

Abstract
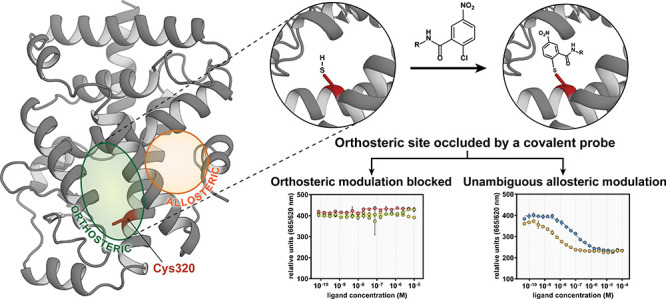
The nuclear receptor RORγt is a key positive regulator in the differentiation and proliferation of T helper 17 (Th17) cells and the production of proinflammatory cytokines like IL-17a. Dysregulation of this pathway can result in the development of various autoimmune diseases, and inhibition of RORγt with small molecules thus holds great potential as a therapeutic strategy. RORγt has a unique allosteric ligand binding site in the ligand binding domain, which is distinct from the canonical, orthosteric binding site. Allosteric modulation of RORγt shows high potential, but the targeted discovery of novel allosteric ligands is highly challenging via currently available methods. Here, we introduce covalent, orthosteric chemical probes for RORγt that occlude the binding of canonical, orthosteric ligands but still allow allosteric ligand binding. Ultimately, these probes could be used to underpin screening approaches for the unambiguous and rapid identification of novel allosteric RORγt ligands.
Keywords: Nuclear receptors, RORγt, covalent probes, allosteric modulators
The retinoic acid receptor-related orphan receptor gamma t (RORγt) is a nuclear receptor (NR) that plays an important regulatory role in the immune system via the Th17/IL-17a pathway.1−3 Inhibition of RORγt by inverse agonists has been shown to be a promising strategy for the treatment of autoimmune diseases and thus has been the focus of several drug discovery programs.4,5 The majority of RORγt ligands bind to a highly conserved canonical binding pocket, termed the orthosteric binding site, within the ligand binding domain (LBD) of RORγt (Figure 1C).6−10 RORγt appears to be transcriptionally active, even in the absence of an agonist ligand.11 However, agonists (e.g., cholesterol and its derivatives) that bind to the orthosteric site enhance RORγt transcriptional activity further by stabilizing the active conformation of helix 12 (H12) in a way that promotes the recruitment of transcriptional coactivators.12 RORγt inverse agonists binding this site (e.g., digoxin) destabilize the active conformation of H12, inhibiting coactivator recruitment.13
Figure 1.

(A) Schematic representation of RORγt orthosteric site occlusion. When both binding sites are available, orthosteric and allosteric ligands are identified in coactivator binding assays. When the orthosteric binding site is occluded, allosteric ligands can be unambiguously identified. (B) Crystal structure of PPARγ ligated to GW9662 (blue) at the Cys285 residue (green) in the orthosteric binding site (PDB: 3B0R). (C) Crystal structure of RORγt (PDB: 6T4I) showing the orthosteric (green) and allosteric (orange) ligand binding site. The Cys320 residue in the orthosteric site is shown in red. (D) Chemical structures of PPARγ covalent ligands GW9662 (1) and SB1404 (2). (E) Enlarged view of GW9662 ligated to PPARγ. (F) The thiol of Cys320 in RORγt is postulated to ligate to the electron-deficient aryl ring of GW9662 or derivatives.
The conserved nature of the RORγt orthosteric binding pocket presents some challenges for drug discovery, for example, because of competition with endogenous ligands.14 Most interestingly, and unique among the NR family, RORγt has a second binding site, termed allosteric binding site (located at a topographically distinct place in the LBD) (Figure 1C).15 This allosteric site offers ample opportunities for innovative NR drug discovery. The indazole MRL-871, thiazole compound 13 (Glenmark), and isoxazole FM26 are examples of highly potent RORγt inverse agonists that bind to this allosteric site.15−19 These ligands exert their effect via a reorientation of H12 into a conformation that prevents coactivator binding.15,18,19
Despite the potential of NR allosteric inverse agonists, the number of examples and their chemical diversity have remained rather limited.15−17,19−22 Furthermore, understanding of the structure–activity relationships (SARs) and scaffold diversity of allosteric RORγt inverse agonists is of importance to tune the potency, selectivity, and pharmacokinetic profiles of potential pharmaceutical lead molecules. However, unambiguous screening for allosteric ligands is challenging, since both orthosteric and allosteric ligands can show an inhibitory response on RORγt (Figure 1A, left). Currently, discrimination between orthosteric and allosteric ligands is not trivial and the orthosteric pocket can host a plethora of chemically diverse compounds. Illustrative of the above, the mode of action for MRL-871 was only described in retrospect.15 Even in a targeted program, the methods currently available require a series of biophysical and structural experiments to discriminate between orthosteric and allosteric inverse agonism.15 Therefore, the development of a molecular approach for the specific identification of allosteric ligands is highly desirable.
A potential strategy to achieve the goal defined above would be via a competitive binding assay, whereby the displacement of a well-characterized allosteric probe ligand can be monitored, such as the one previously reported.15,19 However, competition assays are notoriously problematic for identification of the weaker binding initial chemical entities. Additionally, orthosteric inverse agonists could potentially lower the allosteric probe affinity, which would lead to false positive results that diminish the reliability of the assay. We therefore postulated that using a molecular probe to occlude the RORγt orthosteric binding site, in combination with an established coactivator recruitment assay, would provide a more robust approach (Figure 1A, right). An important feature of the prospective orthosteric probe is that it should have minimal effect on the characteristics of the allosteric pocket and on RORγt coactivator binding.
Our approach took inspiration from studies of the structurally related NR peroxisome proliferator-activated receptor γ (PPARγ). PPARγ contains a cysteine residue within the orthosteric ligand binding pocket (Cys285) (Figure 1B), which has previously been targeted by covalent ligands (Figure 1E).23−25 In particular, covalent modification of Cys285 by the electron deficient aryl chlorides GW9662 (1) and SB1404 (2) (Figure 1D) occlude the binding, and thus also the activity, of certain PPARγ agonists.24−28 RORγt contains an analogous cysteine residue within the orthosteric ligand binding pocket (Cys320, RORγ numbering; see Figures 1C and S1). Therefore, we were interested to determine if this residue could also be targeted with covalent molecular probes that would block ligand binding to the orthosteric site without significantly affecting coactivator binding. Examples of RORγt covalent inverse agonists have been reported, but they, thus far, target other cysteine residues that are not in proximity to the orthosteric binding pocket.22
GW9662 and SB1404 were synthesized via an amide coupling reaction between the commercially available 2-chloro-5-nitrobenzoyl chloride and the appropriate amine (Scheme S1).24,27 A single GW9662 molecule fully ligated to RORγt, following optimization of the ligation conditions24 (Table S1), as shown by quadrupole time-of-flight mass spectrometry (Q-TOF MS) (Figure S2A). In order to prove binding to Cys320, a RORγt Cys320Ala mutant was generated. No ligation of GW9662 to this RORγt mutant was observed which verified the expected Cys320 ligation site (Figure S2B). Surprisingly, SB1404, which has a smaller methyl group instead of the phenyl substituent, did not show any ligation to RORγt, while it did fully ligate to PPARγ. This might be because SB1404 has a lower affinity for the RORγt LBP compared to GW9662. This observation highlights the difference between the binding pockets of the two NRs.
A TR-FRET coactivator recruitment assay29 was used to investigate the effect of GW9662 on the activity of RORγt, i.e., the ability of RORγt to recruit coactivators (Figure 2D). The dose–response curve in Figure 2A shows inhibition of coactivator recruitment upon titration of GW9662 to the protein (similar behavior to the full allosteric inverse agonist MRL-871), demonstrating that the probe acts as a full inverse agonist for RORγt with an IC50 value of 86 ± 5 nM. A possible reason for this inverse agonistic character could be derived from the docking pose, which shows a conformation of GW9662 where the warhead (nitro-phenyl moiety) points toward H11 (Figure S3A), which could result in destabilization of the active conformation of H12, inhibiting coactivator recruitment.30 Alignment of the docking pose of GW9662 to the crystal structure of RORγt bound to the known RORγt orthosteric inverse agonist T090131731,32 shows that the nitro moiety of GW9662 and the CF3 groups of T0901317 have the same orientation and distance toward H11, which could explain the inverse agonistic behavior of GW9662 (Figure S4).
Figure 2.

(A–C) Dose–response curves of a TR-FRET coactivator recruitment assay with RORγt by titration of MRL-871 and different orthosteric covalent probes. Data recorded in triplicate from two independent experiments (one representative data set shown). Error bars represent the SD of the mean. (D) Schematic representation of the TR-FRET coactivator recruitment assay. When RORγt is in its apo state, the coactivator binds to the LBD, resulting in FRET pairing from an anti-His terbium cryptate donor to the d2-labeled coactivator. When the covalent orthosteric probe ligates to the protein, coactivator recruitment is blocked by its (partial) inverse agonistic character.
A ligand binding TR-FRET coactivator recruitment assay was used to determine if GW9662 ligation effectively occluded orthosteric ligand binding to RORγt, without affecting allosteric ligand binding (see Figure 3F/G). To probe this, three orthosteric agonists (cholesterol (CHL), 20α-hydroxycholesterol (20-OH), and desmosterol (DSM)), an orthosteric inverse agonist (digoxin), and two allosteric inverse agonists (MRL-871 and FM26) were tested (see chemical structures in Figure 3H).12,13,15,19 For the apo RORγt protein, the cholesterol derivatives show agonistic character, increasing coactivator recruitment to the LBD (Figure 3A), consistent with literature reports.2,12 The orthosteric inverse agonist digoxin and the allosteric inverse agonists reduced coactivator recruitment in a dose-dependent manner as expected (Figure 3A).13,19 These titration experiments were then repeated with the GW9662-ligated RORγt (Figure 3B). In this experiment, the orthosteric agonists did not cause any increase in coactivator recruitment to RORγt, indicating occlusion of the orthosteric binding site. However, the allosteric ligands MRL-871 and FM26 also did not demonstrate a change in coactivator recruitment, due to the full inverse agonistic behavior of GW9662. Because of its full inverse agonistic behavior, GW9662 is thus not suitable for the screening of allosteric inverse agonists. We therefore set out to identify a GW9662 analogue that would act either as true covalent antagonist (/agonist) or as a partial covalent inverse agonist, that would maintain some RORγt sensitivity to allosteric ligand binding.
Figure 3.

TR-FRET coactivator recruitment ligand binding assay with RORγt (unligated and ligated) by titration of various orthosteric and allosteric ligands. (A) Apo (unligated) RORγt protein. (B) GW9662-ligated RORγt. (C) Compound 9-ligated RORγt. (D) Compound 19-ligated RORγt. (E). Compound 20-ligated RORγt. Data recorded in triplicate from two independent experiments (one representative data set shown). Error bars represent the SD of the mean. Abbreviations: n.a., not active. (F/G) Schematic representation of the TR-FRET coactivator recruitment assay, using the ligated protein. (F) Orthosteric ligand binding will be occluded, showing no effect on the initial coactivator recruitment capacity. (G) Allosteric ligand binding will result in reduced coactivator binding and therefore a lower FRET pairing. (H) Chemical structures of orthosteric agonists (cholesterol (CHL), desmosterol (DSM), and 20α-hydroxycholesterol (20-OH)), orthosteric inverse agonist digoxin, and allosteric inverse agonists MRL-871 and FM26.
An initial library of 12 GW9662-derivatives was synthesized (compounds 3–14, Table 1), containing the same warhead, but with modifications in place of the phenyl moiety, to investigate its influence on the ligation efficiency and activity of RORγt. These modifications were made with the aim to identify a probe that ligates to the RORγt Cys320 but does not behave as a full inverse agonist. The modifications were varied in terms of bulkiness, π–π stacking capacity, aromaticity, and substitution pattern. The compounds were synthesized in a similar manner to GW9662 (Scheme S1B).24,33−35 First, the ligation efficiency was explored for all derivatives via Q-TOF analysis of the reacted protein. Although GW9662 fully ligated to the protein (Figure S6), its derivatives ligated with varying efficiency (Table 1, Figures S7 and S8) (ligation conditions shown in Table S1). From the ligation data, it can be concluded that a ring system (preferably aromatic) is necessary at the phenyl position of GW9662 in order to obtain full ligation to the protein. A methyl substitution on the phenyl ring is only fully tolerated at the ortho position (compound 9), indicating that bulk at meta/para positions lowers ligation (compound 7 and 8). Furthermore, an electron-withdrawing substituent on the phenyl ring improves the ligation efficiency compared to an electron-donating group (compound 13 vs 10), most probably caused by the higher electrophilicity of the warhead. Interestingly, all probes (except for compounds 12 and 14) also showed full covalent attachment to PPARγ, indicating that PPARγ shows less differentiation in the ligation of the compounds than RORγt, which is probably due to the larger size of the PPARγ binding pocket.
Table 1. Chemical Structures of GW9662 Derivatives and Their RORγt Ligation.

The compounds that fully ligated to RORγt (9 and 11) were taken forward for evaluation of the binding behavior in a TR-FRET dose–response assay. Compound 11 showed the same full inverse agonistic behavior as GW9662 but was slightly less potent (IC50 value of 122 ± 6 nM) (Figure 2A, Table 2). More interestingly, in contrast to the other two probes, compound 9 showed partial inverse agonistic behavior, thus not completely blocking coactivator recruitment to the LBD, with 55% remaining activity (Figure 2A, Table 2). The difference in activity between GW9662 and compound 9 could be explained by an inverse binding conformation of 9 (with the warhead pointing toward the orthosteric pocket instead of toward H11) which was supported by an in silico docking experiment (Figure S3B).
Table 2. Chemical Structures of GW9662 Derivatives, Their RORγt Ligation, IC50 ± SD (nM) (Data Recorded in Triplicate in Two Independent Experiments) And Full/Partial Inverse Agonistic Charactera.

Abbreviations: inv. ago., inverse agonist; rem. act., remaining activity; n.d., not determined.
A focused SAR study was performed around compound 9 to obtain more insight into the desirable partial behavior of this probe. A library of nine compound 9 derivatives (compounds 15–23, Table 2) was designed, by varying the size, polarity, and electron density of the ortho-substituent. The probes were synthesized as described above (Scheme S1B). All the probes from this set of derivatives fully ligated with RORγt (Table 2, Figures S9–S16), except for compound 16 that has a tert-butyl substituent. Although an ethyl substituent and a naphthalene moiety appeared to be tolerated (15 and 21), the tert-butyl moiety of 16 is likely too bulky for optimal binding. Furthermore, the polarity of the substituent (22 and 23) did not affect the ligation behavior and even a double-ortho methyl substitution was tolerated (19).
All probes (except 16) were tested in the TR-FRET dose–response assay and showed varying inverse agonistic behavior (Figure 2B/C). A partial inverse agonistic behavior, similar to compound 9, was observed for probes 15 and 18 (increased size of the substituent), 19 (bis-ortho substitution), and 20 (electron-deficient substituent) (Figure 2B). Compound 17 with a fluoro substituent also demonstrated partial behavior, however inducing a greater decrease in coactivator recruitment than the previous four probes (Figure 2C), which might be due to its size being more comparable to GW9662. Compound 23 (hydroxyl modification) resulted in full inverse agonism, similar to GW9662, and 22 (amine modification) also approached the bottom plateau, but with a lower potency than 23. Compound 21, with the large naphthalene substituent, showed a significantly lower potency than the other probes (IC50 = 1080 ± 153 nM), not reaching the bottom plateau, which makes it hard to confidently characterize it as a partial or full inverse agonist.
The modifications with a similar size to the methyl group in compound 9 (e.g., ethyl, methoxy, bismethyl, trifluoromethyl) appear to result in a partial inverse agonistic behavior, while smaller or polar substituents (e.g., fluoro, hydroxyl, amine) show a full inverse agonistic character. A structural explanation could be derived from the docking studies, where the partial inverse agonists generally show a docking pose similar to compound 9 (warhead pointing toward the orthosteric site), while for the full inverse agonists a docking pose similar to GW9662 was observed. Combined, compound 9 and four derivatives were found to be covalent partial inverse agonists and suitable candidates as covalent orthosteric probes.
The TR-FRET ligand binding assay was used to investigate the combination of occlusion of the orthosteric site and potential for allosteric binding. With the partial inverse agonist probes, a screening window is expected to be preserved to detect allosteric inverse agonist binding. Clear occlusion of the orthosteric site for agonist binding was observed for RORγt ligated to 9, 19, and 20 (Figure 3C–E), in comparison to the apo protein (Figure 3A). In addition, the orthosteric inverse agonist digoxin was also ineffective. In contrast, and most importantly, the allosteric inverse agonists MRL-871 and FM26 still induced a clear inverse agonistic response in RORγt. Also, the potency of MRL-871 and FM26 was not negatively affected by the occlusion of the orthosteric site, revealing that the partial inverse agonism of the covalent probes translates to responsiveness of the allosteric site for ligand binding. The other partial inverse agonistic probes 15 and 18 show similar occlusion of the orthosteric site while still allowing allosteric binding, albeit with a smaller assay window (Figure S5). Interestingly, in all cases, the IC50 values for the allosteric ligands MRL-871 and FM26 were actually decreased to different extents (3-fold and 13-fold, respectively) in comparison to the data for the apo protein (Figure 3A/C–E). This increased potency of the allosteric ligands in the presence of an orthosteric probe is likely caused by a cooperative effect between both binding sites, as observed in previous studies.18,19,36
A thermal shift assay (TSA) was used as an orthogonal method to confirm occlusion of orthosteric ligand binding by the most interesting probes (9, 19, and 20) without affecting allosteric modulation. With the apo RORγt protein, all orthosteric ligands showed a significant thermal stabilization (ΔTm between 2.5 and 5.8 °C), except for cholesterol (Figure 4A). In contrast, for RORγt ligated to 9, 19, and 20, a thermal stabilization effect was not observed anymore for these orthosteric ligands and they even caused a small destabilization effect (ΔTm between −0.1 and −2.7 °C) (Figure 4B–D). These results again demonstrate orthosteric site occlusion by the covalent probes. The allosteric ligands MRL-871 and FM26 show a moderate to high thermal stabilization effect for the apo protein (respectively ΔTm of 7.5 and 2.1 °C) (Figure 4A). When RORγt was ligated to a covalent probe, this thermal stabilization effect by the allosteric ligands was preserved and even higher ΔTm values were observed in all cases (Figure 4B–D). These results again prove that the allosteric binding is still functional with the orthosteric site blocked and that affinity is even slightly enhanced. This increased stabilization by the allosteric ligands in the presence of the orthosteric covalent probes is consistent with the cooperative behavior observed in the TR-FRET ligand binding assays. Although the observed cooperative behavior might question the usefulness of the probes at first sight, it will not be an issue from a screening perspective, since it will not lead to false hits but will just enhance an allosteric IC50 value by a limited degree.
Figure 4.

Thermal shift assay performed for Apo RORγt (A), compound 9-ligated RORγt (B), compound 19-ligated RORγt (C), and compound 20-ligated RORγt (D), for ligands cholesterol (CHL), 20α-hydroxycholesterol (20-OH), desmosterol (DSM), digoxin, MRL-871 and FM26 (2 equiv compound relative to protein). The melting temperatures (ΔTm in °C) are shown. Data recorded in triplicate from two independent experiments (one representative data set shown). Error bars represent the SD of the mean.
Although allosteric RORγt inverse agonists have high potential for NR drug discovery, the number of examples and chemical diversity have remained limited. An enlargement of the allosteric RORγt ligand library is therefore essential in order to enhance understanding of the SAR and to tune potency and selectivity. Unambiguous screening for allosteric ligands is challenging, since both orthosteric and allosteric ligands will result in an inhibitory response on the protein and discrimination between them is not trivial.
Here, we introduced a method for occlusion of the RORγt orthosteric binding site via the ligation of covalent chemical probes to a native cysteine residue. This allows for the unambiguous targeting of the allosteric binding site, which has the potential to facilitate the rapid identification of allosteric inverse agonists. The reference compound GW9662 showed full ligation to Cys320 of RORγt but acts as a full inverse agonist and completely inhibits coactivator binding, preventing the detection of allosteric inverse agonists. From a small library of GW9662 derivatives, the methyl-substituted compound 9 was identified as a covalent partial inverse agonist. Further SAR studies around compound 9 resulted in the discovery of four additional covalent probes with a partial inverse agonistic character, for which 19 and 20 appeared to be the most promising probes (containing a bis-ortho-methyl and trifluoromethyl modification). The partial character of these probes can most probably be explained by an inverse binding conformation compared to GW9662 as was supported by docking experiments. Co-crystallization attempts of the probes with RORγt were unsuccessful but could provide more structural evidence. TR-FRET and thermal shift assays revealed complete occlusion of the orthosteric binding site with the covalent probes, while allosteric ligand binding was not inhibited and even occurred with enhanced affinity. This cooperative behavior of the orthosteric inverse agonistic covalent probes with the allosteric ligands is an interesting observation, since these cooperative effects had previously only been observed with orthosteric agonists. The covalent probes are excellent tools that could underpin an assay format that unambiguously screens for allosteric RORγt modulators. Additionally, these covalent orthosteric ligands could be used as inspiration for the development of covalent orthosteric inverse agonists for RORγt, for which future studies could focus on the efficacy and toxicity of covalent RORγt targeting.
Acknowledgments
We thank Rens M.J.M. de Vries for his efforts in crystallography experiments and Joost L.J. van Dongen for performing HRMS measurements.
Glossary
Abbreviations
- 20-OH
20α-hydroxycholesterol
- CHL
cholesterol
- DSM
desmosterol
- H12
helix 12
- IPTG
isopropyl-b-d-thiogalactoside
- mAb
monoclonal antibody
- LBD
ligand binding domain
- NR
nuclear receptor
- PPARγ
peroxisome proliferator-activated receptor γ
- Q-TOF
quadrupole time-of-flight
- RORγt
retinoic acid receptor-related orphan receptor γ t
- SAR
structure–activity relationship
- Th17
T helper 17
- Tm
melting temperature
- TR-FRET
time-resolved FRET
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00029.
Author Contributions
§ F.A.M. and M.C.M.O. contributed equally. M.C.M.O. and E.N.R.S. performed synthesis; F.A.M., M.C.M.O., and E.N.R.S. performed biochemical studies; F.A.M., M.C.M.O., R.G.D., and L.B. designed the studies. The manuscript was written through contributions of all authors.
This work was supported by The Netherlands Organization for Scientific Research through Gravity program 024.001.035 and VICI Grant 016.150.366 and the European Union through a MSCA Individual Fellowship (R.G.D., H2020-MSCA-IEF-2016, Grant Number 705188). This work was carried out on the Dutch national e-infrastructure with the support of SURF Cooperative.
The authors declare no competing financial interest.
Supplementary Material
References
- Ivanov I. I.; McKenzie B. S.; Zhou L.; Tadokoro C. E.; Lepelley A.; Lafaille J. J.; Cua D. J.; Littman D. R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Solt L. A.; Burris T. P. Action of RORs and Their Ligands in (Patho)Physiology. Trends Endocrinol. Metab. 2012, 23, 619–627. 10.1016/j.tem.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel N.; Unutmaz D.; Littman D. R. The Differentiation of Human TH-17 Cells Requires TGF-β and Induction of the Nuclear Receptor RORγt. Nat. Immunol. 2008, 9, 641–649. 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P.; Kolls J. K. Targeting IL-17 and TH17 Cells in Chronic Inflammation. Nat. Rev. Drug Discovery 2012, 11, 763–776. 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- Yang J.; Sundrud M. S.; Skepner J.; Yamagata T. Targeting Th17 Cells in Autoimmune Diseases. Trends Pharmacol. Sci. 2014, 35, 493–500. 10.1016/j.tips.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Fauber B. P.; Magnuson S. Modulators of the Nuclear Receptor Retinoic Acid Receptor-Related Orphan Receptor-γ (RORγ or RORc). J. Med. Chem. 2014, 57, 5871–5792. 10.1021/jm401901d. [DOI] [PubMed] [Google Scholar]
- Cyr P.; Bronner S. M.; Crawford J. J. Recent Progress on Nuclear Receptor RORγ Modulators. Bioorg. Med. Chem. Lett. 2016, 26, 4387–4393. 10.1016/j.bmcl.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Pandya V. B.; Kumar S.; Sachchidanand; Sharma R.; Desai R. C. Combating Autoimmune Diseases with RORγ (or RORc) Inhibitors: Hits and Misses. J. Med. Chem. 2018, 61, 10976–10995. 10.1021/acs.jmedchem.8b00588. [DOI] [PubMed] [Google Scholar]
- Bronner S. M.; Zbieg J. R.; Crawford J. J. RORgamma Antagonists and Inverse Agonists: A Patent Review. Expert Opin. Ther. Pat. 2017, 27, 101–112. 10.1080/13543776.2017.1236918. [DOI] [PubMed] [Google Scholar]
- Sun N.; Guo H.; Wang Y. RORγt Inhibitors in Clinical Development for the Treatment of Autoimmune Diseases: A Patent Review (2016-Present). Expert Opin. Ther. Pat. 2019, 29, 663–674. 10.1080/13543776.2019.1655541. [DOI] [PubMed] [Google Scholar]
- Li X.; Anderson M.; Collin D.; Muegge I.; Wan J.; Brennan D.; Kugler S.; Terenzio D.; Kennedy C.; Lin S.; Labadia M. E.; Cook B.; Hughes R.; Farrow N. A. Structural Studies Unravel the Active Conformation of Apo RORγt Nuclear Receptor and a Common Inverse Agonism of Two Diverse Classes of RORγt Inhibitors. J. Biol. Chem. 2017, 292, 11618–11630. 10.1074/jbc.M117.789024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Wang Y.; Hao L.-Y.; Liu X.; Lesch C. A.; Sanchez B. M.; Wendling J. M.; Morgan R. W.; Aicher T. D.; Carter L. L.; Toogood P. L.; Glick G. D. Sterol Metabolism Controls TH17 Differentiation by Generating Endogenous RORγ Agonists. Nat. Chem. Biol. 2015, 11, 141–147. 10.1038/nchembio.1714. [DOI] [PubMed] [Google Scholar]
- Huh J. R.; Leung M. W. L.; Huang P.; Ryan D. A.; Krout M. R.; Malapaka R. R. V.; Chow J.; Manel N.; Ciofani M.; Kim S. V.; Cuesta A.; Santori F. R.; Lafaille J. J.; Xu E.; Gin D. Y.; Rastinejad F.; Littman D. R. Digoxin and Its Derivatives Suppress TH17 Cell Differentiation by Antagonizing RORγt Activity. Nature 2011, 472, 486–490. 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer F. A.; Leijten-van de Gevel I. A.; de Vries R. M. J. M.; Brunsveld L. Allosteric Small Molecule Modulators of Nuclear Receptors. Mol. Cell. Endocrinol. 2019, 485, 20–34. 10.1016/j.mce.2019.01.022. [DOI] [PubMed] [Google Scholar]
- Scheepstra M.; Leysen S.; van Almen G. C.; Miller J. R.; Piesvaux J.; Kutilek V.; van Eenennaam H.; Zhang H.; Barr K.; Nagpal S.; Soisson S. M.; Kornienko M.; Wiley K.; Elsen N.; Sharma S.; Correll C. C.; Trotter B. W.; van der Stelt M.; Oubrie A.; Ottmann C.; Parthasarathy G.; Brunsveld L. Identification of an Allosteric Binding Site for RORγt Inhibition. Nat. Commun. 2015, 6, e8833 10.1038/ncomms9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karstens W. F. J.; van der Stelt M.; Cals J.; Azevedo R. C. R. G.; Barr K. J.; Zhang H.; Beresis R. T.; Zhang D.; Duan X.. RORgammaT Inhibitors. PCT Int. Appl. WO 2012/106995, 2012.
- Chaudari S. S.; Thomas A.; Dhone S. V.; Khairatkar-Joshi N.; Bajpai M.. Bicyclic Heterocyclic Compounds as ROR Gamma Modulators. PCT Int. Appl. WO 2015/008234, 2015.
- de Vries R. M. J. M.; Meijer F. A.; Doveston R. G.; Brunsveld L. Elucidation of an Allosteric Mode of Action for a Thienopyrazole RORγt Inverse Agonist. ChemMedChem 2020, 15, 561–565. 10.1002/cmdc.202000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer F. A.; Doveston R. G.; De Vries R. M. J. M.; Vos G. M.; Vos A. A. A.; Leysen S.; Scheepstra M.; Ottmann C.; Milroy L.-G.; Brunsveld L. Ligand-Based Design of Allosteric Retinoic Acid Receptor-Related Orphan Receptor γt (RORγt) Inverse Agonists. J. Med. Chem. 2020, 63, 241–259. 10.1021/acs.jmedchem.9b01372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; Gobbi A.; Robarge K.; Zhou A.; Barnard A.; Cao J.; Deng Y.; Eidenschenk C.; Everett C.; Ganguli A.; Hawkins J.; Johnson A. R.; La H.; Norman M.; Salmon G.; Summerhill S.; Ouyang W.; Tang W.; Wong H. Discovery of Imidazo[1,5-a]Pyridines and -Pyrimidines as Potent and Selective RORc Inverse Agonists. Bioorg. Med. Chem. Lett. 2015, 25, 2907–2912. 10.1016/j.bmcl.2015.05.055. [DOI] [PubMed] [Google Scholar]
- Ouvry G.; Bouix-Peter C.; Ciesielski F.; Chantalat L.; Christin O.; Comino C.; Duvert D.; Feret C.; Harris C. S.; Lamy L.; Luzy A.; Musicki B.; Orfila D.; Pascau J.; Parnet V.; Perrin A.; Pierre R.; Polge G.; Raffin C.; Rival Y.; Taquet N.; Thoreau E.; Hennequin L. F. Discovery of Phenoxyindazoles and Phenylthioindazoles as RORγ Inverse Agonists. Bioorg. Med. Chem. Lett. 2016, 26, 5802–5808. 10.1016/j.bmcl.2016.10.023. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Dulubova I.; Reisman S. A.; Hotema M.; Lee C.-Y. I.; Liu L.; McCauley L.; Trevino I.; Ferguson D. A.; Eken Y.; Wilson A. K.; Wigley W. C.; Visnick M. A Novel Series of Cysteine-Dependent, Allosteric Inverse Agonists of the Nuclear Receptor RORγt. Bioorg. Med. Chem. Lett. 2020, 30, 126967 10.1016/j.bmcl.2020.126967. [DOI] [PubMed] [Google Scholar]
- Brust R.; Lin H.; Fuhrmann J.; Asteian A.; Kamenecka T. M.; Kojetin D. J. Modification of the Orthosteric PPARγ Covalent Antagonist Scaffold Yields an Improved Dual-Site Allosteric Inhibitor. ACS Chem. Biol. 2017, 12, 969–978. 10.1021/acschembio.6b01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leesnitzer L. M.; Parks D. J.; Bledsoe R. K.; Cobb J. E.; Collins J. L.; Consler T. G.; Davis R. G.; Hull-Ryde E. A.; Lenhard J. M.; Patel L.; Plunket K. D.; Shenk J. L.; Stimmel J. B.; Therapontos C.; Willson T. M.; Blanchard S. G. Functional Consequences of Cysteine Modification in the Ligand Binding Sites of Peroxisome Proliferator Activated Receptors by GW9662. Biochemistry 2002, 41, 6640–6650. 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- Ohtera A.; Miyamae Y.; Yoshida K.; Maejima K.; Akita T.; Kakizuka A.; Irie K.; Masuda S.; Kambe T.; Nagao M. Identification of a New Type of Covalent PPARγ Agonist Using a Ligand-Linking Strategy. ACS Chem. Biol. 2015, 10, 2794–2804. 10.1021/acschembio.5b00628. [DOI] [PubMed] [Google Scholar]
- Seargent J. M.; Yates E. A.; Gill J. H. GW9662, a Potent Antagonist of PPARgamma, Inhibits Growth of Breast Tumour Cells and Promotes the Anticancer Effects of the PPARgamma Agonist Rosiglitazone, Independently of PPARgamma Activation. Br. J. Pharmacol. 2004, 143, 933–937. 10.1038/sj.bjp.0705973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae H.; Jang J. Y.; Choi S. S.; Lee J. J.; Kim H.; Jo A.; Lee K. J.; Choi J. H.; Suh S. W.; Park S. B. Mechanistic Elucidation Guided by Covalent Inhibitors for the Development of Anti-Diabetic PPARγ Ligands. Chem. Sci. 2016, 7, 5523–5529. 10.1039/C6SC01279E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehringer M.; Laufer S. A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Degorce F. HTRF: A Technology Tailored for Drug Discovery - A Review of Theoretical Aspects and Recent Applications. Curr. Chem. Genomics 2009, 3, 22–32. 10.2174/1875397300903010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallen J.; Izaac A.; Be C.; Arista L.; Orain D.; Kaupmann K.; Guntermann C.; Hoegenauer K.; Hintermann S. Structural States of RORgammat: X-Ray Elucidation of Molecular Mechanisms and Binding Interactions for Natural and Synthetic Compounds. ChemMedChem 2017, 12, 1014–1021. 10.1002/cmdc.201700278. [DOI] [PubMed] [Google Scholar]
- Kumar N.; Solt L. A.; Conkright J. J.; Wang Y.; Istrate M. A.; Busby S. A.; Garcia-Ordonez R. D.; Burris T. P.; Griffin P. R. The Benzenesulfoamide T0901317 is a Novel RORα/γ Inverse Agonist. Mol. Pharmacol. 2010, 77, 228–236. 10.1124/mol.109.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; de Leon Boenig G.; Burton B.; Eidenschenk C.; Everett C.; Gobbi A.; Hymowitz S. G.; Johnson A. R.; Liimatta M.; Lockey P.; Norman M.; Ouyang W.; René O.; Wong H. Structure-Based Design of Substituted Hexafluoroisopropanol-Arylsulfonamides as Modulators of RORc. Bioorg. Med. Chem. Lett. 2013, 23, 6604–6609. 10.1016/j.bmcl.2013.10.054. [DOI] [PubMed] [Google Scholar]
- Henke A.; Srogl J. Thioimides: New Reagents for Effective Synthesis of Thiolesters from Carboxylic Acids. J. Org. Chem. 2008, 73, 7783–7784. 10.1021/jo801319x. [DOI] [PubMed] [Google Scholar]
- Amemiya Y.; Wakabayashi K.; Takaishi S.; Fukuda C.. PPARgamma Modulators, PCT Int. Appl. WO 2001/083427, 2001.
- D’Silva C.; Iqbal R. A New Method to N-Arylmethylenepyrroles from N-Acylpyrroles. Synthesis 1996, 1996, 457–458. 10.1055/s-1996-4247. [DOI] [Google Scholar]
- de Vries R. M. J. M.; Meijer F. A.; Doveston R. G.; Leijten-van de Gevel I. A.; Brunsveld L. Cooperativity between the Orthosteric and Allosteric Ligand Binding Sites of RORγt. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2021287118 10.1073/pnas.2021287118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.