

A ketogenic diet lowered benzodiazepine use during withdrawal in patients with AUD and blunted alcohol administration in rats.
Abstract
Individuals with alcohol use disorder (AUD) show elevated brain metabolism of acetate at the expense of glucose. We hypothesized that a shift in energy substrates during withdrawal may contribute to withdrawal severity and neurotoxicity in AUD and that a ketogenic diet (KD) may mitigate these effects. We found that inpatients with AUD randomized to receive KD (n = 19) required fewer benzodiazepines during the first week of detoxification, in comparison to those receiving a standard American (SA) diet (n = 14). Over a 3-week treatment, KD compared to SA showed lower “wanting” and increased dorsal anterior cingulate cortex (dACC) reactivity to alcohol cues and altered dACC bioenergetics (i.e., elevated ketones and glutamate and lower neuroinflammatory markers). In a rat model of alcohol dependence, a history of KD reduced alcohol consumption. We provide clinical and preclinical evidence for beneficial effects of KD on managing alcohol withdrawal and on reducing alcohol drinking.
INTRODUCTION
Alcohol use disorder (AUD) is a chronic, relapsing brain disorder that accounts for 5% of deaths globally (1). Chronic alcohol consumption in AUD is associated with impairments in multiple cognitive domains, including executive, perceptuomotor, and visuospatial functions, and with disrupted brain structure and function (2–6). Alcohol withdrawal in AUD involves distressing symptoms such as dysphoria, tremor, anxiety, restlessness, and insomnia and can be neurotoxic and life threatening (7). The motivational and somatic signs of alcohol withdrawal are hypothesized to perpetuate alcohol drinking and favor relapse. Hence, there is an urgent need to identify interventions that can minimize withdrawal symptoms and promote abstinence.
Targeting the effects of alcohol on switching brain energetics may have potential as a promising therapeutic intervention. Acute alcohol ingestion decreases glucose metabolism in the human brain and increases the metabolism of acetate, a metabolite of alcohol (4, 8), and this effect is accentuated in individuals with AUD (8). With chronic alcohol drinking as in AUD, there is an increase in brain use of acetate and ketone bodies [i.e., acetoacetate (AcAc), acetone, and beta-hydroxybutyrate (BHB)] as energy substrates (4, 9).
The shift from glucose to acetate metabolism in AUD persists beyond the acute intoxication state. During sobriety, patients with AUD show higher brain acetate metabolism and lower brain glucose metabolism compared with controls (4, 9), which was also observed in preclinical studies (10). Thus, we hypothesize that a paradoxical energy-deficit state in the brain emerges during alcohol detoxification when acetate levels in plasma decrease and that this contributes to withdrawal symptoms and neurotoxicity in patients with AUD (4).
Ketogenic diets (KDs; high fat, low carbohydrate, and protein) increase ketone bodies (i.e., AcAc, acetone, and BHB) in plasma and brain by breaking down fat in the liver (11). This state of “metabolic ketosis” is similar to a fasting state, and ketone bodies can be used by the body and brain as alternative energy source to glucose (11). In healthy controls, a dual-tracer positron emission tomography brain imaging study showed that KD reduced brain glucose metabolism and increased brain ketone metabolism (11) similar to what is observed in patients with AUD (3, 7). Clinically, KDs are used therapeutically to reduce seizures in patients with epilepsy (12). KD has other health benefits, including reductions in appetite and food intake (13). In obese individuals, a 4-month KD lowered food craving and notably craving for alcohol (13), suggesting that KD may also have beneficial effects on alcohol craving and consumption. In laboratory rodents, KD decreased symptoms of acute alcohol withdrawal as compared to regular chow (14, 15). However, whether a KD could be therapeutically beneficial for treating alcohol-related withdrawal symptoms in humans with AUD undergoing detoxification remains to be determined.
Here, we evaluated whether metabolic ketosis has a therapeutic benefit in managing withdrawal and craving in inpatients with AUD undergoing detoxification. For this purpose, we studied 33 treatment-seeking patients with AUD during detoxification and randomized them into a KD (n = 19) or a standard American (SA) diet (n = 14) for 3 weeks. Patients underwent weekly 1H magnetic resonance spectroscopy (1H-MRS) scanning to measure levels of brain ketones, amino acids relevant for the tricarboxylic acid (TCA) cycle [glutamate (Glu) and glutamine] (16, 17), and markers of neuroinflammation [myo-inositol (mI) and choline (Cho)] (18). Serum ketone levels were measured once per week, and urine AcAc levels were assessed daily. We aimed to test the effect of KD in AUD participants on (i) acute withdrawal symptoms and need for benzodiazepines within the first week of study; (ii) alcohol craving and brain reactivity to alcohol cues using functional magnetic resonance imaging (fMRI) during the 3 weeks of study; and (iii) brain ketones, mI, Glu, and Cho during the 3 weeks of study (three scans). The dorsal anterior cingulate cortex (dACC) was our main region of interest (ROI) for both fMRI and MRI, given its involvement in inhibitory control, decision making, reward prediction, control of alcohol craving, and cue reactivity (19–21). Likewise, in the ACC (including the dACC), volumetric deviations (21), reactivity to alcohol cues, and gamma wave activity have been associated with higher alcohol consumption and with relapse (22–24). In addition, chronic alcohol use is associated with reduced glucose metabolic cost in dACC (25), and the dACC provides good-quality 1H-MRS spectra (26). We hypothesized that KD would reduce acute withdrawal symptoms, the need for benzodiazepines to manage withdrawal symptoms, and alcohol craving during detoxification in patients with AUD. Because KD increases ketone body AcAc in the brain (11), we also predicted that dACC ketone levels of AcAc, acetone, and BHB would increase with KD.
In a parallel study using a validated rodent model of alcohol dependence that produces symptoms similar to those observed in humans with AUD (27, 28), we aimed to assess the effects of a KD in alcohol self-administration. However, uncovering that the KD markedly increased blood alcohol levels (BALs) compared to animals on a regular diet (>5-fold; fig. S1), we tested the animals instead shortly after KD discontinuation. We hypothesized that a history of a KD would decrease alcohol self-administration during withdrawal as compared to a history of a regular rodent diet. Overall, the main aims of the present study were to (i) test the effects of KD on alcohol withdrawal and craving in human inpatients with AUD during detoxification and (ii) the effects of a KD on alcohol consumption in a rodent model of alcohol dependence, which we used as a preclinical surrogate for the effects of KD on alcohol craving and wanting. Together, these studies provide evidence for clinical efficacy of metabolic ketosis for both alcohol withdrawal in AUD and alcohol consumption in rats.
RESULTS
Clinical studies
Urine and blood ketones
Table 1 provides demographics and clinical characteristics of the KD and SA groups. A 4:1 (grams of fat–to–grams of carbohydrate and protein ratio) KD significantly increased urinary AcAc and blood BHB (n = 19) compared to SA (n = 14; group × time interaction: AcAc: F18,558 = 11.6, P < 0.0001, η2 = 0.27; BHB: F3,93 = 67.0, P < 0.0001, η2 = 0.68), with plasma BHB levels of >4 mM. Group differences in urinary ketones, which did not differ on the morning after diet initiation (day 2; t31 = 1.2, P = 0.2), were significant on day 3 (t31 = 2.9, P = 0.009) and thereafter (all P < 0.001). Blood ketones did not differ between groups before diet initiation (t31 = 1.4, P = 0.2) and became significantly elevated in the KD group in week 1 and thereafter (all P < 0.001; Fig. 1).
Table 1. Demographics and clinical characteristics of the KD and SA groups.
ADS, Alcohol Dependence Scale; BRAC, Breath Alcohol Concentration; CIWA-Ar, Clinical Institute Withdrawal Assessment (revised version); IQ, intelligence quotient; TLFB, Timeline Followback; WASI, Wechsler Abbreviated Scale of Intelligence.
| KD (n = 19) | SA (n = 14) | P value | |
| Age (years) | 39.3 (11.2) | 44.2 (16.4) | 0.311 |
| Sex | 7 females (37%) | 3 females (21%) | 0.341 |
| 12 males (63%) | 11 males (79%) | ||
| BMI admission | 24.5 (3.4) | 27.6 (5.4) | 0.051 |
| Study weight loss (kg) |
1.4 (2.8) | 1.8 (2.2) | 0.692 |
| Mean calories per day during study (kcal) |
2674.3 (469.2) | 2632.1 (261.5) | 0.764 |
| Ethnicity | 6 Black/ African-American 11 White, 2 Multiracial |
6 Black/ African-American 6 White, 2 Multiracial |
0.694 |
| Years of education |
12.7 (2.8) | 14.2 (2.5) | 0.110 |
| WASI IQ | 99.2 (16.3) | 98.3 (22.5) | 0.892 |
| Smoking status | 10 smokers | 9 smokers | 0.503 |
| 9 nonsmokers | 5 nonsmokers | ||
| LDH (kg) | 984.9 (945.5) | 1414.4 (1462.6) | 0.320 |
| TLFB drinks/day | 15.2 (8.4) | 17.0 (9.8) | 0.584 |
| TLFB no. of drinking days |
73.0 (21.1) | 79.7 (13.0) | 0.299 |
| ADS | 21.0 (9.8) | 23.6 (7.2) | 0.399 |
| Days since last drink admission |
0.58 (1.0) | 0.9 (1.4) | 0.514 |
| BRAC at admission |
0.10 (0.12) | 0.10 (0.12) | 0.920 |
| CIWA-Ar score at admission |
6.4 (4.9) | 5.4 (4.2) | 0.540 |
| Benzodiazepines (mg) at admission |
49.0 (41.2) | 34.3 (36.9) | 0.300 |
| Benzodiazepines at admission (n) |
13 oxazepam, 3 diazepam, and 3 none |
10 oxazepam and 4 none |
0.375 |
| MADRS depression at baseline |
18.2 (9.0) | 21.7 (12.4) | 0.355 |
| Anxiety at baseline |
12.7 (6.4) | 15.6 (9.1) | 0.305 |
Fig. 1. Urine and blood ketone levels of KD and SA participants.
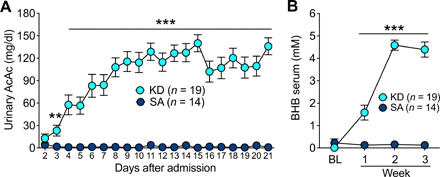
(A) Urinary levels of AcAc and (B) serum levels of BHB in KD and SA. **P < 0.01 and ***P < 0.001, different from SA. The slight decrease in week 3 compared to week 2 for urine and blood measures reflected two participants who were not compliant with the KD in week 3, one for 1 day and the other for 2 days.
Alcohol withdrawal and benzodiazepine use
At admission (before diet initiation), 16 of 19 patients who would be subsequently randomized to KD received benzodiazepines (3 diazepam, 13 oxazepam, and 3 none), and 10 of 14 patients who would be randomized to SA received oxazepam (0 diazepam and 4 none). Diazepam doses were multiplied by 2 for the relative dose of oxazepam (clincalc.com/benzodiazepine). After randomization and diet initiation (within 2 days of admission), there was a significant effect of group × time on benzodiazepine cumulative dose in milligrams (F3,96 = 4.8, P = 0.004, η2 = 0.13), with the KD group requiring less benzodiazepines than the SA group (Fig. 2A); groups differed on day 2 (t31 = 2.2, P = 0.057) and day 3 (t31 = 2.6, P = 0.013). Table S2 lists mean dosages of benzodiazepines for each day. While the cumulative dose of benzodiazepines in KD decreased by 39% (from 86.8 ± 92.7 SD before diet initiation to 34.2 ± 79.4 SD after diet initiation), the cumulative dose in SA increased by 187% (from 83.6 ± 78.2 before diet initiation to 156.4 after diet initiation) (F1,31 = 10.9, P = 0.002, η2 = 0.26). The group × time effect on benzodiazepine use remained significant with a 1:3 conversion of diazepam:oxazepam, which is an alternative conversion equivalency (globalrph.com/medcalcs/benzodiazepine-converter-dosage-conversions), or when removing those three participants who received diazepam (F3,86 = 4.8, P = 0.004, η2 = 0.15). Although the groups did not differ at admission (Fig. 2B), after diet initiation, there was a trend for fewer patients with AUD needing benzodiazepines in the KD compared to SA group (χ1,332 = 3.2, P = 0.07; Fig. 2C).
Fig. 2. Benzodiazepine use during the first week of detoxification.
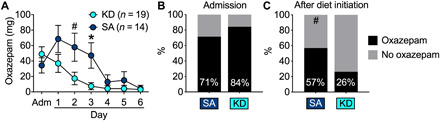
(A) KD participants needed significantly less benzodiazepines during the first week of withdrawal. (B) KD and SA patients needed benzodiazepines at similar levels at admission before diet initiation. (C) After diet initiation, fewer patients needed benzodiazepines for the KD than the SA group. *P < 0.05, different from KD; #P < 0.09, a trend effect compared with KD.
The group × time effect on the rating for maximum withdrawal from the Clinical Institute Withdrawal Assessment–Alcohol revised (CIWA-Ar) (29) showed a trend for significance (F3,88 = 2.5, P = 0.067, η2 = 0.08; fig. S2). Group differences, however, were not significant for any day before or after diet initiation. The lack of a significant effect probably reflects the clinical standard use of benzodiazepines on an “as needed” basis to reduce moderate to severe withdrawal symptoms. The average maximum CIWA score was strongly associated with cumulative benzodiazepine use (r33 = 0.646, P < 0.0001), both in KD (r19 = 0.72, P < 0.001) and in SA (r14 = 0.58, P = 0.029). Moreover, the maximum CIWA for each day correlated with the benzodiazepine dose for each day (admission: r33 = 0.39, P = 0.023; day 1: r33 = 0.78, P < 0.0001; day 2: r33 = 0.78, P < 0.0001; day 3: r33 = 0.83, P < 0.0001; day 4: r33 = 0.72, P < 0.0001; day 5: r33 = 0.86, P < 0.0001; day 6: r33 = 0.98, P < 0.0001).
Alcohol cue reactivity
Mean ratings of “wanting” with exposure to alcohol cues relative to the neutral cues used in the fMRI cue reactivity task decreased over time in the KD (F2,32 = 6.6, P = 0.004, η2 = 0.29) but not in the SA group (F2,24 = 0.5, P = 0.62, η2 = 0.04), and the group × time interaction effect was significant (F2,56 = 4.9, P = 0.048, η2 = 0.20; Fig. 3A). There were no significant group effects for wanting ratings when exposed to food cues versus neutral cues or for valence ratings of alcohol cues or food cues, relative to neutral cues (Fig. 3B).
Fig. 3. Weekly alcohol cue reactivity task.
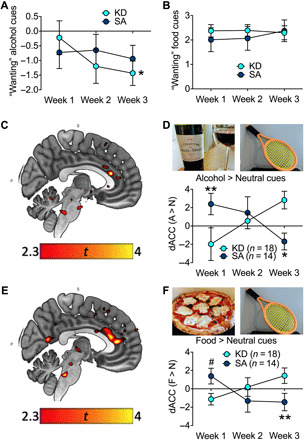
(A) Alcohol wanting ratings of alcohol cues were lower in KD than SA after diet initiation. (B) There were no group differences for wanting ratings caused by food cues. (C) There was a significant group × time for brain reactivity to alcohol > neutral cues in dACC (PFWE < 0.05) with (D) increased dACC reactivity in KD > SA in week 3 > week 1. (E) There was a significant group × time for brain reactivity to food > neutral cues in dACC (PFWE < 0.05) with (F) elevated dACC reactivity in KD > SA in week 3 > week 1. *P < 0.05 and **P < 0.01, difference between KD and SA; #P < 0.09, a trend effect compared with KD. Photo credit: Corinde Wiers, NIH/NIAAA.
The fMRI cue-reactivity paradigm revealed a main effect for alcohol > neutral cues with activation of clusters in superior temporal and frontal gyrus, as well as precuneus/posterior cingulate cortex (all PFWE < 0.05), and deactivation in cerebellum, occipital cortex, and fusiform gyrus (PFWE < 0.05) in both groups (table S3 lists the main effects and interaction effects for alcohol > neutral and food > neutral contrasts at P < 0.005, k ≥ 50). The group × time interaction effect for alcohol > neutral cues showed elevated dACC activation in the KD compared to the SA group over time {peak Montreal Neurological Institute (MNI) space = [4, 30, 18], t = 4.02, PFWE = 0.028 [small volume corrected (SVC)]; Fig. 3, C and D}. The dACC blood-oxygen-level-dependent (BOLD) responses to alcohol cues did not correlate with the scores for alcohol craving or wanting following alcohol cues exposure (all P > 0.1).
The food > neutral cues also showed the main effects of activation in precuneus/posterior cingulate cortex, superior temporal and superior frontal gyrus, and deactivation in cerebellum and inferior and middle frontal gyrus (PFWE < 0.05). The group × time interaction showed greater dACC activation in KD compared to SA over time (peak MNI = [4, 28, 12], t = 3.66, PFWE = 0.003; SVC; Fig. 3, E and F). The dACC cluster also comprised the medial prefrontal cortex and was significant at the whole-brain level along with the bilateral insula (see table S3).
Brain energetics and neuroinflammation markers
The 1H-MRS measures in dACC (Fig. 4, A and B) revealed significant group × metabolite [multivariate analysis of variance (MANOVA) F5,25 = 7.2, P < 0.001, η2 = 0.59] and group × time (F5,28 = 3.5, P = 0.044, η2 = 0.20) interaction effects (Fig. 4, C to H). Creatine (Cr) levels did not differ between groups or over time and was hence used to normalize the other spectroscopic measures. BHB levels could not be accurately assessed in our 1H-MRS. Levels of acetone (2.22 ppm) and AcAc (2.29 ppm) (Cr normalized) increased in KD versus SA over time (group × time acetone: F2,58 = 3.4, P = 0.04, η2 = 0.11; AcAc: F2,58 = 6.3, P = 0.004, η2 = 0.18), and the effect of group (acetone: F1,29 = 12.3, P = 0.001, η2 = 0.30; AcAc: F1,29 = 8.7, P = 0.006, η2 = 0.23) revealed higher levels in KD than SA [acetone, week 1: t18.0 = 2.4, P = 0.016; week 2: t19.5 = 4.3, P = 0.0004; week 3: t17.3 = 0.001; AcAc, weeks 1 and 2, not significant; week 3: t23.3 = 5.4, P = 0.00002 (Fig. 4, C and D)]. Figure S3 provides summed spectra of the KD and SA groups.
Fig. 4. Weekly 1H-MRS metabolites in dACC.
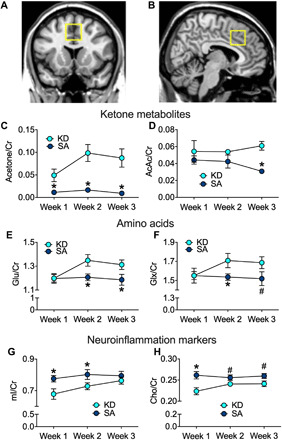
(A) Coronal and (B) sagittal views of dACC 1H-MRS voxel position. Cr-normalized ketone body metabolites (C) acetone and (D) AcAc were higher in the KD than the SA group. Amino acids (E) glutamate (Glu) and (F) Glu and glutamine (Glx) levels were higher in KD than SA. Neuroinflammation markers (G) mI and (H) Cho levels were lower in KD than SA. Group differences, *P < 0.05, and trends, #P < 0.09.
The KD group, compared to SA, showed higher glutamate (Glu) (group effect: F1,29 = 4.7, P = 0.039, η2 = 0.14), which was significant in week 2 (t30 = 2.2, P = 0.034) and week 3 (t29 = 2.1, P = 0.047) (Fig. 4E), and higher Glu and glutamine (Glx) at trend level (group: F1,29 = 3.0, P = 0.095, η2 = 0.09; increased in week 2: t24.8 = 2.1, P = 0.045; and at trend level in week 3: t29 = 1.8, P = 0.09; Fig. 4F). However, the KD group showed lower mI levels than the SA group (group: F1,29 = 4.3, P = 0.047, η2 = 0.13), which was significant in week 1 (t30 = 2.3, P = 0.028) and week 2 (t30 = 2.3, P = 0.031) but not in week 3 (t29 = 0.86, P = 0.4; Fig. 3G). Cho is also lower in KD than SA (group effect: F1,29 = 7.4, P = 0.011, η2 = 0.20; week 1: t30 = 3.1, P = 0.004; week 2: t30 = 1.9, P = 0.063; week 3: t29 = 2.0, P = 0.056; Fig. 4H).
Craving, mood ratings, and neuropsychological testing
There was a main effect of time in self-reports for the “Desire for alcohol questionnaire” (DAQ) (25) at trend level (F2,58 = 2.5, P = 0.092, η2 = 0.08), and DAQ craving reduced over time in KD at trend level (F2,32 = 3.2, P = 0.056, η2 = 0.17) but not in SA (F2,26 = 0.5, P = 0.64, η2 = 0.03). The group × time interaction effect was not significant (F2,58 = 0.5, P = 0.6, η2 = 0.02; fig. S4). There were no significant group or group × time effects for daily mood or hunger ratings (fig. S5). Despite significant main effects of abstinence/training on various cognitive tasks, there were no significant group or group × time interaction effects (table S4).
Study blinding
There were no significant group effects on diet expectation (KD or SA) on the surveys obtained after study completion (χ2 = 0.14, P = 0.71) or on the subjective ratings for diet “pleasantness” (t28 = 1.0, P = 0.32) or “improved health” (t28 = 0.12, P = 0.90) (fig. S6), indicating successful study blinding.
Preclinical studies
Preclinical studies were conducted to assess whether a KD would have similar effects in a validated animal model of excessive drinking associated with alcohol withdrawal in rats made dependent on alcohol. Blood glucose and ketone levels were measured before and during the diet intervention, and the consequences of the chow and KD were assessed on alcohol self-administration during alcohol vapor exposure after diet discontinuation (KD or chow). The reason why we exposed the rats to alcohol vapor and measured alcohol self-administration after diet discontinuation instead of during the diet exposure was because animals on KD when exposed to alcohol vapor had fivefold higher BALs than those in the chow diet (fig. S1). Instead, exposing them to alcohol after diet discontinuation ensured equivalent BAL during vapor exposure.
Blood glucose and ketone levels
Adult, male, Wistar rats on a chow diet (n = 36) were trained to press a lever for access to alcohol associated with a 0.5-s cue light [0.1 ml of 10% (w/v) alcohol; right lever] or water (0.1 ml; left lever) in 30-min operant sessions. We used a fixed-ratio one schedule of reinforcement, in which each lever press was reinforced with liquid delivery. Upon stable levels of alcohol versus water self-administration, the rats were exposed to a KD (KD rats; n = 18) or chow diet (chow rats; n = 18). The blood glucose and ketone levels were measured at baseline (prediet), when all rats were maintained on their regular chow diet, and after 1 and 2 months on the intervention diet (KD or chow). The rats were not fasted for these measurements. A repeated-measure analysis of variance (ANOVA) showed a significant diet × time interaction for glucose levels (F2,68 = 43.09, P < 0.0001) and for ketone levels (F2,68 = 107.30, P < 0.0001). Post hoc comparisons indicated that KD rats, compared to chow rats, had lower glucose levels (P < 0.0001; Fig. 5A) and higher ketone levels (P < 0.0001; Fig. 5B). KD rats weighed significantly less than chow rats (fig. S7A).
Fig. 5. Blood glucose and ketone levels in rats maintained on a KD (KD rats) or chow diet (chow rats) for 2 months.
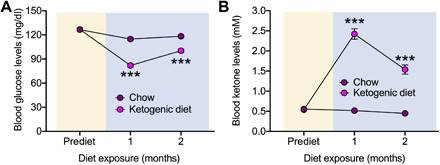
KD rats exhibited (A) decreased glucose and (B) increased ketone levels in blood compared with chow rats. ***P < 0.0001, different from chow at the same time points.
Alcohol self-administration and BALs
After 2 months of KD (or chow), all rats were given free access to chow for the remainder of the experiment. Half of the rats in each group were exposed to alcohol vapor for 7 weeks to produce alcohol dependence, and the other half were exposed to air without alcohol to serve as nondependent controls. The alcohol concentration in the air was progressively increased to target BALs of 175 to 225 mg/dl. Figure 6A shows the timeline of the experiment. Figure 6B shows BALs produced by alcohol vapor exposure over the course of the self-administration experiment. Figure 6C shows the number of alcohol reinforcers that the rats earned in 30-min operant self-administration sessions that were conducted during acute withdrawal, twice a week, for 7 weeks. The two-way repeated-measure ANOVA yielded significant group (F3,32 = 4.54, P = 0.0092, η2 = 0.30) and session (F13,416 = 3.10, P = 0.0002, η2 = 0.09) effects but not a significant session × group interaction (F39,416 = 1.01, P = 0.46, η2 = 0.08). The post hoc comparisons on the overall group effect indicated that alcohol vapor–chow rats self-administered significantly more alcohol than alcohol vapor–KD rats (P = 0.0452, η2 = 0.25) regardless of session. No significant differences were found between air-chow and air-KD rats. When we compared the average of the last four alcohol self-administration sessions, when BALs during alcohol vapor exposure approached our target (200 mg/dl), the one-way ANOVA yielded a significant group effect (F3,32 = 5.71, P = 0.0030). Post hoc comparisons indicated that alcohol vapor–chow self-administered significantly more alcohol compared with all the other groups (P < 0.05) during the last four self-administration sessions during withdrawal (data are shown in the inset of Fig. 6C). Note that these differences could not be explained by differential dependence induction in that BALs during alcohol vapor exposure were the same in both groups, as shown in Fig. 6B. There were no group differences in blood glucose and ketone levels or in body weight during alcohol vapor exposure when all of the rats were maintained on chow (fig. S7).
Fig. 6. Operant alcohol self-administration and BALs in rats with a history of KD or chow diet.
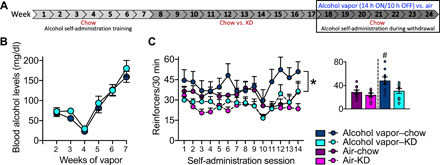
(A) The rats were trained to lever press (fixed-ratio 1) for alcohol or water in 30-min operant sessions. Next, the rats were exposed to a KD or chow diet for 8 weeks. All rats were then given free access to chow for the remainder of the experiment. Half of the rats (alcohol vapor) were exposed to chronic (daily), intermittent alcohol vapor (14 hours of alcohol on followed by 10 hours off, alcohol withdrawal) for 7 weeks to produce alcohol dependence; the other half (air) were exposed to air without alcohol to serve as nondependent controls. The black rectangular box indicates when data shown in (B) and (C) were collected. (B) Rats with a history of KD or chow did not differ in BAL over the course of the alcohol vapor exposure. (C) Alcohol vapor–exposed rats with a history of chow diet (alcohol vapor–chow) self-administered significantly more alcohol during twice weekly self-administration sessions during withdrawal (6 to 8 hours after vapor turned OFF) than alcohol vapor–exposed rats with a history of KD (alcohol vapor–KD). *P < 0.05, alcohol vapor–chow > alcohol vapor–KD, regardless of self-administration session. Alcohol self-administration did not differ between the groups of air-exposed rats with a history of chow or KD (air-chow not different from air-KD). The inset bar figure shows the average of alcohol reinforcers that the rats earned in the last four self-administration sessions that corresponded to weeks 6 and 7 of alcohol vapor exposure. #P < 0.05, alcohol vapor–chow self-administered significantly more alcohol compared with all of the other groups.
DISCUSSION
Here, we provide clinical and preclinical evidence for the potential therapeutic benefit of a KD in reducing the severity of alcohol withdrawal symptoms during early alcohol detoxification in treatment-seeking inpatients with AUD and in reducing alcohol drinking during acute withdrawal in a rat model of alcohol dependence. In the clinical findings, there was a significant decrease in the need for benzodiazepines to manage acute alcohol withdrawal, an overall lowering of alcohol withdrawal scores (trend level), and reduced alcohol craving and wanting ratings with exposure to alcohol cues. Because benzodiazepines were prescribed when patients’ withdrawal scores were high (i.e., CIWA-Ar scores of >8) and benzodiazepines significantly reduce withdrawal symptoms, the lack of group effect on CIWA-Ar scores very likely reflects the higher doses of benzodiazepines prescribed in the SA compared to the KD group. We also show preliminary support of beneficial brain effects of KD in AUD including reduced neuroinflammation, as shown by low mI and Cho. Moreover, because the dACC is a key node of the executive function network (30), we speculate that the increased reactivity of the dACC to alcohol and food cues in the KD versus SA group may be related to increased top-down control of craving. In the preclinical model, we document that chow rats escalated alcohol self-administration during acute withdrawal following chronic, intermittent alcohol vapor exposure and withdrawal (dependent rats), whereas rats with a history of KD did not and instead maintained alcohol intake at the levels of nondependent, air-exposed rats.
We hypothesized that providing ketone bodies via KD during detoxification would attenuate the emergence of withdrawal symptoms. We reasoned that the abrupt transition from the brain’s consumption of ketone bodies, which occurs in AUD as an adaptation to repeated alcohol intake, to the use of glucose as energy source, which reemerges with detoxification, may contribute to the alcohol withdrawal syndrome. Notably, BHB, which is a major ketone body is thought to be a more efficient “fuel” source than glucose. Therefore, inducing ketosis with a KD during detoxification would bolster or halt such an abrupt transition (11), thereby reducing withdrawal. Disorders of brain glucose metabolism are associated with disrupted neuronal excitability, including epilepsy (31), and are ameliorated by KD. Thus, it is possible that the reduction of withdrawal symptoms by KD reflects an attenuation of neuronal excitability following alcohol discontinuation that at extremes can result in seizures and death (32). The following mechanism(s) have been proposed to be implicated in KD-induced reduction of seizures, including (i) restoration of glutamatergic neurotransmission and enhancement of γ-aminobutyric acid (GABA) synthesis, (ii) circumvention of glycolysis and providing acetyl–coenzyme A for the TCA cycle through fatty acid oxidation, (iii) activation of adenosine triphosphate (ATP)–sensitive potassium channels, and (iv) inhibition of voltage-dependent Ca+ channels (33, 34). Glucose metabolism, through the TCA cycle, is not only the main source of energy to the brain but is also the main carbon source for synthesis of glutamate and GABA (16, 34). In preclinical models, reduced flux through the TCA results in reduced glutamate brain content and elicits epileptiform discharges, which are ameliorated by acetate administration (35). Thus, the increases in Glu and Glx that we observed with the KD are consistent with it helping to restore TCA flux and balancing excitatory and inhibitory neurotransmission. A recent study in a mouse model of Alzheimer’s disease (triple transgenic Alzheimer’s 3xTgAD, which shows reduced brain glucose utilization) demonstrated that BHB ketone ester intervention, compared to a regular chow diet, elevated hippocampal glutamate and α-glutarate (a precursor of glutamate), and glutamate and α-glutarate levels intercorrelated positively in both groups. This may indicate that metabolic ketosis furnishes mitochondria with TCA cycle substrates (17). Furthermore, a 4-month KD elevated Glu and glutamine in young adult rats (36), and although human patients with epilepsy did not show differences in posterior cingulate cortical Glu measures compared to controls, elevated Glu concentrations predicted short-term freedom from seizures, hence indicating clinical relevance of elevated Glu in epilepsy (37). In the meantime, a recent study found that acetone and BHB acted as inhibitors of Glu at the excitatory NMDA (N-methyl-d-aspartate) receptors (38). These studies highlight the diverse roles of glutamate in extracellular versus intracellular processes, which 1H-MRS studies cannot differentiate and that have reported both elevated (39) and reduced (40) brain Glu in AUD during withdrawal. KD is efficacious in the treatment of epilepsy, and seizures can occur during severe alcohol withdrawal. However, the use of benzodiazepines for managing severe alcohol withdrawal also prevents seizures (41), and thus, it is not unexpected that none of our patients with AUD experienced seizures. It is therefore possible that the KD reduced overall neuronal excitability, hence reducing the need of benzodiazepine during acute withdrawal. However, future studies are needed to investigate the effect of KD on alcohol-induced seizures in rodent models that mimic human alcohol withdrawal–induced seizures (41).
Another possible explanation of our results is that the KD ameliorated withdrawal symptoms by minimizing neuroinflammatory changes associated with alcohol withdrawal (42). BHB was shown to influence inflammatory processes through its inhibition of the NLRP3 inflammasome (18). In our study, we observed that the patients on KD compared to SA showed lower levels of mI and total Cho (phosphatidylcholine + glycerylphosphorylcholine), which are markers of neuroinflammation (43). Specifically, mI is primarily localized in glial cells and is considered a marker of gliosis (43), whereas elevated Cho has been detected in several inflammatory brain disorders (44, 45). Thus, our findings could be interpreted to reflect a reduction of neuroinflammation in patients with AUD maintained on a KD compared to SA. Reduced mI with KD is consistent with prior brain 1H-MRS studies reporting higher mI in AUD compared to healthy controls (46), predominantly during early detoxification with recovery after protracted abstinence, presumably as neuroinflammation subsides. However, 1H-MRS findings on Cho and alcohol exposures are less clear. Elevated levels of brain Cho have been reported during periods of active drinking (47), whereas lower levels have been reported during early abstinence that recovered with protracted abstinence (48), although one study did not show recovery (49). Elevated Cho during acute exposure and decreased Cho during chronic exposure and early detoxification are reminiscent of the pattern observed for the translocator protein, which has also been used as a marker of neuroinflammation in AUD (50, 51) and altered in alcohol-dependent rats (50, 52). Although Cho can recover over time, further research is required to investigate its significance in AUD and the effects of detoxification and KD on this metabolite.
Patients with AUD on KD also reported lower alcohol craving and wanting ratings of alcohol cues compared to SA. These effects could be a consequence of reduced withdrawal symptoms and/or reflect a potentially beneficial effect of KD in disrupting conditioning to alcohol through its provision of acetate and other ketone bodies that can serve as energy substrates and their subsequent effects on neurotransmission. Although most of the research studying conditioned effects of alcohol link it to its reinforcing effects mediated by its enhancement of GABAergic, opioid, and dopaminergic systems, it is possible that its bioenergetic switch is also relevant to its reinforcing effects, particularly given that energy-rich stimuli engender strong conditioned responses (53, 54). In this respect, we speculate that KD would satisfy the altered energy requirements associated with chronic alcohol consumption through its metabolism of acetate and BHB.
The beneficial effects of KD on alcohol withdrawal symptoms could also reflect the enhanced reactivity of the dACC that we observed in patients with AUD on the KD when exposed to alcohol and food cues. Activation of ACC is necessary to inhibit prepotent responses to reinforcing stimuli in addiction (19, 20). However, increased ACC responses to alcohol cues have also been associated with higher alcohol use and craving (22) and higher rates of relapse (23); thus, more research is warranted into the contribution of specific subsections of the ACC in alcohol cue reactivity and its associations with clinical characteristics in AUD.
A limitation of our clinical study was the relatively small sample size, which was constrained by the complexity of the study that required 3 weeks of hospitalization and compliance with the diet protocol. Nevertheless, the effect sizes of our findings were either large (i.e., MRS metabolites and rat alcohol consumption), medium large (i.e., effect on alcohol wanting ratings), or medium (i.e., effects on benzodiazepines, alcohol withdrawal, and craving). Moreover, the effects on benzodiazepines, MRS metabolites, and rat alcohol consumption remained significant after testing for multiple comparisons. Note also that while 57% of SA subjects needed benzodiazepines after diet initiation to manage withdrawal, only 26% of KD subjects required these. The absolute risk reduction was thus 31%, and the number needed to treat (NNT) was 3.2 (i.e., one in three AUD are expected to benefit from KD compared to SA), which is comparable to the NNT of antidepressant medications for reducing depression scores by 50% in major depressive disorder and of lithium to prevent relapse in bipolar disorder (55). Nonetheless, because of the small sample size of our study and the failure to detect significant correlations between bioenergetics/neuroinflammatory markers and the scores for alcohol craving/wanting, we interpret our clinical findings as preliminary. Although there were no group differences in KD versus SA before diet initiation and no group differences in study withdrawal rates (table S1), the sample may still be biased to only those who could tolerate a liquid diet or a KD, which may have led to an overestimation of treatment effects relative to the general population. The limited sample size did not allow us to assess characteristics that might have predicted better responses or to investigate whether there were sex differences. Although there were 7 females of 19 (37%) in the KD group and only 3 females of 14 (21%) in the SA group, the group difference in sex distribution was not significant. The limited sample size, as well as our standard 1H-MRS point-resolved spectroscopy (PRESS) sequence, might have reduced the sensitivity of 1H-MRS in detecting BHB. In addition, in our study, the first MRI was performed 4.0 ± 1.7 (SD) days after admission (no group differences) at a time when the KD participants were already in mild ketosis (blood BHB = 1.6 ± 1.5 mM). Therefore, an absolute baseline is missing. Historical data on diet before entering the treatment regimen were not collected, thus we do not know whether past diet history influenced the results of our study. However, this is unlikely because the variables of interest did not differ at baseline (i.e., benzodiazepine use, CIWA-Ar scores, or alcohol craving) and showed no association with the body mass index (BMI). We are therefore confident that the effects were due to the dietary intervention rather than past diet histories. Although we had hypothesized that KD would be associated with improved cognitive performance and mood compared to SA, we could not corroborate this, which could reflect inadequate sensitivity for the cognitive tests that were selected, limited statistical power, or lack of effect. Our preclinical study showed effects of a history of KD on alcohol self-administration, which was different from our human study in that the rats were not in ketosis when outcome measures were assessed. The reason for this design was that rats on a KD were unable to tolerate alcohol drinking during alcohol vapor exposure, likely due to the fact that they had much higher BAL (>5-fold) than rodents on the chow diet (fig. S1). To our knowledge, the marked increase in BAL has not been previously reported but could underlie web reports of alcohol intolerance when on a KD (e.g., www.menshealth.com/nutrition/a20046779/keto-diet-side-effects-alcohol/). Our preclinical finding that a history of KD decreased alcohol consumption, however, merits further investigation for we do not have clinical data on long-lasting effects of KD on alcohol abstinence. Although preclinical studies have not been reported on long-lasting effects of KD on brain glucose metabolism, there are reports of long-lasting changes in brain structure and neurochemistry following a KD, including elevated glutamate and glutamine (36). Thus, future studies are needed to assess whether there are long-lasting changes in brain glucose metabolism that persist after KD discontinuation. Last, our preclinical study was performed in male rats only, which is also a limitation.
Our study has strong clinical implications as it offers a potential new treatment for AUD that is diet based. Although not all patients with AUD were able to comply with the predominantly liquid diet protocol (see table S1 for details), it is possible that the use of ketone supplements or a less stringent diet (e.g., 3:1 rather than 4:1 ratio of KD and/or 100% solid diet) might make it easier for patients to comply. Translating this protocol into an outpatient clinic will make it more accessible as a treatment option that does not require hospitalization. However, more research is needed to determine whether there is any risk of hepatoxicity with the KD in AUD. In our study, we excluded patients with current or past liver disease, and liver function, platelet count, and coagulation were monitored weekly and showed no evidence that KD elevated hepatic or coagulation risks in patients with AUD (fig. S8). Moreover, the promising results obtained from the preclinical model of alcohol dependence that showed the successful reduction in alcohol intake 6 to 7 weeks after KD termination hint at the possibility that the KD might have long-lasting beneficial effects, which merits further clinical corroboration. In sum, this study documents (i) beneficial effects from a KD in the management of acute alcohol withdrawal in AUD and (ii) long-lasting protective effects from KD on alcohol self-administration in alcohol-dependent rats.
MATERIALS AND METHODS
Clinical studies
See Fig. 7 for the overview of human study procedures.
Fig. 7. Overview of human study procedures.
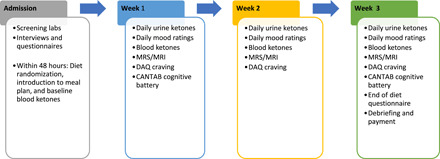
Participants and screening
Within 2 days of admission at the National Institute on Alcohol Abuse and Alcoholism inpatient unit, n = 53 inpatients were randomized into the study while receiving treatment as usual. Thirty-three patients completed 3 weeks of diet intervention (n = 19 KD, n = 14 SA), and 20 patients withdrew. There were no significant group demographic or clinical differences between those that completed the study and those that withdrew (χ2 = 0.82, P = 0.37; see table S1 for details on study withdrawal). There were also no group differences in demographics, clinical characteristics, or dietary caloric intake on the diet intervention between diet groups (Table 1), although BMI tended to be higher in SA than KD. However, exploratory analyses showed that BMI did not correlate with any variable of interest (e.g., benzodiazepine use, withdrawal scores, alcohol craving and wanting, and MRS metabolites).
All patients were medically screened to exclude ferromagnetic implants, major medical problems, chronic use of psychoactive medications, neurological problems, liver disease, kidney stones, head trauma, or milk or soy allergy and current diagnosis of a substance use disorder (other than AUD or nicotine) that was severe or any other major psychiatric disorder that needed treatment for longer than a month in the past year as assessed by the Structured Clinical Interview for the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) or DSM-5 (56). Women were neither pregnant nor breastfeeding. AUD participants were admitted for detoxification and had at least 5 years of history of heavy drinking (i.e., the Substance Abuse and Mental Health Services Administration (SAMHSA) criteria for heavy drinking: for men, five or more drinks/day on at least five different days per month, and for women, four or more drinks/day on at least five different days per month). Alcohol had to be specified as the preferred drug. All participants were free of psychoactive medications within 24 hours of study procedures (except benzodiazepines as needed for detoxification) and had a negative urine drug screen on days of testing [except for benzodiazepines or 11-Nor-9-carboxy-Δ9-tetrahydrocannabinol (THC-COOH)]. If THC-COOH in urine was positive, then a saliva drug screen (Dräger DrugTest 5000; Draeger Safety Diagnostics, Lübeck, Germany) was used to rule out recent cannabis use on study days (cutoff, 5 μg/liter). Liver function, platelet count, and coagulation were monitored weekly, and patients were removed if values were >3× the upper bound limit. Moreover, coagulation and platelet counts were monitored weekly. Patients provided written informed consent to participate in the study, which was approved by the Institutional Review Board at the National Institutes of Health (Combined Neurosciences White Panel). Participants were scanned between October 2017 and February 2020. The study was registered at ClinicalTrials.gov (NCT03255031).
KD randomization
Patients were randomized to a meal plan of a KD or SA at the time they signed the consent (within 2 days of admission) for 3 weeks. KD was randomized within 0.95 ± 0.23 SD days after admission and SA within 0.78 ± 0.42 SD days, which did not significantly differ between groups (t = 1.3, P = 0.21). The randomization scheme was preset at a 3:2 KD:SA ratio for every five consecutive enrollments to account for an expected higher dropout for the KD group. The KD and SA meals were eucaloric. The KD provided a classic 4:1 ratio of grams of fat:grams of carbohydrate and protein (i.e., 80% fat, 15% protein, and 5% carbohydrates). The SA corresponded to 50% calories from carbohydrates, 15% protein, and 35% fat. For each meal at breakfast, lunch, and dinner, the diets consisted of a shake (either KD or SA) that provided 75% of meal calories and a ketogenic solid snack (e.g., scrambled egg, yogurt with nuts, salad, chicken salad, and broth) that provided 25% of meal calories, which ensured double blinding. Patients were allowed to drink water ad libitum and were given the option to drink tea or coffee with or without stevia and/or diet ginger ale with their meals. Meals were provided from the Nutrition Department’s Metabolic Kitchen with all foods and beverages weighed on a gram scale. Initial calorie provision was determined using Mifflin St. Jeor equation with standard activity factor with intention to maintain stable weight. Daily weight was measured blindly to the participant’s diet, and if weight loss greater than 1 kg was detected, then daily calorie provision was increased until baseline weight was achieved.
Ketone testing and study compliance
To measure the effectiveness of KD in increasing ketone bodies, ketones in urine (AcAc) were measured every morning before breakfast (Ketostix, Bayer Vital GmbH, Leverkusen, Germany). Blood measures of BHB were assessed on a weekly basis using a finger prick (Precision Xtra). To ensure subject blinding, the reading and recording of these measures were performed by an independent research nurse and in the absence of the participant.
Urine and blood results were also used to assess diet compliance. If, after the first 5 days of KD initiation, a participant showed three negative urine tests for ketone bodies, then this participant was removed from the study based on an assumption of dietary noncompliance. In this case, the independent person who monitored daily urine testing informed the study staff about the compliance breach. In addition, if after the first 3 days of diet initiation (KD or SA) a participant reported noncompliance for 4 days, the participant was removed from the study (n = 3 KD; see table S1).
Alcohol withdrawal and benzodiazepine use
All patients were assessed with the CIWA-Ar (29) at admission and then approximately every 2 hours until withdrawal ceased. If the CIWA-Ar scores were ≥8 on admission, then patients were provided with benzodiazepines (oxazepam or diazepam) to treat withdrawal symptoms. The CIWA-Ar assessments and benzodiazepine prescriptions were performed by medical staff blind to the study randomization. At admission, n = 16 KD patients received benzodiazepines (3 diazepam, 13 oxazepam, and 3 none) and n = 10 SA patients received oxazepam (0 diazepam and 4 none). To compare cumulative doses, diazepam doses were multiplied by 2 for the relative dose of oxazepam, as per https://clincalc.com/Benzodiazepine. We also report a 1:3 conversion as per https://globalrph.com/medcalcs/benzodiazepine-converter-dosage-conversions. Neither CIWA-Ar scores nor benzodiazepine dose in mg at admission differed between groups (Table 1).
Questionnaires, craving, mood ratings, and neuropsychological testing
Participants completed the Wechsler Abbreviated Scale of Intelligence, Second Edition (WASI-II) subtests Matrix Reasoning and Vocabulary as a proxy for general intelligence (57), the Timeline Followback (TLFB) to assess daily alcohol consumption in 90 days before the study (58), the Lifetime Drinking History (LDH) to assess lifetime alcohol consumption (59), the Alcohol Dependence Scale (ADS) to assess severity of dependence (60), and the Fagerström test was used as a measure of nicotine dependence. On days 2, 9, 16, and 23 after admission, participants completed the comprehensive psychopathological rating scale (61), which provides scores for the Montgomery-Åsberg Depression Rating Scale (MADRS) (62) and the brief scale for anxiety (63). MADRS depression ratings at baseline ranged from 2 to 31, indicating no (<7), mild (7 to 19), and moderate (30 to 34) depression. Scores did not significantly differ between groups.
On a weekly basis, participants rated their alcohol craving on the DAQ (64). Before and after breakfast (7:30 a.m.) and dinner (5 p.m.), participants were asked to rate how alert, tired, hungry, friendly, happy, sad, anxious, irritable, social, confused, bored, comfortable, energetic, craving sugar, and craving alcohol they felt, from 1 to 10 (not at all to extremely). Two DAQ craving measures were missing in week 3, leaving 17 KD for the repeated-measure ANOVA. After study participation and before transitioning into a regular diet, patients filled out a questionnaire regarding their diet expectations [i.e., “Which diet did you think you were on?” (KD/SA), “How pleasant was the diet?” (0 to 10), “Do you think your diet improved your health? (0 to 10)].
Participants performed a Cambridge neuropsychological test automated battery (CANTAB) in weeks 1 and 3 with the following tasks: Emotion Recognition, Task Pattern Recognition Memory, Cambridge Gambling Task, Reaction Time, Stockings of Cambridge, Spatial Working Memory, Stop Signal Task, Intra-Extra Dimensional Set Shift, Spatial Span, and Affective Go/No-Go. Details about the tasks can be found in table S4.
MRI acquisition and preprocessing
Patients underwent MRI on a 3.0T Magnetom Prisma scanner (Siemens Medical Solutions USA Inc., Malvern, PA) equipped with a 32-channel head coil. T1-weighted three-dimensional magnetization-prepared rapid gradient-echo MP-RAGE [repetition time/echo time (TR/TE) = 2200/4.25 ms; flip angle (FA) = 9°, 1-mm isotropic resolution] and T2-weighted multislice spin-echo (TR/TE = 8000/72 ms; 1.1-mm in-plane resolution; 94 slices, 1.7-mm slice thickness; matrix = 192) pulse sequences were used to acquire high-resolution anatomical brain images. One participant did not complete session 3 due to scheduling problems. For functional MRI, a 32-channel head coil and a standard echo planar imaging sequence were used: sequential interleaved acquisition, repetition time of 1.5 s, echo time of 30 ms, flip angle α = 70°, 64 × 64 pixels in-plane resolution, 36 slices, slice thickness of 4 mm, voxel dimensions of 3 mm by 3 mm by 4 mm, and field of view of 192 mm × 192 mm. Stimuli were presented on a black background under dimmed room lighting using a liquid-crystal display screen (BOLDscreen 32, Cambridge Research Systems, UK).
fMRI cue reactivity
A total of 40 alcohol, 40 appetitive food, and 40 neutral images were randomly presented in an event-related design using E-Prime software [see (65) for details]. There were three runs showing 40 cues that included all three categories of cues (alcohol, food, and neutral). Cues were presented at 750 ms. Pictures were selected from the International Affective Picture System and from in-house picture libraries. To ensure that participants were paying attention, they were instructed to press a button if they saw a bicycle. Total task duration was 13 min and 30 s. After completion of the MRI sessions, participants were asked to rate the cues for their subjective valence [“How negative/positive do you find the picture?” from −3 to 3 (very negative to very positive)] and wanting [“How much do you want to consume this right now?” from 0 to 6 (not at all to extremely)], on a seven-point Likert scale.
The fMRI data analysis was performed with SPM8 (Wellcome Department of Cognitive Neurology, London, UK). During preprocessing, scans were slice-time corrected, spatially realigned, coregistered to the T1 structural images, and normalized to the MNI template with a final 2-mm isotropic resolution. Smoothing was performed with a 6-mm full width at half-maximum Gaussian kernel.
1H magnetic resonance spectroscopy
Localized proton 1H-MRS was performed in an ROI located in the dACC (2 cm × 2 cm × 2 cm). A PRESS pulse sequence (TE/TR = 30/3000 s, 64 averages) was used to acquire water-suppressed spectra. Spectral fitting of the 1H-MRS datasets was carried with the LCModel program (66) to determine absolute and relative metabolite concentrations for total Cr, total Cho (phosphocholine and glycerophosphocholine), mI, Glu, and Glx, and we used a modified basis set for ketone bodies (67). The results from LCModel spectral analysis were inspected for nonrandom residuals as well as baseline fitting. Spectra with signal-to-noise ratio < 15 and linewidth > 0.1 ppm were excluded from further analyses. A Cramér-Rao lower bound of 20% for each individual peak was used as a quality criterion (66). On the basis of these criteria, one participant per session was removed for all metabolites (i.e., week 1: n = 1 KD; week 2: n = 1 SA; week 3: n = 1 SD). Means ± SD signal-to-noise ratios did not differ between groups.
Statistical analysis
For all ketone, behavioral, and 1H-MRS measures, we performed mixed ANOVAs with group (KD/SA) as between-group factor and time as within-subjects factor using SPSS. We used the Greenhouse-Geisser correction if the Mauchly’s test of sphericity was significant. Effect sizes are reported in η2. Post hoc t tests were performed with a significance threshold of α < 0.05, and t values were corrected if Levene’s test for equality of variance was P < 0.05. χ2 tests were used for sample distributions.
For fMRI, three fMRI regressors, food, alcohol, and neutral events, were created and then convolved with the hemodynamic response function with default temporal filtering of 128 s. Six realignment parameters were included as regressors of no interest. The following contrasts were calculated at the single-subject level: alcohol > neutral, food > neutral. At the second level, a repeated measures factorial design was used for the contrast alcohol > neutral and food > neutral, with group (KD/SA) as between-subject factors and time (weeks 1, 2, and 3) as within-subject factors. We performed whole-brain analyses and tested (i) the main effects of alcohol > neutral and food > neutral and (ii) the interaction effects of group × time alcohol > neutral and food > neutral. For whole-brain analyses, we used a combined threshold of P < 0.005 uncorrected at the voxel level, with a family-wise error (FWE)–corrected threshold of PFWE < 0.05 at the cluster level (minimum cluster size k > 50). Coordinates are reported in MNI space. For the dACC ROI, we used SVC using the coordinates of a previously described dACC ROI [0, 24, 26] (68) with a box of 2 cm by 2 cm by 2 cm representative of the dACC 1H-MRS voxel. We performed exploratory Pearson’s correlations among 1H-MRS metabolites, BOLD responses, and behavioral measures, both in week 1 and for the differences between weeks 3 and 1.
Preclinical studies
See Fig. 8 for the overview of rat study procedures.
Fig. 8. Overview of rat study procedures.
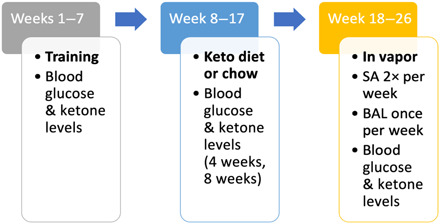
SA, alcohol self-administration session; BAL, blood alcohol level.
Rats
Thirty-six male Wistar rats (Charles River Laboratories), weighing 470 g on average, were group-housed three per cage with water and standard rodent chow (Teklad diet, Envigo, Madison, WI) available ad libitum, except during operant testing and when noted. Rats were kept in a temperature (22° ± 2°C)– and humidity (50 to 60%)–controlled environment on a reverse 12/12-hour light/dark cycle (lights on at 6:00). All of the procedures were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the National Institute on Drug Abuse, Intramural Research Program, Animal Care and Use Committee.
Alcohol self-administration
Rats were trained on an operant self-administration task that involved a concurrent, two-lever choice that delivered 0.1 ml of 10% alcohol (w/v) paired with a cue light or water (no cue light), as previously reported (27). We used a fixed ratio one schedule of reinforcement, in which each lever press was reinforced with alcohol or water. They were typically tested twice a week in 30-min sessions. Once stable operant responding was reached, the rats were split into two groups either to receive a high-fat, low-carbohydrate KD or to remain on the control chow diet. During the diet phase of the experiment, four baseline self-administration sessions were performed during the last 2 weeks of diet exposure.
Diets
All of the rats were initially fed with standard laboratory chow ad libitum. After the rats had been trained on the operant task described above, they were split into two groups. One group remained on the control chow diet (Teklad, Envigo, Madison, WI), the other group received a high-fat, low-carbohydrate KD (Bio-Serv, Flemington, NJ). The caloric content of the chow diet was as follows: 30% of calories from protein, 13% from fat, and 57% from carbohydrates. In the KD, 5% of calories was from protein, 2% was from carbohydrates, and 93% was from fat. Rats were fed their respective diets for 9 weeks, during which time body weight was measured weekly and blood glucose and blood ketone levels were measured after 4 and 8 weeks on the diets.
Alcohol vapor exposure
Alcohol vapor exposure was conducted as previously reported (27). Following 9 weeks of chow or KD exposure, all rats were placed back on the chow diet and split in two groups. Half of the rats out of each diet group were placed into alcohol vapor chambers to produce alcohol dependence. The other half of rats were equally treated throughout the experiment, but they were exposed to air without alcohol. Thus, we had four groups of rats: alcohol vapor–chow (n = 9), alcohol vapor–KD (n = 9), air-chow (n = 9), and air-KD (n = 9). All groups were exposed to the alcohol/air vapor for 8 weeks. We were not able to test the effect of continuing the KD on alcohol drinking during alcohol vapor exposure because the rats on the KD were more sensitive to the sedative effects of alcohol (fig. S1). This effect was unlikely due to differences in body weight because the KD rats that transitioned back to chow before alcohol vapor exposure did not show signs of increased alcohol-induced sedation. Moreover, BAL did not differ between chow and KD rats.
During alcohol vapor exposure, rats were housed in routine husbandry plastic cages while being exposed to alcohol vapor in the air supply. The cages had hardwood chip, chow diet, food, and water ad libitum identical to rats exposed to air. The rats underwent daily cycles of intoxication and withdrawal (14 hours on/10 hours off). Over several days, the concentration of alcohol vapor in the chambers was gradually increased. BALs were accurately adjusted by changing the concentration of alcohol in the chambers. Blood was collected as described below for weekly BAL measurements. Operant alcohol self-administration experiments were conducted twice a week, 6 to 8 hours into acute withdrawal. After the alcohol self-administration sessions, the rats were returned to the vapor chambers. Air-exposed rats were not exposed to alcohol vapor but were concomitantly tested with alcohol vapor–exposed rats.
Blood collection
Rats were lightly restrained, and the tip of the tail was nicked to collect a few drops of blood. Blood glucose and ketone levels were analyzed using a glucometer (Tyson Bioresearch Inc., Zhunan, Taiwan) and BHB ketone levels with the Precision Xtra Blood Ketone Monitoring System (Abbott, Alameda, CA). Measurements were taken before diet exposure, after 4 and 8 weeks of diet exposure, and weekly during alcohol vapor or air exposure. BALs were analyzed using an Analox Alcohol Analyzer instrument (Analox Technologies North America, Toronto, Ontario, Canada) at the end of the 14 hours of alcohol vapor exposure, when peak alcohol vapor levels are achieved.
Statistical analysis
Blood glucose, ketones, and alcohol levels were analyzed with a repeated-measure two-way ANOVA with diet as the between-subjects factor and time as the within-subjects factor. Alcohol self-administration data were analyzed with a two-way ANOVA with group (alcohol-chow, alcohol-KD, air-chow, and air-KD) as the between-subject factor and session as the within-subjects factor. The Tukey’s multiple comparisons test was used for post hoc comparisons. Statistical significance was set at P < 0.05. We calculated η2 to determine effect sizes.
Acknowledgments
We thank K. Torres, M. McFarland, L. Talagala, M.-V. Yonga, Y. Horneffer, D. George, T. Vinson, D. Goldman, L. Leggio, J. Burns, C. K. Liu, A. Zehra, C. Freeman, C. Wong, S. Kim, S. B. Demiral, T. Wu, D. Picchioni, T. Ernst, L. Chang, J. Gomez, M. Michaelides, P. Joseph, V. Darcey, T. King, and R. Veech for contributions and discussions. We also thank C. Smith for technical assistance in the preclinical studies. Funding: This work was accomplished with support from the National Institute on Alcohol Abuse and Alcoholism (Y1AA-3009) and the NIDA-IRP. Author contributions: C.E.W. and N.D.V. designed the clinical studies, and C.E.W., N.D.V., J.-W.v.d.V., G.-J.W., P.M., E.S.-K., D.S.K., D.E.F., K.L.M., C.L.B., R.Z., K.H., S.A.T., S.Y., M.S., D.T., M.C.C., A.F.-J., H.B., and N.D. participated in the collection, analyses, and interpretation of clinical findings. L.F.V., J.C.M.V., S.K.E., and G.F.K. designed the preclinical studies and analyzed the results. C.E.W., N.D.V., and L.F.V. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabf6780/DC1
REFERENCES AND NOTES
- 1.Rehm J., Imtiaz S., A narrative review of alcohol consumption as a risk factor for global burden of disease. Subst. Abuse Treat. Prev. Policy 11, 37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutwinski S., Schreiter S., Priller J., Henssler J., Wiers C. E., Heinz A., Drink and think: Impact of alcohol on cognitive functions and dementia - evidence of dose-related effects. Pharmacopsychiatry 51, 136–143 (2018). [DOI] [PubMed] [Google Scholar]
- 3.de la Monte S. M., Kril J. J., Human alcohol-related neuropathology. Acta Neuropathol. 127, 71–90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volkow N. D., Wiers C. E., Shokri-Kojori E., Tomasi D., Wang G.-J., Baler R., Neurochemical and metabolic effects of acute and chronic alcohol in the human brain: Studies with positron emission tomography. Neuropharmacology 122, 175–188 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Zehra A., Lindgren E., Wiers C. E., Freeman C., Miller G., Ramirez V., Shokri-Kojori E., Wang G.-J., Talagala L., Tomasi D., Volkow N. D., Neural correlates of visual attention in alcohol use disorder. Drug Alcohol Depend. 194, 430–437 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Tomasi D. G., Wiers C. E., Shokri-Kojori E., Zehra A., Ramirez V., Freeman C., Burns J., Liu C. K., Manza P., Kim S. W., Wang G.-J., Volkow N. D., Association between reduced brain glucose metabolism and cortical thickness in alcoholics: Evidence of neurotoxicity. Int. J. Neuropsychopharmacol. 22, 548–559 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muncie H. L. Jr., Yasinian Y., Oge L., Outpatient management of alcohol withdrawal syndrome. Am. Fam. Physician 88, 589–595 (2013). [PubMed] [Google Scholar]
- 8.Volkow N. D., Kim S. W., Wang G. J., Alexoff D., Logan J., Muench L., Shea C., Telang F., Fowler J. S., Wong C., Benveniste H., Tomasi D., Acute alcohol intoxication decreases glucose metabolism but increases acetate uptake in the human brain. Neuroimage 64, 277–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang L., Gulanski B. I., De Feyter H. M., Weinzimer S. A., Pittman B., Guidone E., Koretski J., Harman S., Petrakis I. L., Krystal J. H., Mason G. F., Increased brain uptake and oxidation of acetate in heavy drinkers. J. Clin. Invest. 123, 1605–1614 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J., Du H., Jiang L., Ma X., de Graaf R. A., Behar K. L., Mason G. F., Oxidation of ethanol in the rat brain and effects associated with chronic ethanol exposure. Proc. Natl. Acad. Sci. U.S.A. 110, 14444–14449 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Courchesne-Loyer A., Croteau E., Castellano C.-A., St-Pierre V., Hennebelle M., Cunnane S. C., Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow Metab. 37, 2485–2493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cervenka M. C., Hocker S., Koenig M., Bar B., Henry-Barron B., Kossoff E. H., Hartman A. L., Probasco J. C., Benavides D. R., Venkatesan A., Hagen E. C., Dittrich D., Stern T., Radzik B., Depew M., Caserta F. M., Nyquist P., Kaplan P. W., Geocadin R. G., Phase I/II multicenter ketogenic diet study for adult superrefractory status epilepticus. Neurology 88, 938–943 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castro A. I., Gomez-Arbelaez D., Crujeiras A. B., Granero R., Aguera Z., Jimenez-Murcia S., Sajoux I., Lopez-Jaramillo P., Fernandez-Aranda F., Casanueva F. F., Effect of a very low-calorie ketogenic diet on food and alcohol cravings, physical and sexual activity, sleep disturbances, and quality of life in obese patients. Nutrients 10, 1348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derr R. F., Draves K., Derr M., Abatement by acetate of an ethanol withdrawal syndrome. Life Sci. 29, 1787–1790 (1981). [DOI] [PubMed] [Google Scholar]
- 15.Dencker D., Molander A., Thomsen M., Schlumberger C., Wortwein G., Weikop P., Benveniste H., Volkow N. D., Fink-Jensen A., Ketogenic diet suppresses alcohol withdrawal syndrome in rats. Alcohol. Clin. Exp. Res. 42, 270–277 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Rothman D. L., De Feyter H. M., de Graaf R. A., Mason G. F., Behar K. L., 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed. 24, 943–957 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pawlosky R. J., Kashiwaya Y., King M. T., Veech R. L., A dietary ketone ester normalizes abnormal behavior in a mouse model of Alzheimer’s disease. Int. J. Mol. Sci. 21, 1044 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamanashi T., Iwata M., Kamiya N., Tsunetomi K., Kajitani N., Wada N., Iitsuka T., Yamauchi T., Miura A., Pu S., Shirayama Y., Watanabe K., Duman R. S., Kaneko K., Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci. Rep. 7, 7677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein R. Z., Alia-Klein N., Tomasi D., Carrillo J. H., Maloney T., Woicik P. A., Wang R., Telang F., Volkow N. D., Anterior cingulate cortex hypoactivations to an emotionally salient task in cocaine addiction. Proc. Natl. Acad. Sci. U.S.A. 106, 9453–9458 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein R. Z., Volkow N. D., Dysfunction of the prefrontal cortex in addiction: Neuroimaging findings and clinical implications. Nat. Rev. Neurosci. 12, 652–669 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck A., Wüstenberg T., Genauck A., Wrase J., Schlagenhauf F., Smolka M.-c. N., Mann K., Heinz A., Effect of brain structure, brain function, and brain connectivity on relapse in alcohol-dependent patients. Arch. Gen. Psychiatry 69, 842–852 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Jasinska A. J., Stein E. A., Kaiser J., Naumer M. J., Yalachkov Y., Factors modulating neural reactivity to drug cues in addiction: A survey of human neuroimaging studies. Neurosci. Biobehav. Rev. 38, 1–16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grüsser S. M., Wrase J., Klein S., Hermann D., Smolka M. N., Ruf M., Weber-Fahr W., Flor H., Mann K., Braus D. F., Heinz A., Cue-induced activation of the striatum and medial prefrontal cortex is associated with subsequent relapse in abstinent alcoholics. Psychopharmacology 175, 296–302 (2004). [DOI] [PubMed] [Google Scholar]
- 24.De Ridder D., Vanneste S., Kovacs S., Sunaert S., Dom G., Transient alcohol craving suppression by rTMS of dorsal anterior cingulate: An fMRI and LORETA EEG study. Neurosci. Lett. 496, 5–10 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Shokri-Kojori E., Tomasi D., Alipanahi B., Wiers C. E., Wang G.-J., Volkow N. D., Correspondence between cerebral glucose metabolism and BOLD reveals relative power and cost in human brain. Nat. Commun. 10, 690 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prisciandaro J. J., Schacht J. P., Prescot A. P., Brenner H. M., Renshaw P. F., Brown T. R., Anton R. F., Intraindividual changes in brain GABA, glutamate, and glutamine during monitored abstinence from alcohol in treatment-naive individuals with alcohol use disorder. Addict. Biol. 25, e12810 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vendruscolo L. F., Estey D., Goodell V., Macshane L. G., Logrip M. L., Schlosburg J. E., McGinn M. A., Zamora-Martinez E. R., Belanoff J. K., Hunt H. J., Sanna P. P., George O., Koob G. F., Edwards S., Mason B. J., Glucocorticoid receptor antagonism decreases alcohol seeking in alcohol-dependent individuals. J. Clin. Invest. 125, 3193–3197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aoun E. G., Jimenez V. A., Vendruscolo L. F., Walter N. A. R., Barbier E., Ferrulli A., Haass-Koffler C. L., Darakjian P., Lee M. R., Addolorato G., Heilig M., Hitzemann R., Koob G. F., Grant K. A., Leggio L., A relationship between the aldosterone-mineralocorticoid receptor pathway and alcohol drinking: Preliminary translational findings across rats, monkeys and humans. Mol. Psychiatry 23, 1466–1473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan J. T., Sykora K., Schneiderman J., Naranjo C. A., Sellers E. M., Assessment of alcohol withdrawal: The revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br. J. Addict. 84, 1353–1357 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Simões-Franklin C., Hester R., Shpaner M., Foxe J. J., Garavan H., Executive function and error detection: The effect of motivation on cingulate and ventral striatum activity. Hum. Brain Mapp. 31, 458–469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pascual J. M., Campistol J., Gil-Nagel A., Epilepsy in inherited metabolic disorders. Neurologist 14, S2–S14 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Long D., Long B., Koyfman A., The emergency medicine management of severe alcohol withdrawal. Am. J. Emerg. Med. 35, 1005–1011 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Lutas A., Yellen G., The ketogenic diet: Metabolic influences on brain excitability and epilepsy. Trends Neurosci. 36, 32–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haslam R. J., Krebs H. A., The metabolism of glutamate in homogenates and slices of brain cortex. Biochem. J. 88, 566–578 (1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jakkamsetti V., Marin-Valencia I., Ma Q., Good L. B., Terrill T., Rajasekaran K., Pichumani K., Khemtong C., Hooshyar M. A., Sundarrajan C., Patel M. S., Bachoo R. M., Malloy C. R., Pascual J. M., Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. 11, eaan0457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gzieło K., Janeczko K., Węglarz W., Jasiński K., Kłodowski K., Setkowicz Z., MRI spectroscopic and tractography studies indicate consequences of long-term ketogenic diet. Brain Struct. Funct. 225, 2077–2089 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonen O. M., Moffat B. A., Desmond P. M., Lui E., Kwan P., O’Brien T. J., Seven-tesla quantitative magnetic resonance spectroscopy of glutamate, γ-aminobutyric acid, and glutathione in the posterior cingulate cortex/precuneus in patients with epilepsy. Epilepsia 61, 2785–2794 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Pflanz N. C., Daszkowski A. W., James K. A., Mihic S. J., Ketone body modulation of ligand-gated ion channels. Neuropharmacology 148, 21–30 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Wiers C. E., Cunningham S. I., Tomasi D. G., Ernst T., Chang L., Shokri-Kojori E., Wang G.-J., Volkow N. D., Elevated thalamic glutamate levels and reduced water diffusivity in alcohol use disorder: Association with impulsivity. Psychiatry Res. Neuroimaging 305, 111185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mon A., Durazzo T. C., Meyerhoff D. J., Glutamate, GABA, and other cortical metabolite concentrations during early abstinence from alcohol and their associations with neurocognitive changes. Drug Alcohol Depend. 125, 27–36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mirijello A., D’Angelo C., Ferrulli A., Vassallo G., Antonelli M., Caputo F., Leggio L., Gasbarrini A., Addolorato G., Identification and management of alcohol withdrawal syndrome. Drugs 75, 353–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitman B. A., Knapp D. J., Werner D. F., Crews F. T., Breese G. R., The cytokine mRNA increase induced by withdrawal from chronic ethanol in the sterile environment of brain is mediated by CRF and HMGB1 release. Alcohol. Clin. Exp. Res. 37, 2086–2097 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quarantelli M., MRI/MRS in neuroinflammation: Methodology and applications. Clin Transl Imaging 3, 475–489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brooks W. M., Stidley C. A., Petropoulos H., Jung R. E., Weers D. C., Friedman S. D., Barlow M. A., Sibbitt W. L. Jr., Yeo R. A., Metabolic and cognitive response to human traumatic brain injury: A quantitative proton magnetic resonance study. J. Neurotrauma 17, 629–640 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Schuhmann M. U., Stiller D., Skardelly M., Bernarding J., Klinge P. M., Samii A., Samii M., Brinker T., Metabolic changes in the vicinity of brain contusions: A proton magnetic resonance spectroscopy and histology study. J. Neurotrauma 20, 725–743 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Schweinsburg B. C., Taylor M. J., Alhassoon O. M., Videen J. S., Brown G. G., Patterson T. L., Berger F., Grant I., Chemical pathology in brain white matter of recently detoxified alcoholics: A 1H magnetic resonance spectroscopy investigation of alcohol-associated frontal lobe injury. Alcohol. Clin. Exp. Res. 25, 924–934 (2001). [PubMed] [Google Scholar]
- 47.Meyerhoff D. J., Blumenfeld R., Truran D., Lindgren J., Flenniken D., Cardenas V., Chao L. L., Rothlind J., Studholme C., Weiner M. W., Effects of heavy drinking, binge drinking, and family history of alcoholism on regional brain metabolites. Alcohol. Clin. Exp. Res. 28, 650–661 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee E., Jang D.-P., Kim J.-J., An S. K., Park S., Kim I.-Y., Kim S. I., Yoon K.-J., Namkoong K., Alteration of brain metabolites in young alcoholics without structural changes. Neuroreport 18, 1511–1514 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Parks M. H., Dawant B. M., Riddle W. R., Hartmann S. L., Dietrich M. S., Nickel M. K., Price R. R., Martin P. R., Longitudinal brain metabolic characterization of chronic alcoholics with proton magnetic resonance spectroscopy. Alcohol. Clin. Exp. Res. 26, 1368–1380 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Kim S. W., Wiers C. E., Tyler R., Shokri-Kojori E., Jang Y. J., Zehra A., Freeman C., Ramirez V., Lindgren E., Miller G., Cabrera E. A., Stodden T., Guo M., Demiral Ş. B., Diazgranados N., Park L., Liow J.-S., Pike V., Morse C., Vendruscolo L. F., Innis R. B., Koob G. F., Tomasi D., Wang G.-J., Volkow N. D., Influence of alcoholism and cholesterol on TSPO binding in brain: PET [11C]PBR28 studies in humans and rodents. Neuropsychopharmacology. 43, 1832–1839 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Carvalho L. M., Wiers C. E., Sun H., Wang G.-J., Volkow N. D., Increased transcription of TSPO, HDAC2, and HDAC6 in the amygdala of males with alcohol use disorder. Brain and behavior 11, e01961 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyler R. E., Kim S. W., Guo M., Jang Y. J., Damadzic R., Stodden T., Vendruscolo L. F., Koob G. F., Wang G.-J., Wiers C. E., Volkow N. D., Detecting neuroinflammation in the brain following chronic alcohol exposure in rats: A comparison between in vivo and in vitro TSPO radioligand binding. Eur. J. Neurosci. 50, 1831–1842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Araujo I. E., Oliveira-Maia A. J., Sotnikova T. D., Gainetdinov R. R., Caron M. G., Nicolelis M. A., Simon S. A., Food reward in the absence of taste receptor signaling. Neuron 57, 930–941 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Rejeski W. J., Blumenthal T. D., Miller G. D., Lobe M., Davis C., Brown L., State craving, food availability, and reactivity to preferred snack foods. Appetite 54, 77–83 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinson L., Gray G. E., Psychopharmacology: Number needed to treat: An underused measure of treatment effect. Psychiatr. Serv. 54, 145–146 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Diagnostic and Statistical Manual of Mental Disorders (American Psychiatric Association, ed. 5, 2013). [Google Scholar]
- 57.D. Wechsler, Wechsler Abbreviated Scale of Intelligence (The Psychological Corporation, 1999). [Google Scholar]
- 58.L. C. Sobell, M. B. Sobell (Addiction Research Foundation, 1996).
- 59.Skinner H. A., Sheu W. J., Reliability of alcohol use indices. The lifetime drinking history and the MAST. J. Stud. Alcohol 43, 1157–1170 (1982). [DOI] [PubMed] [Google Scholar]
- 60.Skinner H. A., Allen B. A., Alcohol dependence syndrome: Measurement and validation. J. Abnorm. Psychol. 91, 199–209 (1982). [DOI] [PubMed] [Google Scholar]
- 61.Asberg M., Montgomery S. A., Perris C., Schalling D., Sedvall G., A comprehensive psychopathological rating scale. Acta Psychiatr. Scand. Suppl. , 5–27 (1978). [DOI] [PubMed] [Google Scholar]
- 62.Montgomery S. A., Åsberg M., A new depression scale designed to be sensitive to change. Br. J. Psychiatry 134, 382–389 (1979). [DOI] [PubMed] [Google Scholar]
- 63.Tyrer P., Owen R. T., Cicchetti D. V., The brief scale for anxiety: A subdivision of the comprehensive psychopathological rating scale. J. Neurol. Neurosurg. Psychiatry 47, 970–975 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love A., James D., Willner P., A comparison of two alcohol craving questionnaires. Addiction 93, 1091–1102 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Wiers C. E., Zhao J., Manza P., Murani K., Ramirez V., Zehra A., Freeman C., Yuan K., Wang G. J., Demiral S. B., Childress A. R., Tomasi D., Volkow N. D., Conscious and unconscious brain responses to food and cocaine cues. Brain Imaging Behav. 15, 311–319 (2021). [DOI] [PubMed] [Google Scholar]
- 66.Provencher S. W., Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med. 30, 672–679 (1993). [DOI] [PubMed] [Google Scholar]
- 67.Cecil K. M., Mulkey S. B., Ou X., Glasier C. M., Brain ketones detected by proton magnetic resonance spectroscopy in an infant with Ohtahara syndrome treated with ketogenic diet. Pediatr. Radiol. 45, 133–137 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Götting F. N., Borchardt V., Demenescu L. R., Teckentrup V., Dinica K., Lord A. R., Rohe T., Hausdörfer D. I., Li M., Metzger C. D., Walter M., Higher interference susceptibility in reaction time task is accompanied by weakened functional dissociation between salience and default mode network. Neurosci. Lett. 649, 34–40 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabf6780/DC1