

PREFACE
Fibrosis can affect any organ and is responsible for up to 45% of all deaths in the industrialized world. Long thought to be relentlessly progressive and irreversible, both pre-clinical models and clinical trials in various organ systems have shown that fibrosis is a highly dynamic process. This has clear implications for therapeutic interventions designed to capitalize on this inherent plasticity. However, despite significant progress in our understanding of the pathobiology of fibrosis, a translational gap remains between identification of putative antifibrotic targets and conversion into effective patient therapies. Here we discuss the transformative experimental strategies that are being leveraged to dissect the key cellular and molecular mechanisms regulating fibrosis, and the translational approaches which are enabling the emergence of precision medicine-based therapies for patients with fibrosis.
Keywords: Fibrosis, single-cell RNA sequencing, single cell genomics, fibroblasts, macrophages, integrins, cytokines, microbiome, metabolomic, antifibrotic therapy
Fibrosis is not a disease but rather an outcome of the tissue repair response that becomes dysregulated following many types of tissue injury, most notably during chronic inflammatory disorders. The formation of fibrotic tissue, which is defined by the excessive accumulation of extracellular matrix (ECM) components such as collagen and fibronectin, is in fact a normal and important phase of tissue repair in all organs. When tissues are injured, local tissue fibroblasts become activated, increasing their contractility, secretion of inflammatory mediators, and synthesis of ECM components that together initiate the wound healing response. When damage is minor or non-repetitive, the wound healing response is efficient, resulting in only a transient accumulation of excess ECM components that is then quickly eliminated, facilitating the restoration of normal tissue architecture. However, when the injury is repetitive or severe, ECM components continue to accumulate, which can lead to disruption of tissue architecture, organ dysfunction and ultimately organ failure. Genetic influences, aging, the response to invading microbes, and the changing character of the inflammatory response over time influence whether wound healing responses lead to progressive fibrosis or end in efficient repair. In this review, we provide an update on recent research investigating the mechanisms of fibrosis and discuss how this information is enabling the development of novel anti-fibrotic treatments.
FIBROSIS DECONVOLVED BY SINGLE CELL GENOMICS
Single cell multi-omics approaches are transforming our understanding of disease pathogenesis across medicine, allowing interrogation of cell populations in health and disease at unprecedented resolution. This ‘resolution revolution’ allows the powerful, unbiased exploration of cell states and types at single-cell level, resulting in unexpected novel insights into tissue biology and disease mechanisms.
These cutting-edge single-cell approaches have already been avidly adopted by the fibrosis research community to deepen our understanding of the complex, multi-cellular interplay driving lung fibrosis (Figure 1)1. Mesenchymal cells are the key source of pathological ECM deposition during lung fibrosis, ultimately leading to architectural disruption and reduced lung function. Spatial transcriptional maps of the mouse lung mesenchyme have been generated by combining single-cell RNA sequencing (scRNAseq) and signaling lineage reporters2. Each mesenchymal lineage demonstrated a distinct spatial address and transcriptome conferring unique niche regulatory functions. Examples include the mesenchymal alveolar niche cells being Pdgfrα+, Wnt responsive, and critical for alveolar epithelial cell growth and self-renewal. In contrast, Axin2+ myofibrogenic progenitor cells preferentially generated pathologically deleterious myofibroblasts following lung injury2. Further studies using the mouse bleomycin-injury model have also identified lung mesenchymal cell heterogeneity in healthy and fibrotic mouse lungs3,4.
Figure 1: Deconvolving fibrosis using multi-modal single-cell approaches.
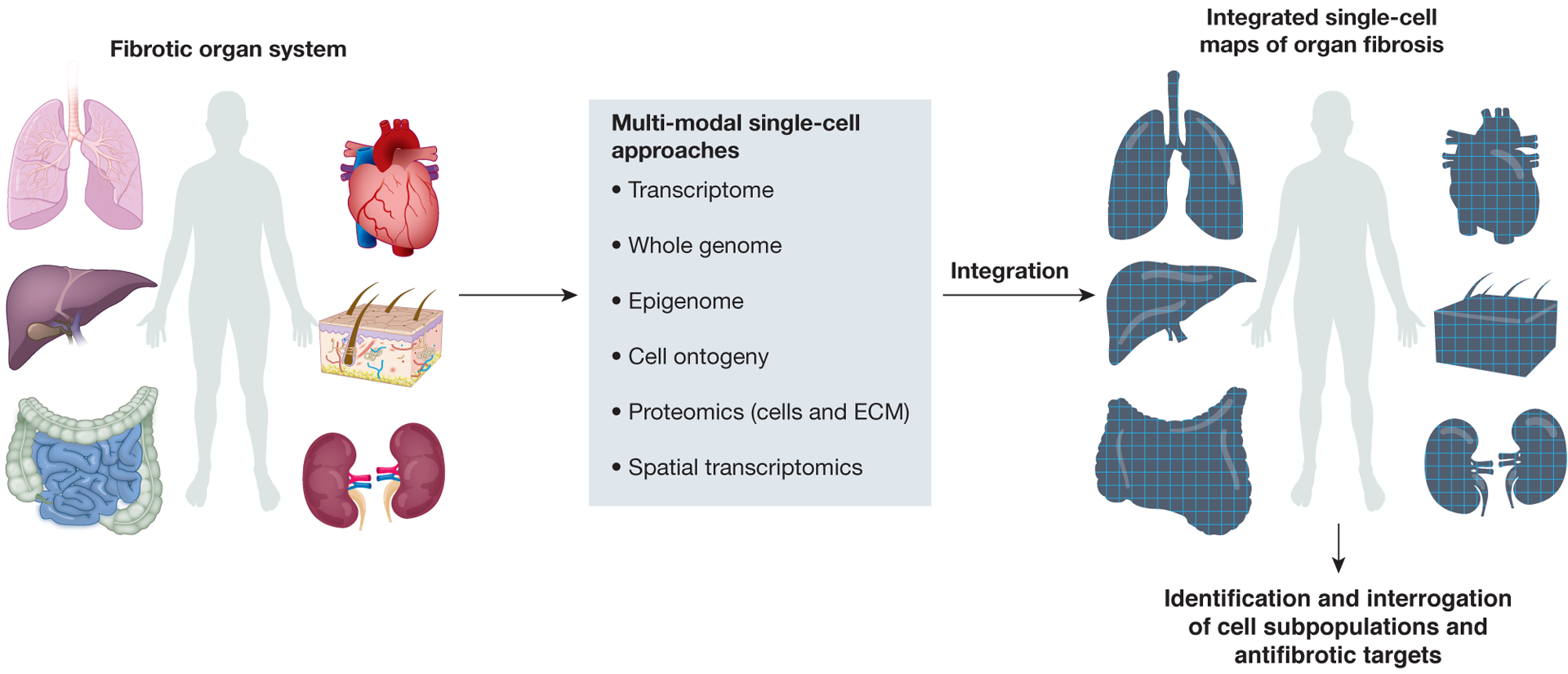
Cutting-edge single-cell approaches are transforming our understanding of the complex cellular and molecular mechanisms regulating fibrosis, allowing assessment of the transcriptome, genome, epigenome and proteome at single-cell level, in addition to spatial profiling. Furthermore, combined readouts from the same single cell are now possible (for example, the simultaneous profiling of transcriptome and chromatin accessibilty), and integration of these multi-modal single-cell omics readouts has allowed ever more powerful, comprehensive assessments of cell state, ontogeny, phenotype and function during human fibrotic disease. The new biological insights gained from these integrated approaches should enable the identification of novel and tractable therapeutic targets to treat patients across a broad range of fibrotic diseases.
Analyzing over 70,000 multi-lineage cells from eight human pulmonary fibrosis lung explants (of varying etiologies) and eight healthy donor lung samples5, identified a distinct, novel population of profibrotic alveolar macrophages restricted to patients with fibrosis which had previously been identified in mice5,6. This study and others5,7 have suggested a pathological role for alveolar type (AT) 2 cells, which secrete pulmonary surfactant and serve as alveolar stem cells, uncovering a distinct AT2 cell population in fibrotic lungs8, and establishing a direct mechanistic link between elevated mechanical tension-activated TGF-β signaling caused by impaired alveolar regeneration and progressive lung fibrosis. Decreasing mechanical tension on alveoli represents a potentially novel therapeutic approach to treat progressive lung fibrosis8. Profiling 312,928 cells from 32 IPF, 29 healthy control and 18 chronic obstructive pulmonary disease (COPD) lungs as a disease-control Adams et.al. identified a novel population of IPF-enriched aberrant basaloid epithelial cells located at the edge of myofibroblast foci in the IPF lung9. Within the vascular endothelial cell compartment an expanded cell population was identified in IPF samples that is transcriptomically identical to vascular endothelial cells normally restricted to the bronchial circulation. Furthermore, diffusion map and pseudotemporal trajectory analyses (novel computational techniques used in single-cell transcriptomics to determine the pattern of a dynamic process experienced by cells, and then arrange cells based on their progression through the process), also allowed inference of activated IPF myofibroblast origins9.
ScRNAseq studies have also comprehensively profiled the cellular and molecular landscape in liver homeostasis and regeneration10–14. Since their discovery as major collagen-producing cells in the liver15, hepatic stellate cells (HSC) have been considered a homogenous population, with the potential to transition to the activated, myofibroblast phenotype thought to be equally distributed across all HSCs. ScRNAseq has uncovered functional zonation of murine HSC, allowing high-resolution identification of the critical pathogenic collagen-producing cells following centrilobular liver injury16. Pseudotemporal trajectory and RNA velocity, another computational approach that predicts the future state of individual cells on a timescale, demonstrated that central vein-associated HSCs are the dominant source of pathogenic collagen-producing cells following centrilobular liver injury16. Furthermore, using scRNAseq to interrogate retinol-positive myofibroblasts isolated from fibrotic mouse liver has also demonstrated heterogeneity and functional diversity within liver myofibroblasts17.
Profiling of more than 100,000 single human liver cells yielded molecular definitions for non-parenchymal cell types found in healthy and cirrhotic human liver, allowing identification of a novel scar-associated triggering receptor expressed on a myeloid cell (TREM)2+CD9+ subpopulation of macrophages which expands in liver fibrosis, differentiates from circulating monocytes and is pro-fibrogenic18. Disease-associated atypical chemokine receptor (ACKR1)+ and plasmalemma vesicle-associated protein (PLVAP)+ endothelial cell subpopulations which are topographically restricted to the fibrotic niche and enhance the transmigration of leucocytes were also defined. Using multi-lineage modelling19,20 of ligand-receptor interactions between the scar-associated macrophages, endothelial cells and PDGFRα+ collagen-producing mesenchymal cells revealed intra-scar activity of several pro-fibrogenic pathways including TNF receptor superfamily (TNFRSF) 12A, PDGFR and NOTCH signalling, providing a conceptual framework for the discovery of rational therapeutic targets in liver cirrhosis18. Of note, murine liver injury-associated macrophages show significant overlap of marker genes with those observed for human scar-associated macrophages, with TREM2 and CD9 demonstrating conservation across species18. Unbiased cross-species mapping of scRNAseq data using canonical correlation analysis (CCA)21 confirmed that murine and human scar-associated macrophages represent corollary populations. This demonstrates the utility of scRNAseq approaches in defining ‘core’ fibrotic injury-induced populations and therapeutic targets across species, thereby increasing precision in the interrogation of putative targets across the translational pipeline, from pre-clinical rodent models to human liver primary cell / organoid-based systems.
In the gastrointestinal tract22, a single-cell census of the colonic mesenchyme revealed four subsets of fibroblasts in addition to pericytes and myofibroblasts, and identification of a fibroblast subpopulation proximal to the colonic crypt niche which expressed SOX6, F3 (CD142), and WNT genes essential for colonic epithelial stem cell function. In colitis, this niche became dysregulated with emergence of an activated mesenchymal population expressing TNFSF14, fibroblastic reticular cell-associated genes, interleukin (IL)-33, and lysyl oxidases (LOX) leading to impaired epithelial proliferation and maturation, thus illustrating how the colonic mesenchyme remodels to drive inflammation and barrier dysfunction in inflammatory bowel disease (IBD)22. In the context of arthritis, deletion of fibroblast activation protein-α (FAPα)+ fibroblasts suppressed inflammation and bone erosions in mouse models of resolving and persistent arthritis, and scRNASeq identified two anatomically distinct fibroblast subsets within the FAPα+ population: FAPα+ thymus cell antigen (THY1)+ immune ‘effector’ fibroblasts located in the synovial sub-lining, and FAPα+THY1− ‘destructive’ fibroblasts restricted to the synovial lining layer. Adoptive transfer of FAPα+THY1− fibroblasts into the joint selectively mediated bone and cartilage damage with little effect on inflammation, whereas transfer of FAPα+ THY1+ fibroblasts resulted in a more severe and persistent inflammatory arthritis, with minimal effect on bone and cartilage. Discovery of these anatomically discrete, functionally distinct fibroblast subsets with non-overlapping functions clearly has important implications in the rational design of therapies aimed at precisely modulating inflammation, fibrosis and tissue repair23.
FIBROBLAST HETEROGENEITY AND PLASTICITY
Functional fibroblast heterogeneity
Increasingly sophisticated experimental approaches have allowed discovery of significant diversity and functional heterogeneity within the fibroblast population during organ fibrosis22–25 . A combination of fate mapping and live imaging, revealed that a specialised subset of fibroblasts, fascia fibroblasts, rise to the surface of the skin following wounding26. These fascia fibroblasts gather their surrounding extracellular matrix (including blood vessels, macrophages and peripheral nerves) to form the provisional matrix, and ablation of these fibroblasts inhibited matrix homing into wounds leading to defective scars. Intriguingly, placement of an impermeable film beneath the skin (preventing upward migration of fascia fibroblasts) led to chronic open wounds. Thus, fascia contains a specialized prefabricated kit of sentry fibroblasts, which are embedded within a movable sealant. Whether similar fibroblast subpopulations exist in other organs and use analogous mechanisms to promote wound healing remains to be determined. Significant myofibroblast functional diversity has also been identified during skin injury and ageing27. Lineage tracing and flow cytometry identified distinct subsets of wound bed myofibroblasts, including CD26-expressing adipocyte precursors and also a CD29High subpopulation. Adipocyte precursors were significantly reduced and CD29High cells more abundant in wound beds from aged mice and bleomycin-induced fibrotic mouse skin, suggesting that the fibrotic microenvironment impacts myofibroblast composition.
Recent studies have implicated a range of mesenchymal progenitor cells (MPs) in the initiation and propagation of fibrosis28–30. In particular two studies, focussing on populations of hypermethylated in cancer protein (Hic1)+Pdgfrα+ lymphocyte antigen 6 complex (LY6A)+ MP populations in heart and skeletal muscle, have elegantly demonstrated MP hierarchy, diversity in the pathophysiological roles of their progeny, and how fate determination of MP is context dependent31,32. Conditional genetic inactivation of Hic1 in mice led to activation and expansion of MPs in both heart and skeletal muscle, demonstrating that Hic1 is required for maintaining MP quiescence. In the heart, Hic1 deficiency (in Pdgfrα-expressing cells) led to activation of MPs and accumulation of cardiac fibroadipogenic progenitor cells, with epicardial thickening, interstitial fibrosis and fibrofatty depositions resulting in major pathological features pathognomonic in arrhythmogenic cardiomyopathy32. However, although Hic1 inactivation in skeletal muscle also resulted in a marked expansion of Pdgfrα+LY6A+ cells during homeostasis, this did not increase skeletal muscle fibrosis and had no substantial effect on skeletal muscle regeneration31, highlighting the diverse pathophysiological roles of these cells in different organs.
Fibrosis, secondary to age-associated chronic low-grade inflammation, is increasingly recognised as an important cause of morbidity and mortality33. Increasing age is a driver of fibroblast functional heterogeneity, with ‘old’ fibroblasts demonstrating variability in their ability to reprogram and heal wounds34 . Old mice exhibited variability in wound healing rates in vivo, and scRNASeq identified distinct subpopulations of fibroblasts with differing cytokine expression profiles in the wounds of old mice with slow versus fast healing rates. This increased variability in wound healing with increasing age may reflect distinct stochastic ageing trajectories between individuals, which will need to be considered when designing personalized antifibrotic therapies for the elderly population34.
Fibroblast reprogramming
Fibroblasts, in addition to displaying significant functional heterogeneity, are capable of remarkable degrees of plasticity and phenotype switching during progression and regression of fibrosis30,35,36. For example, the transcription factor PU.1, whose expression is silenced in fibroblasts during tissue homeostasis but upregulated in fibrotic disease, has been shown to play a major role in fibroblast polarization and fibrogenesis37. PU.1 both polarized resting fibroblasts and repolarized ECM-degrading inflammatory fibroblasts to an ECM-producing fibrotic phenotype. Furthermore, genetic and pharmacological inactivation of PU.1 enabled reprogramming of fibrotic fibroblasts into resting fibroblasts, leading to regression of fibrosis in several organs37. In addition, a remarkable inter-lineage plasticity between myofibroblasts and other cell types during fibrosis exists. During cutaneous wound healing in mice, adipocytes were regenerated from myofibroblasts. This reprogramming required neogenic hair follicles, which triggered bone morphogenetic protein (BMP) signaling and activation of adipocyte transcription factors expressed during development38. Furthermore, adipocytes were generated from human keloid fibroblasts when treated with BMP in vitro, or when placed with human hair follicles38. In the liver, viral vector-mediated expression of specific transcription factors in myofibroblasts has been used to reprogram myofibroblasts into hepatocyte-like cells in fibrotic mouse livers thereby reducing liver fibrosis and increasing liver function39,40. The ability to selectively target scar-producing myofibroblasts during fibrosis and reprogram these cells into other lineages which support organ function opens up exciting new avenues for antifibrotic and pro-regenerative therapies.
Taken together, these studies highlight that profound fibroblast diversity, functional heterogeneity and plasticity exists during fibrosis, both within and between organs (Figure 2). More precise delineation of fibroblast heterogeneity and phenotype in different disease settings should facilitate the design of rational, highly-targeted antifibrotic therapies, ultimately allowing specific inhibition and reprogramming of pathologic fibroblast subpopulations whilst preserving essential, homeostatic fibroblast function. One of the first trials to explore fibroblast cellular therapy was a phase 2 study using allogeneic human dermal fibroblasts to remodel contracted scars (NCT01564407; TABLE 1).
Figure 2: Functional fibroblast heterogeneity and plasticity.

Recent studies have uncovered significant functional heterogeneity and plasticity within fibroblast populations during fibrosis. In the context of arthritis, scRNASeq combined with adoptive transfer experiments identified two anatomically distinct fibroblast subsets within the FAPα+ population: FAPα+ thymus cell antigen (THY1)+ immune ‘effector’ fibroblasts located in the synovial sub-lining, and FAPα+THY1− ‘destructive’ fibroblasts restricted to the synovial lining layer. Studies of mesenchymal progenitor (MP) populations (Hic1+Pdgfrα+ LY6A+) in heart and skeletal muscle have demonstrated MP hierarchy, diversity in the pathophysiological roles of their progeny, and how fate determination of MP is tissue-dependent. Fibroblasts are also capable of remarkable degrees of plasticity and phenotype switching during progression and regression of fibrosis. Myofibroblasts can revert to a quiescent state in the absence of ongoing injury, or may undergo full lineage switching with adipocytes which is observed during cutaneous wound healing in mice. Furthermore, genetic and pharmacological inactivation of the transcription factor PU.1 can reprogramme fibrotic fibroblasts into resting fibroblasts, resulting in regression of fibrosis in several organs. Finally, viral vector-mediated expression of specific transcription factors in myofibroblasts in the liver has been used to reprogram myofibroblasts into hepatocyte-like cells in fibrotic mouse livers, thereby reducing liver fibrosis and increasing liver function.
Table 1.
Drugs tested in phase 2 or phase 3 clinical trials for antifibrotic therapy with relevance to this review article.
| Anti αvβ6 | Small molecule | GSK3008348 | Glaxo Smith Kline | IPF | 2 | 03069989 |
| Anti αvβ6 | Antibody | STX-100 | Biogen | IPF | 2 | 01371305 |
| Anti αvβ1/αvβ6 | Small molecule | PLN-74809 | Pliant | IPF | 2 | 04072315 |
| Anti αvβ1/αvβ3/αvβ6 | Small molecule | IDL-2965 | Indalo | IPF | 2 | 03949530 |
| Anti-IL-6 | Antibody | Tocilizumab | Genentech | Scleroderma | 3 | 02453256 |
| Anti IL-13 | Antibody | QAX576 | Novartis | IPF, skin keloids | 2 | 01266135; 0987545 |
| Anti IL-13 | Antibody | Tralokinumab | AstraZeneca | IPF | 2 | 01629667 |
| Anti IL-13 | Antibody | Lebrikizumab | Roche | IPF | 2 | 01872689 |
| Anti IL-4 / Anti IL-13 | Antibody | SAR156597 | Sanofi | IPF, scleroderma | 2 | 01529853 |
| Anti CCL2 | Antibody | Carlumab | Centocor | IPF | 2 | 0786201 |
| Anti CCR2 / Anti CCR5 | Antibody | Cenicriviroc | Allergan | NASH, liver cirrhosis | 3 | 02217475; 03028740; 03059446 |
| Anti TLR4 | Antibody | NI-0101 | Light Chain Bioscience | RA | 2 | 03241108 |
| Pentraxin 2 | Recombinant protein | PRM-151 | Promedior | IPF, myelofibrosis | 2 | 02550873; 01981850 |
| Allogeneic dermal fibroblasts | Allogeneic fibroblasts | ICX-RHY-013 | Intercytex | Skin scars | 2 | 01564407 |
Abbreviations: IPF: Idiopathic pulmonary fibrosis; NASH: Non-alcoholic steatohepatitis; RA: Rheumatoid arthritis.
METABOLIC REGULATION OF MESENCHYMAL CELLS
It is now apparent that cells involved in the progression and resolution of fibrosis are metabolically ‘reprogrammed’ to perform distinct functions during tissue repair. The impact of metabolism has been explored extensively in the context of NASH-driven fibrosis, in which dysregulated hepatic lipid metabolism serves as a key driver of liver injury and cirrhosis41. As discussed earlier, fibroblasts are the key source of ECM deposition during fibrosis. Hence, metabolic alterations of local tissue mesenchymal cells may offer future therapeutic avenues that include the major carbohydrate, amino acid and lipid metabolism pathways (Figure 3).
Figure 3: Metabolomic reprogramming of activated fibroblasts.

Profibrotic fibroblasts increase glycolysis via hexokinase leading to ehanced pyruvate and lactate. Lactate decreases extracellular pH and activates latent TGF-β1. Pyruvate also feeds the TCA cycle after conversion to acetyl-CoA increasing succinate. Both mechanisms lead to increase in a-SMA, collagen production and proliferation. Glutaminase activity is increased converting glutamate to glutamine which gets converted into α-KG via the TCA cycle and decreases apoptosis and enhances collagen stabilization. Abbreviations: α-KG: alpha-ketoglutarate; a-SMA: alpha smooth muscle actin; LDH: Lactate hydrogenase; P: Phosphate; R: Receptor; TCA:tricarboxylic acid; TGF: Transforming growth factor.
Following tissue injury, mesenchymal cells undergo profound metabolic changes in order to facilitate energy consuming cellular functions such as proliferation and protein synthesis42. Fibroblast aerobic glycolysis is increased by upregulating rate-limiting glycolytic enzymes43. In addition to providing a rapid energy-generating mechanism compared to oxidative phosphorylation, glycolysis produces by-products such as lactate that are also important in the context of fibrosis. Lowering extracellular pH combined with an increase in lactic acid promotes myofibroblast differentiation via activation of TGF-β1 and lactate itself may serve as an additional energy source promoting a synthetic phenotype in mesenchymal cells44–46. Fibroblast activation also involves increased activity of key glycolytic enzymes, such as hexokinase 2 and lactate dehydrogenase (LDH)47, which in turn increase cell proliferation47 and collagen synthesis48. During enhanced glycolysis, increased amounts of pyruvate are converted to acetyl-CoA in the mitochondrial matrix47 before entering the citric acid cycle. This yields intermediate metabolites, such as succinate, which promote fibrosis49. Increased glycolysis has been implicated in experimental lung, liver and kidney fibrosis models and inhibition of glycolysis reduces ECM accumulation50–52. During fibrogenesis, mesenchymal cells also exploit changes in amino acid metabolism through glutaminolytic reprogramming. Glutaminolysis and levels of the key enzyme glutaminase are increased in TGF-β1 stimulated fibroblasts53. This leads to enhanced conversion from glutamine to glutamate, which confers resistance to apoptosis54 and regulates collagen production by promoting its stabilization53 via mTOR signaling. In vivo, inhibition of glutaminase 1 ameliorates bleomycin- and TGF-β1 induced pulmonary fibrosis55. As a further mechanism, changes in fatty acid oxidation have also been linked to fibrogenesis. Intracellular fatty acid oxidation is downregulated in tubulointerstitial fibrosis in mice and humans, and its restoration was fibroprotective56. Of note, upregulation of glycolysis has been reported as a compensatory mechanism for reduced fatty acid oxidation during kidney injury, which could result in enhanced progression to fibrosis52 (Figure 3).
Taken together, key metabolic pathways such as increased glycolysis, upregulation of glutaminolysis and enhanced fatty acid oxidation are emerging as important drivers of fibroblast activation. To date no drugs targeting these metabolic pathways have reached the clinic as antifibrotic therapies. However drugs targeting metabolism pathways are approved or in clinical trials for treatment of cancer with known safety profiles, fueling the hope for future application in fibrotic diseases57.
MACROPHAGE-MEDIATED REGULATION OF FIBROSIS
Inflammatory monocytes and tissue-resident macrophages are key regulators of tissue fibrosis, playing major roles in the initiation, maintenance and resolution of tissue injury58–60. Furthermore, monocytes and macrophages are capable of remarkable functional plasticity, displaying diverse phenotypes during wound healing dependent on multiple cues including the environmental niche61,62 and the temporal stage of tissue injury and repair63–66. Tissue macrophages are also important producers of T cell- and fibroblast-recruiting chemokines that orchestrate the development of the fibrotic niche67.
Functional Heterogeneity of Monocytes and Macrophages
Several recent studies have identified subpopulations of monocytes and macrophages which have the capacity to regulate fibrosis and tissue remodelling18,61,68–71. For example, a newly identified population of atypical monocytes carcinoembryonic antigen-related adhesion molecules (Ceacam)1+Msr1+Ly6C−F4/80−Mac1+ monocytes)68 termed segregated-nucleus-containing atypical monocytes (SatM) and sharing granulocyte characteristics, have been shown to play a key role in lung fibrogenesis. SatM were found to be regulated by CCAAT/enhancer binding protein β (C/EBPβ), with Cebpb deficiency leading to a complete deficiency of SatM. Bleomycin-induced fibrosis, but not inflammation, was inhibited in chimaeric mice with Cebpb−/− haematopoietic cells, and adoptive transfer of SatM into Cebpb−/− mice resulted in fibrosis. Interestingly, SatM were derived from Ly6C−FcεRI+ granulocyte/macrophage progenitors, but not from macrophage/dendritic cell progenitors. ScRNAseq approaches have also been used to investigate macrophage heterogeneity and function in the context of lung fibrosis. Although the major tissue-resident macrophage populations have been intensively studied, much less is known regarding the role of interstitial macrophages (IMs) during tissue homeostasis and injury. Two independent IM subpopulations that are conserved across lung, fat, heart, and dermis were identified: (Lyve1loMHCIIhiCX3CR1hi (Lyve1loMHCIIhi) and Lyve1hiMHCIIloCX3CR1lo (Lyve1hiMHCIIlo) monocyte-derived IMs. Using a novel mouse model of inducible macrophage depletion (Slco2b1flox/DTR), demonstrated that the absence of Lyve1hiMHCIIlo IMs exacerbates experimental lung fibrosis, thereby showing that two independent populations of IMs coexist across tissues with conserved niche-dependent functional programs61. In addition, a pathological subgroup of transitional macrophages that are required for the fibrotic response to injury was indentified in murine bleomycin-induced lung fibrosis. Using a novel computational approach which allows annotation of scRNAseq by reference to bulk transcriptomes (SingleR) enabled macrophage subclustering and uncovered a disease-associated subpopulation with a transitional gene expression profile intermediate between monocyte-derived and alveolar macrophages. These CX3CR1+SiglecF+ transitional macrophages localized to the fibrotic niche and were profibrotic in vivo. This has been linked to human disease because human orthologs of genes expressed by these transitional macrophages were upregulated in samples from patients with idiopathic pulmonary fibrosis70.
Research examining the regulatory roles of monocytes and macrophages during tissue injury and repair has largely focussed on blood-derived monocytes and macrophages. However, emerging evidence has also identified resident cavity macrophages as key contributors to fibrosis and tissue remodelling. Previous work demonstrated that a reservoir of mature F4/80HiGata6+ peritoneal cavity macrophages rapidly invade the liver via direct (avascular) recruitment across the mesothelium in response to sterile liver injury72. These recruited macrophages dismantle necrotic cell nuclei, releasing DNA and forming a cover across the site of injury72. Building on this observation, the same group has investigated the contribution of resident cavity macrophages located in the pericardial space adjacent to the site of cardiac injury. Following myocardial infarction in mice, Gata6+ macrophages in mouse pericardial fluid invaded the epicardium and lost Gata6 expression but maintained anti-fibrotic properties, and loss of this macrophage population enhanced interstitial fibrosis post-ischemic injury. Gata6+ macrophages were also present in human pericardial fluid, suggesting that this immune cardioprotective role for the pericardial tissue compartment may be relevant in human disease73.
Macrophage and fibroblast cross-talk
Irrespective of the mode of monocyte and macrophage recruitment into areas of tissue injury, pro-fibrotic macrophages commonly coordinate scar formation via a range of interactions with fibroblasts, the major cellular source of pathological extracellular matrix deposition during fibrosis18,27,74–78. For example, macrophage-derived amphiregulin (an epidermal growth factor receptor ligand) has recently been shown to induce the differentiation of mesenchymal stromal cells into myofibroblasts via integrin αv-mediated activation of TGF-β79. Previous work has shown that proximity is crucial to allow crosstalk between macrophages and contractile fibroblasts27,77,78, however until recently it remained elusive as to how proximity between these two cell types is established. In an elegant study, contracting fibroblasts were shown to generate deformation fields in fibrillar collagen matrix that provided far-reaching physical cues to macrophages80. Within the collagen deformation fields created by fibroblasts or actuated microneedles, macrophages migrated towards the force source from distances of several hundred micrometres, and the presence of a dynamic force source within the matrix was critical to initiate and direct macrophage migration. Interestingly, and disrupting traditional views on how macrophages migrate within fibrotic tissues, the authors proposed that macrophages mechanosense the velocity of local displacements of their substrate, allowing contractile fibroblasts to attract macrophages over distances that exceed the range of chemotactic gradients80.
INTEGRIN MEDIATED TGF-BETA ACTIVATION
Secreted TGF-β1 is a major pro-fibrogenic cytokine81, and therefore potentially represents an attractive anti-fibrotic target. Sustained systemic inhibition of TGF-β1, however, has been shown to have undesired effects including cardiac valve problems, and TGF-β1 knockout mice develop systemic autoimmunity82. This has relevance to all mucosal surfaces, especially the intestine, where TGF-β1 activity is believed to control tissue homeostasis83–85. In addition, pan-TGF-β1 blockade has been found to induce carcinogenesis, perhaps owing to the role of TGF-β1 as an anti-proliferative mediator for most epithelial cell types. Strategies to avoid these deleterious effects could invovle choosing the correct magnitude or duration of inhibition, following the yet to be proven hypothesis that low levels of TGF-β1 control immune homeostasis while high levels mediate fibrosis, co-administering anti-inflammatory therapies, or inhibiting TGF-β1 locally at specific sites in the tissue by blocking integrins and other locally acting mediators that activate latent TGF-β1.
The pericellular fibrotic matrix is a remarkably dynamic environment, which exerts profound influences on cell behavior, and many of the key cell–cell and cell–matrix interactions which regulate fibrosis are mediated by members of the integrin family (noncovalent α/β heterodimers from 18 different α subunits and 8 β subunits resulting in 24 known members in humans)86,87. Importantly, integrins can mediate the translation of spatially-fixed extracellular signals into a wide variety of changes in cell behavior including cell adhesion, migration, proliferation, differentiation and apoptosis86,87. Of key relevance to fibrosis, integrins can also potentiate signals from soluble pro-fibrogenic growth factors such as TGF-β1. Nearly all TGF-β1 is secreted and bound to the ECM in a latent form, and therefore the majority of the regulation of TGF-β function during fibrosis is dependent on site-specific regulation of TGF-β activation, rather than synthesis or secretion88.
The most intensively studied activation mechanism for TGF-β1 with demonstrated relevance in vivo has been the interaction of the TGF-β1 latent complex with the αv-containing subset of integrins. Specifically, the integrins αvβ1, αvβ3, αvβ5, αvβ6 and αvβ8 have all been shown to bind to an N-terminal fragment of the TGFβ1 gene product called the latency associated peptide (LAP), which normally forms a noncovalent complex (latent complex) with the active cytokine, preventing TGF-β from binding to its cognate receptors and inducing biological effects89–93. When a mechanical force is applied to the latent complex by contraction of αvβ6 integrin-expressing cells, the resultant conformational change results in release of active TGF-β194–96. Interestingly, a recent study has shown that in contrast to this αvβ6-mediated mechanism of TGF-β activation, αvβ8-dependent TGFβ activation can occur independently of actin-cytoskeletal force and does not require release of mature TGF-β97, further highlighting the complexity of αv integrin-mediated TGF-β activation and signaling in different contexts.
There is now abundant pre-clinical data across a range of fibrotic disease models demonstrating critical regulatory roles for αv-containing integrins expressed on various different cell lineages. Mice lacking the αvβ6 integrin are protected in mouse models of lung, kidney and biliary fibrosis90,98–102. This protection appears to be secondary to local inhibition of TGF-β, and antibody-mediated inhibition of αvβ6-mediated TGF-β1 activation decreased lung fibrosis in pre-clinical models99,103. αvβ8-dependent TGF-β1 activation represents a further potential therapeutic target104,105. Conditional depletion of lung fibroblast αvβ8 inhibited experimental airway fibrosis105, and mice genetically engineered to replace murine β8 with its human ortholog demonstrated that a blocking antibody against human αvβ8 blocks TGF-β1 activation and protects against allergic airway inflammation and remodeling induced by cigarette smoke106. Furthermore, depletion of the αv integrin subunit on mesenchymal cells has also proven effective in inhibiting fibrosis in models of liver, lung and kidney fibrosis107. Using Pdgfrβ-Cre mice to deplete αv integrins on hepatic myofibroblasts protected mice from carbon tetrachloride-induced hepatic fibrosis, whereas global loss of β3, β5 or β6 integrins or conditional loss of β8 integrins in myofibroblasts did not, highlighting context dependency of the various αv-containing integrins in the regulation of fibrosis across different organs. Pharmacological blockade of αv-containing integrins by a small molecule (CWHM 12) attenuated both liver and lung fibrosis, even when fibrosis was already established107. Tissue fibroblasts can express four αv-containing integrins, αvβ1, αvβ3, αvβ5 and αvβ8. However, as the αvβ1 integrin is composed of α and β subunits that are both present in multiple heterodimers, it has been challenging to generate heterodimer-specific antibodies or to deduce function from gene knockout studies. Therefore, small molecule inhibitors of αvβ1 have been developed to help interrogate the role of this integrin in fibrosis, with recent studies demonstrating an anti-fibrotic effect of αvβ1 blockade in models of lung and liver fibrosis89.
Given the abundance of pre-clinical data, this remains a very active area of research and development in the fibrosis field, with multiple small molecule and antibody-based approaches undergoing assessment in clinical trials, including inhibitors designed to selectively target multiple αv-containing integrins simultaneously. This includes phase 2 trials inhibiting αvβ6 (NCT01371305), αvβ1/αvβ6 (NCT04072315) and αvβ1/αvβ3/αvβ6 (NCT03949530), all in pulmonary fibrosis (TABLE 1). Patient safety will be an important consideration in these trials as Biogen recently terminated their IPF trial of a selective anti-αvβ6 antibody due to unspecified safety concerns.
CYTOKINE-MEDIATED REGULATION OF FIBROSIS
Other than TGF-β several additional cytokines secreted from multiple cellular sources have been identified as triggers of fibrosis108. The pro-inflammatory cytokine IL-17A was identified as a key inducer of fibrosis in several different organ systems, including the lung, liver, kidney, heart, and skin109–114. In a study of bleomycin-induced pulmonary fibrosis, IL-17A produced by γδ and CD4+ T cells induced severe lung inflammation, neutrophil recruitment, and production of the pro-fibrotic cytokine TGF-β110. Neutrophils and mast cells were also identified as important sources of IL-17A115. Experiments with IL-17A- and IL-17RA-deficient mice, as well as therapeutic studies with IL-17A neutralizing monoclonal antibodies confirmed a role for IL-17A signaling in the development of fibrosis in multiple tissues110–112,116–118. In addition to promoting TGF-β production116,119, IL-17A increases and stabilizes TGF-βRII expression on fibroblasts, thereby enhancing their sensitivity to TGF-β120. The Th17-associated cytokine IL-22 similarly enhanced TGF-β signaling in fibroblasts115. TGF-β in turn induced the expression of IL-17A when produced concurrently with the pro-inflammatory cytokines IL-1, IL-6, or TNF110,121,122, suggesting a feedforward mechanism involving acute phase cytokines, IL-17A, and TGF-β is responsible for the development of fibrosis following acute tissue injury109,110,123 (Figure 4). Importantly, IL-17A exhibited similar pro-fibrotic activity in both animal studies and in human cells124,125.
Figure 4: Divergent cytokine pathways drive fibrosis.
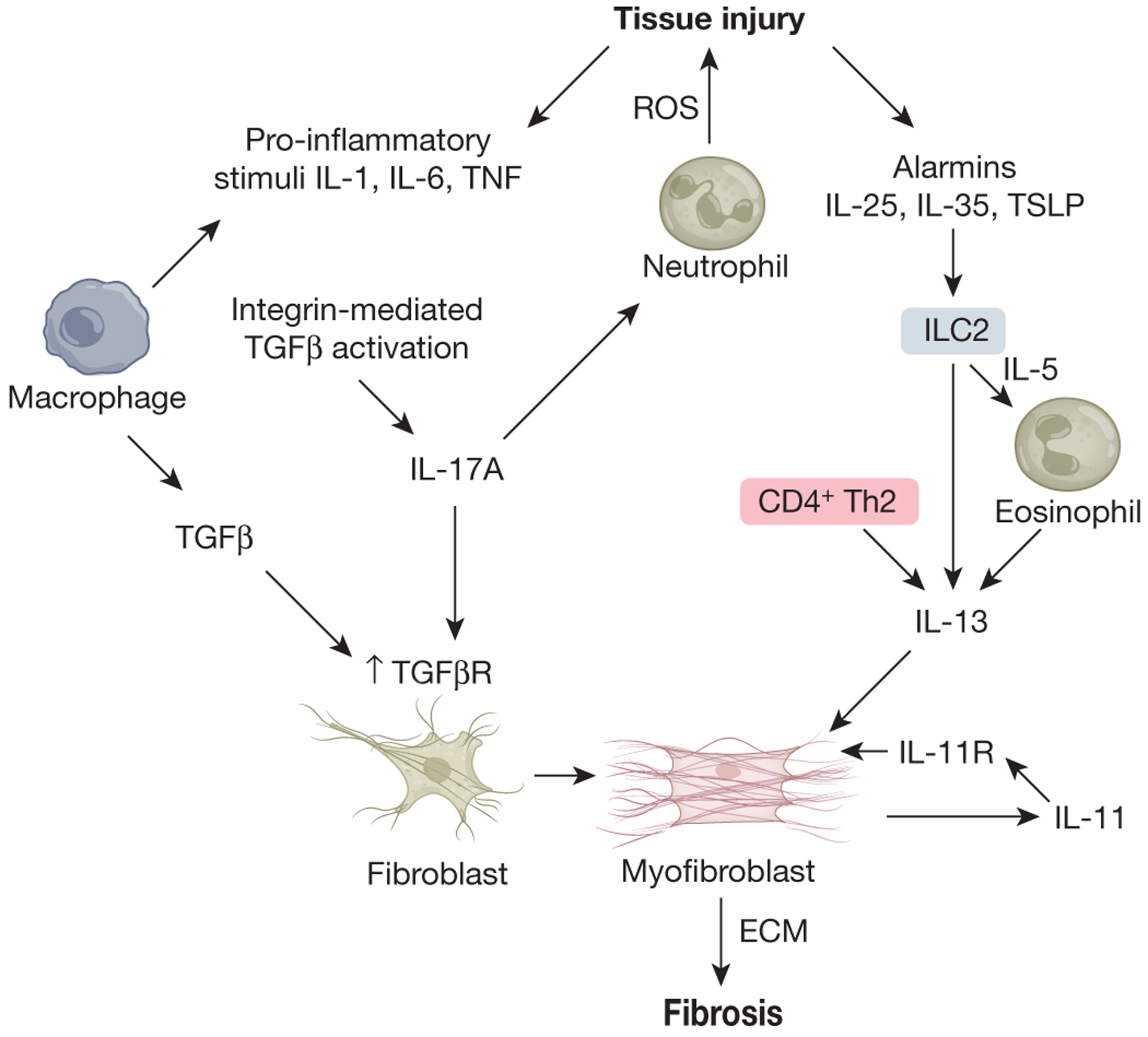
The Innate acute phase pro-inflammatory cytokines IL-1, IL-6, and TNF, together with TGF beta promote the development of IL-17 secreting cells. IL-17A potentiates neutrophil responses that contribute to tissue injury through ROS production, while increasing TGF-beta receptor expression on fibroblasts that facilitates ECM production in response to TGF-beta. TGF-beta, activated locally through integrin-mediated mechanisms, serves as a key driver of fibrosis. A second and distinct cytokine-mediated pathway that can promote fibrosis independently of TGF-beta is the type-2 cytokine axis. Here, the alarmin cytokines IL-25, IL-35, and TSLP, secreted by epithelial cells and other damaged tissues drives the expansion and activation of type-2 innate lymphoid cells (ILC2) that secrete large amounts of IL-5 and IL-13. IL-5 in turn drives the recruitment and activation of local tissue eosinophils, which provide an additional source of type 2 cytokines and other pro-fibrotic mediators. IL-13, derived from eosinophils, CD4+ Th2 cells, and ILC2s exhibits potent pro-fibrotic activity that is TGF-beta independent. Finally, the cytokine IL-11, produced by activated myofibroblasts has been found to stimulate ECM production by myofibroblasts in response to multiple pro-fibrotic mediators, including TGF beta and IL-13.
Caspase-1, NOD, LRR and pyrin domain-containing (NLRP) 3 inflammasome, and NFkB were identified as important upstream mediators contributing to the activation of the IL-17A-TGF-β axis122. The exact mechanisms responsible for the sustained activation of NFkB and NLRP3 inflammasome signaling remain unclear, although commensal microbe stimulation of Toll-like receptors on myeloid cells and tissue fibroblasts has been hypothesized as an important activating mechanism, with the resulting pro-inflammatory cytokine and chemokine production exacerbating inflammation and the progression of fibrosis126,127. Of note, stimulation of TLR4/NFkB in hepatic stellate cells enhances TGF-β signaling by directly downregulating the TGF-β pseudoreceptor BMP and the activin membrane-bound inhibitor Bambi127. A related study showed that sustained activation of the NLRP3 inflammasome is associated with increased chemokine expression, recruitment of neutrophils and macrophages, and persistent production of IL-17A and TNF122. Thus, activation of the pro-fibrotic TGF-β signaling pathway is driven by several collaborating mechanisms, with the pro-inflammatory cytokine IL-17A playing a prominent role.
While the TGF-β signaling pathway is a well-known driver of fibrosis in many organs and tissues, the type 2-associated cytokines IL-4 and IL-13 have also emerged as distinct but important inducers of fibrosis. Here, the fibrotic response is associated with a predominant eosinophil and M2-like macrophage infiltrate, rather than the neutrophil and M1-like monocyte/macrophage phenotype that characterizes the IL-1-IL-17A-TGF-β axis128. Also, instead of acute phase cytokines serving as co-inducers, the alarmin cytokines thymic stromal lymphopoietin, IL-25, and IL-33 function as key initiators of type 2-dependent fibrosis by triggering the production of IL-4 and IL-13 in innate lymphoid cells, T cells, eosinophils, and other type 2-associated leukocytes129–132. Although IL-13 can induce and activate TGF-β in macrophages133, it may promote fibrosis independently of TGF-β134 in part by directly targeting stromal and parenchymal cells, including epithelial populations and collagen-producing myofibroblasts135. IL-13, IL-4R, and IL-13Rβ1-deficient mice as well as animals treated with neutralizing antibodies to IL-13 or IL-4R display reduced fibrosis following many different types of tissue injury136–140, confirming a critical role for type 2 cytokine signaling in the progression of fibrosis (Figure 4).
The mechanisms that dictate whether the IL-1-IL-17A-TGF-β axis or type 2 cytokine response dominate as the key driver of fibrosis remain unclear although the type of cellular damage or duration of the injury likely play key roles. For example, studies with the commonly used “single hit” bleomycin pulmonary fibrosis model revealed a prominent role for the IL-1-IL-17A-TGF-β axis but little to no contribution for type 2 cytokine signaling, despite significant upregulation of IL-4 and IL-13 in the lung110. Nevertheless, a modified version of this model in which bleomycin is injected intra-dermally rather than intra-tracheally over several weeks uncovered a substantial role for IL-4R signaling in the development of pulmonary fibrosis141. Different stimuli or types of injuries that lead to the preferential production of alarmin cytokines versus activation of NF-kB and inflammasome signaling likely play decisive roles as well. For example, integrin receptors that interact with the extracellular matrix were recently shown to preferentially activate TGF-β signaling and production of IL-17A while antagonizing the production of type 2 cytokines142. Consistent with these observations, several studies have revealed substantial cross-regulation between IL-17A and IL-13110,143, with marked upregulation of the opposing pathway when only one mechanism was targeted therapeutically136,142,144,145. Consequently, a successful anti-fibrotic strategy will either target the dominant mechanism or reduce both pathways simultaneously. Interestingly, IL-11, a member of the IL-6/gp130 cytokine family, may be one such target as it was recently shown to integrate pro-fibrotic signals emanating from both pathways146–149.
Not surprisingly, given the robust preclinical data, inhibitors to IL-13 alone (NCT01266135; NCT00987545; NCT00581997; NCT01872689; NCT01629667) or the combination of IL-4/IL-13 (NCT02921971; NCT01529853) were tested in phase 2 trials for pulmonary fibrosis, skin keloids, and systemic sclerosis. Additional cytokine, chemokine or growth factor inhibitors in development for fibrosis are anti-CCR2/CCR5 (NCT02217475; NCT03028740; NCT03059446; NCT02330549) for liver fibrosis and NASH, anti IL-1 (NCT01538719) for systemic sclerosis, anti IL-6 (NCT02453256) for scleroderma, and anti CCL2 (NCT00786201) for pulmonary fibrosis (Table 1).
CONTRIBUTION OF THE MICROBIOME TO FIBROSIS
Environmental factors such as pollutants, chemicals, and microbes can trigger or protect from fibrosis. While the exact environmental agents remain poorly characterized, recent work has focused on host / microbiome interactions as some microbe populations have been found to regulate the progression of fibrosis. Immune and non-immune cells sense microbe-derived, pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs), including Nod-like receptors (NLRs) and Toll-like receptors (TLRs), driving innate and adaptive immune responses150. Extensive studies have investigated microbial sensing in the context of immunity and epithelial cell biology, but studies reporting a direct link between the microbiota and fibrogenesis are now emerging.
The vast majority of human-associated microbes are found in the gut, and during homeostasis these microbial populations are essential in maintaining gut health. However, when the balance between healthy and pathogenic microbes shifts towards pathogenic subsets disease can ensue. Inflammatory bowel disease comprising Crohn’s disease (CD) and ulcerative colitis (UC)) represents a prototypical pathology where dysbiosis is thought to be a key driver of disease pathogenesis, with evidence suggesting a strong link between the microbiota and the development of fibrosis. For example, CD patients carrying variants of the NOD2 gene, an intracellular pattern recognition receptor, are at increased risk of stricture formation which is the major manifestation of intestinal fibrosis151. Furthermore, serologic anti-microbial antibodies are common in patients with CD and are associated with and predictive of intestinal strictures152,153, and almost all murine models of intestinal fibrosis are influenced by the microbiota154. For instance, global deletion of the bacterial signaling adaptor molecule MyD88 (a critical signaling adaptor molecule for all TLRs, except TLR3155) reduced intestinal fibrosis in a model of Salmonella-induced colitis156. Microbiota driven intestinal fibrosis may be mediated by induction of the IL-33 receptor ST2 on epithelial cells157 or the pro-fibrotic action of tumor necrosis factor-like cytokine 1A (TL1A)158. On a cellular level, TLR2 or TLR4 ligands induce cytokine and chemokine secretion from cultured intestinal myofibroblasts159. Interestingly, although intestinal mesenchymal cells express multiple TLRs and NLRs, a pro-fibrogenic phenotype was triggered exclusively by flagellin, a broad activator of innate and adaptive immunity and a TLR5 ligand. This occurred in a TGF-β1 independent manner and via post-transcriptional regulation160. The role of myofibroblasts directly sensing PAMPs in intestinal fibrosis has been confirmed in vivo, as selective deletion of MyD88 in αSMA positive cells ameliorated intestinal fibrosis160.
Dysbiosis in the gut has been shown to influence liver fibrosis as well. Translocation of bacteria and their products across the intestinal barrier due to intestinal barrier disruption is common in patients with chronic liver disease. Increased levels of the microbe-derived ligand lipopolysaccaride (LPS) in the portal vein or translocation of whole bacteria or their products to the liver activates inflammation that leads to fibrosis161,162. Blocking TLR4 signaling in mice or reducing hepatic exposure to intestinal microbes by reducing microbial load with antibiotics ameliorates experimental liver fibrosis127. HSCs express all known human TLRs and respond to TLR4 ligands127,163, which also downregulate a TGF-β1 decoy receptor sensitizing HSC to the action of TGF-β1127. A comparable mechanism has been described in rat pancreatic fibrosis164. TLR4 signaling includes an additional signaling adaptor, called TRIF. Deletion of TRIF in a diet-induced NASH mouse model reduced hepatic steatosis but increased hepatic fibrosis, and TRIF−/− HSCs expressed higher levels of CXCL1 and C-C motif chemokine ligands in response to LPS, highlighting a potential mechanism for this unexpected effect165. Conversely, distinct gut microbiota may be hepatoprotective in the context of liver fibrosis. Increased liver ECM deposition was observed in germ free mice compared to conventionally housed mice166, which was also observed in MyD88/TRIF deficient mice. In the kidney, pericytes, a myofibroblast precursor, activate a TLR2/TLR4/MyD88-dependent proinflammatory program in response to tissue injury167. The downstream kinase IRAK4 controlled pericyte myofibroblast conversion in vitro and pharmacological inhibition of MyD88 signaling with an IRAK4 inhibitor reduced fibrosis by attenuating tissue injury in vivo167. Global TLR4 knockout168 and a small molecule inhibiting MyD88 ameliorated renal fibrosis in mice169. In systemic sclerosis, TLR4 and its co-receptors lymphocyte antigen 96 (MD2) and CD14 are overexpressed in lesional skin and chronic dermal LPS exposure led to overexpression of TGF-β signature genes170.
Of note, several TLRs are promiscuous and can also sense damage associated molecular patterns, lipids or ECM. This has been elegantly shown in lung fibrosis, where TLR4 and the glycosaminoglycan hyaluronan are important for type 2 alveolar epithelial cell renewal, limiting lung injury and fibrosis171. Interestingly, TLR4 was protective in the lung, as opposed to its pathogenic effect in gut and liver fibrosis127,159. Nasal polyposis is a disease characterized by remodeling of the sinonasal mucosa. Short single stranded DNA molecules, so called CpG oligonucleotides, can activate fibroblasts derived from nasal polyposis patients via TLR9 stimulation, providing an additional example whereby multiple PRRs, activated by distinct ligands, can contribute to aberrant wound healing and fibrosis.
The pathophysiologic relevance of TLR4 in inflammation has led to clinical development programs for rheumatoid arthritis using a TLR4 inhibitor (NCT03241108). Furthermore, pentraxin 2 (PTX2), also known as serum amyloid protein 2, has demonstrated anti-inflammatory and anti-fibrotic properties in multiple preclinical fibrosis models172,173 and recombinant PTX2 (PRM-151) has entered phase 2 trials for pulmonary fibrosis and myelofibrosis (NCT02550873; NCT01981850) (Table 1).
FUTURE DIRECTIONS
Fibrosis is a major global healthcare burden. Consequently, the discovery of key therapeutic targets with high relevance to human fibrotic disease and the subsequent development of effective antifibrotic therapies directed against these targets continues to be a research priority. Single-cell genomics methodologies have already yielded multiple new discoveries in fibrotic disease that would previously have been unattainable. This field continues to evolve rapidly, and emerging technologies are now able to measure multiple “omic” readouts (genomes, epigenomes, transcriptomes and proteomes) in single cells174–176. Spatially resolved molecular profiling is expanding our understanding of how these unique populations interact in situ177–179. The convergence and integration of these multi-modal single-cell technologies20,180, alongside global initiatives such as the Human Cell Atlas181, represent an extraordinary opportunity to decode the cellular and molecular mechanisms of fibrosis at unprecedented resolution, which should in turn help drive a new era of precision medicine in the treartment of fibrotic disease. Novel therapeutics developed for one fibrotic disorder may be applicable to a wide range of fibrotic diseases due to shared pathways across organs that are uncovered by this work. Drug repositioning efforts may also be impacted by these studies182.
Despite impressive progress over the past few years in our understanding of the pathogenesis of fibrosis, multiple challenges need to be overcome to translate this information to effective anti-fibrotic therapies (Figure 5). Prognostic animal models and ex vivo primary human tissue culture systems need to be developed that allow better translation of novel mechanisms from the bench to the bedside. Accurate and validated predictors of fibrotic disease progression are needed to stratify patients into high-risk populations. Patient heterogeneity together with the fact that fibrosis progression is typically slow makes selection of patients for clinical trials difficult. Hence, accurate and validated predictors of fibrotic disease progression are needed to stratify patients into high-risk populations prior to inclusion. Currently used trial endpoints are highly variable and often lack the sensitivity needed to predict favorable responses over a short period of time, which necessitates the inclusion of large numbers of patients in clinical trials. Consequently, endpoints for fibrosis clinical trials continue to evolve and may require a more global approach involving scientists, industry leaders, patients and regulatory partners, as recently shown for liver fibrosis and intestinal fibrosis183,184. Ideally, non-invasive endpoints that better correlate with clinically meaningful outcomes are needed. Recent research in the field has been fueled by the discovery of robust biomarkers and cutting-edge imaging modalities such as PET imaging of collagen and molecular imaging of fibrosis185,186, which allows fast, non-invasive, and whole-organ-quantitative and longitudinal readouts of drug efficacy in antifibrotic clinical trials. Furthermore, combining molecular imaging of fibrosis185 with cutting edge omic approaches such as single cell genomics174–176 could markedly improve patient diagnostics, staging, prognostication, stratification and cohort enrichment, which would in turn optimize clinical trial design and maximize the number of trials that could be run quickly and efficiently177–179. Innovative approaches to trial design are being developed that allow incorporation of adaptive strategies and use of ‘bucket’ trials that include patients with different types of fibrosis, as well as inclusion of ‘real-world’ evidence into the regulatory approval process.
Figure 5:
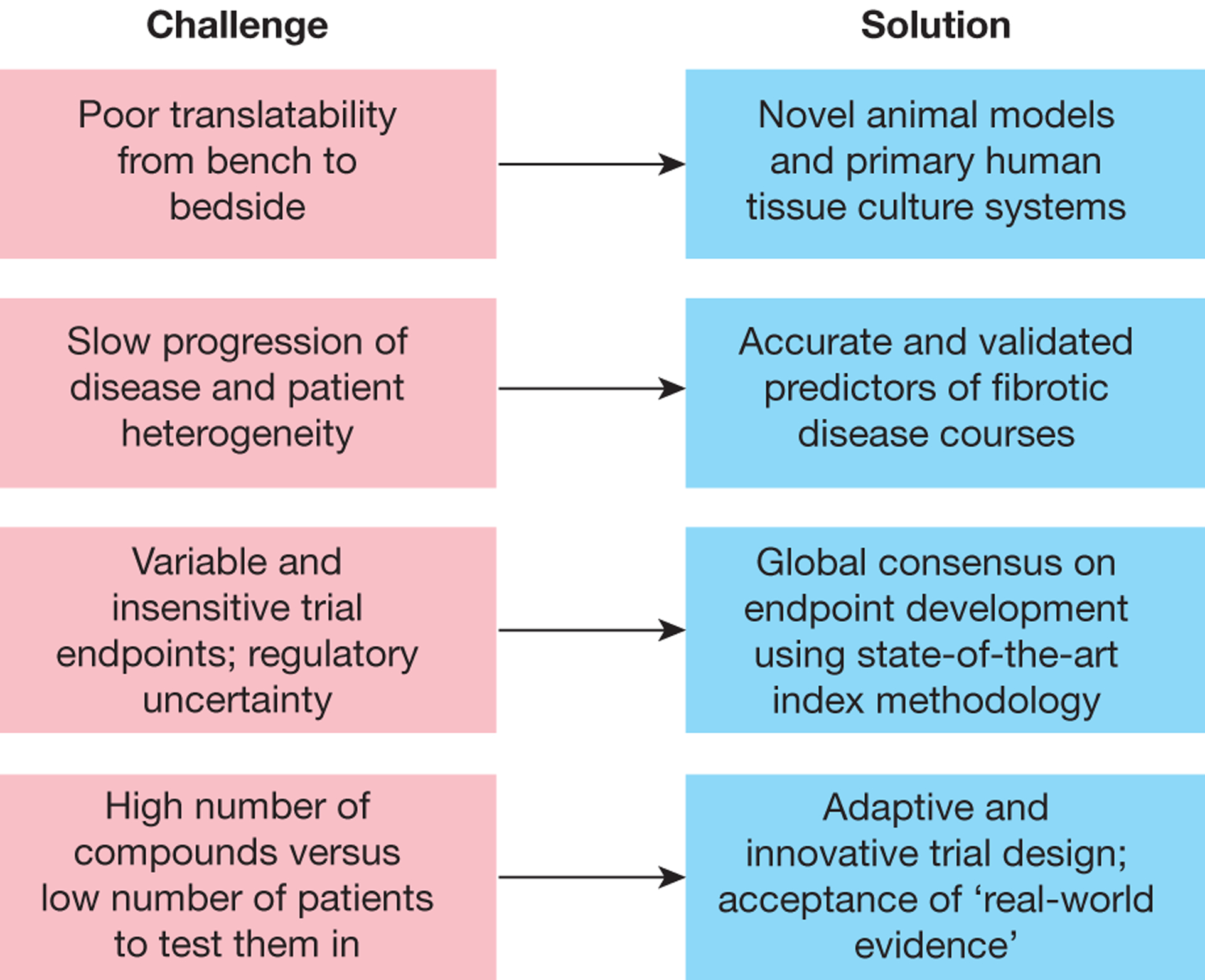
Challenges and solutions in the translation of antifibrotic mechanisms into drugs.
Similar to the major advances seen in cancer therapy or the successful treatment of HIV and viral hepatitis, we will very likely see increasing numbers of clinical trials testing combinations of drugs, as fibrosis is increasingly recognized as a highly complex disorder, with multiple mechanisms collaborating to drive disease progression. These antifibrotic drug cocktails will likely target a variety of orthogonal mechanisms, including a range of receptors, signaling pathways, and cell types that have been shown to function as core drivers of fibrosis in multiple disease states. These multi-faceted approaches should pave the way towards the delivery of effective antifibrotic therapies in the future.
Acknowledgments:
N.C.H. is supported by a Wellcome Trust Senior Research Fellowship in Clinical Science (Ref. 103749), Medical Research Council, Chan Zuckerberg Initiative Seed Network Grant and British Heart Foundation grants (RM/17/3/33381; RE/18/5/34216). F.R. is supported by grants from the National Institutes of Health (T32DK083251, P30DK097948 Pilot, K08DK110415), Crohn’s and Colitis Foundation, Cleveland Clinic, Rainin Foundation and the Helmsley Charitable Trust through the Stenosis Therapy and Anti-Fibrotic Research (STAR) Consortium.
Competing interests:
N.C.H. has received research funding from AbbVie, Pfizer, Inc., Gilead and Galecto, and is a scientific advisor for Galecto and Indalo Therapeutics. FR is on the advisory board or consultant for AbbVie, Allergan, Celgene, Gilead, Genentech, Gossamer, Receptos, Thetis, UCB, Samsung, Koutif, Pliant, Boehringer-Ingelheim, Metacrine, Takeda, Pfizer, Agomab, Allergan, Helmsley, RedX and Roche. T.A.W. is employed by Pfizer, Inc.
Abbreviations
- ACKR
Atypical chemokine receptor
- AT
Alveolar type
- BMP
Bone morphogenetic protein
- CCA
Canonical correlation analysis
- CD
Crohn’s disease
- CEACAM
Carcinoembyonic antigen-related adhesion molecules
- C/EBPβ
CCAAT/enhancer binding protein β
- ECM
Extracellular matrix
- FAP
Fibroblast activation protein
- Hic
Hypermethylated in cancer protein
- HSC
Hepatic stellate cells
- Ly
Lymphocyte antigen
- Lyve 1
Lymphatic vessel endothelial hyaluronan receptor 1
- IL
Interleukin
- IBD
Inflammatory Bowel Disease
- LOX
Lysyl oxidases
- LPS
lipopolysaccaride
- MP
Mesenchymal progenitor cells
- NLR
Nod-like receptors
- NLRP
NOD, LRR and pyrin domain-containing protein 3
- NOD
Nucleotide-binding oligomerization domain-containing protein
- PAMP
Pathogen-associated molecular patterns
- PDGF
Platelet derived growth factor
- PLVAP
Plasmalemma vesicle-associated protein
- PRR
Pattern recognition receptors
- scRNAseq
Single cell RNA sequencing
- SatM
Segregated-nucleus-containing atypical monocytes
- Siglec
Sialic acid-binding immunoglobulin-type lectins
- SMA
Smooth muscle actin
- SOX
SRY-box
- TGF
Transforming growth factor
- THY
Thymus cell antigen
- TLR
Toll-like receptors
- TNFRSF
Tumor necrosis factor receptor superfamily
- TREM
Triggering receptor expressed on myeloid cells
- UC
Ulcerative colitis
REFERENCES
- 1.Schiller HB et al. The Human Lung Cell Atlas: A High-Resolution Reference Map of the Human Lung in Health and Disease. Am J Respir Cell Mol Biol 61, 31–41, doi: 10.1165/rcmb.2018-0416TR (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zepp JA et al. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 170, 1134–1148 e1110, doi: 10.1016/j.cell.2017.07.034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie T et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep 22, 3625–3640, doi: 10.1016/j.celrep.2018.03.010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peyser R et al. Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing. Am J Respir Cell Mol Biol 61, 74–85, doi: 10.1165/rcmb.2018-0313OC (2019). [DOI] [PubMed] [Google Scholar]
- 5.Reyfman PA et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am J Respir Crit Care Med 199, 1517–1536, doi: 10.1164/rccm.201712-2410OC (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misharin AV et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214, 2387–2404, doi: 10.1084/jem.20162152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 1, e90558, doi: 10.1172/jci.insight.90558 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 180, 107–121 e117, doi: 10.1016/j.cell.2019.11.027 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Adams TS et al. Single Cell RNA-seq reveals ectopic and aberrant lung resident cell populations in Idiopathic Pulmonary Fibrosis. BioRxiv https://www.biorxiv.org/content/10.1101/759902v1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halpern KB et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356, doi: 10.1038/nature21065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacParland SA et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 9, 4383, doi: 10.1038/s41467-018-06318-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aizarani N et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572, 199–204, doi: 10.1038/s41586-019-1373-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halpern KB et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat Biotechnol 36, 962–970, doi: 10.1038/nbt.4231 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pepe-Mooney BJ et al. Single-Cell Analysis of the Liver Epithelium Reveals Dynamic Heterogeneity and an Essential Role for YAP in Homeostasis and Regeneration. Cell Stem Cell 25, 23–38 e28, doi: 10.1016/j.stem.2019.04.004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman SL, Roll FJ, Boyles J & Bissell DM Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proceedings of the National Academy of Sciences of the United States of America 82, 8681–8685, doi: 10.1073/pnas.82.24.8681 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobie R et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep 29, 1832–1847 e1838, doi: 10.1016/j.celrep.2019.10.024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krenkel O, Hundertmark J, Ritz TP, Weiskirchen R & Tacke F Single Cell RNA Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis. Cells 8, doi: 10.3390/cells8050503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramachandran P et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518, doi: 10.1038/s41586-019-1631-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study dissected unanticipated aspects of the cellular and molecular basis of human liver fibrosis at a single-cell level, providing a framework for the discovery of rational therapeutic targets in liver cirrhosis.
- 19.Vento-Tormo R et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353, doi: 10.1038/s41586-018-0698-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efremova M & Teichmann SA Computational methods for single-cell omics across modalities. Nat Methods 17, 14–17, doi: 10.1038/s41592-019-0692-4 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420, doi: 10.1038/nbt.4096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinchen J et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 175, 372–386 e317, doi: 10.1016/j.cell.2018.08.067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Croft AP et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251, doi: 10.1038/s41586-019-1263-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uncovered anatomically discrete, functionally distinct fibroblast subsets in the context of arthritis.
- 24.Driskell RR et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281, doi: 10.1038/nature12783 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rinkevich Y et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151, doi: 10.1126/science.aaa2151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Correa-Gallegos D et al. Patch repair of deep wounds by mobilized fascia. Nature 576, 287–292, doi: 10.1038/s41586-019-1794-y (2019). [DOI] [PubMed] [Google Scholar]; This work identified a specialised subset of fibroblasts, fascia fibroblasts, which gather the surrounding extracellular matrix and then rise to the surface of the skin following wounding.
- 27.Shook BA et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 362, doi: 10.1126/science.aar2971 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramann R et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16, 51–66, doi: 10.1016/j.stem.2014.11.004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider RK et al. Gli1(+) Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 23, 308–309, doi: 10.1016/j.stem.2018.07.006 (2018). [DOI] [PubMed] [Google Scholar]
- 30.El Agha E et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 20, 261–273 e263, doi: 10.1016/j.stem.2016.10.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott RW, Arostegui M, Schweitzer R, Rossi FMV & Underhill TM Hic1 Defines Quiescent Mesenchymal Progenitor Subpopulations with Distinct Functions and Fates in Skeletal Muscle Regeneration. Cell Stem Cell 25, 797–813 e799, doi: 10.1016/j.stem.2019.11.004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soliman H et al. Pathogenic Potential of Hic1-Expressing Cardiac Stromal Progenitors. Cell Stem Cell 26, 459–461, doi: 10.1016/j.stem.2020.01.023 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Franceschi C & Campisi J Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69 Suppl 1, S4–9, doi: 10.1093/gerona/glu057 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Mahmoudi S et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 574, 553–558, doi: 10.1038/s41586-019-1658-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kisseleva T et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proceedings of the National Academy of Sciences of the United States of America 109, 9448–9453, doi: 10.1073/pnas.1201840109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Troeger JS et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 143, 1073–1083 e1022, doi: 10.1053/j.gastro.2012.06.036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wohlfahrt T et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566, 344–349, doi: 10.1038/s41586-019-0896-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plikus MV et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 355, 748–752, doi: 10.1126/science.aai8792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song G et al. Direct Reprogramming of Hepatic Myofibroblasts into Hepatocytes In Vivo Attenuates Liver Fibrosis. Cell Stem Cell 18, 797–808, doi: 10.1016/j.stem.2016.01.010 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Rezvani M et al. In Vivo Hepatic Reprogramming of Myofibroblasts with AAV Vectors as a Therapeutic Strategy for Liver Fibrosis. Cell Stem Cell 18, 809–816, doi: 10.1016/j.stem.2016.05.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwabe RF, Tabas I & Pajvani UB Mechanisms of Fibrosis Development in NASH. Gastroenterology, doi: 10.1053/j.gastro.2019.11.311 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernard K et al. Metabolic Reprogramming Is Required for Myofibroblast Contractility and Differentiation. J Biol Chem 290, 25427–25438, doi: 10.1074/jbc.M115.646984 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie N et al. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am J Respir Crit Care Med 192, 1462–1474, doi: 10.1164/rccm.201504-0780OC (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L et al. Lactate Promotes Synthetic Phenotype in Vascular Smooth Muscle Cells. Circ Res 121, 1251–1262, doi: 10.1161/CIRCRESAHA.117.311819 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faubert B et al. Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359, doi: 10.1016/j.cell.2017.09.019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kottmann RM et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am J Respir Crit Care Med 186, 740–751, doi: 10.1164/rccm.201201-0084OC (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu G & Summer R Cellular Metabolism in Lung Health and Disease. Annu Rev Physiol 81, 403–428, doi: 10.1146/annurev-physiol-020518-114640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nigdelioglu R et al. Transforming Growth Factor (TGF)-beta Promotes de Novo Serine Synthesis for Collagen Production. J Biol Chem 291, 27239–27251, doi: 10.1074/jbc.M116.756247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park SY, Le CT, Sung KY, Choi DH & Cho EH Succinate induces hepatic fibrogenesis by promoting activation, proliferation, and migration, and inhibiting apoptosis of hepatic stellate cells. Biochem Biophys Res Commun 496, 673–678, doi: 10.1016/j.bbrc.2018.01.106 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Lian N et al. Curcumin regulates cell fate and metabolism by inhibiting hedgehog signaling in hepatic stellate cells. Lab Invest 95, 790–803, doi: 10.1038/labinvest.2015.59 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Ding H et al. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol 313, F561–F575, doi: 10.1152/ajprenal.00036.2017 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Wei Q et al. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am J Physiol Renal Physiol 316, F1162–F1172, doi: 10.1152/ajprenal.00422.2018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ge J et al. Glutaminolysis Promotes Collagen Translation and Stability via alpha-Ketoglutarate-mediated mTOR Activation and Proline Hydroxylation. Am J Respir Cell Mol Biol 58, 378–390, doi: 10.1165/rcmb.2017-0238OC (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bai L et al. Glutaminolysis Epigenetically Regulates Antiapoptotic Gene Expression in Idiopathic Pulmonary Fibrosis Fibroblasts. Am J Respir Cell Mol Biol 60, 49–57, doi: 10.1165/rcmb.2018-0180OC (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui H et al. Inhibition of Glutaminase 1 Attenuates Experimental Pulmonary Fibrosis. Am J Respir Cell Mol Biol 61, 492–500, doi: 10.1165/rcmb.2019-0051OC (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang HM et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21, 37–46, doi: 10.1038/nm.3762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This publication elegantly links abnormal fatty acid oxidation to fibrogenesis
- 57.Luengo A, Gui DY & Vander Heiden MG Targeting Metabolism for Cancer Therapy. Cell Chem Biol 24, 1161–1180, doi: 10.1016/j.chembiol.2017.08.028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wynn TA & Vannella KM Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462, doi: 10.1016/j.immuni.2016.02.015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vannella KM & Wynn TA Mechanisms of Organ Injury and Repair by Macrophages. Annu Rev Physiol 79, 593–617, doi: 10.1146/annurev-physiol-022516-034356 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Krenkel O & Tacke F Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17, 306–321, doi: 10.1038/nri.2017.11 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Chakarov S et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, doi: 10.1126/science.aau0964 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Guilliams M, Thierry GR, Bonnardel J & Bajenoff M Establishment and Maintenance of the Macrophage Niche. Immunity 52, 434–451, doi: 10.1016/j.immuni.2020.02.015 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Miron VE et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16, 1211–1218, doi: 10.1038/nn.3469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Epelman S et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104, doi: 10.1016/j.immuni.2013.11.019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lavine KJ et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proceedings of the National Academy of Sciences of the United States of America 111, 16029–16034, doi: 10.1073/pnas.1406508111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duffield JS et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. The Journal of clinical investigation 115, 56–65, doi: 10.1172/JCI22675 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borthwick LA et al. Macrophages are critical to the maintenance of IL-13-dependent lung inflammation and fibrosis. Mucosal Immunol 9, 38–55, doi: 10.1038/mi.2015.34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Satoh T et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541, 96–101, doi: 10.1038/nature20611 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Bajpai G et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 24, 1234–1245, doi: 10.1038/s41591-018-0059-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aran D et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172, doi: 10.1038/s41590-018-0276-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dick SA et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20, 29–39, doi: 10.1038/s41590-018-0272-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J & Kubes P A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell 165, 668–678, doi: 10.1016/j.cell.2016.03.009 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Deniset JF et al. Gata6(+) Pericardial Cavity Macrophages Relocate to the Injured Heart and Prevent Cardiac Fibrosis. Immunity 51, 131–140 e135, doi: 10.1016/j.immuni.2019.06.010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Henderson NC et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 172, 288–298, doi: 10.2353/ajpath.2008.070726 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pakshir P & Hinz B The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol 68–69, 81–93, doi: 10.1016/j.matbio.2018.01.019 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Pradere JP et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473, doi: 10.1002/hep.26429 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou X et al. Circuit Design Features of a Stable Two-Cell System. Cell 172, 744–757 e717, doi: 10.1016/j.cell.2018.01.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lodyga M et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-beta. Sci Signal 12, doi: 10.1126/scisignal.aao3469 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Minutti CM et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 50, 645–654 e646, doi: 10.1016/j.immuni.2019.01.008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pakshir P et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat Commun 10, 1850, doi: 10.1038/s41467-019-09709-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wynn TA & Ramalingam TR Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature medicine 18, 1028–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diebold RJ et al. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proceedings of the National Academy of Sciences of the United States of America 92, 12215–12219 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Babyatsky MW, Rossiter G & Podolsky DK Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 110, 975–984 (1996). [DOI] [PubMed] [Google Scholar]
- 84.McEntee CP, Gunaltay S & Travis MA Regulation of barrier immunity and homeostasis by integrin-mediated transforming growth factor beta activation. Immunology, doi: 10.1111/imm.13162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelly A et al. Human monocytes and macrophages regulate immune tolerance via integrin alphavbeta8-mediated TGFbeta activation. J Exp Med 215, 2725–2736, doi: 10.1084/jem.20171491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hynes RO Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Barczyk M, Carracedo S & Gullberg D Integrins. Cell Tissue Res 339, 269–280, doi: 10.1007/s00441-009-0834-6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Robertson IB & Rifkin DB Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb Perspect Biol 8, doi: 10.1101/cshperspect.a021907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reed NI et al. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med 7, 288ra279, doi: 10.1126/scitranslmed.aaa5094 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Munger JS et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328, doi: 10.1016/s0092-8674(00)80545-0 (1999). [DOI] [PubMed] [Google Scholar]
- 91.Mu D et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol 157, 493–507, doi: 10.1083/jcb.200109100 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wipff PJ, Rifkin DB, Meister JJ & Hinz B Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol 179, 1311–1323, doi: 10.1083/jcb.200704042 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Asano Y et al. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J Immunol 175, 7708–7718, doi: 10.4049/jimmunol.175.11.7708 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Shi M et al. Latent TGF-beta structure and activation. Nature 474, 343–349, doi: 10.1038/nature10152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dong X et al. Force interacts with macromolecular structure in activation of TGF-beta. Nature 542, 55–59, doi: 10.1038/nature21035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dong X, Hudson NE, Lu C & Springer TA Structural determinants of integrin beta-subunit specificity for latent TGF-beta. Nat Struct Mol Biol 21, 1091–1096, doi: 10.1038/nsmb.2905 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Campbell MG et al. Cryo-EM Reveals Integrin-Mediated TGF-beta Activation without Release from Latent TGF-beta. Cell 180, 490–501 e416, doi: 10.1016/j.cell.2019.12.030 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma LJ et al. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(−/−) mice. Am J Pathol 163, 1261–1273, doi: 10.1016/s0002-9440(10)63486-4 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Puthawala K et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med 177, 82–90, doi: 10.1164/rccm.200706-806OC (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hahm K et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol 170, 110–125, doi: 10.2353/ajpath.2007.060158 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang B et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology 46, 1404–1412, doi: 10.1002/hep.21849 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peng ZW et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 63, 217–232, doi: 10.1002/hep.28274 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Horan GS et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med 177, 56–65, doi: 10.1164/rccm.200706-805OC (2008). [DOI] [PubMed] [Google Scholar]
- 104.Araya J et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. The Journal of clinical investigation 117, 3551–3562, doi: 10.1172/JCI32526 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kitamura H et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin alphavbeta8-mediated activation of TGF-beta. The Journal of clinical investigation 121, 2863–2875, doi: 10.1172/JCI45589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Minagawa S et al. Selective targeting of TGF-beta activation to treat fibroinflammatory airway disease. Sci Transl Med 6, 241ra279, doi: 10.1126/scitranslmed.3008074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Henderson NC et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19, 1617–1624, doi: 10.1038/nm.3282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barron L & Wynn TA Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. American journal of physiology 300, G723–728, doi: 10.1152/ajpgi.00414.2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park MJ et al. IL-1-IL-17 Signaling Axis Contributes to Fibrosis and Inflammation in Two Different Murine Models of Systemic Sclerosis. Front Immunol 9, 1611, doi: 10.3389/fimmu.2018.01611 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wilson MS et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207, 535–552, doi: 10.1084/jem.20092121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang BZ et al. Interleukin-17A antagonist attenuates radiation-induced lung injuries in mice. Exp Lung Res 40, 77–85, doi: 10.3109/01902148.2013.872210 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Meng F et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 143, 765–776 e761–763, doi: 10.1053/j.gastro.2012.05.049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun B et al. Role of interleukin 17 in TGF-beta signaling-mediated renal interstitial fibrosis. Cytokine 106, 80–88, doi: 10.1016/j.cyto.2017.10.015 (2018). [DOI] [PubMed] [Google Scholar]
- 114.Feng W et al. IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp Mol Pathol 87, 212–218, doi: 10.1016/j.yexmp.2009.06.001 (2009). [DOI] [PubMed] [Google Scholar]
- 115.Fabre T et al. Type 3 cytokines IL-17A and IL-22 drive TGF-beta-dependent liver fibrosis. Sci Immunol 3, doi: 10.1126/sciimmunol.aar7754 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Tan Z et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol 191, 1835–1844, doi: 10.4049/jimmunol.1203013 (2013). [DOI] [PubMed] [Google Scholar]
- 117.Zhang S et al. Neutralization of Interleukin-17 Attenuates Cholestatic Liver Fibrosis in Mice. Scand J Immunol 83, 102–108, doi: 10.1111/sji.12395 (2016). [DOI] [PubMed] [Google Scholar]
- 118.Zhang XW et al. Antagonism of Interleukin-17A ameliorates experimental hepatic fibrosis by restoring the IL-10/STAT3-suppressed autophagy in hepatocytes. Oncotarget 8, 9922–9934, doi: 10.18632/oncotarget.14266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hara M et al. Interleukin-17A plays a pivotal role in cholestatic liver fibrosis in mice. The Journal of surgical research 183, 574–582, doi: 10.1016/j.jss.2013.03.025 (2013). [DOI] [PubMed] [Google Scholar]
- 120.Fabre T, Kared H, Friedman SL & Shoukry NH IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-beta receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol 193, 3925–3933, doi: 10.4049/jimmunol.1400861 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oh K et al. Epithelial transglutaminase 2 is needed for T cell interleukin-17 production and subsequent pulmonary inflammation and fibrosis in bleomycin-treated mice. The Journal of experimental medicine 208, 1707–1719, doi: 10.1084/jem.20101457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wree A et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 67, 736–749, doi: 10.1002/hep.29523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gasse P et al. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One 6, e23185, doi: 10.1371/journal.pone.0023185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lemmers A et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 49, 646–657, doi: 10.1002/hep.22680 (2009). [DOI] [PubMed] [Google Scholar]
- 125.Macek Jilkova Z et al. Progression of fibrosis in patients with chronic viral hepatitis is associated with IL-17(+) neutrophils. Liver Int 36, 1116–1124, doi: 10.1111/liv.13060 (2016). [DOI] [PubMed] [Google Scholar]
- 126.Yang D et al. Dysregulated Lung Commensal Bacteria Drive Interleukin-17B Production to Promote Pulmonary Fibrosis through Their Outer Membrane Vesicles. Immunity 50, 692–706 e697, doi: 10.1016/j.immuni.2019.02.001 (2019). [DOI] [PubMed] [Google Scholar]
- 127.Seki E et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nature medicine 13, 1324–1332 (2007). [DOI] [PubMed] [Google Scholar]
- 128.Gieseck RL 3rd, Wilson MS & Wynn TA Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 18, 62–76, doi: 10.1038/nri.2017.90 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Hams E et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proceedings of the National Academy of Sciences of the United States of America 111, 367–372, doi: 10.1073/pnas.1315854111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jessup HK et al. Intradermal administration of thymic stromal lymphopoietin induces a T cell- and eosinophil-dependent systemic Th2 inflammatory response. J Immunol 181, 4311–4319, doi: 10.4049/jimmunol.181.6.4311 (2008). [DOI] [PubMed] [Google Scholar]
- 131.McHedlidze T et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 39, 357–371, doi: 10.1016/j.immuni.2013.07.018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vannella KM et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med 8, 337ra365, doi: 10.1126/scitranslmed.aaf1938 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Lee CG et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 194, 809–821, doi: 10.1084/jem.194.6.809 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaviratne M et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol 173, 4020–4029, doi: 10.4049/jimmunol.173.6.4020 (2004). [DOI] [PubMed] [Google Scholar]
- 135.Gieseck RL 3rd et al. Interleukin-13 Activates Distinct Cellular Pathways Leading to Ductular Reaction, Steatosis, and Fibrosis. Immunity 45, 145–158, doi: 10.1016/j.immuni.2016.06.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hart KM et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-beta. Sci Transl Med 9, doi: 10.1126/scitranslmed.aal3694 (2017). [DOI] [PubMed] [Google Scholar]; This study identified opposing roles for type 2 immunity in metabolic syndrome and liver fibrosis in an experimental model of NASH.
- 137.Chiaramonte MG, Donaldson DD, Cheever AW & Wynn TA An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. The Journal of clinical investigation 104, 777–785, doi: 10.1172/JCI7325 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xue J et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun 6, 7158, doi: 10.1038/ncomms8158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu L et al. CD4+ T Lymphocytes, especially Th2 cells, contribute to the progress of renal fibrosis. Am J Nephrol 36, 386–396, doi: 10.1159/000343283 (2012). [DOI] [PubMed] [Google Scholar]
- 140.Chung SI et al. IL-13 is a therapeutic target in radiation lung injury. Sci Rep 6, 39714, doi: 10.1038/srep39714 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Singh B, Kasam RK, Sontake V, Wynn TA & Madala SK Repetitive intradermal bleomycin injections evoke T-helper cell 2 cytokine-driven pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 313, L796–L806, doi: 10.1152/ajplung.00184.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sciurba JC et al. Fibroblast-specific integrin-alpha V differentially regulates type 17 and type 2 driven inflammation and fibrosis. J Pathol 248, 16–29, doi: 10.1002/path.5215 (2019). [DOI] [PubMed] [Google Scholar]
- 143.Wang M et al. Cross-talk between TH2 and TH17 pathways in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 144, 1254–1264, doi: 10.1016/j.jaci.2019.06.023 (2019). [DOI] [PubMed] [Google Scholar]
- 144.Choy DF et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 7, 3011ra129, doi: 10.1126/scitranslmed.aab3142 (2015). [DOI] [PubMed] [Google Scholar]
- 145.Ramalingam TR et al. Enhanced protection from fibrosis and inflammation in the combined absence of IL-13 and IFN-gamma. J Pathol 239, 344–354, doi: 10.1002/path.4733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tang W et al. Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. The Journal of clinical investigation 98, 2845–2853, doi: 10.1172/JCI119113 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Widjaja AA et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 157, 777–792 e714, doi: 10.1053/j.gastro.2019.05.002 (2019). [DOI] [PubMed] [Google Scholar]
- 148.Schafer S et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552, 110–115, doi: 10.1038/nature24676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identified autocrine IL-11/IL-11R signaling in fibroblasts as a key mechanism driving cardiovascular fibrosis in response to a variety of stimuli.
- 149.Ng B et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci Transl Med 11, doi: 10.1126/scitranslmed.aaw1237 (2019). [DOI] [PubMed] [Google Scholar]
- 150.Takeuchi O & Akira S Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010). [DOI] [PubMed] [Google Scholar]
- 151.Abreu MT et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology 123, 679–688 (2002). [DOI] [PubMed] [Google Scholar]
- 152.Rieder F et al. Association of the novel serologic anti-glycan antibodies anti-laminarin and anti-chitin with complicated Crohn’s disease behavior. Inflamm Bowel Dis 16, 263–274, doi: 10.1002/ibd.21046 (2010). [DOI] [PubMed] [Google Scholar]
- 153.Rieder F et al. Serum anti-glycan antibodies predict complicated Crohn’s disease behavior: a cohort study. Inflamm Bowel Dis 16, 1367–1375, doi: 10.1002/ibd.21179 (2010). [DOI] [PubMed] [Google Scholar]
- 154.Rieder F, Kessler S, Sans M & Fiocchi C Animal models of intestinal fibrosis: new tools for the understanding of pathogenesis and therapy of human disease. American journal of physiology 303, G786–801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Moresco EM, LaVine D & Beutler B Toll-like receptors. Curr Biol 21, R488–493, doi: 10.1016/j.cub.2011.05.039 (2011). [DOI] [PubMed] [Google Scholar]
- 156.Mansson LE et al. MyD88 signaling promotes both mucosal homeostatic and fibrotic responses during Salmonella-induced colitis. American journal of physiology 303, G311–323 (2012). [DOI] [PubMed] [Google Scholar]
- 157.Imai J et al. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal immunology, doi: 10.1038/s41385-019-0138-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jacob N et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal immunology 11, 1466–1476, doi: 10.1038/s41385-018-0055-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Otte JM, Rosenberg IM & Podolsky DK Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology 124, 1866–1878 (2003). [DOI] [PubMed] [Google Scholar]
- 160.Zhao S et al. Selective deletion of MyD88 signaling in alpha-SMA positive cells ameliorates experimental intestinal fibrosis via post-transcriptional regulation. Mucosal Immunol, doi: 10.1038/s41385-020-0259-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This publication highlights a selective mechanism on how bacteria can activate myofibroblast through flagellin.
- 161.Lin RS et al. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol 22, 165–172 (1995). [DOI] [PubMed] [Google Scholar]
- 162.Chan CC et al. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scandinavian journal of gastroenterology 32, 942–946 (1997). [DOI] [PubMed] [Google Scholar]
- 163.Seki E & Brenner DA Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48, 322–335 (2008). [DOI] [PubMed] [Google Scholar]
- 164.Sun L et al. Lipopolysaccharide enhances TGF-beta1 signalling pathway and rat pancreatic fibrosis. J Cell Mol Med 22, 2346–2356, doi: 10.1111/jcmm.13526 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang L et al. TRIF Differentially Regulates Hepatic Steatosis and Inflammation/Fibrosis in Mice. Cell Mol Gastroenterol Hepatol 3, 469–483, doi: 10.1016/j.jcmgh.2016.12.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mazagova M et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J 29, 1043–1055, doi: 10.1096/fj.14-259515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Leaf IA et al. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. The Journal of clinical investigation 127, 321–334, doi: 10.1172/JCI87532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jialal I, Major AM & Devaraj S Global Toll-like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. J Diabetes Complications 28, 755–761, doi: 10.1016/j.jdiacomp.2014.07.003 (2014). [DOI] [PubMed] [Google Scholar]
- 169.Liu JH et al. A Novel Inhibitor of Homodimerization Targeting MyD88 Ameliorates Renal Interstitial Fibrosis by Counteracting TGF-beta1-Induced EMT in Vivo and in Vitro. Kidney Blood Press Res 43, 1677–1687, doi: 10.1159/000494745 (2018). [DOI] [PubMed] [Google Scholar]
- 170.Stifano G et al. Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther 16, R136, doi: 10.1186/ar4598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liang J et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nature medicine 22, 1285–1293, doi: 10.1038/nm.4192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work discovered an anti-fibrotic mechanism of hyaluronan in pulmonary fibrosis via TLR4 providing a novel function of this toll-like receptor.
- 172.Pilling D et al. Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J Immunol 179, 4035–4044 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nakagawa N et al. Pentraxin-2 suppresses c-Jun/AP-1 signaling to inhibit progressive fibrotic disease. JCI Insight 1, e87446, doi: 10.1172/jci.insight.87446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385, doi: 10.1126/science.aau0730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 35, 936–939, doi: 10.1038/nbt.3973 (2017). [DOI] [PubMed] [Google Scholar]
- 176.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868, doi: 10.1038/nmeth.4380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rodriques SG et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467, doi: 10.1126/science.aaw1219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vickovic S et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods 16, 987–990, doi: 10.1038/s41592-019-0548-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Eng CL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239, doi: 10.1038/s41586-019-1049-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821, doi: 10.1016/j.cell.2019.05.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Regev A et al. The Human Cell Atlas. Elife 6, doi: 10.7554/eLife.27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dudley JT et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med 3, 96ra76, doi: 10.1126/scitranslmed.3002648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Torok NJ, Dranoff JA, Schuppan D & Friedman SL Strategies and endpoints of antifibrotic drug trials: Summary and recommendations from the AASLD Emerging Trends Conference, Chicago, June 2014. Hepatology 62, 627–634, doi: 10.1002/hep.27720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rieder F et al. An expert consensus to standardise definitions, diagnosis and treatment targets for anti-fibrotic stricture therapies in Crohn’s disease. Aliment Pharmacol Ther 48, 347–357, doi: 10.1111/apt.14853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Montesi SB, Desogere P, Fuchs BC & Caravan P Molecular imaging of fibrosis: recent advances and future directions. The Journal of clinical investigation 129, 24–33, doi: 10.1172/JCI122132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Montesi SB et al. Type I Collagen-targeted Positron Emission Tomography Imaging in Idiopathic Pulmonary Fibrosis: First-in-Human Studies. Am J Respir Crit Care Med 200, 258–261, doi: 10.1164/rccm.201903-0503LE (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]