

Abstract
BACKGROUND:
Pancreatic cancer is characterized by extensive metastasis. EMT plasticity plays a critical role in tumor progression and metastasis by maintaining the transition between EMT and MET states. Our aim is to understand the molecular events regulating metastasis and EMT plasticity in pancreatic cancer.
METHODS:
The interactions between a cancer promoting zinc transporter ZIP4, a zinc dependent EMT transcriptional factor ZEB1, a co-activator YAP1, and integrin α3 (ITGA3) were examined in human pancreatic cancer cells, clinical specimens, spontaneous mouse models (KPC and KPCZ) and orthotopic xenografts, and 3D spheroid and organoid models. Correlations between ZIP4, miR-373, and its downstream targets were assessed by RNA in situ hybridization and IHC staining. The transcriptional regulation of ZEB1, YAP1, ITGA3 by ZIP4 was determined by ChIP, Co-IP and luciferase reporter assays.
RESULTS:
The Hippo pathway effector YAP1 is a potent transcriptional co-activator and forms a complex with ZEB1 to activate ITGA3 transcription through the YAP1/TEAD binding sites in human pancreatic cancer cells and KPC derived mouse cells. ZIP4 upregulated YAP1 expression via activation of miR-373 and inhibition of the YAP1 repressor LATS2. Furthermore, upregulation of ZIP4 promoted EMT plasticity, cell adhesion, spheroid formation and organogenesis both in human pancreatic cancer cells, 3D spheroid model, xenograft model, and spontaneous mouse models (KPC and KPCZ) through ZEB1/YAP1-ITGA3 signaling axis.
CONCLUSION:
We demonstrated that ZIP4 activates ZEB1 and YAP1 through distinct mechanisms. The ZIP4-miR-373-LATS2-ZEB1/YAP1-ITGA3 signaling axis has a significant impact on pancreatic cancer metastasis and EMT plasticity.
Keywords: Transcription co-activation, zinc homeostasis, post-transcriptional regulation
Lay Summary
This study defines a novel ZIP4-miR-373-LATS2-ZEB1/YAP1-ITGA3 signaling pathway that meditates pancreatic cancer metastasis and EMT plasticity. The findings are highly translational to develop new personalized therapy for pancreatic cancer.
Graphical Abstract
Introduction
Pancreatic cancer is the third leading cause of cancer-related death in the United States, and has an overall survival rate of only 9%1. Over 60% of pancreatic cancer patients had distant metastasis within the first 24 months after surgery2. Pancreatic cancer metastasis requires cell detachment, migration, invasion and adhesion. Epithelial-mesenchymal transition (EMT) is an essential process for tumor metastasis, and is activated by pathogenic stimuli and promotes the whole course of early to late stage metastasis. EMT plasticity is a dynamic program which transits between EMT and MET phenotypes to facilitate tumor metastasis3. At the same time adhesion is required for endothelial cells to form stronger bonds, attach at the new location, and proliferate to produce the secondary tumor4. Integrins are transmembrane receptors which are upregulated on cell surface and correlate with increased metastatic capacity of tumor cells5. Control of EMT and cell adhesion is essential for regulation of pancreatic cancer metastasis.
EMT is mediated by a set of EMT-activating transcription factors (EMT-TFs) including Snail, Slug, Twist1, ZEB1 and ZEB2. Most EMT TFs except Twist1 are zinc finger proteins6. These factors are overexpressed in many cancers including bladder, breast, colon and pancreatic cancer. EMT TFs act as transcription repressors by reducing epithelial markers during embryonic development and tumor progression7–9. ZEB1 is the most important EMT-TFs in pancreatic cancer which promotes stemness, invasion and metastasis in pancreatic cancer as shown in the KPC and KPC ZEB1 knockout mouse models10. ZEB1 normally acts as a transcription repressor to inhibit downstream target transcription including E-cadherin and miR-200 family members11 through binding to E-box motif at their promoter regions. However, ZEB1 can also switch from a transcription repressor to transcription activator upon interacting with co-activators such as Lef1, YAP1, P300 and Smad, which belong to other regulatory and cancer promoting pathways such as Hippo and Wnt pathway12–14. Hippo pathway controls cell shape, organ size, tissue regeneration, tumorigenesis, EMT and cell adhesion. YAP1 is a major downstream effector of Hippo pathway and activation of YAP1 leads to lung, colon and pancreatic cancer carcinogenesis and tumor progress15–17. However, how these two pathways interact and what functional impact this interaction has remains unknown in pancreatic cancer. The large tumor suppressor 2 kinase (LATS2) is one of the Hippo pathway kinases which directly phosphorylates YAP1 and inhibits YAP1 activity by protein degradation, thereby inhibiting transcription of YAP1’s downstream targets. Moreover, inhibition of LATS2 increases the YAP1/TEAD interaction, leading to YAP1/TEAD transcriptional activation18. YAP1/TEAD complex activates their oncogenic downstream genes such as CTGF and c-Myc leading to gastric cancer carcinogenesis19. Several studies show that miRNAs including let-7, miR-10b, and miR-183, are involved in the regulation of the Hippo pathway20, 21. LATS2 is a direct target of miR-373 in pancreatic cancer and inhibition of LATS2 promoted tumor growth22. The specific mechanism of miRNA-LATS2 regulation in pancreatic cancer also remain to be elucidated.
In the current study, we identified a novel function of a cancer promoting zinc transporter ZIP4 in EMT plasticity and cell-ECM adhesion induced by ZEB1-integrin in both 2D and 3D culture conditions. We found that ZIP4 promoted EMT plasticity and pancreatic cancer cell-laminin adhesion and enhanced spheroid and organoid formation through activating integrins. We also found that knock down of ZEB1 significantly inhibited organogenesis and apical to basal polarity in a 3D organoid model derived from KPC ZEB1 knock out cell lines. We further elucidated the mechanism showing that ZEB1 activated ITGA3 through binding to the YAP1/TEAD binding sites at ITGA3 promoter region and ZIP4 upregulated YAP1 expression via miR-373-LATS2 axis. These findings show a novel molecular link between ZIP4 and EMT, cell-ECM adhesion, and pancreatic cancer organogenesis, this essential link may serve as the foundation for the development of new therapeutic strategies for this devastating disease.
Materials and Methods
Cell lines, siRNAs and plasmids.
Human pancreatic cancer cell lines AsPC-1 and MIA PaCa-2 were purchased from American Type Culture Collection (ATCC, Rockville, MD), and were cultured in RPMI 1640 medium or DMEM medium with 10% fetal bovine serum (FBS). KPC and KPC-ZEB1 knockout (KPCZ) cells were kindly provided by Dr. Thomas Brabletz, University of Erlangen-Nürnberg, Germany. All cell lines were authenticated and verified to be mycoplasma free using MycoAlert™ kit (Lonza). ITGA3, YAP1, LATS2 siRNAs were purchased from Santa Cruz, CA. Mimic and inhibitor of miR-373–3p were purchased from Ambion. hZEB1 and mYAP1 ORF plasmids were purchased from Genecopoeia, while the hYAP1 ORF plasmid was obtained from Addgene (catalog number 42555).
Human tissue samples.
Human pancreatic cancer tissue specimens were obtained from the University of Oklahoma Health Sciences Center (OUHSC). This study was approved by the Institutional Review Board (IRB) at OUHSC. Banked de-identified tissues were used. Written consent from all subjects was obtained.
Stable cell line construction.
Stable pancreatic cancer cell lines MIA-V, MIA-ZIP4, AsPC-shV, AsPC-shZIP4, MIA-ZIP4-Pre-C, MIA-ZIP4-Pre-miR-373, AsPC-shZIP4-Anti-C, AsPC-shZIP4-Anti-miR-373 cells were generated as previously described22–24. The MIA-ZEB1/YAP1 overexpression stable cell line was selected using the lentivirus vector from Genecopoeia and Addgene following the manufacturer’s instructions. Briefly, human ZEB1 ORF was cloned into the pEZ-Lv207 vector and human YAP1 ORF was cloned into pLX304 vector. Viral supernatants were collected and transduced to the target cells. Stable cell lines expressing ZEB1 were selected using 200μg/ml Hygromycin B and stable cell lines expressing YAP1 were selected with 10μg/ml Blasticidin.
Spheroid formation assay.
Cells were resuspended in culture medium containing 0.24% methylcellulose (Sigma, M0512). And then were seeded onto inside of the lid of 10 cm2 dishes with 20 μl/drop. Lids were inverted over dishes containing 10 ml PBS. Spheroids were cultured at 37 °C and then imaged using a light microscope at 2, 4, and 24 h post seeding. 10 spheroids were included and analyzed in each group.
Three-dimensional organoid culture.
Matrigel embedded organoid: The chamber slides were pre-coated with growth factor reduced Matrigel (Corning) at 37°C for 2 h. After rinsing with PBS, the single cell suspension mixed with 10 ng/ml EGF, 10μg/ml insulin and 2% Matrigel was seeded onto the chamber slide at 8×103 cells/well. And the slides were incubated at 37 °C overnight. The cells were cultured with fresh medium containing 10 ng/ml EGF and 10μg/ml insulin and replenished every three days. The cells formed clusters by day 6 to day 8.
Matrigel suspended organoid: The organoids were also generated by seeding the cell suspension with 50% Matrigel onto the 24 well plate with 5×105 cells/ml. And the 24 well plate was incubated at 37 °C overnight. The cells were refed as above and cells formed clusters at multiple layers by day 6 to day 8.
Chromatin Immunoprecipitation assay.
The chromatin immunoprecipitation (ChIP) assay was performed in AsPC-1 cells by using the anti-ZEB1, YAP1 and TEAD antibody (Cell Signaling Technology) with the MAGnif Chromatin Immunoprecipitation System (Life Technologies) following the standard protocol. After the antibody pulling down, the target DNA fragment was amplified and determined by PCR. Primers were designed within YAP1/TEAD binding sites at the ITGA3 promoter region.
Promoter activity assay.
ITGA3 promoter sequence was generated from USCS genomic browser. The 930 bp promoter regions of ITGA3 were cloned into pGL4.10-basic reporter vector as previously described25. Promoter sequence of ITGA3 was uploaded to JASPAR database. The threshold was set to 70%. YAP1/TEAD binding sites were predicted through JASPAR database26. The mutant ITGA3 promoter vector was constructed with YAP1/TEAD binding sites mutated at ITGA3 promoter region. The wild type (WT) and mutant ITGA3 promoter vector were co-transfected with control plasmid pRL-TK into AsPC-1 and MIA PaCa-2 cells. The promoter activity was determined by a Dual-Luciferase Reporter Assay (Promega).
Co-Immunoprecipitation.
MIA-ZEB1/YAP1 cells were lysed, and anti-ZEB1, YAP1 and TEAD (Cell Signaling Technology) antibodies (1–10 μg) were diluted in 200 μl of Ab binding & washing buffer and incubated with rotation for 10 min at room temperature and the magnetic bead-Ab complex was washed with washing buffer. The magnetic bead-Ab-Ag complex was incubated with rotation for 20 min at room temperature to allow the antigen to bind to the magnetic bead-Ab complex. 20 μl elution buffer and 10 μl of pre-mixed NuPAGE™ LDS sample buffer and NuPAGE sample reducing agent were added to the complex and heated for 10 min at 70°C. The supernatant containing eluted Ab and Ag was transferred to a clean tube and loaded onto the SDS gel for Western Blotting.
RNA Basescope in situ staining.
Pancreatic cancer tissues were freshly sectioned at 4–5μm. miR-373 BaseScope LS Reagents (Advanced Cell Diagnostics Inc., Newark, CA) were loaded onto the Staining System. Tissue sections were deparaffinized on the instrument, followed by epitope retrieval, miR-373 probe hybridization, signal amplification, colorimetric detection, and counterstaining. Dihydrodipicolinate reductase (DapB) and peptidyl-prolyl cis-trans isomerase B (PPIB) were used as the negative and positive control probes, respectively.
Pancreatic cancer xenograft mouse model.
MIA-ZIP4-Anti-C, MIA-ZIP4-Anti-miR-373, AsPC-shZIP4-Pre-C, AsPC-shZIP4-Pre-miR-373 MIA-ZIP4 shV, MIA-ZIP4 sh-ITGA3 stable cell lines were used to generate the orthotopic xenograft tumor model as previously described22, 25. Briefly stable pancreatic cancer cells were harvested and resuspended in DMEM or RPMI medium. 3×106 cells in 50 μl culture medium were injected into the pancreases of 5-to-6-week-old nude mouse. The peritoneum and skin were closed with 4.0 surgical sutures. All mice were cared for in accordance with the Office for Protection from Research Risks (OPRR) and Animal Welfare Act Guidelines under an animal protocol approved by the Animal Welfare Committee at OUHSC. After 5–6 weeks, all surviving mice were euthanized by CO2 asphyxiation and tumor tissues were collected and fixed in formalin.
Statistical analysis.
Quantitative results are shown as means ± SD. Overall difference among groups were assessed by ANOVA and post-hoc Dunnett’s multiple comparison tests were used to compare data from control and each treated groups. Two-group comparisons were analyzed by Student’s t-tests. A P value of < .05 was considered statistically significant. All tests were two-sided. Further details of the Materials and Methods will be found in the Supplementary Materials and Methods.
Results
YAP1 is a potent co-activator of ZEB1 to enhance ITGA3 transcription in pancreatic cancer cells.
Previously we have shown that a zinc dependent activation of ZEB1 promotes pancreatic cancer growth and chemoresistance25. We thus sought to determine how ZEB1 activates the integrin pathway and promotes pancreatic cancer progression, metastasis, and EMT plasticity. ZEB1 switches from a transcription repressor to a transcription activator when co-activators are recruited to the same locus in the promoter region, therefore we examined the potential co-activators of ZEB1 in human pancreatic cancer cells. YAP1, Lef1, CAF, and P300 have been reported to serve as transcriptional co-activators in several cancers12–14, and were upregulated in pancreatic cancer compared with normal pancreas as shown in TCGA and GTEx database (Fig. S1A). We knocked down all four co-activators; only YAP1 knockdown significantly inhibited the promoter activity and reduced the mRNA level of ITGA3 in MIA PaCa-2 and AsPC-1cells (Fig. 1A–1B, Fig. S1B–S1C). The TCGA database also showed that YAP1 positively correlates with ZIP4, ZEB1, and ITGA3 (Fig. S1D–S1F). ZIP4 also upregulated YAP1 both in human pancreatic cancer cell lines and xenograft tumor tissues (Fig. S1G–S1J). We thus concluded that YAP1 is the potential co-activator of ZEB1, and can be upregulated by ZIP4 in pancreatic cancer. We then performed gene set enrichment analysis (GSEA) of ZEB1 and YAP1 expression profiles in the TCGA database and found that gene sets of YAP1 are significantly enriched in ZEB1 highly expressed group (Fig. S1K). We analyzed the promoter region of ITGA3 and found YAP1 was enriched and shared the transcriptional peak call with ZEB1 at the same locus of ITGA3 promoter according to the ChIP sequencing data (Fig. S1L). These data show that ZEB1-mediated upregulation of ITGA3 is dependent on the YAP1 status in pancreatic cancer.
Fig. 1. ZEB1 activates ITGA3 transcription through YAP1’s binding sites at ITGA3 promoter.
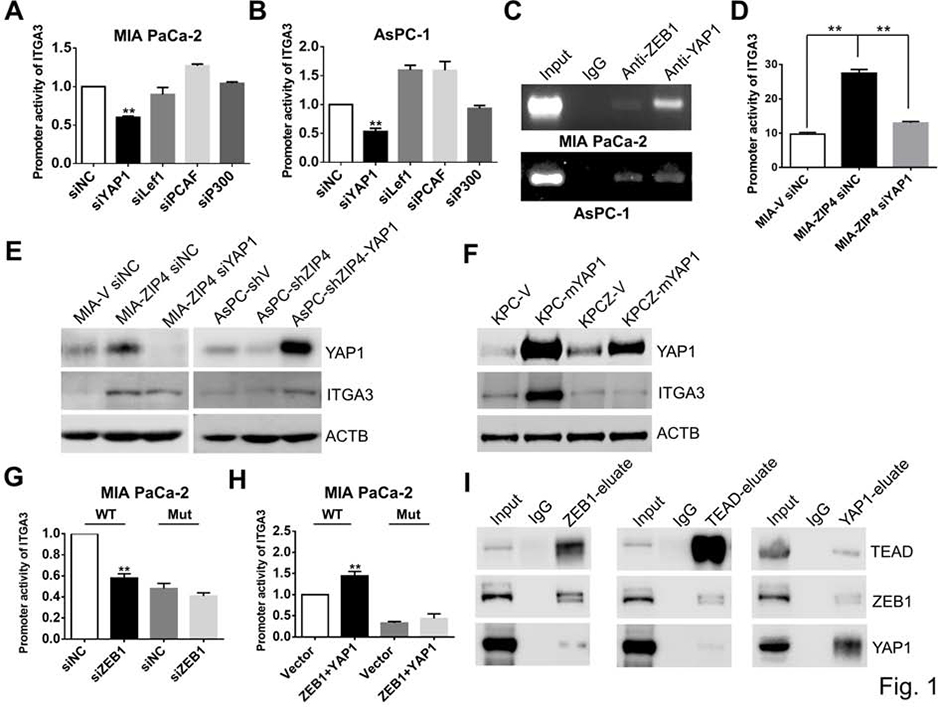
(A). Promoter activity of ITGA3 in MIA PaCa-2 siYAP1, siLef1, siCAF and siP300 cells. (B). Promoter activity of ITGA3 in AsPC-1 siYAP1, siLef1, siCAF and siP300 cells. (C). ChIP-PCR. ChIP binding assay with anti-ZEB1 and anti-YAP1 in MIA PaCa-2 and AsPC-1 cells confirms the binding of ZEB1and YAP1 to the ITGA3 promoter region. (D). Promoter activity of ITGA3 in MIA-V siNC, MIA-ZIP4 siNC and MIA-ZIP4 siYAP1 cells. ** means P< .01. (E). Expression of YAP1 and ITGA3 in PC cells with YAP1 high or low. (F). Expression of YAP1 and ITGA3 in KPC and KPCZ cells with mYAP1 overexpressed. Empty vector and mYAP1 ORF vector were transiently transfected into KPC and KPCZ cells. (G). Promoter activity of ITGA3 in MIA PaCa-2 cell transfected with WT or mutation vector (YAP1 binding sites). siRNA of ZEB1 was co-transfected with ITGA3 promoter vector. ** means P< .01. (H). Promoter activity of ITGA3 in MIA PaCa-2 cell transfected with WT or mutation vector (YAP1 binding sites). ORF vectors of ZEB1/YAP1 was co-transfected with ITGA3 promoter vector. ** means P< .01. (I). Co-IP in MIA PaCa-2-ZEB1/YAP1 cells.
ZEB1 activates ITGA3 transcription through YAP1/TEAD binding sites.
To further investigate whether YAP1 directly activates ITGA3 in pancreatic cancer, we performed ChIP-PCR assay; ZEB1 and YAP1 bound to the same region of the ITGA3 promoter (Fig. 1C). Based on the luciferase reporter assay results we found that knockdown of YAP1 inhibited the promoter activity of ITGA3 even when ZIP4 was overexpressed (Fig. 1D), indicating that the transcriptional activation of ITGA3 may require the presence of YAP1. We also found blocking of YAP1 through siRNA decreased ITGA3 expression and overexpression of YAP1 rescued the inhibition of ZIP4 on ITGA3 expression (Fig. 1E, Fig. S2A–S2B). We also overexpressed mouse YAP1 (mYAP1) in wild type KPC and ZEB1 knockout KPCZ mouse cell lines; overexpression of mYAP1 increased ITGA3 level in KPC cells but not in KPCZ cells (Fig. 1F, Fig. S2C–S2D), suggesting that the YAP1 activated ITGA3 requires ZEB1. Although YAP1 is a transcriptional co-activator, it does not have DNA-binding domains but regulates gene expression through TEAD1–427. The JASPAR database26 predicts there are 10 TEAD1–4 binding sites in the ITGA3 promoter. We generated double mutations at the binding sites which are with the highest scores at JASPAR database (Fig. S2E), and found that sequential mutation of the TEAD binding sites inhibited co-stimulation of the ITGA3 promoter by ZEB1 and YAP1 (Fig. 1G–1H, Fig. S2F–S2G). This indicate that the YAP1/TEAD binding sites are essential for ZEB1 stimulation of ITGA3 transcription. We further investigated whether ZEB1 and YAP1 directly interact to co-activate ITGA3. We performed Co-IP assays and as expected YAP1 and TEAD directly interacted with each other; ZEB1 and TEAD also interacted in the pancreatic cancer cells (Fig. 1I). It is established that the protein structure of YAP1 contains several domains including a WW domain, a TEAD transcription factor interacting domain, a transcription activation domain and a PDZ domain-binding motif28. We sought to investigate which domain contributes to ITGA3 transcriptional activation. We transfected MIA PaCa-2 cells with the empty vector, WT YAP1 vector, YAP1-TEAD TID mutated vector, YAP1-PDZ deletion vector, YAP1-WW mutated vector, and YAP1-phosphorylation mutated vector respectively (Fig. S3A); only mutation of TID domain decreased ITGA3 expression but did not affect YAP1 level (Fig. S3B) comparing with the WT YAP1 vector. Mutation of the TID domain significantly reduced ITGA3 transcription (Fig. S3C), while mutation of other three domains had no significant impact on the transcription of ITGA3 in pancreatic cancer cells (Fig. S3D–S3F). Additionally the YAP1 inhibitor verteporfin which is known to reduce YAP1/TEAD activity and deactivate YAP1 targets29 was used to treat MIA-ZIP4 cells and inhibited expression of ITGA3 (Fig. S3G). In total, these data demonstrated that the transcription co-activator YAP1 is required for ITGA3 transcriptional activation and that ZEB1 activates ITGA3 through YAP1/TEAD binding sites.
ZIP4 upregulates YAP1 through miR-373-LATS2 in pancreatic cancer cells.
These results above showed that ZEB1 and YAP1 cooperatively stimulate ITGA3 transcription. ZEB1 can be activated by ZIP4 in a zinc dependent manner25, however, whether YAP1 is also regulated by ZIP4 in pancreatic cancer is unknown. In this study, we found that ZIP4 significantly upregulated YAP1 expression in pancreatic cancer cells, whereas knock down of ZIP4 decreased YAP1 (Fig. 2A). Having shown an impact of ZIP4 on YAP1 levels we sought to elucidate how ZIP4 upregulates YAP1 in pancreatic cancer. YAP1 is known to be inhibited by large tumor suppressor 1/2 (LATS2) via its effects on YAP1 phosphorylation and cytoplasmic localization, ubiquitination, and degradation30. We found that YAP1 inversely correlated with LATS2 in human pancreatic cancer cells (Fig. 2A, Fig. S4A), and that knock down of ZIP4 inhibited YAP1 expression but further blocking of LATS2 with siRNA rescued YAP1 level (Fig. 2B, Fig. S4B). Knock down of ZIP4 also decreased YAP1 nuclear translocation (Fig. S4C). We have previously shown that ZIP4 downregulated LATS2 via activating a CREB-miR-373 signaling axis22, we sought to determine whether ZIP4 upregulates YAP1 through a miR-373-LATS2 signaling pathway. Overexpression of miR-373 inhibited LATS2 but increased YAP1 and ITGA3 expression, while blocking miR-373 increased LATS2 level but inhibited YAP1 and ITGA3 expression (Fig. 2C–2F, Fig. S4D–S4H). To assess the impact of miR-373 on YAP1 and ITGA3 in vivo, we used orthotopic xenograft tumor models; either AsPC-shZIP4-pre-miR-373 or MIA-ZIP4-Anti-373 stable cell lines were injected into the pancreas of the nude mouse (Fig. S4I). To validate this apparent correlation between ZIP4 and miR-373, we assessed miR-373 and ZIP4 levels using RNA Basescope in situ and IHC staining in 35 human pancreatic cancer tissues. miR-373 co-localized with ZIP4 in the tumor tissue, and when expression of ZIP4 was high, miR-373 levels were also high (Fig. 3A). We determined protein levels of LATS2, YAP1 and ITGA3 in the xenograft tumor tissues from both models. In the miR-373 overexpressed tumor model LATS2 levels were lower and YAP1 and ITGA3 expression was higher, while in the miR-373 inhibited animals, LATS2 levels increased but YAP1 and ITGA3 levels decreased (Fig. 3B). We also assessed correlations between LATS2, YAP1 and ITGA3 in the human pancreatic cancer tissues and adjacent normal pancreatic tissues, we found correlations in levels of these proteins, when YAP1 and ITGA3 were high, the expression of LATS2 was low (Fig. 3C and Fig. S5A–S5B). Additionally, we found that YAP1 and ZEB1 co-localized in these human pancreatic cancer tissues (Fig. 3C, Fig. S5B). These findings indicate that ZIP4 upregulates YAP1 by activating a miR-373-LATS2 signaling pathway in pancreatic cancer.
Fig. 2. ZIP4 upregulates YAP1 through miR-373-LATS2 in pancreatic cancer cells.

(A). Expression of YAP1 and LATS2 in MIA-V, MIA-ZIP4, AsPC-shV and AsPC-shZIP4 cells. (B). Expression of YAP1 and LATS2 in AsPC-shZIP4 cells with LATS2 knock down. (C). Protein levels of LATS2, YAP1, and ITGA3 in AsPC-shV miR-C, AsPC-shZIP4 miR-C, AsPC-shZIP4 miR-373, MIA-V Anti-C, MIA-ZIP4 Anti-C, and MIA-ZIP4 Anti-miR-373 cells. (D-F). mRNA levels of LATS2, YAP1 and ITGA3 in AsPC-shV miR-C, AsPC-shZIP4 miR-C, AsPC-shZIP4 miR-373, MIA-V Anti-C, MIA-ZIP4 Anti-C, and MIA-ZIP4 Anti-miR-373 cells. * means P< .05, ** means P< .01.
Fig. 3. miR-373 upregulates ITGA3 through LATS2-YAP1 in pancreatic cancer in vivo.

(A). RNA Basescope in situ staining with miR-373 and ZIP4 IHC staining in human pancreatic cancer tissues indicated the co-localization of miR-373 and ZIP4. The scale bar is 50 μm. (B). IHC staining of Ki67, LATS2, YAP1 and ITGA3 in the xenograft tumor tissues. The scale bar is 50 μm. (C). IHC staining of ZIP4, LATS2, YAP1 and ITGA3 in human pancreatic cancer tissues. The scale bar is 50 μm.
ZIP4 promotes cell adhesion, spheroid formation and organogenesis through its downstream target ITGA3.
Organogenesis is important for tumor initiation and cell adhesion and it supports metastatic colonization in the late stage of metastasis. To further understand the underlying mechanism of tumor progression and metastasis, we interrogated the effect of ZIP4 on organogenesis and cell adhesion. We have shown that ZIP4 is vital for pancreatic cancer cell proliferation, metastasis and chemoresistance22–25, 31, 32, in addition, our GO analysis of the TCGA database showed that ZIP4 also contributes to pancreatic cancer cell motility and cell-matrix adhesion. To determine the function of ZIP4 in modulating cell adhesion, we first performed adhesion assays and found that ZIP4 increased laminin induced cell adhesion in pancreatic cancer cell lines (Fig. 4A). However, the lack of structural architecture in a 2D model makes difficult to assess the specific role of ZIP4 in cell adhesion. The recent development of 3D tumor spheroid and organoid cultures allow for this type of study because they better simulate and recapitulate the biology of tumors than 2D cell monolayers33. We analyzed the morphology of spheroids following 2, 4 and 24 hours of spheroid culture. Spheroids aggregated more rapidly when ZIP4 was overexpressed but blocking of ZIP4 inhibited the spheroid formation (Fig. 4B). We additionally investigated the effect of ZIP4 on organogenesis using 3D organoid culture following 8 days of organoid culture. ZIP4 overexpressing cells showed more organoid clusters of larger size, and with increased density and a more integrated structure (Fig. 4C–4E). However, when ZIP4 was knocked down in AsPC-1 cells, the organoid size was decreased in both Matrigel embedded and suspended models (Fig. 4C–4E). Similar results were also observed in KPC and KPCZ models, where knockout of ZEB1 decreased the size of organoids (Fig. 4C–4D). Integrins play an essential role on cell to cell and cell to ECM adhesion, and we found ITGA3 knockdown inhibited cell to ECM adhesion and spheroid formation in pancreatic cancer cells (Fig. S5C–S5D). Inhibition of ITGA3 also suppressed tumor growth in the xenograft tumor model (Fig. S6A) and increased cell to cell connections through upregulating F-actin level (Fig. S6B). Overall, these findings identify ZIP4-promoted organogenesis and cell to ECM adhesion via upregulation of ITGA3 in pancreatic cancer.
Fig. 4. ZIP4 enhances cell-ECM adhesion and spheroid/organoid formation.

(A). Adhesion assay. 96 well plates were pre-coated with 500μg/ml Laminin for 2h. MIA-V, MIA-ZIP4, AsPC-shV, AsPC-shZIP4 cells were seeded in 96 well plate for 30 min at 37 °C, then cells were gently washed with PBS for two times. Absorbance was recorded at 490 nm to determine the percentage of cells attached to the bottom of 96 well plate. The scale bar is 500 μm. ** means P< .01. (B). Spheroid formation assay. The solidified spheroid drops (generated from MIA-V, MIA-ZIP4, AsPC-shV, AsPC-shZIP4 cells) were kept on the dish lid for 2, 4 and 24 h at 37 °C and then were imaged using a light microscope.10 spheroids were included and analyzed in each group. The scale bar is 200 μm. (C). Matrigel embedded organoids. The chamber slide was pre-coated with Matrigel at 37 °C for 2h. Human and mouse pancreatic cancer cells were resuspended in the culture medium containing 2% Matrigel and cultured on the chamber slide for 8 days. The scale bar is 50 μm. (D). Matrigel suspended organoids. Cells were resuspended in the culture medium containing 50% Matrigel in the 24 well plate for 8 days. The scale bar is 200 μm. (E). H&E staining of 3D organoids established from MIA-V, MIA-ZIP4, AsPC-shV and AsPC-shZIP4 cells. The scale bar is 50 μm.
ZIP4 promoted EMT in pancreatic cancer 3D spheroid and organoid model.
Having shown that ZIP4 is important for organogenesis, we sought to determine the mechanism underlying ZIP4-mediated promotion of organogenesis in pancreatic cancer using our organoid model. Integrins have a profound role on pancreatic organogenesis and tissue maintenance and promote pancreas development. Thus, we first examined the expression profile of ITGA3 in the 3D organoid model. ITGA3 signal was significantly higher in the organoid derived from cells overexpressing ZIP4 (Fig. 5A). Similarly ITGA3 level was higher in the 3D spheroid model generated from ZIP4 overexpressing cells (Fig. 5C). In contrast, ITGA3 signal was decreased in the organoids and spheroids derived from cell lines with ZIP4 knock down (Fig. 5A, 5C, Fig. S6C–S6D). We also determined the level of cytoskeletal protein F-actin in the 3D organoids and spheroids and found it to be highly expressed between cell to cell junctions and connection edges in the organoids and spheroids with lower levels of ZIP4 (Fig. 5A, 5C). We also assessed the ITGA3 expression in 2D cell cultures, the differences were not as significant as that in the 3D organoid model (Fig. 5B). We also noted that knockout of ZEB1 resulted in decreased ITGA3 in organoids derived from KPC cells, while leading to increased F-actin expression (Fig. 5D).
Fig. 5. Expression of ITGA3 in the 3D spheroid/organoid model.

(A). Images were taken using 3D confocal microscopy. ITGA3 was stained and F-actin was labeled in 3D organoid Matrigel embedded model derived from AsPC-shV, AsPC-shZIP4, MIA-V and MIA-ZIP4 cells. The scale bar is 50 μm. (B). Images were taken using 3D confocal microscopy. ITGA3 was stained and F-actin was labeled with 2D AsPC-shV, AsPC-shZIP4, MIA-V and MIA-ZIP4 cells. The scale bar is 10 μm. (C). Images were taken using 3D confocal microscopy. ITGA3 was stained and F-actin was labeled in the 3D spheroid sectional slides. The scale bar is 50 μm. (D). Images were taken using 3D confocal microscopy. ITGA3 was stained and F-actin was labeled with 3D organoid Matrigel embedded model derived from KPC and KPCZ cells. The scale bar is 50 μm.
Several studies indicated that EMT contributes to normal organogenesis during the development of epithelial cell34. In 2D culture, planar attachment inhibits cell geometry resulting in limited cell motility. In contrast 3D systems are more appropriate for EMT studies because cancer cells acquire morphological and cellular characteristics reminiscent of in vivo tumors35. Thus, we assessed the morphology of spheroid sections using H&E staining; when ZIP4 was overexpressed, cell to cell connections in the spheroid appeared less tight (Fig. 6A) but blocking of ZIP4 increased cell to cell connections (Fig. S6E). We additionally assessed expression of EMT markers in both the 2D cell lines and the 3D spheroid model. Blocking ZIP4 in the 3D spheroid model significantly decreased Vimentin and N-cadherin level but increased E-cadherin level (Fig. 6B). When ZIP4 was overexpressed EMT was enhanced via upregulation of vimentin and N-cadherin paired with downregulation of claudin-1 (Fig. 6C). However, there were no significant changes in EMT marker expression in the 2D system when ZIP4 was knocked down (Fig. 6B, 6C, Fig. S7A–S7B). We additionally determined levels of the ZIP4 downstream markers LATS2, YAP1 and ITGA3 in the 3D spheroid model and found significant positive correlations of YAP1 and ITGA3 levels with ZIP4, and reverse correlation of LATS2 level with ZIP4 (Fig. 6D–6E, Fig. S7C–S7D). And in the 3D organoid model it’s also shown that knocking down ZIP4 decreased YAP1, Vimentin and N-cadherin expression (Fig. S7E). Our results indicate that ZIP4 promotes EMT and organogenesis in the 3D organoid system via activation of ITGA3.
Fig. 6. ZIP4 promotes EMT in pancreatic cancer.

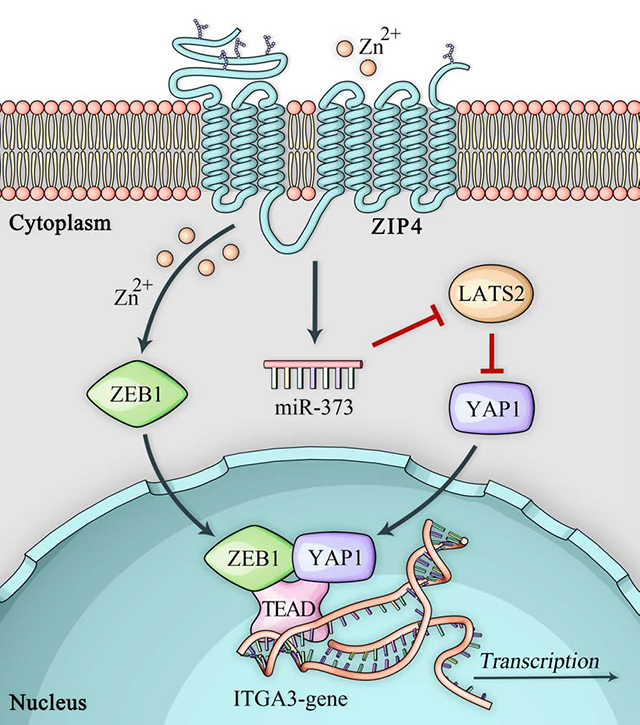
(A). H&E staining of 3D tumor spheroid generated from MIA-V and MIA-ZIP4 cells. Spheroids were collected after 3 days of spheroid culture and processed tissue embedding and sectioning. The scale bar is 100 μm. (B). Expression of EMT markers Vimentin, N-cadherin, E-cadherin or claudin-1 was examined in AsPC-shV and AsPC-shZIP4 cells cultured under 2D and 3D conditions. (C). Expression of EMT markers in MIA-V and MIA-ZIP4 cells. (D). Protein levels of LATS2, YAP1 and ITGA3 in AsPC-shV and AsPC-shZIP4 cells cultured under 2D and 3D conditions. (E). Protein levels of LATS2, YAP1 and ITGA3 in MIA-V and MIA-ZIP4 cells. (F). Schematic diagram of the signaling axis of ZIP4-miR-373-LATS2-ZEB1/YAP1/TEAD-ITGA3.
Discussion
In this study we demonstrated a novel ZIP4-miR-373-LATS2-ZEB1/YAP1-ITGA3 signaling pathway in pancreatic cancer EMT plasticity, cell-ECM adhesion, organogenesis and metastasis (Fig. 6F). ZEB1 and YAP1 interacted with each other to co-activate ITGA3 transcription and ZIP4 upregulated ZEB1/YAP1 and contributes to ITGA3 expression and EMT plasticity. At the same time, we found in the KPC mouse cells, knocking out ZEB1 inhibited organogenesis and apical to basal polarity.
The role of EMT in cancer metastasis is well appreciated, and EMT-induced cellular phenotype change enables cancer cells to experience dissociation, intravasation, extravasation, and macrometastasis formation9. EMT plasticity prompts cells to transit between EMT and MET states allowing the cells migrate from the primary tumor and colonize at the secondary tumor3. Kilinc et al36 reported that TGF-β induced EMT in murine epithelial mammary carcinoma cells, but this reverted to the epithelial phenotype when TGF-β was depleted. Therefore, EMT plasticity is a dynamic transiting between EMT and MET phenotypes reversibly. Brabletz has recently proposed an exciting concept of partial EMT6, 10, in which EMT program can be transient and reversible between epithelial and mesenchymal states. Partial EMT may have been activated even no significant EMT morphologies are identified in cells. In our study, we found that there were significant differences of EMT markers between cells with ZIP4 high or low, but no morphology changes were identified between the cells. The partial EMT may help to explain this finding that EMT is still active and is not needed to experience the full EMT program. Previously we have shown ZIP4 promoted pancreatic cancer migration and invasion through downregulation of tight junction protein ZO-1 and claudin-1, important intermediate molecules in EMT program32. However, little is known about the effect of ZIP4 on EMT program in pancreatic cancer. In this study, our data indicated ZIP4 promoted EMT plasticity in pancreatic cancer cells through a zinc dependent regulation of EMT TFs and co-activation. That also explained why the structure of spheroids derived from ZIP4 highly expressed cells was looser since ZIP4 promoted EMT and disrupted cell to cell connections in the spheroid model.
ZEB1 is a key EMT TF in pancreatic cancer. Krebs et al identified genetic depletion of ZEB1 in KC mouse model reduced ADM and PanIN-precursor lesions formation, tumorigenesis and in the KPC mouse model, they found depletion of ZEB1 inhibited cell plasticity, invasion and metastasis10. However genetic depletion of other EMT-activators such as Snai1 or Twist1 had no effect on tumor invasion or metastasis in KPC model10, 37. Liu et al elucidated effects of ZEB1 on tumor initiation and EMT/metastasis can be separated based on different levels of ZEB138. It’s indicated that lower level of ZEB1 contributes to tumor initiation, however further induction of ZEB1 is required for tumor metastasis which explained why genetic depletion of Snail1 in the KPC mice had no effect on tumor invasiveness and metastasis10. Even though Snail1 was knocked out, ZEB1 was only partial depleted which is not sufficient to abolish tumor metastasis. Brabletz et al demonstrated that depletion of ZEB1 inhibited tumorigenic, metastasis and cell plasticity in the KPC mouse model ZEB110, 12. ZEB1 is an important EMT activator in several different cancers, but less is known on how ZEB1 is activated and upregulated in pancreatic cancer. Our findings showed that as a zinc dependent transcription factor, ZEB1 can be activated by a cancer promoting zinc transporter ZIP4 through phosphorylation of STAT3. ZEB1 was found to directly repress miR-200 family and promoted breast cancer EMT and progress11. Other than transcriptional repression, ZEB1 also act as a transcriptional activator. The dual function of ZEB1 depends on which co-transcription factor does ZEB1 interacts with. Lehmann et al showed ZEB1 activated CTGF and AXL through interaction with YAP1/TEAD in breast cancer12. ZEB1 can also interact with factor Lef/Tcf to activate downstream targets Nrp2 and Prex1 transcription in glioblastoma13. However, little is known about how ZEB1 transcriptionally activates ITGA3 in pancreatic cancer. We found in pancreatic cancer cells, among many co-activators such as Lef1, CAF and P300, YAP1 is the only potent co-activator of ZEB1 which significantly activates ITGA3 transcription. ZEB1 usually binds to the promoter of candidate target genes through E-box known as the ZEB1 binding motif39. But we found after the mutation of E-box binding sites at ITGA3 promoter, when ZEB1 was blocked, the promoter activity of ITGA3 was increased. Therefore, it seemed that ZEB1 didn’t activate ITGA3 transcription through E-box binding sites. Later we found ZEB1 interacted with YAP1/TEAD and formed a complex to co-activate ITGA3 through YAP1/TEAD binding sites at ITGA3 promoter. Our data explained how ZEB1 activates ITGA3 through its co-activator YAP1 in pancreatic cancer. ZEB1 is a zinc dependent transcription factor and previously we have shown ZIP4 upregulates ZEB1 through phosphorylation of STAT3. But it’s unclear how ZIP4 regulates YAP1 in pancreatic cancer. Aberrant activated signaling by Hippo pathway has been reported in lung, breast, colon, and pancreatic cancer40. YAP1 is the downstream effector of Hippo pathway and Meng at al indicated Hippo pathway regulator LATS2 inhibits YAP1 activity through phosphorylation, cytoplasmic retention and protein degradation41. Previously we have identified LATS2 is the downstream target of miR-373 which can be downregulated by ZIP4 in pancreatic cancer22. Based on GO analysis from TCGA pancreatic cancer database, it’s shown ZIP4 is linked to Hippo pathway. For the first time we demonstrated ZIP4 is positively correlated with YAP1 in pancreatic cancer and ZIP4 upregulated YAP1 through miR-373-LATS2 signaling axis. We further identified upregulation of ZEB1 and YAP1 through separate signaling pathway by ZIP4 contributes to ITGA3 expression in pancreatic cancer.
Cell adhesion plays a critical role in the late stage of metastasis and the secondary tumor production. It helps establish tight connections between cells to cells and cells to the matrix when the circulating cancer cells reach the distal organ sites4. And cell adhesion not only links cells to ECM but also activates cell proliferation, modulates tumor microenvironment in order to facilitate EMT plasticity and the secondary tumor formation. However, the mechanism how ZIP4 promotes cell adhesion in pancreatic cancer was not clear. There are increasing evidence shows that activated integrin signaling contributes to cell-cell adhesion, cell proliferation and EMT. Activated α5β1 integrin increased cell adhesion to fibronectin during EMT42. Recently Li at al summarized some clinical trials targeting integrins in cancer and indicated integrin antagonists such as α vβ3 and αvβ5 integrins are applied more frequently in lung, liver and prostate cancer. In our study we showed among the integrin families, integrin α3β1 can be activated by ZIP4 and both high level of ZIP4 and integrin α3β1 predicted poor overall survival of pancreatic cancer patients. And blocking of integrin α3β1 attenuated the effect of ZIP4 on pancreatic cancer progression and metastasis both in vitro and in vivo. It suggested ITGA3 may serve as a good candidate for pancreatic cancer therapy.
In summary, we have shown that ZIP4 upregulated YAP1 through the Hippo pathway and that ZIP4 promotes pancreatic cancer cell to ECM adhesion, organogenesis and EMT plasticity by upregulating ITGA3 which is directly activated by ZEB1/YAP1 complex. Furthermore, we elucidated a novel signaling axis ZIP4-miR-373-LATS2-YAP1-ITGA3 in pancreatic cancer. Our data suggested that ZIP4 is a critical regulator of this signaling pathway and the phenotypes of EMT and tumor metastasis; thus, ZIP4 represents a novel and potentially effective therapeutic target in pancreatic cancer.
Supplementary Material
What You Need to Know.
BACKGROUND AND CONTEXT
Pancreatic cancer is characterized by extensive metastasis. EMT plasticity plays a critical role in tumor progression and metastasis by maintaining the transition between EMT and MET states. Our aim is to understand the molecular events regulating metastasis and EMT plasticity in pancreatic cancer.
NEW FINDINGS
The Hippo pathway effector YAP1 is a potent transcriptional co-activator and forms a complex with ZEB1 to activate ITGA3 transcription through the YAP1/TEAD binding sites in human pancreatic cancer cells and KPC derived mouse cells. ZIP4 upregulated YAP1 expression via activation of miR-373 and inhibition of the YAP1 repressor LATS2. Furthermore, upregulation of ZIP4 promoted EMT plasticity, cell adhesion, spheroid formation and organogenesis both in human pancreatic cancer cells, 3D spheroid model, xenograft model, and spontaneous mouse models (KPC and KPCZ) through ZEB1/YAP1-ITGA3 signaling axis.
LIMITATIONS
The role of other EMT transcription factors were not included in this study.
IMPACT
We demonstrated that ZIP4 activates ZEB1 and YAP1 through distinct mechanisms. The ZIP4-miR-373-LATS2-ZEB1/YAP1-ITGA3 signaling axis has a significant impact on pancreatic cancer metastasis and EMT plasticity.
Acknowledgements
We thank the Peggy and Charles Stephenson Cancer Center (SCC) at the University of Oklahoma Health Sciences Center for the use of Histology and Immunohistochemistry Core, which provided RNA Basescope in situ staining, immunohistochemistry and image analysis services. We also thank TCGA pancreatic cancer database and our results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Grant support: This work was supported in part by the National Institutes of Health (NIH) grants R01 CA186338-01A1, R01 CA203108, R01 CA247234-01, the William and Ella Owens Medical Research Foundation (Li M), and the NIH/NCI award P30CA225520.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer 2013;13:858–70. [DOI] [PubMed] [Google Scholar]
- 3.Chin VL, Lim CL. Epithelial-mesenchymal plasticity-engaging stemness in an interplay of phenotypes. Stem Cell Investig 2019;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan X Cancer metastases: challenges and opportunities. Acta Pharm Sin B 2015;5:402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selivanova G, Ivaska J. Integrins and mutant p53 on the road to metastasis. Cell 2009;139:1220–2. [DOI] [PubMed] [Google Scholar]
- 6.Stemmler MP, Eccles RL, Brabletz S, et al. Non-redundant functions of EMT transcription factors. Nat Cell Biol 2019;21:102–112. [DOI] [PubMed] [Google Scholar]
- 7.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 2002;3:155–66. [DOI] [PubMed] [Google Scholar]
- 8.Nieto MA, Huang RY, Jackson RA, et al. Emt: 2016. Cell 2016;166:21–45. [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Antin P, Berx G, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020;21:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krebs AM, Mitschke J, Lasierra Losada M, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 2017;19:518–529. [DOI] [PubMed] [Google Scholar]
- 11.Burk U, Schubert J, Wellner U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008;9:582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann W, Mossmann D, Kleemann J, et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun 2016;7:10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosmaninho P, Mukusch S, Piscopo V, et al. Zeb1 potentiates genome-wide gene transcription with Lef1 to promote glioblastoma cell invasion. EMBO J 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JH, Cho EJ, Kim ST, et al. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat Struct Mol Biol 2005;12:423–8. [DOI] [PubMed] [Google Scholar]
- 15.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016;29:783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park J, Kim DH, Shah SR, et al. Switch-like enhancement of epithelial-mesenchymal transition by YAP through feedback regulation of WT1 and Rho-family GTPases. Nat Commun 2019;10:2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nardone G, Oliver-De La Cruz J, Vrbsky J, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun 2017;8:15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moroishi T, Park HW, Qin B, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 2015;29:1271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Zhang J, Li H, et al. AMOTL1 enhances YAP1 stability and promotes YAP1-driven gastric oncogenesis. Oncogene 2020;39:4375–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori M, Triboulet R, Mohseni M, et al. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014;156:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diez-Cunado M, Wei K, Bushway PJ, et al. miRNAs that Induce Human Cardiomyocyte Proliferation Converge on the Hippo Pathway. Cell Rep 2018;23:2168–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Yang J, Cui X, et al. A novel epigenetic CREB-miR-373 axis mediates ZIP4-induced pancreatic cancer growth. EMBO Mol Med 2013;5:1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, Zhang Y, Liu Z, et al. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc Natl Acad Sci U S A 2007;104:18636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M, Zhang Y, Bharadwaj U, et al. Down-regulation of ZIP4 by RNA interference inhibits pancreatic cancer growth and increases the survival of nude mice with pancreatic cancer xenografts. Clin Cancer Res 2009;15:5993–6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Zhang Y, Yang J, et al. ZIP4 Increases Expression of Transcription Factor ZEB1 to Promote Integrin alpha3beta1 Signaling and Inhibit Expression of the Gemcitabine Transporter ENT1 in Pancreatic Cancer Cells. Gastroenterology 2020;158:679–692 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 2020;48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22:1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sudol M, Shields DC, Farooq A. Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin Cell Dev Biol 2012;23:827–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kandasamy S, Adhikary G, Rorke EA, et al. The YAP1 Signaling Inhibitors, Verteporfin and CA3, Suppress the Mesothelioma Cancer Stem Cell Phenotype. Mol Cancer Res 2020;18:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Du J, Wang S, et al. OTUB2 Promotes Cancer Metastasis via Hippo-Independent Activation of YAP and TAZ. Mol Cell 2019;73:7–21 e7. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Zhang Z, Zhang Y, et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019;156:722–734 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Yang J, Zhang Y, et al. ZIP4 Promotes Pancreatic Cancer Progression by Repressing ZO-1 and Claudin-1 through a ZEB1-Dependent Transcriptional Mechanism. Clin Cancer Res 2018;24:3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreira L, Bakir B, Chatterji P, et al. Pancreas 3D Organoids: Current and Future Aspects as a Research Platform for Personalized Medicine in Pancreatic Cancer. Cell Mol Gastroenterol Hepatol 2018;5:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell K Contribution of epithelial-mesenchymal transitions to organogenesis and cancer metastasis. Curr Opin Cell Biol 2018;55:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tevis KM, Colson YL, Grinstaff MW. Embedded Spheroids as Models of the Cancer Microenvironment. Adv Biosyst 2017;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nihan Kilinc A, Sugiyama N, Reddy Kalathur RK, et al. Histone deacetylases, Mbd3/NuRD, and Tet2 hydroxylase are crucial regulators of epithelial-mesenchymal plasticity and tumor metastasis. Oncogene 2020;39:1498–1513. [DOI] [PubMed] [Google Scholar]
- 37.Zheng X, Carstens JL, Kim J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015;527:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Lu X, Huang L, et al. Different thresholds of ZEB1 are required for Ras-mediated tumour initiation and metastasis. Nat Commun 2014;5:5660. [DOI] [PubMed] [Google Scholar]
- 39.Manshouri R, Coyaud E, Kundu ST, et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat Commun 2019;10:5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Xu X, Maglic D, et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep 2018;25:1304–1317 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev 2016;30:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desgrosellier JS, Barnes LA, Shields DJ, et al. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med 2009;15:1163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.