

Abstract
Protein lysine methylation is a crucial post-translational modification that regulates the functions of both histone and non-histone proteins. Deregulation of the enzymes or’writers’ of protein lysine methylation, lysine methyltransferases (KMTs), is implicated in the cause of many diseases, including cancer, mental health disorders and developmental disorders. Over the past decade, significant advances have been made in developing drugs to target KMTs that are involved in histone methylation and epigenetic regulation. The first of these inhibitors, tazemetostat, was recently approved for the treatment of epithelioid sarcoma and follicular lymphoma, and several more are in clinical and preclinical evaluation. Beyond chromatin, the many KMTs that regulate protein synthesis and other fundamental biological processes are emerging as promising new targets for drug development to treat diverse diseases.
Covalent post-translational modifications (PTMs) of proteins are a major source of molecular functional diversity in mammalian cells, and their aberrant regulation is a common feature of human diseases. Lysine methylation is a prevalent PTM that influences many cellular pathways but for which drug development is in a relatively early stage compared with, for example, a classic PTM such as phosphorylation. Indeed, kinase inhibitors are widely used in the clinic, with approximately 70 FDA-approved drugs to date and dozens more being evaluated in clinical trials. Described in the late 1950s, phosphorylase kinase was the first biochemically characterized kinase (reviewed in REF.1). In 2001, more than 40 years later, imatinib (Gleevec), which selectively blocks the BCR–ABL fusion created by the Philadelphia chromosome in chronic myelogenous leukaemia, was the first kinase inhibitor to receive FDA approval2. By comparison, the first biochemically characterized lysine methyltransferase (KMT) was described in 1995 (REF.3), and the first (and to date only) FDA approval of a KMT inhibitor (tazemetostat for epithelioid sarcoma4 and subsequently follicular lymphoma) occurred in 2020. Thus, there is tremendous potential in targeting lysine methylation pathways as a therapeutic strategy to treat diverse diseases.
The lysine methylation chemical reaction is the reversible addition of one, two or three methyl groups to the ε-nitrogen of a lysine side chain, forming monomethylated, dimethylated and trimethylated derivatives (referred to here as ‘Kme1’, ‘Kme2’ and ‘Kme3’, respectively; FIG. 1a). The addition of methyl groups to lysine residues is catalysed by KMTs, and removal is catalysed by protein lysine demethylases (FIG. 1a). In the human genome, there are predicted to be in excess of 100 KMTs, and mass spectrometry-based studies suggest that thousands of human proteins harbour lysine methylation5. The addition of methyl moieties to lysine has only a subtle impact on the primary structure of the modified polypeptide. Nonetheless, the signalling potential associated with methylation is extensive as each methyl state at a specific lysine — from Kme0 to Kme3 — can be linked to unique activities6.
Fig. 1 |. overview of lysine methylation.
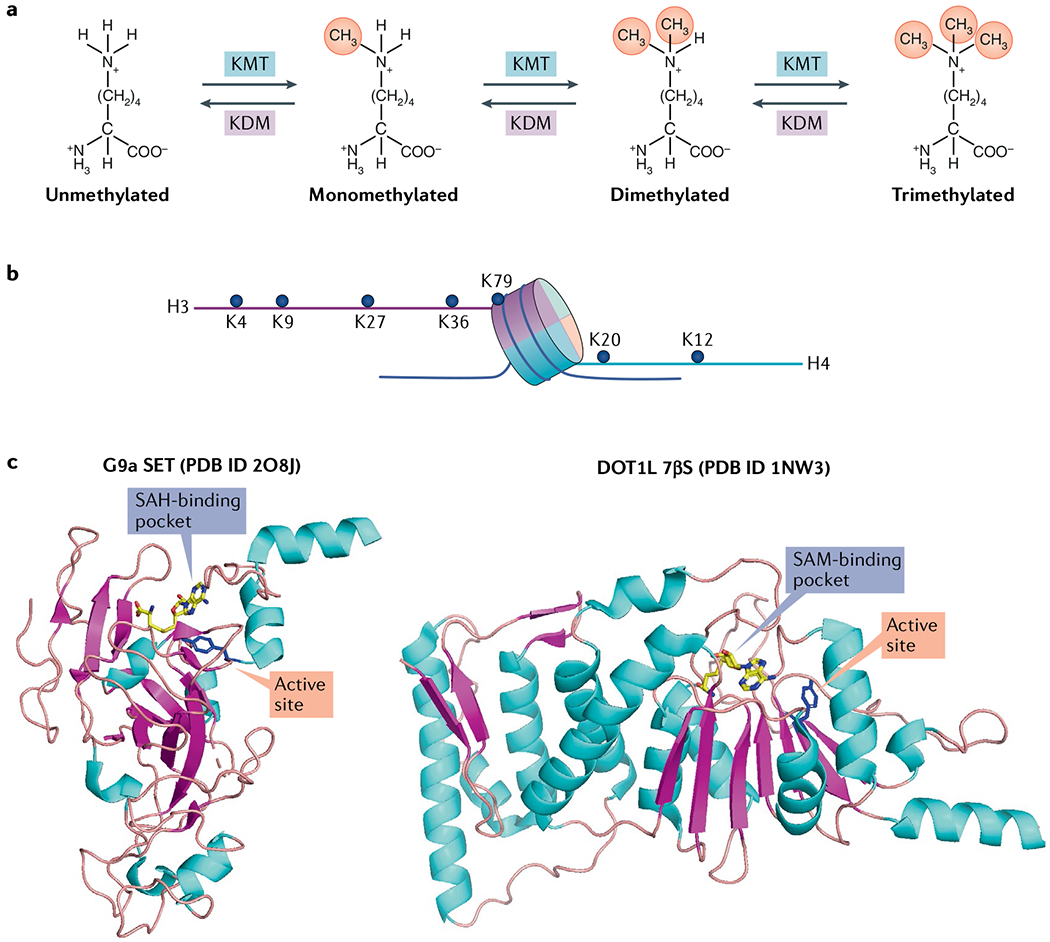
a | The lysine methylation reaction is catalysed by lysine methyltransferase (KMT) and reversed by lysine demethylase (KDM), and results in monomethylation, dimethylation and trimethylation of lysine residues. b | The main lysine residues on histones H3 and H4 that are methylated and/or clinically relevant and discussed in this Review are shown. c | Protein Data Bank (PDB) structures of G9a (SET domain family) and disruptor of telomeric silencing 1-like protein (DOT1L) (7β-strand (7βS) domain family) methyltransferases as representative examples of the two known KMT catalytic families. The conserved tyrosine residue in the catalytic pocket in both structures is shown in blue. The cofactor S-adenosyl methionine (SAM) and its by-product S-adenosyl homocysteine (SAH) are shown bound to DOT1L and G9a, respectively.
The identification of methyllysine was first described on a bacterial flagellar protein in 1959 (REF.7) and soon thereafter was identified on histone proteins8. In 1995, Rubisco large subunit methyltransferase (RLSMT) was described as the first KMT3, although the molecular functions of the methylation of Rubisco (a photosynthetic enzyme) remain poorly understood. In the early years of the first decade of the twenty-first century, several discoveries helped establish lysine methylation as a dynamic PTM with fundamental roles in chromatin biology, epigenetics and human disease. Examples of the landmark findings include the identification of SUV39H1 as the first human KMT, which catalyses histone H3 K9 (H3K9) methylation9, the demonstration that the chromodomain of HP1 selectively binds methylated H3K9 (REFS10,11] and the identification of LSD1 as the first lysine demethylase12. Accordingly, over the past two decades, the role of methylation in chromatin biology has been the main focus of researchers, although as discussed herein, recent investigations of lysine methylation functions outside histones have uncovered important biology and identified new therapeutic targets (see REF.13 for a recent review).
At the molecular level, the addition of a methyl moiety to a protein is best characterized as a signal that directly regulates modular protein–protein interactions (see, for example, REFS10,11,14,15). Lysine methylation can also regulate protein function in cis (that is, it can have autoregulatory activities)16. Furthermore, although there are no examples to date, a methylation event on lysine could in theory influence interactions between the modified protein and molecules such as nucleic acids. In the chromatin biology lexicon, the proteins and protein motifs that recognize histone lysine methylation (see FIG. 1b for the main modified residues) are referred to as ‘reader domains’ (reviewed in REF.17). Indeed, histone methylation has been clearly linked through the action of specific readers to diverse functions, including transcriptional regulation, DNA repair, DNA replication and DNA recombination (see, for example, REFS14,15,18–21). However, there are clinically important methylations, such as dimethylation of H3K79 (to give H3K79me2), for which a reader is yet to be found. From a therapeutic perspective, small-molecule inhibitors of reader domains are a promising strategy for targeting lysine methylation signalling pathways. Indeed, as described herein, clinical trials are under way with drugs that attenuate H3K27 methylation signalling through pharmacological blockade of a reader domain.
The role of reader domain functions in non-histone methylation signalling is a relatively unexplored area that we anticipate may uncover compelling therapeutic opportunities. Overall, given the number of proteins that are regulated by lysine methylation and the complexity associated with sensing and transducing these molecular events, there is tremendous untapped potential in selectively targeting components of this network to treat human disease.
In this Review, we focus on the disease relevance of KMTs, the writers of protein lysine methylation on both histone and non-histone proteins. We begin with a discussion of the biology of writers and general considerations for therapeutic targeting of this class of proteins. We then focus on the two KMTs that have been most extensively characterized as drug targets and for which inhibitors have been tested in the clinic — namely enhancer of zeste homologue 2 (EZH2) and disruptor of telomeric silencing 1-like protein (DOT1L). We review biological and pathological features of EZH2 and DOT1L in the context of therapeutic applications and discuss the chemistry, current clinical trials and results for the inhibitors that target these enzymes. We also discuss inhibitors that target writers that have shown promise in preclinical studies as well as the development of drugs to target KMTs for which inhibition has clear therapeutic potential.
Writers of lysine methylation
Two protein domain families are known to have KMT activity: the SET domain (named for three Drosophila proteins originally recognized to contain the domain: Su(var)3-9, Enhancer of zeste and Trithorax) and the 7β-strand (7βS) domain (FIG. 1c; Supplementary Fig. 1). Approximately 60% of the 55 SET domain-containing proteins in humans have well-documented methylation activity on histone and/or non-histone proteins6. One protein, SETD3, is a SET domain enzyme that catalyses histidine, rather than lysine, methylation22,23. The enzymatic activity for the other ~20 SET domain proteins is unclear.
In humans, approximately 150 proteins catalysing diverse chemistries comprise the 7βS family. Two members of this family, DOT1L and KMT9, catalyse lysine methylation of histones24–28, and about a dozen additional enzymes lysine methylate cytoplasmic proteins involved in processes ranging from protein synthesis and proteostasis to signal transduction and metabolism29. There are also a number of 7βS enzymes that methylate various RNA species, such as the METTL3–METTL14 complex, which catalyses methylation of N6-adenosine (see Supplementary Fig. 1), and which have fundamental roles in development and diseases such as leukaemia30,31.
Clinically relevant histone methylation sites in humans include H3K4, H3K9, H3K27, H3K36, H3K79, histone H4 K20 (H4K20) and H4K12 (REF.6) (FIG. 1b). The non-histone methylated proteins and the specific sites of methylation are too extensive to list here but include several clinically relevant targets in the oncology space32 (see Supplementary Fig. 1 for examples).
Rationale for therapeutic targeting of writers.
There are several general structural and biological characteristics of KMTs that make them promising candidate targets for drug development efforts. First, all KMTs have at least two distinct pockets on their surfaces that are amenable to chemical targeting: the cofactor-binding site for the methyl donor S-adenosyl methionine (SAM) and the lysine substrate-binding pocket (FIG. 1c). Notably, unlike several other types of PTMs (for example, serine/threonine phosphorylation), most KMTs exhibit a high degree of substrate specificity. For example, NSD2, a histone KMT, methylates H3K36 only in the context of a nucleosome substrate33,34. Moreover, while kinases typically phosphorylate a nearby serine if the main substrate site is mutated, mutation of K36 on H3 abrogates NSD2 activity on chromatin33,34; that is, the enzyme cannot methylate H3 on a different lysine residue. Thus, KMT inhibitors can be designed with selectivity for the target enzyme, mitigating off-target toxicity. The high degree of substrate specificity is a common feature of KMTs but does carry the biochemical cost of the methylation reaction generally having slow kinetics relative to other PTM reactions. Finally, several writers are associated with recurrent chromosomal translocations, gain-of-function (GOF) mutations and gene amplifications in distinct cancer populations, which can help focus drug development efforts and optimize patient selection for trials35.
General chemical considerations and strategies for developing KMT inhibitors.
Of the two targetable surfaces for small-molecule engagement on KMTs, the substrate-binding site is naturally more structurally diverse (given the diversity of substrates) compared with the SAM-binding site (FIG. 1c) and therefore offers greater opportunity to selectively target KMT subtypes. Consequently, many of the existing KMT inhibitors are substrate competitors. Nonetheless, the side chains in the SAM-binding pockets are poorly conserved among KMTs — despite SAM being universally used as the methyl donor. Therefore, even close analogues of SAM have been designed and shown to achieve excellent selectivity as inhibitors. The major issue relating to SAM-competitive inhibitors is not their selectivity but rather the hydrophilic nature required to efficiently exploit the SAM-binding pocket. The potential poor cell permeability can be overcome; SAM-competitive, clinical-grade inhibitors of the KMTs EZH2 and DOT1L have been developed. For both enzymes, drug design efforts used unique hydrophobic pockets that arise as a result of subunit interactions (for EZH2) or by inducing a conformational change (for DOT1L)36.
Most KMT inhibitors discovered to date were identified through high-throughput screening (HTS) campaigns followed by medicinal chemistry optimization to improve key drug-like features, ranging from selectivity to pharmacokinetic properties. Structure-based drug design strategies have also been successfully used, particularly when the co-crystal structures of KMT inhibitor complexes are available. Moreover, an iterative process that combines HTS and structure-based optimization has been highly effective, with versions of these strategies underlying the design of many of the SAM-mimetic inhibitors. Structural information in the form of high-resolution complexes of enzymes bound to substrates and/or tool compounds is also providing indispensable molecular insights to understand the basis for enzyme selectivity for rational drug design. We anticipate that resolving high-resolution structures of known and less studied KMTs in complex with a ligand will propel the discovery of new clinical candidate inhibitors.
EZH2 inhibitors
Tazemetostat, which recently became the first FDA-approved KMT-inhibitory drug4 (discussed below), is an EZH2 inhibitor. To date, the indications for this drug include a blood malignancy and a solid tumour, highlighting the broad potential of KMT inhibitors to be efficacious in treating diverse types of cancer.
EZH2 is the main catalytic subunit of Polycomb repressive complex 2 (PRC2), an epigenetic regulatory complex that methylates H3K27 to repress gene transcription (FIG. 2). Targeting PRC2 activity, either directly through inhibiting its catalytic activity or by disrupting its interaction with histones or with other PRC2 proteins, is the most developed clinical translation strategy in the KMT inhibitor space. The excitement around EZH2 and H3K27me3 is driven by the important, if complex, roles this pathway plays in cancer.
Fig. 2 |. eZH2, H3K27 methylation and tumorigenesis.

a | Histone H3 K27 (H3K27) methylation activity relative to processivity for wild-type (WT) enhancer of zeste homologue 2 (EZH2) and mutant (MUT) EZH2. b | EZH2 forms a complex with SUZ12, EED and other subunits of Polycomb repressive complex 2 (PRC2) to catalyse H3K27 trimethylation. Normal PRC2 activity is critical for gene regulation during development, and deregulation of PRC2 activity can promote tumorigenesis by pathological silencing of key genes. Inhibitors of EZH2 (EZH2i) or EED (EEDi) block PRC2-mediated methylation in cancer to attenuate tumour development and progression. c | Structures of EZH2 inhibitors, including S-adenosyl methionine (SAM)-competitive PRC2-EZH2 inhibitors in clinical trials, and allosteric inhibitors that disrupt the EED–H3K27me3 interaction. The EZH2 inhibitor tazemetostat is approved by the FDA for treating epithelioid sarcoma and follicular lymphoma after at least two prior systemic therapies.
EZH2 belongs to the SET family of KMTs (Supplementary Fig. 1); however, unlike most SET proteins, it adopts an autoinhibited conformation so that it is not active in isolation37–39. The stability and activity of EZH2 are dependent on its interaction with two other core members of PRC2, EED (which binds to H3K27me3 to stimulate EZH2 activity) and SUZ12 (REFS40–43). In the context of PRC2, EZH2 catalyses monomethylation, dimethylation and trimethylation of H3K27, a key epigenetic silencing modification (FIG. 2a). EZH2 also undergoes automethylation to regulate its activity and can methylate non-histone substrates44–46. Finally, EZH2 is often replaced by its closely related homologue EZH1 in terminally differentiated and quiescent cells47. As discussed later, EZH2 inhibitors target EZH1 to various degrees, which can make them more toxic but, depending on the clinical context, also increase their therapeutic efficacy48.
As early as 2002, gene expression studies linked high EZH2 expression to cellular proliferation and poor prognosis in prostate cancer49. EZH2 overexpression was subsequently shown to contribute to oncogenic transformation in cellular and mouse xenograft models of human cancer50. EZH2 overexpression has now been linked to a wide range of cancer types51. Further evidence linking H3K27 methylation to cancer came from the observation that the H3K27 demethylase UTX (also called KDM6A) is often lost or inactivated in cancer52. Thus, initial studies implicated EZH2 and H3K27 methylation function in oncogenesis but a specific genetic lesion linking EZH2 activity to a disease state was lacking.
Clinical contexts.
In 2010, recurrent, heterozygous mutations at Y641 of EZH2 were found in ~20% of patients with germinal centre B cell-like diffuse large B cell lymphoma (DLBCL) and 10% of patients with follicular lymphoma53. On the basis of enzymatic assays, substitutions at Y641 were initially thought to be inactivating, which was surprising for a mutation that is both recurrent and heterozygous53,54. This paradox was resolved when it was discovered that Y641 mutations uniquely alter EZH2’s substrate methyl state preference relative to the wild-type enzyme (FIG. 2a). Wild-type EZH2 is most active in converting non-methylated H3K27 to the monomethylated state, with progressively lower activity in transitioning it to the dimethyl and trimethyl states. By contrast, Y641 mutants are almost entirely unable to methylate unmodified H3K27 but have enhanced activity on H3K27me1 and H3K27me2 to generate H3K27me2 and, in particular, H3K27me3 (REFS55,56). Thus, the concerted activities of wild-type and mutant EZH2 yield excessive H3K27me3 and dysregulated silencing of PRC2-target genes (FIG. 2b). This synergy explains why heterozygous Y641 mutations are pathological as the activity of the wild-type allele is required55,56. Additional EZH2 variants found in DLBCL and follicular lymphoma, most notably mutations at A677, also drive H3K27me3 hypermethylation57–59. Beyond GOF mutations, several other mechanisms increase EZH2 activity in tumour cells, including gene amplification, deregulation of EZH2-regulatory micro-RNAs and transcriptional upregulation of EZH2 (REF51). Regardless of the mechanism, enhanced EZH2 activity with elevated H3K27me3 levels promotes tumorigenesis via gene silencing51 (FIG. 2b).
In addition to mechanisms that directly alter EZH2 activity, other genetic lesions render certain cancer types reliant on elevated H3K27me3 levels and hence vulnerable to EZH2 inhibitors. The ~20% of human cancers harbouring mutations in subunits of the SWI/SNF (BAF) ATP-dependent chromatin-remodelling complexes have, to various extents, developed H3K27me3 addiction35 (that is, dependence on H3K27me3 generation). The human SWI/SNF complex antagonizes PRC2 activity, and loss-of-function mutations in SWI/SNF components are frequently associated with increased H3K27me3 levels and sensitivity to EZH2 inhibition60–62. For example, EZH2 inactivation in mouse models abrogates lymphomagenesis due to deletion of the SWI/SNF component INI1 (also named SMARCB1 and SNF5)62. INI1 loss of function, or less frequently, mutations in the related protein SMARCA4, is a defining aetiologic characteristic of rhabdoid tumours, a rare malignant paediatric cancer63,64. Loss of INI1 expression is also a driver of epithelioid sarcoma, another rare and highly aggressive tumour in young adults65. Mutations of INI1 and SMARCA4, which also occur with lower frequency in other solid tumours61, define a clinically actionable genetic signature for EZH2 inhibitor application61,62. Synthetic lethal relationships between mutations in other components of the SWI/SNF family and EZH2 inhibition have also been described60,66. Exploiting these relationships to treat a variety of cancer types with EZH2 inhibitors is presently being explored in clinical trials (for example, NCT03213665), and the EZH2 inhibitor tazemetostat has received FDA approval for treating epithelioid sarcoma4 and follicular lymphoma.
Despite the role of EZH2 in cancer pathogenesis, EZH2 deletion and loss-of-function mutations have also been found to contribute to myeloid malignancies67, possibly through deregulation of the Notch/Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway genes51. Furthermore, dominant negative K27M ‘oncomutations’ in two H3 variants (H3.1K27M and H3.3K27M, respectively) drive tumorigenesis via depletion of H3K27me3 levels68,69; counterintuitively, EZH2 inhibition in this context is therapeutically beneficial, as H3K27me3, while largely depleted at a global level, accumulates aberrantly at specific genes to facilitate cellular transformation70,71. Overall, much remains to be learnt about the molecular contexts in which alterations in EZH2 activity contribute to tumorigenesis, highlighting the importance of careful patient stratification for application of EZH2 inhibitors in the clinic.
Chemical and structural considerations.
Numerous potent and selective inhibitors of PRC2 have been reported since the description of the first selective EZH2 inhibitor in 2012 (REFS72–74). Most PRC2-EZH2 inhibitors share a pyridone core and are SAM-competitive inhibitors. In addition to tazemetostat, three of these inhibitors, GSK2816126 (hereafter GSK126), CPI-1205 and PF-06821497 (FIG. 2c), have advanced into clinical evaluation.
Drug development efforts have benefited greatly from structural information. The first PRC2–substrate complex structure was the yeast Chaetomium thermophilum PRC2 (containing EZH2, EED and the VEFS (Vrn2–Emf2–Fis2–Su(z)12) domain of SUZ12) bound to H3K27M peptide and S-adenosyl homocysteine (SAH; the cofactor product that is formed by demethylation of SAM during the methylation reaction)75. Subsequently, the crystal structure of the human PRC2 (REF76) and the structure of a PF-06821497 analogue bound to wild-type PRC2 and Y641N-mutated PRC2 (REF.77) were determined. Together, these structures revealed that EZH2 wraps around EED, with SUZ12 sandwiched between the SET domain of EZH2 and EED. The selective recognition of H3K27me3 by EED results in stabilization of the stimulation-responsive motif (SRM) helix of EZH2 to increase methyltransferase activity, an interaction that is both fundamental for the cellular function of PRC2 and targetable. There is also an EZH2 loop region that moves away from the EED surface and extends to the SET domain. This loop is referred to as the ‘SET activation loop’ (SAL), which together with the SET domain constitutes the catalytically active domain of EZH2. The co-crystal structures of PRC2 in complex with small-molecule analogues of PF-06821497 (Protein Data Bank (PDB) ID 5IJ7) and CPI-1205 (PDB ID 5LS6) further revealed the central role of the pyridone motif, which forms two hydrogen bonds with the protein backbone and fits in an aromatic cage, where it overlaps with the cofactor SAM. Further insight into the molecular basis of PRC2 activity comes from recent cryo-electron microscopy structures of PRC2 incorporating EZH2, EED, SUZ12, RBBP4, AEBP2 and JARID2 subunits78,79.
The main PRC2-EZH2 inhibitors potently affect both the wild-type and the GOF-mutant forms of EZH2, have lower activity against EZH1 and show no significant affinity against a panel of other methyltransferases and other standard targets. Three of the four clinical candidate EZH2 inhibitors (GSK126, tazemetostat and CPI-1205 (FIG. 2c)) are chemically similar in structure. GSK126 was discovered through an HTS campaign and potently inhibits wild-type and mutant forms of EZH2 (REF74). GSK126 is 150-fold more selective for EZH2 than EZH1, despite the high sequence similarity (about 96%) between the SET domains of the two enzymes. A concurrent effort yielded EPZ005687, a highly potent EZH2 inhibitor73, which was further optimized to increase its potency and improve its pharmacokinetic profile to yield tazemetostat61. While tazemetostat contains the same pyridone core as other EZH2 inhibitors in the class, it lacks the indole or indazole moiety (FIG. 2c). Tazemetostat potently inhibits wild-type and mutant EZH2 variants, with ~35-fold selectivity over EZH1. The third EZH2 inhibitor in this class, CPI-1205, was discovered through HTS and optimized to be a highly potent EZH2 inhibitor with excellent selectivity (inhibits EZH1 with more than 250-fold lower potency than EZH2)80. The fourth EZH2 inhibitor being evaluated in clinical trials is PF-06821497, which features a bicyclic ring in the middle of the molecule joined to a pyridone moiety, making it distinct from the other three inhibitors81 (FIG. 2c). PF-06821497 was discovered by the identification of a novel series of lactam-containing EZH2 inhibitors with use of computational torsional angle analysis coupled with a ligand cyclization strategy81.
MS1943 was recently developed as a first-in-class EZH2-selective degrader that reduces EZH2 levels in cells82. This compound was identified from a series of bivalent compounds by connecting the piperazine group of the EZH2 inhibitor C24, via a linker, to various hydrophobic groups, such as an adamantyl group83. C24, a close analogue of the dual EZH2 and EZH1 inhibitor UNC1999 (REF84), was selected for this study as it combined high potency and selectivity for EZH2 (REF83). Notably, in contrast to EZH2 inhibitors that target the EZH2 catalytic activity and effectively reduce the H3 K27 trimethylation mark but fail to block proliferation of triple-negative breast cancer cells, MS1943 kills multiple triple-negative breast cancer cell lines, with little effect on normal cells82. Furthermore, this compound is orally bioavailable in mice and has shown in vivo efficacy in xenograft models82. Additional methylation-independent functions of EZH2 are reported in prostate cancer and for facilitating immune evasion in brain metastases85–87. Thus, pharmacological degradation of EZH2 may have advantages over chemical inhibition in particular disease scenarios. We further speculate that pharmacological degradation may result in a more sustained suppression of EZH2 function and be useful in the settings of combination therapies as it eliminates both catalytic and non-catalytic activities important for oncogenesis and immunosuppression, as discussed in detail later.
Results in the clinic.
Epizyme’s tazemetostat was approved in January 2020 for the treatment of epithelioid sarcoma, becoming the first FDA-approved KMT inhibitor4. Besides this rare cancer, tazemetostat received approval in June 2020 for treatment of EZH2 mutant-positive follicular lymphoma after at least two prior systemic therapies. Further, EZH2 inhibitors are being or have been evaluated for safety and efficacy in several clinical trials covering a wide range of other cancer types (TABLE 1). Together, these trials have focused on testing EZH2 inhibition as a monotherapy or in combination with other drugs in molecularly defined patient populations.
Table 1 |.
Selected clinical trials of KMT inhibitors
| NCT identifier | Drug | Indications | Design | Phase | Current status |
|---|---|---|---|---|---|
| EZH2 inhibitors | |||||
| NCT04104776 | CPI-0209 | Advanced solid tumours | CPI-0209 with irinotecan | I/II | Recruiting |
| NCT02395601 | CPI-1205 | B cell lymphomas | Single agent | I | Completed |
| NCT03525795 | CPI-1205 | Advanced solid tumours Selected tumour types previously treated with PD1 or PDL1 inhibitors |
Single-agent CPI-1205 and ipilimumab |
I II |
Unknown |
| NCT03480646 | CPI-1205 | Metastatic castration-resistant prostate cancer | CPI-1205 (or placebo) in combination with enzalutamide or abiraterone/prednisone | Ib/II | Active, not recruiting |
| NCT02601950 | Tazemetostat (EPZ-6438) | 7 cohorts: R/R SNF5-negative tumours; any solid tumour with EZH2 gain of function; rhabdoid tumours; synovial sarcomas; epithelioid sarcomas; poorly differentiated chordomas; renal medullary carcinoma | Single agent | II | Recruiting |
| NCT02860286 | Tazemetostat | Malignant mesothelioma with BAP1 loss of function | Single agent | II | Completed |
| NCT03456726 | Tazemetostat | R/R B cell non-Hodgkin lymphoma with EZH2 mutation | Single agent | II | Active, not recruiting |
| NCT02875548 | Tazemetostat | DLBCL, FL, rhabdoid tumours, synovial sarcoma, epithelioid sarcoma, mesothelioma and advanced solid tumours | Single agent (rollover study) | II | Recruiting |
| NCT03213665 | Tazemetostat | R/R advanced solid tumours, non-Hodgkin lymphoma or histiocytic disorders with EZH2, SMARCB1, or SMARCA4 mutations | Single agent | II | Temporarily suspended (scheduled interim monitoring) |
| NCT03348631 | Tazemetostat | Recurrent ovarian or endometrial cancer | Single agent | II | Temporarily suspended (scheduled interim monitoring) |
| NCT04204941 | Tazemetostat | Advanced epithelioid or soft tissue sarcoma | Tazemetostat and doxorubicin | Ib/III | Recruiting |
| NCT04224493 | Tazemetostat | R/RFL | Tazemetostat (or placebo) in combination with lenalidomide and rituximab | Ib/III | Recruiting |
| NCT01897571 | Tazemetostat | Advanced solid tumours or B cell lymphomas DLBCL | Single agent Tazemetostat and prednisolone |
I/II | Active, not recruiting |
| NCT03854474 | Tazemetostat | Locally advanced or metastatic urothelial carcinoma | Tazemetostat and pembrolizumab | I/II | Recruiting |
| NCT02889523 | Tazemetostat | Newly diagnosed DLBCL with poor prognosis | Tazemetostat and Epi-RCHOP | Ib/II | Recruiting |
| NCT04179864 | Tazemetostat | Chemotherapy-naive metastatic castration-resistant prostate cancer | Tazemetostat (or placebo) in combination with enzalutamide or abiraterone/prednisone | Ib/II | Recruiting |
| NCT02220842 | Tazemetostat | R/R FL or DLBCL | Tazemetostat (or placebo) in combination with atezolizumab and obinutuzumab | Ib | Completed |
| NCT02082977 | GSK2816126 | R/R DLBCL, transformed FL, other non-Hodgkin lymphomas, solid tumours and multiple myeloma | Single agent | I | Terminated (see text for details) |
| NCT03460977 | PF-06821497 | R/R small cell lung cancer, castration-resistant prostate cancer and FL | Single agent | I | Recruiting |
| NCT03603951 | SHR2554 | R/R mature lymphoid neoplasms | Single agent | I | Unknown |
| NCT03741712 | SHR2554 | Metastatic castration-resistant prostate cancer | SHR2554 and SHR3680 | I/II | Recruiting |
| Combined EZH1 and EZH2 inhibitors | |||||
| NCT04102150 | DS-3201b | R/R adult T cell leukaemia/lymphoma | Single agent | II | Active, not recruiting |
| NCT03110354 | DS-3201b | AML or acute lymphoblastic leukaemia | Single agent | I | Recruiting |
| NCT02732275 | DS-3201b | Lymphoma | Single agent | I | Recruiting |
| NCT04388852 | DS-3201b | Metastatic prostate, urothelial and renal cell cancers | DS-3201b and ipilimumab | Ib | Recruiting |
| NCT03879798 | DS-3201b | Recurrent small cell lung cancer | DS-3201b and irinotecan | I/II | Recruiting |
| NCT04390737 | HH2853 | R/R non-Hodgkin lymphomas or advanced solid tumours | Single agent | I | Recruiting |
| PRC2-EED inhibitors | |||||
| NCT02900651 | MAK683 | Advanced malignancies, including DLBCL, solid tumours and nasopharyngeal carcinoma | Single agent | I/II | Recruiting |
| DOT1L inhibitors | |||||
| NCT02141828 | EPZ-5676 | R/R leukaemias bearing MLL-r (paediatric) | Single agent | I | Completed |
| NCT01684150 | EPZ-5676 | Leukaemias involving MLL-r or advanced haematologic malignancies | Single agent | I | Completed |
| NCT03701295 | EPZ-5676 | R/R or newly diagnosed MLL-r AML | Pinometostat and azacytidine | Ib/II | Active, not recruiting |
| NCT03724084 | EPZ-5676 | Newly diagnosed AML with MLL-r | Pinometostat and standard chemotherapy | Ib/II | Recruiting |
| Menin–MLL protein inhibitors | |||||
| NCT04067336 | KO-539 | R/R AML | Single agent | I | Recruiting |
| NCT04065399 | SNDX-5613 | R/R leukaemias 3 cohorts: MLL-r ALL or MPAL; MLL-r AML; NPM1c AML |
SNDX-5613 and placebo or CYP3A4 inhibitors SNDX-5613 |
I II |
Recruiting |
AML, acute myeloid leukaemia; DLBCL, diffuse large B cell lymphoma; DOT1L, disruptor of telomeric silencing 1-like protein; Epi-RCHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone; EZH2, enhancer of zeste homologue 2; FL, follicular lymphoma; KMT, lysine methyltransferase; MLL, mixed-lineage leukaemia; MLL-r, rearrangements in mixed-lineage leukaemia genes; NPM1c, nucleophosmin 1 mutation that causes cytoplasmic localization; PRC2, Polycomb repressive complex 2; R/R, relapsed or refractory.
Trials with single-agent use of EZH2 inhibitors have largely focused on patient populations with cancers that are predicted to be most sensitive to inhibition of EZH2, including mutant-EZH2 follicular lymphoma/DLBCL and INI1-negative solid tumours, including rhabdoid tumours and synovial sarcomas (TABLE 1). Promising interim data have come from various trials, including one of tazemetostat in patients with relapsed or refractory (R/R) follicular lymphoma (NCT01897571). In this study, encouraging antitumour activity was observed in patients regardless of EZH2 mutation status, although the objective response rate was more pronounced in GOF-positive EZH2 relative to wild-type EZH2 (69% and 35%, respectively) as was progression-free survival (11.1 and 5.7 months, respectively)88. Tazemetostat recently received FDA approval for treating patients with R/R follicular lymphoma who have received at least two prior lines of systemic therapy. Data from the same trial on the activity of tazemetostat for treating R/R DLBCL are less clear, and it is too early to draw a conclusion about potential clinical use.
EZH2 inhibitors have also been evaluated in solid tumours (TABLE 1). While the complete results are not yet available, interim data from a phase II trial (NCT02860286) in patients with R/R malignant mesothelioma with BAP1 inactivation showed promising antitumour activity for tazemetostat, including sustained long-term disease control in 25% of the patients89. Interim data from a separate phase II trial with tazemetostat in adults with epithelioid sarcoma (NCT02601950) showed a 15% partial response rate and a 26% disease control rate90. The duration of response ranged from 7.3 to 103 weeks, with the median not reached, with 67% of patients having a response of at least 6 months, a significant improvement compared with the standard-of-care treatment. On the basis of these data, the FDA granted accelerated approval of tazemetostat for the treatment of patients 16 years or older with metastatic or locally advanced epithelioid sarcoma not eligible for surgical intervention. Notably, one patient from this cohort had an exceptional response (durable response exceeding 2 years), possibly through upregulation of an antitumour immune response91. These and other data provide a rationale for testing EZH2 inhibitors in combination with immune checkpoint inhibitors. Indeed, trials of tazemetostat in combination with the standard-of-care chemotherapeutic regime in DLBCL (NCT02889523) and with immunomodulators (NCT03854474, NCT02220842 and NCT04224493) are ongoing (TABLE 1).
Constellation Pharmaceuticals’s EZH2 inhibitor CPI-1205 is being tested in a two-arm, open-label phase Ib/II study in combination with enzalutamide or abiraterone/prednisone for metastatic castration-resistant prostate cancer (CRPC) (NCT03480646). This trial has progressed to phase II on the basis of encouraging phase Ib safety and efficacy data, including several patients showing declining prostate-specific antigen levels92. CPI-1205 dosage is also being evaluated in a phase I/II, multicentre, open-label study for use in combination with the checkpoint drug ipilimumab in patients with advanced solid tumours (NCT03525795). CPI-0209, a second-generation, higher-potency inhibitor, is being evaluated for safety and dosage as a monotherapy and in combination with the cytotoxic drug irinotecan in solid tumours (NCT04104776).
Dose escalation studies are ongoing for Pfizer’s EZH2 inhibitor PF-06821497 in follicular lymphoma, DLBCL, CRPC and R/R small cell lung cancer (NCT03460977). This trial will expand to investigate PF-06821497 as a monotherapy and/or in combination with standard-of-care treatments depending on the disease type. An additional drug, SHR2554 (Jiangsu HengRui Medicine Co.), for which the structure is not publicly available, is currently being evaluated in phase I studies either as a monotherapy for R/R lymphomas (NCT03603951) or in combination with an androgen receptor antagonist in a phase I/II trial for metastatic CRPC (NCT03741712). The EZH2 inhibitor HH2853 (Haihe Biopharma; no publicly available structural information) will be evaluated for dosage, safety and tolerability in a phase I trial (NCT04390737), while the GlaxoSmithKline EZH2 inhibitor GSK126 showed modest antitumour activity due to a short half-life, and the trial has been terminated93 (TABLE 1).
Given that EZH1 and EZH2 can both catalyse H3K27 methylation, dual EZH1 and EZH2 inhibitors such as UNC1999 have been developed84. Indeed, dual EZH1 and EZH2 inhibitors suppress H3K27 methylation more strongly than EZH2 inhibitors alone and have higher antitumour activity against several haematologic malignancies in preclinical models94,95. The most clinically advanced dual inhibitor is DS-3201b, also named ‘valemetostat’, from Daiichi Sankyo. Valemetostat is effective in cells that overexpress EZH2 or are vulnerable to H3K27me3 depletion due to secondary mutations in chromatin factors such as SWI/SNF components and UTX96. It is being evaluated in five ongoing clinical trials in different malignancies, including a phase II for patients with R/R adult T cell leukaemia/lymphoma (TABLE 1).
Disrupting reader functions as an alternative therapeutic strategy.
All the drugs discussed so far are SAM-competitive inhibitors that target EZH2 catalytic activity. An alternative strategy to interfere with EZH2 activity is to target other functionally important and druggable components of PRC2 (REFS43,97–99). In 2017, two compounds, EED226 and A-395, were described that selectively block the interaction between the PRC2 subunit EED and H3K27me3 (REFS100,101) (FIG. 2b,c). The recognition of H3K27me3 by EED, which is mediated by the β-propeller WD40 domain of EED, triggers allosteric modulation of PRC2 to facilitate methylation catalysis43,97,99. Accordingly, A-395 and EED226, despite their divergent chemotypes, are selective inhibitors of PRC2-catalysed methylation of H3K27 in vitro on nucleosome substrates and in cells100,101. The high-resolution crystal structures of EED bound to A-395 and EED226 (PDB IDs 5K0M and 5WUK, respectively) revealed that both molecules bind to the H3K27me3-recognition pocket of EED and cause significant conformational changes in the side chains of key residues. The reorganization caused by ligand binding disrupts the methyllysine-binding aromatic cage in EED and creates a deeper and larger aromatic pocket that accommodates the pyrrolidine core of A-395 and the triazolopyrimidine of EED226.
A-395 and EED226 treatments inhibit proliferation of cancer cells similarly to SAM-competitive inhibitors of EZH2, but importantly are also effective against cells that have acquired resistance to EZH2 inhibitors100,101. Specifically, EZH2 mutations arise that confer resistance to SAM-competitive EZH2 inhibitors. However, in vitro methylation by PRC2 containing EZH2 with these resistance mutations is still inhibited by EED226 (REFS100,101). Indeed, in addition to EED226 and A-395 phenocopying SAM-competitive EZH2 inhibitors in xenograft models of DLBCL, both compounds were also effective in cellular models of mutant EZH2 (Y111 and I109 point mutations) that are insensitive to tazemetostat100,101. Thus, pharmacological targeting of EED can be used to overcome cancer cell resistance to EZH2 inhibitors. Furthermore, as discussed earlier, it may be advantageous in certain clinical contexts to inhibit EZH1 and EZH2, although potentially at the cost of increased toxicity35. Given that EED is present in both EZH2-containing PRC2 and EZH1-containing PRC2, EED226 also inhibits EZH1-mediated H3K27 methylation. Accordingly, Novartis’s MAK683, a molecule evolved from EED226 (REF.100), is currently being evaluated in a phase I/II trial for multiple EZH2 inhibitor-indicated cancers, including DLBCL, prostate cancer and sarcomas (NCT02900651) (TABLE 1).
Potential future applications and challenges.
One of the ongoing challenges with drugging EZH2 has been the context-dependent biology; EZH2 is oncogenic in several cancers but tumour suppressive in other cancer types. Furthermore, in a mouse model study of acute myeloid leukaemia (AML), EZH2 was tumour suppressive in the early disease stage but promoted oncogenesis as the disease progressed102. The dichotomous nature of EZH2 is also evident in preclinical studies investigating the effects of EZH2 inhibition on tumour immune evasion and acquired resistance to immunotherapy. EZH2 is required for differentiation and plasticity of various T cell populations, which is naturally important for an effective antitumour immune response (reviewed in REFS103,104). EZH2-mediated gene silencing is also important for direct regression of tumours by macrophages in a mesothelioma model due to suppression of PD1 expression105. At the same time, EZH2 plays important roles in tumour immunosuppression; EZH2, via H3K37me3-mediated silencing, suppresses expression of PDL1 in hepatocellular carcinoma and head and neck cancer106,107. EZH2 activity also renders melanoma cells less immunogenic, and inhibition of EZH2 increases the efficacy of anti-CTLA4 therapy in different cancer models108,109. Finally, EZH2, via a non-catalytic mechanism, promotes infiltration of immunosuppressive neutrophils that facilitate brain metastatic disease; thus, degraders of EZH2 could be effective in this clinical context87. The intricate functions of EZH2 in distinct cell types, particularly in the immune system and its interaction within the tumour microenvironment, pose a challenge and an opportunity in using EZH2 inhibitors in combination with immunotherapy to treat cancer104.
The clinical indications for using EZH2 inhibition have expanded to include both haematologic and solid tumours104. In addition, selective targeting of EZH2 in tumours without impacting the immune system may be a powerful adjuvant of immune checkpoint blockade treatments104. As cancer treatments evolve to include combination therapies that target synergistic pathways, we speculate that a newer generation of EZH2 inhibitors with better pharmacokinetic and pharmacodynamic properties may facilitate studies testing epigenetic drugs with various immune-based and targeted therapies. Looking forward, EZH2 inhibition has shown promise in models of paediatric diffuse intrinsic pontine glioma, a devastating disease with no cure70,71. We hope that brain-penetrable EZH2 inhibitory analogues will be developed to directly test the clinical efficacy of EZH2 inhibitors in this and other brain cancers.
DOT1L inhibitors
H3K79 methylation is a modification conserved from yeast to humans and linked to transcriptional activation110. DOT1L, a 7βS enzyme, is the only known KMT in the human (or any) genome that catalyses H3K79 methylation24–27, which in humans is primarily H3K79 dimethylation (FIG. 3a). H3K79 methylation, unlike the other main histone methylation events, occurs within the histone globular region rather than the unstructured amino-terminal (N-terminal) tails. Therefore, DOT1L has in vitro activity only on nucleosome substrates and does not methylate H3 alone24,26,111. Additionally, for H3K79 methylation, there are no validated ‘erasers’ or clear ‘readers’ of this modification. Thus, the underlying molecular mechanism by which methylation at H3K79 promotes transcription is not understood110.
Fig. 3 |. DoT1L, H3K79 methylation and MLL-r leukaemia.
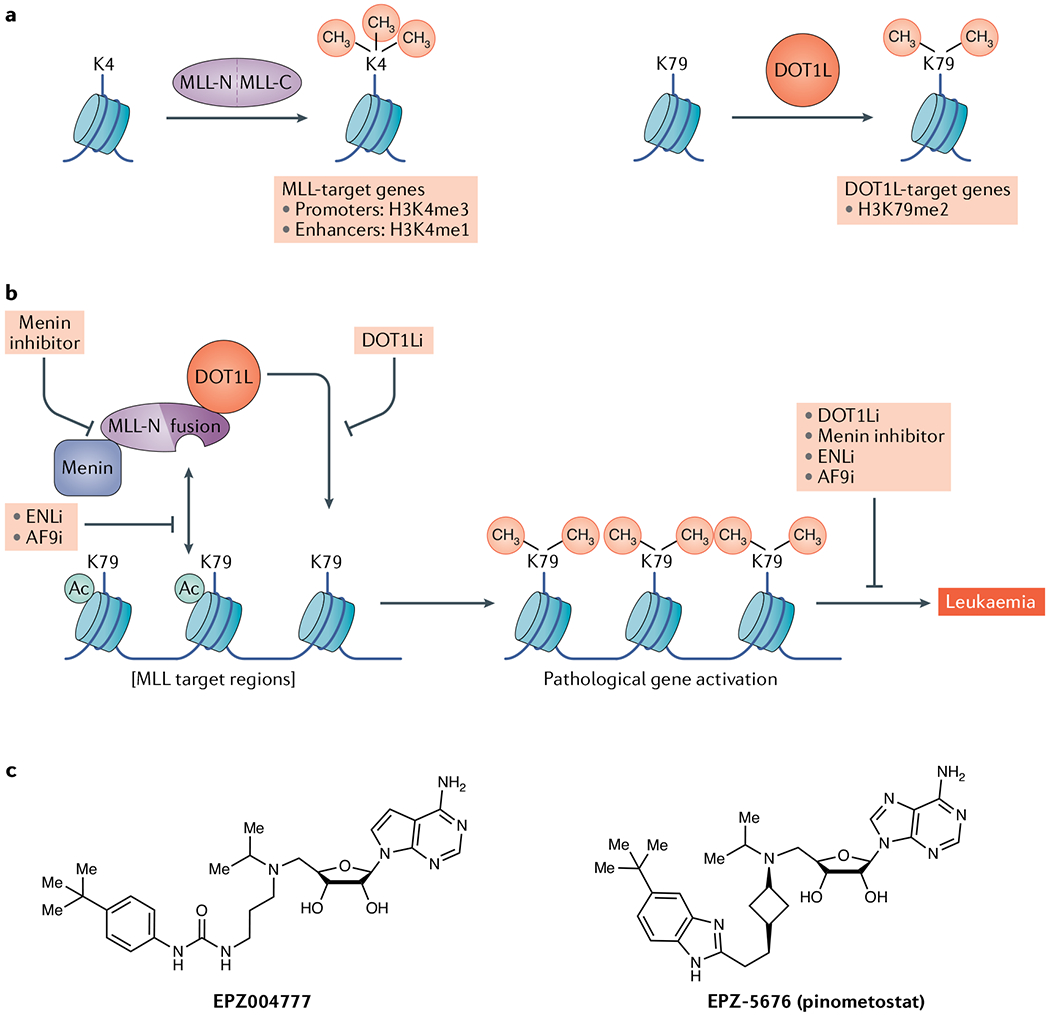
a | Principal catalytic functions of mixed-lineage leukaemia (MLL) proteins and disruptor of telomeric silencing 1-like protein (DOT1L). b | DOT1L is mislocalized by MLL fusion proteins to catalyse histone H3 K79 dimethylation at non-physiologic loci. Inhibitors of DOT1L, menin–MLL protein interaction and reader domains in the DotCom complex block this activity and could have therapeutic utility. See the main text for details of mechanisms. c | Structures of DOT1L catalytic inhibitors. AF9i, AF9 inhibitor; DOT1Li, DOT1L inhibitor; ENLi, ENL inhibitor; H3K4me1, K4-monomethlylated histone H3; H3K4me3, K4-trimethlylated histone H3; H3K79me2, K79-dimethlylated histone H3; MLL-C, carboxy-terminal side of MLL protein; MLL-N, amino-terminal side of MLL protein; MLL-r, rearrangements in mixed-lineage leukaemia genes.
Human DOT1L is implicated in several processes, most notably transcription activation (reviewed in REF.110). Physiologically, DOT1L is essential for proper embryonic development in mice, regulating haematopoiesis and the cardiovascular system112,113. Dysregulation of DOT1L also drives a subset of childhood leukaemia114–116. Mechanistically, DOT1L forms a large and variable multimeric complex referred to as ‘DotCom’, which contains several transcriptional elongation factors117. Many of these proteins harbour reader domains that stabilize DOT1L complexes at specific genomic regions through recognition of distinct chromatin signatures. Examples of DotCom proteins that harbour these reader domains include the acetyllysine-binding YEATS domain proteins AF9 and ENL, and the H3K27me0-sensing PZP domain protein AF10 (REFS118–121). Notably, all three of these proteins are fusion partners with the mixed-ineage leukaemia (MLL) genes and link chromatin-reading functions to DOT1L activity in leukaemogenesis115. Under both normal and pathological conditions, DOT1L, via H3K79me2 generation, is thought to promote gene expression by regulating transcriptional elongation117 and a subset of enhancers122.
H3K79 methylation catalysis by DOT1L is dependent on histone H2B K120 ubiquitylation25,123,124 and a basic region on the H4 tail125–127. However, it was not well understood how the relatively inaccessible K79 residue enters the DOT1L active site. A series of recent cryo-electron microscopy studies of DOT1L bound to the H2B ubiquitylated nucleosome posited that DOT1L exists in two states — ‘poised’ and ‘active’128–132. In the poised state, DOT1L interacts with the ubiquitin bound on H2B through a carboxy-terminal (C-terminal) motif. In addition, it interacts with the conserved acidic patch on H2A/H2B through residue R282. These two interactions anchor DOT1L, allowing the N-terminal domain to sample a large area of the nucleosome. In the active state, the N terminus rotates and moves closer to the nucleosome, facilitating insertion of the H4 tail into a groove formed by the N-terminal domain of DOT1L. The interaction of the H4 tail with DOT1L induces a conformational change in H3 that allows the ‘pinching’ of K79 from the backbone. This in turn reorients the K79 side chain by 90°, allowing insertion into the enzyme active site. These new structures, in addition to elucidating how K79 is accessed, will aid future development of inhibitors.
Clinical context.
DOT1L is an unusual drug target because it is not directly affected by oncogenic mutations or aberrant expression in cancer. Instead, DOT1L activity and H3K79 dimethylation are important drivers of leukaemogenesis in the subset of haematologic malignancies caused by rearrangements in the MLL genes (referred to as MLL-r leukaemia)114,116,133–135 (FIG. 3b). MLL1 (also known as KMT2A) was originally cloned as the gene associated with recurrent translocations of chromosome band 11.q23 in a wide range of leukaemias and is similar to the key developmental Trithorax gene in Drosophila136,137. Such translocations occur in 5–10% of acute lymphoblastic leukaemias and AML, and almost all cases of MLL (reviewed in REF.115). There are four MLL genes (MLL1, MLL2 (also known as KMT2D), MLL3 (also known as KMT2C) and MLL4 (also known as KMT2B)) that encode H3K4 methyltransferases and promote transcription. The MLL proteins have a similar basic structure, with an N-terminal domain important for genomic localization and a C-terminal catalytic SET domain (FIG. 3a). The most common rearrangements result in an MLL1 N-terminal fusion protein coupled to one of more than 70 different C-terminal partners138. These translocations often involve members of the AF and ENL protein families, notably AF4, AF6, AF9, AF10, ELL and ENL. The causal role of MLL gene fusions in leukaemia was demonstrated when knockins of Kmt2a–AF9 (also known as MLLT3) fusion alleles were found to cause leukaemia in mice139. These fusion alleles contain the DNA-binding regions of MLL genes but have lost the catalytic SET domain.
In 2005, DOT1L was shown to interact with AF10, one of the most common MLL protein fusion partners, and a mechanism was proposed in which DOT1L is recruited via MLL–AF10 to activate target genes116 (FIG. 3b). The study authors demonstrated that an artificial MLL–DOT1L fusion protein immortalizes mouse bone marrow progenitor cells and that the catalytic activity of DOT1L is required for leukaemic transformation by the MLL–AF10 fusion protein116. It is now known that MLL protein fusions retain the chromatin-targeting domains of MLL protiens, causing inappropriate localization of DOT1L, which in turn leads to aberrant H3K79 methylation and increased expression of key development genes, including HOXA7, HOXA9 and MEIS1 (REFS135,140,141) (FIG. 3b). Preclinical studies demonstrated that DOT1L activity is required for the oncogenic effect in most MLL-r leukaemias, and that these cancers are exquisitely sensitive to DOT1L inhibition133,142,143. For example, the first potent and specific DOT1L inhibitor, EPZ004777, selectively killed MLL-r leukaemia cells in culture and was able to prolong the survival of mice in a leukaemia xenograft model144,145 (FIG. 3b). Preclinical studies with an improved inhibitor, pinometostat (EPZ-5676), that has superior pharmacokinetic properties observed sustained regression in an MLL–AF4-driven rat xenograft model134.
Chemical and structural considerations.
EPZ004777 and the clinical candidate pinometostat were designed and synthesized on the basis of the DOT1L cofactor product SAH and the crystal structure of the enzyme active site134,144,146 (FIG. 3c). EPZ004777 was highly potent in vitro (half-maximal inhibitory concentration in the picomolar range) and displayed greater than 1,000-fold selectivity for DOT1L over nine other methyltransferases, despite the similar mode of SAM binding. Not surprisingly, EPZ004777 was competitive with SAM and non-competitive with the peptide substrate. EPZ004777 exhibited picomolar binding affinity and an unusually long residence time (~1 h) on DOT1L. For all the compounds reported in the EPZ004777 series, as well as for SAH, the association rate was quite slow and invariant. It was around 100-fold slower than the expected rate for a diffusion-controlled binding event, suggesting that a slow conformational change of DOT1L was required for inhibitor binding. The clinical candidate pinometostat displayed an even higher binding affinity for DOT1L and a longer residence time (more than 24 h) than EPZ004777. The crystal structures of the DOT1L–EPZ004777 and DOT1L–pinometostat (PDB IDs 4ER3 and 4HRA, respectively) complexes reveal that these inhibitors bind to DOT1L through the 5-aminoisopropyl group, engaging a region that is occupied by the methyl group of the thiomethyl on SAM. Furthermore, the proximal nitrogen atom of the urea of EPZ004777 and the benzimidazole of pinometostat form hydrogen bonds with DOT1L. Importantly, the steric bulk of the tert-butyl phenyl groups of the two inhibitors open up a novel hydrophobic pocket on DOT1L by changing the side chain conformation, including moving the L10–L11 loop between the β-strands away from the SAM-binding pocket. Interactions within this newly formed hydrophobic pocket result in the high potency and longer residence times of EPZ004777 and pinometostat. This also accounts for the remarkable selectivity of these inhibitors for DOT1L over other methyltransferases. Several other preclinical inhibitors of DOT1L have been reported that are structurally similar to SAH48. In addition, a new series of DOT1L inhibitors that differ structurally from all previously published SAM-based inhibitors have recently been reported. These non-SAM/SAH inhibitors have been shown to interact with an induced pocket adjacent to the SAM-binding site — without interacting with the SAM-binding site147,148. While EPZ004777 and pinometostat are not orally bio-available, these non-SAM/SAH inhibitors could potentially be optimized into orally bioavailable DOT1L inhibitors for clinical studies.
Results in the clinic.
On the basis of the promising preclinical data with pinometostat, phase I clinical trials were initiated in R/R adult and paediatric MLL-r leukaemias (NCT01684150 and NCT02141828, respectively) (TABLE 1). These trials observed acceptable safety and pharmacodynamics, as well as a moderate reduction in H3K79me2 at genes targeted by MLL fusion proteins. However, the responses to pinometostat monotherapy were somewhat limited, with objective responses observed in a small number of adult patients followed by development of resistance and progressive disease149,150. At the time of writing, there are no active clinical trials of pinometostat monotherapy. There have been preclinical studies supporting the use of pinometostat as a combination therapy with existing standard-of-care drugs for AML, including DNA methyltransferase inhibitors151. Additionally, there is preclinical evidence that pinometostat might be effective in AML with mutations in DNA methyltransferase DNMT3A152. Indeed, there are two active trials evaluating pinometostat in combination with either the DNA methyltransferase inhibitor azacytidine153 (NCT03701295) or with standard-of-care chemotherapy (NCT03724084) to treat R/R AML with MLL-r (TABLE 1). Furthermore, recent preclinical studies have observed synergistic effects of pinometostat in combination with PRMT5 inhibitors in cell lines with MLL–AF4 or MLL–AF9 fusions154 or with SETD2 loss155. Thus, a detailed understanding of how the genetic landscape influences leukaemia sensitivity to DOT1L inhibition may reveal more precise therapeutic opportunities.
Alternative therapeutic strategies.
As with targeting PRC2, inhibitors that disrupt interactions of DOT1L with its binding partners may be a viable and clinically actionable approach, particularly in MLL-r leukaemia involving fusion partners that interact with DOT1L (for example, AF9, ENL, AF10 and AF17). In addition, compelling data suggest that targeting the acetyllysine-binding YEATS domain of ENL might be therapeutic in leukaemia beyond the MLL-r subset120,121 (FIG. 3b). An additional strategy is to block the protein-protein interaction between menin and MLL proteins (FIG. 3b). Menin plays a role in MLL chromatin docking, including localization of MLL fusions to chromatin. Notably, inhibitors that block the MLL–menin interactions downregulate differentiation of leukaemic blasts and prolong the survival of mouse models of MLL-r leukaemia without impairing murine haematopoiesis156,157. Two clinical-grade inhibitors of the menin–MLL protein interaction have been developed: Syndax’s SNDX-5613 and Kuro Oncology’s KO-539. SNDX-5613 is being evaluated in a phase I/II trial in acute leukaemias, and the phase II trial will focus on efficacy in patients with MLL-r leukaemia and patients with AML with mutant NPM1 (which encodes a nucleus–cytoplasm shuttling protein), a common genetic alteration in this disease (NCT04065399). KO-539 is being tested in a phase I trial (NCT04067336) (TABLE 1).
Inhibitors of KMTs in preclinical investigation
Beyond EZH2 and DOT1L, there are selective inhibitors of several other KMTs, all at the preclinical evaluation stage. In contrast to EZH2 and DOT1L inhibitors, which are SAM competitive, most inhibitors of other enzymes are substrate competitive.
G9a and GLP.
G9a and the closely related G9a-like protein (GLP) (also known as EHMT2 and EHMT2, respecively) were initially discovered as H3K9 mono-methyltransferases and dimethyltransferases that are required for early development and are responsible for generating the bulk of H3K9me1 and H3K9me2 in most mammalian cell lines158–160. H3K9me1/H3K9me2 is bound by a number of reader domain-containing proteins, including the ankyrin repeat domain of G9a and GLP and different chromodomain-containing proteins, that together link H3K9 methylation to transcriptional repression and gene silencing17,161,162. Elevated G9a expression is observed in several cancer types (for example, breast and lung cancer) and is associated with metastatic disease and an overall poor prognosis. For example, G9a interacts with MYC to repress transcription and promote oncogenesis in breast cancer cells163. Notably, oncogenic GOF mutations and gene amplification in G9A were recently identified in melanoma, and G9a-mediated H3K9 methylation is linked to the pathogenesis of this disease in preclinical studies164. G9a and GLP have also been implicated in the development of adaptive resistance to targeted therapy in pancreatic and ovarian cancers165,166. Finally, G9a activity is associated with other diseases, including addiction and psychiatric disorders167,168. On the other hand, in certain cancer contexts, such as lung cancer, the long-term inhibition of G9a/GLP can promote tumour progression169,170.
The G9a/GLP inhibitor BIX-01294 is one of the first examples of a selective, peptide-competitive KMT inhibitor171 (FIG. 4a). Despite promising results in cell culture and in vivo models172, the cellular toxicity of BIX-01294 limited its utility. Leveraging of the co-crystal structure of the GLP SET domain bound by BIX-01294 fuelled the discovery of more potent quinazoline-based G9a/GLP inhibitors, including the cellular chemical probe UNC0638 (REFS173,174) and the in vivo chemical probe UNC0642 (REF.175) (FIG. 4a). UNC0642 has greater selectivity and potency and lower toxicity than BIX-01294 and is bioavailable and efficacious in vivo, making it a promising potential candidate for clinical development. Indeed, in therapy-resistant pancreatic tumours in mouse or human models, combined treatment with UNC0642 and the HDAC3 inhibitor RGFP966 resensitized these tumours to the MEK1/MEK2 inhibitor trametinib165. Another G9a- and GLP-selective inhibitor is A-366, which has a scaffold that differs from the quinazoloine inhibitors176 (FIG. 4a). A potent GLP-selective inhibitor, MS012, which is 140-fold selective for GLP over G9a, was also developed177 (FIG. 4a). Notably, co-crystal structures of this substrate-competitive inhibitor in complex with GLP or G9a revealed virtually identical binding modes, highlighting the challenges in structure-based design of inhibitors selective for one of these two highly homologous enzymes. Although such selectivity may be unnecessary from a therapeutic perspective, selective small-molecule inhibitors may be valuable tools to distinguish physiologic functions between G9a and GLP.
Fig. 4 |. Selective inhibitors of lysine methyltransferases in preclinical development.

Chemical structures of compounds targeting G9a/G9a-like protein (GLP) (part a), SETD8 (part b), SUV420H1/SUV420H2 (part c), SETD7 (part d) and SMYD2 (part e) are shown.
One challenge in translating the promising preclinical data with G9a/GLP inhibitors into the clinic is that the two enzymes methylate substrates besides H3K9. For example, there is convincing evidence that G9a and GLP physiologically methylate LIG1, p53, WIZ, Reptin, ACINUS, CDYL1 and other substrates (see32), with LIG1 being a particularly high-affinity substrate178. Thus, in progressing G9a/GLP inhibitors into the clinic, it will be important to evaluate which substrates of G9a and GLP contribute to the enzymes’ physiologic effects as well as the contribution of such activities to G9a/GLP-linked diseases.
SETD8.
SETD8 (also known as SET8 and PR-Set7) is the only known physiologic H4K20 monomethyltransferase in metazoan systems6,179,180. SETD8 and H4K20me1 regulate several cancer-associated cellular processes, including DNA repair, cell cycle regulation, chromatin condensation and transcriptional regulation (reviewed in REF.181). In addition to H4K20me1, SETD8 monomethylates p53 at K382 and other substrates32,182,183. In Drosophila, deletion of SETD8 is lethal, whereas an alanine substitution at H4K20 causes developmental delay but is otherwise tolerated, indicating a broader role for SETD8 in this organism beyond H4 modification184. As SETD8 regulates several important pathways (see REF.185) and there was available structural insight186,187, inhibitors of this enzyme have been developed.
Several of the early SETD8 inhibitory compounds decreased H4K20me1 levels in cells but also inhibited other KMTs188. Recently, the selective SETD8 inhibitor UNC0379 was discovered via a cross-screen of a quinazoline-based library of more than 150 compounds that were originally prepared for the development of G9a/GLP inhibitors189,190 (FIG. 4b). UNC0379 was selective for SETD8 over 15 other methyltransferases, including G9a and GLP, and phenocopied SETD8 knockdown in cells191,192. The strategy of using a chemical scaffold that inhibits one KMT to develop a selective inhibitor for another, as was successfully done for SETD8, should be broadly applicable for targeting KMTs. Subsequent optimization of UNC0379 led to a new compound, MS2177, with increased potency for SETD8, enabling the generation of the first crystal structure of SETD8 in complex with a small-molecule inhibitor193. The co-crystal structure of the SETD8–MS2177 complex revealed a cysteine residue (C311) that was near the inhibitor-binding site, which led to the design of the C311 covalent modifying inhibitor MS453 (FIG. 4b). MS453 did not covalently modify other KMTs such as EZH2, SMYD2 and SMYD3, indicating specificity for SETD8. Despite the availability of effective SETD8 inhibitors, the appropriate clinical application of these inhibitors is at present obscure; SETD8 deletion causes early embryonic lethality in mice194, and thus a defined disease state is needed to justify advancing any compound that targets SETD8 as a therapeutic.
SUV420H1 and SUV420H2.
SUV420H1 and SUV420H2 are related enzymes that use SETD8-generated H4K20me1 as the substrate to synthesize H4K20me2 and H4K20me3 in cells and multiple organisms21,195–197. While they have similar kinetics and substrate preferences in vitro196,198,199, in mouse embryonic fibroblasts, SUV420H1 is responsible for most of the H4K20me2 and SUV420H2 is more responsible for H4K20me3 (REF.197). SUV420H1 and SUV420H2 are linked to transcriptional silencing196–200, chromatin compaction201, DNA replication21,202 and DNA repair. Indeed, one of the most well-characterized functions of H4K20me2 is in the maintenance of genome integrity and recruitment of the double-strand break repair factor 53BP1 (REFS18,197,203,204)
A-196 (FIG. 4c) was the first potent, selective and cell-active inhibitor of these two highly homologous KMTs. It was discovered via HTS followed by medicinal chemistry optimization205 and is a substrate-competitive inhibitor with more than 100-fold selectivity for SUV420H1 and SUV420H22 over other methyltransferases and a broad range of non-epigenetic targets. Despite being a substrate-competitive inhibitor, it exhibits high cooperativity with SAM binding. In cells, A-196 reduces H4K20me3 and H4K20me2 levels and attenuates the formation of 53BP1 foci, and thus is a valuable tool for advancing the understanding of the cellular roles of SUV420H1 and SUV420H2.
SETD7.
SETD7 (also known as SET7 and SET9), one of the first characterized KMTs, was initially identified as an H3K4 monomethyltransferase206–207. However, H3K4me1 levels are unchanged in SETD7-deleted mouse embryonic fibroblasts208. SETD7 is also reported to monomethylate numerous other proteins, including p53 and the maintenance DNA methyltransferase DNMT1 (REFS13,32). A potent, selective and cell-active small-molecule inhibitor of SETD7, (R)-PFI-2 (FiG. 4d) was developed via HTS followed by several rounds of structure-guided medicinal chemistry optimization209. Importantly, (R)-PFI-2 is not purely a substrate-competitive inhibitor; SAM binding to SETD7 plays a significant role in the binding of (R)-PFI-2, rendering it a cofactor-dependent and substrate-competitive inhibitor. In a small molecule-based proteomic strategy, this inhibitor was used to identify the ribosomal regulatory protein RPL29 as a major, physiologic target of SETD7 (REF.210). Overall, SETD7 is potentially involved in several biological processes, but a clear activity that would be therapeutically beneficial to inhibit is yet to be established.
SMYD2.
SMYD2, another monomethyl KMT, is overexpressed in several types of cancer, and its expression is associated with poor clinical prognoses211,212. Consistent with a potential role in tumorigenesis, knockdown of SMYD2 affects proliferation of different cancer cell types211,212. SMYD2, like G9a/GLP and SETD7, is one of the more promiscuous KMTs6. Various studies have claimed that SMYD2 methylates histones, but this activity is not specific for a distinct histone lysine and does not occur on nucleosomes, and SMYD2 depletion does not have a clear impact on histone methylation levels213. Notable non-histone substrates of SMYD2 include p53 at K370, retinoblastoma protein (RB), HSP90, oestrogen receptor-α, PARP1, and phosphatase and tensin homologue (PTEN) (see REFS13,32). Moreover, recent proteomics studies identified additional candidate SMYD2 targets, including the proteins AHNAK and AHNAK2, which are implicated in cell migration and invasion214 and the stress kinase MAPKAPK3 (REF.213). It is unclear how these various activities are integrated to contribute to SMYD2 behaviour under physiologic and disease conditions. In vivo, SMYD2 deletion modestly attenuates KRAS-driven pancreatic cancer in mouse models213. In addition, studies in mouse models of AML suggested that SMYD2 is a MYC target that plays a role in MLL-r-driven leukaemogenesis215.
SMYD2 inhibitors have been developed largely via HTS followed by structure-based medicinal chemistry optimizations. AZ-505 and A-893 (FiG. 4e) share a scaffold, with the latter displaying some cellular activities, such as inhibition of SMYD2 methylation activity216–218. LLY-507 was the first cell-active, selective inhibitor of SMYD2. In cells, it reduces p53 K370me1 levels and inhibits cell proliferation in a concentration-dependent manner219. However, this compound inhibits other enzymes, complicating interpretation of its cellular phenotype. Another screening campaign followed by structure-activity relationship studies led to the development of BAY-598, a cell-active inhibitor of SMYD2 suitable for in vivo studies220. Treatment with this compound decreased p53 K370me1 levels but had no effect on cellular proliferation, possibly because K370me1 is thought to repress p53 function in a context-dependent manner221.
In contrast to the SMYD2-inhibitory compounds described above, which are substrate competitive, EPZ033294 and EPZ032597, two non-substrate-competitive inhibitors with high biochemical potency and selectivity, representing a novel scaffold, were recently discovered222 (FiG. 4e). Similarly to BAY-598, these inhibitors had little effect on cellular proliferation despite blocking SMYD2 activity222, suggesting that the antiproliferative effects associated with SMYD2 depletion may be due to off-target effects, cell-specific differences in SMYD2 requirement or non-catalytic functions of the protein.
SMYD3.
SMYD3 is a trimethyl KMT overexpressed in several cancers, including pancreas, lung, liver, colon and breast cancer223–225. Several studies have linked SMYD3 to oncogenic functions, including stimulation of proliferation, adhesion and migration, and tumorigenesis in in vivo mouse models223,224. While SMYD3 was initially claimed to be an H3K4me3 KMT225, this study was conducted when characterization of lysine methylation activities was a relatively nascent field. Several subsequent studies demonstrated that SMYD3 does not methylate H3K4 on peptides, histones or nucleosomes in vitro or on chromatin in vivo223,226,227, although SMYD3 may bind to H3K4me3 (REF.224). A weak SMYD3 trimethylation activity on H4 and nucleosomes was detected at H4K5 in vitro and in cells223,227. Another weak substrate of SMYD3 is vascular endothelial growth factor receptor 1 (VEGFR1), although the functional consequences of this methylation event are unclear228.
The RAS–mitogen-activated protein kinase (MAPK) pathway is frequently activated in SMYD3-overexpressed cancers, in particular pancreatic and lung cancers (FIG. 5a). In a proteome-level protein array activity-based screen, the cytoplasmic protein mitogen-activated protein kinase kinase kinase 2 (MAP3K2), a kinase within the MAPK signalling module, was established as a robust, physiologic substrate of SMYD3 (90-fold higher catalytic activity on MAP3K2 vs H4)223. SMYD3 trimethylation of MAP3K2 at K260 does not affect MAP3K2’s intrinsic kinase activity but rather blocks protein phosphatase 2A (PP2A) from engaging with and thereby inactivating MAP3K2 via dephosphorylation. SMYD3-mediated trimethylation of MAP3K2 therefore results in sustained MAP3K2 activation, ultimately leading to increased extracellular signal-regulated kinase 1 (ERK1)/ERK2 activation, which in turn promotes RAS-driven tumorigenesis in mouse and human pancreatic and lung cancer models223 (FIG. 5a). While the SMYD3–K260-trimethylated MAP3K2 axis regulates pancreatic and lung cancer, MAP3K2 is not expressed in all types of cancers, in all cancers that carry KRAS mutations or in all cancers that overexpress SMYD3. Thus, it may be important to elucidate SMYD3 mechanisms of action for targeting specific cancer contexts. Finally, SMYD3 may have roles outside oncology, such as in inflammation, but more work is required to judge potential therapeutic benefits of SMYD3 inhibitors outside cancer applications.
Fig. 5 |. SMyD3 promotes RAS-driven tumorigenesis.

a | Basic schematic of the RAS–mitogen-activated protein kinase (MAPK) signalling cascade. Under normal conditions, growth factor activation of a receptor tyrosine kinase (RTK) induces RAS to switch from the GDP-bound inactive state to the GTP-bound active state. Constitutively active oncogenic mutant RAS bypasses normal induction, resulting in increased downstream signalling. Mitogen-activated protein kinase kinase kinase 2 (MAP3K2) feeds into RAS–MAPK signalling by phosphorylating and activating MEK1/2. Dephosphorylation of MAP3K2 by protein phosphatase 2A (PP2A) renders it inactive. SMYD3 trimethylation (me3) of MAP3K2 repels PP2A to prevent dephosphorylation, resulting in increased MAP3K2–MEK1/2–extracellular signal-regulated kinase 1/2 (ERK1/2) signalling and promotion of RAS-driven tumorigenesis. b | Structures of SMYD3 inhibitors.
A SAM mimetic, GSK2807 (FIG. 5b), was discovered as an SMYD3 inhibitor via structure-based design229, but displayed poor cell permeability. Two potent, selective, cell-active and reversible small-molecule inhibitors of SYMD3, EPZ0330456 and EPZ031686 (FIG. 5b), based on oxindole sulfonamide or sulfamide scaffolds, respectively, were identified via HTS and medicinal chemistry optimization230. Further structure–activity relationship studies of these inhibitors yielded the isoxazole sulfonamide scaffold-based EPZ028862, which displayed high potency and selectivity for SMYD3 in biochemical assays with peptides and activity in cellular assays222, and should be suitable for in vivo studies. A new class of inhibitors that covalently modify SMYD3 (at the C186 residue) via a nucleophilic aromatic substitution reaction also potently inhibit SMYD3 (REF.231). These inhibitors are antiproliferative in HepG2 colonies grown in 3D culture and cause a decrease in K260-trimethylated MAP3K2 levels. However, their selectivity needs to be more thoroughly investigated. Overall, SMYD3 inhibitors may have potential in combination regimens to treat RAS-driven cancers, but more preclinical evaluation is required.
KMT targets for future drug development
Beyond the KMTs discussed so far, there are many others with links to human disease, ranging from cancer to intellectual disabilities to metabolic syndromes, and the dozens of other known and candidate KMTs in the human genome6. Thus, there is tremendous potential for developing new precision-based medicines as the field develops new biological and chemical understanding of these enzymes.
Histone KMTs.
There are several histone KMTs with clear links to disease but for which selective inhibitors have not been developed. For example, there are four enzymes — NSD1, NSD2, NSD3 and ASH1L — that are H3K36 dimethyltransferases, and all are excellent candidate oncology targets for drug development (reviewed in REFS6,232) (see Supplementary Fig. 1). Alterations in these genes (GOF mutations, gene amplifications and translocations) are aetiologically linked to cancers ranging from multiple myeloma and paediatric acute lymphoblastic leukaemia to diverse solid tumours6,232. Thus, substantial efforts have been made to develop compounds that selectively inhibit these enzymes. One major challenge towards this goal is that all of the H3K36me2-generating enzymes adopt an autoinhibitory state in the apo (unbound) form that is relieved only on nucleosome engagement6. Thus, the development of inhibitors may necessitate screening in the presence of nucleosomes. Nonetheless, while there has been little success to date, a recent provisional patent (20190183865) and progress in structures (see, for example, REF.233) suggest that drugs blocking the activity of these enzymes may soon be available for clinical investigation. Moreover, a drug that binds to a reader domain present within NSD3 and blocks NSD3–chromatin interactions offers another strategy to target this class of KMTs234.
Another promising histone KMT target is SETDB1, an enzyme that methylates H3K9 and is overexpressed in several cancers235,236. SETDB1 was also recently shown to have a role in the pathology of various neuropsychiatric disorders237 and in Prader–Willi syndrome238, suggesting potential broader applications for SETDB1 inhibitors. Another candidate is KMT9, a heterodimer of C21orf127 (also known as N6AMT1) and TRMT112 that catalyses monomethylation of H4K12 and regulates genes involved in cell cycle control. KMT9 depletion selectively leads to decreased growth of prostate cancer cells and xenografts, and thus inhibitors may offer hope for treatment of CRPC28. KMT9 also catalyses other reactions, such as glutamine methylation of eukaryotic release factor 1 (REFS239–241), and the consequences of inhibiting these activities should be considered in any drug development efforts28.
On the basis of expression patterns in cancer, PRDM9 is another histone KMT worth further preclinical investigation, and for which a first-in-class tool compound was recently developed242. PRDM9 trimethylates H3K4 and H3K36 and is a key meiosis recombination factor that is not expressed in somatic cells but becomes overexpressed due to gene amplification in squamous cell lung cancer and testicular cancer243,244. PRDM9 belongs to the 17-member PR domain subfamily of SET domain proteins (reviewed in REF245; see Supplementary Fig. 1). To date, the only proteins that have a SET-like PR domain with a clearly demonstrated methylation activity are PRDM9 and the closely related PRDM7, but several of the other PR domain factors play major roles in development and in cancer. For example PRDM1 (also known as BLIMP1) and PRDM2 (also known as RIZ1) are potent tumour suppressors, whereas PRDM3, PRDM14, PRDM15 and PRDM16 have all been linked to oncogenesis245. Understanding the enzymatic nature of the PR domain in these proteins — whether they are active KMTs, catalyse a different type of chemistry or are catalytically inert — will be key to leveraging their roles in disease for therapeutic purposes.
In this regard, SETDB2, MLL5 and SETD5 are candidate KMTs that are incorrectly annotated as methylating histones (see Supplementary Fig. 1). SETDB2, a regulator of fibrotic diseases, is presumed to have H3K9 methylation activity due to homology to SETDB1, but no such activity has been rigorously demonstrated246. SETD5 was recently claimed to be an H3K36 KMT247, but data from others have failed to reproduce these results165. Instead, SETD5 scaffolds a co-repressor complex containing G9a and HDAC3, which can epigenetically regulate adaptive targeted therapy resistance to MAPK/ERK kinase (MEK) inhibitors in pancreatic cancer165. A related enzyme, MLL5, was initially misidentified as an H3K4 methyltransferase, but like SETD5 is almost certainly catalytically inactive248. Nevertheless, MLL5 expression is associated with several cancers, although the mechanisms and whether it is clinically actionable remain unclear.
Non-histone KMTs.
Mechanisms to increase protein synthesis are crucial in tumours driven by oncogenic growth signalling pathways (for example, RAS–MAPK, MYC, PI3K–AKT andmechanistic target of rapamycin (mTOR)). While most oncogenic pathways target the translation initiation machinery, the elongation step of translation is also an important regulatory node (reviewed in REF249). The GTPase and eukaryotic elongation factor EEF1A is a fundamental, non-ribosomal component of the translational machinery. The canonical function of EEF1A is to deliver with fidelity aminoacylated tRNAs to the A-site of translating ribosomes during the elongation step of protein synthesis (FIG. 6). In humans, there are five EEF1A KMTs, all from the 7βS family, that methylate five distinct lysine residues250 (FIG. 1e). One of these KMTs, METTL13, dimethylates EEF1A at K55 (REFS16,250). This methylation accelerates translation elongation kinetics and enhances protein synthesis to promote oncogenesis16 (FIG. 6). Deletion of METTL13 strongly inhibits RAS-driven pancreatic and lung cancers in mouse and human models16. METTL13 does not appear to affect non-transformed cells, suggesting that enhancement of translation elongation by METTL13-mediated K55-dimethylation of EEF1A becomes rate limiting only in growth signal-driven tumours such as pancreatic ductal adenocarcinoma and lung adenocarcinoma, which could potentially render these lethal cancers vulnerable to METTL13 inhibition16 (FIG. 6b). Besides METTL13, the EEF1A KMT named ‘EEF1AKMT4’ (previously incorrectly annotated as ECE2 (REF.251)) is upregulated in different cancers. Another 7βS KMT, FAM86A, which methylates EEF2 (REFS252,253), is amplified in cancers. However, apart from METTL13, direct clinically relevant functions for KMTs of translational factors have yet to be investigated in detail. By analogy to the kinase field, it is reasonable to expect that future work on these enzymes and many other poorly characterized or uncharacterized KMTs may uncover important new targets for drug development.
Fig. 6 |. RAS-driven cancer dependency on MeTTL13-mediated protein synthesis.

a | METTL13-mediated dimethylation of EEF1A on K55 increases EEF1A’s intrinsic GTPase activity, resulting in accelerated translation elongation and increased protein synthesis. b | Mutant KRAS-driven cancers become dependent on the METTL13–K55-dimethylated EEF1A axis to maintain high translational rates required for malignant growth. The vulnerability of tumours to METTL13 depletion suggests potential efficacy of METTL13 inhibitors (METTL13i) to treat lethal malignancies. PI3K/mTORi, dual inhibitor of PI3K and mechanistic target of rapamycin.
Summary and outlook.
The development of advanced proteomic and structural techniques has fuelled several important advances in the lysine methylation field, accelerating understanding of the basic biology of these enzymes and aiding drug discovery efforts. Furthermore, the past decade of work has provided an impressive expansion of our understanding of how KMTs are engaged from the chemical and molecular perspectives. We anticipate the application of this knowledge will overcome many existing challenges in drug-hunting efforts and lead to the discovery of new inhibitors of some of the most promising targets in the field. We anticipate that revisiting programmes that previously failed to yield drug candidates for high-value KMT clinical targets such as NSD2 should now find success. Furthermore, we predict that the knowledge gained during this past decade will pay off in the coming years through the progression of current inhibitors of targets such as PRDM9 to clinical candidates and in the development of inhibitors of non-histone KMTs with promising preclinical data, such as METTL13. Finally, we anticipate discoveries of synergies between KMT inhibitors and targeted and immune-based therapies that will mitigate resistance development and toxic effects associated with individual treatment regimens. With an EZH2 inhibitor approved for treating both solid tumours and blood cancer and inhibitors that block the PRC2 and MLL protein complexes advancing in clinical trials, we expect the excitement and drug discovery efforts targeting KMTs to rapidly increase over the next decade, and to ultimately benefit patients with diverse diseases through the creation of many new medicines.
Supplementary Material
Acknowledgements
This work was supported in part by grants from the NIH to K.P.B. (K00CA212435), O.G. (R01 CA236118) and J.J. (R01CA218600, R01CA230854, R01GM122749 and R01HD088626).
Glossary
- Reader domains
Modules on proteins that bind to post-translationally modified lysine residues on other proteins in a manner that is dependent on the immediate surrounding sequence and the state of methylation on the lysine.
- Writers of protein lysine methylation
Enzymes that catalyse the addition of one, two or three methyl moieties to the ε-nitrogen of lysine residues.
- Nucleosome
The fundamental building block of chromatin, which consists of ~ 146 base pairs of DNA wrapped around a protein core unit made of two copies each of histones H2A, H2B, H3 and H4.
- Chromatin-remodeling complexes
Large ATP-dependent multisubunit protein complexes that evict, load, alter or otherwise move nucleosomes on DNA to control DNA accessibility.
- Transcriptional elongation factors
Proteins that regulate the elongation step in gene transcription, which occurs after transcription is stably initiated and before transcription termination.
- Mixed-lineage leukaemia (MLL) genes
MLL1, MLL2, MLL3 and MLL4 encode four distinct lysine methyltransferases that catalyse methylation at histone H3 K4; MLL1 was originally identified as a gene involved in a recurrent chromosomal translocation in the neoplasm mixed-lineage leukaemia.
- Fusion protein
Chimeric proteins that result from the fusion of genes from different chromosomes during chromosomal translocations. They often have a new, non-physiologic activity that can unbalance cells and drive cancer pathogenesis.
- A-site
The aminoacyl site, or A-site, on the ribosome is the entry site for amino acid–tRNA molecules to bind and for proper base pairing between the mRNA codon and the tRNA anticodon during the elongation step of protein synthesis.
Footnotes
Competing interests
O.G. is a co-founder of EpiCypher and Athelas Therapeutics. J.J. and H.Ü.K. are inventors of patent applications filed by the Icahn School of Medicine at Mount Sinai. J.J. is a consultant of Cullgen and Accent Therapeutics, a scientific advisory board member of Petra Pharma Corporation and an equity shareholder of Cullgen. K.P.B. is an employee at Genentech.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41573-020-00108-x.
References
- 1.Fischer EH Phosphorylase and the origin of reversible protein phosphorylation. Biol. Chem 391, 131–137 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Chandra HS et al. Philadelphia chromosome symposium: commemoration of the 50th anniversary of the discovery of the Ph chromosome. Cancer Genet. 204, 171–179 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang P, Royer M & Houtz RL Affinity purification of ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit epsilon N-methyltransferase. Protein Expr. Purif 6, 528–536 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Rothbart SB & Baylin SB Epigenetic therapy for epithelioid sarcoma. Cell 181, 211 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Cao XJ & Garcia BA Global proteomics analysis of protein lysine methylation. Curr. Protoc. Protein Sci 10.1002/cpps.16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Husmann D & Gozani O Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol 26, 880–889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ambler RP & Rees MW Epsilon-N-methyl-lysine in bacterial flagellar protein. Nature 184, 56–57 (1959). [DOI] [PubMed] [Google Scholar]
- 8.Murray K The occurrence of epsilon-N-methyl lysine in histones. Biochemistry 3, 10–15 (1964). [DOI] [PubMed] [Google Scholar]
- 9.Rea S et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000). [DOI] [PubMed] [Google Scholar]; This study identfied the first histone lysine methyltransferase and established a connection between histone methylation and epigenetic mechanisms.
- 10.Bannister AJ et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Lachner M, O’Carroll D, Rea S, Mechtler K & Jenuwein T Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Shi Y et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Cornett EM, Ferry L, Defossez PA & Rothbart SB Lysine methylation regulators moonlighting outside the epigenome. Mol. Cell 75, 1092–1101 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi X et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wysocka J et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442, 86–90 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Liu S et al. METTL13 methylation of eEF1A increases translational output to promote tumorigenesis. Cell 176, 491–504.e421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study established a role for non-histone methylation by METTL13 in the regulation of translational elongation to promote RAS-driven cancers.
- 17.Andrews FH, Strahl BD & Kutateladze TG Insights into newly discovered marks and readers of epigenetic information. Nat. Chem. Biol 12, 662–668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Botuyan MV et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews AG et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450, 1106–1110 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vermeulen M et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Kuo AJ et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484, 115–119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilkinson AW et al. SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 565, 372–376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwiatkowski S et al. SETD3 protein is the actin-specific histidine N-methyltransferase. eLife 7, e37921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacoste N, Utley RT, Hunter JM, Poirier GG & Côte J Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J. Biol. Chem 277, 30421–30424 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Ng HH, Xu RM, Zhang Y & Struhl K Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem 277, 34655–34657 (2002). [DOI] [PubMed] [Google Scholar]
- 26.van Leeuwen F, Gafken PR & Gottschling DE Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109, 745–756 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Feng Q et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol 12, 1052–1058 (2002). [DOI] [PubMed] [Google Scholar]; This study, along with Lacoste, N. et al., Ng, H. H. et al. & van Leeuwen, F. et al. identified yeast Dot1 as the H3K79 methyltransferase.
- 28.Metzger E et al. KMT9 monomethylates histone H4 lysine 12 and controls proliferation of prostate cancer cells. Nat. Struct. Mol. Biol 26, 361–371 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Falnes P, Jakobsson ME, Davydova E, Ho A & Małecki J Protein lysine methylation by seven-β-strand methyltransferases. Biochem. J 473, 1995–2009 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Liu J et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbieri I et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552, 126–131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biggar KK, Wang Z & Li SS SnapShot: lysine methylation beyond histones. Mol. Cell 68, 1016–1016.e1 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Li Y et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem 284, 34283–34295 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study established the biochemical activity of NSD1, NSD2, NSD3 and SETD2 as H3K36 methyltransferases that have a strong preference for nucleosomal substrates.
- 34.Kuo AJ et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfister SX & Ashworth A Marked for death: targeting epigenetic changes in cancer. Nat. Rev. Drug Discov 16, 241–263 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Schapira M Chemical inhibition of protein methyltransferases. Cell Chem. Biol 23, 1067–1076 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P & Reinberg D Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16, 2893–2905 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao R et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Müller J et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208 (2002). [DOI] [PubMed] [Google Scholar]; This study, along with Kuzmichev, A. et al. & Cao, R. et al. (2002), provided the first descritpion of EZH2 as the H3K27 methyltransferase.
- 40.Cao R & Zhang Y SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15, 57–67 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E & Helin K Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23, 4061–4071 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tie F, Stratton CA, Kurzhals RL & Harte PJ The N terminus of Drosophila ESC binds directly to histone H3 and is required for E(Z)-dependent trimethylation of H3 lysine 27. Mol. Cell. Biol 27,2014–2026 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Margueron R et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee CH et al. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev 33, 1428–1440 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X et al. Regulation of histone methylation by automethylation of PRC2. Genes Dev. 33, 1416–1427 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ardehali MB et al. Polycomb repressive complex 2 methylates elongin A to regulate transcription. Mol. Cell 68, 872–884.e876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Margueron R et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32, 503–518 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaniskan H, Martini ML & Jin J Inhibitors of protein methyltransferases and demethylases. Chem. Rev 118, 989–1068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varambally S et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Kleer CG et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl Acad. Sci. USA 100, 11606–11611 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim KH & Roberts CW Targeting EZH2 in cancer. Nat. Med 22, 128–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Haaften G et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet 41, 521–523 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morin RD et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet 42, 181–185 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bödör C et al. EZH2 Y641 mutations in follicular lymphoma. Leukemia 25, 726–729 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Sneeringer CJ et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl Acad. Sci. USA 107, 20980–20985 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yap DB et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bödör C et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 122, 3165–3168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Majer CR et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 586, 3448–3451 (2012). [DOI] [PubMed] [Google Scholar]
- 59.McCabe MT et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl Acad. Sci. USA 109, 2989–2994 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim KH et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med 21, 1491–1496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knutson SK et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl Acad. Sci. USA 110, 7922–7927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first publication describing the EZH2 inhibitor EPZ-6438 (tazemetostat) that subsequently gained FDA approval.
- 62.Wilson BG et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18, 316–328 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Versteege I et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394, 203–206 (1998). [DOI] [PubMed] [Google Scholar]
- 64.Hasselblatt M et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am. J. Surg. Pathol 35, 933–935 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Modena P et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 65, 4012–4019 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Bitler BG et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med 21, 231–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ernst T et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet 42, 722–726 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Wu G et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and nonbrainstem glioblastomas. Nat. Genet 44, 251–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lewis PW et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study described the dominant negative activity of histone oncomutations, with significant mechanistic implications for pediatric cancers.
- 70.Mohammad F et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med 23, 483–492 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Piunti A et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med 23, 493–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaniskan H, Konze KD & Jin J Selective inhibitors of protein methyltransferases. J. Med. Chem 58, 1596–1629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Knutson SK et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol 8, 890–896 (2012). [DOI] [PubMed] [Google Scholar]
- 74.McCabe MT et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012). [DOI] [PubMed] [Google Scholar]; This study, along with Knutson, S. K. et al. (2012), describes the discovery of the first EZH2 inhibitors, which set the stage for tazemetostat to ultimately receive FDA approval as the first KMT inhibitory medicine.
- 75.Jiao L & Liu X Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350, aac4383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Justin N et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun 7, 11316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brooun A et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat. Commun 7, 11384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kasinath V et al. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 359, 940–944 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ciferri C et al. Molecular architecture of human polycomb repressive complex 2. eLife 1, e00005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vaswani RG et al. Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl) ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J. Med. Chem 59, 9928–9941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kung PP et al. Design and synthesis of pyridonecontaining 3,4-dihydroisoquinoline-1(2H)-ones as a novel class of enhancer of zeste homolog 2 (EZH2) inhibitors. J. Med. Chem 59, 8306–8325 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Ma A et al. Discovery of a first-in-class EZH2 selective degrader. Nat. Chem. Biol 16, 214–222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang X et al. Structure-activity relationship studies for enhancer of zeste homologue 2 (EZH2) and enhancer of zeste homologue 1 (EZH1) inhibitors. J. Med. Chem 59, 7617–7633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Konze KD et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol 8, 1324–1334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao Y et al. EZH2 cooperates with gain-of-function p53 mutants to promote cancer growth and metastasis. EMBO J. 38, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim J et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25, 2808–2820 e2804 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang L et al. Blocking immunosuppressive neutrophils deters pY696-EZH2-driven brain metastases. Sci. Transl Med 12, eaaz5387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morschhauser F et al. Phase 2 multicenter study of tazemetostat, an EZH2 inhibitor, in patients with relapsed or refractory follicular lymphoma blood. Blood 134 (Suppl. 1), 123 (2019).30862646 [Google Scholar]
- 89.Zauderer MG et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with relapsed or refractory (R/R) malignant mesothelioma (MM) with BAP1 inactivation. J. Clin. Oncol 36 (Suppl. 15), 8515 (2018). [Google Scholar]
- 90.Stacchiotti S et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES) (NCT02601950). J. Clin. Oncol 37 (Suppl. 15), 11003 (2019). [Google Scholar]
- 91.Gounder MM et al. Immunologic correlates of the abscopal effect in a SMARCB1/INI1-negative poorly differentiated chordoma after EZH2 inhibition and radiotherapy. Clin. Cancer Res 25, 2064–2071 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Taplin M-E et al. Phase Ib results of ProSTAR: CPI-1205, EZH2 inhibitor, combined with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration-resistant prostate cancer (mCRPC). Cancer Res. 79 (Suppl. 13), CT094 (2019). [Google Scholar]
- 93.Yap TA et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin. Cancer Res 25, 7331–7339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu B et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 125, 346–357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Honma D et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 108, 2069–2078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yamagishi M et al. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 29, 2321–2337.e2327 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Hansen KH et al. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol 10, 1291–1300 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Kim W et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol 9, 643–650 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xu C et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc. Natl Acad. Sci. USA 107, 19266–19271 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qi W et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol 13, 381–388 (2017). [DOI] [PubMed] [Google Scholar]
- 101.He Y et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol 13, 389–395 (2017). [DOI] [PubMed] [Google Scholar]; This study, along with Qi, W. et al., describes the discovery of the first protein–protein interaction disrupting EED inhibitors that function as allosteric inhibitors of PRC2.
- 102.Basheer F et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J. Exp. Med 216, 966–981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karantanos T, Chistofides A, Barhdan K, Li L & Boussiotis VA Regulation of T cell differentiation and function by EZH2. Front. Immunol 7, 172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qiu J, Sharma S, Rollins RA & Paul TA The complex role of EZH2 in the tumor microenvironment: opportunities and challenges for immunotherapy combinations. Future Med. Chem 10.4155/fmc-2020-0072 (2020). [DOI] [PubMed] [Google Scholar]
- 105.Hamaidia M et al. Inhibition of EZH2 methyltransferase decreases immunoediting of mesothelioma cells by autologous macrophages through a PD-1-dependent mechanism. JCI Insight 4, e128474 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xiao G et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J. Immunother. Cancer 7, 300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou L, Mudianto T, Ma X, Riley R & Uppaluri R Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents Anti-PD-1 resistance in head and neck cancer. Clin. Cancer Res 26, 290–300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goswami S et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Invest 128, 3813–3818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zingg D et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 20, 854–867 (2017). [DOI] [PubMed] [Google Scholar]
- 110.Vlaming H & van Leeuwen F The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma 125, 593–605 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Min J, Feng Q, Li Z, Zhang Y & Xu RM Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723 (2003). [DOI] [PubMed] [Google Scholar]
- 112.Jones B et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 4, e1000190 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deshpande AJ et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell 26, 896–908 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bernt KM et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krivtsov AV, Hoshii T & Armstrong SA Mixed-lineage leukemia fusions and chromatin in leukemia. Cold Spring Harb. Perspect. Med 7, a026658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Okada Y et al. hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178 (2005). [DOI] [PubMed] [Google Scholar]; This study identified a role for human DOT1L, via H3K79 methylation, in promoting MLL-driven leukaemia.
- 117.Mohan M et al. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 24, 574–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen S et al. The PZP domain of AF10 senses unmodified H3K27 to regulate DOT1L-mediated methylation of H3K79. Mol. Cell 60, 319–327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li Y et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Erb MA et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wan L et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Godfrey L et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat. Commun 10, 2803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Briggs SD et al. Gene silencing: trans-histone regulatory pathway in chromatin. Nature 418, 498 (2002). [DOI] [PubMed] [Google Scholar]
- 124.McGinty RK, Kim J, Chatterjee C, Roeder RG & Muir TW Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 453, 812–816 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Altaf M et al. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol. Cell 28, 1002–1014 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fingerman IM, Li HC & Briggs SD A chargebased interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: identification of a new trans-histone pathway. Genes Dev. 21, 2018–2029 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McGinty RK et al. Structure-activity analysis of semisynthetic nucleosomes: mechanistic insights into the stimulation of Dot1L by ubiquitylated histone H2B. ACS Chem. Biol 4, 958–968 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Anderson CJ et al. Structural basis for recognition of ubiquitylated nucleosome by Dot1L methyltransferase. Cell Rep. 26, 1681–1690.e1685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jang S et al. Structural basis of recognition and destabilization of the histone H2B ubiquitinated nucleosome by the DOT1L histone H3 Lys79 methyltransferase. Genes Dev. 33, 620–625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Valencia-Sánchez MI et al. Structural basis of Dot1L stimulation by histone H2B lysine 120 ubiquitination. Mol. Cell 74, 1010–1019.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Worden EJ, Hoffmann NA, Hicks CW & Wolberger C Mechanism of cross-talk between H2B ubiquitination and H3 methylation by Dot1L. Cell 176, 1490–1501.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yao T et al. Structural basis of the crosstalk between histone H2B monoubiquitination and H3 lysine 79 methylation on nucleosome. Cell Res. 29, 330–333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Deshpande AJ et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 121, 2533–2541 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Daigle SR et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Krivtsov AV et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14, 355–368 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gu Y et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 71, 701–708 (1992). [DOI] [PubMed] [Google Scholar]
- 137.Tkachuk DC, Kohler S & Cleary ML Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 71, 691–700 (1992). [DOI] [PubMed] [Google Scholar]
- 138.Meyer C et al. The MLL recombinome of acute leukemias in 2017. Leukemia 32, 273–284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Corral J et al. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell 85, 853–861 (1996). [DOI] [PubMed] [Google Scholar]
- 140.Ayton PM & Cleary ML Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 17, 2298–2307 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zeisig BB et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol. Cell. Biol 24, 617–628 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brzezinka K et al. Functional diversity of inhibitors tackling the differentiation blockage of MLL-rearranged leukemia. J. Hematol. Oncol 12, 66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nguyen AT, Taranova O, He J & Zhang Y DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 117, 6912–6922 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Daigle SR et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides the first example of a highly selective histone lysine methyltransferase inhibitor with in vivo efficacy in a cancer model.
- 145.Chen L et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 27, 813–822 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Basavapathruni A et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem. Biol. Drug Des 80, 971–980 (2012). [DOI] [PubMed] [Google Scholar]
- 147.Chen C et al. Discovery of novel Dot1L inhibitors through a structure-based fragmentation approach. ACS Med. Chem. Lett 7, 735–740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Scheufler C et al. Optimization of a fragment-based screening hit toward potent DOT1L inhibitors interacting in an induced binding pocket. ACS Med. Chem. Lett 7, 730–734 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shukla N et al. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-r acute leukemia. Blood 128, 2780 (2016). [Google Scholar]
- 150.Stein EM et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131, 2661–2669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Klaus CR et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther 350, 646–656 (2014). [DOI] [PubMed] [Google Scholar]
- 152.Rau RE et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 128, 971–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Menghrajani K et al. A phase Ib/II study of the histone methyltransferase inhibitor pinometostat in combination with azacitidine in patients with 11q23-rearranged acute myeloid leukemia. Blood 134, 2655 (2019). [Google Scholar]
- 154.Secker KA et al. Inhibition of DOT1L and PRMT5 promote synergistic anti-tumor activity in a human MLL leukemia model induced by CRISPR/Cas9. Oncogene 38, 7181–7195 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Skucha A et al. MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nat. Commun 9, 1983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Borkin D et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 27, 589–602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Borkin D et al. Complexity of blocking bivalent protein-protein interactions: development of a highly potent inhibitor of the menin-mixed-lineage leukemia interaction. J. Med. Chem 61, 4832–4850 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tachibana M, Sugimoto K, Fukushima T & Shinkai Y Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem 276, 25309–25317 (2001). [DOI] [PubMed] [Google Scholar]
- 159.Tachibana M et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tachibana M et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liu N et al. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. 29, 379–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Collins RE et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol 15, 245–250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tu WB et al. MYC Interacts with the G9a histone methyltransferase to drive transcriptional repression and tumorigenesis. Cancer Cell 34, 579–595.e578 (2018). [DOI] [PubMed] [Google Scholar]
- 164.Kato S et al. Gain-of-function genetic alterations of G9a drive oncogenesis. Cancer Discov. 10.1158/2159-8290.CD-19-0532 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang Z et al. SETD5-coordinated chromatin reprogramming regulates adaptive resistance to targeted pancreatic cancer therapy. Cancer Cell 37, 834–849.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Watson ZL et al. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin. Epigenet 11, 165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Maze I et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Anderson EM et al. Knockdown of the histone di-methyltransferase G9a in nucleus accumbens shell decreases cocaine self-administration, stress-induced reinstatement, and anxiety. Neuropsychopharmacology 44, 1370–1376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Avgustinova A et al. Loss of G9a preserves mutation patterns but increases chromatin accessibility, genomic instability and aggressiveness in skin tumours. Nat. Cell Biol 20, 1400–1409 (2018). [DOI] [PubMed] [Google Scholar]
- 170.Rowbotham SP et al. H3K9 methyltransferases and demethylases control lung tumor-propagating cells and lung cancer progression. Nat. Commun 9, 4559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kubicek S et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481 (2007). [DOI] [PubMed] [Google Scholar]; This study provides the first example of a substrate-competitive, selective lysine methyltransferase inhibitor.
- 172.Dong C et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Invest 122, 1469–1486 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chang Y et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol 16, 312–317 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vedadi M et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol 7, 566–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided the first example of a selective, cellular chemical probe of G9a/GLP.
- 175.Liu F et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem 56, 8931–8942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sweis RF et al. Discovery and development of potent and selective inhibitors of histone methyltransferase g9a. ACS Med. Chem. Lett 5, 205–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xiong Y et al. Discovery of potent and selective inhibitors for G9a-like protein (GLP) lysine methyltransferase. J. Med. Chem 60, 1876–1891 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ferry L et al. Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol. Cell 67, 550–565.e555 (2017). [DOI] [PubMed] [Google Scholar]
- 179.Fang J et al. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol 12, 1086–1099 (2002). [DOI] [PubMed] [Google Scholar]
- 180.Nishioka K et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213 (2002). [DOI] [PubMed] [Google Scholar]
- 181.van Nuland R & Gozani O Histone H4 lysine 20 (H4K20) methylation, expanding the signaling potential of the proteome one methyl moiety at a time. Mol. Cell Proteom 15, 755–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shi X et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kudithipudi S, Dhayalan A, Kebede AF & Jeltsch A The SET8 H4K20 protein lysine methyltransferase has a long recognition sequence covering seven amino acid residues. Biochimie 94, 2212–2218 (2012). [DOI] [PubMed] [Google Scholar]
- 184.McKay DJ et al. Interrogating the function of metazoan histones using engineered gene clusters. Dev. Cell 32, 373–386 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Milite C et al. The emerging role of lysine methyltransferase SETD8 in human diseases. Clin. Epigenet 8, 102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Couture JF, Collazo E, Brunzelle JS & Trievel RC Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 19, 1455–1465 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xiao B et al. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 19, 1444–1454 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Blum G et al. Small-molecule inhibitors of SETD8 with cellular activity. ACS Chem. Biol 9, 2471–2478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ma A et al. Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem 57, 6822–6833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ma A et al. Structure-activity relationship studies of SETD8 inhibitors. Medchemcomm 5, 1892–1898 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Veschi V et al. Epigenetic siRNA and chemical screens identify SETD8 inhibition as a therapeutic strategy for p53 activation in high-risk neuroblastoma. Cancer Cell 31, 50–63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wu J et al. Downregulation of histone methyltransferase SET8 inhibits progression of hepatocellular carcinoma. Sci. Rep 10, 4490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Butler KV et al. Structure-based design of a covalent inhibitor of the SET domain-containing protein 8 (SETD8) lysine methyltransferase. J. Med. Chem 59, 9881–9889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Oda H et al. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell. Biol 29, 2278–2295 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pannetier M et al. PR-SET7 and SUV4–20H regulate H4 lysine-20 methylation at imprinting control regions in the mouse. EMBO Rep. 9, 998–1005 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schotta G et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251–1262 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Schotta G et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 22, 2048–2061 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Southall SM, Cronin NB & Wilson JR A novel route to product specificity in the Suv4-20 family of histone H4K20 methyltransferases. Nucleic Acids Res. 42, 661–671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wu H et al. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep. 5, 13–20 (2013). [DOI] [PubMed] [Google Scholar]
- 200.Gonzalo S et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat. Cell Biol 7, 420–428 (2005). [DOI] [PubMed] [Google Scholar]
- 201.Lu X et al. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol 15, 1122–1124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Long H et al. H2A.Z facilitates licensing and activation of early replication origins. Nature 577, 576–581 (2020). [DOI] [PubMed] [Google Scholar]
- 203.Sanders SL et al. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119, 603–614 (2004). [DOI] [PubMed] [Google Scholar]
- 204.Yang H et al. Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J. Biol. Chem 283, 12085–12092 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bromberg KD et al. The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat. Chem. Biol 13, 317–324 (2017). [DOI] [PubMed] [Google Scholar]
- 206.Wang H et al. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 8, 1207–1217 (2001). [DOI] [PubMed] [Google Scholar]
- 207.Nishioka K et al. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 16, 479–489 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lehnertz B et al. p53-dependent transcription and tumor suppression are not affected in Set7/9-deficient mice. Mol. Cell 43, 673–680 (2011). [DOI] [PubMed] [Google Scholar]
- 209.Barsyte-Lovejoy D et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl Acad. Sci. USA 111, 12853–12858 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hamidi T et al. Identification of Rpl29 as a major substrate of the lysine methyltransferase Set7/9. J. Biol. Chem 293, 12770–12780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Komatsu S et al. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br. J. Cancer 112, 357–364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Komatsu S et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 30, 1139–1146 (2009). [DOI] [PubMed] [Google Scholar]
- 213.Reynoird N et al. Coordination of stress signals by the lysine methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev. 30, 772–785 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Olsen JB et al. Quantitative profiling of the activity of protein lysine methyltransferase SMYD2 Using SILAC-based proteomics. Mol. Cell Proteom 15, 892–905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bagislar S et al. Smyd2 is a Myc-regulated gene critical for MLL-AF9 induced leukemogenesis. Oncotarget 7, 66398–66415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cowen SD et al. Design, synthesis, and biological activity of substrate competitive SMYD2 inhibitors. J. Med. Chem 59, 11079–11097 (2016). [DOI] [PubMed] [Google Scholar]
- 217.Ferguson AD et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 19, 1262–1273 (2011). [DOI] [PubMed] [Google Scholar]
- 218.Sweis RF et al. Discovery of A-893, a new cell-active benzoxazinone inhibitor of lysine methyltransferase SMYD2. ACS Med. Chem. Lett 6, 695–700 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Nguyen H et al. LLY-507, a cell-active, potent, and selective inhibitor of protein-lysine methyltransferase SMYD2. J. Biol. Chem 290, 13641–13653 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Eggert E et al. Discovery and characterization of a highly potent and selective aminopyrazoline-based in vivo probe (BAY-598) for the protein lysine methyltransferase SMYD2. J. Med. Chem 59, 4578–4600 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Huang J et al. Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632 (2006). [DOI] [PubMed] [Google Scholar]
- 222.Thomenius MJ et al. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS ONE 13, e0197372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mazur PK et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified a role for protein lysine methylation in integrating MAP kinase signaling in the cytoplasm to promote pancreatic and lung cancer.
- 224.Sarris ME, Moulos P, Haroniti A, Giakountis A & Talianidis I Smyd3 Is a transcriptional potentiator of multiple cancer-promoting genes and required for liver and colon cancer development. Cancer Cell 29, 354–366 (2016). [DOI] [PubMed] [Google Scholar]
- 225.Hamamoto R et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol 6, 731–740 (2004). [DOI] [PubMed] [Google Scholar]
- 226.Fabini E et al. Unveiling the biochemistry of the epigenetic regulator SMYD3. Biochemistry 58, 3634–3645 (2019). [DOI] [PubMed] [Google Scholar]
- 227.Van Aller GS et al. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 7, 340–343 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kunizaki M et al. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 67, 10759–10765 (2007). [DOI] [PubMed] [Google Scholar]
- 229.Van Aller GS et al. Structure-based design of a novel SMYD3 inhibitor that bridges the SAM- and MEKK2-binding pockets. Structure 24, 774–781 (2016). [DOI] [PubMed] [Google Scholar]
- 230.Mitchell LH et al. Novel oxindole sulfonamides and sulfamides: EPZ031686, the first orally bioavailable small molecule SMYD3 inhibitor. ACS Med. Chem. Lett 7, 134–138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Huang C et al. Discovery of irreversible inhibitors targeting histone methyltransferase, SMYD3. ACS Med. Chem. Lett 10, 978–984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bennett RL, Swaroop A, Troche C & Licht JD The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harb. Perspect. Med 7, a026708 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yang S et al. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 30, 1611–1616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Böttcher J et al. Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat. Chem. Biol 15, 822–829 (2019). [DOI] [PubMed] [Google Scholar]
- 235.Sun Y et al. Histone methyltransferase SETDB1 is required for prostate cancer cell proliferation, migration and invasion. Asian J. Androl 16, 319–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wong CM et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology 63, 474–487 (2016). [DOI] [PubMed] [Google Scholar]
- 237.Zhu Y, Sun D, Jakovcevski M & Jiang Y Epigenetic mechanism of SETDB1 in brain: implications for neuropsychiatric disorders. Transl Psychiatry 10, 115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kim Y, Wang SE & Jiang YH Epigenetic therapy of Prader-Willi syndrome. Transl Res. 208, 105–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Woodcock CB, Yu D, Zhang X & Cheng X Human HemK2/KMT9/N6AMT1 is an active protein methyltransferase, but does not act on DNA in vitro, in the presence of Trm112. Cell Discov 5, 50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kusevic D, Kudithipudi S & Jeltsch A Substrate specificity of the HEMK2 protein glutamine methyltransferase and identification of novel substrates. J. Biol. Chem 291, 6124–6133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Figaro S, Scrima N, Buckingham RH & Heurgué-Hamard V HemK2 protein, encoded on human chromosome 21, methylates translation termination factor eRF1. FEBS Lett. 582, 2352–2356 (2008). [DOI] [PubMed] [Google Scholar]
- 242.Allali-Hassani A et al. Discovery of a chemical probe for PRDM9. Nat. Commun 10, 5759 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the first inhibitor of PRDM9, a potential target in multiple cancers.
- 243.Houle AA et al. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. 28, 1611–1620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Powers NR et al. The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLoS Genet. 12, e1006146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mzoughi S, Tan YX, Low D & Guccione E The role of PRDMs in cancer: one family, two sides. Curr. Opin. Genet. Dev 36, 83–91 (2016). [DOI] [PubMed] [Google Scholar]
- 246.Roqueta-Rivera M et al. SETDB2 links glucocorticoid to lipid metabolism through Insig2a regulation. Cell Metab 24, 474–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sessa A et al. SETD5 regulates chromatin methylation state and preserves global transcriptional fidelity during brain development and neuronal wiring. Neuron 104, 271–289.e213 (2019). [DOI] [PubMed] [Google Scholar]
- 248.Mas YMS et al. The human mixed lineage leukemia 5 (MLL5), a sequentially and structurally divergent set domain-containing protein with no intrinsic catalytic activity. PLoS ONE 11, e0165139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Robichaud N, Sonenberg N, Ruggero D & Schneider RJ Translational control in cancer. Cold Spring Harb. Perspect. Biol 11, 254–266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Jakobsson ME, Małecki J & Falnes P Regulation of eukaryotic elongation factor 1 alpha (eEF1A) by dynamic lysine methylation. RNA Biol. 15, 314–319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Jakobsson ME et al. Methylation of human eukaryotic elongation factor alpha (eEF1A) by a member of a novel protein lysine methyltransferase family modulates mRNA translation. Nucleic Acids Res. 45, 8239–8254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Davydova E et al. Identification and characterization of a novel evolutionarily conserved lysine-specific methyltransferase targeting eukaryotic translation elongation factor 2 (eEF2). J. Biol. Chem 289, 30499–30510 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhang L, Hamey JJ, Hart-Smith G, Erce MA & Wilkins MR Elongation factor methyltransferase 3 – a novel eukaryotic lysine methyltransferase. Biochem. Biophys. Res. Commun 451, 229–234 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.