

Abstract
Glycoproteins secreted by cells play essential roles in the regulation of extracellular activities. Secreted glycoproteins are often reflective of cellular status, and thus glycoproteins from easily accessible bodily fluids can serve as excellent biomarkers for disease detection. Cultured cells have been extensively employed as models in the research fields of biology and biomedicine, and global analysis of glycoproteins secreted from these cells provides insights into cellular activities and glycoprotein functions. However, comprehensive identification and quantification of secreted glycoproteins is a daunting task because of their low abundances compared with the high-abundance serum proteins required for cell growth and proliferation. Several studies employed serum-free media to analyze secreted proteins, but it has been shown that serum starvation, even for a short period of time, can alter protein secretion. To overcome these issues, we developed a method to globally characterize secreted glycoproteins and their N-glycosylation sites from cultured cells by combining selective enrichment of secreted glycoproteins with a boosting approach. The results demonstrated the importance of the boosting sample selection and the boosting-to-sample ratio for improving the coverage of secreted glycoproteins. The method was applied to globally quantify secreted glycoproteins from THP-1 monocytes and macrophages in response to lipopolysaccharides (LPS) and from Hep G2 cells treated with TGF-β without serum starvation. We found differentially secreted glycoproteins in these model systems that showed the cellular response to the immune activation or the epithelial-to-mesenchymal transition. Benefiting from the selective enrichment and the signal enhancement of low-abundance secreted glycoproteins, this method can be extensively applied to study secreted glycoproteins without serum starvation, which will provide a better understanding of protein secretion and cellular activity.
Graphical Abstract
INTRODUCTION
Cells secrete many glycoproteins to regulate extracellular activities, including cell–cell communication and manipulation of the cellular microenvironment.1–3 Secreted glycoproteins, including cytokines, antibodies, growth factors, hormones, and enzymes, from easily accessible and noninvasive sources such as blood and urine contain much valuable information about cellular development and disease statuses, and thus they can serve as effective biomarkers for early disease detection.4–8 For example, secreted prostate-specific antigen (PSA) is a well-known glycoprotein used in prostate cancer diagnosis.9 Recent studies even indicated that measuring different urinary PSA glycoforms may improve the diagnostic accuracy in the “grey zone” concentration range (4.0–10.0 ng/mL PSA).10,11 Cells in the immune system such as monocytes and macrophages secrete glycoproteins during the infection to fight against foreign pathogens. For instance, once stimulated with lipopolysaccharides (LPS) after a bacterial infection, macrophages trigger nuclear factor kappa-B (NF-κB) to migrate into the nucleus, resulting in the secretion of cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), and chemokines that attract other white blood cells such as neutrophils to the vicinity.2,12,13
Cultured cells have been extensively used as models in the fields of biology and biomedicine, and global analysis of glycoproteins secreted from these cells can aid in a better understanding of their functions and extracellular events. Despite their importance, secreted glycoproteins cannot be easily analyzed at the global level because many of them are present in an extremely low abundance. Furthermore, detection of secreted glycoproteins is often hindered by many other proteins from fetal bovine serum (FBS) including growth factors and proteins necessary for cell growth and proliferation used in the culture medium. Compared with serum proteins, the abundance of secreted glycoproteins is much lower.14 While the analysis of secreted proteins can be performed using antibody-based methods such as Western blotting and ELISA, the antibody availability and specificity may be an issue. More importantly, it is almost impossible to use these methods for global analysis of secreted glycoproteins, and they cannot provide site-specific glycosylation information.
Mass spectrometry (MS)-based proteomics has also been used for comprehensive analysis of secreted glycoproteins.15–19 To overcome the challenges in secreted glycoprotein analysis, extensive fractionation may be performed. Yet, this method is not very effective and time-consuming. Furthermore, it does not result in the high coverage of secreted glycoproteins. Protein depletion is also commonly employed to remove some highest abundant proteins from the samples, but the reproducibility and the loss of proteins of interest are problematic.20 Alternatively, serum-free media (SFM) have been employed to avoid the interference from high-abundance serum proteins. While this approach increases the coverage of secreted proteins detected, studies have shown that serum starvation dramatically affects cell growth and proliferation and thus results in protein secretion alterations.14,21 Taking these issues into consideration, global analysis of secreted glycoproteins and their glycosylation sites under the serum-containing conditions is much more challenging and remains to be further explored.
In this study, in order to increase the coverage of secreted glycoproteins, we integrated a selective enrichment method with a signal boosting approach for comprehensive analysis of secreted glycoproteins from cells cultured in serum-containing media. Since low-abundance secreted glycoproteins are buried among many extremely high-abundance FBS proteins in the medium, selective enrichment is critical for their global analysis. However, the enrichment alone is not sufficient because, besides nonspecific binding, the detection limit of LC-MS is also unfavorable for low-abundance secreted glycoproteins. We first evaluated different glycoprotein sources to serve as the signal boosting sample. Furthermore, the boosting-to-sample ratio as another important parameter was carefully chosen to maximize the boosting effect and increase the coverage of glycoproteins. The method allowed us to quantify hundreds of secreted glycoproteins with the glycosylation sites from THP-1 monocytes and M0 macrophages in response to LPS and from Hep G2 treated with TGF-β. Several secreted glycoproteins were found to be regulated in these models, including those involved in the immune and inflammatory responses from THP-1 monocytes and macrophages treated with LPS, and those participated in the extracellular matrix organization and the epithelial-to-mesenchymal transition (EMT) from Hep G2 cells. Global quantification of secreted glycoproteins from cultured human cells without serum starvation provides valuable information to the scientific community, and the developed method can be extensively applied for secreted glycoprotein analysis.
EXPERIMENTAL SECTION
Cell Culture, Treatments, and Metabolic Labeling of Glycoproteins.
Reagents were purchased from Sigma-Aldrich unless noted otherwise. For the experiment with THP-1 (ATCC), the cells were maintained in RPMI-1640 medium (Gibco) containing 10% FBS (Corning) and 1% penicillin-streptomycin (P–S) in a humidified incubator with 5% carbon dioxide at 37 °C. Once the density reached ~8 × 105 cells/mL, the cells were centrifuged at 500g for 5 min, washed twice with warm PBS, and resuspended in the corresponding medium before metabolic labeling. For the serum-containing medium (SCM), the original medium with 10% FBS and 1% P–S was used. For the serum-free medium (SFM), 10% FBS was not added. Depending on the experiment, the cells were prelabeled with 100 μM N-azidoacetylgalactosamine-tetraacylated (Ac4GalNAz; Click Chemistry Tools) in SCM or SFM in the incubator. After 12 h, either 1 μg/mL LPS from E. coli O111:B4 or PBS at the same volume was added to the culture flasks and then incubated for 12 h. For the experiments with M0 macrophages, THP-1 monocytes were differentiated as reported previously using 100 ng/mL phorbol 12-myristate 13-acetate (PMA) in the SCM for 48 h, rested for 24 h in the SCM without PMA, and washed with warm PBS twice before adding the corresponding medium containing Ac4GalNAz.22,23 For the experiments with Hep G2 (ATCC), the cells were grown similarly in high-glucose DMEM containing 10% FBS and 1% P–S. Once the confluency reached ~50%, the cells were switched to the serum-free or serum-containing conditions, prelabeled with 100 μM Ac4GalNAz for 12 h, and treated with 10 ng/mL human transforming growth factor β1 (TGF-β, Cell Signaling Technology) for 48 h before medium collection.
Medium Collection, Cell Lysis, and Click-Chemistry Reaction.
The cells and the media were collected by centrifugation at 500 g for 5 min. The cells were washed with ice-cold PBS twice and the wash solution was pooled with the media. For the experiments with the cell part, proteins were extracted using a lysis buffer containing 50 mM N-(2-hydroxyethyl)piperazine-N′−2-ethanesulfonic acid (HEPES; pH 8.8), 150 mM sodium chloride (NaCl), 10% sodium deoxycholate (SDC), 1 tablet/50 mL protease inhibitors (Roche), and 20 units/mL universal nuclease for cell lysis (Pierce) at 4 °C with end-over-end rotation for 1 h. The lysates were collected by centrifugation at 25 830 g for 10 min. For the medium part, the same protease inhibitors were added to the final concentration of 1 tablet/50 mL before centrifugation at 5000g for 10 min to remove any debris. The media were concentrated using an Amicon Ultra-15 centrifugal filter unit with a 3000 Da molecular weight cutoff (Millipore) at 4000g until the volume was reduced to ~5 mL. For comparison purposes, the amounts of HEPES, NaCl, SDC, protease inhibitors, and universal nuclease were adjusted to the same final concentrations as those in the cell part. The final volume of the lysates or the concentrated media was adjusted with PBS to 10 mL before the click reaction. Similar steps were performed in the experiments with M0 macrophages and Hep G2 cells, except that the media were collected and the cells were washed directly from the culture flasks. Glycoproteins containing the azide group from Ac4GalNAz labeling were tagged with 100 μM dibenzocyclooctyne (DBCO)-biotin (Click Chemistry Tools) at 37 °C with shaking for 1 h.
Protein Purification and Digestion and Peptide Purification.
After the click-chemistry reaction, the proteins were reduced with 5 mM dithiothreitol at 56 °C for 30 min, alkylated with 14 mM iodoacetamide at room temperature in the dark for 30 min, and purified with the methanol-chloroform precipitation method. Proteins were digested with sequencing-grade modified trypsin (Promega) in the buffer containing 1.6 M urea, 50 mM HEPES pH 8.8, and 5% acetonitrile (ACN), which lasted for 16 h with shaking at 37 °C. The digestion was quenched with 0.4% trifluoroacetic acid (Millipore), and the pH was checked to be lower than 2. The peptides were desalted using a tC18 Sep-Pak Vac Cartridge (Waters) and dried using a vacuum concentrator.
Enrichment of Secreted Glycopeptides.
The dried peptides were resuspended in PBS and glycopeptides were enriched using 150 μL of NeutrAvidin Agarose Resins (Pierce) for 1 h at 37 °C with end-over-end rotation. The beads were subsequently washed 10 times with PBS. The enriched glycopeptides were eluted three times with 8 M guanidine hydrochloride pH 1.5 (Promega) at 56 °C for 2 min each. The eluted glycopeptides were desalted using the Sep-Pak cartridge.
TMT Labeling and High-pH Reverse-Phase Fractionation.
For the quantification experiment using the boosting method, the dried peptides were resuspended in 33 μL of 200 mM HEPES pH 8.5 and 10 μL of ACN. The TMT 6-plex or TMT 10-plex reagents (Thermo) were dissolved in 41 μL of anhydrous ACN, and 5 μL was added to the solution containing the peptides. The reaction was performed at room temperature with shaking for 1 h and subsequently quenched with 8 μL of 5% hydroxylamine hydrochloride in 200 mM HEPES pH 8.5 at room temperature with shaking for 15 min. The peptides from all TMT channels were combined, purified, and dried using a vacuum concentrator. The mixed peptide samples were fractionated using high-pH reverse-phase HPLC with an XBridge C18 3.5 μm, 4.6 × 250 mm column (Waters) over a 40 min gradient of 16–60% ACN in 10 mM ammonium formate, pH 10. The peptides were collected every 2 min and consolidated into 5 fractions.
PNGase F Treatment.
The dried peptides were resuspended in 40 μL of 50 mM ammonium bicarbonate (pH 9) in heavy-oxygen water (H218O) and deglycosylated using 3 units of PNGase F at 37 °C for 3 h with shaking. The reaction was quenched with 1% formic acid (FA) and the pH was adjusted to be ~2. The peptides were purified using the StageTip method described previously and dried before LC-MS/MS analysis.24 For the optimization experiments, each sample was eluted into three fractions with 20%, 50%, and 80% ACN containing 1% acetic acid, respectively. For the quantification experiments with the boosting channel, each of the five fractions was eluted with 50% ACN containing 1% acetic acid. The purified peptides were dried using a vacuum concentrator.
LC-MS/MS Analysis.
The dried peptides were resuspended in a solution containing 5% ACN and 4% FA and analyzed using an online LC-MS/MS system. The separation was performed using a Dionex UltiMate 3000 UHPLC system (Thermo). The microcapillary column was packed in-house (Magic C18AQ, 3 μm, 200 Å, 75 μm × 16 cm). For the optimization experiment, the peptides from the 20%, 50%, and 80% fractions were separated using 112 min gradients of 3–22%, 6–30%, and 8–35% ACN containing 0.125% FA, respectively. For the quantification experiments with the boosting channel, each of the five fractions was separated with a 112 min gradient of 4–17% ACN with 0.125% FA. The LC is connected to an LTQ Orbitrap Elite hybrid mass spectrometer (Thermo). For each cycle, a full MS spectrum was recorded in the Orbitrap cell at the resolution of 60 000 with automatic gain control (AGC) of 1 × 106. For the optimization experiment, the peptides were fragmented in the LTQ using a data-dependent Top20 method where a full MS scan in the Orbitrap is followed by up to 20 MS/MS in the LTQ for the most intense ions. Selected ions were excluded from further sequencing for 90 s. Ions with singly or unassigned charge were not sequenced. Maximum ion accumulation times were 1000 ms for each full MS scan and 50 ms for each MS/MS scan. For the quantification experiments, peptide fragmentation was performed by higher-energy collisional dissociation (HCD) with the normalized collisional energy of 40% using a Top15 method, i.e., up to 15 MS/MS of the most abundant precursor ions were recorded in the Orbitrap cell at the resolution of 15 000.
Data Analysis and Bioinformatics Analysis.
The raw files were converted into an mzXML format and searched using the SEQUEST algorithm.25 The spectra were matched against the human proteome database downloaded from UniProt (www.uniprot.org). In the optimization experiment, the peptide mass tolerance was 20 ppm and the fragment ion mass tolerance was 1.0 Da. The maximum number of missed cleavages was three, and the maximum number of differential modifications per peptide was four. Differential modifications included +15.9949 Da for oxidation of methionine and +2.9883 Da for glycosylation on asparagine, which was deglycosylated with PNGase F in H218O. Static modification included +57.0215 Da for carbamidomethylation of cysteine. Similar parameters were used in the quantification experiment except that the fragment ion mass tolerance was 0.025 Da and the static modification at lysine and the peptide N-terminus (+229.1629 Da) for the TMT labeling was added. The target-decoy method was used to estimate the false discovery rates (FDRs) of peptide and protein identification.26 Linear discriminant analysis (LDA) was applied to control the quality of peptide identification using multiple parameters such as XCorr, ppm, peptide length, and charge state. The FDR was controlled to <1% at the deglycosylated peptide level, and an additional filter was also applied to control the FDR to <1% at the glycoprotein level. The data set was limited to only deglycosylated peptides or glycoproteins when calculating and controlling the FDRs at both levels. Note that only for the secretome quantification with THP-1 monocytes without the boosting channel, one of the five fractions did not pass the LDA, and the results from four fractions were included.
The confidence of the glycosylation site localization was calculated using an algorithm similar to Ascore called ModScore.27 A ModScore of >13 represents the site being well-localized (P < 0.05). Reverse hits and contaminants were removed. The deglycosylated peptides were filtered so that each sequence contained the N-!P–S/T/C motif for N-linked glycosylation. For the quantification, the signal-to-noise ratio of the TMT reporter ions was used. Peptides with a zero S/N were removed. The abundance of each unique glycopeptide was calculated from the sum S/N of all peptide copies detected and the one with the highest ModScore is reported. The final protein abundance change is the sum S/N ratios of all peptides for the protein. Statistical analysis was performed using Perseus,28 including the one-sample t test where the significance change is defined for those with a minimum fold change of 1.5 and the P-value less than 0.05. Phobius was employed to predict the transmembrane region and the signal peptide in proteins.29 SecretomeP was used to predict nonclassical secretion.30 Protein clustering was performed using the Database for Annotation Visualization and Integrated Discovery (DAVID) version 6.8.31 The raw files can be accessed on ftp://massive.ucsd.edu/MSV000086461/.
RESULTS AND DISCUSSION
Principle of Enhancing the Detection of Secreted Glycoproteins with Low Abundances.
Cells secrete glycoproteins into the extracellular environment to communicate with other cells and manipulate the cellular microenvironment. Many secreted glycoproteins have a very low abundance (below ng/mL) among the high-abundance background proteins in the mg/mL range. Methods to enrich secreted proteins from cell culture media were previously reported.14,21,32,33 However, they generally do not target secreted glycoproteins and/or the analysis does not reveal the glycosylation site information. While glycoprotein enrichment is imperative for their global analysis, it is not always sufficient for comprehensive analysis of secreted glycoproteins due to the high-abundance protein background in the culture media. Furthermore, even after they are enriched, the amount of many low-abundance glycoproteins may still be below the detection limit of LC-MS.
Multiplexed proteomics using the tandem mass tag (TMT) allows for the quantification of proteins from multiple samples simultaneously. This increases the reproducibility and shortens the analysis time. Through this approach, the same peptide from different samples is labeled with each channel of the TMT reagents and then combined. The resulting peptides have the same mass-to-charge ratio and appear as a single peak in MS1. It not only increases the analysis efficiency but also improves the peptide signal in MS1, allowing for the better detection and isolation of the precursor ion for MS2. More importantly, much higher intensities of fragments in MS2 enable us to more confidently identify the peptide, while the reporter ions from the TMT tags allow for quantification of the peptides from different samples. Benefiting from this signal enhancement, the approach has been applied for post-translational modification and single-cell analyses by dedicating a separate TMT channel for peptide carriers that will result in the higher total intensity in MS1. Previously, Budnik et al. developed a method called SCoPE-MS to study single-cell proteomics,34 and Yi et al. reported the BASIL method for phosphopeptide identification and quantification.35
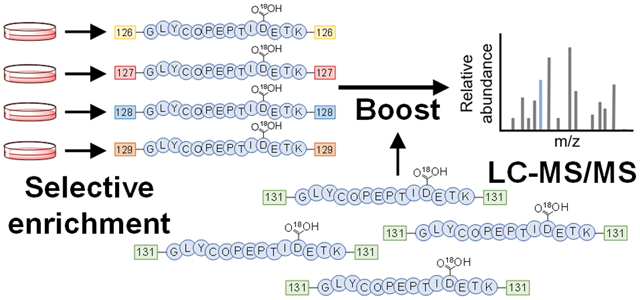
To overcome the problems in secreted glycoprotein analysis, we envisioned that the boosting approach in combination with a glycoprotein enrichment method could be very useful to uncover low-abundance secreted glycoproteins (Figure 1). In previous studies, the boosting sample (channel) generally has the same composition as the quantification samples (channels), e.g., for the BASIL method, the boosting sample is generated by combining the quantification samples; in SCoPE-MS, the number of cells used in the boosting channel is much higher compared with the real single-cell samples. In this work, secreted glycoproteins are among the highly abundant FBS proteins. Using the combined media containing the highly abundant FBS proteins from the samples as the boosting channel may not work well because it further increases the abundance of FBS proteins and will not facilitate the global analysis of secreted proteins. Therefore, a different and more effective boosting sample must be used.
Figure 1.
Principle of the glycopeptide signal boosting for secreted glycoprotein analysis. Enriched glycopeptides from secreted glycoproteins in the cell culture media (shown in the deglycosylated form) are labeled with the TMT reagents. When the boosting sample is added, the signal of the deglycosylated peptide in MS1 is increased, thus facilitating the precursor ion selection for MS/MS fragmentation. Higher intensities of the fragments also allow for confident identification of the glycopeptide.
Previously, we specifically analyzed cell-surface glycoproteins by integrating metabolic labeling with a sugar analog, bioorthogonal chemistry, MS-based proteomics, and studied cell surface glycoproteins in different cell types and their dynamics.36–41 Briefly, cells are labeled using Ac4GalNAz, which is incorporated into the glycan part of glycoproteins in the endoplasmic reticulum or the Golgi apparatus through the classical secretory pathway. In cells, Ac4GalNAz is deacetylated and activated into UDP-GalNAz, which is converted to UDP-GlcNAz or remained as UDP-GalNAz. Many N-glycans can be labeled with the azido sugar. It has also been shown that UDP-GlcNAc can be converted into ManNAz for the labeling of sialic acid-containing glycans.42,43 Because secreted glycoproteins are normally exported through the classical secretory pathway, they can be labeled with the sugar analog and enriched from the culture medium. It is expected that coupling the boosting approach with selective enrichment of secreted glycoproteins will be highly effective to cover low-abundance glycoproteins secreted in the medium.
Comparison of Different Secreted Glycoprotein Sources to Most Effectively Boost the Signals of Secreted Glycopeptides from SCM.
While the cells from which the medium was collected present an attractive choice as a boosting sample, the abundances of secreted glycoproteins inside of the cells could be low and the inferences from many other highly abundant intracellular proteins, especially house-keeping ones, may pose a problem. To make sure that the boosting channel is appropriate and the highest coverage of secreted glycoproteins is obtained, we first compared theoretical annotated secreted glycoproteins that can be found from different sources, including the cells and the media under serum-free or serum-containing conditions.
THP-1 monocytes were first cultured in 25 mL of SCM until the density reached ~8 × 105 cells/mL. The cells were harvested, washed twice with warm PBS to remove FBS proteins, and passaged to either SFM or SCM with 100 μM Ac4GalNAz. After being labeled for 24 h, the cells from both conditions were harvested and separated from the medium, resulting in four sources of glycoproteins: (1) the cell part, SFM; (2) the medium part, SFM; (3) the cell part, SCM; and (4) the medium part, SCM. The cell parts were lysed, and the lysates were collected. The medium parts were concentrated using a centrifugal filter. Proteins containing the azide groups from both the cell lysates and the media were then tagged with DBCO-sulfo-biotin, followed by digestion with trypsin. The resulting glycopeptides containing the biotin group were enriched using NeutrAvidin beads, deglycosylated with PNGase F in H218O, and analyzed with LC-MS/MS.
In the comparison experiments, different numbers of glycoproteins and deglycosylated peptides were detected from the cells and the media under serum-free or serum-containing conditions (Figure 2A,B and Table S1). As expected, the results from the cell parts are very similar, i.e., 989 deglycosylated peptides from 455 glycoproteins and 952 deglycosylated peptides from 443 glycoproteins in the serum-free or serum-containing media, respectively. The number of glycoproteins from the SFM is slightly lower; 698 deglycosylated peptides from 341 glycoproteins. Yet, the number of glycoproteins detected from the SCM is the lowest, with only 54 deglycosylated peptides from 41 glycoproteins found. In a separate search from this experiment where the FDR is not restricted to only glycopeptides, peptides with the highest number of hits are from bovine albumin (62 unique peptides and 330 total peptides) and fetuin-A (19 unique peptides and 219 total peptides), while the human protein detected with the highest number of peptides is progranulin (GRN) with only 4 unique peptides and 17 total peptides (all contain a glycosylation site). This demonstrated that the enrichment alone is not enough to overcome high-abundance FBS proteins left in the final sample through nonspecific binding.
Figure 2.
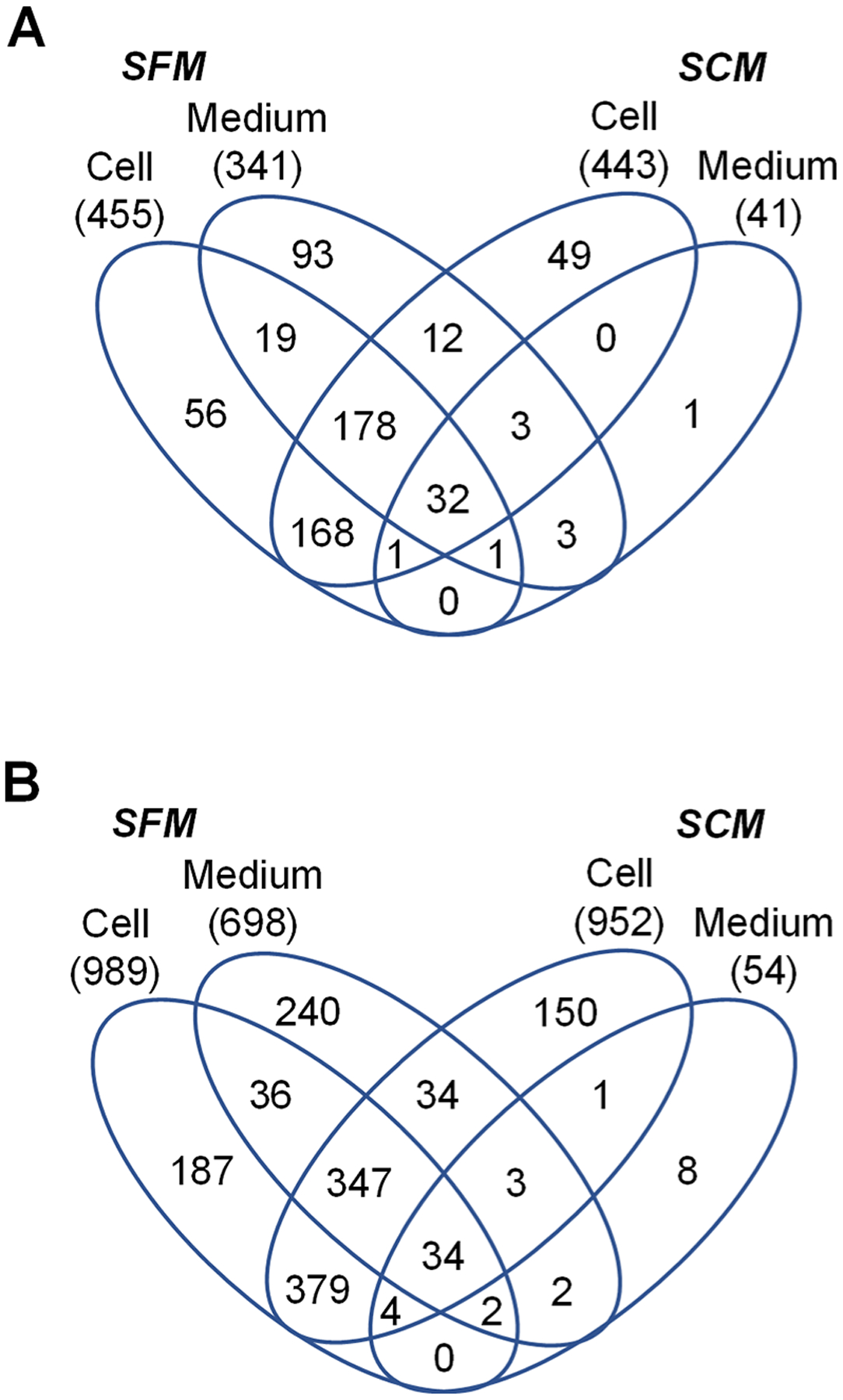
Comparison of glycoproteins and deglycosylated peptides from different boosting samples. The number of glycoproteins (A) or deglycosylated peptides (B) detected from the cells or the media from serum-free or serum-containing conditions. The number in parentheses is the number of glycoproteins or unique deglycosylated peptides identified.
At first glance, the number of glycoproteins and deglycosylated peptides detected from the cell part grown in SFM is the highest and could serve as the boosting channel during the quantification. We performed several bioinformatic analyses to determine the fraction of theoretical secreted glycoproteins among these different samples. Based on the criteria used in previous studies, secreted proteins should contain a signal peptide sequence but not a transmembrane region.14,21 We used Phobius to predict these two components and also compared with the UniProt database for proteins that contain the keyword “Signal” or “Secreted”.29 Due to its coverage, the medium part of the SCM condition has the lowest number of glycoproteins with the transmembrane region, with the signal peptide, without the transmembrane region but with the signal peptide, or containing the keywords “Signal” or “Secreted” (Figure 3A). Those from the cell parts in both the SFM and SCM conditions have the highest numbers of glycoproteins that matched these criteria, except for proteins that contain a signal peptide but not a transmembrane region, which is characteristic for secreted proteins and the highest in the medium part of the SFM (Figure 3A). On the contrary, the percentages of proteins that match these criteria revealed that, as expected, all glycoproteins (100%) enriched from the medium part of the SCM are annotated with the keywords “Signal” or “Secreted” (Figure 3B). Additionally, 68% have a signal peptide sequence but not the transmembrane region, 95% have a signal peptide (highest among the four conditions), and 32% have the transmembrane region (the lowest among the four). While the percentages are not as high as those from the medium part of SCM, the medium part of SFM still has 46% with a signal peptide sequence but not a transmembrane region. Those enriched from the cell parts have the highest number of proteins with the transmembrane region, the lowest for proteins containing a signal peptide sequence, or containing a signal peptide sequence but not the transmembrane region. Nonclassical secretory pathways where glycoproteins are secreted through are also predicted (Table S1). Additionally, we clustered the secreted glycoproteins based on their gene-ontology (GO) terms. Glycoproteins detected from the SFM resulted in the highest number of proteins annotated in the extracellular vesicle (secreted proteins from databases are also clustered into this GO) and the lowest P-values (highest −log(P-value); as shown in Figure 3C). The number of proteins that belong to the integral component of membrane is also the lowest when the glycoproteins from the SCM are not considered.
Figure 3.
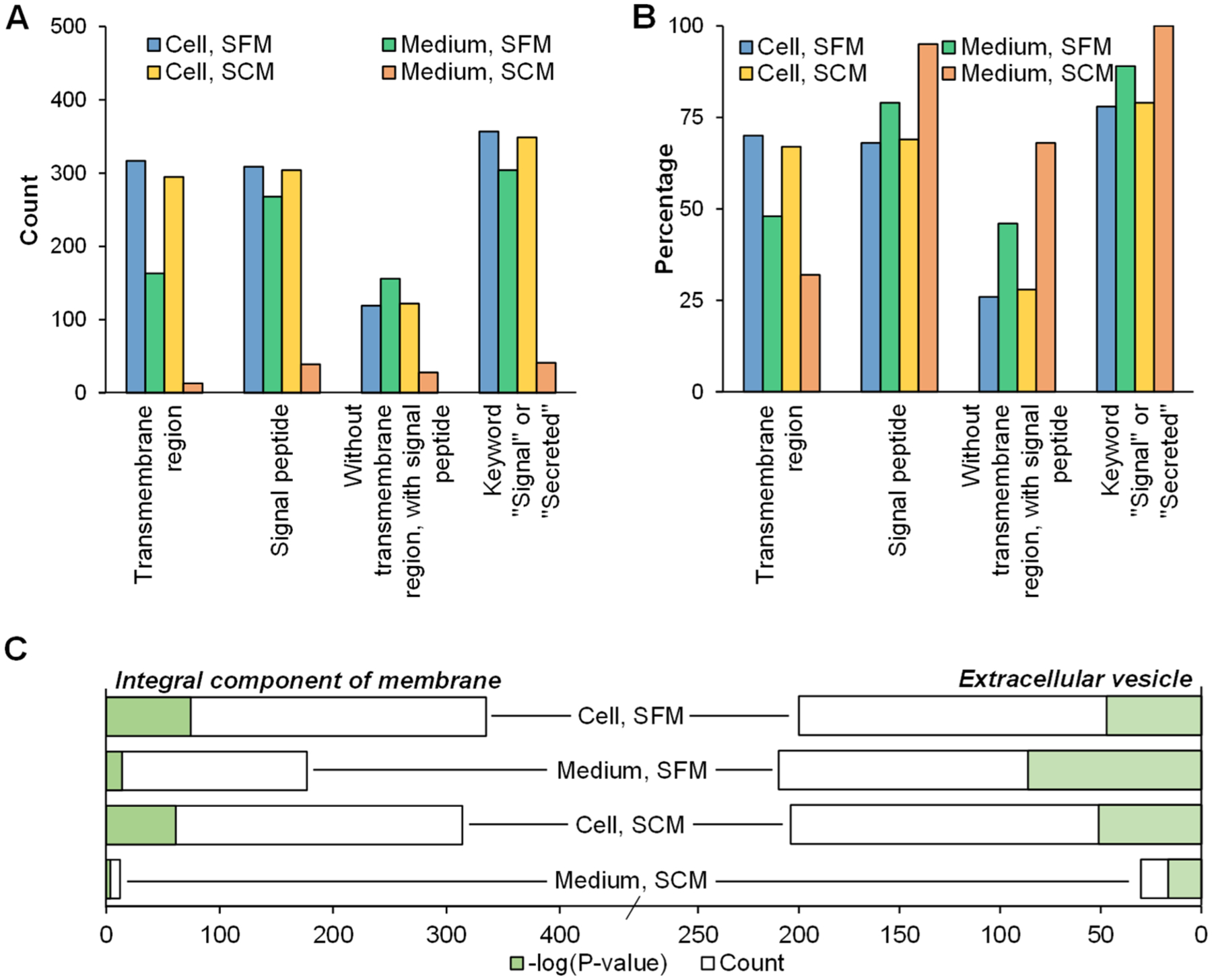
Analysis of secreted glycoproteins from different boosting samples. (A) The number of glycoproteins from Figure 2 that contain the transmembrane region, have a signal peptide sequence, does not contain the transmembrane region, but contain a signal peptide sequence, and are annotated as the keywords “signal” or “secreted” from UniProt. (B) The percentage of glycoproteins from (A) to the total number of detected glycoproteins for each sample. (C) Gene ontology (GO) based clustering of glycoproteins related to the integral component of membrane or extracellular vesicle in different samples based on cellular components.
Taking these together, SFM resulted in the highest number of theoretically secreted glycoproteins, and the coverage is much higher compared with that from the SCM. The SFM contains almost all glycoproteins detected from the SCM except two glycoproteins, i.e., C–C motif chemokine 24 (CCL24) and cathepsin Z (CTSZ). It also contains the lowest number of proteins with the transmembrane region, which is not characteristic of typical secreted glycoproteins. Additionally, considering that glycosylation is an important step in the classical secretory pathway, almost all secreted proteins may be glycosylated and could possibly be used as the boosting sample without the enrichment. We grew the cells in the SFM, omitted the enrichment with NeutrAvidin beads, and directly performed the deglycosylation with PNGase F for glycopeptides prior to LC-MS analysis. With this approach, only 83 deglycosylated peptides from 66 glycoproteins were detected (Table S1). The results are worse than expected. Therefore, we concluded that the enriched secreted glycoproteins in the SFM are best suited for boosting the signals of those from cells grown in the SCM. While previous studies showed that cells grown in the SFM may alter the abundance of secreted proteins, the glycoproteins here are merely used to boost the signal while the true quantification results will still be obtained from the cells grown under the SCM conditions.
Optimization of the Ratio between the Boosting and the Quantification Channels.
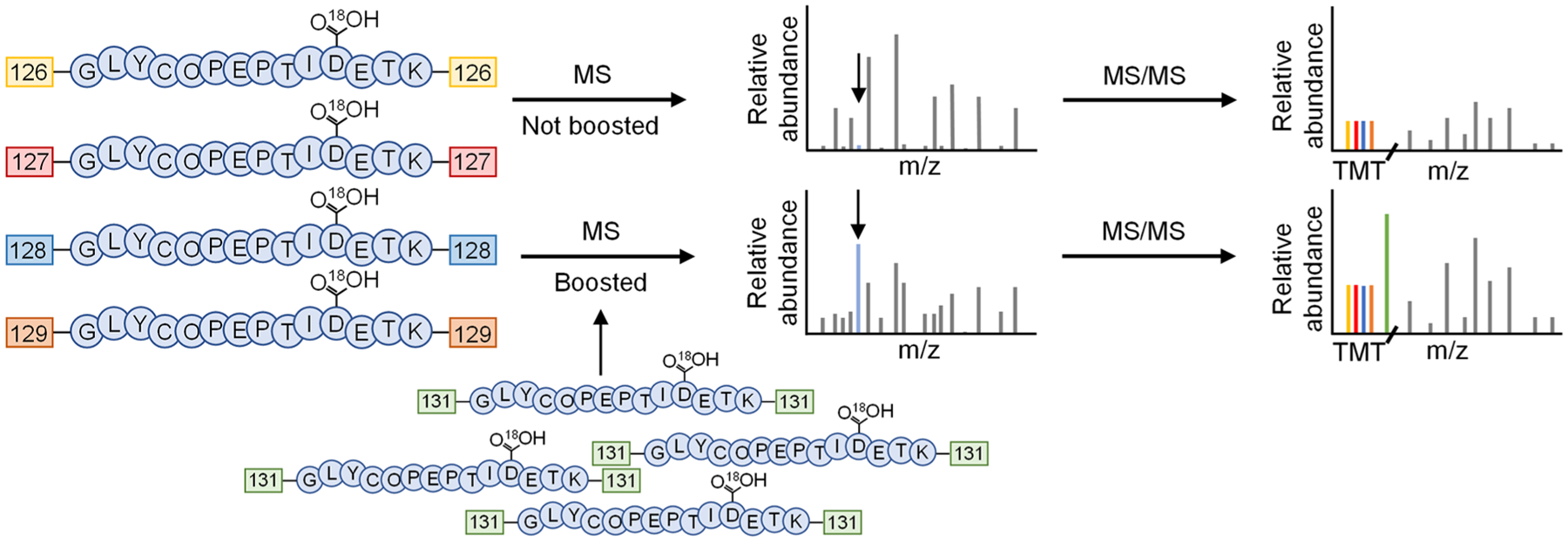
To evaluate the boosting effect, we performed the quantification experiments without the boosting channel and with different boosting-to-sample ratios. The cells were prelabeled with Ac4GalNAz and treated with LPS or PBS (as a control) for 12 h. The media were collected, and secreted glycoproteins were tagged. After the enrichment, the glycopeptides were labeled with the TMT 6-plex reagents (Figure 4). In the experiment without the boosting sample, channels 126–129 of the TMT reagents were used to label glycopeptides from the biological duplicate experiments of cells treated with LPS or PBS, while channels 130 and 131 were left blank. With this approach, 103 unique deglycosylated peptides were detected from 71 glycoproteins (Figure 5A and Table S2). With the boosting approach, channel 131 was dedicated for the boosting sample. Since the cells treated with LPS or PBS are expected to secrete glycoproteins differently, we combined the SFM collected from both the cells treated with LPS or PBS as a boosting sample. Channel 130 was left blank. With the boosting sample, the number of glycoproteins and deglycosylated peptides identified markedly increased. For the boosting-to-sample ratio of 2:1, i.e., the volume of the SCM for each channel is 5 mL and that of the boosting channel is 10 mL (5 mL from the SFM of cells treated with LPS and 5 mL from cells treated with PBS), 179 unique deglycosylated peptides were quantified from 107 secreted glycoproteins (Figure 5A and Table S2). With the ratio of 10:1 where 5 mL of the SCM for each channel and 50 mL of the SFM for the boosting channel were used (25 mL from the SFM of cells treated with LPS and 25 mL of cells treated with PBS), the number further increased to 308 deglycosylated peptides from 178 glycoproteins (Figure 5A and Table S2). Compared with the results from the identification in the comparison experiment, the secreted glycoproteins quantified at the 10-to-1 ratio cover 89% of the secreted glycoproteins (36 of 41) detected from the SCM. Nonetheless, the coverage is lower than the identification experiment in the SFM (599 deglycosylated peptides and 341 glycoproteins). One major reason is that the ion trap used for MS2 in the identification experiment is much more sensitive than the Orbitrap cell employed for MS2 in this quantification experiment because of the detection of the TMT reporter ions.
Figure 4.
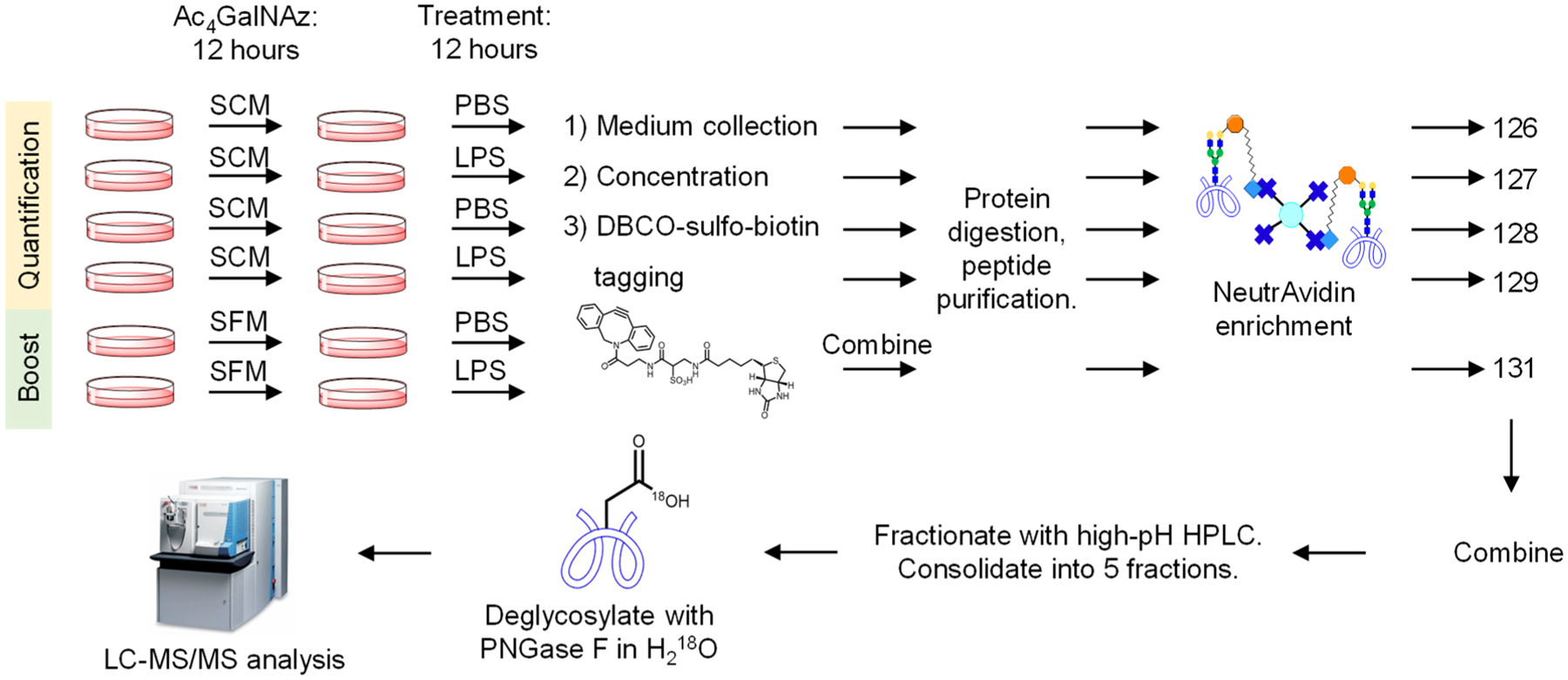
Experimental procedure for quantitative analysis of secreted glycoproteins using a boosting channel. The cells are prelabeled with Ac4GalNAz for glycoprotein enrichment in either SCM (quantification channels) or SFM (boosting channel). The media are collected and glycoproteins are tagged with DBCO-sulfo-biotin. The enriched glycopeptides are labeled with the TMT 6-plex reagents, deglycosylated with PNGase F, and analyzed with LC-MS/MS.
Figure 5.
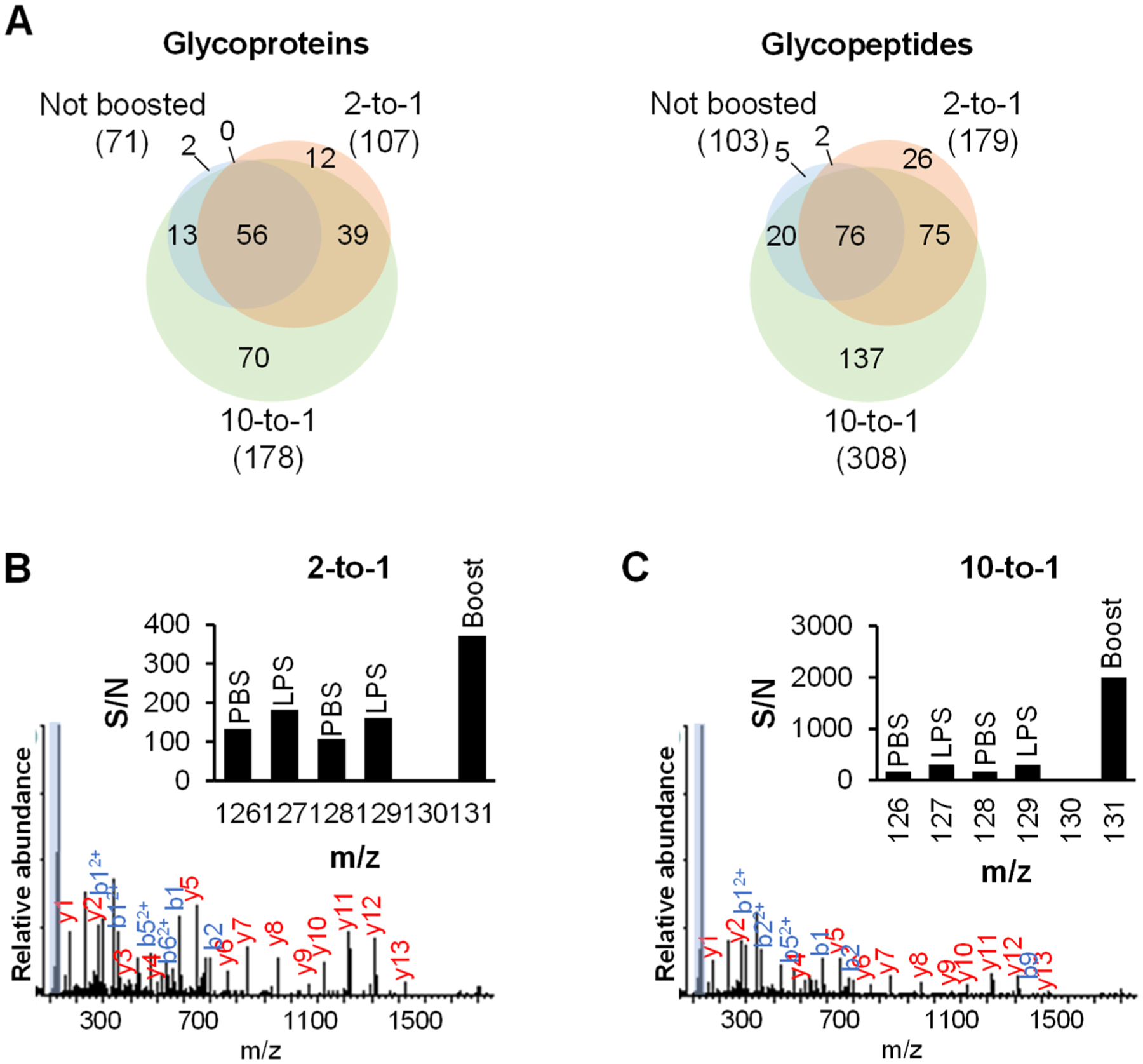
Optimizing the boosting-to-sample ratio. (A) The number of secreted glycoproteins or deglycosylated peptides from the experiments without the boosting channel, boosted at the 2-to-1 ratio, or boosted at the 10-to-1 ratio. (B and C) Example tandem mass spectra of the deglycosylated peptide KLPPGLLAN#FTLLR from LRG1 from the experiments boosted at the 2-to-1 ratio (B) or boosted at the 10-to-1 ratio (C). The TMT reporter ion region is highlighted and shown in the inset.
In one example, the deglycosylated peptide KLPPGLLAN#FTLLR, where # represents the glycosylation site, was quantified in the boosting experiments with the 2:1 and 10:1 boosting ratios (Figure 5B,C). The peptide was confidently identified with the XCorr values of 4.86, and 4.93, respectively. The glycosylation site is well-localized at N186 with the ModScore of 1000 in all experiments. The peptide is from leucine-rich alpha-2-glycoprotein (LRG1). As the boosting-to-sample ratio increases, the signal at the channel of 131 also increases. Note that the abundance of this glycoprotein is high enough so that it can be detected even without the boosting channel. It might also be due to the LPS treatment that increased the glycoprotein abundance in the secretome. For other glycopeptides that were not detected from the quantification experiment without the boosting channel, their abundances may be too low to be selected for further fragmentation, or not enough fragments are produced for glycopeptide identification in MS2. The addition of the boosting channel increases the signals in MS1 and MS2 and improves the glycopeptide coverage.
Quantification of Secreted Glycoproteins from THP-1 Monocytes in Response to LPS.
In the experiment with THP-1 monocytes, we quantified 309 deglycosylated peptides from 179 glycoproteins (Figure 6C and Table S2). The reproducibility from two biological duplicate experiments is reasonably high at both the deglycosylated peptide and glycoprotein levels (Figure 6A,B). GO analysis demonstrates that over one hundred glycoproteins are related to the extracellular vesicle (P = 8.5 × 10–50), 170 are annotated with the keyword “Secreted” or “Signal”, while 82 are predicted to not have the transmembrane region but contain a signal peptide sequence. At the peptide level, the ratios of 66 unique deglycosylated peptides from secreted glycoproteins are ≥1.5 from the cells treated with LPS compared with the control sample (Figure S2A). Clustering of 27 glycoproteins that were up-regulated demonstrated that they are involved in biological processes such as positive regulation of programmed cell death, extracellular matrix organization, inflammatory response, and response to cytokine, which are the expected responses (Figure 6D). Several deglycosylated peptides with the glycosylation sites of N118 and N258 from tumor necrosis factor-inducible gene 6 protein (TNFAIP6) were up-regulated by ~16 times. TNFAIP6 has been associated with several inflammatory diseases and was expressed in response to TNF-α, one of the known cytokines secreted from cells treated with LPS.44 Another study showed that TNFAIP6 inhibited pro-inflammatory proteins while increasing the anti-inflammatory ones. Among these, TNFAIP6 inhibited toll-like receptor 4 (TLR4), the direct receptor of LPS, from associating with myeloid differentiation primary response protein MyD88 (MYD88) to suppress NF-κB activation.45 This suggests that after the inflammation induced by LPS, cells may secrete more TNFAIP6 to counter this inflammation. Surprisingly, only one glycoprotein containing the N170 site from angiotensinogen (AGT) was down-regulated. AGT normally has roles in blood pressure regulation and bodily fluid and electrolyte homeostasis, but a recent study showed its involvement in the inflammatory response.46 If the abundance of a secreted glycoprotein is already very low and the even lower amount of the glycoprotein is secreted from cells with the LPS treatment, the boosting samples may not be able to effectively improve its detection in the secretome. This potentially results in the underestimated number of down-regulated glycoproteins and glycopeptides with very low abundances from the LPS-treated cells. Some exemplary regulated glycosylation sites are shown in Table 1.
Figure 6.
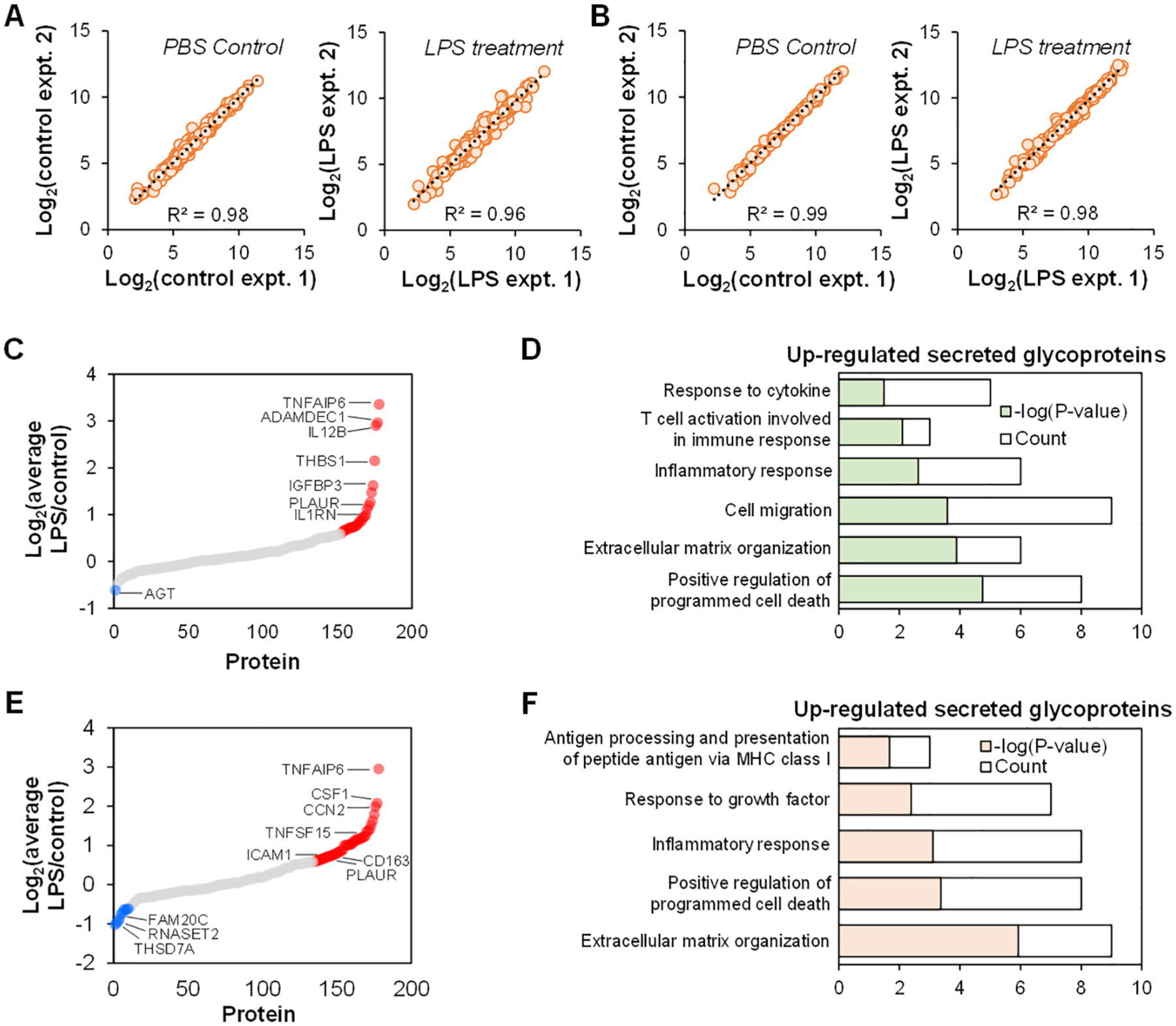
Quantification of secreted glycoproteins from THP-1 monocytes and M0 macrophages treated with LPS. (A-B) Correlation of deglycosylated peptides (A) and glycoproteins (B) quantified from THP-1 monocytes. (C) The abundance changes of secreted glycoproteins from THP-1 monocytes stimulated with LPS. The cutoff for up-or down-regulation is the fold change of 1.5. (D) GO clustering of up-regulated secreted glycoproteins from THP-1 monocytes after the LPS treatment based on biological process. (E) The abundance changes of secreted glycoproteins from M0 macrophages stimulated with LPS. (F) GO clustering of up-regulated secreted glycoproteins from M0 macrophages after the LPS treatment based on biological process.
Table 1.
Example Deglycosylated Peptides from Secreted Glycoproteins Quantified from THP-1 Monocytes and Macrophages in Response to LPS or from Hep G2 Cells Treated with TGF-β
| cell | UniProt ID | gene | deglycosylated peptide | site | ModScore | ratio | annotation |
|---|---|---|---|---|---|---|---|
| THP-1 Monocytes + LPS | P98066 | TNFAIP6 | K.N#TSTTSTGNK.N | 258 | 150.77 | 23.10 | tumor necrosis factor-inducible gene 6 protein |
| O15204 | ADAMDEC1 | R.EIKNN#QTEK.H | 61 | 17.01 | 9.58 | ADAM DEC1 | |
| P29460 | IL12B | R.KN#ASISVR.A | 303 | 1000 | 7.51 | interleukin-12 subunit beta | |
| P07996 | THBS1 | K.KVSCPIMPCSN#ATVPD GECCPR.C | 360 | 1000 | 4.11 | thrombospondin-1 | |
| P05362 | ICAM1 | R.LNPTVTYGN#DSFSAK.A | 267 | 127.53 | 2.46 | intercellular adhesion molecule 1 | |
| THP-1 M0 Macrophages + LPS | Q01151 | CD83 | R.N#TTSCNSGTYR.C | 96 | 100.21 | 8.94 | CD83 antigen |
| P98066 | TNFAIP6 | K.N#TSTTSTGNK.N | 258 | 150.77 | 8.34 | tumor necrosis factor-inducible gene 6 protein | |
| P09603 | CSF1 | K.NVFN#ETK.N | 154 | 73.31 | 4.23 | macrophage colony-stimulating factor 1 | |
| P17936 | IGFBP3 | K.VDYESQSTDTQN#FSS ESKR.E | 199 | 1000 | 4.06 | insulin-like growth factor-binding protein 3 | |
| Q9BXX0 | EMILIN2 | K.SLN#DTMHRK.F | 587 | 1000 | 0.62 | EMILIN-2 | |
| Hep G2 + TGF-β | Q14767 | LTBP2 | R.DECWCPAN#STGK.F | 421 | 1000 | 10.60 | latent-transforming growth factor beta-binding protein 2 |
| P19883 | FST | R.CVCAPDCSN#ITWK.G | 124 | 1000 | 3.65 | Follistatin | |
| P15018 | LIF | K.LN#ATADILR.G | 138 | 1000 | 2.12 | leukemia inhibitory factor |
refers to the N-glycosylation site.
Five cytokines were also quantified despite their abundances at the ng/mL level, including interleukin-12 subunit beta (IL12B), macrophage colony-stimulating factor 1 (CSF1), growth/differentiation factor 11 (GDF11), bone morphogenetic protein 1 (BMP1), and progranulin (GRN). Among these, the deglycosylated peptide with the site of N303 from IL12B was up-regulated by 7.5 times, and this has been reported previously.47,48 We also found that the two deglycosylated peptides with the site of N183 or N267 from intercellular adhesion molecule 1 (ICAM1) were up-regulated by 2.1 and 2.5 times, respectively. ICAM1 is a known cell-surface glycoprotein involved in leukocyte adhesion and is often up-regulated under inflammatory conditions.49 ICAM1 can be shed and was found in a study that the LPS challenge increased its concentration in the culture medium.50 This may explain the detection of some glycoproteins with the transmembrane region and a signal peptide in the current experiment, despite that they are generally considered as being localized at the cell surface but not in the secretome.
Analysis of Glycoproteins Secreted from THP-1 Macrophages Treated with LPS.
THP-1 cells were further differentiated into M0 macrophages using PMA similar to the previously reported protocols.22,23,51 The cells were subsequently labeled with Ac4GalNAz and treated with LPS as the above experiment with THP-1 monocytes. In total, 400 deglycosylated peptides were quantified from 178 glycoproteins (Table S3). The results from both replicates indicate the relatively high R2 values (Figure S1A,B). More than one hundred glycoproteins are related to the extracellular vesicle (P = 2.9 × 10–37), and 170 are annotated with the keyword “Secreted” or “Signal”. Moreover, 81 are predicted to not have the transmembrane region but contain a signal peptide. The results at the protein level are shown in Figure 6E. Forty-four glycoproteins were up-regulated while ten were down-regulated. Clustering of up-regulated secreted glycoproteins revealed that they are involved in extracellular matrix organization (P = 1.2 × 10–6), inflammation response (P = 7.8 × 10–4), and response to growth factor (P = 4.1 × 10–3; Figure 6F). Among the quantified deglycosylated peptides, 101 were up-regulated in the cells treated with LPS while 9 were down-regulated compared with the control group (Figure S2B). The deglycosylated peptide with the highest fold change is the one containing the N96 site from CD83 antigen (CD83), a well-known surface glycoprotein, which was up-regulated by 9.2-fold in cells treated with LPS. In dendritic cells, CD83 was reported to be preformed inside the cells and transported to the surface once the cells are activated.52 The soluble form of CD83 has been well-documented and may interact with the TLR4/MD-2 complex (receptor for LPS) to reduce the inflammation.53 The peptide containing the glycosylation site was also quantified from the experiment with THP-1 monocytes, but the secretion was not regulated (fold change = 1.0), suggesting that the genetically identical cells responded differently to LPS. Five cytokines were quantified, including CSF1, tumor necrosis factor ligand superfamily member 15 (TNFSF15), GRN, BMP1, and CCL24. Among these, the deglycosylated peptide containing the N154 site from CSF1 was up-regulated by over 4 times, in contrast to the experiment with THP-1 monocytes where the peptide was not regulated. Yet, some deglycosylated peptides, such as those with the N258 site of TNFAIP6, were also up-regulated from cells treated with LPS, which is similar to the experiment with THP-1 monocytes. Some examples of deglycosylated peptides are displayed in Table 1.
Analysis of Secreted Glycoproteins from Hep G2 Cells Treated with TGF-β.
To further demonstrate the effectiveness of the current method, we applied it to study secreted glycoproteins from Hep G2 cells treated with TGF-β. TGF-β has been reported to induce the epithelial-to-mesenchymal transition (EMT) in Hep G2 cells, which is a process involved in cancer metastasis where epithelial cells become more invasive and mobile.54 The cells were prelabeled with 100 μM Ac4GalNAz for 12 h and then treated with 10 ng/mL TGF-β for 48 h before medium collection. We expanded the number of samples analyzed by employing the TMT 10-plex reagents and performing the experiment in biological triplicates (Figure 7A). We quantified 531 unique deglycosylated peptides from 236 glycoproteins (Figure 7B and Table S4). The glycoproteins related to the extracellular vesicle are highly enriched with a very low P value (P = 8.8 × 10–73), and 231 proteins are annotated with the keyword “Secreted” or “Signal”. Furthermore, 147 are predicted to not have the transmembrane region but contain a signal peptide. We found 21 secreted glycoproteins being up-regulated and 6 being down-regulated in response to TGF-β. The proteins that were up-regulated are involved in processes such as extracellular matrix organization (Figure 7C). Latent-transforming growth factor beta-binding protein 2 (LTBP2) was up-regulated by over 6 times (Figure 7B). The protein was reported to be up-regulated at both the mRNA and protein levels after TGF-β stimulation,55 corroborating the results from this experiment. Other glycoproteins that have also been reported to be up-regulated after TGF-β stimulation are also shown in the figure, including metalloproteinase inhibitor 1 (TIMP1) and lysyl oxidase homologue 4 (LOXL4).56,57 Several examples of quantified deglycosylated peptides are listed in Table 1.
Figure 7.
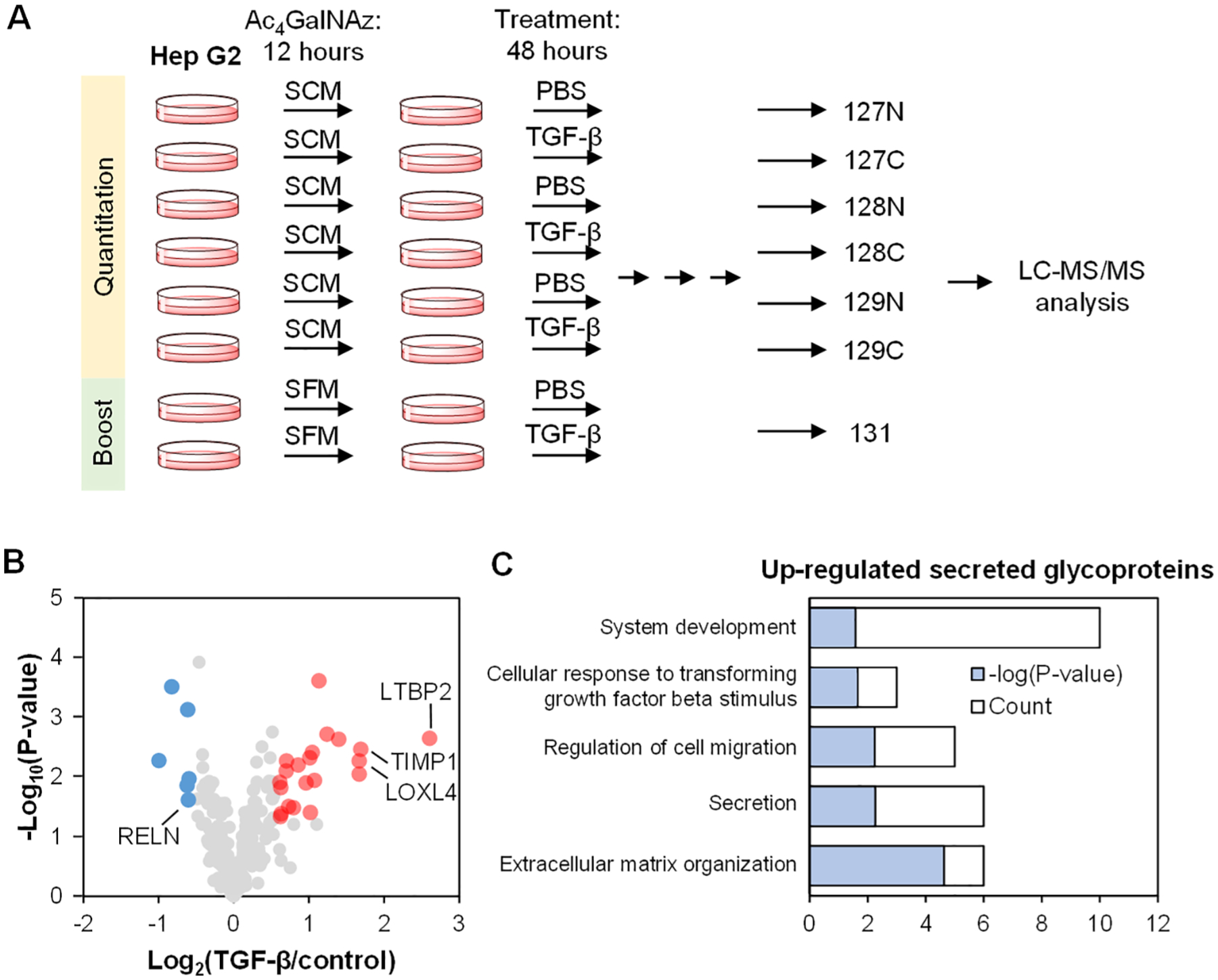
Quantification of secreted glycoproteins from Hep G2 cells treated with TGF-β. (A) Experimental scheme using the TMT 10-plex platform. (B) Glycoprotein abundance changes after the TGF-β treatment from the triplicate experiments. A t test was used to determine the significance (shown with P < 0.05 and the ratio cutoff of 1.5). (C) GO clustering of secreted glycoproteins that are up-regulated after the TGF-β treatment based on biological process.
CONCLUSIONS
In this work, we coupled the selective enrichment of glycopeptides with a signal boosting approach to enhance the coverage of secreted glycoproteins with low abundances among many extremely high-abundance FBS proteins. This method was applied to globally quantify glycoproteins secreted from THP-1 monocytes and macrophages in response to LPS and from Hep G2 cells treated with TGF-β. While the interference from highly abundant FBS proteins on the analysis of secreted proteins can be eliminated by using serum-free media, the serum starvation alters protein secretion even after a short period of time, and thus the quantification of secreted proteins is questionable. Here, we aimed to quantify secreted glycoproteins when cells are grown under the normal conditions, i.e., the serum-containing media. The enrichment of secreted glycopeptides alone is not sufficient for their comprehensive analysis because some high-abundance peptides are still present in the enriched samples due to nonspecific binding. Moreover, LC-MS has its detection limit, which is biased against the analysis of low-abundance glycoproteins. To overcome these issues, we combined the selective enrichment of glycopeptides from secreted glycoproteins with a boosting approach for global analysis of secreted glycoproteins by LC-MS. The boosting sample was carefully chosen by comparing the coverage of theoretical secreted glycoproteins from the cells or the media under serum-free or serum-containing conditions. The results demonstrated that the serum-free medium as a glycoprotein source can best serve as the boosting sample due to the higher coverage and the selectivity of secreted glycoproteins. Nonetheless, the quantitative information on secreted glycoproteins was obtained solely from the cells grown under the serum-containing conditions. We first applied this integrated method to globally quantify secreted glycoproteins in THP-1 monocytes and macrophages in response to LPS. Almost 400 deglycosylated peptides from ~200 secreted glycoproteins were quantified from THP-1 monocytes and macrophages treated with LPS, respectively. We further expanded the number of samples quantified by using the TMT 10-plex reagents to determine secreted glycoproteins from Hep G2 cells treated with TGF-β in the EMT context, in which we quantified over 200 secreted glycoproteins related to the extracellular matrix organization. Here, the glycosylation site information was also obtained, which provides the solid experimental evidence for the identification of glycoproteins, minimizing the false-positive rates. Considering the importance of secreted glycoproteins, this method can be extensively applied in the biological and biomedical research fields.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R01GM127711) and the National Science Foundation (2003597).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c05126.
Quantification of glycoproteins and deglycosylated peptides from THP-1 M0 macrophages treated with LPS (Figure S1) and quantification deglycosylated peptides from THP-1 monocytes and M0 macrophages treated with LPS (Figure S2) (PDF)
Deglycosylated peptides and glycoproteins detected from each potential source for signal boosting (Table S1) (XLSX)
Deglycosylated peptides and glycoproteins quantified from THP-1 monocytes treated with LPS (Table S2) (XLSX)
Deglycosylated peptides and glycoproteins quantified from THP-1 M0 macrophages treated with LPS (Table S3) (XLSX)
Deglycosylated peptides and glycoproteins quantified from Hep G2 cells treated with TGF-β (Table S4) (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c05126
The authors declare no competing financial interest.
REFERENCES
- (1).Seiradake E; Zhao Y; Lu W; Aricescu AR; Jones EY Methods Mol. Biol 2015, 1261, 115–27. [DOI] [PubMed] [Google Scholar]
- (2).Turner MD; Nedjai B; Hurst T; Pennington DJ Biochim. Biophys. Acta, Mol. Cell Res 2014, 1843 (11), 2563–2582. [DOI] [PubMed] [Google Scholar]
- (3).Wahl-Jensen V; Kurz SK; Hazelton PR; Schnittler HJ; Stroher U; Burton DR; Feldmann HJ Virol. 2005, 79 (4), 2413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chandler K; Goldman R Mol. Cell. Proteomics 2013, 12 (4), 836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Thaysen-Andersen M; Thogersen IB; Lademann U; Offenberg H; Giessing AMB; Enghild JJ; Nielsen HJ; Brunner N; Hojrup P Biochim. Biophys. Acta, Proteins Proteomics 2008, 1784 (3), 455–463. [DOI] [PubMed] [Google Scholar]
- (6).Song E; Mechref Y Biomarkers Med. 2015, 9 (9), 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kailemia MJ; Park D; Lebrilla CB Anal. Bioanal. Chem 2017, 409 (2), 395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhu JH; Warner E; Parikh ND; Lubman DM Mass Spectrom. Rev 2019, 38 (3), 265–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lilja H; Ulmert D; Vickers AJ Nat. Rev. Cancer 2008, 8 (4), 268–78. [DOI] [PubMed] [Google Scholar]
- (10).Llop E; Ferrer-Batalle M; Barrabes S; Guerrero PE; Ramirez M; Saldova R; Rudd PM; Aleixandre RN; Comet J; de Llorens R; Peracaula R Theranostics 2016, 6 (8), 1190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hsiao C-J; Tzai T-S; Chen C-H; Yang W-H; Chen C-H Dis. Markers 2016, 2016, 8915809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Popa C; Netea MG; van Riel PL; van der Meer JW; Stalenhoef AF J. Lipid Res 2007, 48 (4), 751–62. [DOI] [PubMed] [Google Scholar]
- (13).Terrando N; Monaco C; Ma D; Foxwell BM; Feldmann M; Maze M Proc. Natl. Acad. Sci. U. S. A 2010, 107 (47), 20518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shin J; Rhim J; Kwon Y; Choi SY; Shin S; Ha CW; Lee C Sci. Rep 2019, 9 (1), 3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Boersema PJ; Geiger T; Wisniewski JR; Mann M Mol. Cell. Proteomics 2013, 12 (1), 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yen TY; Haste N; Timpe LC; Litsakos-Cheung C; Yen R; Macher BA J. Proteomics 2014, 96, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yang W; Zhou JY; Chen L; Ao MH; Sun SS; Aiyetan P; Simmons A; Zhang H; Jackson JB Clin. Proteom 2014, 11, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Smeekens JM; Xiao H; Wu RJ Proteome Res. 2017, 16 (2), 1039–1049. [DOI] [PubMed] [Google Scholar]
- (19).Suttapitugsakul S; Sun F; Wu R Anal. Chem 2020, 92 (1), 267–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bjorhall K; Miliotis T; Davidsson P Proteomics 2005, 5 (1), 307–17. [DOI] [PubMed] [Google Scholar]
- (21).Eichelbaum K; Winter M; Diaz MB; Herzig S; Krijgsveld J Nat. Biotechnol 2012, 30 (10), 984–90. [DOI] [PubMed] [Google Scholar]
- (22).Chanput W; Mes J; Vreeburg RA; Savelkoul HF; Wichers HJ Food Funct. 2010, 1 (3), 254–61. [DOI] [PubMed] [Google Scholar]
- (23).Chanput W; Mes JJ; Savelkoul HF; Wichers HJ Food Funct. 2013, 4 (2), 266–76. [DOI] [PubMed] [Google Scholar]
- (24).Rappsilber J; Mann M; Ishihama Y Nat. Protoc 2007, 2 (8), 1896–906. [DOI] [PubMed] [Google Scholar]
- (25).Eng JK; McCormack AL; Yates JR J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–89. [DOI] [PubMed] [Google Scholar]
- (26).Elias JE; Gygi SP Nat. Methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- (27).Beausoleil SA; Villen J; Gerber SA; Rush J; Gygi SP Nat. Biotechnol 2006, 24 (10), 1285–92. [DOI] [PubMed] [Google Scholar]
- (28).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J Nat. Methods 2016, 13 (9), 731–40. [DOI] [PubMed] [Google Scholar]
- (29).Kall L; Krogh A; Sonnhammer EL J. Mol. Biol 2004, 338 (5), 1027–36. [DOI] [PubMed] [Google Scholar]
- (30).Bendtsen JD; Jensen LJ; Blom N; Von Heijne G; Brunak S Protein Eng., Des. Sel 2004, 17 (4), 349–56. [DOI] [PubMed] [Google Scholar]
- (31).Huang DW; Sherman BT; Lempicki RA Nat. Protoc 2009, 4 (1), 44–57. [DOI] [PubMed] [Google Scholar]
- (32).Roper SM; Zemskova M; Neely BA; Martin A; Gao P; Jones EE; Kraft AS; Drake RR Proteomics: Clin. Appl 2013, 7 (5–6), 367–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Witzke KE; Rosowski K; Muller C; Ahrens M; Eisenacher M; Megger DA; Knobloch J; Koch A; Bracht T; Sitek BJ Proteome Res. 2017, 16 (1), 137–146. [DOI] [PubMed] [Google Scholar]
- (34).Budnik B; Levy E; Harmange G; Slavov N Genome Biol. 2018, 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yi L; Tsai CF; Dirice E; Swensen AC; Chen J; Shi T; Gritsenko MA; Chu RK; Piehowski PD; Smith RD; Rodland KD; Atkinson MA; Mathews CE; Kulkarni RN; Liu T; Qian WJ Anal. Chem 2019, 91 (9), 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen W; Smeekens JM; Wu R Chem. Sci 2015, 6 (8), 4681–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Smeekens JM; Chen W; Wu RJ Am. Soc. Mass Spectrom 2015, 26 (4), 604–14. [DOI] [PubMed] [Google Scholar]
- (38).Xiao H; Tang GX; Wu R Anal. Chem 2016, 88 (6), 3324–3332. [DOI] [PubMed] [Google Scholar]
- (39).Xiao H; Wu R Chem. Sci 2017, 8 (1), 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Xiao H; Suttapitugsakul S; Sun F; Wu R Acc. Chem. Res 2018, 51 (8), 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Suttapitugsakul S; Ulmer LD; Jiang C; Sun F; Wu R Anal. Chem 2019, 91 (10), 6934–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Laughlin ST; Bertozzi CR Nat. Protoc 2007, 2 (11), 2930–44. [DOI] [PubMed] [Google Scholar]
- (43).Shajahan A; Supekar NT; Wu H; Wands AM; Bhat G; Kalimurthy A; Matsubara M; Ranzinger R; Kohler JJ; Azadi P ACS Chem. Biol 2020, 15 (10), 2692–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Milner CM J. Cell Sci 2003, 116 (10), 1863–1873. [DOI] [PubMed] [Google Scholar]
- (45).Mittal M; Tiruppathi C; Nepal S; Zhao YY; Grzych D; Soni D; Prockop DJ; Malik AB Proc. Natl. Acad. Sci. U. S. A 2016, 113 (50), E8151–E8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Benigni A; Cassis P; Remuzzi G EMBO Mol. Med 2010, 2 (7), 247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Peng JC; Abu Bakar S; Richardson MM; Jonsson JJ; Frazer IH; Nielsen LK; Morahan G; Thomas R Immunol. Cell Biol 2006, 84 (2), 227–32. [DOI] [PubMed] [Google Scholar]
- (48).Zhao Q; Du Q; Wei F; Xie J; Ma XJ Immunol. 2017, 198 (7), 2935–2942. [DOI] [PubMed] [Google Scholar]
- (49).Frank PG; Lisanti MP Am. J. Physiol. Heart Circ. Physiol 2008, 295 (3), H926–H927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Videm V; Albrigtsen M Scand. J. Immunol 2008, 67 (5), 523–31. [DOI] [PubMed] [Google Scholar]
- (51).Starr T; Bauler TJ; Malik-Kale P; Steele-Mortimer O PLoS One 2018, 13 (3), e0193601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Cao W; Lee SH; Lu J Biochem. J 2005, 385 (1), 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Horvatinovich JM; Grogan EW; Norris M; Steinkasserer A; Lemos H; Mellor AL; Tcherepanova IY; Nicolette CA; DeBenedette MA J. Immunol 2017, 198 (6), 2286–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lin XL; Liu M; Liu Y; Hu H; Pan Y; Zou W; Fan X; Hu X Int. J. Mol. Med 2017, 41 (1), 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Enomoto Y; Matsushima S; Shibata K; Aoshima Y; Yagi H; Meguro S; Kawasaki H; Kosugi I; Fujisawa T; Enomoto N; Inui N; Nakamura Y; Suda T; Iwashita T Clin. Sci 2018, 132 (14), 1565–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Park SA; Kim MJ; Park SY; Kim JS; Lim W; Nam JS; Sheen YY Sci. Rep 2015, 5, 16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Busnadiego O; Gonzalez-Santamaria J; Lagares D; Guinea-Viniegra J; Pichol-Thievend C; Muller L; Rodriguez-Pascual F Mol. Cell. Biol 2013, 33 (12), 2388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.