

Abstract
The development of osteoarthritis (OA) correlates with a rise in the number of senescent cells in joint tissues, and the senescence-associated secretory phenotype (SASP) has been implicated in cartilage degradation and OA. Age-related mitochondrial dysfunction and associated oxidative stress might induce senescence in joint tissue cells. However, senescence is not the only driver of OA, and the mechanisms by which senescent cells contribute to disease progression are not fully understood. Furthermore, it remains uncertain which joint cells and SASP-factors contribute to the OA phenotype. Research in the field has looked at developing therapeutics (namely senolytics and senomorphics) that eliminate or alter senescent cells to stop disease progression and pathogenesis. A better understanding of how senescence contributes to joint dysfunction may enhance the effectiveness of these approaches and provide relief for patients with OA.
Osteoarthritis (OA), the most common form of arthritis, is a disease of the synovial joints that is characterized by cartilage degradation and bony overgrowth in the form of osteophytes and subchondral thickening1. OA is also associated with varying degrees of synovitis and damage to other joint structures, including ligaments and the menisci in the knee1. OA progresses gradually and eventually leads to debilitating pain and loss of mobility, especially in older adults2. Although risk factors such as obesity, joint injury and genetics have all been linked to OA, the most prevalent risk factor is age3. With the ageing baby boomer generation (that is, individuals born between 1946 and 1964), the number of people in the USA afflicted with OA is estimated to rise from 30 million to 67 million by the year 2030, with over half of those cases predicted to be in individuals aged 65 years and older4,5. Along with the burden of pain and disability suffered by patients with OA, treatment and care for this disease was estimated in 2013 to cost the US healthcare system $27 billion annually6 and even more in lost workforce productivity. Accordingly, researchers in the ageing and pharmaceutical fields have taken great interest in designing novel therapeutics to alleviate the symptoms of OA and slow its progression.
Within the past 5 years, researchers have begun to explore a novel approach to treating OA through the targeting of chondrocytes and other joint tissue cells that have undergone cellular senescence. Senescence, one of the hallmarks of ageing7, is a cell fate characterized by permanent cell cycle arrest and the release of harmful pro-inflammatory molecules into the surrounding microenvironment, a feature known as the senescence-associated secretory phenotype (SASP). Senescent cells accumulate as an organism ages, resulting in reduced cellular proliferation and impaired tissue regeneration and function8. For these reasons, senescence has been implicated in the pathogenesis and progression of a myriad of ageing-associated diseases, including OA9,10. Although age correlates with both OA and cellular senescence, the exact mechanism linking senescence to OA pathology remains unclear. Nevertheless, clinical trials are underway to test a pharmacotherapeutic approach to treating OA by eliminating senescent cells using senolytics, a class of drugs that selectively induce the death of senescent cells. This approach has shown promising early results by amelio-rating other ageing-related diseases in murine models, such as idiopathic pulmonary fibrosis, atherosclerosis and cancer11. Additionally, enzymes linked to the progression of OA have been identified as SASP factors, and the selective inhibition of these factors with therapeutics called senomorphics (also known as SASP inhibitors and senostatics) could one day provide relief for patients with OA. However, evidence of the benefit of senomorphics in treating OA is currently limited by a lack of studies testing the specificity and efficacy of these drugs for treating joint diseases.
In this Review, we explore several common phenotypes associated with cellular senescence and their links to OA pathology. Additionally, we examine several therapeutic strategies that target senescent cells directly and are being tested as a means of preventing the disease or improving patient outcomes.
Cellular senescence and the SASP
Since its discovery by Hayflick and Moorhead over a half century ago12, cellular senescence has been commonly defined as irreversible cell cycle arrest in response to replicative stress and ageing. However, studies from the past decade have expanded this definition beyond simply a reduction in proliferative capacity. For example, senescent phenotypes have been detected in postmitotic cells, such as damaged neurons and aged osteocytes13,14. Furthermore, senescence can be induced independently of replicative stress and ageing, such as by DNA damage, oncogenic signalling and oxidative stress15–17. Senescence is best described as a complex process involving the metabolic, morphological, and physiological transformation of cells in response to a multitude of cellular stresses18. Additionally, this process can affect neighbouring cells by altering paracrine signalling pathways, a discovery that has compelled researchers to investigate how senescent cells transform their microenvironments, a process that can have systemic effects on the entire organism19.
Much of the research on senescence has been devoted to understanding its pleiotropic role as both a tumour suppressor and a driver of ageing-related disease. In its role as a tumour suppressor, senescence involves the upregulation of cell cycle inhibitor genes in response to oncogenic signals, resulting in permanent growth arrest and the prevention of neoplastic proliferation20. In its role as a driver of disease, senescence hinders long-term tissue regeneration and normal cell function and has been linked to pathologies such as sarcopenia, osteoporosis, macular degeneration, neurodegeneration and OA21. Furthermore, novel roles for senescence include critical functions in the early stages of wound healing and in embryogenesis22,23.
Although senescent cells live in a state of permanent growth arrest, they are not dormant within tissues. Instead, senescent cells remain metabolically active and undergo dynamic transformations in their physiology, which can include alterations to paracrine signalling. The SASP is characterized by the increased secretion of particular bioactive molecules by senescent cells, including chemokines, cytokines, proteases and growth factors; these molecules can induce a range of physiological responses in the surrounding microenvironment, including inflammation, growth arrest and tumorigenesis24. Mechanistically, mTOR is a key regulator of the SASP owing to its ability to differentially regulate the translation of MAP kinase-activated protein kinase 2 (MAPKAPK2, also known as MK2)25 and IL-1α26. MK2 is phosphorylated by p38 and deactivates ZFP36L1, a zinc-finger protein that degrades the mRNA of many pro-inflammatory SASP factors. IL-1α promotes NFκB signalling, which has been linked to the upregulation of many SASP genes. Accordingly, inhibition of mTOR by rapamycin reduces SASP factor expression25,26.
Furthering the complexity of this phenotype, different senescence-inducing stimuli produce distinct secretory proteomes that can result in different biological outcomes depending on the tissues affected24. Much of the research on the SASP has focused on its role in disease pathogenesis and progression and on how SASP factors might be targeted for therapeutic intervention27. Diseases linked to the expression of SASP factors include atherosclerosis, cancer, cardiac dysfunction, myeloid skewing, kidney dysfunction, OA and a general decrease in health span. Identifying how specific SASP factors contribute to different pathological outcomes in patients with ageing-related diseases could help further the development of therapeutics that attenuate disease development. To this end, repositories such as the SASP Atlas24 are helpful tools that allow researchers to search and catalogue the discovery of novel SASP factors and their contextual effects on tissue phenotypes.
Cellular senescence and OA
Although chondrocytes are hypo-replicative during homeostasis, they do maintain the potential to proliferate in some settings. For example, chondrocytes proliferate in the form of ‘clusters’ during the early stages of OA, which is commonly viewed as an attempt to repair damaged matrix28. Chondrocytes also initiate cell division when plated in tissue culture28. The relationship between quiescence (that is, reversible cell cycle arrest) and senescence is complex, with evidence that mitogenic stimulation of damaged, quiescent cells can actually contribute to the induction of senescence upon re-entry into the cell cycle29.
Like other organs, joint tissues are subject to senescence and decay over time, and the number of senescent chondrocytes and synovial fibroblasts correlates strongly with age30,31. Given the important role of bone–cartilage crosstalk, increased osteocyte senescence during ageing might also contribute to OA14. Senescence is also a feature of post-traumatic OA, as joint injury can accelerate chondrocyte senescence and stimulate cartilage degradation32. Abnormal mechanical loading could be one cause of premature senescence after injury, as catabolic shear stress has been found to initiate senescence in young cartilage explants33. Additionally, lifestyle factors that increase susceptibility to OA have been found to overlap with cellular senescence. For example, mice placed on calorie-dense and nutrient-poor diets exhibited increased senescence in adipose tissue, while exercise reduced this outcome34. Furthermore, OA can induce phenotypic changes in joint cells that correlate with senescent signatures. For example, the cell surface protein urokinase plasminogen activator surface receptor (uPAR) is induced broadly in senescent cells35, as well as in chondrocytes derived from osteoarthritic cartilage36.
Senescence induces metabolic reconfigurations in cells that, over time, can contribute to the pathogenesis of OA. In fact, the transplantation of senescent fibroblasts into the knee joints of mice induced cartilage erosion, osteophyte formation and loss of mobility, suggesting that senescent cells alter the synovial microenvironment and induce OA-like arthropathy37. Senescent joint cells exhibit common hallmarks, such as telomere erosion, increased expression of p53 and of the cyclin-dependent kinase (CDK) inhibitors p21 and p16INK4a (p16), enhanced generation of reactive oxygen species (ROS) via mitochondrial dysfunction, and increased senescence-associated heterochromatin38. Notably, chondrocytes, osteocytes and synovial fibroblasts can also exhibit the SASP14,30,31. As noted above, a hallmark of the SASP is the secretion of pro-inflammatory cytokines, such as IL-6, IL-17, IL-1β, oncostatin M and TNF19,24, and several SASP factors induce OA-related changes, including inflammation, bone growth and degradation of the extracellular matrix (ECM) (FIG. 1). Therefore, a better understanding of OA pathogenesis will include identifying the phenotypic consequences of SASP factors in joint tissues.
Fig. 1 |. Associations between age-related stress, senescence and OA.
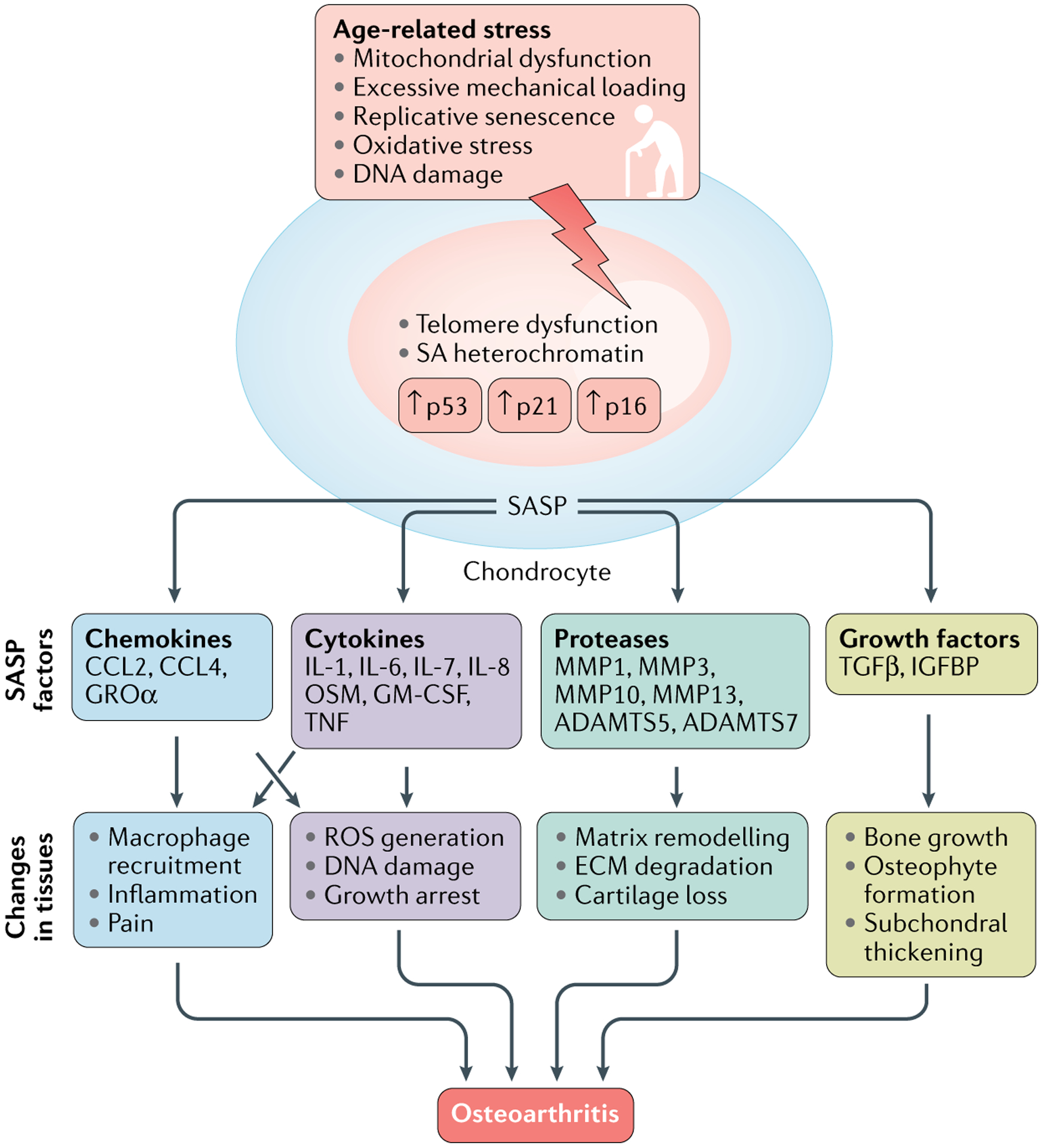
Multiple age-related stresses converge on the induction of senescent hallmarks in articular joint cells. These cells can exhibit the senescence-associated secretory phenotype (SASP) and secrete factors (including chemokines, cytokines, proteases and growth factors) that act independently or together to induce changes commonly found in osteoarthritic tissues. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; CCL, CC-chemokine ligand; ECM, extracellular matrix; GM-CSF, granulocyte–macrophage colony-stimulating factor; GRO, growth-regulated alpha protein; IGFBP, insulin-like growth factor binding protein; MMP, matrix metalloproteinase; OA, osteoarthritis; OSM, oncostatin M; ROS, reactive oxygen species; SA heterochromatin, senescence-associated heterochromatin; TGFβ, transforming growth factor-β.
Cytokines such as IL-6 are elevated in the synovial fluid of patients with OA39. The IL-6–STAT3 signalling pathway induces premature senescence in normal human fibroblasts, suggesting that these cells might trigger a bystander effect that drives further senescence and SASP in surrounding cells40,41. Furthering this hypothesis in cartilage, chondrocytes have been shown to facilitate intercellular communication via the production of extracellular vesicles (EVs), the levels of which were greatly upregulated in patients with OA compared with those in healthy individuals and resulted in the induction of a senescent state in nearby cells42. The role of EVs in cellular senescence and OA is discussed in more detail later in this Review.
Cytokines can upregulate the expression of a family of enzymes known as matrix metalloproteinases (MMPs)1. Like cytokines, MMPs, such as MMP13 (also known as collagenase-3), and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), such as ADAMTS-5, are secreted by cells into the ECM. The catabolic activity of MMPs and ADAMTS can degrade ECM proteins in cartilage, including sulfated proteoglycans, collagen and fibronectin1. Loss of cartilage ECM is a key early feature of OA, which further implicates the senescence of chondrocytes and other cells of the synovial joints as drivers of OA pathogenesis.
Senescence processes and biomarkers
Several phenotypic transformations occur during cellular senescence (Box 1). Here we discuss three of them (senescence-associated β-galactosidase (SA-β-gal) production, p16 expression and EV secretion), and their relevance to osteoarthritis pathogenesis.
Box 1 |. Common changes seen in cellular senescence.
Increased production of β-galactosidase
Increased expression of p16INK4a
Irreversible growth arrest
Increased secretion of extracellular vesicles (EVs)
Alterations in the microRNA content of EVs
Genomic instability
Increased levels of heterochromatin
Telomere attrition
Loss of proteostasis
Dysregulated nutrient sensing
Mitochondrial dysfunction
Increased production of reactive oxygen species and reactive nitrogen species
Increased secretion of senescence-associated secretory phenotype factors
Upregulation of urokinase plasminogen activator surface receptor (uPAR)
Senescence-associated β-galactosidase
The cytochemical staining of β-galactosidase activity to detect senescent cells, known as SA-β-gal staining, is one of the most commonly used techniques in both cell culture and tissue samples43,44. Positive staining is caused by the upregulation of β-galactosidase activity in lysosomes, which is optimally detected at pH 4.0 but detectable in senescent cells at pH 6.0 (REF.45). In articular cartilage, the number of SA-β-gal-positive chondrocytes was higher in old mice than in young mice30. However, a few precautions must be observed when using SA-β-gal as a marker for senescent cells in the joint. First, the enzymatic activity of lysosomes is regulated by the autophagy pathway, and isolating and culturing primary cells in monolayer can increase both basal autophagy46 and senescent phenotypes47; thus, SA-β-gal staining in cultured chondrocytes can represent an increase in autophagy rather than a senescent state. Second, silencing GLB1, the gene encoding β-galactosidase, eliminates SA-β-gal staining but does not inhibit senescence, demonstrating an indirect link between positive staining for SA-β-gal and senescence45. Third, senescence-independent β-galactosidase staining was observed in vivo in the neurons of young rodents and correlated with increased expansion of lysosomes during cell growth48. Finally, fibroblasts from patients with autosomal recessive G(M1)-gangliosidosis, a disease in which lysosomes are dysfunctional, were negative for SA-β-gal after undergoing replicative senescence45. Taken together, these studies suggest that changes to autophagy and the lysosomal activity of a cell, rather than senescence, determine the results of SA-β-gal staining.
Accordingly, changes in autophagy that occur with ageing and OA should be considered when performing SA-β-gal staining. Autophagy and lysosomal function decrease in patients with OA, whereas stimulation of autophagy (for example, with rapamycin), can confer protective homeostatic effects on normal human cartilage49–51. Autophagy can also be stimulated by a multitude of cellular stresses that can occur independently of senescence, including nutrient deprivation, hypoxia, ROS, DNA damage, protein aggregates, damaged organelles or intracellular pathogens52,53. Hypoxia-induced autophagy is of particular concern because chondrocytes reside naturally in low oxygen conditions due to a lack of blood vessels in cartilage54. Metabolism and homeostasis in this environment are maintained through autophagy, which recycles intracellular amino acids and clears dysfunctional mitochondria. Consequently, chondrocytes express the autophagy markers ULK1, Beclin1 and LC3 under normal physiological conditions, suggesting that autophagy is constitutively active in these cells50.
For these reasons, in studies using cellular senescence as an indicator of OA progression, SA-β-gal experiments should ideally be performed on joint tissues rather than on cultured cells, and the studies should incorporate one or more additional biomarkers of senescence. Furthermore, when inducing or treating OA-like phenotypes, careful consideration should be given to how the treatment being applied effects autophagy.
p16
p16 induces cellular senescence by binding CDK4 and CDK6 and preventing the downstream inhibition of the cell cycle repressor protein retinoblastoma-associated protein (Rb). p16 is upregulated in response to cellular stress, such as DNA damage from radiation or telomere shortening, ROS or oncogenic stress55. As a tumour suppressor, p16 mutations have been linked to an increased risk of several cancers, including cutaneous malignant melanoma and pancreatic cancer56,57. Notably, p16 expression is highly correlated with age, and measuring cellular p16 levels has been proposed as a biomarker both for cellular senescence and for determining the biological age of an organism58. In addition to its role as a biomarker, the selective removal of p16-high cells can extend the lifespan and healthspan of mice, demonstrating that p16-expressing cells influence the onset of ageing-related pathologies59.
Importantly, higher p16 expression was found to correlate with age in murine and human articular chondrocytes30. Chondrocytes expressing high levels of p16 also displayed lower expression of cartilage-related ECM proteins, such as aggrecan, but increased expression of ECM-degrading SASP factors such as MMP13 and MMP1. These initial results suggest that chondrocyte senescence not only correlates strongly with age, but also results in a metabolic transformation that contributes to the further destruction of cartilage. Given that p16 and the SASP can be independent arms of the senescence phenotype60, the group also assessed whether p16 itself contributed to OA pathology and found that it did not30. Indeed, somatic inactivation of p16 in chondrocytes of adult mice did not inhibit the SASP, nor did it alter the rate at which OA occurred in response to physiological ageing or induced joint injury. Together, these results demonstrate that p16 can be utilized as a biomarker of chondrocyte ageing but chondrocyte p16 does not appear to play a causal role in OA.
Extracellular vesicles
Understanding how ageing contributes to changes in tissue structure is a major focus of ageing research, but how ageing affects circulating factors, which are crucial for maintaining tissue homeostasis and function, is also important. In a landmark study, aged mice exposed to factors present in young mice through parabiosis exhibited restored regenerative capacity in skeletal muscle progenitor cells61. Moreover, a study in which young mice were exposed to the blood of aged mice resulted in impaired tissue function and repair62. Parabiosis has not been widely used to study cartilage function, but an experiment described this year demonstrated that mice had less severe OA if they shared circulation with young mice as opposed to older mice for the past 4 months before they were killed63. Further experiments in this study showed that daily systemic injection with the rejuvenating factor growth/differentiation factor 11 increased chondrocyte proliferation and protected mice from joint tissue degradation. Given these results, identifying specific circulating factors that contribute to the promotion or deterioration of joint tissue health could be important for understanding the mechanisms underlying OA as well as other age-related diseases.
EVs such as exosomes are small lipid membrane-bound particles that facilitate intercellular communication via the transport of proteins and RNA64. Like SASP factors, EV secretion is upregulated in senescent cells65,66, which can induce premature senescence in neighbouring cells, for example, through the transfer of microRNAs that activate senescence pathways67. Interestingly, a cross-sectional and longitudinal study found that EV concentration in plasma decreases with advancing age68. However, this decrease was accompanied by increased vesicle internalization and activation of B cells and monocytes, suggesting that EVs might enhance pro-inflammatory immune responses with age. Together, these studies highlight the emerging role of EVs in cellular and organismal senescence.
In another study, both senescent chondrocytes and EV concentrations were enriched in cartilage from patients with OA relative to cartilage from healthy individuals42. Furthermore, exposing non-senescent chondrocytes to EVs derived from patients with OA increased senescent phenotypes and decreased proteoglycan production. Fluorescent labelling and tracking of EVs confirmed that these vesicles were internalized by chondrocytes within 6 h of exposure. MicroRNAs were also differentially expressed between senescence-associated EVs and EVs not associated with senescence; the former displayed a decrease in miR-140–3p, the depletion of which was associated with cartilage dysfunction69, and an increase in miR-34a-5p that was linked to the upregulation of senescence-associated proteins70. The selective removal of senescent cells using the senolytic compound UBX0101 (see below) reduced the number of EVs in cultured chondrocytes from patients with OA, and EVs isolated from the synovial fluid of UBX0101-treated mice contained features associated with cartilage growth, such as increased aggrecan and decreased proteases42. Together, this work suggests that increased EV secretion and internalization, along with changes to vesicular RNA and protein content, should be investigated as potential biomarkers for both chondrocyte senescence and OA. Importantly, the authors of this study also found differences in the expression of microRNAs in EVs from the synovial fluid between aged healthy donors and donors with clinical OA42. Examining EV microRNA profiles could help distinguish cartilage loss caused by OA and other arthropathies rather than by ageing.
Oxidative stress drives OA and senescence
Another hallmark of ageing is mitochondrial dysfunction, which causes oxidative stress by increasing cellular levels of ROS. ROS-induced DNA damage has been linked to the pathogenesis of many age-related conditions, including cardiovascular, pulmonary, kidney and neurodegenerative diseases71. Additionally, increased oxidative stress and a decrease in the antioxidant capacity of mitochondria can disrupt physiological cell signal transduction, which might promote ageing by gradually causing loss of cellular integrity and tissue homeostasis72,73.
With regard to OA, oxidative stress has been proposed as a driver of the catabolic and anabolic signalling imbalance in cartilage that results in progressive matrix degradation74 (FIG. 2). For example, survival and tolerance of oxidative stress is regulated by members of the mitogen-activated protein kinase (MAPK) pathway, such as c-Jun N-terminal kinases (JNKs) and p38. It has been suggested that cytokine-mediated activation of JNK signalling worsens OA-associated phenotypes by activating pro-inflammatory and ECM degradation pathways in joint tissue cells75. However, oxidative stress in cultured human chondrocytes inactivated JNKs while p38 remained active76. Deletion of JNK1 and JNK2 in mice resulted in more severe age-related OA than in wild-type mice, as well as increased senescence in cartilage and particularly in the synovium77, suggesting that JNK is a negative regulator of joint senescence.
Fig. 2 |. Model for oxidative stress-induced senescence in joint cells.
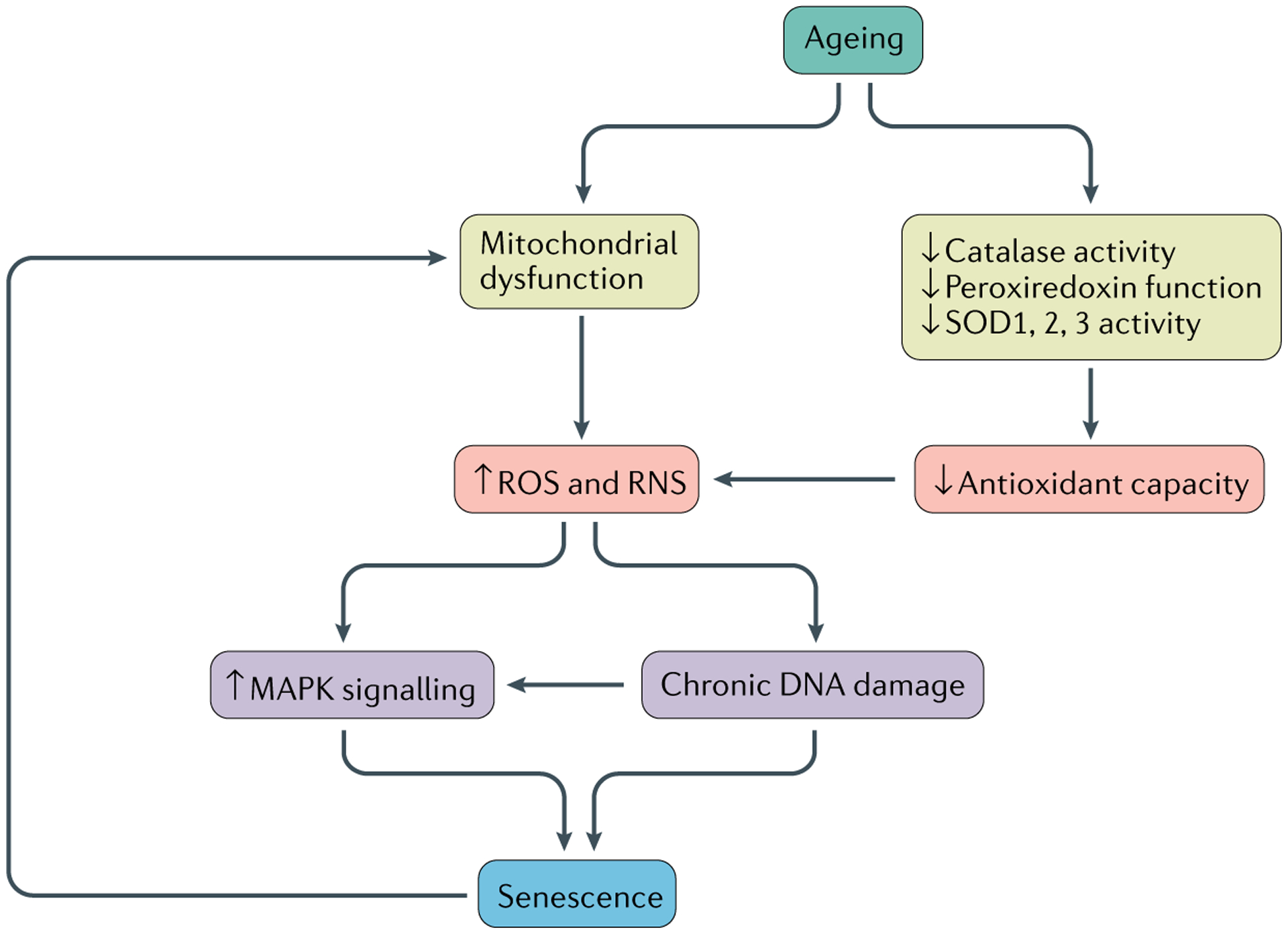
Aged chondrocytes and synovial cells exhibit mitochondrial dysfunction, as well as a reduction in antioxidant capacity, via a decrease in the activity of catalase and superoxide dismutase (SOD) and decreased peroxiredoxin function. These phenotypes increase the generation of reactive oxidative species (ROS) and reactive nitrogen species (RNS), which induce chronic DNA damage and increase MAPK stress signalling, both of which can act independently or together to induce senescence. Senescence itself can cause further mitochondrial damage, causing positive feedback.
In addition to ageing, senescence itself has been shown to induce mitochondrial dysfunction and stimulate ROS production78. Overproduction of hydrogen peroxide and reactive nitrogen species, including nitric oxide (NO), has been detected in aged cartilage and OA cartilage from both humans and monkeys79. Cells from human cartilage explants cultured in the presence of hydrogen peroxide exhibited hallmarks of chondrocyte senescence, including shortened telomeres, reduced replicative capacity and lower production of glycosaminoglycan80. Loss of antioxidant enzymes, such as superoxide dismutase (SOD), is known to correlate with premature senescence and accelerated ageing phenotypes81,82. All three SOD family members (SOD1, SOD2 and SOD3) are abundantly expressed in human articular cartilage, but their activity is markedly decreased in cartilage from patients with OA83,84. Similarly, peroxiredoxins and catalases are antioxidants that are critical in the regulation of redox signalling and the protection against oxidative stress by controlling levels of H2O2 (REF.85). Chondrocytes isolated from older adults were noted to have hyperoxidized (and thus inactive) peroxiredoxins, whereas overexpression of catalase targeted to the mitochondria reduced the severity of OA in 24-month-old mice86. Together, these results suggest a correlation between increased oxidative stress and the induction of senescence in cartilage, which might drive OA. They also support the strategy of using antioxidants to prevent ROS-induced senescence, which could be a useful approach to the treatment of OA.
Senolytics and senomorphics for OA
Senolytics and senomorphics are two classes of therapeutics that have been reported to alleviate ageing-associated pathologies in murine models and are currently being investigated in trials in humans. Senolytics induce apoptosis preferentially in senescent cells, whereas senomorphics inhibit the SASP factors linked to pro-inflammatory paracrine signalling and tissue damage87 (FIG. 3). Given the correlations between senescence, SASP and OA, these drugs are attractive candidates for targeting OA pathogenesis and slowing its progression (TABLE 1).
Fig. 3 |. Model for cellular senescence in joint tissue and potential treatments.
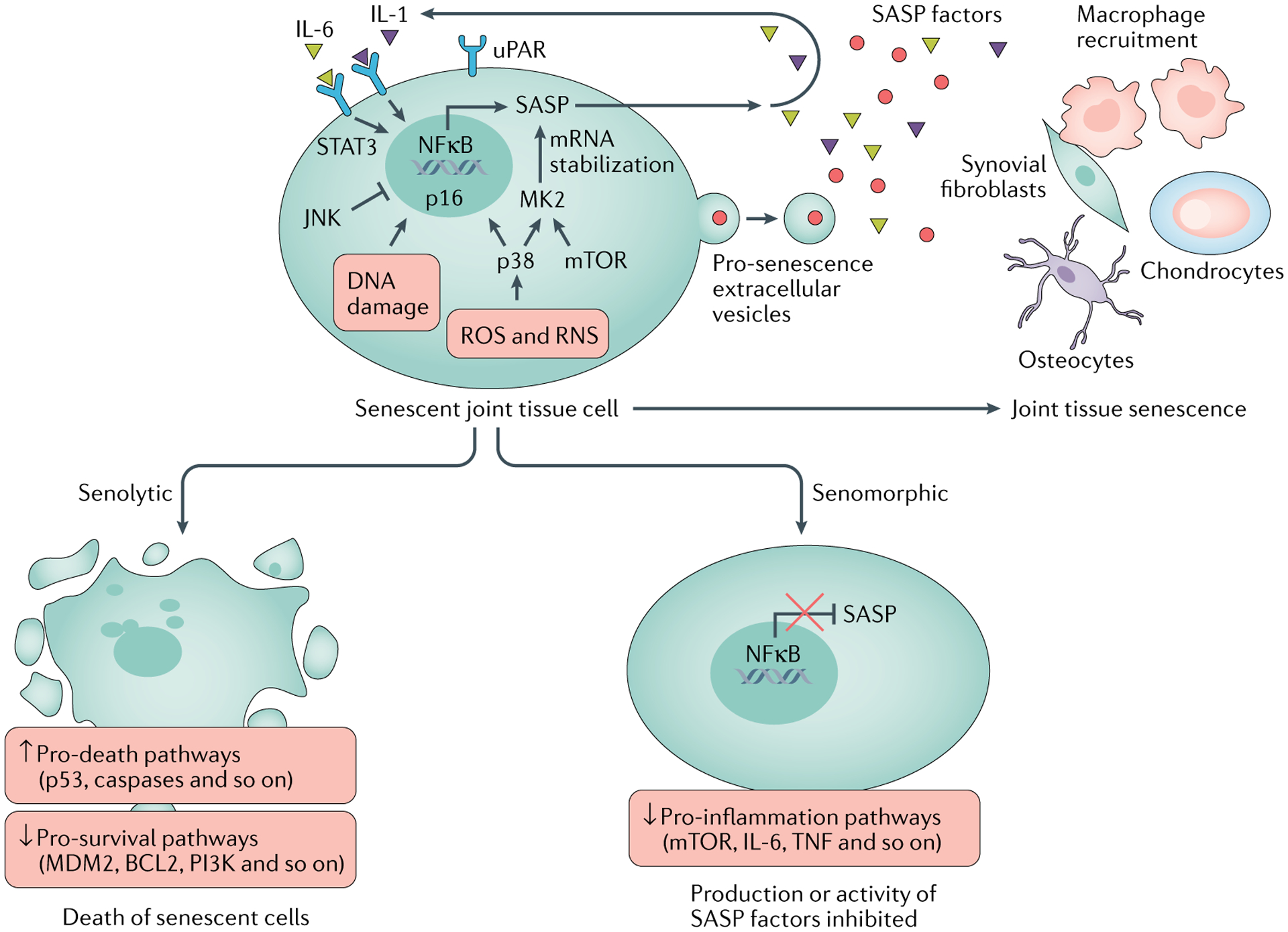
Cytokines such as IL-6 promote senescence via the transcription factor STAT3, and IL-1 can induce NFκB-driven expression of genes encoding senescence-associated secretory phenotype (SASP) factors. Senescent joint cells are characterized by increased oxidative stress (owing to the generation of reactive oxidative species (ROS) and reactive nitrogen species (RNS)), DNA damage, increased expression of urokinase-type plasminogen activator surface receptor (uPAR), and upregulation of stress proteins such as p38, c-Jun N-terminal kinase (JNK) and mTOR. p38 induces senescence and the expression of p16, while JNK negatively regulates senescence in cells in joint tissue. mTOR and p38 promote the SASP by upregulating the translation of (mTOR) and phosphorylating (p38) MK2 (also known as MAPKAPK2), which stabilizes mRNA transcripts encoding SASP factors. SASP factors (including IL-1 and IL-6) and senescence-inducing extracellular vesicles are secreted by these cells into the extracellular matrix, promoting macrophage recruitment to, and driving further senescence in, the surrounding joint tissue. Senolytic drugs aim to prevent senescence-associated disease by inducing apoptosis specifically in senescent cells via the upregulation of p53, caspases and other proteins in death-associated pathways, while repressing pathways associated with cell survival (for example, pathways involving MDM2, BCL2 and PI3K). Senomorphic drugs do not kill senescent cells, but repress the SASP by inhibiting the activity of proteins related to inflammation, such as mTOR, or by directly inhibiting the activity or production of SASP factors such as IL-6 and TNF.
Table 1 |.
Senolytics and senomorphics with potential as therapeutics for OA
| Drug name | Target of action | Refs |
|---|---|---|
| Senolytics | ||
| Dasatinib | BCR-ABL, SRC, c-KIT, ephrin A receptor | 92–96 |
| Quercetin | PI3K and serpins | 89,92,93,95,96 |
| Fenofibrate | PPARα | 102 |
| Fisetin | SIRT1, IL-1β | 103–105 |
| UBX0101 | MDM2 | 32,97–100 |
| Navitoclax (ABT-263) | BCL-2, BCL-XL | 90,91,100 |
| Senomorphics | ||
| Lutikizumab | IL-1α, IL-1β | 120 |
| Canakinumab | IL-1β | 122 |
| Tocilizumab | IL-6 receptors | 123,124 |
| Etanercept | TNF | 121 |
| CL82198 | MMP13 | 130 |
MDM2, mouse double minute 2 homologue; MMP13, matrix metalloproteinase 13; PPARα, peroxisome proliferator-activated receptor alpha; SIRT1, NAD-dependent deacetylase sirtuin-1.
Senolytics
Development of senolytics for OA.
In pioneering preclinical studies, an inducible transgene was developed that allowed the targeted killing and clearance of senescent cells expressing high levels of p16 (REFS59,88). Mice expressing this transgene demonstrated increased median lifespan and delayed onset of ageing-associated pathologies compared with wild-type mice. When a similar transgenic technique was used to clear senescent cells locally in mouse articular cartilage, the development of post-traumatic OA was substantially decreased32.
Although these experiments utilized transgenic mice to induce apoptosis in cells undergoing senescence, other studies have tested whether senolytics can mimic this effect therapeutically. In one study, which compared the gene expression profiles of senescent cells and proliferating cells, senescence was found to upregulate genes encoding proteins in anti-apoptotic signalling networks, such as BCL-2 family members and proteins in the PI3K–AKT pathway89. Many senolytics induce apoptosis selectively in senescent cells by suppressing pro-survival pathways that are activated in senescent, but not healthy, cells. For example, treatment of irradiated or normally aged mice with navitoclax (ABT-263), a BCL-2 and BCL-X L dual inhibitor, depleted senescent haematopoietic stem cells in bone marrow and senescent muscle stem cells, and promoted cellular rejuvenation90. Furthermore, in mouse cartilage explants, navitoclax reduced the senescence burden by eliminating chondrocytes expressing high levels of p16 through apoptosis91. Another example is the senolytic cocktail of dasatinib and quercetin, which effectively eliminates senescent cells and is being investigated in clinical trials for treating idiopathic pulmonary fibrosis, a potentially fatal disease associated with senescence92,93. Dasatinib inhibits multiple tyrosine kinases, including BCR-ABL, SRC, c-KIT, ephrin A receptor and platelet-derived growth factor-β receptor kinases94, whereas quercetin is a plant flavonol that inhibits PI3K and inhibitors of serine proteinases called serpins89. In another study, senescent cells were transplanted into young and old mice, and caused physical dysfunction and decreased lifespan95. However, treating these mice with dasatinib and quercetin attenuated the harmful effects of senescence and increased healthspan and lifespan. Similarly, treating aged mice with dasatinib and quercetin reduced the number of senescent osteocytes in bone, decreased osteoclast formation and bone loss, improved mineral reabsorption and thickness, and substantially improved the trabecular and cortical bone microarchitecture96.
Although these drugs have yet to be tested in humans for the treatment of joint tissue disease, several other senolytics are currently being investigated in clinical trials for OA, including UBX0101 (REFS97–99), which inhibits the interaction between p53 and mouse double minute 2 homologue (MDM2), the E3 ubiquitin protein ligase that targets p53 for degradation. Local intra-articular injection of UBX0101 in mice with post-traumatic OA selectively cleared senescent cells, decreased proteoglycan loss, and alleviated OA-related disease outcomes of pain and articular cartilage degradation32. In another study, pro-inflammatory stress in chondrocytes induced cathepsin B-mediated cleavage of the NAD-dependent deacetylase sirtuin-1 (SIRT1)100, an enzyme that was found to play a critical role in chondrocyte survival and ECM homeostasis101. Cleavage of SIRT1 resulted in an N-terminal fragment that lacks deacetylase activity, and an elevated ratio of N-terminal to C-terminal SIRT1 fragments in serum correlated with both early-stage OA and chondrosenescence100. The researchers demonstrated that anterior cruciate ligament transection increased the ratio of N-terminal to C-terminal SIRT1 in serum and that clearance of senescent cells by the combined application of systemic navitoclax and intra-articular UBX0101 lowered this ratio.
High-throughput drug screening can be utilized to find new senolytics that work on chondrocytes and synovial cells, as well as to discover novel mechanisms that contribute to OA pathology. For example, in one study, over 1,000 compounds were screened for senolytic activity in a human chondrocyte cell line102. Fenofibrate, a flavonoid and agonist of peroxisome proliferator-activated receptor-α (PPARα) that is used to treat dyslipidaemias, was found to induce apoptosis in SA-β-gal-positive chondrocytes. This discovery led the authors to investigate PPARα expression in the context of OA, and they found that it was reduced in the blood and knee cartilage of patients with OA102. Flavonoids that activate sirtuins, such as fisetin, are linked to longevity and inhibit IL-1β-induced inflammation in osteoarthritic chondrocytes103,104. Fisetin is currently being evaluated in clinical trials for efficacy in alleviating OA symptoms by reducing senescence burden in cartilage105.
Concerns associated with the use of senolytics in OA.
Although pharmacological approaches to treating age-related diseases appear promising, the potential for side effects and disparities in drug potency remain a concern. Regarding the treatment of joint disease, it is unknown if promoting cell death with senolytics will compromise tissue integrity and exacerbate cartilage and bone loss seen in patients with OA. Interestingly, killing chondrocytes in the superficial zone of articular cartilage in mice, using diphtheria toxin produced by cells expressing proteoglycan 4 (also known as superficial zone proteoglycan), did not induce further cartilage damage106. In fact, the death of chondrocytes in the superficial zone appeared to improve injury outcomes following surgical destabilization of the medial meniscus. The authors proposed that catabolism from intact chondrocytes, rather than chondrocyte death, drives further cartilage loss following joint injury. Given that senescence is a feature of post-traumatic OA, this evidence suggests that killing senescent chondrocytes with senolytics might help to prevent injury-induced cartilage loss caused by catabolic SASP factors that are secreted by senescent chondrocytes. It will be important to perform similar studies in patients with age-related OA to determine the capacity of cartilage to maintain long-term homeostasis after cell death is induced.
Another consideration for the use of senolytics in OA strategies is that, while a plethora of evidence implicates cellular senescence as a driver of ageing and disease pathology, some studies have suggested a beneficial role for senescence in various physiological processes, including tissue remodelling and wound healing107. For example, senescence was found to be induced during the intermediate stages of limb regeneration in salamanders108. After amputation, senescent cells accumulated in the cartilage and muscles of the developing limb but were subsequently cleared naturally by macrophages before full regrowth. Macrophage depletion prevented the clearance of senescent cells108, and was found, in another study, to stunt regeneration109. Importantly, the proportion of cells that became senescent after amputation was not influenced by age, suggesting an ageing-independent role of senescence in tissue repair. Although more research into this concept is needed, the authors of this study postulated that efficient immunosurveillance of senescent cells might have allowed macrophages to be recruited to areas of damaged tissue, which was necessary for regeneration. In a study in mice, senescent fibroblasts and endothelial cells were found to accumulate near sites of cutaneous wounds and to accelerate healing through the secretion of platelet-derived growth factor AA (PDGF-AA; that is, PDGF composed of two A subunits), which induced myofibroblast differentiation110. This study suggests that secretion of growth factors and remodelling enzymes by the SASP might help to stimulate cell growth, which can aid tissue renewal and wound closure. Accordingly, more research is needed to establish if the wholesale elimination of senescent cells from joints causes side effects that could further contribute to tissue loss in OA.
Senomorphics
Overview of senomorphic candidates.
The therapeutic targeting of pathways and molecules linked to inflammation and disease is not a new strategy, and a wide array of senomorphic candidates have been shown to inhibit pathways linked to the SASP without inducing apoptosis. These senomorphic candidates include inhibitors of IκB kinase and NFκB (such as NEMO-binding domain peptides)111, inhibitors of the tyrosine protein kinase JAK (such as ruxolitinib)112, ATM inhibitors (such as KU-60019)113, compounds that block progerin–lamin A/C binding (such as JH4)114, activators of PDGF and fibroblast growth factor signalling (for example, conditioned medium from embryonic stem cells)115, inhibitors of TGFβ receptor type 2 and p21 (such as Mmu-miR-291a-3p)116, and more117. Given the correlation between the expression of SASP factors and OA-like pathology, the inhibition of these factors is an attractive treatment approach. However, choosing the right target is necessary to ensure therapeutic efficacy and specificity.
Cytokine inhibition.
In cartilage, TNF combined with the release of other SASP factors such as IL-1β stimulates the production of damaging MMPs and inhibits tissue repair118,119. Clinical trials of TNF or IL-1 inhibition for the treatment of OA have been somewhat disappointing. For example, in a phase II trial of lutikizumab, a dual inhibitor of IL-1α and IL-1β, in patients with knee OA and synovitis, lutikizumab treatment led to a very limited improvement in pain and had no effect on synovitis120, and in a trial of etanercept, a TNF inhibitor, in patients with inflammatory hand OA, etanercept treatment failed to improve pain and had a limited effect on structure121. However, a recent exploratory analysis of data from a trial designed to examine the efficacy of the anti-IL-1β antibody canakinumab on cardiac events in an at-risk population (that is, patients with previous myocardial infarction and elevated C-reactive protein) found a lower incidence of knee and hip replacement in the canakinumab-treated groups than in a placebo-treated control group122.
IL-6 has been implicated in the pathogenesis of rheumatoid arthritis (RA), and the IL-6 receptor inhibitor tocilizumab is effective in clinical therapy for RA123 and is currently in phase III trials for hand OA124. Although RA is an autoimmune disease, it shares common features with OA, including the release of pro-inflammatory cytokines and degradation of the cartilage matrix. Surprisingly, however, Il6 knockout mice exhibit more severe OA in response to physiological ageing than wild-type mice125, suggesting that OA pathogenesis is complex and requires a multifaceted approach to treatment.
Targeting MMPs.
MMPs are another class of SASP factors to consider as targets for pharmacological intervention due to their known catabolic effects on cartilage. Specifically, MMP13 is the most highly expressed MMP in connective tissue126 and the most specific enzyme for the degradation of type-II collagen found in articular cartilage127. Human chondrocytes from patients with OA were found to express higher levels of MMP13 than chondrocytes from donors with healthy cartilage128. Furthermore, postnatal overexpression of MMP13 in transgenic mice induced OA-like arthropathy, implicating MMP13 as a primary driver of OA pathogenesis129. In another study, chondrocyte-specific deletion of MMP13 reduced the severity of OA induced by meniscal-ligamentous injury (MLI)130. To test the effects of senomorphics on OA progression, the researchers also treated wild-type mice with CL82198, a selective inhibitor of MMP13, after MLI. CL82198 treatment reduced OA severity, increased levels of type II collagen and inhibited chondrocyte death.
Together, these data suggest that the inhibition of SASP factors via senomorphics might be a promising therapeutic approach to treating OA. However, more research is needed to determine precisely which SASP factors contribute to OA pathology, and if their inhibition slows or prevents disease progression.
Conclusions
The evidence implicating cellular senescence in joint tissues as a primary driver of OA pathogenesis and progression is compelling, but further investigation is needed to identify the precise mechanisms by which senescence causes specific disease phenotypes. Most likely, the thread tying ageing, senescence and OA pathology together is the accumulation of senescent cells over time combined with gradual changes in cellular metabolism, morphology and function, all of which contribute to loss of joint tissue homeostasis and integrity. Effective OA treatment strategies will require first establishing the underlying mechanisms that drive these changes to cell physiology, and then designing therapies directed towards these mechanisms.
Additionally, the common biomarkers used to identify senescence are insufficient for diagnosing OA. SA-β-gal staining is not necessarily an indicator of chondrocyte senescence and can be influenced by changes in autophagy and lysosome function, both of which are reduced in OA49,50. Also, the expression of p16 in chondrocytes, which is used in many studies using senolytics to identify senescent cells, is not required for the SASP or OA pathogenesis30. Therefore, other biomarkers should be considered for the therapeutic targeting of cells involved in OA to ensure specificity and prevent unintended effects. Recent evidence has demonstrated that chondrocyte senescence and OA are linked to changes in the secretion of EVs and their cargo42. Accordingly, EVs, as well as the expression of uPAR (which is present on senescent chondrocytes36), should be further investigated to determine if they are accurate clinical markers for joint disease.
Senescence in joint tissues is driven by several stress-related pathways that converge on the SASP, and techniques that suppress inflammatory cytokines or selectively eliminate senescent cells while leaving healthy cells unharmed are attractive candidates for use in anti-ageing strategies (FIG. 3). However, although the preclinical evidence for using senolytics and senomorphics to treat OA phenotypes looks promising, these approaches have not yet demonstrated efficacy in eliminating or preventing the disease. Additionally, although SASP inhibitors, such as CL82198, have been proven effective in reducing the severity of post-traumatic OA in mice130, the same effect has yet to be demonstrated on aged or diseased chondrocytes and other synovial joint cells in humans.
Furthermore, the progression of these therapies from the laboratory to the clinic is hindered by the lack of evidence implicating a specific cell type as the primary driver of OA. Chondrocytes, synovial fibroblasts, osteocytes and probably other joint tissue cells not yet studied, are all capable of becoming senescent and secreting SASP factors into the joint environment. Without knowing which cells are responsible for each OA phenotype, drug specificity for disease treatment will be difficult to evaluate.
Finally, further investigation is needed into the potential harmful effects of killing or altering senescent cells in an organ. Recent studies have demonstrated that senescence stimulates early wound healing and tissue regeneration via macrophage recruitment108–110. Even if senescent cells are responsible for the progression of OA after injury, eliminating these cells or preventing paracrine signalling too early might prevent the initial healing of damaged cartilage and other tissues, which could have devastating consequences for the entire joint. For this reason, studies using senolytics and senomorphics must include comparisons of disease outcomes from different treatment timings to ensure that drug efficacy can be properly inferred.
Key points.
Osteoarthritis (OA) pathology overlaps with the senescence of cells in joint tissue and the senescence-associated secretory phenotype.
Several hallmarks of senescence are associated with OA, but it is unclear which of these cause disease progression.
Ageing, DNA damage and oxidative stress can induce senescence in cells in joint tissue.
The complexity of the senescent cellular phenotype necessitates the careful use of biomarkers to identify senescent cells.
Targeting senescence for OA therapy is a promising new approach that deserves further investigation.
Acknowledgements
The authors’ research work is funded by grants from the National Institute on Aging (RO1 AG044034) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R37 AR049003).
Footnotes
Competing interests
R.F.L. has consulted for Unity Biotechnology (<$1,000). The other authors declare no competing interests.
Peer review information
Nature Reviews Rheumatology thanks F. Blanco, P. van der Kraan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Loeser RF, Goldring SR, Scanzello CR & Goldring MB Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loeser RF, Collins JA & Diekman BO Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol 12, 412–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson VL & Hunter DJ The epidemiology of osteoarthritis. Best Pract. Res. Clin. Rheumatol 28, 5–15 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Cisternas MG et al. Alternative methods for defining osteoarthritis and the impact on estimating prevalence in a US population-based survey. Arthritis Care Res. 68, 574–580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hootman JM & Helmick CG Projections of US prevalence of arthritis and associated activity limitations. Arthritis Rheum. 54, 226–229 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Losina E et al. Lifetime medical costs of knee osteoarthritis management in the United States: impact of extending indications for total knee arthroplasty. Arthritis Care Res. 67, 203–215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He S & Sharpless NE Senescence in health and disease. Cell 169, 1000–1011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCulloch K, Litherland GJ & Rai TS Cellular senescence in osteoarthritis pathology. Aging Cell 16, 210–218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeon OH, David N, Campisi J & Elisseeff JH Senescent cells and osteoarthritis: a painful connection. J. Clin. Invest 128, 1229–1237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Childs BG et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov 16, 718–735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayflick L & Moorhead PS The serial cultivation of human diploid cell strains. Exp. Cell Res 25, 585–621 (1961). [DOI] [PubMed] [Google Scholar]
- 13.Jurk D et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farr JN et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res 31, 1920–1929 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker DJ, Alimirah F, van Deursen JM, Campisi J & Hildesheim J Oncogenic senescence: a multi-functional perspective. Oncotarget 8, 27661–27672 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki M, Kajiya H, Ozeki S, Okabe K & Ikebe T Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16(INK4a.). Biochem. Biophys. Res. Commun 452, 622–628 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Sanokawa-Akakura R, Akakura S, Ostrakhovitch EA & Tabibzadeh S Replicative senescence is distinguishable from DNA damage-induced senescence by increased methylation of promoter of rDNA and reduced expression of rRNA. Mech. Ageing Dev 183, 111149 (2019). [DOI] [PubMed] [Google Scholar]
- 18.van Deursen JM The role of senescent cells in ageing. Nature 509, 439–446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppe JP, Desprez PY, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sager R Senescence as a mode of tumor suppression. Environ. Health Perspect 93, 59–62 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campisi J Aging, cellular senescence, and cancer. Annu. Rev. Physiol 75, 685–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campisi J Cellular senescence: putting the paradoxes in perspective. Curr. Opin. Genet. Dev 21, 107–112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Storer M et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Basisty N et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herranz N et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol 17, 1205–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laberge RM et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol 17, 1049–1061 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tchkonia T, Zhu Y, van Deursen J, Campisi J & Kirkland JL Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest 123, 966–972 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lotz MK et al. Cartilage cell clusters. Arthritis Rheum. 62, 2206–2218 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogrodnik M, Salmonowicz H, Jurk D & Passos JF Expansion and cell-cycle arrest: common denominators of cellular senescence. Trends Biochem. Sci 44, 996–1008 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Diekman BO et al. Expression of p16INK 4a is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 17, e12771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Rey MJ et al. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun. Ageing 16, 29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon OH et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin JA, Brown T, Heiner A & Buckwalter JA Post-traumatic osteoarthritis: the role of accelerated chondrocyte senescence. Biorheology 41, 479–491 (2004). [PubMed] [Google Scholar]
- 34.Schafer MJ et al. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes 65, 1606–1615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amor C et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwab W et al. Interleukin-1β-induced expression of the urokinase-type plasminogen activator receptor and its co-localization with MMPs in human articular chondrocytes. Histol. Histopathol 19, 105–112 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Xu M et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J. Gerontol. A Biol. Sci. Med. Sci 72, 780–785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loeser RF Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil 17, 971–979 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearson MJ et al. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep 7, 3451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kojima H, Inoue T, Kunimoto H & Nakajima K IL-6-STAT3 signaling and premature senescence. JAKSTAT 2, e25763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson G et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeon OH et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 4, e125019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Debacq-Chainiaux F, Erusalimsky JD, Campisi J & Toussaint O Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc 4, 1798–1806 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Dimri GP et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee BY et al. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5, 187–195 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Baisantry A et al. The impact of autophagy on the development of senescence in primary tubular epithelial cells. Cell Cycle 15, 2973–2979 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berkenkamp B et al. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 9, e88071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piechota M et al. Is senescence-associated β-galactosidase a marker of neuronal senescence? Oncotarget 7, 81099–81109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeon H & Im GI Autophagy in osteoarthritis. Connect. Tissue Res 58, 497–508 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Carames B, Taniguchi N, Otsuki S, Blanco FJ & Lotz M Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 62, 791–801 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carames B et al. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis 71, 575–581 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coryell PR et al. Autophagy regulates the localization and degradation of p16ink4a. Aging Cell 19, e13171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kroemer G, Marino G & Levine B Autophagy and the integrated stress response. Mol. Cell 40, 280–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schipani E et al. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 15, 2865–2876 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serrano M The tumor suppressor protein p16ink4a. Exp. Cell Res 237, 7–13 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Goldstein AM et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16ink4 mutations. N. Engl. J. Med 333, 970–974 (1995). [DOI] [PubMed] [Google Scholar]
- 57.Goldstein AM & Tucker MA Screening for CDKN2A mutations in hereditary melanoma. J. Natl Cancer Inst 89, 676–678 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Krishnamurthy J et al. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest 114, 1299–1307 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baker DJ et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coppe JP et al. Tumor suppressor and aging biomarker p16(Ink4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem 286, 36396–36403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conboy IM et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Rebo J et al. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nat. Commun 7, 13363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li L et al. Positive effects of a young systemic environment and high growth differentiation factor 11 levels on chondrocyte proliferation and cartilage matrix synthesis in old mice. Arthritis Rheumatol. 72, 1123–1133 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Valadi H et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 9, 654–659 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Lehmann BD et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 68, 7864–7871 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Effenberger T et al. Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J. 28, 4847–4856 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Overhoff MG et al. Cellular senescence mediated by p16INK4A-coupled miRNA pathways. Nucleic Acids Res. 42, 1606–1618 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eitan E et al. Age-related changes in plasma extracellular vesicle characteristics and internalization by leukocytes. Sci. Rep 7, 1342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miyaki S et al. Microrna-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 60, 2723–2730 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feliciano A, Sanchez-Sendra B, Kondoh H & Lleonart ME Micrornas regulate key effector pathways of senescence. J. Aging Res 2011, 205378 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Venkataraman K, Khurana S & Tai TC Oxidative stress in aging–matters of the heart and mind. Int. J. Mol. Sci 14, 17897–17925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones DP Redox theory of aging. Redox Biol. 5, 71–79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hui W et al. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis 75, 449–458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loeser RF The role of aging in the development of osteoarthritis. Trans. Am. Clin. Climatol. Assoc 128, 44–54 (2017). [PMC free article] [PubMed] [Google Scholar]
- 75.Ismail HM et al. Interleukin-1 acts via the JNK-2 signaling pathway to induce aggrecan degradation by human chondrocytes. Arthritis Rheumatol. 67, 1826–1836 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Nelson KJ et al. H2O2 oxidation of cysteine residues in c-Jun N-terminal kinase 2 (JNK2) contributes to redox regulation in human articular chondrocytes. J. Biol. Chem 293, 16376–16389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Loeser RF et al. Deletion of c-Jun N-terminal kinase enhances senescence in joint tissues and increases the severity of age-related osteoarthritis in mice. Arthritis Rheumatol. 72, 1679–1688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Passos JF et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol 6, 347 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loeser RF, Carlson CS, Del Carlo M & Cole A Detection of nitrotyrosine in aging and osteoarthritic cartilage: correlation of oxidative damage with the presence of interleukin-1β and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 46, 2349–2357 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Yudoh K et al. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res. Ther 7, R380–R391 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blander G, de Oliveira RM, Conboy CM, Haigis M & Guarente L Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J. Biol. Chem 278, 38966–38969 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y et al. A new role for oxidative stress in aging: the accelerated aging phenotype in Sod1−/− mice is correlated to increased cellular senescence. Redox Biol. 11, 30–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Regan E et al. Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis Rheum. 52, 3479–3491 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scott JL et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis 69, 1502–1510 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rhee SG, Woo HA & Kang D The role of peroxiredoxins in the transduction of H2O2 signals. Antioxid. Redox Signal 28, 537–557 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Collins JA et al. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signaling in aging chondrocytes. J. Biol. Chem 291, 6641–6654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang C Senolytics and senostatics: a two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol. Cell 42, 821–827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baker DJ et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu Y et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang J et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 22, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sessions GA et al. Controlled induction and targeted elimination of p16(INK4a)-expressing chondrocytes in cartilage explant culture. FASEB J. 33, 12364–12373 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hickson LJ et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Justice JN et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Keam SJ Dasatinib: in chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. BioDrugs 22, 59–69 (2008). [DOI] [PubMed] [Google Scholar]
- 95.Xu M et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med 24, 1246–1256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Farr JN et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med 23, 1072–1079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03513016 (2020). [DOI] [PubMed]
- 98.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04229225 (2020). [DOI] [PubMed]
- 99.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04349956 (2020). [DOI] [PubMed]
- 100.Batshon G et al. Serum NT/CT SIRT1 ratio reflects early osteoarthritis and chondrosenescence. Ann. Rheum. Dis 79, 1370–1380 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Matsuzaki T et al. Disruption of SIRT1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis 73, 1397–1404 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Nogueira-Recalde U et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 45, 588–605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yousefzadeh MJ et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zheng W et al. Fisetin inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes through activating sirt1 and attenuates the progression of osteoarthritis in mice. Int. Immunopharmacol 45, 135–147 (2017). [DOI] [PubMed] [Google Scholar]
- 105.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04210986 (2020). [DOI] [PubMed]
- 106.Zhang M et al. Induced superficial chondrocyte death reduces catabolic cartilage damage in murine posttraumatic osteoarthritis. J. Clin. Invest 126, 2893–2902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yun MH Cellular senescence in tissue repair: every cloud has a silver lining. Int. J. Dev. Biol 62, 591–604 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Yun MH, Davaapil H & Brockes JP Recurrent turnover of senescent cells during regeneration of a complex structure. eLife 4, e05505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Godwin JW, Pinto AR & Rosenthal NA Macrophages are required for adult salamander limb regeneration. Proc. Natl Acad. Sci. USA 110, 9415–9420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Demaria M et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tilstra JS et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest 122, 2601–2612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu M et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kang HT et al. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol 13, 616–623 (2017). [DOI] [PubMed] [Google Scholar]
- 114.Lee SJ et al. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J. Clin. Invest 126, 3879–3893 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bae YU, Choi JH, Nagy A, Sung HK & Kim JR Antisenescence effect of mouse embryonic stem cell conditioned medium through a PDGF/FGF pathway. FASEB J. 30, 1276–1286 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Bae YU et al. Embryonic stem cell-derived mmu-miR-291a-3p inhibits cellular senescence in human dermal fibroblasts through the TGF-β receptor 2 pathway. J. Gerontol. A Biol. Sci. Med. Sci 74, 1359–1367 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Kim EC & Kim JR Senotherapeutics: emerging strategy for healthy aging and age-related disease. BMB Rep. 52, 47–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stadler J et al. Articular chondrocytes synthesize nitric oxide in response to cytokines and lipopolysaccharide. J. Immunol 147, 3915–3920 (1991). [PubMed] [Google Scholar]
- 119.Pelletier JP, Roughley PJ, DiBattista JA, McCollum R & Martel-Pelletier J Are cytokines involved in osteoarthritic pathophysiology? Semin. Arthritis Rheum 20, 12–25 (1991). [DOI] [PubMed] [Google Scholar]
- 120.Fleischmann RM et al. A phase II trial of lutikizumab, an anti-interleukin-1α/β dual variable domain immunoglobulin, in knee osteoarthritis patients with synovitis. Arthritis Rheumatol. 71, 1056–1069 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Kloppenburg M et al. Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis 77, 1757–1764 (2018). [DOI] [PubMed] [Google Scholar]
- 122.Schieker M et al. Effects of interleukin-1β inhibition on incident hip and knee replacement: Exploratory analyses from a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med 173, 509–515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Smolen JS et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet 371, 987–997 (2008). [DOI] [PubMed] [Google Scholar]
- 124.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02477059 (2019). [DOI] [PubMed]
- 125.de Hooge AS et al. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthr. Cartil 13, 66–73 (2005). [DOI] [PubMed] [Google Scholar]
- 126.Vincenti MP & Brinckerhoff CE Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 4, 157–164 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shiomi T, Lemaitre V, D’Armiento J & Okada Y Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol. Int 60, 477–496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roach HI et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 52, 3110–3124 (2005). [DOI] [PubMed] [Google Scholar]
- 129.Neuhold LA et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest 107, 35–44 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang M et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther 15, R5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]