

Abstract
Cell iron uptake in mammals is commonly distinguished by whether the iron is presented to the cell as transferrin-bound or not: TBI or NTBI. This generic perspective conflates TBI with canonical transferrin receptor, endosomal iron uptake, and NTBI with uptake supported by a plasma membrane-localized divalent metal ion transporter, most often identified as DMT1. In fact, iron uptake by mammalian cells is far more nuanced than this somewhat proscribed view suggests. This view fails to accommodate the substantial role that ZIP8 and ZIP14 play in iron uptake, while adhering to the traditional premise that a relatively high endosomal [H+] is thermodynamically required for release of iron from holo-Tf. The canonical view of iron uptake also does not encompass the fact that plasma membrane electron transport – PMET – has long been linked to cell iron uptake. In fact, the known mammalian metallo-reductases – Dcytb and the STEAP proteins – are members of this cohort of cytochrome-dependent oxido-reductases that shuttle reducing equivalents across the plasma membrane. A not commonly appreciated fact is the reduction potential of ferric iron in holo-Tf is accessible to cytoplasmic reducing equivalents – reduced pyridine and flavin mono- and di-nucleotides and dihydroascorbic acid. This allows for the reductive release of Fe2+ at the extracellular surface of the PM and subsequent transport into the cytoplasm by a neutral pH transporter – a ZIP protein. What this perspective emphasizes is that there are two TfR-dependent uptake pathways, one which does and one which does not involve clathrin-dependent, endolysosomal trafficking. This raises the question as to the selective advantage of having two Tf, TfR-dependent routes of iron accumulation. This review of canonical and non-canonical iron uptake uses cerebral iron trafficking as a point of discussion, a focus that encourages inclusion also of the importance of ferritin as a circulating ‘chaperone’ of ferric iron.
Introduction
If one wants to catch up with current thinking on iron uptake in mammalian cells, scanning through contemporary reviews on the subject confirms one’s general knowledge of transferrin receptor-mediated iron acquisition from transferrin-bound Fe(iii) (TBI). One also finds that divalent metal ion transporter 1 – DMT1 – serves not only in the TfR/endosomal uptake pathway of TBI but supports the uptake of ‘aqueous’ Fe2+ – non-transferrin bound iron, NTBI – as well. For example, in a summary of fluorescent probes for Fe(ii), one reads “The cellular iron uptake machinery primarily utilizes divalent metal transporter 1 (DMT1)”.1 Or, in a review on DMT1, while there is a paragraph reviewing “Other iron transporters” all of the highly illustrative figures feature DMT1 as the sole representative, an unfortunately limited representation of cellular iron metabolism.2 And then there’s the statement from the review, Mechanisms of Brain Iron Transport: “Most (italics added) other mammalian cell types recruit the cellular iron transport machinery that is used by enterocytes and erythroid precursor cells.”3 There is also a general emphasis on TBI as the major source of iron in the brain’s abluminal space for uptake into neurons: “It is proposed that neurons take up iron through the transferrin receptor and DMT1, as described for iron uptake in enterocytes and endothelial cells…”4 In this Perspective the focus is on the misapprehension that when it comes to cell iron accumulation the TfR, DMT1 pathway is the only game in town. As this commentary will argue, while TfR is a key player in cell iron accumulation, in many cell types the role it often plays is unrelated to the mechanism alluded to in the quotes above.
Physiologic handling of the differing aqueous properties of Fe2+ and Fe3+
Systemic and abluminal (interstitial brain) iron is generally described as being in either the ferrous or ferric form and this redox speciation is thought to reflect the ligand to which the metal ion is bound. Ferric iron, Fe3+, is found as transferrin-bound iron (TBI).5–7 Circulating ferritin (Ft) also contributes to this iron pool, again present as Fe3+.8 That systemic, extra-cellular Fe3+ is exclusively protein-bound to Tf or Ft reflects the essential insolubility of trivalent iron at physiologic pH.9 Ferrous iron, Fe2+, is referred to as non-transferrin iron (NTBI) and can be thought of as ‘aqueous’ Fe2+ in that irrespective of other ligation, at least one water ligand will be present in the metal ion’s inner coordination sphere. However, the reader should not think of this as ‘free, non-liganded’ Fe2+, Fe(H2O)62+, Ferrous iron is far less electropositive than ferric; Fe2+-bound waters do not deprotonate thus the precipitation of ferrousoxide polymers is a far slower kinetic process than the precipitation of ferri-oxo ones.9
This is not to exclude a likely redox speciation of circulating NTBI. Given the dissolved [O2] in the serum (~50 μM) and at neutral pH, redox-cycling of iron is likely supported by dioxygen and a physiologic reductant such as dihydroascorbic acid.9 One essential aspect of iron redox cycling in this environment is the fact that both ferric iron hydrolysis – rust formation – and reduction by ascorbate require an ‘open’ or exchangeable coordination site. By definition, ‘hydrolysis’ requires water coordination, while a coordination site occupied by water is de facto ‘open.’ The reduction potential of plasma – ~−140 mV – is set by the GSH/GSSG buffer.10 Given that the reduction potential of ascorbic acid at pH 7.0 is −80 mV, it circulates primarily in its reduced form.11 Work by the Cabantchik group indicated that the ~50 μM ascorbate found in serum supported a robust redox cycling of low micromolar Fe2+/3+. Note, however, that this group’s experiments were performed under normobaric conditions, [O2]dissolved ≅ 250 μM.12 In contrast, Osaki, Johnson and Frieden carried out comparable experiments but at [O2]dissolved typical of plasma, ~50 μM. Given that ferrous iron redox cycling is linearly-dependent on [O2] it is not surprising that the latter group found a cycling rate ~1/5th the rate found by the former.13 Thus, except for iron found in very oxygen-rich coordination spheres (e.g. as in Tf), ferrous iron is thermodynamically quite comfortable in plasma. Professor Cabantchik has authored a thorough summary of chelate-accessible systemic iron – essentially all Fe2+ – that is an excellent introduction to the topic.14
Ferric and ferrous iron uptake: the malleable reduction potential of transferrin-bound Fe3+
Conceptually, cell utilization of these two disparate, oxidation states of iron would require equally disparate pathways. In fact, the two pathways differ only in one way, underscoring an inherent similarity that is seldom noted and that is the point of this commentary. Accumulation of TBI is initiated by binding of iron-bound Tf (holo-Tf) to the transferrin receptor, TfR, a dimeric transmembrane glycoprotein.15 (As noted below, ferritin – Ft – is also taken up by cells via a receptor-mediated process.16,17) NTBI cell uptake involves no such ‘docking protein’ but the following chemical step in cell iron accumulation of the two ‘forms’ of iron is essentially the same and is simple: ferri-reduction if this cell-associated iron is Fe3+ (as in Fe-Tf), followed by ferrous iron transport into the cytoplasmic compartment via a divalent metal ion transporter.18,19 The paradigmatic Tf-TfR-DMT1 pathway was, in part, a conflation of two separate experimental trajectories. First, the DMT1/Dcytb one that was proposed by McKie et al. with their cloning and expression of intestinal Dcytb (Cybrd1).20 Dcytb is one member of the family of mammalian Fe3+ and Cu2+ metallo-reductases.21 The second was the roundabout association with iron metabolism of the ‘natural resistance-associated macrophage protein 2′ – NRAMP222,23 – rechristened DMT124 that, it turned out, is essential to the mobilization of endosomal, Tf-delivered iron for efflux into the cytoplasm. The fact that the macrophage phagolysosome is an acidic compartment, and studies that linked iron release from holo-Tf to an acidic pH25 established the link between capture of the holo-Tf·TfR complex in an endolysosome and the DMT1-dependent efflux of Fe2+ from this compartment.
What has increasingly become apparent is that these canonical iron uptake pathways, both of which are DMT1-dependent, are strongly represented only by the index cell types in which they were first identified: enterocytes, macrophages and cells of the reticuloendothelial cohort. For example, while Dcytb is clearly the essential ferric and cupric reductase in the intestinal lumen,26 STEAP family member metallo-reductases have been associated with the equivalent metal trafficking function in a variety of if not all other cell types.27–29 As for alternatives to DMT1, the divalent metal ion transporters ZIP8 and ZIP14 show a strong kinetic preference for Fe2+ and Mn2+.30–37 The physiologic necessity for having two classes of reductase/ferrous iron uptake pairs is obvious: the disparate pH of the duodenum and endolysosome, versus essentially every other physiologic compartment, whether at the organ or cell level. Among other factors is simply the strongly differing aqueous, redox chemistry of iron at pH 5.5 versus 7.4, i.e. the ~120 mV lower iron reduction potential at the higher pH (lower [H+]) requiring a stronger driving force for reduction of ferric to ferrous iron.
The iron in ferric-Tf is bound in a very oxygen-rich coordination sphere. As is characteristic of most transition metals, oxygen ligation (in contrast to nitrogen, for example) supports a more negative reduction potential, that is, makes the metal a reductant, not oxidant. If one searches for “reduction potential of ferric transferrin” the leading hit tells you it is ‘below −500 mV.”38 This is a potential far too low for this ferric iron to serve as electron acceptor (as an oxidant) from any physiologic reductant, e.g. from a reduced pyridine or flavin nucleotide.38,39 However, as discussed below, physiologically this potential is much higher, in the range of −240 mV. This is significant since, overall, the cytoplasmic reduction potential is ~−240 mV due primarily to the GSH/GSSH ratio much like in the plasma.10,40,41 Thus, Tf is a perfect ferric iron chaperone stabilizing Fe3+ from water hydrolysis while poising it for physiologic ferri-reduction. This is key to the cell utilization of Tf-iron since the stability of the ferric complex is 1014 greater than the ferrous one.42
Mobilization of iron from ferric transferrin does not require the endolysosome
However, reduction of the Fe3+ in ferric-Tf is not necessary to mobilize the iron. Protein allostery has been employed by both pro- and eukaryotes (vertebrates) to pry the iron out of holo-Tf without change in redox state. Thus, how is it that Neisseria can scavenge ferric iron from transferrin? By allostery: using the ‘work’ made available in TbpA binding to TbpB and then to Fe3+-Tf to ‘loosen’ the hold that Tf has on Fe3+ thus ‘catalyzing’ an innersphere, non-dissociative transfer of Fe3+ from Tf to TbpA.43,44 As for vertebrates, the equivalent ‘TbpA’ is the transferrin receptor, TfR. Not widely appreciated is that the Fe3+-Tf iron reduction potential increases by >200 mV simply by holo-Tf binding to TfR, bringing the Fe3+ into ‘redox equilibrium’ with typical biologic electron donors as noted above.38 This increase is found at pH 7.0, i.e. it is independent of the increase in reduction potential that follows Pourbaix behavior: increasing [H+], increasing E1/2. These studies, reported by Aisen and Crumbliss in 2004, used spectroelectrochemical analysis of the visible absorption spectrum unique to Fe(iii)-Tf that, when analyzed by a classic Nernst analysis revealed a shift in the E1/2 value from −501 mV for Fe(iii)-Tf to −285 mV for Fe(iii)-Tf·TfR.38
The structural explanation for this change in iron reduction potential is found in the conformation change in holo-Tf upon binding to TfR that is linked to a repositioning of a histidine residue required for the ‘catalysis’ of ferri-reduction and ferrous iron release. In short, TfR can support the reductive mobilization of iron – as Fe2+ – from Fe3+-Tf at neutral pH.45 The overall ‘reaction’ chemistry is the same as that envisioned in the endolysosome, but it can take place entirely on the extra-cytoplasmic face of the plasma membrane: holo-Tf binding to TfR making the Tf Fe3+ a better oxidant, followed by reductive Fe2+ release. This labilized ferrous iron then serves as ligand for a plasma membrane-localized divalent metal ion transporter for delivery to the cytoplasm. To emphasize: this “NTBI-like” uptake of TBI is TfR dependent but does not involve the canonical, clathrin-dependent endosomal-trafficking TBI pathway.18 If nothing else, this is a far more energetically-efficient utilization of TBI, avoiding the multiple nucleotide triphosphate-driven steps involved in endosome budding and transport. These two TfR-dependent pathways available for Fe-uptake from holo-Tf are illustrated in Fig. 1; the ‘non-canonical’ TfR-dependent pathway is given in panel A.
Fig. 1.
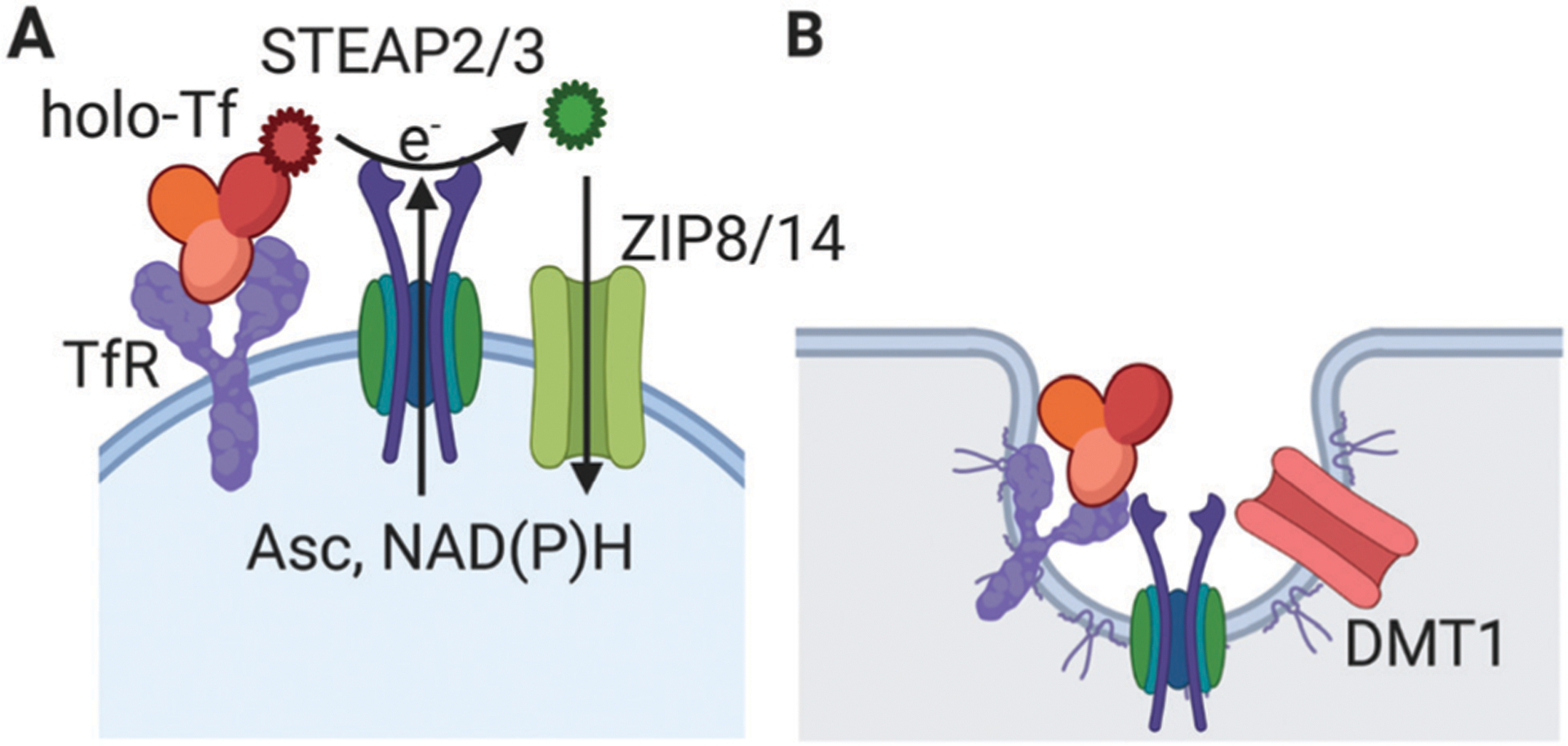
Transferrin receptor-mediated cell iron uptake from holo-transferrin. (A) The cohort of receptor (TfR), ligand (holo-Tf), ferri-reductase (STEAP2 or 3) and plasma membrane, neutral pH ferrous iron transporter (ZIP8 or 14) are illustrated. As noted in the text, this uptake pathway is blocked by (1) a TfR-specific antibody; (2) an inhibitor or knock-down of ferri-reductase activity; (3) trapping of the reductase-generated Fe2+ with a membrane-impermeant ferrous iron chelator; or (4) knock-down of surface abundance of the ZIP transporter.18,31 Fe3+ is illustrated in red; Fe2+ is illustrated in green. (B) The ‘canonical’ TfR-mediated endolysosomal iron uptake from holo-transferrin. The key difference is the specific role played by the H+-coupled ferrous iron transporter, DMT1. These two pathways are not mutually exclusive; their function in a given cell type could be dependent solely on the relative expression of the two transporter types.
This ‘ectodomain’ model of reductive, TfR-mediated Tf-iron uptake builds upon the long-standing experimental evidence for the extra-cytoplasmic ferri-reduction of Tf-bound ferric iron. One can read in the abstract of a paper in Biochimica Biophysica Acta (1983): “These observations suggest that most if not all iron is loosened from transferrin upon interaction of transferrin with the hepatocyte membrane.”46 Or in the abstract of another paper, “We propose that the NADH diferric transferrin reductase in plasma membranes measures the activity of the enzyme that causes the reduction of diferric transferrin by intact cells. This transmembrane electron transport system requires the transferrin receptor for diferric transferrin reduction.”47 Not surprisingly, there was considerable ‘discussion’ about this ‘reductive’ mechanism for accumulation of iron from TBI. A summary of these discussions was provided in 1990 and while correctly summarizing the data that supported this ‘non-canonical’ pathway, noted also that48
A serious obstacle to the model of reductive release of iron from transferrin is the fact that at neutral pH the reduction potential for transferrin iron is much more negative than that of NADH.
This was a solid thermodynamic argument in 1990, but loses strength in light of the determination of the considerably less negative E1/2 for TfR·Tf-Fe3+ provided by Professors Aisen, Crumbliss and their co-workers in 2004.38
PMET and other thermodynamic considerations
Eukaryotic cells express a fairly well-studied plasma membrane electron transport (PMET) activity driven by NADPH; ascorbic acid also can contribute the reducing equivalents needed to drive this flux.49–54 Indeed, the structure of Dcytb has identified a cytoplasm-localized ascorbate binding site indicating the likelihood that dihydroascorbic acid is the electron donor for this metallo-reductase.55 PMET has been linked by redox chemistry to ferri-Tf reduction in cells and in purified mammalian cell plasma membranes primed with NADH. Based on the substrates that support this ET, this system has been given the complementary names diferric transferrin reductase47,56 or diferric transferrin oxidase57 activity in the sense that holo-Tf is acting an ‘oxidase’ of cytoplasmic reducing equivalents. Another relevant finding is that among fungi, Candida albicans can reductively assimilate Tf iron with the iron being taken into the cell by the canonical ferro-oxidase, ferri-permease complex found in fungi.58 One can argue about who’s copying whom here, but the point is that holo-Tf is substrate for “NTBI” iron accumulation machinery.
The concept of ‘nutritional immunity’ highlights the fact that all the organisms with which mammals commonly interact have an equally essential need for iron.59–61 How both host and visitor scavenge this nutrient are reflections of one another, e.g. how Neisseria TbpA mimics TfR’s binding of holo-Tf and using a linked thermodynamic equilibrium ‘pries’ Fe3+ out of Tf non-reductively. Note that the increase in the ferric iron reduction potential in holo-Tf upon binding to TfR is simply the electrochemical manifestation of a decrease in Tf’s relative affinity for ferric versus ferrous iron: higher reduction potential equates to less affinity for Fe3+, more affinity for Fe2+.38,42 Looked at in this way, one can appreciate the idea of an ‘oxidase’ function for holo-Tf.57
When it comes to ‘iron’, vertebrates are all the same: Tf, TfR, Dcytb, DMT1, ZIP etc. What is the selective advantage of this iron trafficking scheme? What is on that list of ‘need to do’, those items checked off with the addition of one of these canonical components of this metabolic pathway? What is the same and what is different about this list from the one free-living organisms needed to fulfill? The same one is the key one: iron’s Pourbaix behavior in the increasingly alkaline ocean under an increasing free oxygen partial pressure that steadily raised the biosphere’s reduction potential past the point at which ferrous iron was spontaneously possible.62 How do you deal with this thermodynamically? Where can one get the ‘work’ to overcome this trend towards ‘rust’? The list is short: live with the ferric iron by sequestering it in a coordination sphere that omits access to H2O thus suppressing hydrolysis; and move the iron and its chaperone into an acidic compartment that favors ferrous rather than ferric iron. Binding energy and proton concentration will do the work.
But clearly you don’t need both. Diferric transferrin is reduced at neutral pH by PMET driven by reduced pyridine nucleotides;47,56 Candida uses this PMET to supply ferrous iron for its canonical ‘NTBI’ uptake pathway, again at neutral pH.58 Tf, TbpA binding energy is sufficient to lower the transition state energy of the inner sphere transfer of Fe3+ from one to the other at neutral pH.43 One could argue that endosomal [H+] was not on this list. By definition, endolysosomes are acidic, and the premise is that this elevated [H+] promotes ligand, receptor dissociation and activation of proteases or other enzymic functions required for the handling of compartment cargo.63,64 An acidic pH also suppresses complicating and generally undesirable adventitious disulfide bond formation or exchange. In this view, the holo-Tf, TfR complex takes advantage of this acidic chemical environment, but an acidic chemical milieu is not an essential thermodynamic component of the release of iron from the complex. The increase iron reduction potential afforded by an increase in [H+] thermodynamically can be supported also by the “work” available from a protein–protein interaction as illustrated by the TfR and TbpA examples above.
Transferrin and brain iron uptake and trafficking
The question of how TBI contributes to systemic iron metabolism is particularly relevant to the trafficking of systemic iron into the brain’s abluminal (interstitial) space, and between the cells of the neurovascular unit. A widely held opinion is that abluminal iron results from holo-Tf transcytosis across the brain microvascular endothelial cells (BMVEC) that constitute the blood–brain barrier. In fact, there is little evidence for this model, and reasonably good data from well-designed experiments that it can’t be demonstrated; these latter data go back as far as 1993.65 As the previous discussion makes clear, the presence of plasma membrane TfR is not a ‘proof’ of an endosomal-dependent pathway for essential iron acquisition by that cell type. That brain microvascular endothelial cells express TfR does not ‘demonstrate’ a transcytosis pathway for Tf-mediated delivery of abluminal iron; it doesn’t even demonstrate that the predominant TBI iron uptake in BMVEC is endosome-mediated. The most recent thorough examination of the contribution of Tf-TfR transcytosis in the transendothelial transport of iron in brain endothelial cells concluded that “TfR was expressed and facilitated luminal (apical, blood side) uptake but not transcellular transport of Tf” in a primary brain microvascular endothelial cell transwell blood–brain barrier model system.66
This result was consistent with similar kinetic studies, again in the transwell paradigm, using an immortalized human brain microvascular endothelial cell line.18 In this study cell iron accumulation from 59Fe-Tf was quantified in the presence of: (1) a TfR antibody that blocks Tf binding to the receptor; (2) an inhibitor of plasma membrane ferri-reductase activity; and (3) a cell-impermeant ferrous iron-specific chelating agent. The key findings were that uptake required TfR (inhibition by the antibody); ferri-reductase activity (inhibition by reductase knock-down); and was inhibited by the ferrous iron chelating agent. Overall, these experiments demonstrated that ferrous iron was extra-cytoplasmically released reductively from holo-Tf bound to the cell-surface TfR and that the ferrous iron was then taken into the cell via a neutral pH transporter, i.e. not DMT1.18 The reader can return to Fig. 1, panel A to see how the model illustrated there coheres with the results of these experiments. More recent work in these brain capillary endothelial cells indicates that this uptake is mediated by a combination of the neutral pH transporters, ZIP8 and ZIP14.67 Indeed, both of these solute carrier transporters have been demonstrated to support ferrous iron uptake in a variety of cell types.30
Three other concerns which weaken the premise that Tf-iron delivery to the abluminal space is via Tf-TfR transcytosis are: (1) how is the canonical endosomal iron-release suppressed on the way across the cell? (2) what is the driving force for the dissociation of holo-Tf from TfR at the basal membrane? and (3) what happened to this systemic Tf presumed delivered to the interstitial space? The latter point is both one of interstitial Tf concentration, which is diminishing small, and the fact that little of this Tf has the glycosylation pattern characteristic of serum Tf; the majority, as assayed in the cerebrospinal fluid (CSF) is ‘brain’ Tf that has its own distinct glycan composition and structure.68 By way of comparison, the concentration of Tf in blood is ~30 μM; the concentration of ‘blood’ Tf in the brain’s interstitium is ~0.04 μM ([Tf]Brain type is ~0.1 μM).68 The other two questions are linked and a possible answer may come from the plethora of studies designed to manipulate this putative TfR transcytosis to deliver a pharmacologic cargo to the abluminal space. Simply stated, cargo that binds more weakly has a higher propensity to be delivered.69 Does this suggest that Tf-TfR transcytosis ‘happens’ but the complex simply cycles back to the apical (blood) membrane since, for some reason, the iron never gets mobilized and so the stability of the Tf·TfR complex is not subject to the negative allosteric regulation that follows from this iron dissociation?
Differentiating ferrous iron transporters and ferric iron reductases: mechanism designed for function
The fact about mechanism that differentiates DMT1 from the ZIP family divalent metal ion transporters not only functionally but metabolically is that DMT1 is a H+ symporter;70 the ZIPs are not. As proton symporter, DMT1 functions best at 5.5 (or lower), ZIPs at 7.4; the activities of the two are readily distinguishable. This property delineates where each transporter type functions; there really is no controversy about this. DMT1 functions in the duodenal and endolysosomal lumen. ZIP transporters operate everywhere else. Unfortunately, one often sees cartoons depicting the iron transport functions in a cell with DMT1 supporting plasma membrane NTBI Fe2+ uptake. Given the descending pH dependence of DMT1 function, this picture pertains only to the intestinal enterocyte and not the brain microvasculature or abluminal space, for example. DMT1 is expressed by brain endothelial, glia and neuronal cells, as are TfR and STEAP family members 2 and 3 but multiple studies indicate iron uptake by the endothelium, for example, is mediated by the neutral pH transporters, ZIP8 and ZIP14. In short, in summarizing cell iron accumulation, and the impact on it resulting from inhibition, up-regulation or knock-down of DMT1 or a ZIP family member, these transporters’ differing pH dependence is an essential consideration and can provide significant insight as to the molecular basis for any phenotype observed. If knockdown of DMT1 in your favorite cell results in a reduced iron accumulation then for this cell under the conditions of the experiment, the dominant mode of iron uptake is endosome-mediated – unless your cell of choice is an intestinal enterocyte.
The six-transmembrane epithelial antigen of the prostate 1 protein – STEAP1 – was the index protein for the STEAP family, STEAP1–4.28 Of the four, STEAP 2–4 have ferric-reductase (and cupric-reductase) activity comparable to that expressed by Dcytb, and, for example, the widely-studied fungal FRE proteins all of which are metallo-reductases (as are the plant FRO gene products).19,21 The physiologic functions of STEAP 2–4 have not been thoroughly studied, although there is good evidence that STEAP3 (not Dcytb, for example) provides the ferri-reductase function in the specialized endosomes found in hematopoietic cells that supply Tf-bound ferric iron – as Fe2+ – to DMT1.71,72 On the other hand, in hippocampal neurons, STEAP2 and not STEAP3 is expressed, and co-localizes with both plasma membrane and endosomal-associated Tf·TfR. But in the hippocampal endosome, it is ZIP8 and not DMT1 that co-localizes with Tf·TfR.73 Given the fact that ZIP8 has limited activity at pH 5.5, it is not surprising that inhibition of clathrin-mediated endocytosis with dynasore fails to inhibit TBI iron uptake in these neurons; that is, cycling occurs but Fe2+ released into the endosomal compartment is recycled back to the extra-cellular space rather than being transported into the cytoplasm.73 The model that emerges about iron uptake in capillary endothelial cells in the brain, and in hippocampal neurons, at the least, is that both TBI and NTBI uptake is supported by PM ferri-reduction and ZIP8/ZIP14 ferro-permeation, with TfR acting as an allosteric modulator of the reduction potential of Tf-bound ferric iron.74
Chaperoning iron – Tf, PCBP1/2 and ferritin
Endosomal trafficking of Tf-delivered iron offered a mechanism for ‘chaperoning’ the metal ion within the cytoplasm. One example of how this model might contribute to delivery of iron for use as a prosthetic group is the ‘kiss-and-run’ notion whereby an endosomal compartment fuses sufficiently with the mitochondrial outer membrane to allow for transfer of luminal iron from one to the other compartment.75 On the other hand, the developing story of how PCBP1 and PCBP2 act as cytoplasmic ferrous iron chaperones76,77 offers another compelling mechanistic paradigm, particularly in those cell types, like capillary endothelial cells and neurons, that appear not to rely on canonical TBI iron accumulation. There is evidence that PCBP2 interacts with DMT1 in a model that has PCBP2 acting as chaperone between iron uptake and efflux via the iron efflux transporter, ferroportin (FPN).78,79 The limitation in this model is the lack of any data indicating a comparable PCBP1/2 interaction with ZIP8 and/or ZIP14; given the likely broader role these transporters play in cellular iron trafficking30 such an interaction would be likely if this model of PCBP1/2 function is correct. Given the negative reduction potential of the cytosol and richness of potential small molecule ferrous iron ligands, a direct interaction between a chaperone like PCBP2 and transporter may not be an essential requirement for efficient handling of cell iron.
The difference between cytoplasmic ‘chaperoning’ of ferrous iron in comparison to cuprous copper is striking. Both low valent metal ions are ligands for the uptake transporters that ferry them into the cell and those that ferry them out. There is little argument that newly arrived Cu1+ is channeled from transporter to chaperone to apo-copper protein by a series of non-dissociative, inner-sphere ligand exchange reactions.80,81 Although the same appears to be true for ferrous iron targeting to apo-iron proteins and ferritin,76,77 there is little evidence for a chaperone function in iron efflux. Furthermore, whereas there is no evidence for ‘free’ cell copper, there is widely accepted evidence that ~20% of cell iron is chelatable, as Fe2+.82–87 From an inorganic chemistry perspective this difference could reflect a subtle difference in the redox chemistry of Cu1+ versus Fe2+ and that is the tendency for cuprous copper to disproportionate: 2Cu1+ → Cu2+ + Cu°, i.e. copper metal.88 Whatever the selective advantage provided by copper chaperones, they clearly reflect a chemistry unique to Cu1+ in comparison to Fe2+ at the pH, reduction potential, and ligand environment in a eukaryotic cell.
This commentary would not be complete if it did not include consideration of ferritin as an iron ‘chaperone’ and not simply as an iron storage depot. There are two aspects of ferritin physiology that deserve some reflection: (1) ferritin (Ft) does circulate, both systemically8,17 and in the abluminal space of the brain as indicated by Ft levels in the cerebrospinal fluid;89 and (2) mobilization of iron from Ft may not require lysosomal degradation,90,91 a mechanism for which there is strong experimental evidence.92,93 Other work has shown that TfR binds and internalizes Ft as do other receptors including TIM1 and 2, and CXCR4.16,17 In the vascular endothelial cells in the brain, there is less clear evidence as to the fate of internalized Ft: is it a source of iron for the cell, or is it transcytosed and delivered to the abluminal compartment, or both? There is evidence for adsorbed Ft following both pathways albeit with limited definition as to the precise mechanisms involved.94 However, TRIM16 acting with Sec22b in combination with plasma membrane syntaxin 3 and syntaxin 4 as well as SNAP-23 and SNAP-29 supports a non-canonical secretion of Ft, by-passing the lysosome.95 Possibly, this pathway could support the transcytosis of H-ferritin demonstrated in induced pluripotent stem cell-derived brain endothelial cells in transwell,94 a robust cell culture model of the blood brain barrier.94,96
This lysosomal by-pass model contrasts with the more thoroughly interrogated one, i.e. that iron recovery from Ft is a degradative process. For example, there is recent, exceptionally clear evidence from studies using HepG2 cells of lysosomal-targeted Ft trafficking.93 In addition is the fact that impaired lysosomal acidification in isolated neurons and a mouse model triggers an iron deficiency resolved by supplementation with iron citrate.97 On the other hand, there are a variety of data that demonstrate iron mobilization from Ft is stimulated also by a reductive process without modification (degradation) of the protein shell. The paradigmatic biologic example of this reductive mobilization is in bacterioferritin with electrons coming from a bound ferredoxin.98 Mammalian ferritins exhibit no such reactivity, most certainly due to the fact that they do not form an electron transfer-competent complex with such an electron transfer protein. In vitro, a variety of physiologic reductants, e.g. FMNH2, ascorbate – without or with an electron mediator – readily mobilize Fe2+ from mammalian ferritins.90,93 Nonetheless, there is no experimental evidence for this type of lysosome/degradation-independent ferritin iron mobilization in cells. A lysosome-dependent mechanism compares to the handling of iron in fungi, for example, where iron is stored in the vacuole – as a ferric poly-phosphate – and is reductively mobilized by a ferri-reductase within this acidic compartment (low pH, accessible E1/2) followed by efflux into the cytosol.19 As far as how iron is handled and then effluxed from lysosomes is concerned, while a comparable mechanism would be thermodynamically robust, it remains to be thoroughly interrogated. Intriguingly, however, an ascorbat-fueled cytochrome b561 has been localized to macrophage endolysosomes that could support a DMT1-facilitated efflux of ferrous iron from these compartments into the cytoplasm.99 DMT1 does localize to lysosomes and some evidence supports its role for mobilization of iron from these organelles.93
Concluding thoughts
In a 1987 review in the European Journal of Biochemistry, Robert Crichton, arguably one of the ‘Deans’ of 20th Century ‘Metals in Biology’ wrote about iron transport and storage: “In the same year (1949) it was established that transferrin iron can be released by acidification of the medium. Subsequent studies have clearly established the importance of this mechanism within the cell.”100 This mechanism cohered well with Professor Crichton’s chemical perspective on metals in biology that with iron you’re always on more solid ground at higher [H+]. This perspective fit well, too, with the subsequent identification of the Nramp family of divalent metal ion transporters found in eukaryotes, first the SMF family in yeast101 and then Nramp1, expressed exclusively in phagocytic cells, that was associated with resistance to mycobacterial infection,102 and Nramp2 – DMT1-associated with microcytic anemia in mk mice and the Belgrade (b/b) rat.22,23 Molecular characterization of the transport activity of these proteins demonstrated the pH dependence characteristic of Nramp family members and the fact that they were H+-coupled transporters. Together with the use of TIBC and UIBC as the standard clinical read-outs for patient iron status, the general emphasis on canonical Tf- and endosome mediated iron trafficking as the de facto systemic – and abluminal – hallmark of mammalian iron metabolism was unremarkable. However, the current appreciation of the roles played by ZIP8 and ZIP14 in the accumulation of iron (and manganese) by a wide variety of cell types – not just enterocytes, macrophages and erythroid cells – requires an expansion of our view of the overall landscape of iron metabolism.30
One fundamental change is to appreciate that a distinction between TBI and NTBI uptake is functionally irrelevant in those cell types that express a robust cell surface reductase activity and a ZIP transporter as illustrated in Fig. 1. Furthermore, arguably more relevant to the clinical assessment of systemic iron status are serum ferritin, hepcidin and erythroferrone rather than fractional Tf iron loading. The latter hormone regulates HAMP expression103 which, in turn, regulates the efflux of iron from all cell types independent of whether the iron ends up in Tf or not.104 As for secretion of Ft, it clearly is an acute phase response to a precipitous decline in systemic iron status or an attack on the body’s circulating iron by an invading pathogen. It certainly is no coincidence that ferritin, an iron carrier for which Neisseria doesn’t have answer, is an acute phase protein.105 Indeed, one might include in this change of perspective the emerging role that ferritin plays in the transport of iron, not just its storage.
Also in order is a fuller appreciation of the WHY of an acid pH, protein-coupled di-valent metal ion transporter family as well as a neutral pH one. As to the former, all eukaryotes have acid compartments; at the cell level – broadly speaking – vacuoles, lysosomes and essentially all of the ‘vesicles’ found in the cytoplasm. At the organismal level, vertebrates have an increasingly specialized acid pH stomach followed by an intestine, differentiated to varying degrees to be sure, but all characterized by a diminishing [H+] from the proximal to distal portion. The same pH gradient is seen in cytoplasm discontinuous vesicular compartments in the cell, from low pH in early endosomes to a mildly acidic pH in the endoplasmic reticulum. These are the environments that selected for the metabolic pathways now illustrated in reviews on them. The same is true for the selection of a pathway that had the same end-point but functioned in a neutral pH environment, that is, essentially everywhere else in the organism. There had to be two functionally disparate iron transporters. And there had to be a ‘chaperone’ for iron in the neutral pH regime found in the circulation. Not only did this prevent rust accumulation on arterial cell walls but also inhibited iron scavenging by invading pathogens. As for TfR selection, how is the homodimeric TfR any different from the heterodimeric TbpA, TbpB complex in Neisseria? Both bind holo-Tf and by classic thermodynamic energy coupling, ‘labilize’ the Fe3+ coordinately and electrochemically at neutral pH.
Which brings us to the Why? of Nramp1 and Nramp2, the endosomal ‘DMT1’ in macrophages and erythroid cells, respectively. If TBI is readily taken up by the cell surface combination of TfR, a STEAP ferri-reductase and ZIP divalent metal ion transporter, why bother with the more energy expensive endosomal cycling pathway? One can only speculate but a good case can be made that both pathways represent a ‘logical’ response to the corresponding selective pressure. Macrophages are where pathogens are ‘quarantined’ by the organism, taken up by the same pathway that retrieves TfR, STEAPs and transporters from the membrane. As part of the ‘nutritional immunity’ circuitry, withholding iron from the pathogen is afforded by pumping iron out of the compartment. Failure to do so leads to a compromised ‘natural resistance’ to pathogens. Macrophages and erythroid cells also handle by far the largest flux of re-cycled iron given their role in clearing senescent red cells and making new ones, respectively. Efficiency in iron handling has clear selective advantage. So, Is the canonical, TfR, endosomal handling of TBI more efficient than the handling of this iron by the ectodomains of TfR, STEAP2 and ZIP8, for example?
The data indicate that the former endosomal pathway is efficient given the known factors that mediate this process, including the differential stability of the TfR·Tf complex for different Fe-bound forms of Tf. Indeed, that cellular retrieval of iron from TfR-bound holo-Tf approached 100% was indicated by the first kinetic analyses of 131I-Tf binding to and 59Fe uptake by reticulocytes conducted in 1963 by Jandl and Katz.106 How does a cell surface reductase, permease pathway compare? In yeast, the relative rates of ferri-reduction, ferro-oxidation and ferri-permeation supported by the coupled activities of Fre1p, Fet3p and Ftr1p are ~200 : 50 : 1 indicating only a small fraction of the exo-cytoplasmic iron that is substrate for this pathway actually is accumulated by the cell.107 The pathway is inefficient; it is ‘leaky’. This is indicated by the observation that a ferrous iron – not ferric iron – chelator inhibits yeast iron uptake when 59Fe3+ is the ‘substrate’ for this process.108 Similarly, in brain microvascular endothelial cells (BMVEC) and hippocampal neurons, reductive iron uptake from TBI – again, Fe3+ as initiating substrate – is inhibited by a ferrous iron chelator like bathophenanthroline disulfonate indicating that the ferrous iron product of the STEAP ferri-reductase reaction is ‘labile’ at the least, if not actually released into the aqueous milieu.18,73 This was illustrated in Fig. 1A. In BMVEC, a comparison of the rate of ‘labile’ (chelatable) Fe2+ production from holo-Tf by ferri-reduction to the rate of 59Fe uptake from TBI shows the former to be 3-fold greater than the latter.18 In short, this cell surface reductive pathway is inefficient, transporting only a fraction of the iron labilized from holo-Tf. One could compare this to the efficiency of Neisseria high-jacking of Tf iron; since this process relies on an associative transfer of Fe3+ from Tf to TpbA, it is essentially stoichiometric.43,44
Which brings us to the last question. What purpose does this seemingly inefficient reductive iron trafficking pathway serve? It has a ‘cost’, requiring cell reducing equivalents, while ‘offering’ a highly bio-available form of iron to circulating pathogens, in conflict with the concept of nutritional immunity. Any answer is speculative but one notion at least can be beta-checked and it follows from the fact that Tf-iron accumulated by canonical endosomal trafficking begins its cellular metabolism in an endolysosome while reductively released iron transported into the cell by a ZIP transporter starts its metabolic pathway in the cytosol. At least in terms of cell compartment these two iron pools will be subject to a different metabolic trajectory in as much as endolysosomal iron will rely on the activity of an efflux transporter to ‘join’ the cytosolic pool (or not, as the case may be), an activity undoubtedly under regulation by any number of pathways. One can reflect again on the ‘standard model’ (to use the ‘loaded’ terminology that separated the Bohr from the Einstein point of view about quantum theory) of Tf iron handling that pictures the release of ferric iron from holo-Tf, its reduction and efflux via DMT1 from the endolysosome as synchronous processes. In fact, there is no evidence for this simultaneity; there is no reason why the ferrous iron is not retained in the endolysosome until needed. The DMT1 efflux machinery in these organelles is regulated by ubiquitination109,110 and phosphorylation;111 these organelles are not colanders. In this model, ZIP-managed iron is for house-keeping, endosome-, DMT1-managed iron is for special purposes. What’s good about this hypothesis is that it is controversial, and it is testable. One hopes that it is the latter that now gets the focus of our attention.
Significance to metallomics.
Mammalian iron uptake has generically been categorized by the chelate in which the iron is presented to the cell, either transferrin bound (TBI) or non-transferrin bound (NTBI). In this historical view, TBI uptake is endolysosome-mediated, while NTBI is supported by plasma membrane divalent metal ion transporters. This view fails to include a variety of generally known but commonly under-appreciated data that together indicate that TBI uptake, while transferrin receptor-dependent, does not require clathrin-dependent endosome trafficking. Rather, relying solely on the thermodynamics of the Tf, TfR interaction that increases the ferric iron reduction potential, plasma membrane electron transfer – PMET – reductively mobilizes ferrous iron as ligand for neutral pH ferrous iron transporters. That is, the other commonly overlooked factor is the striking mechanistic difference between the two classes of divalent metal ion transporters. DMT1 is a proton-symporter, exhibiting a strong descending pH dependence; ZIP family members function optimally at neutral – systemic – pH. DMT1 functions solely in the intestinal lumen and the lumen of the acid pH endosomal compartments found in reticulocytes and macrophages. ZIP proteins function on the plasma membranes of all other cells. The reductive release of ferrous iron from holo-Tf – and from ferritin – is thermodynamically accessible and kinetically demonstrable. This is the Perspective provided in this manuscript.
Acknowledgements
The work in KosmanLab is supported by a research grant from the National Institute of Neurologic Disorders and Stroke of the Unites States, RO1NS102337.
Abbreviations
- Tf
Transferrin
- TfR
Transferrin receptor
- TBI
Transferrin-bound iron
- NTBI
Non-transferrin-bound iron
- DMT1
Divalent metal ion transporter 1 (NRAMP2, SLC11A2)
- ZIP
Znt/Irt-like proteins (SLC39A8 and SLC39A14)
- Dcytb
Duodenal cytochrome b (CYBRD1)
- STEAP
Six-transmembrane epithelial antigen of the prostate 1–4 (STEAP1–4)
- PMET
Plasma membrane electron transport
- E°′
Reduction potential (at pH 7.0)
- PCBP1/2
Poly(rC)-binding protein (PCBP1, PCBP2)
- GSH/GSSG
Glutathione (reduced)/glutathione (oxidized)
- BMVEC
Brain microvascular endothelial cell
Footnotes
Conflicts of interest
The author has no conflicts nor competing interests to declare.
References
- 1.Hirayama T, Fluorescent probes for the detection of catalytic Fe(II) ion, Free Radical Biol. Med, 2019, 133, 38–45. [DOI] [PubMed] [Google Scholar]
- 2.Yanatori I and Kishi F, DMT1 and iron transport, Free Radical Biol. Med, 2019, 133, 55–63. [DOI] [PubMed] [Google Scholar]
- 3.Mills E, Dong XP, Wang F and Xu H, Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders, Future Med. Chem, 2010, 2, 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Routhe LJ and Moos T, Handling iron in restorative neuroscience, Neural Regener. Res, 2015, 10, 1558–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crichton RR, Wilmet S, Legssyer R and Ward RJ, Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells, J. Inorg. Biochem, 2002, 91, 9–18. [DOI] [PubMed] [Google Scholar]
- 6.Crichton RR and Ward RJ, Iron homeostasis, Met. Ions Biol. Syst, 1998, 35, 633–665. [PubMed] [Google Scholar]
- 7.Crichton RR, Proteins of iron storage and transport, Adv. Protein Chem, 1990, 40, 281–363. [DOI] [PubMed] [Google Scholar]
- 8.Yang Z, Dewey KG, Lonnerdal B, Hernell O, Chaparro C, Adu-Afarwuah S, McLean ED, Cohen RJ, Domellof M, Allen LH and Brown KH, Comparison of plasma ferritin concentration with the ratio of plasma transferrin receptor to ferritin in estimating body iron stores: results of 4 intervention trials, Am. J. Clin. Nutr, 2008, 87, 1892–1898. [DOI] [PubMed] [Google Scholar]
- 9.Kosman DJ, Iron metabolism in aerobes: Managing ferric iron hydrolysis and ferrous iron autoxidation, Coord. Chem. Rev, 2013, 257, 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ and Sternberg P, Redox state of glutathione in human plasma, Free Radical Biol. Med, 2000, 28, 625–635. [DOI] [PubMed] [Google Scholar]
- 11.Fruton JS, Oxidation-reduction potentials of ascorbic acid, J. Biol. Chem, 1934, 105, 79–85. [Google Scholar]
- 12.Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C and Cabantchik ZI, Labile plasma iron in iron overload: redox activity and susceptibility to chelation, Blood, 2003, 102, 2670–2677. [DOI] [PubMed] [Google Scholar]
- 13.Osaki S, Johnson DA and Frieden E, The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum, J. Biol. Chem, 1966, 241, 2746–2751. [PubMed] [Google Scholar]
- 14.Cabantchik ZI, Labile iron in cells and body fluids: physiology, pathology, and pharmacology, Front. Pharmacol, 2014, 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ponka P and Lok CN, The transferrin receptor: role in health and disease, Int. J. Biochem. Cell Biol, 1999, 31, 1111–1137. [DOI] [PubMed] [Google Scholar]
- 16.Montemiglio LC, Testi C, Ceci P, Falvo E, Pitea M, Savino C, Arcovito A, Peruzzi G, Baiocco P, Mancia F, Boffi A, des Georges A and Vallone B, Cryo-EM structure of the human ferritin-transferrin receptor 1 complex, Nat. Commun, 2019, 10, 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakamoto S, Kawabata H, Masuda T, Uchiyama T, Mizumoto C, Ohmori K, Koeffler HP, Kadowaki N and Takaori-Kondo A, H-Ferritin Is Preferentially Incorporated by Human Erythroid Cells through Transferrin Receptor 1 in a Threshold-Dependent Manner, PLoS One, 2015, 10, e0139915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarthy RC and Kosman DJ, Mechanistic analysis of iron accumulation by endothelial cells of the BBB, Biometals, 2012, 25, 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosman DJ, Redox cycling in iron uptake, efflux, and trafficking, J. Biol. Chem, 2010, 285, 26729–26735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW and Simpson RJ, A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation, Mol. Cell, 2000, 5, 299–309. [DOI] [PubMed] [Google Scholar]
- 21.Kosman DJ, The teleos of metallo-reduction and metalo-oxidation in eukaryotic iron and copper trafficking, Metallomics, 2018, 10, 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD and Andrews NC, Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport, Proc. Natl. Acad. Sci. U. S. A, 1998, 95, 1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleming MD, Trenor CC 3rd, Su MA, Foernzler D, Beier DR, Dietrich WF and Andrews NC, Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene, Nat. Genet, 1997, 16, 383–386. [DOI] [PubMed] [Google Scholar]
- 24.Andrews NC, The iron transporter DMT1, Int. J. Biochem. Cell Biol, 1999, 31, 991–994. [DOI] [PubMed] [Google Scholar]
- 25.Sipe DM and Murphy RF, Binding to cellular receptors results in increased iron release from transferrin at mildly acidic pH, J. Biol. Chem, 1991, 266, 8002–8007. [PubMed] [Google Scholar]
- 26.Latunde-Dada GO, Van der Westhuizen J, Vulpe CD, Anderson GJ, Simpson RJ and McKie AT, Molecular and functional roles of duodenal cytochrome B (Dcytb) in iron metabolism, Blood Cells, Mol., Dis, 2002, 29, 356–360. [DOI] [PubMed] [Google Scholar]
- 27.Grunewald TG, Bach H, Cossarizza A and Matsumoto I, The STEAP protein family: versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions, Biol. Cell, 2012, 104, 641–657. [DOI] [PubMed] [Google Scholar]
- 28.Knutson MD, Steap proteins: implications for iron and copper metabolism, Nutr. Rev, 2007, 65, 335–340. [DOI] [PubMed] [Google Scholar]
- 29.Ohgami RS, Campagna DR, McDonald A and Fleming MD, The Steap proteins are metalloreductases, Blood, 2006, 108, 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knutson MD, Non-transferrin-bound iron transporters, Free Radical Biol. Med, 2019, 133, 101–111. [DOI] [PubMed] [Google Scholar]
- 31.Steimle BL, Smith FM and Kosman DJ, The solute carriers ZIP8 and ZIP14 regulate manganese accumulation in brain microvascular endothelial cells and control brain manganese levels, J. Biol. Chem, 2019, 294, 19197–19208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheiber IF, Alarcon NO and Zhao N, Manganese Uptake by A549 Cells is Mediated by Both ZIP8 and ZIP14, Nutrients, 2019, 11, 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon SJV, Fenker DE, Vest KE and Padilla-Benavides T, Manganese influx and expression of ZIP8 is essential in primary myoblasts and contributes to activation of SOD2, Metallomics, 2019, 11, 1140–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B and Knutson MD, ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading, J. Biol. Chem, 2012, 287, 34032–34043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nebert DW, Galvez-Peralta M, Hay EB, Li H, Johansson E, Yin C, Wang B, He L and Soleimani M, ZIP14 and ZIP8 zinc/bicarbonate symporters in Xenopus oocytes: characterization of metal uptake and inhibition, Metallomics, 2012, 4, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Girijashanker K, He L, Soleimani M, Reed JM, Li H, Liu Z, Wang B, Dalton TP and Nebert DW, Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter, Mol. Pharmacol, 2008, 73, 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He L, Girijashanker K, Dalton TP, Reed J, Li H, Soleimani M and Nebert DW, ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties, Mol. Pharmacol, 2006, 70, 171–180. [DOI] [PubMed] [Google Scholar]
- 38.Dhungana S, Taboy CH, Zak O, Larvie M, Crumbliss AL and Aisen P, Redox properties of human transferrin bound to its receptor, Biochemistry, 2004, 43, 205–209. [DOI] [PubMed] [Google Scholar]
- 39.Harris DC, Rinehart AL, Hereld D, Schwartz RW, Burke FP and Salvador AP, Reduction potential of iron in transferrin, Biochim. Biophys. Acta, 1985, 838, 295–301. [DOI] [PubMed] [Google Scholar]
- 40.Jones DP and Sies H, The Redox Code, Antioxid. Redox Signaling, 2015, 23, 734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schafer FQ and Buettner GR, Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple, Free Radical Biol. Med, 2001, 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- 42.Terpstra T, McNally J, Han TH, Ha-Duong NT, El-Hage-Chahine JM and Bou-Abdallah F, Direct thermodynamic and kinetic measurements of Fe(2)(+) and Zn(2)(+) binding to human serum transferrin, J. Inorg. Biochem, 2014, 136, 24–32. [DOI] [PubMed] [Google Scholar]
- 43.Noinaj N, Easley NC, Oke M, Mizuno N, Gumbart J, Boura E, Steere AN, Zak O, Aisen P, Tajkhorshid E, Evans RW, Gorringe AR, Mason AB, Steven AC and Buchanan SK, Structural basis for iron piracy by pathogenic Neisseria, Nature, 2012, 483, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noinaj N, Buchanan SK and Cornelissen CN, The transferrin-iron import system from pathogenic Neisseria species, Mol. Microbiol, 2012, 86, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giannetti AM, Halbrooks PJ, Mason AB, Vogt TM, Enns CA and Bjorkman PJ, The molecular mechanism for receptor-stimulated iron release from the plasma iron transport protein transferrin, Structure, 2005, 13, 1613–1623. [DOI] [PubMed] [Google Scholar]
- 46.Cole ES and Glass J, Transferrin binding and iron uptake in mouse hepatocytes, Biochim. Biophys. Acta, 1983, 762, 102–110. [DOI] [PubMed] [Google Scholar]
- 47.Sun IL, Navas P, Crane FL, Morre DJ and Low H, NADH diferric transferrin reductase in liver plasma membrane, J. Biol. Chem, 1987, 262, 15915–15921. [PubMed] [Google Scholar]
- 48.Thorstensen K and Romslo I, The role of transferrin in the mechanism of cellular iron uptake, Biochem. J, 1990, 271, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lane DJ and Lawen A, Ascorbate and plasma membrane electron transport--enzymes vs. efflux, Free Radical Biol. Med, 2009, 47, 485–495. [DOI] [PubMed] [Google Scholar]
- 50.Lane DJ and Lawen A, Transplasma membrane electron transport comes in two flavors, BioFactors, 2008, 34, 191–200. [DOI] [PubMed] [Google Scholar]
- 51.Kennett EC and Kuchel PW, Redox reactions and electron transfer across the red cell membrane, IUBMB Life, 2003, 55, 375–385. [DOI] [PubMed] [Google Scholar]
- 52.Baker MA and Lawen A, Plasma membrane NADH-oxidoreductase system: a critical review of the structural and functional data, Antioxid. Redox Signaling, 2000, 2, 197–212. [DOI] [PubMed] [Google Scholar]
- 53.Lesuisse E and Labbe P, Iron Reduction and Trans Plasma Membrane Electron Transfer in the Yeast Saccharomyces cerevisiae, Plant Physiol, 1992, 100, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lane DJ and Richardson DR, The active role of vitamin C in mammalian iron metabolism: much more than just enhanced iron absorption!, Free Radical Biol. Med, 2014, 75, 69–83. [DOI] [PubMed] [Google Scholar]
- 55.Ganasen M, Togashi H, Takeda H, Asakura H, Tosha T, Yamashita K, Hirata K, Nariai Y, Urano T, Yuan X, Hamza I, Mauk AG, Shiro Y, Sugimoto H and Sawai H, Structural basis for promotion of duodenal iron absorption by enteric ferric reductase with ascorbate, Commun. Biol, 2018, 1, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun IL, Navas P, Crane FL, Morre DJ and Low H, Diferric transferrin reductase in the plasma membrane is inhibited by adriamycin, Biochem. Int, 1987, 14, 119–127. [PubMed] [Google Scholar]
- 57.Crane FL and Low H, The oxidative function of diferric transferrin, Biochem. Res. Int, 2012, 2012, 592806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Knight SA, Vilaire G, Lesuisse E and Dancis A, Iron acquisition from transferrin by Candida albicans depends on the reductive pathway, Infect. Immun, 2005, 73, 5482–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nunez G, Sakamoto K and Soares MP, Innate Nutritional Immunity, J. Immunol, 2018, 201, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Malavia D, Crawford A and Wilson D, Nutritional Immunity and Fungal Pathogenesis: The Struggle for Micronutrients at the Host-Pathogen Interface, Adv. Microb. Physiol, 2017, 70, 85–103. [DOI] [PubMed] [Google Scholar]
- 61.Crawford A and Wilson D, Essential metals at the host-pathogen interface: nutritional immunity and micronutrient assimilation by human fungal pathogens, FEMS Yeast Res, 2015, 15, fov071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams RJP, Iron in evolution, FEBS Lett, 2012, 586, 479–484. [DOI] [PubMed] [Google Scholar]
- 63.Wang C, Zhao T, Li Y, Huang G, White MA and Gao J, Investigation of endosome and lysosome biology by ultra pH-sensitive nanoprobes, Adv. Drug Delivery Rev, 2017, 113, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scott CC, Vacca F and Gruenberg J, Endosome maturation, transport and functions, Semin. Cell Dev. Biol, 2014, 31, 2–10. [DOI] [PubMed] [Google Scholar]
- 65.Roberts RL, Fine RE and Sandra A, Receptor-mediated endocytosis of transferrin at the blood-brain barrier, J. Cell Sci, 1993, 104(Pt 2), 521–532. [DOI] [PubMed] [Google Scholar]
- 66.Hersom M, Helms HC, Pretzer N, Goldeman C, Jensen AI, Severin G, Nielsen MS, Holm R and Brodin B, Transferrin receptor expression and role in transendothelial transport of transferrin in cultured brain endothelial monolayers, Mol. Cell. Neurosci, 2016, 76, 59–67. [DOI] [PubMed] [Google Scholar]
- 67.Song Y, Bailey DK and Kosman DJ, 2020, to be published.
- 68.Murakami Y, Saito K, Ito H and Hashimoto Y, Transferrin isoforms in cerebrospinal fluid and their relation to neurological diseases, Proc. Jpn. Acad., Ser. B, 2019, 95, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bien-Ly N, Yu YJ, Bumbaca D, Elstrott J, Boswell CA, Zhang Y, Luk W, Lu Y, Dennis MS, Weimer RM, Chung I and Watts RJ, Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants, J. Exp. Med, 2014, 211, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mackenzie B, Ujwal ML, Chang MH, Romero MF and Hediger MA, Divalent metal-ion transporter DMT1 mediates both H+-coupled Fe2+ transport and uncoupled fluxes, Pflugers Arch, 2006, 451, 544–558. [DOI] [PubMed] [Google Scholar]
- 71.Lambe T, Simpson RJ, Dawson S, Bouriez-Jones T, Crockford TL, Lepherd M, Latunde-Dada GO, Robinson H, Raja KB, Campagna DR, Villarreal G Jr., Ellory JC, Goodnow CC, Fleming MD, McKie AT and Cornall RJ, Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism, Blood, 2009, 113, 1805–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang F, Tao Y, Zhang Z, Guo X, An P, Shen Y, Wu Q, Yu Y and Wang F, Metalloreductase Steap3 coordinates the regulation of iron homeostasis and inflammatory responses, Haematologica, 2012, 97, 1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ji C and Kosman DJ, Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons, J. Neurochem, 2015, 133, 668–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCarthy RC and Kosman DJ, Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier, Front. Mol. Neurosci, 2015, 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hamdi A, Roshan TM, Kahawita TM, Mason AB, Sheftel AD and Ponka P, Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism, Biochim. Biophys. Acta, 2016, 1863, 2859–2867. [DOI] [PubMed] [Google Scholar]
- 76.Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M and Philpott CC, PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis, J. Clin. Invest, 2017, 127, 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, Ghosh MC, Lee J, Rouault TA, Park MH and Philpott CC, Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, 8031–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yanatori I, Richardson DR, Imada K and Kishi F, Iron Export through the Transporter Ferroportin 1 is Modulated by the Iron Chaperone PCBP2, J. Biol. Chem, 2016, 291, 17303–17318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yanatori I, Yasui Y, Tabuchi M and Kishi F, Chaperone protein involved in transmembrane transport of iron, Biochem. J, 2014, 462, 25–37. [DOI] [PubMed] [Google Scholar]
- 80.Magistrato A, Pavlin M, Qasem Z and Ruthstein S, Copper trafficking in eukaryotic systems: current knowledge from experimental and computational efforts, Curr. Opin. Struct. Biol, 2019, 58, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hatori Y and Lutsenko S, The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution, Antioxidants, 2016, 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greenberg GR and Wintrobe MM, A labile iron pool, J. Biol. Chem, 1946, 165, 397. [PubMed] [Google Scholar]
- 83.Muir RK, Zhao N, Wei J, Wang YH, Moroz A, Huang Y, Chen YC, Sriram R, Kurhanewicz J, Ruggero D, Renslo AR and Evans MJ, Measuring Dynamic Changes in the Labile Iron Pool in Vivo with a Reactivity-Based Probe for Positron Emission Tomography, ACS Cent. Sci, 2019, 5, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krijt M, Jirkovska A, Kabickova T, Melenovsky V, Petrak J and Vyoral D, Detection and quantitation of iron in ferritin, transferrin and labile iron pool (LIP) in cardiomyocytes using (55)Fe and storage phosphorimaging, Biochim. Biophys. Acta, Gen. Subj, 2018, 1862, 2895–2901. [DOI] [PubMed] [Google Scholar]
- 85.Chutvanichkul B, Vattanaviboon P, Mas-Oodi S, U-pratya Y and Wanachiwanawin W, Labile iron pool as a parameter to monitor iron overload and oxidative stress status in beta-thalassemic erythrocytes, Cytometry, Part B, 2018, 94, 631–636. [DOI] [PubMed] [Google Scholar]
- 86.Pai AB, Meyer DE, Bales BC, Cotero VE, Pai MP, Zheng N and Jiang W, Performance of Redox Active and Chelatable Iron Assays to Determine Labile Iron Release From Intravenous Iron Formulations, Clin. Transl. Sci, 2017, 10, 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abbate V, Reelfs O, Kong X, Pourzand C and Hider RC, Dual selective iron chelating probes with a potential to monitor mitochondrial labile iron pools, Chem. Commun, 2016, 52, 784–787. [DOI] [PubMed] [Google Scholar]
- 88.Johnson DK, Stevenson MJ, Almadidy ZA, Jenkins SE, Wilcox DE and Grossoehme NE, Stabilization of Cu(I) for binding and calorimetric measurements in aqueous solution, Dalton Trans., 2015, 44, 16494–16505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ayton S, Faux NG, Bush AI and Alzheimer’s Disease Neuroimaging I, Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE, Nat. Commun, 2015, 6, 6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bou-Abdallah F, Paliakkara JJ, Melman G and Melman A, Reductive Mobilization of Iron from Intact Ferritin: Mechanisms and Physiological Implication, Pharmaceuticals, 2018, 11, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Melman G, Bou-Abdallah F, Vane E, Maura P, Arosio P and Melman A, Iron release from ferritin by flavin nucleotides, Biochim. Biophys. Acta, 2013, 1830, 4669–4674. [DOI] [PubMed] [Google Scholar]
- 92.Linder MC, Mobilization of stored iron in mammals: a review, Nutrients, 2013, 5, 4022–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.La A, Nguyen T, Tran K, Sauble E, Tu D, Gonzalez A, Kidane TZ, Soriano C, Morgan J, Doan M, Tran K, Wang CY, Knutson MD and Linder MC, Mobilization of iron from ferritin: new steps and details, Metallomics, 2018, 10, 154–168. [DOI] [PubMed] [Google Scholar]
- 94.Chiou B, Neal EH, Bowman AB, Lippmann ES, Simpson IA and Connor JR, Endothelial cells are critical regulators of iron transport in a model of the human blood-brain barrier, J. Cereb. Blood Flow Metab, 2019, 39, 2117–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, Dupont N, Jiang S, Peters R, Farzam F, Jain A, Lidke KA, Adams CM, Johansen T and Deretic V, Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy, EMBO J, 2017, 36, 42–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stone NL, England TJ and O’Sullivan SE, A Novel Transwell Blood Brain Barrier Model Using Primary Human Cells, Front. Cell. Neurosci, 2019, 13, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yambire KF, Rostosky C, Watanabe T, Pacheu-Grau D, Torres-Odio S, Sanchez-Guerrero A, Senderovich O, Meyron-Holtz EG, Milosevic I, Frahm J, West AP and Raimundo N, Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo, eLife, 2019, 8, e51031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bradley JM, Svistunenko DA, Moore GR and Le Brun NE, Tyr25, Tyr58 and Trp133 of Escherichia coli bacterioferritin transfer electrons between iron in the central cavity and the ferroxidase centre, Metallomics, 2017, 9, 1421–1428. [DOI] [PubMed] [Google Scholar]
- 99.Zhang DL, Su D, Berczi A, Vargas A and Asard H, An ascorbate-reducible cytochrome b561 is localized in macrophage lysosomes, Biochim. Biophys. Acta, 2006, 1760, 1903–1913. [DOI] [PubMed] [Google Scholar]
- 100.Crichton RR and Charloteaux-Wauters M, Iron transport and storage, Eur. J. Biochem, 1987, 164, 485–506. [DOI] [PubMed] [Google Scholar]
- 101.Cohen A, Nelson H and Nelson N, The family of SMF metal ion transporters in yeast cells, J. Biol. Chem, 2000, 275, 33388–33394. [DOI] [PubMed] [Google Scholar]
- 102.Govoni G, Vidal S, Cellier M, Lepage P, Malo D and Gros P, Genomic structure, promoter sequence, and induction of expression of the mouse Nramp1 gene in macrophages, Genomics, 1995, 27, 9–19. [DOI] [PubMed] [Google Scholar]
- 103.Coffey R and Ganz T, Erythroferrone: An Erythroid Regulator of Hepcidin and Iron Metabolism, Hemasphere, 2018, 2, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sangkhae V and Nemeth E, Regulation of the Iron Homeostatic Hormone Hepcidin, Adv. Nutr, 2017, 8, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thachil J, The beneficial effect of acute phase increase in serum ferritin, Eur. J. Intern. Med, 2016, 35, e16–e17. [DOI] [PubMed] [Google Scholar]
- 106.Jandl JH and Katz JH, The plasma-to-cell cycle of transferrin, J. Clin. Invest, 1963, 42, 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kosman DJ, Molecular mechanisms of iron uptake in fungi, Mol. Microbiol, 2003, 47, 1185–1197. [DOI] [PubMed] [Google Scholar]
- 108.Hassett RF, Romeo AM and Kosman DJ, Regulation of high affinity iron uptake in the yeast Saccharomyces cerevisiae. Role of dioxygen and Fe, J. Biol. Chem, 1998, 273, 7628–7636. [DOI] [PubMed] [Google Scholar]
- 109.Howitt J, Putz U, Lackovic J, Doan A, Dorstyn L, Cheng H, Yang B, Chan-Ling T, Silke J, Kumar S and Tan SS, Divalent metal transporter 1 (DMT1) regulation by Ndfip1 prevents metal toxicity in human neurons, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 15489–15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Foot NJ, Dalton HE, Shearwin-Whyatt LM, Dorstyn L, Tan SS, Yang B and Kumar S, Regulation of the divalent metal ion transporter DMT1 and iron homeostasis by a ubiquitin-dependent mechanism involving Ndfips and WWP2, Blood, 2008, 112, 4268–4275. [DOI] [PubMed] [Google Scholar]
- 111.Seo YA, Kumara R, Wetli H and Wessling-Resnick M, Regulation of divalent metal transporter-1 by serine phosphorylation, Biochem. J, 2016, 473, 4243–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]