

Abstract
Background and Aims:
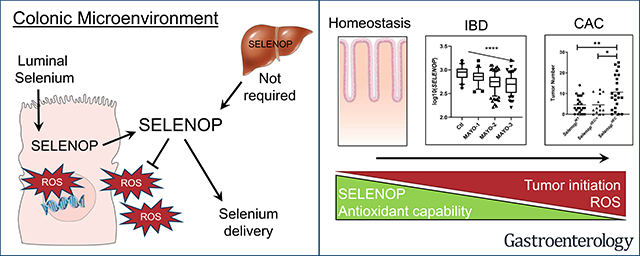
Patients with inflammatory bowel disease (IBD) demonstrate nutritional selenium deficiencies and are at greater risk for colon cancer. Previously, we determined that global reduction of the secreted antioxidant selenium-containing protein, Selenoprotein P (SELENOP), substantially increased tumor development in an experimental colitis-associated cancer (CAC) model. We next sought to delineate tissue-specific contributions of SELENOP to intestinal inflammatory carcinogenesis and define clinical context.
Methods:
Selenop floxed mice crossed with Cre driver lines to delete Selenop from the liver, myeloid lineages, or intestinal epithelium were placed on an azoxymethane/dextran sodium sulfate (AOM/DSS) experimental CAC protocol. SELENOP loss was assessed in human ulcerative colitis (UC) organoids, and expression was queried in human and adult UC samples.
Results:
Although large sources of SELENOP, both liver- and myeloid-specific Selenop deletion failed to modify AOM/DSS-mediated tumorigenesis. Instead, epithelial-specific deletion increased CAC tumorigenesis, likely due to elevated oxidative stress with a resulting increase in genomic instability and augmented tumor initiation. SELENOP was downregulated in UC colon biopsies and levels were inversely correlated with endoscopic disease severity and tissue S100A8 (calprotectin) gene expression.
Conclusions:
While global selenium status is typically assessed by measuring liver-derived plasma SELENOP levels, our results indicate that the peripheral SELENOP pool is dispensable for CAC. Colonic epithelial SELENOP is the main contributor to local antioxidant capabilities. Thus, colonic SELENOP is the most informative means to assess selenium levels and activity in IBD patients and may serve as a novel biomarker for UC disease severity and identify patients most predisposed to CAC development.
Keywords: Colitis-associated cancer, selenium, selenoproteins, reactive oxygen species
LAY SUMMARY
Selenoprotein P, a key antioxidant enzyme reduced in ulcerative colitis and produced by the colon epithelium, protects against genomic instability and colitis-associated cancer.
Graphical Abstract
Introduction
Discovered by J.J. Berzelius in 1817, selenium is a micronutrient essential for human health. Selenium deficiency is implicated in etiology of the cardiomyopathy, Keshan disease, and the osteochondropathy, Kashin-Beck disease1. Deficiency has also been shown to result in more severe viral infections, including HIV and coxsackie virus, as well as more virulent viral mutation, which may explain why the etiological origins of many recent pandemic viral diseases correlate with geologic regions of poor selenium bioavailability2–6. Selenium’s biologic activity is in part derived from its incorporation into one of 25 selenium-containing selenoproteins as the specialized amino acid, selenocysteine7. Many selenoproteins, such as glutathione peroxidases (GPx) and thioredoxin reductases, function as antioxidants which utilize selenocysteine within an enzymatic center to catalyze oxidation-reduction reactions8, 9. Thus, selenium, through selenoproteins, has long been thought to confer protection against oxidative stress.
Patients with the inflammatory bowel diseases (IBD), such as Crohn’s disease (CD) and ulcerative colitis (UC), are at increased risk for developing colitis-associated cancer (CAC). CAC risk is increased by colitis severity and duration, with up to an 18% risk in IBD patients with longstanding, severe disease10. While CAC development is likely multifactorial, increased oxidative stress from chronic inflammation is believed to be a key driver of tumor formation11. This is likely due to its ability to induce DNA damage via multiple mechanisms, including replication stress, oxidative base lesions, DNA crosslinking, and strand breaks, which ultimately result in genomic instability and tumor inititiation12. Indeed, analysis of inflammation-linked cancers has revealed elevated levels of reactive oxygen species (ROS) and oxidative DNA damage specifically at sites of tumor development13. However, because CAC can be difficult to detect and has a high overall mortality rate, understanding mechanisms which contribute to CAC development are of paramount importance to patients with IBD.
Because of their antioxidant roles, selenium and selenoproteins are thought to protect against IBD and CAC development. Indeed, selenium deficiency is often observed in IBD patients1, and animal models have provided further functional evidence that low selenium levels can augment the development and severity of colitis and CAC14–16. Other studies have demonstrated that loss of selenoproteins themselves can augment intestinal inflammation and CAC development17, 18. One selenoprotein previously implicated in colitis and CAC is selenoprotein P (SEPP1/SELENOP). SELENOP is unique among selenoproteins in that, while it can function as an extracellular antioxidant, its primary function is theorized to be selenium transport19. SELENOP is predominantly expressed in the liver and secreted into the plasma, effectively delivering selenium to other tissues via a total of 10 selenocysteine residues incorporated into its structure20. Once there, SELENOP is internalized and degraded to provide selenocysteines for production of other selenoproteins. Because of this role, levels of plasma SELENOP often complement direct selenium and GPx measurements to assess overall selenium status. Further, as observed with selenium, plasma SELENOP is decreased in CD patients, although studies have not identified similar reductions in UC1.
Recently, using SELENOP global knockout mice, we reported that SELENOP reduction (via heterozygosity) increased inflammatory tumor development21. SELENOP reduction altered behavior in multiple cell types, with Wnt pathway activation and proliferation increases in epithelial cells, as well as greater macrophage infiltration, all of which may contribute to augmented tumor development11. Thus, the cellular SELENOP source most responsible for attenuating CAC development is unknown. Furthermore, it is unclear whether liver-derived SELENOP, most often used as a clinical marker for selenium bioavailability, contributes to antioxidant capability in the colonic microenvironment. This work aimed to dissect tissue-specific contributions of SELENOP and provide further clinical context for SELENOP in IBD.
Methods
Murine inflammatory carcinogenesis protocol
Both male and female, cohoused littermate mice were used for all experiments. To minimize differences in gut microbiota which may contribute to inflammatory tumor development, soiled bedding was collected from all cages, mixed, and redispersed two weeks prior to ΔMye and ΔIEC experiments and repeated every two weeks throughout the duration of the studies22, 23. After selenium supplementation, cohorts of 20–22 week-old liver-specific (n = 16 WT, 17 ΔHep), myeloid-specific (n = 23 WT, 25 ΔMye/+, 29 ΔMye) and intestinal epithelial-specific (n = 23 WT, 14 ΔIEC/+, 23 ΔIEC) Selenop mice were placed on an inflammatory tumorigenesis protocol (Figure 1A). Mice were given a single intraperitoneal (IP) injection of 12.5 mg/kg azoxymethane (AOM, Sigma Aldrich) and three days post-injection the animals were started on the first of three cycles of either 3% dextran sodium sulfate (DSS) (Affymetrix, used for ΔHep and ΔIEC experiments) or 2% DSS (Gojira, used for ΔMye experiments) ad libitum. While different percentages of DSS were used from each stock, these percentages induced similar weight loss and intestinal damage in separate pilot experiments. Each DSS cycle lasted 4 days and was followed by a 16-day recovery period. During the second cycle of recovery (on day 37), colonoscopy (KARL STORZ) was performed to assess tumor development. Mice were sacrificed at the conclusion of the third recovery period. Histological analysis was performed by an experienced pathologist blinded to animal genotypes. All in vivo procedures were carried out in accordance with protocols approved by the Vanderbilt Institutional Animal Care and Use Committee and standards articulated in Animal Research: Reporting of In Vivo Experiments.
Figure 1. Liver-specific loss of SELENOP does not modify inflammatory tumorigenesis.

(A) Cohorts of SelenopWT and SelenopΔHep mice were placed on an AOM/DSS inflammatory colitis protocol consisting of 3 cycles of 4-day DSS treatment followed by 16 days of recovery. (B) Total SELENOP protein was assessed from plasma of SelenopWT and SelenopΔHep mice (n=10/genotype). (C) Total selenium (n=6/genotype) and (D) GPx activity (n=15 SelenopWT and n=8 SelenopΔHep) measured from non-tumor colonic tissue. (E) Colon length, (F) gross tumor number, and (G) gross tumor size (n=16 SelenopWT and 17 SelenopΔHep). Tumor size was measured with calipers and numbers represent tumor length × tumor width averaged per mouse. *P<0.05, ****P<0.0001, Student’s t test.
Human tissues
We analyzed previously published rectal tissue RNA-sequencing (RNA-seq) data from 206 UC patients enrolled in the Predicting Response to Standardized Pediatric Colitis Therapy (PROTECT) Study and from 20 non-IBD controls enrolled at Cincinnati Children’s Hospital Medical Center24, 25. PROTECT was a multicenter inception cohort study of treatment-naïve pediatric patients aged 4 to 17 years with a new diagnosis of UC. RNA isolation, library preparation, 75 base pair paired-end sequencing, and data cleaning were performed as described24, 25. Differentially expressed genes between UC versus non-IBD and endoscopically severe (Mayo 3) versus mild (Mayo 1) UC were identified in GeneSpring® software with fold change differences (FC) ≥ 1.5 and a Benjamini–Hochberg false discovery rate correction (FDR, 0.001) for all analyses. To assess associations between SELENOP expression and colitis severity, Log-transformed SELENOP expression in transcripts per million (TPM) was correlated to log-transformed S100A8 (component of the inflammatory marker calprotectin) using Pearson’s correlation coefficient. Log-transformed SELENOP expression was also compared ordinally among non-IBD patients and UC patients with endoscopic severity of Mayo 1, 2 and 3 using one-way ANOVA followed by post-test for linear trend.
For adult tissues, SELENOP was detected via RNAscope and in situ staining intensity was quantified on a 0–4 scale in a blinded fashion. Deidentified normal and UC tissues were obtained from colonic resections with permission from the Vanderbilt Institutional Review Board. Tissue microarrays (TMAs) prepared from deidentified normal and UC tissues with and without dysplasia and CAC have been described previously26, 27. As a positive control, samples were also stained for PPIB and samples without detectable staining were excluded from the analysis.
Statistics
Unless noted, statistical analysis comparing two or four groups was performed in Graphpad Prism 8 Software using Student’s t-test (unpaired, two-tailed) or one-way ANOVA and Tukey’s Multiple Comparison post-test, respectively. Samples were excluded if determined to be statistical outliers based on “robust regression and outlier removal” (ROUT) analysis. For all studies, center values represent experimental mean, error is represented by standard error of the mean, and P<0.05 is considered significant.
Please refer to the online Supplemental Materials for additional detailed methods.
Results
Hepatic SELENOP is dispensable in colon inflammatory tumorigenesis.
Hepatocytes produce ~90% of plasma SELENOP and liver-specific Selenop deletion greatly disrupts whole body selenium metabolism28. Therefore, we hypothesized that loss of liver SELENOP would be the primary driver of inflammatory tumorigenesis phenotypes induced by global Selenop reduction. To test this, Selenop was deleted from the liver by crossing Selenop floxed mice with the albumin-cre driver to yield WT (Selenop+/+; alb-cre) and ΔHep (Selenopf/f; alb-cre) cohorts. Mice were then placed on the AOM/DSS CAC protocol illustrated in Figure 1A.
Consistent with the role of hepatocytes in SELENOP production, ΔHep mice displayed substantial decreases in plasma SELENOP at the end of the AOM/DSS protocol (Figure 1B). However, while previous reports indicated that ΔHep mice display a 90% reduction in plasma selenium at baseline28, only a modest 18% reduction was observed in colonic selenium levels (Figure 1C), suggesting another selenium source in the colon. In support of this, we were unable to detect a significant reduction in colonic GPx activity in ΔHep mice. (Figure 1D). In response to AOM/DSS, ΔHep mice showed no increase in intestinal injury or inflammatory tumorigenesis as assessed via colon length (Figure 1E), tumor number (Figure 1F), or tumor size (Figure 1G) as compared to WT cohorts.
SELENOP is expressed in colonic macrophages and alters migratory ability.
ΔHep mice showed no change in development of inflammatory tumorigenesis, thus implicating a local source of SELENOP in modifying response to the AOM/DSS protocol. Visualization of Selenop-expressing cells in the normal colon revealed highest expression in stromal cells, as well as intestinal epithelial cells (Figure 2A). As query of Selenop expression in murine immune cells from the Immunological Genome Project public data sets identified highest expression in macrophage populations (Supplemental Figure 1)29, we next assessed Selenop expression in bone marrow-derived macrophages (BMDM). Interestingly, Selenop expression was higher in BMDM than in brain tissue, which is also known to express high amounts of Selenop19 (Figure 2B). Finally, to confirm lineage of the Selenop-high stromal cells, we co-labeled Selenop with the macrophage marker Cd68, finding that the majority of Selenop-expressing stromal cells express Cd68 (Figure 2C). Thus, colonic macrophages highly express SELENOP and may serve as a local source of SELENOP to protect against inflammatory tumorigenesis.
Figure 2. SELENOP is expressed in colonic macrophages and modifies migration.

(A) Selenop mRNA was detected by RNAscope. Negative control probe shown on left. Inset areas marked by dotted lines. Scale bar = 50μm. (B) qPCR of Selenop expression in BMDM (n=3) and whole colon tissue (n=3) as compared to liver (n=4) and brain tissue (n=4). Selenop levels were normalized to Gapdh and represented as fold change over liver expression. (C) Representative image (left) and quantification (right) of multiplex RNAscope for Selenop (green) and Cd68 (red) in murine colon tissue. Results represented as percent co-labeled Selenop (+) stromal cells per high powered field (n=15). Scale bar = 25μm. (D) Selenop expression was assessed by qPCR in BMDM established from SelenopWT, SelenopΔMye/+, and SelenopΔMye mice (n=2/genotype with 3 technical replicates). (E) BMDM were plated on transwell filters and allowed to migrate over 72 hours. Representative images (left) and quantification (right). For quantification, the number of migrated cells was assessed over 5 low power images per well and combined to yield the number of migrated cells. Results are combined data points from 3 independent experiments, each containing 3 technical replicates, represented as fold change over SelenopWT. (F) qPCR analysis of matrix-associated genes, biglycan/Bgn, transgelin/Tagln, and tissue inhibitor of metalloproteinases 3/Timp3 and (G) Gpx1 and Gpx3 (n=4/genotype with 3 technical replicates). (H) GPx1 protein assessed from BMDM. Representative results (left) and quantification normalized to tubulin and represented as fold change over SelenopWT (right) (n=5 SelenopWT, 4 SelenopΔMye/+, and 5 SelenopΔMye). *P<0.05, **P<0.01, ****P<0.0001, Student’s t test (C) or one-way ANOVA with Tukey’s multiple comparisons test (D-H). For all qPCR analysis, results were normalized to Gapdh and represented as fold change over SelenopWT.
To first specifically test the role of SELENOP in macrophages, we used the LysM-cre mouse line to target myeloid lineage cells, including macrophages30. LysM-cre mice were intercrossed with the Selenop floxed line to yield WT (Selenop+/+; LysMcre/cre), ΔMye/+ (Selenopf/+; LysMcre/cre), and ΔMye (Selenopf/f; LysMcre/cre) cohorts. This model efficiently deleted Selenop, as mRNA analysis of BMDM demonstrated ~50% reduction in ΔMye/+ cells and near complete reduction in ΔMye cells (Figure 2D). Because we previously identified increased macrophage infiltration in AOM/DSS tumors from the Selenop global model, which was hypothesized to contribute to tumorigenesis21, we first assessed whether Selenop loss modified migratory potential. However, upon plating WT, ΔMye/+, and ΔMye BMDM on transwell filters, we instead noted significantly attenuated migration in Selenop ΔMye cells (Figure 2E). While surprising in light of our previous data, this result phenocopied studies which depleted all selenoproteins from macrophages31. In these studies, the authors observed increased expression of extracellular matrix-associated genes, which may contribute to the reduction in BMDM migration. Assessment of the same extracellular matrix-associated genes, including biglycan (Bgn), transgelin (Tagln), and tissue inhibitor of metalloproteinases 3 (Timp3) (Figure 2F), revealed these were also upregulated upon Selenop loss. Thus, SELENOP is likely a key driver of selenoprotein-mediated changes in macrophages, particularly in modifying migratory potential.
Loss of SELENOP in macrophages phenocopies loss of all selenoproteins in macrophages, and SELENOP is a key facilitator of selenium transport7. Therefore, we next assessed whether SELENOP loss broadly altered selenoprotein expression. We focused on the GPx family of selenoproteins as many are ubiquitously expressed and believed to be a major contributor to selenium’s antioxidant effects32. Here, we observed increased mRNA expression of Gpx1 and Gpx3 (Figure 2G), as well as increased protein expression of GPx1 (Figure 2H, Supplemental Figure 2) in ΔMye BMDM as compared to either WT or ΔMye/+ cells. Thus, in addition to migration, SELENOP loss alters selenoprotein homeostasis in macrophages, and together these data suggest macrophage SELENOP expression may have broad implications beyond IBD and inflammatory carcinogenesis.
Inflammatory tumorigenesis is not altered by myeloid-specific Selenop deletion.
Next, we assessed the contribution of myeloid-derived SELENOP to inflammatory tumorigenesis by placing cohorts of WT, ΔMye/+, and ΔMye mice on the AOM/DSS protocol. Importantly, stromal Selenop staining was essentially absent in ΔMye colons, further confirming high SELENOP-expressing cells were of the myeloid lineage and likely macrophages (Figure 3A). However, there were no overall changes in tumor incidence (Figure 3B), tumor number (Figure 3C), or tumor size (Figure 3D) between AOM/DSS-treated WT, ΔMye/+, and ΔMye mice. Interestingly, while there was a trend toward reduced macrophage infiltration into adjacent non-tumor tissues (Figure 3E), tumors from ΔMye mice had no alterations in total numbers of infiltrating macrophages (Figure 3F), or neutrophils (Figure 3G), and total histologic injury was unchanged between genotypes (Figure 3H). Thus, while SELENOP appears to have a cell-autonomous role in regulating macrophage phenotypes, particularly migration, macrophage-derived SELENOP is not a significant contributor to the development of intestinal inflammatory tumorigenesis in this model.
Figure 3. Inflammatory tumorigenesis is not altered by myeloid-specific Selenop knockout.

(A) Selenop RNAscope in SelenopWT and SelenopΔMye mice. Scale bar = 50μm. (B) Cohorts of SelenopWT (n=23), SelenopΔMye/+ (n=25) and SelenopΔMye (n=29) mice were placed on the AOM/DSS protocol. Tumor incidence, (C) gross tumor number, and (D) gross tumor size as measured with calipers and shown as tumor length × width (n=91 SelenopWT, 126 SelenopΔMye/+, and 121 SelenopΔMye tumors). (E) Non-tumor AOM/DSS tissues were stained with F4/80 to label macrophages, and infiltrated cells were quantified per crypt. (F) Tumor sections were stained for F4/80 to label macrophages or (G) Ly6B.2 to label neutrophils. Intratumoral immune cells were quantified per high powered field (HPF). (H) Quantification of histologic injury scores accounting for depth and percent of colonic involvement (n=7 SelenopWT, 8 SelenopΔMye/+, and 8 SelenopΔMye mice). n≥24 fields for all imaging studies. **P<0.01, one-way way ANOVA with Tukey’s multiple comparisons test.
Selenop is expressed in differentiated enterocytes but loss does not alter baseline homeostasis.
In addition to stromal expression, Selenop was also highly expressed in differentiated intestinal epithelial cells (Figure 2A) and query of publicly available single cell RNA-sequencing data confirmed expression was highest in colonic enterocytes (Figure 4A)33. Using patient-derived normal colonic organoids (i.e., “colonoids”), colonocytes were further confirmed to secrete SELENOP (Figure 4B). Interestingly, absorption of dietary selenium is primarily thought to occur in the small intestine, where it is then delivered to the liver for SELENOP production and systemic distribution7. However, it was unknown whether human colonocytes can obtain selenium for selenoprotein production via direct uptake of dietary selenium. Indeed, we determined colonoid treatment with sodium selenite (Na2SeO3) increased SELENOP protein levels (Figure 4B). These results were confirmed with Na2SeO3 treatment of polarized Caco-2 BBe1 cells on transwell filters, which indicated predominantly basolateral secretion of SELENOP (Figure 4C). Together, our results suggest that differentiated colonocytes may uptake selenium from the gut lumen and use it to synthesize SELENOP, which can then be secreted basolaterally into the colonic microenvironment.
Figure 4. SELENOP is expressed in intestinal epithelial cells but its loss does not modify baseline intestinal cell homeostasis.

(A) Selenop expression queried from the Tabula Muris public single cell sequencing data set. tSNE plot with outlines of intestinal cell populations (left) and mean Selenop expression levels of each cell population (right). (B) Human patient-derived colonoids were treated overnight with 0.5μM sodium selenite (Na2SeO3) and SELENOP was detected from concentrated culture media. n=2 independent colonoid lines, run in duplicate. (C) Caco-2 BBe1 cells were polarized on transwell filters and SELENOP was measured from apical (A) and basolateral (BL) medias with and without overnight stimulation with 0.5μM sodium selenite. n=2 independent experiments, run in triplicate. (D) Organoids were established from colonic tissue of SelenopWT and SelenopΔIEC mice and Selenop expression was queried by qPCR. (E) Plating efficiency as calculated by dividing the numbers of colonoids formed by number of crypts plated (left) and viability of established colonoids at day 4 post-plating (right). (F) Colonoid Gpx1, Gpx2, and GPx3 mRNA expression quantified via qPCR. Results were normalized to Gapdh and represented as fold change over SelenopWT. (G) Colonoid GPx1 and GPx2 protein expression with β-actin provided as loading control (left) and quantification following normalization (right). n≥3 independent colonoid lines/genotype for D-G. *P<0.05, ****P<0.0001, one-way ANOVA with Tukey’s multiple comparisons test (B, C) or Student’s t test (E-G).
Next, we tested the contribution of intestinal epithelial-derived SELENOP by crossing Selenop floxed mice with the TAM-inducible Vil1-cre/ERT2+ driver line. TAM injection yielded cohorts of WT (Selenop+/+; Vil1-cre/ERT2+), ΔIEC/+ (Selenopf/+; Vil1-cre/ERT2+), and ΔIEC (Selenopf/f; Vil1-cre/ERT2+) mice. To investigate whether Selenop loss induced baseline epithelial cell autonomous changes, as observed in BMDM, colonic organoids were established from WT and ΔIEC mice. Despite near complete Selenop loss in ΔIEC colonoids (Figure 4D), no changes were observed in metrics commonly used to assess epithelial stem cell growth and survival, such as plating efficiency or colonoid viability post-plating (Figure 4E). Additionally, there was no significant change in Gpx1, Gpx2, or Gpx3 mRNA levels (Figure 4F) nor GPx2 protein (Figure 4G). Furthermore, while only lowly expressed in colonoids, GPx1 was not upregulated as previously observed in ΔMye BMDM (Figure 4G, Supplemental Figure 3). In vivo analysis of ΔIEC colons also indicated no change in baseline levels of proliferation or apoptosis as assessed by immunohistochemistry for phospho-histone H3 and cleaved caspase-3, respectively (Supplemental Figure 4). Thus, while Selenop is highly expressed in intestinal epithelial cells, it is dispensable for baseline intestinal homeostasis.
Epithelial-specific Selenop loss augments inflammatory tumorigenesis.
Cohorts of WT, ΔIEC/+, and ΔIEC mice were then placed on the AOM/DSS protocol. ΔIEC mice maintained stromal staining but displayed widespread epithelial Selenop loss in both tumor-adjacent and tumor tissues (Figure 5A and 5B). Despite the lack of baseline phenotypes induced in Selenop null epithelial cells, ΔIEC mice had increased AOM/DSS-induced tumorigenesis. Here, we noted over a 2-fold increase in tumor number in ΔIEC mice as compared to WT littermates (Figure 5C and 5D). Tumors were also significantly larger in ΔIEC mice (Figure 5E) and showed a higher incidence of high-grade dysplasia (Figure 5F and 5G). Thus, unlike myeloid or hepatic deletion, loss of SELENOP from intestinal epithelial cells greatly augments inflammatory tumorigenesis.
Figure 5. Epithelial-specific Selenop loss augments inflammatory tumorigenesis.

(A) Cohorts of SelenopWT (n=23), SelenopΔIEC/+ (n=15), and SelenopΔIEC (n=23) mice were placed on the AOM/DSS inflammatory tumorigenesis protocol. Selenop mRNA expression was detected in adjacent non-tumor (left) and tumor tissues (right) by RNAScope. (B) Selenop qPCR (n=6 non-tumor and 5 tumor samples/genotype with 3 technical replicates). (C) Gross tumor images, (D) tumor number, and (E) tumor size as measured with calipers and represented as length × width. (F) Degree of tumor dysplasia as assessed by pathologist. Data represented as percentage of mice with at least one tumor showing high-grade dysplasia (n=14 SelenopWT, 11 SelenopΔIEC/+, and 16 SelenopΔIEC samples). (G) Representative hematoxylin and eosin stained images. Area of enlargement noted by dotted lines. Scale bar = 50μm. *P<0.05, **P<0.01, ***P<0.001, one-way ANOVA with Tukey’s multiple comparisons test (B, D, E) or chi-squared test (F, WT vs. ΔIEC).
Epithelial Selenop loss increases DNA damage and tumor initiation.
We next aimed to determine how SELENOP loss contributed to tumor development in the AOM/DSS model. Previously, we noted that global Selenop reduction increased macrophage infiltration21. However, quantification of immune cell infiltration by immunohistochemistry and assessment of histological inflammatory scores did not identify broad inflammation-associated alterations in ΔIEC tissues (Supplemental Figure 5). Using immunohistochemistry, we assessed proliferation and apoptosis in ΔIEC tumors. Despite the larger tumors in ΔIEC mice, we were surprised to observe no increase in proliferation (Figure 6A) and increased apoptosis (Figure 6B) in ΔIEC tumors as compared with those from WT or ΔIEC/+ mice. As oxidative stress can induce DNA double-strand breaks identifiable by focal localization DNA damage response proteins, such as phospho-γH2AX, we next assessed phospho-γH2AX staining by immunohistochemistry34, 35. While both WT and ΔIEC tumors had equivalent numbers of phospho-γH2AX-positive cells (Supplemental Figure 6), ΔIEC tumor cells displayed increased numbers of γH2AX foci, which are widely used to determine the extent of DNA damage and repair36. This data provides evidence for increased genomic instability and oxidative stress (Figure 6C), which was not observed in tumors from the ΔMye model (Supplemental Figure 7).
Figure 6. Epithelial Selenop loss increases DNA damage and tumor initiation.

(A) Representative images (left) and quantification (right) of proliferative tumor cells by staining for phospho-histone H3 (pH3, red). Cells were co-labeled with E-cadherin (green) and DAPI (blue). (B) Representative images (left) and quantification (right) of intratumoral apoptosis by staining for cleaved caspase 3 (CC3, red) co-labeled with β-catenin (green) and DAPI (blue). n=29 SelenopWT, 8 SelenopΔIEC/+, and 35 SelenopΔIEC HPFs for A and B. Scale bar = 50μm. (C) Representative images (arrowheads, left) and quantification (right) of intratumoral DNA damage by staining for phospho-γH2AX (pγH2AX, red) co-labeled with E-cadherin (green) and DAPI (blue). Quantification is represented as number of staining puncta per positive cell (n>200 cells/genotype). Scale bar = 25μm. (D) Representative images (arrowheads, left) and quantification (right) of phospho-γH2AX staining in adjacent non-tumor tissue represented as positive cells per crypt (n=74 SelenopWT, 28 SelenopΔIEC/+, and 65 SelenopΔIEC crypts). Scale bar = 25μm. (E) Representative endoscopy stills (left) and quantification (right) of tumor number after the second DSS cycle (n=19 SelenopWT, 16 SelenopΔIEC/+, and 27 SelenopΔIEC). (F) SELENOP knockdown (shSELENOP) and control (shNontarget) human UC organoids were stained with CellROX Orange to detect ROS. Representative images (left) and quantification (right) of mean and maximum intensities by ImageJ, represented as arbitrary units (n=3 independent experiments, 39 images total). Scale bar = 50μm. (G) Nontargeted and shSELENOP UC organoids were treated overnight with H2O2. Viability was assessed after 24 hours by CellTiter-Blue and normalized to pre-treatment readings (n=2 independent experiments with 3 technical replicates). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, one-way ANOVA with Tukey’s multiple comparisons test (A-E), Student’s t test (F), or two-way ANOVA (G).
We hypothesized that the increased DNA damage in ΔIEC tumors may lead to the observed increases in tumor number and size via accelerating mutation and tumor initiation. Consistent with this hypothesis, greater numbers of phospho-γH2AX-positive cells were observed in tumor-adjacent tissues (Figure 6D). To more accurately assess the timing of tumor development, we next analyzed endoscopy videos of AOM/DSS-treated mice taken following the second cycle of DSS. While few tumors were observed in WT mice, tumors were already much more numerous in ΔIEC mice at this earlier timepoint (Figure 6E). Taken together, these data support a model in which epithelial SELENOP protects from excessive oxidative stress, mutation, and tumor initiation in the setting of colitis.
To next assess whether SELENOP loss similarly augments oxidative stress in human colitis, organoids were generated from adult UC patients and SELENOP was reduced via lentiviral-mediated shRNA expression. Following SELENOP knockdown (Supplemental Figure 8A), ROS levels were measured by staining with CellRox Orange, which only fluoresces upon oxidation by ROS, and both staining intensity and maximum fluorescence were greatly increased upon SELENOP reduction (Figure 6F). SELENOP knockdown organoids were also more sensitive to hydrogen peroxide than matched nontargeted shRNA control organoids (Figure 6G), again suggesting heightened levels of oxidative stress and resultant cytotoxicity. Interestingly, as observed in ΔIEC tumors, SELENOP loss also induced a pronounced growth defect in SELENOP knockdown organoids, while SELENOP knockdown in Caco-2 BBe1 cells induced a dose-dependent increase in cell death (Supplemental Figure 8). Combined with our results from AOM/DSS inflammatory tumorigenesis models, these data indicate that reduced SELENOP increases oxidative stress, resulting in DNA damage and cytotoxicity.
SELENOP is reduced in human UC and modulates epithelial oxidative stress.
SELENOP plasma levels are frequently used as a biomarker for total body selenium status and have been reported to be reduced in CD patients37–39. However, our results indicate that plasma SELENOP is likely dispensable for the colonic microenvironment. Therefore, we next assessed expression in normal colon and UC biopsy samples using RNA-sequencing data from rectal tissues of treatment-naïve pediatric UC patients within the Predicting Response to Standardized Pediatric Colitis Therapy (PROTECT) cohort24, 25. Here, we determined that SELENOP was significantly downregulated in samples from UC patients compared to non-IBD controls, while genes for other antioxidant selenoproteins, such as those in the GPx family or thioredoxin reductase family, were not differentially expressed (Figure 7A). Furthermore, SELENOP expression was inversely associated with disease severity, as determined by either endoscopic disease activity (Mayo Endoscopic Score) (Figure 7B), or in relation to expression of tissue S100A8, the gene for the established IBD fecal biomarker, calprotectin (Figure 7C).
Figure 7. SELENOP is reduced in human UC and is inversely associated with disease severity.

(A) Expression of SELENOP and other antioxidant selenoproteins queried from the PROTECT cohort. (B) Log-transformed SELENOP expression stratified by clinical severity scores (n=20 control, 54 mild UC, and 152 moderate-severe UC samples). (C) Log-transformed SELENOP expression in transcripts per million (TPM) correlated to log-transformed expression of a component of the inflammatory marker calprotectin, S100A8. (D) SELENOP expression was visualized by RNAscope in resected normal (n=6) and UC colonic tissues (n=5). Staining intensity was scored from 0–4 in both epithelial and myeloid populations. Quantification and (E) representative images. Dotted lines = inset area, red arrowheads = differentiated epithelium, blue arrowheads = myeloid cells, scale bars = 100μm. (F) SELENOP expression was visualized by RNAscope in a TMA containing UC tissues without active disease (uninvolved, n=8) or colitis with no dysplasia (colitis, n=51), low-grade dysplasia (LGD, n=74), high-grade dysplasia (HGD, n=24), and cancer (CAC, n=28). Staining was scored separately in epithelial (left) and myeloid populations (right). *P<0.05, **P<0.01, ****P<0.0001, one-way ANOVA followed by post-test for linear trend (B), Pearson’s correlation coefficient (C), Mann-Whitney test (D) or one-way ANOVA with Tukey’s multiple comparisons test (F). For F, * = significance vs. normal, § = significance vs. colitis, and # = significance vs. LGD.
Next, we aimed to determine whether similar changes were also observed in tissues from adult UC patients. First, we assessed SELENOP levels via in situ staining in a cohort of normal colonic and UC tissues collected from colonic resections. In agreement with the PROTECT cohort, lower staining scores were observed in UC tissues as compared to normal tissues (Figure 7D). Interestingly, SELENOP downregulation was only observed in intestinal epithelial cells, particularly in the differentiated epithelium (Figure 7E, red arrows), with no reduction in myeloid cell populations (Figure 7E, blue arrows). Next, we assessed SELENOP levels in a previously described tissue microarray (TMA) consisting of UC patient tissues without active disease (uninvolved), or colitis with no dysplasia, low-grade dysplasia (LGD), high-grade dysplasia (HGD), or CAC26, 27. Again, epithelial SELENOP levels were significantly lower in areas of active inflammation and LGD as compared to uninvolved tissues, and were further downregulated in samples with HGD and CAC (Figure 7F, left). In contrast, myeloid SELENOP expression was unchanged in the setting of colitis and LGD, although downregulation was observed in severe lesions (Figure 7F, right). Interestingly, in other tissues, SELENOP is reported to be regulated by a number of inflammatory cytokines, including interleukin 6 (IL6), interleukin 10 (IL10), and interferon-γ (IFN-γ)40–42. Therefore. we next assessed whether these cytokines may contribute to SELENOP’s downregulation in UC and determined SELENOP was significantly downregulated following treatment with IL6 (Supplemental Figure 9). Thus, while this does not rule out contribution from other mechanisms, SELENOP reduction may be driven by the inflammatory microenvironment. Taken together, these results indicate that SELENOP expression, particularly in epithelial cells, is inversely correlated with pediatric and adult disease and may constitute a novel biomarker for IBD severity and risk of CAC development.
Discussion
SELENOP is a key antioxidant protein which helps maintain antioxidant defenses by both providing selenium for the translation of other antioxidant selenoproteins as well as catalyzing redox reactions20. Previously, we determined that global reductions in Selenop increased inflammatory tumor formation in the AOM/DSS model21. However, as SELENOP is a secreted protein and expressed in several different cell types, these results did not indicate which cells produce SELENOP and/or respond to SELENOP reduction. Of the three tissue-specific models tested, here we show that only SELENOP loss in intestinal epithelial cells impacted inflammatory tumorigenesis, thus identifying a tissue-specific protective function. Collectively, our data indicate that intestinal epithelial cells are responsible for both the production of SELENOP and the pro-tumorigenic response to its loss.
Our previous experiments with Selenop+/− mice identified several mechanisms by which SELENOP reduction may augment inflammatory tumorigenesis. First, increased intra-tumoral macrophage infiltration was observed, which was concurrent with increased polarization into a protumorigenic “M2” phenotype21. Yet myeloid-specific loss did not impact inflammatory tumorigenesis in the AOM/DSS model. However, numerous studies have identified that selenium and selenoproteins often “boost” overall immunity and play key roles in immune cell regulation and function43. Indeed, loss of all selenoproteins attenuated migration and induced expression of matrix-associated genes in macrophages31, which was phenocopied in our ΔMye model ex vivo and in non-tumor AOM/DSS tissues. While no decrease was observed in tumor tissues, this may be due to altered polarization or response to tumor-derived cytokines. Interestingly, Selenop is one of the genes most highly expressed in M2 macrophages as compared to M1 macrophages in response to African trypanosomiasis parasitic infection41. Thus, myeloid-produced SELENOP loss may have a more pronounced effect in other disease systems and may have a contextual role in immune cell function. Because immune cells often generate ROS as part of their antimicrobial functions, they also have robust systems in place to buffer against increases in oxidative stress, which are likely less developed in intestinal epithelial cells44. This may explain why ΔMye BMDM were able to upregulate both GPx1 and GPx3 in response to SELENOP loss, and this compensation may contribute to the lack of phenotype in ΔMye mice.
As epithelial oxidative stress was increased in both the global and ΔIEC models, this likely represents the key driver of tumorigenesis in both models. Yet, it is important to note that the level of oxidative stress can differentially affect tumor formation. In our previous AOM/DSS study, tumor number was reduced in the global Selenop null mice and tumors were smaller, with increased apoptosis and oxidative stress. The difference between homozygous and heterozygous Selenop loss is likely due to a “double-edged sword” effect of oxidative stress, in which modestly increased oxidative stress promotes mutagenesis and tumor formation, while higher levels induce critical genomic instability/cellular toxicity culminating in cell death32. In our current study, ΔIEC mice displayed increased tumor formation concurrent with modestly increased oxidative stress, although at the time of sacrifice the tumors themselves displayed higher apoptosis and lower proliferation. Because tumor cells have higher levels of oxidative stress than normal tissues45, and SELENOP levels are low in tumors at baseline, it is likely that complete loss of epithelial SELENOP promotes tumorigenesis at early stages, but tips the scale in favor of cytotoxicity at later stages.
While we demonstrate that SELENOP loss increases inflammatory tumor initiation, the precise reason why loss of only epithelial-produced SELENOP alters tumorigenesis remains unclear. Interestingly, ΔIEC mice express a known SELENOP receptor, ApoER2, at levels similar to WT mice in the colon (Supplemental Figure 10). Therefore, Selenop null IECs should have no defect endocytosing and processing SELENOP secreted by other cell types (e.g., hepatocytes and myeloid cells) to provide selenocysteine for production of other selenoproteins. In support of this, GPx protein expression was unchanged in ΔIEC organoids. Thus, our data indicate that the protumorigenic effects in ΔIEC mice are not due to global changes in selenium transport or reduced production of other SePs. In fact, colonic selenium levels were unusually resistant to depletion in ΔHep mice as compared to other organ systems; here we report only an 18% reduction in colonic selenium as compared to the reductions in the kidney (65%) and muscle (42%) previously reported for ΔHep mice28. Instead of reliance on liver-derived SELENOP pools, our data suggest the colonic epithelium is instead able to source selenium from the diet and use this to produce SELENOP, which acts as an antioxidant in the intestinal microenvironment. Furthermore, it is interesting to note that while SELENOP is most highly expressed in differentiated colonocytes, we have noted an inverse expression pattern in ApoER2 (Supplemental Figure 10). As we also observed primarily basolateral SELENOP secretion, it is possible that enterocyte-produced SELENOP may also deliver selenium and/or signal in a paracrine fashion to other IECs, including Lgr5+ stem cells, and future studies investigating the SELENOP/ApoER2 interaction may uncover interesting roles in intestinal stem cell biology.
If the colon does indeed maintain selenium homeostasis without plasma SELENOP, our results have profound implications for clinical assessment of selenium status. Because SELENOP is central to selenium homeostasis, SELENOP plasma levels are often measured as an index of whole body selenium levels and selenoprotein activity46. In the case of IBD, decreased SELENOP was observed in the plasma of CD patients; however, these authors identify no change in patients with UC, nor are SELENOP levels correlated with CD disease activity37, 39. However, because the plasma SELENOP pool is likely dispensable for intestinal phenotypes, this may not be sufficient for assessing disease risk within the gastrointestinal tract and instead, tissue-specific measurements should be obtained. Here, for the first time, we assessed SELENOP mRNA levels in UC samples and determined that SELENOP is downregulated in these tissues. Furthermore, colonic SELENOP showed a strong inverse correlation with disease severity, suggesting it may constitute a novel biomarker. SELENOP downregulation may also increase the risk of CAC development, and thus help stratify patients most at risk for tumor initiation. It is also noteworthy that decreased selenium and/or SELENOP has been observed in sporadic colorectal cancer (CRC), and dietary selenium reduced tumor number in the ApcMin/+ model1. While the role of SELENOP in sporadic CRC has not yet been established, these reports suggest our results are not limited to inflammatory tumorigenesis. Together, these data strongly support an indispensable antioxidant role for epithelial-produced SELENOP in the colonic microenvironment and add additional context to the relationship of selenium, selenoproteins, and inflammatory bowel disease.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Inflammatory bowel disease (IBD) patients are at increased risk for colitis-associated carcinoma (CAC). Oxidative stress is a key driver of genomic instability, tumor formation, and cancer progression. The roles of antioxidant defense systems at play in the epithelium and infiltrating macrophage populations are not well understood.
NEW FINDINGS
Selenoprotein P (SELENOP) is downregulated in ulcerative colitis and its loss promoted oxidative stress. Using genetically engineered mouse models, we discovered that epithelial SELENOP loss increased tumor burden and genomic instability in a CAC experimental model, identifying a key colon-specific role for SELENOP in protection against CAC development.
LIMITATIONS
While these experiments relied on human, mouse and organoid systems, further investigation in larger patient cohorts will be important to contextualize SELENOP as a potential IBD biomarker.
IMPACT
These studies identify a key epithelial-specific antioxidant system that protects from inflammatory carcinogenesis and may represent a biomarker or therapeutic/dietary target in IBD.
Acknowledgements:
The authors thank all members of the Williams and collaborating laboratories for thoughtful discussions about this research project. We would also like to thank the Vanderbilt Translational Pathology Shared Resource for aid with histology and the Cooperative Human Tissue Network for tissue procurement.
Grant Support: Financial support includes National Institutes of Health (R01DK099204 to CSW, R01AT004821 and P30DK058404 to KTW, 1F31CA167920 to CWB, F32DK108492 to SPS, F30DK103498 to VKR, F31CA232272 to JMP, R01DK117119 to MJR, U01DK095745 to JSH and LAD); Office of Medical Research, Department of Veterans Affairs (1I01BX001426 to CSW and 1I01BX001453 to KTW); Crohn’s and Colitis Foundation (623541 to CSW and 662877 to SPS).
Abbreviations:
- SELENOP
Selenoprotein P
- CAC
colitis-associated cancer
- AOM
azoxymethane
- DSS
dextran sodium sulfate
- UC
ulcerative colitis
- IBD
inflammatory bowel disease
- CD
Crohn’s disease
- SeP
selenoprotein
- ROS
reactive oxygen species
- GPx
glutathione peroxidase
- IP
intraperitoneal
- TAM
tamoxifen
- BMDM
bone marrow derived macrophages
Footnotes
Disclosures: The authors declare no competing interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Short SP, Pilat JM, Williams CS. Roles for selenium and selenoprotein P in the development, progression, and prevention of intestinal disease. Free Radic Biol Med 2018;127:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baum MK, Shor-Posner G, Lai S, et al. High risk of HIV-related mortality is associated with selenium deficiency. J Acquir Immune Defic Syndr Hum Retrovirol 1997;15:370–4. [DOI] [PubMed] [Google Scholar]
- 3.Gladyshev VN, Stadtman TC, Hatfield DL, et al. Levels of major selenoproteins in T cells decrease during HIV infection and low molecular mass selenium compounds increase. Proc Natl Acad Sci U S A 1999;96:835–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck MA, Williams-Toone D, Levander OA. Coxsackievirus B3-resistant mice become susceptible in Se/vitamin E deficiency. Free Radic Biol Med 2003;34:1263–70. [DOI] [PubMed] [Google Scholar]
- 5.Beck MA, Shi Q, Morris VC, et al. Rapid genomic evolution of a non-virulent coxsackievirus B3 in selenium-deficient mice results in selection of identical virulent isolates. Nat Med 1995;1:433–6. [DOI] [PubMed] [Google Scholar]
- 6.Harthill M Review: micronutrient selenium deficiency influences evolution of some viral infectious diseases. Biol Trace Elem Res 2011;143:1325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burk RF, Hill KE. Regulation of Selenium Metabolism and Transport. Annu Rev Nutr 2015;35:109–34. [DOI] [PubMed] [Google Scholar]
- 8.Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta 2013;1830:3289–303. [DOI] [PubMed] [Google Scholar]
- 9.Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol 2006;16:420–6. [DOI] [PubMed] [Google Scholar]
- 10.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 2001;48:526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol 2004;287:G7–17. [DOI] [PubMed] [Google Scholar]
- 12.Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawanishi S, Hiraku Y, Pinlaor S, et al. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem 2006;387:365–72. [DOI] [PubMed] [Google Scholar]
- 14.Barrett CW, Singh K, Motley AK, et al. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumorigenesis. PLoS One 2013;8:e67845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaushal N, Kudva AK, Patterson AD, et al. Crucial role of macrophage selenoproteins in experimental colitis. J Immunol 2014;193:3683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JH, Hue JJ, Kang BS, et al. Effects of selenium on colon carcinogenesis induced by azoxymethane and dextran sodium sulfate in mouse model with high-iron diet. Lab Anim Res 2011;27:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esworthy RS, Aranda R, Martin MG, et al. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol 2001;281:G848–55. [DOI] [PubMed] [Google Scholar]
- 18.Barrett CW, Ning W, Chen X, et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res 2013;73:1245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burk RF, Hill KE. Selenoprotein P-expression, functions, and roles in mammals. Biochim Biophys Acta 2009;1790:1441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr 2005;25:215–35. [DOI] [PubMed] [Google Scholar]
- 21.Barrett CW, Reddy VK, Short SP, et al. Selenoprotein P influences colitis-induced tumorigenesis by mediating stemness and oxidative damage. J Clin Invest 2015;125:2646–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uronis JM, Muhlbauer M, Herfarth HH, et al. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One 2009;4:e6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyoshi J, Leone V, Nobutani K, et al. Minimizing confounders and increasing data quality in murine models for studies of the gut microbiome. PeerJ 2018;6:e5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haberman Y, Karns R, Dexheimer PJ, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 2019;10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyams JS, Davis Thomas S, Gotman N, et al. Clinical and biological predictors of response to standardised paediatric colitis therapy (PROTECT): a multicentre inception cohort study. Lancet 2019;393:1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Means AL, Freeman TJ, Zhu J, et al. Epithelial Smad4 Deletion Up-Regulates Inflammation and Promotes Inflammation-Associated Cancer. Cell Mol Gastroenterol Hepatol 2018;6:257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh K, Coburn LA, Asim M, et al. Ornithine Decarboxylase in Macrophages Exacerbates Colitis and Promotes Colitis-Associated Colon Carcinogenesis by Impairing M1 Immune Responses. Cancer Res 2018;78:4303–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill KE, Wu S, Motley AK, et al. Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J Biol Chem 2012;287:40414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heng TS, Painter MW, Immunological Genome Project C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 2008;9:1091–4. [DOI] [PubMed] [Google Scholar]
- 30.Shi J, Hua L, Harmer D, et al. Cre Driver Mice Targeting Macrophages. Methods Mol Biol 2018;1784:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson BA, Yoo MH, Sano Y, et al. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol 2009;10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett CW, Short SP, Williams CS. Selenoproteins and oxidative stress-induced inflammatory tumorigenesis in the gut. Cell Mol Life Sci 2017;74:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaum N, Karkanias J, Neff NF, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z, Yang J, Huang H. Oxidative stress induces H2AX phosphorylation in human spermatozoa. FEBS Lett 2006;580:6161–8. [DOI] [PubMed] [Google Scholar]
- 35.Zhao H, Traganos F, Albino AP, et al. Oxidative stress induces cell cycle-dependent Mre11 recruitment, ATM and Chk2 activation and histone H2AX phosphorylation. Cell Cycle 2008;7:1490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 2012;920:613–26. [DOI] [PubMed] [Google Scholar]
- 37.Andoh A, Hirashima M, Maeda H, et al. Serum selenoprotein-P levels in patients with inflammatory bowel disease. Nutrition 2005;21:574–9. [DOI] [PubMed] [Google Scholar]
- 38.Xia Y, Hill KE, Li P, et al. Optimization of selenoprotein P and other plasma selenium biomarkers for the assessment of the selenium nutritional requirement: a placebo-controlled, double-blind study of selenomethionine supplementation in selenium-deficient Chinese subjects. Am J Clin Nutr 2010;92:525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barros SEL, Dias T, Moura MSB, et al. Relationship between selenium status and biomarkers of oxidative stress in Crohn disease. Nutrition 2020;74:110762. [DOI] [PubMed] [Google Scholar]
- 40.Martitz J, Becker NP, Renko K, et al. Gene-specific regulation of hepatic selenoprotein expression by interleukin-6. Metallomics 2015;7:1515–21. [DOI] [PubMed] [Google Scholar]
- 41.Bosschaerts T, Guilliams M, Noel W, et al. Alternatively activated myeloid cells limit pathogenicity associated with African trypanosomiasis through the IL-10 inducible gene selenoprotein P. J Immunol 2008;180:6168–75. [DOI] [PubMed] [Google Scholar]
- 42.Dreher I, Jakobs TC, Kohrle J. Cloning and characterization of the human selenoprotein P promoter. Response of selenoprotein P expression to cytokines in liver cells. J Biol Chem 1997;272:29364–71. [DOI] [PubMed] [Google Scholar]
- 43.Huang Z, Rose AH, Hoffmann PR. The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 2012;16:705–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Virag L, Jaen RI, Regdon Z, et al. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol 2019;26:101261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sosa V, Moline T, Somoza R, et al. Oxidative stress and cancer: an overview. Ageing Res Rev 2013;12:376–90. [DOI] [PubMed] [Google Scholar]
- 46.Hill KE, Xia Y, Akesson B, et al. Selenoprotein P concentration in plasma is an index of selenium status in selenium-deficient and selenium-supplemented Chinese subjects. J Nutr 1996;126:138–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.