

Abstract
Epigenetic mechanisms, like those involving DNA methylation, are thought to mediate the relationship between chronic cocaine dependence and molecular changes in addiction-related neurocircuitry but have been understudied in human brain. We initially used reduced representation bisulfite sequencing (RRBS) to generate a methylome-wide profile of cocaine dependence in human post-mortem caudate tissue. We focused on the Iroquois Homeobox A (IRXA) gene cluster, where hypomethylation in exon 3 of IRX2 in neuronal nuclei was associated with cocaine dependence. We replicated this finding in an independent cohort and found similar results in dorsal striatum from cocaine self-administering mice. Using epigenome editing and 3C assays, we demonstrated a causal relationship between methylation within the IRX2 gene body, CTCF protein binding, 3D chromatin interaction, and gene expression. Together, these findings suggest that cocaine-related hypomethylation of IRX2 contributes to the development and maintenance of cocaine dependence through alterations in 3D chromatin structure in the caudate nucleus.
Keywords: cocaine, addiction, epigenetics, DNA methylation, 3C, 3D chromatin structure, psychiatry, post-mortem brain
Introduction
Like other drug use disorders, cocaine dependence is characterized by cycles of binging, preoccupation and compulsive drug seeking behaviors despite negative outcomes[1]. The development and maintenance of dependence-related behaviors, in both humans and animal models, is accompanied by profound alterations in gene expression and lasting changes in cellular plasticity in the mesolimbic dopamine neurocircuitry [2–7]. Accordingly, multiple targets of midbrain dopamine projections display widespread epigenetic alterations, particularly histone post translational modifications, that may mark distinct phases of dependence and withdrawal[7]. DNA methylation may act to stabilize dependence-related gene expression programs and it has become an active area of research in cocaine addiction neurobiology.
Cocaine-related changes in DNA methylation have primarily been measured at the level of individual gene promoters, although methylome-wide studies have begun to appear in animal models (for review, see [8]). Furthermore, research is just beginning to investigate how changes in methylation are likely to contribute to maladaptive behavioral phenotypes [9, 10]. However, the relationship between DNA methylation and cocaine dependence has been understudied in the human brain. Of particular interest is the caudate nucleus, as it appears to be necessary for the development of addiction related drug cravings, and has been implicated in the transition from recreational drug use to dependence [11–14]. Here, we report on the findings of the first methylome-wide study of cocaine dependence in the human caudate nucleus, using post-mortem tissue samples. We also present supporting evidence for a role of cocaine-related gene body methylation of the IRX2, a gene located in a region containing the largest cluster of differentially methylated CpGs from our methylome-wide analysis, in regulating local chromatin architecture and expression of two genes in the IRXA neurodevelopmental gene cluster.
Results
Chronic cocaine dependence in humans is associated with genome-wide changes in DNA methylation in the caudate nucleus
To understand how DNA methylation patterns in chronic cocaine users may differ from unaffected non-cocaine users in the caudate nucleus (Figure 1a), we performed reduced representation bisulfite sequencing (RRBS) from 25 cases who died from cocaine intoxication that had a lifetime history of cocaine dependence, but no other diagnosed psychopathology, and 25 psychiatrically healthy drug-free controls who died suddenly. RRBS allowed us to interrogate the methylation status of genome-wide loci, while enriching for CpG islands[15]. The groups were matched for commonly confounding factors such as age, post-mortem interval, and tissue pH, with small effect sizes on comparison (ps > 0.1, d < 0.3; Supplemental Table 1), and sequencing statistics were not different between the groups (ps > 0.1; Supplemental Table 2). Since DNA methylation across a region of CpGs is more likely to be biologically relevant than methylation at a single nucleotide, we combined all CpGs within 50bp of another into functional regions. We detected 6712 CpG regions containing at least 2 CpGs (Supplemental Figure 1), and 173 clusters were differentially methylated between groups when correcting for ethnicity, age, smoker status and ethanol toxicology (FDR corrected q<0.05; Figure 1b; Supplemental Table 3).
Figure 1. Widespread changes in DNA methylation associated with chronic cocaine dependence in the human caudate nucleus.
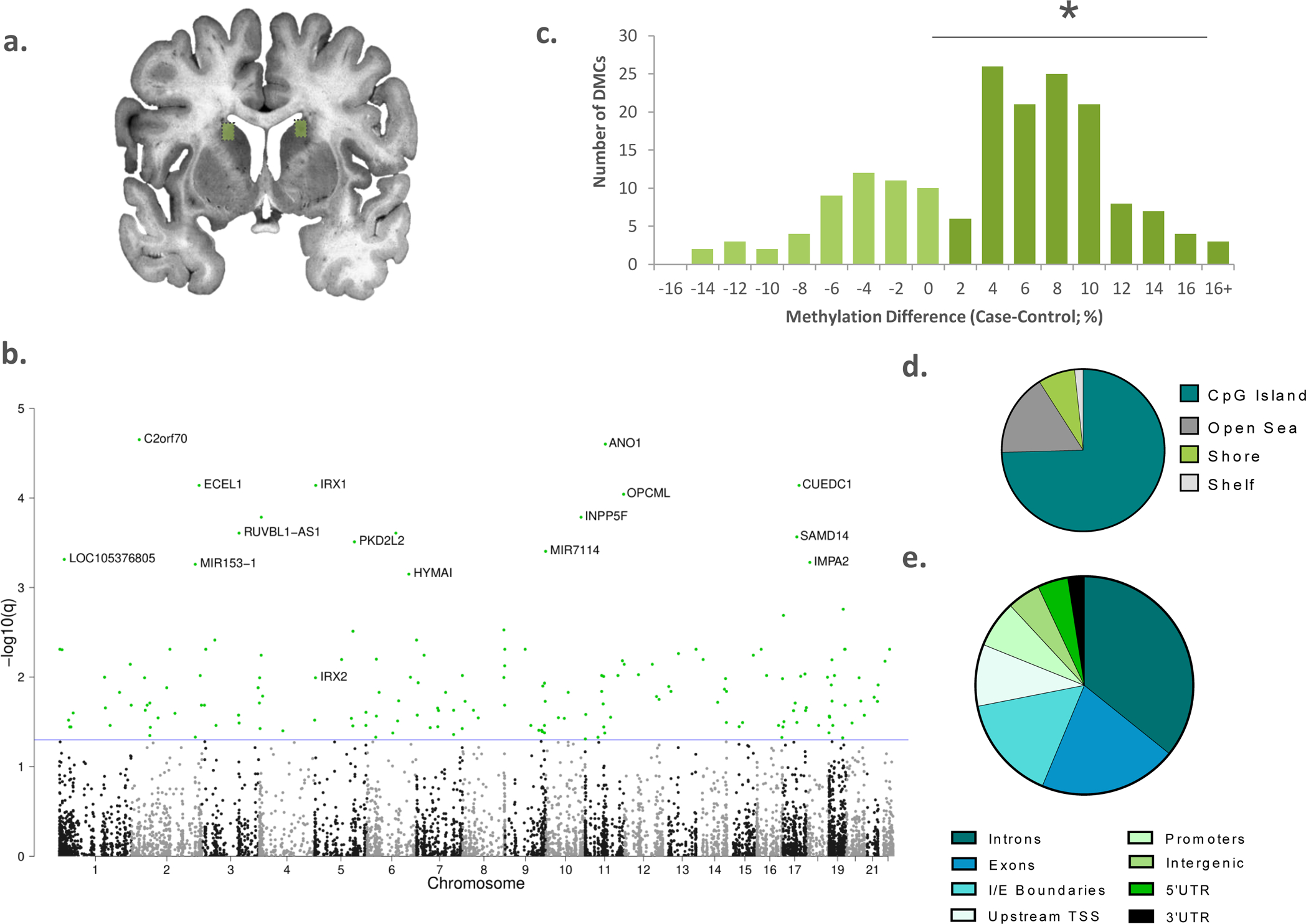
a) Dissections from dorsolateral caudate nucleus were used for reduced representation bisulfite sequencing (n=25 per group, boxed). b) Manhattan plot showing the chromosomal location of all significantly differentially methylated CpG regions (DMRs); blue line represents FDR q < 0.05. c) Although hyper- and hypomethylated DMRs were identified, there was a significant bias towards clusters with increased methylation in the cocaine group χ2= 26.575; * p < 0.05. d)Most of the DMRs overlap with known CpG islands and e) annotated introns, exons and intron-exon boundaries.
Although we detected differentially methylated regions (DMRs) that were both hyper- and hypomethylated in the cocaine group, there were significantly more hypermethylated regions than hypomethylated (Chi Squared Goodness of Fit Tests, χ2= 26.575; p < 0.05, Figure 1c). Using RNA sequencing data from the same subjects and brain nuclei, we found that transcription of the de novo methyltransferase DNMT3a is increased in the cocaine group (t=2.628, df=42, p=0.0120, Supplemental Figure 2a). This finding is in-line with animal studies that have shown that cocaine exposure can induce de novo methyltransferase expression in the striatum[10]. We found no differences in expression of the two other DNA methyltransferase genes, DNMT1 and DNMT3b (ps > 0.05, Supplemental Figure 2b, c). To determine if the corrected DMRs were functionally related, we performed PANTHER gene ontology analysis of genes that either overlapped with, or were in the closest proximity to, the differential methylation signal [16]. Although we found no enrichment for genes belonging to any particular cellular component or biological process, which is likely due to unknown long-range target genes, we found a significant enrichment for genes involved in regulatory sequence-specific DNA binding and transcriptional activation (fold enrichment=4.38, FDR=0.0253).
Most of the regions, including those which were differentially methylated, mapped onto known CpG islands (Figure 1d; Supplemental Figure 3a), and although they were highly present within gene bodies (introns, exons and intron-exon boundaries), they were significantly enriched for intergenic regions (Figure 1e; Supplemental Figure 3b, Fisher’s exact test q=7.10 × 10−4). We used data from the 15-state core model of chromatin states from the Roadmaps Epigenome Consortium[17], which was generated from human caudate nucleus tissues, to annotate the hypothetical chromatin status of our CpG clusters. We found that DMRs were significantly enriched for enhancers (Fisher’s exact test q=1.76 ×10−10), regions flanking active transcription start sites (Fisher’s exact test q=2.69 × 10−2), and weakly transcribed and quiescent regions (Fisher’s exact test q= 5.26 × 10−6 and 1.76 × 10−7 respectively) when compared against the list of all CpG clusters (Supplemental Figure 3c, d). When we assessed the hyper- and hypomethylated DMRs separately, we found that this effect was driven by the hypermethylated loci which were enriched in the same context as the overall list (qs = 1.30 5 × 10−4 − 2.46 × 10−8).
DNA methylation related to gene expression in cis.
To determine whether differentially methylated regions were related to transcriptomic changes in cis, we generated RNA-seq data from caudate tissue from the same subjects. We calculated the fold change of all genes within 5kb of a DMR, and took a liberal approach to generate a list of putative DMR-gene pairs with a nominally significant expression difference (uncorrected p < 0.1) and statistically significant differences in methylation (q value < 0.05). This analysis identified 23 DMR-gene pairs (Table 1).
Table 1.
Differentially methylated regions with nominally differential gene expression
| Chr | From | To | Gene | Number of CpGs | Methylation Difference | RNAseq Fold Change | RNAseq p-value |
|---|---|---|---|---|---|---|---|
| 5 | 2748781 | 2748955 | IRX2 | 21 | −3.06% | 1.20 | 8.17E-02 |
| 9 | 126776177 | 126776282 | LHX2 | 12 | −8.22% | 1.22 | 1.46E-02 |
| 5 | 3599609 | 3599704 | IRX1 | 9 | 11.05% | 1.39 | 1.21E-03 |
| 4 | 53474 | 53566 | ZNF595 | 9 | 7.67% | 0.79 | 1.08E-02 |
| 17 | 1960987 | 1961029 | HIC1 | 8 | −11.51% | 1.25 | 7.09E-03 |
| 2 | 26785211 | 26785290 | C2orf70 | 7 | 7.44% | 1.29 | 1.31E-02 |
| 1 | 17215449 | 17215492 | CROCC | 7 | −5.45% | 1.24 | 1.85E-03 |
| 11 | 132812684 | 132812729 | OPCML | 7 | 15.93% | 0.86 | 7.03E-02 |
| 2 | 63274825 | 63274901 | OTX1 | 6 | 9.92% | 1.25 | 2.53E-02 |
| 18 | 77918229 | 77918253 | PARD6G | 6 | −10.83% | 1.51 | 3.98E-05 |
| 5 | 131607235 | 131607278 | PDLIM4 | 6 | 8.28% | 1.36 | 2.78E-03 |
| 7 | 150002 | 150037 | AC093627.10 | 5 | 8.95% | 0.84 | 2.14E-02 |
| 12 | 120654707 | 120654747 | PXN | 5 | 10.21% | 1.17 | 5.55E-02 |
| 19 | 18980163 | 18980188 | UPF1 | 5 | 10.03% | 1.07 | 2.35E-02 |
| 2 | 63275003 | 63275040 | OTX1 | 4 | 7.49% | 1.25 | 2.53E-02 |
| 17 | 76172805 | 76172850 | TK1 | 4 | 7.10% | 0.87 | 8.77E-02 |
| 8 | 37556087 | 37556121 | ZNF703 | 4 | 12.14% | 1.20 | 1.41E-02 |
| 6 | 32165134 | 32165176 | NOTCH4 | 3 | 8.58% | 1.14 | 9.81E-02 |
| 3 | 8799985 | 8800008 | OXTR | 3 | 12.55% | 1.29 | 1.35E-02 |
| 19 | 47220817 | 47220856 | PRKD2 | 3 | 5.76% | 1.33 | 3.70E-04 |
| 5 | 176877611 | 176877636 | PRR7 | 3 | 11.64% | 1.53 | 4.52E-11 |
| 4 | 6273547 | 6273577 | WFS1 | 3 | 21.42% | 1.12 | 7.12E-02 |
| 9 | 136654410 | 136654426 | VAV2 | 2 | 12.46% | 1.17 | 3.08E-02 |
Given our analysis strategy, we rationalized that regions with more CpGs would be most likely to represent strong, biologically meaningful signals. Interestingly, the largest and third largest DMRs overlapped with members of the Iroquois Homeobox (IRXA) gene family that are grouped in a highly conserved cluster within vertebrate genomes and were both upregulated according to RNA-seq (Table 1). Iroquois Homeobox 2 (IRX2) and Iroquois Homeobox 1 (IRX1) are head-to-head neighbours on chromosome 5, and code for transcription factors that are involved in embryonic patterning during neural development [18]. Our RRBS analysis identified 21 CpGs within the third exon of IRX2 that were 3% less methylated (Figure 2a, Table 1), and 9 CpGs within the second exon of IRX1 that were 11% more methylated in the cocaine group (Supplemental Figure4a; Table 1).
Figure 2. IRX2 is hypomethylated in the caudate nucleus.
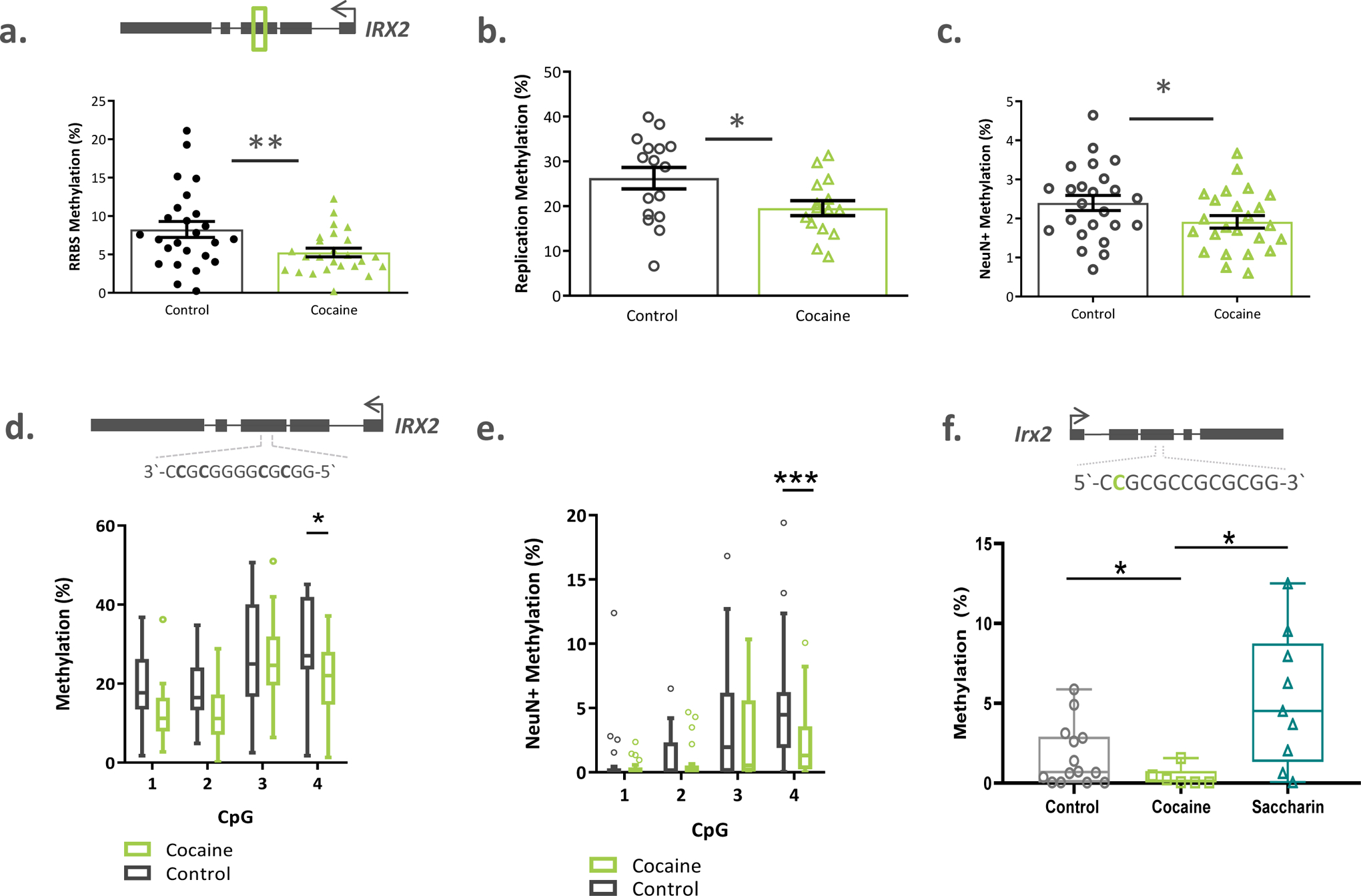
a) RRBS analysis identified a cluster of 21 CpGs within the third exon of IRX2 that were less methylated in the cocaine group of the discovery cohort (n=25 per group). b) This data was replicated in an independent cohort of caudate samples (n=15 cases and n=20 controls). c) Hypomethylation was specific to neuronal (NeuN+) nuclei (n=24 cases and n=24 controls; discovery cohort). d) The 5’ CpG within the CTCF binding site (exon 3) was hypomethylated in the cocaine group in caudate tissue homogenate (n= 20 controls and 17 cases, replication cohort) and e) neuronal nuclei (n=24 controls and n=23 cases). f) The 5’ most CpG in the mouse CTCF site was significantly less methylated after cocaine self-administration (n=6), compared to non-drug reward self-administration (n=9) or controls (n=14). Box plots indicate mean and range of data. Bar data represented as mean ± s.e.m. * p < 0.05; ** q val < 0.02
Decreased gene body methylation of IRX2 is associated with chronic cocaine dependence
We turned to an independent cohort of dorsal caudate tissue samples from individuals with cocaine dependence (who died by causes other than cocaine overdose) and unaffected controls to replicate the methylation findings (Supplementary Table 4). Using bisulfite amplicon sequencing, we found a significant decrease in methylation of the same region within exon 3 of IRX2 in the cocaine group compared to controls (5.5%, t=1.908, df=31.83, p=0.033, d=0.631; Figure 2b), but no differences in methylation of the IRX1 region (t=1.149, df=33, p=0.13, d=0.397; Supplemental Figure 4b). Based on this study, we chose to focus on IRX2 for further analyses.
It is well known that DNA methylation patterns are cell-type specific, and are particularly important in the distinct functions of neuronal and non-neuronal cell types within the central nervous system [19, 20]. Thus, we investigated our findings in distinct populations of nuclei, separated from samples of the human caudate from our discovery cohort, using fluorescence activated nuclei sorting (FANS). We separated intact nuclei based on DRAQ5 DNA stain fluorescence, and neuronal nuclei (including D1- and D2-medium spiny neurons, as well as GABAergic and cholinergic interneurons) from non-neuronal nuclei (glial and epithelial cells) based on the nuclear marker NeuN (Supplemental Figure 5a–e). We found the cocaine-associated decrease in IRX2 methylation to be specific to neuronal nuclei (t=1.923, df=46, p=0.03; Figure 2c). There was no group-wise difference in methylation in non-neuronal nuclei (t=0.3254, df=48, p=0.37; Supplemental Figure 6a).
Hypomethylation of a putative CTCF binding site
Expression of IRX2, as well as other members of the IRXA family, is known to be regulated by a large evolutionarily conserved group of enhancers that form a three-dimensional chromatin loop in animals[21]. We hypothesized that cocaine-related methylation might interfere with this regulatory framework, perhaps by impairing the binding of CCCTC-binding factor (CTCF) —a well-studied transcriptional repressor involved in anchoring three dimensional chromatin structures, that can be inhibited by methylation at its binding site [22]. We searched our target sequence for putative CTCF binding sites using its consensus sequence[23], as well as the most highly enriched motifs from chromatin immunoprecipitation data generated by the ENCODE consortium (accessed through FactorBook[24]). We found a known peak motif (3’-AGGGGGCG-5’) 96 base pairs upstream of our DMR and a putative CTCF consensus sequence (3’-CCGCGGGGCGCGG-5’) spanning 4 CpGs within the DMR itself. When considering the methylation state of the consensus sequence separately from the overall region, we found a main effect of cocaine status in both whole tissue homogenates from the replication cohort (F(1, 35)=4.333, p=0.045, Figure 2d)) and in neuronal nuclei (F(1,46)=6.284, p=0.016, Figure 2e)), but not in non-neuronal nuclei from the discovery cohort (F(1,48)=0.072, p=0.78, Supplemental Figure 6b)). Post hoc comparisons showed the fourth CpG to be significantly less methylated in the cocaine group in the tissue (t=2.55, df=140, p=0.012, Figure 2d) and in neuronal nuclei (t=3.73, df=184, p < 0.001, Figure 2e), but not in the non-neuronal fraction (Supplemental Figure 6b).
Next, we sought to corroborate our findings in a well-studied mouse model of self administration (Supplemental Figure 7). After removing outliers using ROUT tests (Q=2%), we found a significant effect of group (W(2,15.7)=6.64, p=0.008, Supplemental Figure 8) where the cocaine group (n=6) was significantly less methylated than the saccharin group (n=9, p=0.029) and nominally significantly hypomethylated compared to controls (n=14. p=0.054). Notably, this sequence contains the only occurrence of the canonical CTCF binding site (5’-CCGCGCCGCGCGGTGG-3’) in the entire 5kB Irx2 gene, and when we examined the methylation status of the upstream-most CpG, we again found methylation to be lower in the cocaine group compared to both control and saccharin animals (W(2, 13.52)=10.06, p= 0.002, cocaine vs. control p=0.032, cocaine vs. saccharin p=0.016, Figure 2f). Importantly, in both analyses, methylation in the control animals did not differ from those who were-trained to self-administer saccharin, which suggests that the cocaine-related hypomethylation is not generalizable to all reward-driven behaviors.
We were able to replicate our initial genome-wide significant finding of decreased methylation within IRX2, across sample cohorts, tissue types, and species, which is suggestive of a conserved and functionally relevant genomic response. As such, we decided to explore the relationship between IRX2 methylation and IRXA cluster gene expression in our sample set.
Intragenic IRX2 methylation is negatively associated with IRXA gene cluster gene expression
Our genome-wide analyses suggested that gene expression might be disrupted in the IRXA gene cluster in relation to cocaine dependence, since the expression of both IRX1 and IRX2 was increased according to RNA-seq (Table 1). In order to validate these findings, we used nanoString technology to count the number of IRX1 and IRX2 transcripts in RNA extracted from samples in our discovery cohort (n=21 cases and n=23 controls). We found significantly higher expression of IRX2 in the cocaine group (Mann-Whitney U = 170; p= 0.019; Figure 3a), and although the increased expression of IRX1 was not statistically significant (t=1.057, df=42; p=0.148; Figure 3b), we found the expression of the two genes to be highly positively correlated in our samples overall (r=0.622; p < 0.0001; Figure3c).
Figure 3. IRX2 expression is increased in cocaine use disorder and is related to exon 3 methylation in cells.
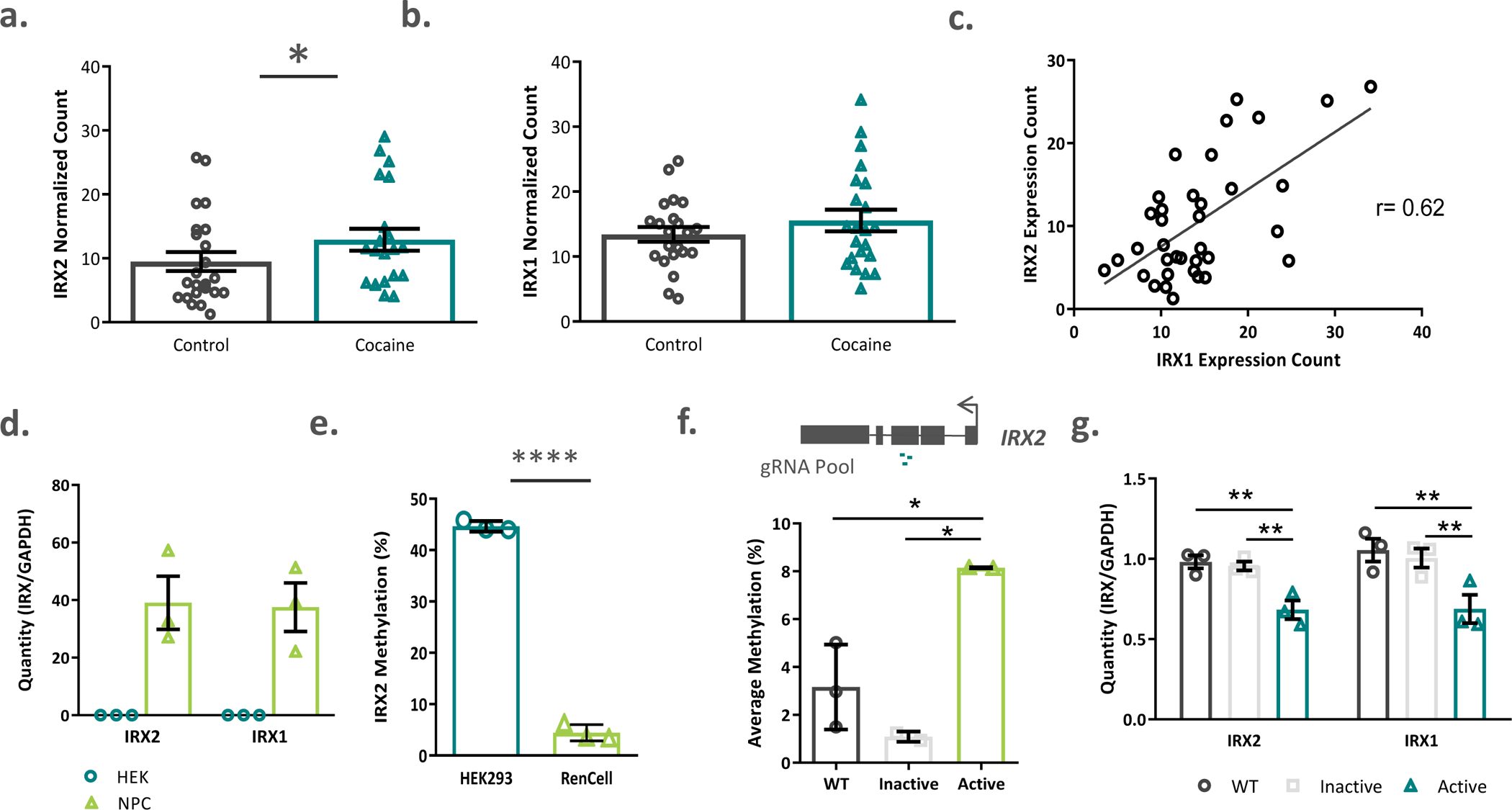
a) IRX2 expression was significantly increased in the caudate nucleus of cocaine dependent subjects (n=21 cases and n=23 controls). b) while no significant increase in IRX1 was detected (n=22 cases and n=22 controls), c) the expression of both IRX1 and IRX2 transcripts was highly correlated (n=36). d) Human neural progenitor cells (RENcells) endogenously expressed IRX1 and IRX2 while kidney epithelial cells (HEK293) did not (n=3 per group). e) Endogenous methylation of IRX2 exon 3 was higher in kidney epithelial cells than in neural progenitor cells (n=3 per group). f) Transfection of an active dCas9-DNMT3A construct, along with a pool of 3 guide RNA constructs significantly increased methylation of IRX2 exon 3 in RenCells, compared to transfection with an inactive construct or wildtype controls. g) Active methylation of IRX2 decreased transcription of both IRX2 and IRX1 compared to inactive or wildtype cells. (n=2–3 replicates per group)WT=wildtype. Data represented as mean ± s.e.m **** p <0.0001, * p < 0.05
We next turned to in vitro modelling to explore the relationship between IRX2 DNA methylation and gene expression of the IRXA gene cluster because homogeneous groups of cells allow better resolution than can be obtained through brain tissue homogenates. We measured endogenous methylation and expression levels in two distinct human cell lines; HEK293 kidney epithelial cells (ATCC, Virginia, US) and RENcell immortalized fetal midbrain cells (Millipore, Burlington, US). Since IRX1 and IRX2 are neurodevelopmental transcription factors, we hypothesized that their expression would be higher in RENcell neural progenitor cells (NPCs) compared to epithelial cells. We found this to be the case, with RENcells expressing both transcripts, whereas neither transcript was detectable in HEK293 samples (Figure 3d). There are likely multiple epigenetically relevant regulatory elements that contribute to the striking dichotomy in gene expression (alternative promoters and enhancers, for example); however, if the region within exon 3 has regulatory potential, we hypothesized that its endogenous methylation level would differ between the cell types. Indeed, HEK293 cells were on average, 40% more methylated within this region than NPCs (t=36.76, df=4, p < 0.0001; Figure 3e).
To determine whether DNA methylation has any causal impact on changes in gene expression, we designed a CRISPR/Cas-9-based epigenome editing experiment in NPCs. We designed three guide RNAs (gRNAs) targeting the region within exon 3, and used a deactivated Cas9 (dCas9) enzyme fused with the active domain of a DNA methyltransferase[25] to experimentally increase methylation and study its regulatory influence on gene expression dynamics. Cells that were transfected with the active construct were on average 4.9% more methylated than wildtype and 7.1% more methylated than the cells that were transfected with a dCas9-DNMT3a plasmid with a mutated methyltransferase domain (inactive) (F(2,4)=16.9, p = 0.011; Active vs WT p = 0.021, Active vs Inactive p = 0.008), Figure 3f). Importantly, methylation was increased across the CpGs within the CTCF binding site (6.7–8.4%, Supplemental Figure 9a) and unchanged within an amplicon in IRX1 that was used as a control for off-target methylation (F(2,6)=3.054, p=0.122, Supplemental Figure 9b). Increased methylation of IRX2 exon 3 resulted in a significant decrease in IRX2 (F(2,6)= 7.928, p=0.021, Active vs. WT p = 0.018, Active vs. Inactive p=0.037)and IRX1 (F(2,6)= 8.417, p=0.018, Active vs. WT p = 0.020, Active vs. Inactive p=0.023) gene expression (Figure 3g).
The three-dimensional chromatin structure of IRXA is associated with gene expression, and is altered by methylation of IRX2 exon 3
It has been shown in animal models that Irx2 and Irx1 share enhancer elements located within the intergenic region between them, and that the two genes are brought closer together to access these enhancers during transcription[21]; but to date, no such regulatory loop has been identified in humans. We designed a 3C assay in human cells to detect the frequency of physical proximity between the promoter of IRX1 (viewpoint, Figure 4a), and the genomic region encompassing IRX2 (test primers, Figure 4a), which are separated by over 850kB of linear genome. We found that, in both neural-progenitor and in kidney epithelial cells, the two genes are in close physical proximity more often than would be expected by chance (dotted grey line, Figure 4a). Interestingly, fragments 2 and 3, which encompass the first two-thirds of IRX2, are physically close to the 5’ end of IRX1 more often in NPCs, where both genes are expressed, than in cells where the genes are not expressed (t=4.41–6.54, df=7, ps < 0.01, Figure 4a). The cocaine-associated hypomethylation that we observed, including the putative CTCF binding site, is located within fragment 2, which is further evidence for a relationship between exon 3 methylation and 3D chromatin structure.
Figure 4. Long range chromatin structure of the IRXA gene cluster is impacted by methylation.
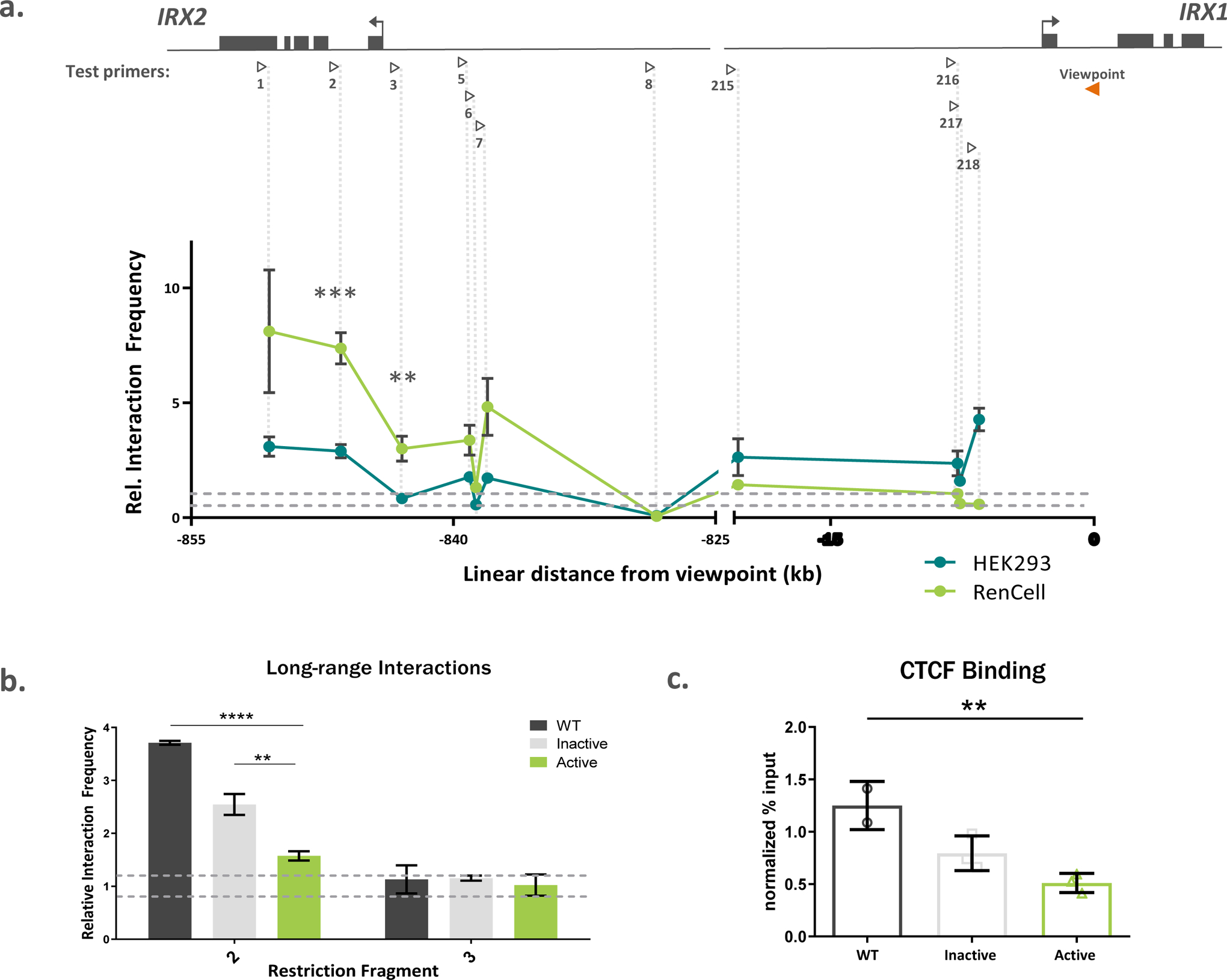
a) Chromatin conformation capture (3C) experimental design contained a standard viewpoint within the first intron of IRX1 (orange arrowhead), and test primers (open arrowheads) tiled across IRX2 and the intergenic region between the genes. The genomic fragments containing IRX2 functionally interacted with the 5’ end IRX1 in two human cell types, with significantly higher rates of interaction observed in RenCells. The noise band, where interaction frequencies would be expected by chance (“random collisions”), is indicated by the horizontal dashed lines. Vertical dashed lines pair restriction fragment with its corresponding data points. N=3 replicates per group. b) Active methylation of HEK293 cells decreased long range interaction between restriction fragment 2 and the IRX1 gene (n=3 replicates of 10 × 106 cells per group). c) The dCas9-DNMT3A transfection significantly (n=3 replicates of 5 × 106 cells per group) decreased CTCF binding to IRX2 exon 3 compared to wildtype cells. WT=wildtype. Data represented as mean ± s.e.m. ** p < 0.01; *** p < 0.0005
We next sought to understand whether methylation of the CpGs within this fragment could directly cause changes in chromatin architecture, and we again turned to epigenome-editing, this time in HEK293 cells which allowed us to transfect and grow the higher amount of cells necessary for 3C (Supplemental Figure 10). The active plasmid increased the methylation of this region by 4.37% compared to the inactive plasmid and by 9.87% compared to untreated cells (F(2,6)=15.28, p = 0.004, WT vs Active p = 0.003, Active vs Inactive p = 0.087, Supplemental Figure 11a). We also found significantly more methylation in the actively transfected cells, compared to WT, when averaging across the entire CTCF binding site (9.91%, F(2,6)=8.78, p=0.017; Active vs WT p = 0.012, Supplemental Figure 11b). There were no significant effects of group on percent methylation of the off-target control (Supplemental Figure 11c).
In order to investigate whether these findings could be translated into alterations in long range chromatin structures, we assayed the local chromatin architecture in cells; particularly the frequency with which restriction fragments 2 and 3 interacted with the viewpoint in the IRX1 gene. Strikingly, we found that methylation of IRX2 exon 3 brought the interaction frequency of fragment 2, which contains the putative CTCF binding site, down to levels near those expected by chance (F(2,6)=71.03, p <0.0001, Active vs WT p <0.0001, Active vs Inactive p = 0.003, Figure 4b). We found no significant effect of methylation on the interaction frequency of fragment 3, which does not contain the putative CTCF binding site, with IRX1 (Figure 4b).
CTCF binds to IRX2 exon 3, and is disrupted by DNA methylation
Finally, to assess whether methylation of exon 3 could alter CTCF protein binding, we performed anti-CTCF ChIP-qPCR on wildtype HEK293 cells and cultures transfected with either the active or inactive dCas9-DNMT3A construct. Importantly, CTCF binding to the fragment of exon 3 containing the putative binding site was experimentally validated to be significantly higher than a non-specific IgG control (F(1,5)=8.329, p = 0.034; Supplemental Figure 11d). Furthermore, methylating the same sequence decreased CTCF binding compared to wildtype cells (F(2,5) = 13.19, p=0.010; Active vs. WT p = 0.007, Figure 4c). These data suggest that exon 3 may indeed contain a functional CTCF binding site that is sensitive to modest changes in cytosine methylation, such as those observed in caudate neurons after chronic cocaine dependence.
Based on the cumulation of data from human, mouse, and cell line experiments, we suggest that exon 3 of IRX2 contains a methylation sensitive CTCF binding site that is disrupted following long-term cocaine exposure and dependence (Figure 5).
Figure 5. A model for cocaine-sensitivity of 3D chromatin organization at the IRXA gene cluster.
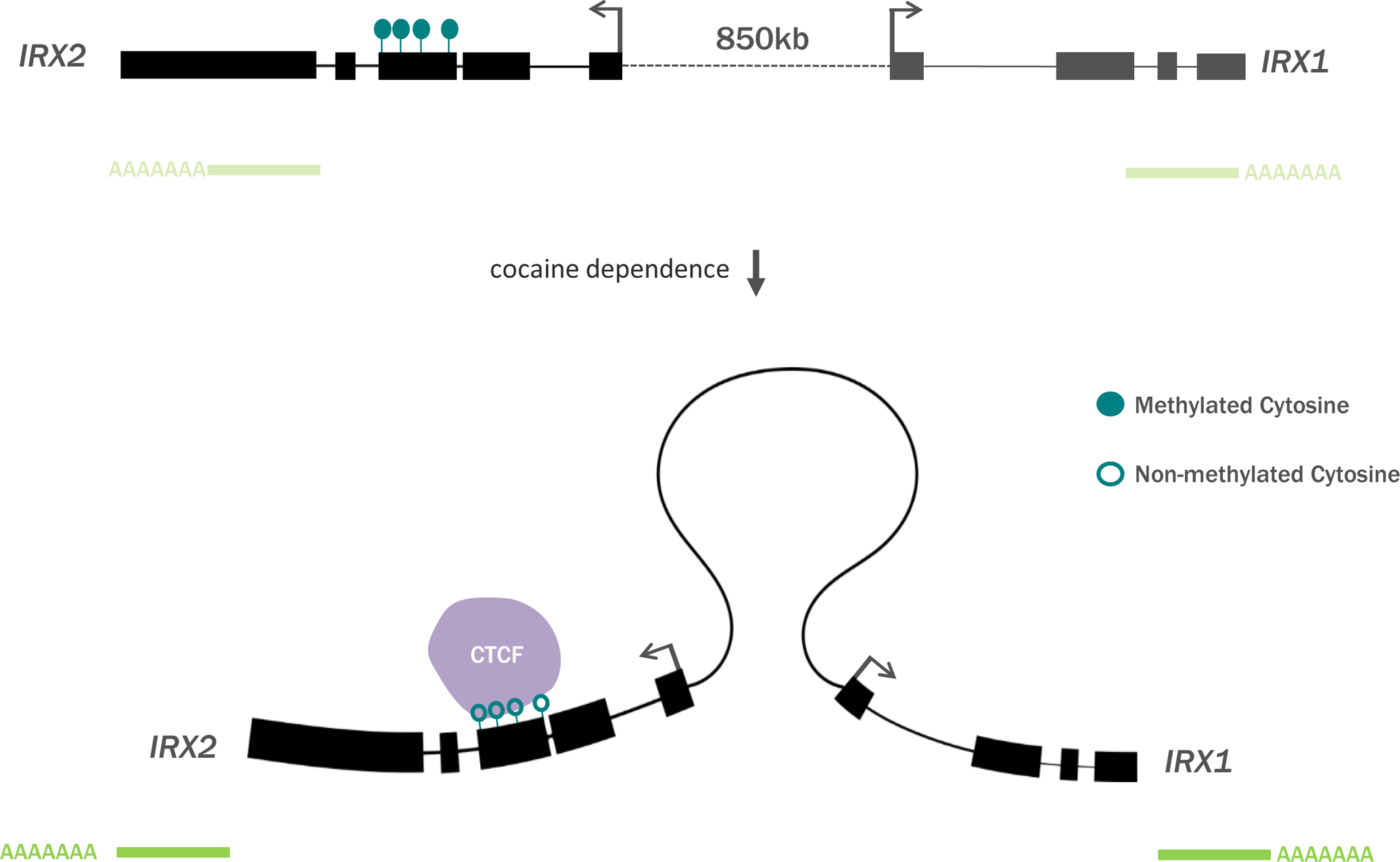
Cocaine dependence is associated with decreased intragenic methylation of IRX2, which may increase IRXA gene expression through CTCF-mediated chromatin architecture.
Discussion
Our experiments show that chronic cocaine dependence in humans is associated with decreased methylation of an intragenic region of CpGs in the IRX2 gene, which overlaps with a novel regulatory site for local gene expression and three-dimensional chromatin structure (Figure 5). This region is one of over 100 DMRs that we have identified in the human caudate nucleus. Although this is the first methylome-wide study of cocaine use disorders using brain tissue from human patients, our work is well-aligned with two decades of studies in animals that have identified regions of both hyper- and hypomethylation in addiction relevant neural circuitry[8].
The caudate nucleus is increasingly implicated in the pathogenesis of drug use disorders as individuals transition from recreational use to compulsive drug seeking behaviors [13]. The neurons within the human caudate are mostly GABAergic medium spiny projection neurons (MSNs) surrounded by at least 4 distinct types of inhibitory interneurons [26, 27]. Striatal MSNs can be classified into two major subgroups, D1 and D2 dopamine receptor expressing cells, which have opposing effects on drug-related behaviors, with D1-MSNs enhancing drug seeking while D2-MSNs inhibit these behaviors in animals[28]. While technical limitations prevent us from discerning the contributions of individual neuronal subtypes, the separation of neuronal from non-neuronal methylation profiles presented here represents the first step towards a human cell-specific cocaine methylome. Future progress in single-cell methylome technologies will undoubtedly guide deconvolution efforts on datasets such as those presented here.
Most of the work in the field, to date, has focused on methylation at specific gene promoters, but intragenic methylation and methylation at distal regulatory elements may have relevance to tissue-specific disease etiology. Indeed, the majority of DMRs identified here do not fall within annotated promoters and may disrupt other regulatory processes that contribute to addiction neurobiology. Additionally, although the DMRs in this study are not enriched for any one particular cellular component or biological process, the effects of human chronic cocaine dependence may not impact all cellular pathways equally; further research into epigenetic alterations of specific processes will be a welcome addition to this work.
Non-promoter elements are enriched for neuropsychiatric heritability factors, and levels of DNA methylation and chromatin accessibility in these regions has been shown to have brain-region specific effects on disease[20, 29, 30]. Thus, although DNA methylation is perturbed in multiple addiction-related brain regions, the exact DMRs are likely to differ between brain nuclei. Furthermore, DNA methylation within gene bodies may directly promote gene expression, direct the use of alternative promoters or regulate alternative splicing events [31–33]. In recent years, it has become clear that these biological processes have important implications for psychiatric phenotypes overall [34], and for cocaine dependence where it has been shown that, in the nucleus accumbens, repeated cocaine exposure can induce genome-wide alternative splicing events that are related to drug seeking behaviors in rodents[35, 36].
Animal work has also identified distinct alterations in methylation and associated machinery that are related to different administration paradigms and exposure time courses. For example, although DNMT3A is initially decreased in the nucleus accumbens during cocaine withdrawal in mice, levels of the de novo methyltransferase becomes significantly increased after 28 days[10]. Indeed, we found increased DNMT3A expression in the caudate nucleus of our cocaine-dependent samples. Similarly, distinct patterns of differential methylation emerge in studies using passive cocaine injection versus self-administration, which are related to long term behavioral changes[37]. Although we are unable to separate the effects of acute and chronic cocaine in our discovery cohort due to positive cocaine toxicology at the time of death, our replication cohort was negative for cocaine metabolites, and suggests that the findings with respect to IRX2 are more likely linked to long-term dependence than to an acute pharmacological effect.
IRX2 is a transcriptional repressor that is highly expressed during neural development and may be related to social behavior in animals[38, 39]. Its expression is known to be regulated by three-dimensional chromatin architecture, which in turn is regulated by the CTCF architectural protein[21, 40]. Although this study is the first to report on the relationship between IRX2 and cocaine dependence, it has already been shown that dependence-related behaviors rely on long-lasting alterations in the expression of transcription factors genes [41, 42] and that genes involved in transcription and chromatin regulation are dysregulated in brain tissue from human patients[43]. Cocaine-related expression of transcription factor genes can be regulated by DNA methylation mechanics[44], and are likely cell-type specific[45], which is in line with what we have shown at this locus, where IRX2 is more highly expressed in the cocaine group. Although the downstream targets of IRX2 regulation have yet to be experimentally identified, target prediction algorithms suggest that it may impact the expression of genes including ADAM10, a metalloprotease that has been linked to multiple psychiatric diseases, and may be involved in the cognitive impairments that can accompany long term psychostimulant use[46–48]. Additionally, little is known about the dynamics of epigenetic regulation in the IRXA gene cluster during neuronal development — future work in animals should identify how the relationship between methylation, expression, and three-dimensional chromatin structures changes throughout development.d
We have shown that cocaine-related methylation of IRX2 exon 3 is negatively associated with gene expression through decreased frequency of three-dimensional chromatin structure. This is in line with evidence that suggests that DNA methylation can compete with CTCF binding, especially at specific CpGs at key regulatory sites[22, 49]. Moreover, repeated cocaine administration has been shown to increase DNA methylation and decrease CTCF-mediated chromatin looping at the Auts2-Caln locus in mice[9].
Like other work using post-mortem samples, this study presents limitations that need to be considered when interpreting the findings. First, the molecular profiles gathered from these tissues highlight the epigenetic landscape immediately prior to death, and although every effort is made to characterize the demographic information of the donors, we are unable to account for corollary factors such as lifestyle, and lifetime history of non-dependent drug exposure that could influence DNA methylation. Complementary evidence from animal models, such as has been presented here, can begin to account for the effect of extraneous factors. Similarly, although we were able to distinguish between broad categories of cell-types (neurons vs. non-neurons), the magnitude of methylation differences observed in our study suggest that the signal is coming from a relatively rare cell type and being masked by cellular diversity. Nonetheless, small changes in methylation have been shown to have physiologically relevant effects on transcription factor binding, and RNA transcription[50, 51], and have previously been associated with cellular and molecular alterations in post mortem psychiatric research[52–54].
Future work on the IRX2 locus, as well as other DMRs identified in this study, should incorporate information about additional levels of epigenetic regulation, including histone modifications as well as DNA modifications outside of the canonical CpG methylation context. For example, non-CpG methylation (CpH), N6-Methyladenosine (m6A) and hydroxymethylation are epigenetic regulators that are highly abundant in the brain and are likely be important mechanisms to drug dependence[55–57]. Additionally, direct manipulation of Irx2 in animals will allow important insight into the behavioral consequences of cocaine-related epigenetic changes. Furthermore, although studies suggest that dependence to other psychostimulants, including amphetamine, associates with DNA methylation changes, direct comparisons between drugs of abuse, and between addiction-related brain regions, will add specificity to epigenome-wide studies. Additionally, as data from single-cell epigenomic experiments continue to become available, researchers will be able to detect differences in rare cell types that are currently masked by bulk and near-bulk tissue experiments[58].
Methods
Subjects
All methods used in this study were approved by the Douglas Hospital Research Ethics Board, and written informed consent was obtained from the next-of-kin for each subject. Autopsy and tissue sampling were performed in accordance to the established standards of the University of Miami Miller School of Medicine, or the Douglas-Bell Canada Brain Bank, depending on cohort source location.
Post-mortem caudate nucleus tissues from our discovery cohort were obtained from the Brain Endowment Bank at the University of Miami Miller School of Medicine (Supplemental Table 1). Samples were dissected from the dorsolateral sector of the caudate from 25 subjects who had long term histories of cocaine dependence as determined by licenced clinicians, and who died from cocaine related complications as determined by forensic pathology and brain and blood toxicology. These subjects were selected based on the absence of toxicology for illicit drugs other than cocaine and were determined to have no other psychiatric diagnoses based on medical records and the reports of next-of kin. Drug-naïve, psychiatrically healthy control subjects (n=25) were selected from accidental or natural deaths. All subjects in this cohort were male, which is reflective of the opportunistic composition of the brain samples available at autopsy. Due to the rarity of these samples, no a priori power analyses were performed in relation to sample sizes.
Caudate tissue from the replication cohort of 15 cases and 21 controls was obtained from the Douglas-Bell Canada Brain Bank (www.douglasbrainbank.ca). Subjects underwent a medical chart review and proxy-based interviews that were used in the characterization of substance use, which was determined through psychological autopsy by the clinical staff of the brain bank. Case status was determined based on these results, as well as toxicology at the time of death. Age, PMI and pH did not significantly differ between groups (t=0.79–1.73, df=34, p=0.09–0.44, Supplemental Table 4). Grey matter was dissected from the left hemisphere of all samples and stored at −80°C until further processing.
Reduced representation bisulfite sequencing (RRBS)
Tissue and Library Preparation
We extracted DNA from 20mg of frozen tissue of all cases and controls from our initial cohort using Qiagen DNA MiniKits as per manufacturer’s instructions. To prepare RRBS libraries, we digested 1ug of genomic DNA with MspI restriction enzyme, repaired the fragment ends and ligated Illumina adapters as described in elsewhere[59]. Purified libraries were treated with EpiTect fast bisulfite conversion kit (QIAGEN, Cat# 59824) according to the standard protocol and indexed through PCR amplification.
Sequencing and Bioinformatic Processing
Final libraries were sequenced on the Illumina HiSeq 2000 platform at the Genome Quebec Innovation Center (Montreal, Canada) using 50bp single end sequencing, and bioinformatics processing was performed in-house, as described[59]. Bisulfite conversion efficiency was determined by the ratio of T to C at the unmethlyated cytosine position added during the end-repair step of library construction. Sequencing data is available under accession number GSE6364.
Differential Methylation Analysis
We defined methylation region as any CpG within 50 bp of another CpG, with no limit on the number of CpGs in a given region, but with a minimum of at least 2 CpGs using the bumphunter 3.5 package for R. For CpGs to be included in the analysis, they must have been present in at least 25 subjects from both cases and controls and have ≥5X coverage, which resulted in 270191 CpGs that went into clustering analysis. CpG regionsthat had a standard deviation <5% methylation across all subjects (i.e., irrespective of status) were removed to avoid comparisons between stable methylation sites. For each cluster, we performed differential analysis using a general linear model with status (cocaine or control) as a fixed factor, and age, ethnicity, smoking status and ethanol toxicology as covariates. We treated CpGs independently in a given regions and used only those regions that had a Benjamini-Hochberg FDR corrected p-value <0.05 and which were <0.05 when calculating a single mean from all CpGs per individual.
Cluster Annotation and Enrichment Analyses
All CpG regions were annotated relative to their genomic context, their CpG island proximity, and their predicted ChromHMM chromatin state using the annotatr 1.10.0 package in R[60]. We calculated enrichment q values for DMRs against all CpG clusters using the LOLA algorithm[61].
Gene Ontological Analysis
We annotated each DMR to its nearest Refseq gene. Gene ontologies were examined with over representation tests in the gene list analysis functions of the PANTHER classification system (www.pantherdb.org). We compared the full DMR list to all human genes with respect to molecular function, biological processes and cellular components and p-values were calculated using Fisher’s Exact tests with FDR correction.
RNA sequencing
RNA was extracted from 100mg dissections of dorsolateral caudate nucleus using RNeasy Lipid Tissue Kits (Qiagen) according to standard procedure. The RNA integrity number (RIN) for the cases was 7.9 ± 1.28 and controls was 8.4 ± 0.78 (mean ± s.d.), and aliquots of 100ng/ul of RNA were sent for library preparation and sequencing at the Broad Institute (Cambridge, MA). Libraries were prepared using a standard non-strand specific protocol (Illumina TruSeq), including poly-A selection, and multiplexed for 50bp paired end sequencing on the Illumina HiSeq 2000 platform. Sequencing data was processed as previously described[62], and fold changes and p-values were generated by student’s t-tests. Uncorrected p-values of <0.1 were used to create DMR-gene pairs for follow-up investigation.
nanoString gene expression validation
For count based RNA quantifications, custom 100bp probe sequences were designed to uniquely capture the majority of transcript variants of the genes indicated in Supplemental Table 5, by nanoString Technologies (Seattle, Washington). Each probe was associated with a unique fluorescent barcode, and 20ng/ul of total RNA, from the same extraction that was used in sequencing, was run on the nCounter system under the high field of view setting at the Lady Davis Institute (Montreal, Quebec). All normalization and statistical analyses were performed with the nSolver software from nanoString technologies. Raw probe counts were normalized to 4 negative control probes and then compared between groups, using two-tailed unpaired student’s t-tests or their non-parametric equivalent when necessary. Two samples were removed from each group due to technical failure, and statistical outliers were removed after ROUT analysis (Q=1%) resulting in 23 controls compared to 21 cases in the IRX2 analysis and 22 cases and controls in the IRX1 comparison.
Fluorescence Activated Nuclei Sorting
Nuclear extraction and labelling
In order to liberate intact nuclei from the caudate nucleus tissue samples, we homogenized 50mg of frozen tissue in nuclei buffer containing 10mM PIPES (pH 7.4), 10mM KCl, 2mM MgCl2, 1mM DTT, 0.1% TritonX-100 and 10X Protease Inhibitor Cocktail (Sigma Aldrich, Darmstadt, Germany). Homogenates were passed through a 30% sucrose gradient in nuclei buffer in order to separate nuclei from cellular debris, then after a wash with nuclei buffer, nuclei pellets were resuspended in blocking buffer containing 0.5% bovine serum albumin in 10X normal goat serum. Each sample was co-incubated with the DNA labelling dye DRAQ5 (1:300) (ThermoFisher, Waltham, MA) and an anti-NeuN-PE antibody (1:300) (cat no. FCMAB317PE, Millipore, Darmstadt, Germany) for 60 min at room temperature, then passed through 40uM filter caps to remove any remaining cellular debris before sorting.
Nuclei Sorting
Labelled nuclear extracts were processed the BDFACSAria III platform (BD Biosciences, San Jose, CA) according to technical specifications provided by the company. We used BD FACSDIVA software (BD Biosciences, San Jose, CA) to first isolate single, intact nuclei based on DRAQ5 fluorescence at the 730/45-A filter (DRAQ5), then to sort neuronal from non-neuronal nuclei based on fluorescence detected by the 585/42 filter (PE). Sorted nuclear fractions were stored at −20°C in sheath fluid (1X PBS) until DNA extraction. On average, we isolated 180 000 NeuN+ nuclei and 492 500 NeuN- nuclei from 50mg of tissue, with roughly 37% NeuN+ in each sample. There were no differences between cases and controls in terms of total nuclei in either fraction or in the ratio of neuronal to non-neuronal nuclei captured per dissection (ps > 0.1, Supplemental Figure 3b–d).
Nuclear DNA extraction and processing
We incubated nuclear fractions with 50X protease (Qiagen, Montreal, Canada) at 56°C for at least 12 hours to ensure thorough digestion of the nuclear membranes. Liberated DNA was precipitated onto 0.2X Agencourt AMPure XP beads (Beckman Coulter, Brea, CA) after adding 20% PEG-8000 2.5M NaCl to increase the final PEG concentration to 10%. The beads were washed twice in a magnetic stand with 70% EthOH, and then DNA was eluted in 50ul MilliQ H2O. We measured the concentration of each DNA sample using Quant-iT PicoGreen dsDNA assays (ThermoFisher, Waltham, MA) according to manufacturer specifications.
Bisulfite amplicon sequencing
DNA extraction and conversion
We obtained genomic DNA from post-mortem, homogenate tissue samples from both the discovery and replication cohorts using QIAmp DNA Mini Kits (Qiagen, Montreal, Canada) as per manufacturer specifications. Notably, DNA for the discovery cohort was extracted from the initial dissections that were used for RRBS library construction. Genomic DNA from both cohorts (2ug/sample), as well as from all sorted nuclear fractions (>100ng/sample) was converted using EpiTect Fast 96 Bisulfite Conversion Kits (Qiagen, Montreal, Canada), diluted to 150ul using MilliQ water, and stored at −20 °C.
Library Preparation
To optimize our ability to cover the desired CpGs within each amplicon, and to increase amplicon diversity for sequencing, we designed three pairs of bisulfite specific primers per DNA strand using Methyl Primer Express Software v1.0 (Applied Biosystems, CA, USA). Redundant primers were designed to be non-overlapping, to have optimal melting temperatures of 60± 2°C and to be between 18 and 24bp long in order to optimize amplification in a multiplexed reaction (primer sequences and PCR conditions in Supplementary Tables 6 and 7). We amplified each sample using 10ul reactions consisting of 5X combined primers (10uM), 3X bisulfite converted DNA and 2X KAPA HiFi HotStart Uracil+ ReadyMix (Kapa Biosystems, MA, USA). Each strand was amplified separately, and after two rounds of paramagnetic bead purification at 0.8X, amplicons from both strands were combined and amplified for 10 additional cycles to add custom primer sequences in 20 ul reactions consisting of 2.5X sample, 5X combined CS1 and CS2 primers (10uM) and 2X KAPA HiFi HotStart ReadyMix (Kapa Biosystems, MA, USA). After an additional round of 0.8X bead purification, we indexed each sample for 10 cycles in a 20ul reaction consisting of 2.5X amplicons, 5X indexing primers (10uM) and 2X KAPA HiFi HotStart ReadyMix (Kapa Biosystems, MA, USA). Each indexed library went through two rounds of double ended bead purification (final ratio 0.8X) to select only those fragments in the predicted range of our amplicons (400–700bp). Final library concentrations and quality control was performed on the Agilent 2200 TapeStation (Agilent Technologies, CA, USA) before samples were pooled and sequenced.
Next Generation Sequencing
We pooled libraries to a final concentration of 2nM and included a 5–10% PhiX spike-in control for each sequencing run. Final libraries were run on the Illumina MiSeq platform (Illumina, San Diego, CA) using customized 300bp paired end sequencing as described elsewhere[63]. All quality control and read alignment, without removing duplicates, were performed in-house and methylation was calculated as the percent of reads containing cytosine rather than thymine at each position.
Statistical Analysis
Samples were removed for poor sequencing (less than 5X coverage of CpGs or less than 80% of CpGs covered within an amplicon) and the number of samples used in each analysis is reflected by the respective degrees of freedom. Methylation at each position was defined as the number of reads called as cytosine, over the number total number of reads. Percent methylation was averaged across all CpGs within an amplicon and compared between groups using unpaired t-tests (with Welch’s correction in cases of unequal variance) or Mann-Whitney U tests where groups were unlikely to be normally distributed as per Shapiro-Wilks test. Two-tailed significance tests were used except for analyses with sorted nuclei fractions and mouse samples, where a priori hypotheses allowed for one-tailed testing. Statistical outliers were removed according to ROUT analysis (Q=1%), and final sample sizes are reported in the corresponding figure legends.
For analysis of methylation at individual CpGs within CTCF binding sites, ordinary two-way ANOVAs were performed with Group and CpG as factors. Main effects of group were dissected using t tests and the Holm-Sidak method for multiple testing corrections. Data are represented as means and error bars as standard error of the means.
Mouse reward self-administration
Jugular catheter implantation:
Animal experiments were approved by the Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai. Male c57BL/6J mice, aged 6–8 weeks at the beginning of the experiment, were randomly assigned to groups, anesthetized with ketamine (100mg/kg) and xylazine (10mg/kg), implanted with chronic indwelling jugular catheters, and trained for i.v. self-administration as previously described[64]. The catheter tubing was passed subcutaneously from the back to the jugular vein and 1.0 cm of tubing was inserted into the vein and secured with silk suture. Catheters were flushed daily with ampicillin (0.5mg/kg) and heparin (10U/mL) solution in sterile saline (0.9% NaCl). Mice recovered >3d before commencing behavioral training. All animals were maintained on a reverse light cycle (7:00am lights off; 7:00 pm lights on) and behavioral training was conducted during the animal’s dark cycle.
Cocaine self-administration
Mice (n=6) were trained to self-administer cocaine as previously described. Briefly, mice were maintained at ~90% of their free-feeding weight and trained in standard mouse operant chambers (Med Associates, St Albans, USA) equipped with a white noise generator and two illuminated nose-pokes. Start of daily sessions (2h) were signaled by white noise. For each task, one nose-poke was designated as the “active poke” and the other designated as the “inactive poke”. Active nose pokes resulted in a cocaine infusion (0.5mg/kg/inj, 3 sec) with a concurrent presentation of the Active nose poke light for 5 seconds. Inactive pokes resulted in no programmed consequences. Mice were trained to self-administer cocaine for 10 consecutive days under a fixed-ratio (FR1) schedule of reinforcement. For cocaine self-administering animals, acquisition (Day 1) was counted when the animal reached 70% responding on the active lever and 10 or more responses. Control animals underwent the same experimental procedures but had access to a saline-paired nose poke (n=7). For both the cocaine and saccharin self-administration groups, most mice reached acquisition criteria on the first day, and since the experimental animals were trained for 10 days, all saline animals underwent 10 days of saline self-administration as controls.
Saccharin Self-Administration
8-week old c57BL/6J mice were ordered from the Jackson Laboratory and were housed in a 12-hour 6:00/6:00 reverse dark/light cycle. Saccharin self-administration was run during the animal’s dark cycle and mice were food restricted to ~90% of free-feeding weight with water provided ad libitum. Mice (n=9) were trained to self-administer saccharin (0.1% solution in water, ~80uL/infusion) or water (n=7) for 1 h over 10 consecutive days. Briefly, activation of white noise signaled the initiation of the (1h) daily session. In each task, one nose-poke was designated the “active” while the other was designated the “inactive”. Active responses initiated a 1 sec saccharin (or water) delivery into an accessible dipper with a concurrent 5 sec presentation of both the nose poke light and dipper light under a Fixed-Ratio 1 (FR1) schedule of reinforcement. Inactive nose pokes had no programmed consequences but were recorded throughout all behavioral sessions.
Tissue preparation
After the 10th test session, animals were euthanized, and brain tissue was removed and flash frozen at −80°C before the caudate-putamen was dissected.
Library Preparation
After standard extraction using, 500ng of genomic DNA from each sample was bisulfite converted using the EZ DNA Methylation Gold kit (Zymo Research, Irvine, California) by a blinded experimenter. Bisulfite specific primers were designed as above, using genomic regions homologous to the hg19 coordinates in the mouse genome (mm10) (Supplemental Table 6). Bisulfite DNA was subjected to the same amplification (Supplemental Table 7) and purification methods as described above, and the final purified libraries were spiked into a 300bp paired end sequencing run containing customized sequencing primers as above. After de-multiplexing and adapter trimming, sequencing reads were aligned to the mm10 mouse genome, and percent methylation at each position was determined as above.
Cell Culture
Human embryonic kidney cells (HEK293) were maintained in eagle’s minimum essential medium (EMEM; ATCC, Virginia, US) supplemented with 10% fetal bovine serum (FBS; Gibco Laboratories, Gaithersburg, MD), 100 I.U./mL penicillin and 100ug/mL streptomycin. Cells were cultured at 37°C in a humidified incubator with 5% CO2.
Human neural progenitor cells (ReNcell; Millipore, Burlington, US) were maintained in STEMdiff neural progenitor medium (STEMCELL Technologies, Vancouver, Canada) supplemented with 100 I.U./mL penicillin and 100ug/mL streptomycin. All cell lines were tested for mycoplasma contamination and authenticated by their respective manufacturers.
Quantitative PCR
The expression of IRX1 and IRX2 in all in vitro experiments was determined using quantitative reverse transcription PCR (RT-qPCR). We used pre-designed probe based assays for IRX1 (Hs.PT.58.2400) and IRX2 (Hs.PT.58.24473971) with FAM-TAMRA dyes (PrimeTime, IDT, Hampton, New Hampshire, Supplemental Table 8), and ran 10ul assays on an Applied Biosystems QuantStudio 6 instrument under default cycling conditions (ThermoFisher, Hampton, New Hampshire). Amplification curves were normalized using QuantStudio Real-Time PCR Software, and the average expression between treatment conditions was compared with two-tailed t tests in GraphPad Prism 6 (www.graphpad.com).
dCas9 epigenome editing
Plasmid preparation
The pdCas9-DNMT3A-PuroR and pdCas9-DNMT3A-Puro (ANV) plasmids were a gift from Vlatka Zoldoš (Addgene plasmids #71667 and #716840). Guide RNA (sgRNA) sequences targeting human IRX2 exon 3 were designed using an online tool (CRISPR gRNA Design Tool, www.ATUM.bio) and purchased from Integrated DNA Technologies (IDT, Iowa, US). All 3 sgRNAs were annealed and cloned into the expression plasmids through the BbsI restriction site. All cloned constructs were confirmed by Sanger sequencing using the U6 sequencing primer (Supplemental Table 9).
Transfection
HEK293 cells were seeded in 10cm culture dishes and transfected the next day, at over 80% confluence, using TransIT-293 transfection reagent (Mirus, Brampton, ON). Transfections were done with a pool of 15ug dCas9-DNMT3A plasmids carrying all three sgRNA targeting IRX2. The experiment was performed in triplicate and constructs carrying inactive DNMT3A were used as negative controls. 48h after transfection, cells were selected with 1.6ug/mL puromycin (Gibco Laboratories, Gaithersburg, MD) for another 48h. Cells were harvested after seven days transfection for DNA and RNA extraction (see above) as well as the nuclei preparation (see below).
RENcells were tranfected by electroporation using the Neon transfection system (Invitrogen, Carlsbad, CA), according to the manufacturer’s standard procedure. Briefly, cells were washed in PBS, detached from the culture vessel using accutase, pelleted by centrifugation and resuspended in Resuspension Buffer R at a final density of 13 × 106 cells/mL. Cells were immediately electroporated three times using 100uL neon tips at voltage 1300, width 20, and pulse 3, giving final 15ug DNA and 4 × 106 cells per 10cm culture dish. After electroporation, cells were immediately transferred into the prepared 10cm culture dish containing prewarmed medium, but without antibiotics. Cells were selected with 0.3ug/mL puromycin with the same time period as in HEK293 and harvested at Day 7. Percent methylation was assessed with bisulfite amplicon sequencing, and gene expression was assessed by qPCR as described above.
Statistical Analyses
Percent methylation, gene expression and interaction frequency were compared between conditions using one-way ANOVAs with Tukey’s post-hoc comparisons.
Chromatin Immunoprecipitation (ChIP-qPCR)
Chromatin Preparation
HEK293 cells were grown, transfected, and nuclei were harvested as above. Aliquots of 5 million nuclei each were prepared from wildtype cells, cells transfected with the active dCas9-DNMT3A + gRNA construct (active group), and cells that were transfected with the mutant dCas9-DNMT3A construct (inactive group). Pelleted nuclei were resuspended with 1mL filtered 1X PBS and 200X Protease Inhibitor Cocktail, and cross-linked with 1% formaldehyde for 7 minutes, at room temperature, with gentle rotation. After the fixation reaction was stopped with 10X glycine, nuclei were pelleted and washed twice with 1XPBS+PIC and resuspended in Sonication Nuclear Lysis Buffer, and chromatin was prepared as per the manufacturer’s protocol (SimpleChIP Plus Sonication Chromatin IP Kit, Cell Signalling Technologies, Massachusetts). Chromatin was sheared on a S220 focused ultrasonicator (Covaris, Massachusetts) under the following conditions: 150 peak power, 200 cycles/burst, duty factor 10, for 15 minutes.
Immunoprecipitation
Diluted “input” chromatin was removed before immunoprecipitation, and anti-IgG was added to chromatin from 1 ×106 nuclei as per manufacturer protocol (SimpleChIP Plus Sonication Chromatin IP Kit, Cell Signalling Technologies, Massachusetts). Anti-CTCF antibody (1:25 dilution, cat no. 2899, Cell Signalling Technologies, Massachusetts) was added to the sonicated chromatin of 4 × 106 nuclei and both anti-IgG and anti-CTCF preparations were precipitated overnight at 4°C with rotation. Chromatin-antibody complexes were separated with magnetic protein G beads, eluted, and de-crosslinked by proteinase K digestion at 65°C overnight (16 hours).
qPCR
DNA from anti-CTCF, anti-IgG and input fractions was purified per manufacturer’s instructions (SimpleChIP® DNA Purification Buffers and Spin Columns Kit, Cell Signalling Technologies, Massachusetts), 10ul SYBRGreen qPCR reactions were prepared using the primers provided in Supplementary Table 10, and 3 technical replicates were amplified using an Applied Biosystems QuantStudio 6 instrument under default cycling conditions (ThermoFisher, Hampton, New Hampshire).
Data normalization and analysis
Ct values were averaged across technical replicates and the amplicon enrichment was calculated with the following formula: E^(Ctinput−CtIP) where E= log(efficiency) of each primer pair. These data were then multiplied by the input dilution factor to obtain “percent of input”. For each sample, the percent input for the test amplicon was normalized to the percent input generated from the amplification of a control amplicon, located upstream of the exon 3 target, by making a ratio. These values were then analysed using Kruskal-Walis test (one outlier was removed from the wildtype group for having a 10-fold difference in normalized percent input) followed by Dunn’s multiple comparisons test, comparing the wildtype and inactive groups to the actively methylated group.
Chromatin Conformation Capture (3C-qPCR)
3C library preparation
In order to assess three dimensional chromatin structure, we used chromatin conformation capture, followed by quantitative PCR (3C-qPCR), using the protocol from Ea and colleagues[65]. Nuclei were extracted from 5–10 million cells using a sucrose gradient, crosslinked in 1% formaldehyde for 10 minutes, and quenched in 125mM glycine before centrifugation and resuspension in a restriction enzyme compatible 3C buffer. Samples were digested overnight at 37°C with 450U of high concentration EcoRI (Promega, Wisconsin) shaking at 200rpm, and then diluted in 4ml of ligation buffer to promote intramolecular ligation and prevent chromatin tangles. We ligated the samples with 195U high concentration T4 ligase (Promega, Wisconsin) overnight at 16°C, and then extracted the ligation products using standard proteinase K digestion, phenol chloroform extraction, and ethanol precipitation with the addition of 1ul glycogen. In order to prevent circularization or coiling of ligation products, we performed a complementary digestion using 100U of BglI (ThermoFisher, Waltham, MA), followed by phenol-chloroform extraction and ethanol precipitation of the final 3C libraries.
3C library quality control
Digestion efficiency can have a crucial impact on the outcome of 3C-based assays and as such, we assessed the digestion efficiency of each cut site within our experiment by comparison with an undigested control (UND) taken from cross linked chromatin, and a digested control (DIG) taken after EcoRI digestion but before ligation. We purified the UND and DIG control samples using proteinase K digestion, phenol chloroform extraction and ethanol precipitation, and then performed a 2-hour BglI digestion at 37°C. Using primers designed to span across each restriction site (R; Supplemental Table 11), we performed SybrGreen qPCR on the UND and DIG fractions for each sample (PowerUp SYBR Green Master Mix, ThermoFisher, Waltham, MA). To control for differences in the amount of starting material, we also amplified each fraction using primers designed for region within GAPDH that does not contain an EcoRI, nor a BglI cut site (C, Supplemental Table 11). We calculated the restriction digestion efficiency for each restriction site using the following formula: % Efficiency = 100 – 100/2^((CtR-CtC)DIG-(CtR-CtC)UND); we excluded all samples whose efficiency, averaged across all restriction sites, was less than 70%.
PCR Control Template Library
In order to determine the minimum concentration of 3C library needed for each qPCR reaction, and to compare relative interaction frequencies between primer pairs, we generated a control template library containing all possible ligation fragments, across our region of interest, in equimolar concentrations. To do this, we obtained two human BACS (RP1182M24 and RP11596I24; ThermoFisher, Waltham, MA) and combined them in equimolar concentration. We digested the BAC pool with EcoRI for two and a half hours at 37°C, and then ligated with T4 ligase overnight at 16°C. After phenol chloroform extraction, we performed BglI digestion at 37°C for two hours and then purified with phenol chloroform and ethanol precipitation for a final time. We made five-fold serial dilutions of this template library in 25ng/ul BglI digested gDNA, starting at a concentration of 25ng/ul to mimic the behavior of the 3C libraries. For each primer pair in our assay, we performed qPCR using these serial dilutions (see below) and obtained a standard curve with a slope (b) and intercept (a) that were used to normalize the Ct values of our samples.
3C-qPCR
Prior to measuring the amount of each ligation product in our 3C libraries, we measured the concentration of each library by SybrGreen PCR using primers for the non-digested site within GAPDH. We then adjusted the concentration of our libraries to 25ng/ul and re-measured the concentration to ensure accuracy. The final concentration values were used as loading control values during normalization.
We designed our qPCR assay to cover the ~1Mb region encompassing both IRX2 and IRX1 (chr5: 2744845–2752662, primers in Supplemental Table 10). We designed a constant reverse primer on the 220th EcoRI digestion fragment, which overlaps the promoter region and TSS of IRX1, as determined by in silico digestion. This fragment also bound to a custom PrimeTime Probe (Integrated DNA Technologies, Iowa) that was positioned between the constant primer and the EcoRI cut site and had a 5’ FAM fluorescent dye and a 3’ TAMRA quencher. We designed 12 test primers, adjacent to the cut sites of fragments concentrated around both IRX genes (Supplemental Table 11), such that the resulting amplicons would be between 100bp and 150bp when paired with the constant reverse primer. We determined the Ct value for each ligation product in 10ul reactions run in triplicate, using 2X TaqMan Master Mix, on a QuantStudio 6 instrument using QuantStudio Real-Time PCR Software (ThermoFisher, Hampton, New Hampshire).
Data Normalization and Analysis
To account for differences in efficiency between primer pairs, we first normalized our experimental Ct values to the standard curve obtained for each reaction using our control template library as follows: Normalized Ct1 = 10^((Ct-b)/a). In order to account for variation in the amount of template, we normalized each Ct1 to the concentration of the input library (Ct2 = Ct1/loading control), and these values were used as the relative interaction frequency between each fragment and the constant. Finally, given that the ends of each hybrid ligation fragment originate from the same DNA molecule, we calculated the basal interaction level (BIL) using the procedure as previously described[66] and defined it as the relative frequency of interaction that would be expected by chance (“random collisions”). Using the BIL for each library, and the standard error of the mean of these values, we determined the “noise band” where any observed interaction within this range would be attributed to chance and not a biologically meaningful effect.
We used student’s t tests to compare the average relative interaction frequencies between groups for each fragment and used the Holm-Sidak method to correct for multiple comparisons across each 3C experiment.
Supplementary Material
Acknowledgments
We are deeply grateful to the families of the subjects used in this study. We would also like to acknowledge the teams of technicians at the Miami Brain Endowment Bank™ and the Douglas Bell Canada Brain Bank, and the bioinformaticians who have worked on these data (A.D., R.P., and A.B.).
This work was supported by a Canadian Institute of Health Research Doctoral Fellowship awarded to K.V., National Institute of Drug Abuse Grants DA033684 awarded to G.T and D.M., R00 DA04211 to ESC, and P01 DA047233 to E.N., and grants from the Whitehall Foundation, the Edward Mallinckrodt Jr, Foundation, and the Brain and Behavior Research Foundation to ESC.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.American Psychiatric Association (APA). Diagnostic and Statistical Manual of Mental Disorders. 4th ed., Washington T:DC: American Psychiatric Association (APA); 2000. [Google Scholar]
- 2.Freeman WM, Lull ME, Patel KM, Brucklacher RM, Morgan D, Roberts DCS, et al. Gene expression changes in the medial prefrontal cortex and nucleus accumbens following abstinence from cocaine self-administration. BMC Neurosci. 2010;11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albertson D, Pruetz B, Schmidt C, Kuhn D, Kapatos G, Bannon M. Gene expression profile of the nucleus accumbens of human cocaine abusers: evidence for dysregulation of myelin. J Neurochem. 2004;88:1211–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannon MJ, Savonen CL, Jia H, Dachet F, Halter SD, Schmidt CJ, et al. Identification of long noncoding RNAs dysregulated in the midbrain of human cocaine abusers. J Neurochem. 2015;135:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mash D, French-Mullen J, Adi N, Qin Y, Buck A, Pablo J, et al. Gene expression in human hippocampus from cocaine abusers identifies genes which regulate extracellular matrix remodeling. {PloS} One. 2007;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Z, Yuan Q, Mash DC, Goldman D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc Natl Acad Sci U S A. 2011;108:6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nestler EJ. Epigenetic mechanisms of drug addiction. Neuropharmacology. 2014;76 Pt B:259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaillancourt K, Ernst C, Mash D, Turecki G. DNA Methylation Dynamics and Cocaine in the Brain: Progress and Prospects. Genes (Basel). 2017;8:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engmann O, Labonté B, Mitchell A, Bashtrykov P, Calipari ES, Rosenbluh C, et al. Cocaine-Induced Chromatin Modifications Associate With Increased Expression and Three-Dimensional Looping of Auts2. Biol Psychiatry. 2017;82:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LaPlant Q, Vialou V, Covington HE, Dumitriu D, Feng J, Warren BL, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garavan H, Pankiewicz J, Bloom A, Cho J-K, Sperry L, Ross TJ, et al. Cue-Induced Cocaine Craving: Neuroanatomical Specificity for Drug Users and Drug Stimuli. Am J Psychiatry. 2000;157:1789–1798. [DOI] [PubMed] [Google Scholar]
- 12.Volkow ND, Wang G-J, Telang F, Fowler JS, Logan J, Childress A-R, et al. Cocaine Cues and Dopamine in Dorsal Striatum: Mechanism of Craving in Cocaine Addiction. J Neurosci. 2006;26:6583–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belin D, Everitt B. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57:432–441. [DOI] [PubMed] [Google Scholar]
- 14.Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: devolving views of their roles in drug addiction. Neurosci Biobehav Rev. 2013;37:1946–1954. [DOI] [PubMed] [Google Scholar]
- 15.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6:468–481. [DOI] [PubMed] [Google Scholar]
- 16.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumoto K, Nishihara S, Kamimura M, Shiraishi T, Otoguro T, Uehara M, et al. The prepattern transcription factor Irx2, a target of the FGF8/MAP kinase cascade, is involved in cerebellum formation. Nat Neurosci. 2004;7:605–612. [DOI] [PubMed] [Google Scholar]
- 19.Kozlenkov A, Roussos P, Timashpolsky A, Barbu M, Rudchenko S, Bibikova M, et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014;42:109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizzardi LF, Hickey PF, Rodriguez DiBlasi V, Tryggvadóttir R, Callahan CM, Idrizi A, et al. Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat Neurosci. 2019;22:307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tena JJ, Alonso ME, de la Calle-Mustienes E, Splinter E, de Laat W, Manzanares M, et al. An evolutionarily conserved three-dimensional structure in the vertebrate Irx clusters facilitates enhancer sharing and coregulation. Nat Commun. 2011;2:310. [DOI] [PubMed] [Google Scholar]
- 22.Hashimoto H, Wang D, Horton JR, Zhang X, Corces VG, Cheng X. Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol Cell. 2017;66:711–720.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, et al. Analysis of the Vertebrate Insulator Protein CTCF-Binding Sites in the Human Genome. Cell. 2007;128:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vojta A, Dobrinić P, Tadić V, Bočkor L, Korać P, Julg B, et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernácer J, Prensa L, Giménez-Amaya JM. Distribution of GABAergic interneurons and dopaminergic cells in the functional territories of the human striatum. PLoS One. 2012;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tepper JM, Tecuapetla F, Koós T, Ibáñez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobo MK, Nestler EJ. The striatal balancing act in drug addiction: Distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat. 2011;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao P, Lin P, Gokoolparsadh A, Assareh A, Thang MWC, Voineagu I. Coexpression networks identify brain region–specific enhancer RNAs in the human brain. Nat Neurosci. 2015;18:1168–1174. [DOI] [PubMed] [Google Scholar]
- 30.Hannon E, Marzi SJ, Schalkwyk LS, Mill J. Genetic risk variants for brain disorders are enriched in cortical H3K27ac domains. Mol Brain. 2019;12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23:1256–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell. 2014;26:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science (80- ). 2018;362:eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng J, Wilkinson M, Liu X, Purushothaman I, Ferguson D, Vialou V, et al. Chronic cocaine-regulated epigenomic changes in mouse nucleus accumbens. Genome Biol. 2014;15:R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cates HM, Heller EA, Lardner CK, Purushothaman I, Peña CJ, Walker DM, et al. Transcription Factor E2F3a in Nucleus Accumbens Affects Cocaine Action via Transcription and Alternative Splicing. Biol Psychiatry. 2018;84:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker-Andresen D, Zhao Q, Li X, Jupp B, Chesworth R, Lawrence AJ, et al. Persistent variations in neuronal DNA methylation following cocaine self-administration and protracted abstinence in mice. Neuroepigenetics. 2015;4:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahn J-I, Lee K-H, Shin D-M, Shim J-W, Lee J-S, Chang SY, et al. Comprehensive transcriptome analysis of differentiation of embryonic stem cells into midbrain and hindbrain neurons. Dev Biol. 2004;265:491–501. [DOI] [PubMed] [Google Scholar]
- 39.Kasper C, Hebert FO, Aubin-Horth N, Taborsky B. Divergent brain gene expression profiles between alternative behavioural helper types in a cooperative breeder. Mol Ecol. 2018;27:4136–4151. [DOI] [PubMed] [Google Scholar]
- 40.Gomez-Velazquez M, Badia-Careaga C, Lechuga-Vieco AV, Nieto-Arellano R, Tena JJ, Rollan I, et al. CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart. PLOS Genet. 2017;13:e1006985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelz MB, Chen J, Carlezon WA, Whisler K, Gilden L, Beckmann AM, et al. Expression of the transcription factor ΔFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–276. [DOI] [PubMed] [Google Scholar]
- 42.Nestler EJ. The neurobiology of cocaine addiction. Sci Pract Perspect. 2005;3:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bannon MJ, Johnson MM, Michelhaugh SK, Hartley ZJ, Halter SD, David J a, et al. A Molecular Profile of Cocaine Abuse Includes the Differential Expression of Genes that Regulate Transcription, Chromatin, and Dopamine Cell Phenotype. Neuropsychopharmacology. 2014;39:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cannella N, Oliveira AMM, Hemstedt T, Lissek T, Buechler E, Bading H, et al. Dnmt3a2 in the Nucleus Accumbens Shell Is Required for Reinstatement of Cocaine Seeking. J Neurosci. 2018;38:7516–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chandra R, Francis TC, Konkalmatt P, Amgalan A, Gancarz AM, Dietz DM, et al. Opposing Role for Egr3 in Nucleus Accumbens Cell Subtypes in Cocaine Action. J Neurosci. 2015;35:7927–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database. 2016;2016:baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saftig P, Lichtenthaler SF. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog Neurobiol. 2015;135:1–20. [DOI] [PubMed] [Google Scholar]
- 48.Shukla M, Maitra S, Hernandez J-F, Govitrapong P, Vincent B. Methamphetamine regulates βAPP processing in human neuroblastoma cells. Neurosci Lett. 2019;701:20–25. [DOI] [PubMed] [Google Scholar]
- 49.Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, et al. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep. 2015;12:1184–1195. [DOI] [PubMed] [Google Scholar]
- 50.Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 2010;30:13130–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang TY, Keown CL, Wen X, Li J, Vousden DA, Anacker C, et al. Environmental enrichment increases transcriptional and epigenetic differentiation between mouse dorsal and ventral dentate gyrus. Nat Commun. 2018;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gross JA, Fiori LM, Labonté B, Lopez JP, Turecki G. Effects of promoter methylation on increased expression of polyamine biosynthetic genes in suicide. J Psychiatr Res. 2013;47:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwata A, Nagata K, Hatsuta H, Takuma H, Bundo M, Iwamoto K, et al. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum Mol Genet. 2014;23:648–656. [DOI] [PubMed] [Google Scholar]
- 54.Lutz PE, Tanti A, Gasecka A, Barnett-Burns S, Kim JJ, Zhou Y, et al. Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: Convergent epigenetic, transcriptional, and morphological evidence. Am J Psychiatry. 2017;174:1185–1194. [DOI] [PubMed] [Google Scholar]
- 55.Guo JU, Su Y, Shin JJH, Shin JJH, Li H, Xie B, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2013;17:215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X, Zhao Q, Wei W, Lin Q, Magnan C, Emami MR, et al. The DNA modification N6-methyl-2’-deoxyadenosine (m6dA) drives activity-induced gene expression and is required for fear extinction. Nat Neurosci. 2019;22:534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng J, Shao N, Szulwach KE, Vialou V, Huynh J, Zhong C, et al. Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat Neurosci. 2015;18:536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science (80- ). 2017;357:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen GG, Diallo AB, Poujol R, Nagy C, Staffa A, Vaillancourt K, et al. BisQC: an operational pipeline for multiplexed bisulfite sequencing. BMC Genomics. 2014;15:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cavalcante RG, Sartor MA. Annotatr: Genomic regions in context. Bioinformatics. 2017;33:2381–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheffield NC, Bock C. LOLA: Enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2016;32:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.GTEx Consortium TGte. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen GG, Gross JA, Lutz P-E, Vaillancourt K, Maussion G, Bramoulle A, et al. Medium throughput bisulfite sequencing for accurate detection of 5-methylcytosine and 5-hydroxymethylcytosine. BMC Genomics. 2017;18:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson AR, Thibeault KC, Lopez AJ, Peck EG, Sands LP, Sanders CM, et al. Cues play a critical role in estrous cycle-dependent enhancement of cocaine reinforcement. Neuropsychopharmacology. 2019. 23 January 2019. 10.1038/s41386-019-0320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ea V, Court F, Forne T. Quantitative Analysis of Intra-chromosomal Contacts: The 3C-qPCR Method. Methods Mol Biol. 2017;1589:75–88. [DOI] [PubMed] [Google Scholar]
- 66.Braem C, Recolin B, Rancourt RC, Angiolini C, Barthès P, Branchu P, et al. Genomic matrix attachment region and chromosome conformation capture quantitative real time PCR assays identify novel putative regulatory elements at the imprinted Dlk1/Gtl2 locus. J Biol Chem. 2008;283:18612–18620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.