDNASP was also not competed by ssDNANS (Figure 3C). Competition experiments monitored by FP corroborated the EMSA results, showing that ssDNASP binding to Csy:F9 or Csy alone was not competed off by ssDNANS, but ssDNANS was competed off by ssDNASP to some extent (Figure 3D). The ability of ssDNASP to only partially displace ssDNANS suggests that these molecules are not binding to completely overlapping sites. These data indicate that in contrast to the case with dsDNA, Csy:F9 binds ssDNASP considerably more strongly than ssDNANS. Using EMSAs, we also observed minimal displacement of pre-saturated ssDNASP when competed with increasing concentrations of dsDNASP from the Csy:F9 complex (Supplementary Figure S6). Overall, these data demonstrate that ssDNASP is unique in its stronger binding to Csy:F9 as compared to any other of the DNA molecules tested, implying that Csy:F9 retains the ability to distinguish specific from non-specific ssDNA.
Positively charged surfaces on AcrIF9 and Cas8f mediate non-specific interaction with DNA
The structure of Csy:F9 bound to dsDNA revealed a positively charged surface on AcrIF9 composed of five conserved residues (K31, R32, K36, K58 and R63) that are in proximity to the DNA, and may stabilize the non-specific interaction of Csy:F9 with DNA (21). An alignment of six diverse AcrIF9 homologues reveals that these positions are always positively charged (Supplementary Figure S1A). The four other positions in the AcrIF9 that display similar levels of conservation play key functional roles: one (A34) is completely buried in the protein core, two (Q38 and W54) are buried in the interface with Cas7f and one (Y5) interacts with Cas8f. Substitutions at Q38 and Y5 abrogate AcrIF9 function (2,27). Thus, the five exposed positively charged positions on AcrIF9 display unusually high conservation equivalent to functionally crucial positions, implying that these residues play a role that has been selected for during evolution. To address this role, we substituted R32 and K36 (Figure 4A), the two residues most intimately associated with the DNA, with alanine individually (F9R32A and F9K36A) and also constructed a double mutant at these positions (F9RA/KA). These mutants were co-expressed with the Csy complex and the mutant complexes were purified (Supplementary Figure S7A). These complexes behaved like wild-type Csy:F9 and remained assembled during purification by gel filtration. Remarkably, EMSA assays revealed that the Csy:F9R32A and Csy:F9K36A complexes still bound to dsDNASP but showed dramatic decreases in binding to dsDNANS (Figure 4B, C). The Csy:F9R32A complex showed the larger change in behavior. This complex bound robustly to dsDNASP, though not as strongly as wild-type Csy:F9 (Figure 1C). However, evidence of non-specific DNA-binding could be discerned only at high complex concentrations. In this case, the non-specific binding manifested as smeared bands moving faster than the Csy:dsDNASP complex rather than the supershifted bands observed for the wild-type Csy:F9 complex. This changed pattern is likely due to weak non-specific DNA-binding mediated by these mutants causing the complexes to dissociate readily during electrophoresis. In contrast with the single mutants, the Csy:F9 RA/KA complex displayed no detectable binding to dsDNANS while maintaining a reduced but easily observable level of binding to dsDNASP (Figure 4D).
Figure 4.

Substitution of positively charged residues on AcrIF9 and Cas8f reduces non-specific dsDNA binding. (A) Model of Csy:F9 bound to dsDNANS (left), AcrIF9 shown in orange, Cas8f in yellow and dsDNA in blue. The mutagenized residues on AcrIF9 and Cas8f are highlighted in red. A detailed view of the non-specific DNA binding sites of AcrIF9.1 and Cas8f is shown in the right panel. Molecular models were made with PyMOL (Schrodinger). EMSA gels show the DNA-binding activity of the WT Csy complex co-expressed with the AcrIF9R32A (B), AcrIF9K36A (C) or AcrIF9RA/KA (D) mutants. The Csy:F9 mutant complexes were incubated in increasing concentrations with dsDNASP and dsDNANS. (E) EMSA gels were used to assess the DNA binding activity of the CsyCas8-RK complex in presence (right two panels) or absence (left panel) of AcrIF9. CsyWT:F9 (4000 nM) binding to dsDNASP was also included for reference (middle panel). Constant concentrations of dsDNASP (left two panels) or dsDNANS (right panel) were mixed with increasing concentrations of the CsyCas8-RK or CsyCas8-RK:F9 complexes. (F) The Csy complex was incubated with AcrIF9, AcrIF2, AcrIF1 or combinations thereof prior to incubation with 50 bp fragments of FAM-6- labeled dsDNASP (left) or Cy5-labeled dsDNANS (right). In cases, where two Acrs were added to one reaction, the Acr added second is indicated by (→). DNA-binding activity was assayed by EMSA.
To ensure that the decreased non-specific binding of the AcrIF9 mutants was not due to disrupted interactions with the Csy complex, we performed a similar competitive binding experiment with AcrIF1 as described above (Supplementary Figure S2). Pre-formed 6xHis-tagged Csy:F9 complexes with the three mutations (F9R32A, F9K36A, F9RA/KA) were incubated in the presence of excess untagged AcrIF1 followed by affinity chromatography to remove unbound Acrs (Supplementary Figure S7B). The results showed no significant competition of the three AcrIF9 mutants by AcrIF1 when compared to the wild-type AcrIF9. This demonstrates that the mutants remain tightly bound to the Csy complex and that the decrease in non-specific DNA binding is due to a disruption in the function of AcrIF9.
Since AcrIF9 does not interact with DNA on its own (Supplementary Figure S3A), we suspected that the positively charged surface on the Acr would not be sufficient on its own to mediate the non-specific DNA binding displayed by the Csy:F9 complex. In the Csy:F9 structure, positively charged residues (R207, K216 and R224) in Cas8f are positioned near one of the AcrIF9:DNA interfaces (Figure 4A). To address the potential role of these residues, we constructed and purified a Csy complex bearing Ala substitutions at these three positions (CsyCas8-RK, Supplementary Figure S7A). The CsyCas8-RK:F9 complex displayed considerably less binding activity towards dsDNANS as compared to dsDNASP, forming a detectable complex in EMSA with dsDNASP at a 125 nM concentration while a complex with dsDNANS was detected only weakly at a complex concentration of 1000 nM (Figure 4E, right panel). The CsyCas8-RK:F9:DNA complexes behaved similarly in EMSA experiments to the complexes involving the AcrIF9 mutants; less supershifting and faster mobility smearing was observed. Although the CsyCas8-RK complex on its own bound to dsDNASP more weakly than wild-type (Figure 4E, left panel), these substitutions in Cas8 clearly manifest their greatest effect on AcrIF9-induced dsDNANS-binding.
To further probe the role of Cas8f in the activity of AcrIF9, we tested the effect AcrIF2, which inhibits the DNA-binding activity of the Csy complex by binding Cas8f. As discussed above, AcrIF2 and AcrIF9 can bind to the Csy complex simultaneously (Supplementary Figure S2). Interestingly, a ternary Csy:F9:F2 complex displayed no binding to either dsDNASP or dsDNANS (Figure 4F). This result supports a role for Cas8f in the non-specific DNA-binding activity mediated by Csy:F9.
Mutant Csy:F9 complexes display decreased in vivo activity but retain specific DNA interaction
The positively charged residues of AcrIF9 implicated in non-specific DNA-binding activity are conserved (Supplementary Figure S1A). Thus, we expected them to play a role in mediating Acr activity. To address this issue, we expressed the AcrIF9 mutants from a plasmid in P. aeruginosa strain PA14 and used a standard phage plaquing assay (9) to assess their Acr activity. As can be seen in (Figure 5A), the AcrIF9R32A, AcrIF9K36A and AcrIF9RA/KA mutants all showed approximately 10-fold reductions in activity.
Figure 5.

Effects of AcrIF9 mutants on the type I-F CRISPR–Cas system in vivo. (A) The relative efficiency of plaquing of phage DMS3m on lawns of P. aeruginosa PA14 cells are shown. This strain possesses a type I-F CRISPR–Cas system targeting this phage. Cells were transformed by plasmids expressing the indicated AcrIF9 mutants. The efficiency of plaquing is calculated by dividing the plaque forming units (p.f.u.) per ml of DMS3m when plated on cells expression a mutant AcrIF9 by the p.f.u./ml of DMS3m obtained on cell where wild-type AcrIF9 was expressed. (B) PA14 WT or PA14 Δcas3 cells were transformed by plasmids expressing the indicated AcrIF9 mutants and a crRNA targeting the promoter region of phzM. The amount of pyocyanin produced was quantitated as a percentage of the same strain with the empty vector (EV) control. Pictures are representative cultures are shown on the right. (C) Growth curves are show for PA14 WT (left), or PA14 Δcas3 (right) cells transformed with the same plasmids used in part (B). Optical densities at 600 nm were measured every 30 min for 6 h. 10 mM arabinose was added to induce expression of crRNA and Acr from the plasmid. In parts (A), (B) and (C), an average of three independent experiments is shown with the error bars representing SD.
To assess the ability of AcrIF9 to block the specific DNA-binding ability of the Csy complex in vivo, we used a previously described transcriptional repression assay (16). Plasmids were constructed that expressed a crRNA targeting the promoter of the phzM gene, which is required for the production of pyocyanin, the distinctive green pigment of P. aeruginosa. The crRNA was expressed on its own or with AcrIF9. In wild-type strain PA14, binding of the phzM promoter by the Csy complex without Acr expression resulted in cell death due to Cas3-mediated cleavage of the bacterial genome. In a Δcas3 strain, binding of the Csy complex to the phzM promoter in the absence of cleavage leads to repression of phzM and no production of pyocyanin (Figure 5B). Simultaneous production of AcrIF9 and crRNA in wild-type PA14 allowed cell growth and wild-type levels of pyocyanin were produced (Figure 5B). The production of pyocyanin indicated that AcrIF9 was preventing the Csy complex from binding its specific target in the phzM promoter. Remarkably, expression of the AcrIF9RA/KA mutant from the same plasmid resulted in almost no production of pyocyanin, while cells expressing of the AcrIF9K36A mutant produced an intermediate level (Figure 5B). These results imply that in the presence of these mutants, the Csy complex is still able to bind to its specific site in the phzM promoter and repress transcription. The degree to which pyocyanin production is reduced in the presence of these mutants parallels our in vitro data showing that Csy:F9RA/KA displays no non-specific DNA-binding while Csy:F9K36A still binds appreciably to non-specific DNA though much less than wild-type.
The plasmids constructed for the phzM repression assay also allowed us to perform a different assay for in vivo Acr activity. Since the crRNA expressed from these plasmids targeted the bacterial genome, cell survival was dependent upon the Acr that is expressed from the plasmid. Cells could be transformed efficiently by all of the plasmids used above, except for the one expressing no Acr. However, the growth rates of cells bearing these plasmids varied widely when plasmid-borne gene expression was induced with arabinose. Most notably, cells expressing the AcrIF9RA/KA mutant grew very slowly compared to cells expressing wild-type AcrIF9 or carrying empty vector. Cells expressing the AcrIF9K36A mutant grew better than those expressing AcrIF9RA/KA but were still much slower than cells expressing wild-type AcrIF9 (Figure 5C, left). Mutant Δcas3 strains carrying these plasmids all grew at the same rates (Figure 5C, right), demonstrating the effects seen in wild-type cell were mediated by the CRISPR system. These experiments emphasize that the AcrIF9 mutants with reduced non-specific DNA-binding activity do not inhibit the CRISPR–Cas system as effectively as wild-type.
Non-specific DNA binding is a conserved property of Csy:F9 complexes
The unusual properties of the Csy:F9 complex led us to question whether this was a conserved feature of this Acr or an unusual coincidence occurring with one particular combination of AcrIF9 homologue and Csy complex. To address this issue, we tested a homologue of AcrIF9 from A. actinomycetemcomitans that is only 33% identical to the homologue from P. penneri used in our other experiments, but still strongly inhibits the type I-F CRISPR–Cas system of P. aeruginosa (Supplementary Figure S1B). Assessment of the DNA binding activity of a Csy:F9Aac complex by EMSA showed that it also bound to dsDNANS in a similar manner as the Csy:F9Ppe complex tested above, including the smeared supershifted band (Supplementary Figure S8).
To further highlight the universality of Csy:F9 behavior, we assessed the activity of the AcrIF9 homologue from V. parahaemolyticus (72% identical to F9Ppe) against the type I-F CRISPR–Cas system from Shewanella baltica (Sba, Cas7f 57% identical to Cas7f of P. aeruginosa). Using an in vivo plasmid transformation assay, it was shown that the S. baltica system was completely inhibited by AcrIF9Vpa, as strains expressing the Acr protein and the complex exhibited transformation efficiencies similar to those carrying a deficient nuclease (Cas2–3 HD domain mutant, Supplementary Figure S9A). The DNA binding properties of CsySba:F9Vpa were investigated using Bio-layer Interferometry (BLI). In this approach, biotinylated dsDNA target oligonucleotides were immobilized on a streptavidin-coated bio-sensor. The CsySba complex was then flowed into the cell and the shift in wavelength of reflected light was measured over time to determine the on-rate (kon) of the reaction. The off-rate (koff) was determined by flowing buffer into the cell after the binding reaction reached equilibrium. The BLI experiments showed that CsySba bound dsDNAsp, but showed weak binding to dsDNANS, similar to the binding of AcrIF9Vpa alone (Figure 6A, B; Supplementary Figure S9B). By contrast, the CsySba:F9Vpa complex was able to bind both dsDNASP and dsDNANS (Figure 6A, B; Supplementary Figure S9B). For the specific target, the CsySba:F9Vpacomplex bound with 14-fold faster kon than the CsySba complex alone (130 050 ± 3674.5 M−1s−1 versus 9023 ± 860.5 M−1s−1), and also displayed an 8-fold faster koff (0.01805 ± 0.0003 s−1 versus 0.0022675 ± 0.0016 s−1).
Figure 6.
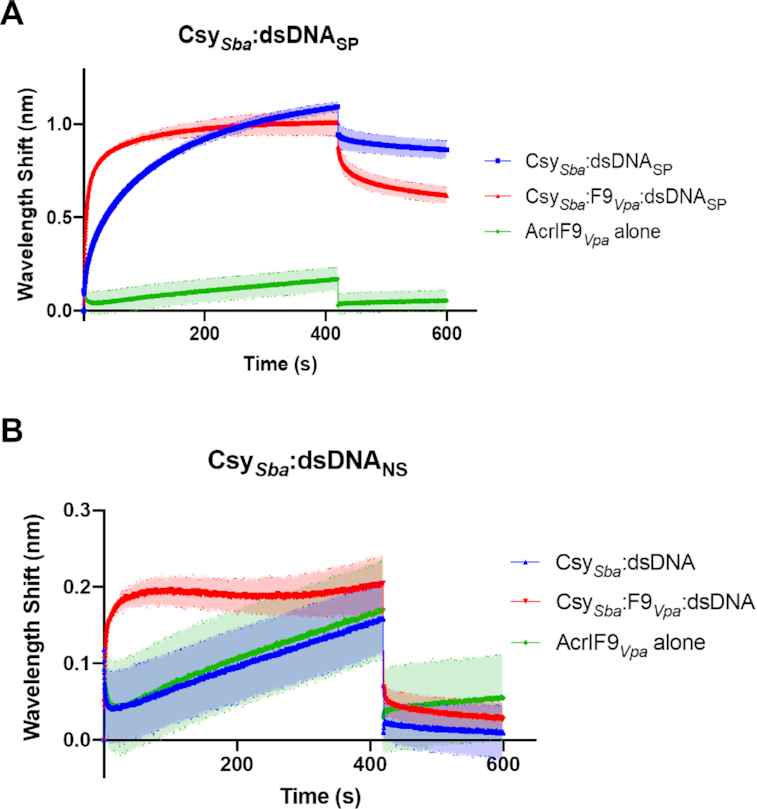
Binding of Type I-F Csy complex from S. baltica (CsySba) to dsDNA measured by Bio-Layer Interferometry (BLI) (A) The wavelength shift (nm) generated by the binding of CsySba complex to immobilized dsDNASP fragments was measured. AcrIF9Vpa was mixed with the Csy complex at a 1:1 molar ratio for 10 min at room temperature prior to the initiation of the BLI assay. The shift elicited by AcrIF9Vpa binding to dsDNASP is shown as a control (AcrIF9Vpa alone). Assays were performed in duplicate, the lighter outline represents the standard error from the mean. (B) The same experiments as described in (A) were performed using dsDNANS.
DISCUSSION
A striking feature of Acrs is their diversity in sequence, structure, and mechanism of action. Here, we characterize the mechanism AcrIF9, the first example of an Acr that induces a CRISPR–Cas system to bind dsDNA independent of sequence complementarity or PAM. Binding of AcrIF9 to the Csy complex renders it incapable of distinguishing dsDNASP from dsDNANS, binding each with similar apparent affinity. Furthermore, the strength of the non-specific DNA-binding induced by AcrIF9 is similar to the specific DNA-binding affinity of the Csy complex on its own, a remarkable property given that non-specific DNA binding activity normally exhibited by the Csy complex is so weak as to be undetectable in our assays.
Our in vitro DNA binding experiments clearly showed that dsDNASP and dsDNANS compete equally for the same site on the Csy:F9 complex, indicating that specific hydrogen bonding with the crRNA is not contributing to the interaction (Figure 3A, B). The non-specific nature of Csy:F9 DNA binding is also supported by the ability of multiple Csy:F9 complexes to bind one molecule of DNA resulting in the supershifted complexes seen in EMSA gels and the increased signal seen in the FP assays (Figures 1C, 2A). Although the affinity of Csy:F9 for non-specific DNA appeared to be similar to that of the Csy complex alone for specific DNA, Csy:F9 binds to DNA much more quickly (Figure 6A, B), but also dissociates more quickly. These properties are consistent with a reaction driven primarily by electrostatics as would be expected for non-specific DNA binding. The smeared appearance the Csy:F9:DNA complexes in EMSA gels is likely a result of the dissociation of these complexes during electrophoresis.
We conclude that wild-type AcrIF9 potently inhibits the CRISPR–Cas system by creating a large positively charged surface on the Csy complex. This surface, which includes the conserved positively charged residues of AcrIF9 and positively charged residues of Cas8, diverts the Csy complex from binding to its specific site by allowing it to bind to any DNA. In addition, even dsDNA bearing a specific site is bound at the same site as non-specific DNA so that the conformational changes required to elicit Cas3 recruitment and cleavage do not occur (AcrIF9 blocks in vitro cleavage activity mediated by the Csy complex and Cas3 (27)). The importance of non-specific DNA-binding in the inhibitory mechanism of AcrIF9 is supported by our data showing that abrogation of this activity by amino acid substitutions on the positively charged surface of AcrIF9 (AcrIF9K36A and AcrIF9RA/KA) led to marked reductions in Acr activity in vivo both in phage plaquing and cell growth assays (Figure 5A, C). These reductions in Acr activity were observed even though these mutants bound to the Csy complex as strongly as wild-type (Supplementary Figure S7B). The centrality of non-specific DNA-binding for AcrIF9 function is reinforced by our finding that the induction of this phenomenon is a conserved property of diverse AcrIF9 homologs, and the effect was manifested on two different CRISPR–Cas systems (Figure 6, Supplementary Figure S8). Furthermore, the five residues comprising the positively charged surface on AcrIF9 that interacts with non-specific DNA-binding are highly conserved (Supplementary Figure S1). The unique nature of AcrIF9 inhibition may partially explain the widespread distribution of members of this Acr family and their ability to block diverse type I-F systems (20). Interestingly, another very widespread Acr, AcrIIA11, possesses DNA binding activity on its own, and this activity is increased in the presence of Cas9 (29).
Despite the reduced Acr activity displayed by the AcrIF9K36A and AcrIF9RA/KA mutants, Csy complexes bound to these mutants could interact with a specific site strongly enough to repress phzM transcription. This was a notable contrast to wild-type AcrIF9, which prevented specific binding in this assay. Since the mutant Csy:F9 complexes could bind to a specific targeted site, why was CRISPR–Cas activity still partially inhibited (i.e. cells still grew though more slowly than in the presence of wild-type AcrIF9)? We speculate that the mutant Csy:F9 complexes bind tightly enough to repress transcription, but that this DNA-binding reaction does not efficiently elicit the conformational changes required for the recruitment and activation of Cas3 to mediate DNA cleavage. From these data, it is evident that AcrIF9 may inhibit the Csy complex by inducing non-specific DNA binding and by inhibiting conformational changes required for Cas3 activity.
Examination of the structure of the Csy:F9 complex suggested that AcrIF9 would sterically block DNA-binding by preventing access to the spacer region of the crRNA (21,27). Supporting this idea, the binding site of AcrIF9 overlaps very closely with that of AcrIF1 (Supplementary Figure S10), and this Acr completely blocks specific binding of both dsDNA and ssDNA. However, in contrast to the Csy:F1 complex, the Csy:F9 complex binds specifically to ssDNA. Furthermore, the Csy:F9R33A, Csy:F9K36A, Csy:F9RA/KA complexes bind specifically to dsDNA despite the presence of bound Acrs that are still able to effectively block binding by AcrIF1 (Supplementary Figure S7B). Thus, binding of Acrs to the same sites on the Csy complex can produce widely divergent effects. What can explain these distinct behaviors? One potential source is differences in the conformational flexibility between these two complexes. The interactions of both Acrs are primarily with one Cas7f subunit to a region known as the thumb (residues 71–95) and to a loop between residues 250 and 261 (Supplementary Figure S10). They contact mostly the same residues in these regions even though their sequences and structures are different. A potentially important distinction is that AcrIF1 contacts residues 251–257 in the neighboring Cas7f subunit forming an additional interface of 275 Å2 (Supplementary Figure S10). This same region was unresolved in the Csy:F9 structure, and AcrIF9 makes minimal contact with adjacent Cas7f subunits. The inter-Cas7 bridging contacts made by AcrIF1 bound to the Csy complex may rigidify the complex and prevent it from undergoing the conformation changes required for specific binding to DNA (7,8). Regardless of the mechanism, these Acrs provide a provocative example where static structures may not fully account for biochemical behavior.
In conclusion, we have shown that AcrIF9 possesses the remarkable ability to convert the normally highly specific dsDNA binding activity of the Csy complex into a non-specific binding activity that is still very strong. This induction of non-specific binding appears to be a fundamental component of the inhibitory mechanism of AcrIF9. The potential to become a strong non-specific DNA binding complex may be inherent in all dsDNA binding CRISPR–Cas systems as they all must form non-specific interactions with the single strand of DNA that is looped out (the R-loop) upon hybridization of the target strand to the crRNA. In the case of the Csy:F9 complex, our mutagenesis experiments imply that Cas8f forms part of the non-specific DNA-binding interface. A partially overlapping region of Cas8f is involved in R-loop binding; thus, AcrIF9 may be taking advantage of the natural function of Cas8f as a non-specific DNA-binding subunit to create a non-specific DNA-binding surface. Further investigation may reveal that other Acrs employ a similar mechanism.
Supplementary Material
Contributor Information
Wang-Ting Lu, Department of Biochemistry, University of Toronto, 661 University Ave, Toronto, ON, M5G 1M1, Canada.
Chantel N Trost, Department of Molecular Genetics, University of Toronto, 661 University Ave, Toronto, ON, M5G 1M1, Canada.
Hanna Müller-Esparza, Faculty of Biology, University of Marburg, Karl-von-Frisch-Straße 1, 35043 Marburg, Germany.
Lennart Randau, Faculty of Biology, University of Marburg, Karl-von-Frisch-Straße 1, 35043 Marburg, Germany; Loewe Center for Synthetic Microbiology (SYNMIKRO), Marburg, Germany.
Alan R Davidson, Department of Biochemistry, University of Toronto, 661 University Ave, Toronto, ON, M5G 1M1, Canada; Department of Molecular Genetics, University of Toronto, 661 University Ave, Toronto, ON, M5G 1M1, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by Canadian Institutes of Health Research (CIHR) grants [MOP-130482 and FDN-15427 to A.R.D.] and German Research Foundation (DFG) grant [SPP 2141 to L.R.] C.N.T. was supported by a CIHR Post-Doctoral Fellowship Award.
Conflict of interest statement. A.R.D. is a scientific advisory board member for Acrigen Biosciences and is an inventor on patents relating to anti-CRISPR proteins. The other authors declare no competing interests.
REFERENCES
- 1. Bondy-Denomy J., Davidson A.R.. To acquire or resist: the complex biological effects of CRISPR–Cas systems. Trends Microbiol. 2014; 22:218–225. [DOI] [PubMed] [Google Scholar]
- 2. Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P.et al.. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020; 18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Makarova K.S., Wolf Y.I., Koonin E.V.. Classification and nomenclature of CRISPR–Cas systems: where from here?. CRISPR J. 2018; 1:325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Duijn E., Barbu I.M., Barendregt A., Jore M.M., Wiedenheft B., Lundgren M., Westra E.R., Brouns S.J., Doudna J.A., van der Oost J.et al.. Native tandem and ion mobility mass spectrometry highlight structural and modular similarities in clustered-regularly-interspaced shot-palindromic-repeats (CRISPR)-associated protein complexes from Escherichia coli and Pseudomonas aeruginosa. Mol. Cell. Proteomics. 2012; 11:1430–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wiedenheft B., van Duijn E., Bultema J.B., Waghmare S.P., Zhou K., Barendregt A., Westphal W., Heck A.J., Boekema E.J., Dickman M.J.et al.. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U. S. A. 2011; 108:10092–10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chowdhury S., Carter J., Rollins M.F., Golden S.M., Jackson R.N., Hoffmann C., Nosaka L., Bondy-Denomy J., Maxwell K.L., Davidson A.R.et al.. Structure reveals mechanisms of viral suppressors that intercept a CRISPR RNA-guided surveillance complex. Cell. 2017; 169:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo T.W., Bartesaghi A., Yang H., Falconieri V., Rao P., Merk A., Eng E.T., Raczkowski A.M., Fox T., Earl L.A.et al.. Cryo-EM structures reveal mechanism and inhibition of DNA targeting by a CRISPR–Cas surveillance complex. Cell. 2017; 171:414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rollins M.F., Chowdhury S., Carter J., Golden S.M., Miettinen H.M., Santiago-Frangos A., Faith D., Lawrence C.M., Lander G.C., Wiedenheft B.. Structure reveals a mechanism of CRISPR-RNA-guided nuclease recruitment and anti-CRISPR viral mimicry. Mol. Cell. 2019; 74:132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bondy-Denomy J., Pawluk A., Maxwell K.L., Davidson A.R.. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013; 493:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bondy-Denomy J., Davidson A.R., Doudna J.A., Fineran P.C., Maxwell K.L., Moineau S., Peng X., Sontheimer E.J., Wiedenheft B.. A unified resource for tracking anti-CRISPR names. CRISPR J. 2018; 1:304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davidson A.R., Lu W.T., Stanley S.Y., Wang J., Mejdani M., Trost C.N., Hicks B.T., Lee J., Sontheimer E.J.. Anti-CRISPRs: protein inhibitors of CRISPR–Cas systems. Annu. Rev. Biochem. 2020; 89:309–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wiegand T., Karambelkar S., Bondy-Denomy J., Wiedenheft B.. Structures and strategies of anti-CRISPR-mediated immune suppression. Annu. Rev. Microbiol. 2020; 74:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang F., Song G., Tian Y.. Anti-CRISPRs: the natural inhibitors for CRISPR–Cas systems. Animal Model Exp Med. 2019; 2:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marino N.D., Pinilla-Redondo R., Csorgo B., Bondy-Denomy J.. Anti-CRISPR protein applications: natural brakes for CRISPR–Cas technologies. Nat. Methods. 2020; 17:471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakamura M., Srinivasan P., Chavez M., Carter M.A., Dominguez A.A., La Russa M., Lau M.B., Abbott T.R., Xu X., Zhao D.et al.. Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun. 2019; 10:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bondy-Denomy J., Garcia B., Strum S., Du M., Rollins M.F., Hidalgo-Reyes Y., Wiedenheft B., Maxwell K.L., Davidson A.R.. Multiple mechanisms for CRISPR–Cas inhibition by anti-CRISPR proteins. Nature. 2015; 526:136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Yao D., Xu J.G., Li A.R., Xu J., Fu P., Zhou Y., Zhu Y.. Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat. Struct. Mol. Biol. 2016; 23:868–870. [DOI] [PubMed] [Google Scholar]
- 18. Niu Y., Yang L., Gao T., Dong C., Zhang B., Yin P., Hopp A.K., Li D., Gan R., Wang H.et al.. A type I-F anti-CRISPR protein inhibits the CRISPR–Cas surveillance complex by ADP-ribosylation. Mol. Cell. 2020; 80:512–524. [DOI] [PubMed] [Google Scholar]
- 19. Gabel C., Li Z., Zhang H., Chang L.. Structural basis for inhibition of the type I-F CRISPR–Cas surveillance complex by AcrIF4, AcrIF7 and AcrIF14. Nucleic. Acids. Res. 2020; 49:584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pawluk A., Staals R.H., Taylor C., Watson B.N., Saha S., Fineran P.C., Maxwell K.L., Davidson A.R.. Inactivation of CRISPR–Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol. 2016; 1:16085. [DOI] [PubMed] [Google Scholar]
- 21. Hirschi M., Lu W.-T., Santiago-Frangos A., Wilkinson R., Golden S.M., Davidson A.R., Lander G.C., Wiedenheft B.. AcrIF9 tethers non-sequence specific dsDNA to the CRISPR RNA-guided surveillance Complex. Nat. Commun. 2020; 11:2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiu D., Damron F.H., Mima T., Schweizer H.P., Yu H.D.. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl. Environ. Microbiol. 2008; 74:7422–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Altschul S.F., Koonin E.V.. Iterated profile searches with PSI-BLAST–a tool for discovery in protein databases. PF115. 1998; 23:444–447. [DOI] [PubMed] [Google Scholar]
- 24. Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J.. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pausch P., Muller-Esparza H., Gleditzsch D., Altegoer F., Randau L., Bange G.. Structural variation of type I-F CRISPR RNA guided DNA surveillance. Mol. Cell. 2017; 67:622–632. [DOI] [PubMed] [Google Scholar]
- 26. Müller-Esparza H., Osorio-Valeriano M., Steube N., Thanbichler M., Randau L.. Bio-layer interferometry analysis of the target binding activity of CRISPR–Cas effector complexes. Front. Microbiol. 2020; 7:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang K., Wang S., Li S., Zhu Y., Pintilie G.D., Mou T.C., Schmid M.F., Huang Z., Chiu W.. Inhibition mechanisms of AcrF9, AcrF8, and AcrF6 against type I-F CRISPR–Cas complex revealed by cryo-EM. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:7176–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anderson B.J., Larkin C., Guja K., Schildbach J.F.. Using fluorophore-labeled oligonucleotides to measure affinities of protein-DNA interactions. Methods Enzymol. 2008; 450:253–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forsberg K.J., Bhatt I.V., Schmidtke D.T., Javanmardi K., Dillard K.E., Stoddard B.L., Finkelstein I.J., Kaiser B.K., Malik H.S.. Functional metagenomics-guided discovery of potent Cas9 inhibitors in the human microbiome. Elife. 2019; 8:e46540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.