

Abstract
ONC201 is the first member of the imipridone family of anticancer drugs to enter the clinic for the treatment of diverse solid and hematologic cancers. A subset of pediatric and adult patients with highly aggressive brain tumors has shown remarkable clinical responses to ONC201, and recently, the more potent derivative ONC206 entered clinical trials as a single agent for the treatment of central nervous system (CNS) cancers. Despite the emerging clinical interest in the utility of imipridones, their exact molecular mechanisms are not fully described. In fact, the existing literature points to multiple pathways (e.g. tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) signaling, dopamine receptor antagonism, and mitochondrial metabolism) as putative drug targets. We have performed a comprehensive literature review and highlighted mitochondrial metabolism as the major target of imipridones. In support of this, we performed a meta-analysis of an ONC201 screen across 539 human cancer cell lines and showed that the mitochondrial caseinolytic protease proteolytic subunit (ClpP) is the most significant predictive biomarker of response to treatment. Herein, we summarize the main findings on the anticancer mechanisms of this potent class of drugs, provide clarity on their role, and identify clinically relevant predictive biomarkers of response.
Keywords: ClpP, imipridone, ONC201, ONC206, ONC212
The anticancer imipridone drug family has emerged as promising candidates for treating a diverse range of solid and hematologic cancers.1,2 ONC201, the parent imipridone (Table 1), exhibits cytotoxicity across a spectrum of preclinical cancer models and has entered phase 1 and 2 clinical trials for treating patients diagnosed with cancers including leukemia, lymphomas, colon, prostate, breast, and central nervous system (CNS) tumors (Table 2). Clinically, ONC201 has demonstrated a favorable safety profile and encouraging performance at prolonging patient survival, even in patients with advanced treatment-refractory solid tumors.3 Moreover, ONC201 demonstrates CNS tumor penetration and encouraging response rates in a subset of both adult and pediatric brain cancer patients,4–7 including children with H3K27M-mutant diffuse midline glioma (DMG),6,7 warranting further clinical study. These positive clinical observations have catalyzed the synthesis of imipridone derivatives that share ONC201’s core chemical structure but harbor modifications conferring enhanced potency and signaling capabilities in preclinical models8–12 (Table 1). However, the precise anticancer mechanisms of imipridones remain elusive. These drugs may selectively antagonize G protein-coupled receptor (GPCR) proteins, most notably the D(2) dopamine receptor (D2R), widely expressed across cancer cells.13 In addition, imipridones may induce tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) signaling, leading to the extrinsic pathway of cell death.1,2 However, emerging data indicate that imipridones' primary mechanism is to potently activate the mitochondrial ATP-dependent caseinolytic protease (Clp)proteolytic subunit (ClpP), leading to enhanced degradation of mitochondrial proteins and impaired tumor cell metabolism.11,14 This observation has reduced support for ONC201’s drug interaction with D2R and has emphasized instead its potential role in targeting disease bioenergetics. In this Review, we focus on the molecular mechanisms of the most well-characterized imipridones ONC201, ONC206 and ONC212. We provide an overview of imipridones’ putative biological targets, highlight data from clinical trials supporting the therapeutic efficacy of these drugs, and note combinatorial approaches in preclinical studies that hold promise for future clinical translation. Finally, we discuss outstanding questions on the molecular mechanisms of imipridones. The encouraging preclinical and clinical findings, which have catalyzed a surge of new clinical trials using ONC201 and the first-in-human ONC206 trial, merit a timely synthesis of the existing literature on the anticancer activity of this novel drug family.
Table 1.
ONC201 Derivativesa
| Properties of imipridone compounds | ||||
|---|---|---|---|---|
| Imipridone | Structure | Properties | Putative targets | Ref |
| ONC201 |
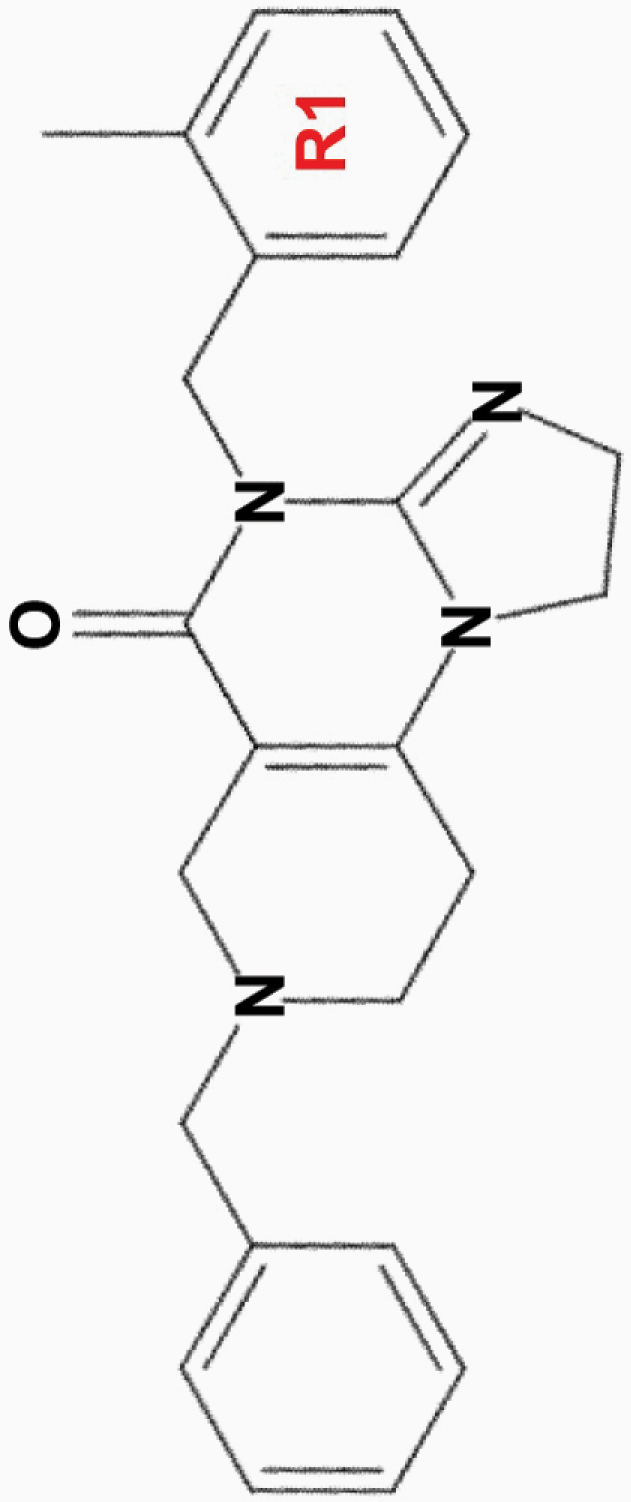
|
• Effective at micromolar (10 µM) concentrations | • Akt, ERK | 1,2,8,10,11,13,15–17 |
| • Anti-proliferative and pro-apoptotic effects | • TRAIL signaling | |||
| • Induces G2/M cell cycle arrest | • D(2) dopamine receptor | |||
| • Inhibits migration and invasion of colon cancer cells in vitro | ||||
| • ClpP | ||||
| • In phase 1/2 clinical trials for CNS, solid and hematologic cancers | • Androgen receptor (AR-V7) signaling (prostate cancer) | |||
|
ONC206
2,4-diF-benzyl |
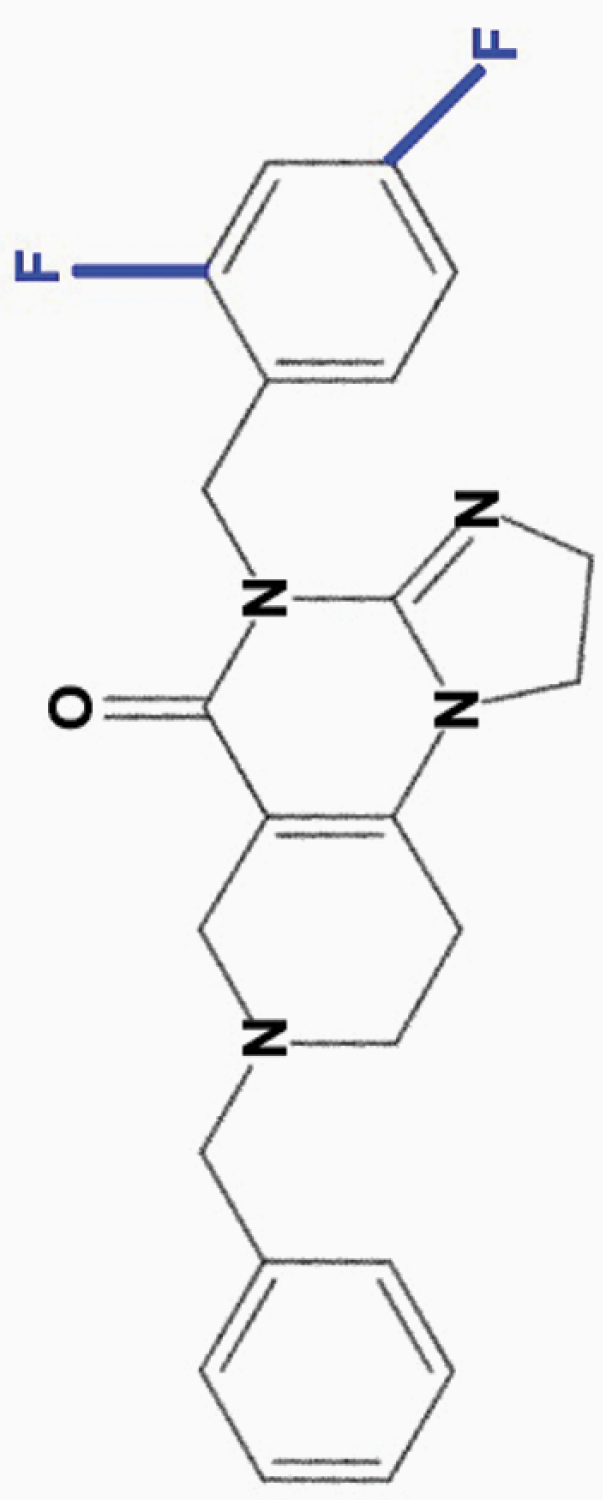
|
• Effective at nanomolar (50 nM) concentrations | 8,10 | |
| • Active against liver, lung, and kidney cancer cell lines | ||||
| • Prolonged efficacy compared to ONC201 and ONC212 | ||||
| • Stronger reduction of ATP levels compared to ONC201 | ||||
| • Inhibits migration and invasion of colon cancer cells in vitro | ||||
| • In phase 1 clinical trial for CNS cancers | ||||
|
ONC212
4-CF3-benzyl |
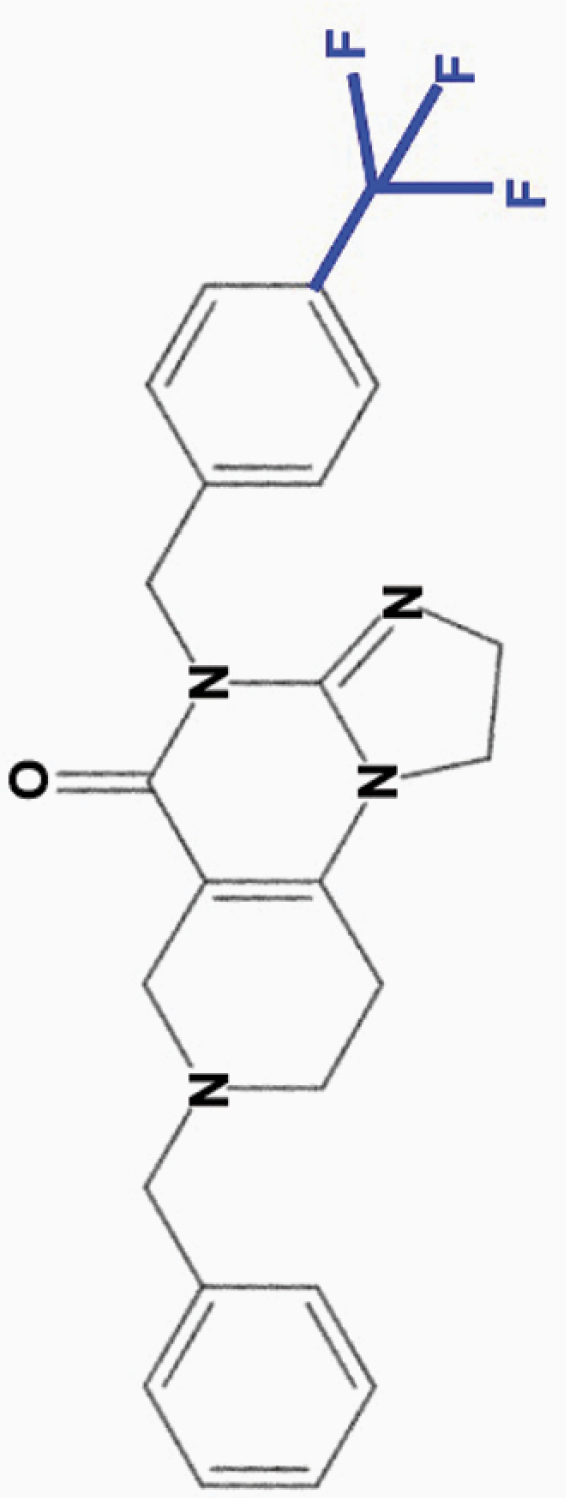
|
• Effective at nanomolar (10 nM) concentrations | • Binds ClpP with higher affinity than ONC201 | 8–12,17 |
| • Wider separation in toxicity between tumor and normal cells compared to ONC201 | ||||
| • Wider therapeutic index than ONC201; active against some, not all, ONC201-resistant cell lines | • Activates GPR132 and Gαq signaling (AML) | |||
| • Highly active against melanoma (including BRAF V600E mutant), prostate, breast, and thyroid cancer cell lines | ||||
| • Faster signaling effects than ONC201 and ONC206 | ||||
| • Shorter half-life in vivo, but prolonged mechanism | ||||
| • Induces cell cycle arrest at both G0/G1 and G2/M transition | ||||
| • Stronger inhibition of OXPHOS and reduction of ATP levels than ONC201 and ONC206 | ||||
| • Inhibits migration, but not invasion, of colon cancer cells in vitro | ||||
|
ONC213
3,4-diF-benzyl |
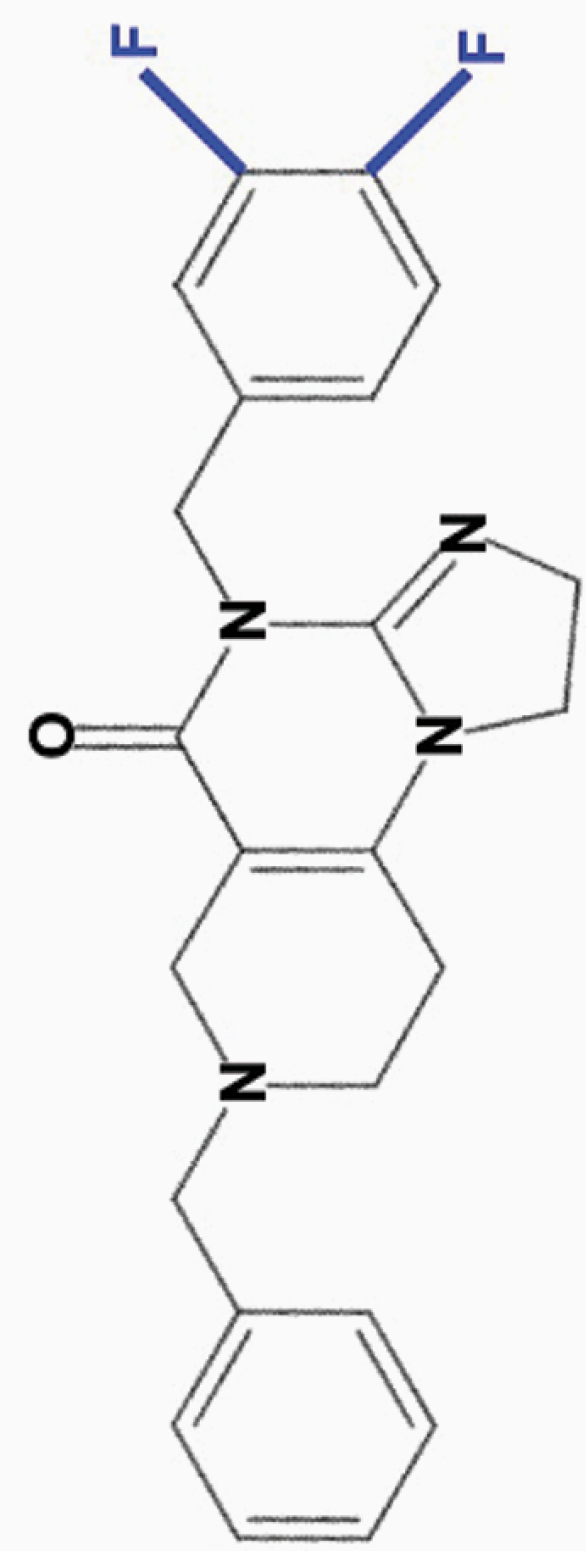
|
• Wide separation in toxicity between tumor and normal cells | 8 | |
|
ONC219
2,4-diCl-benzyl |
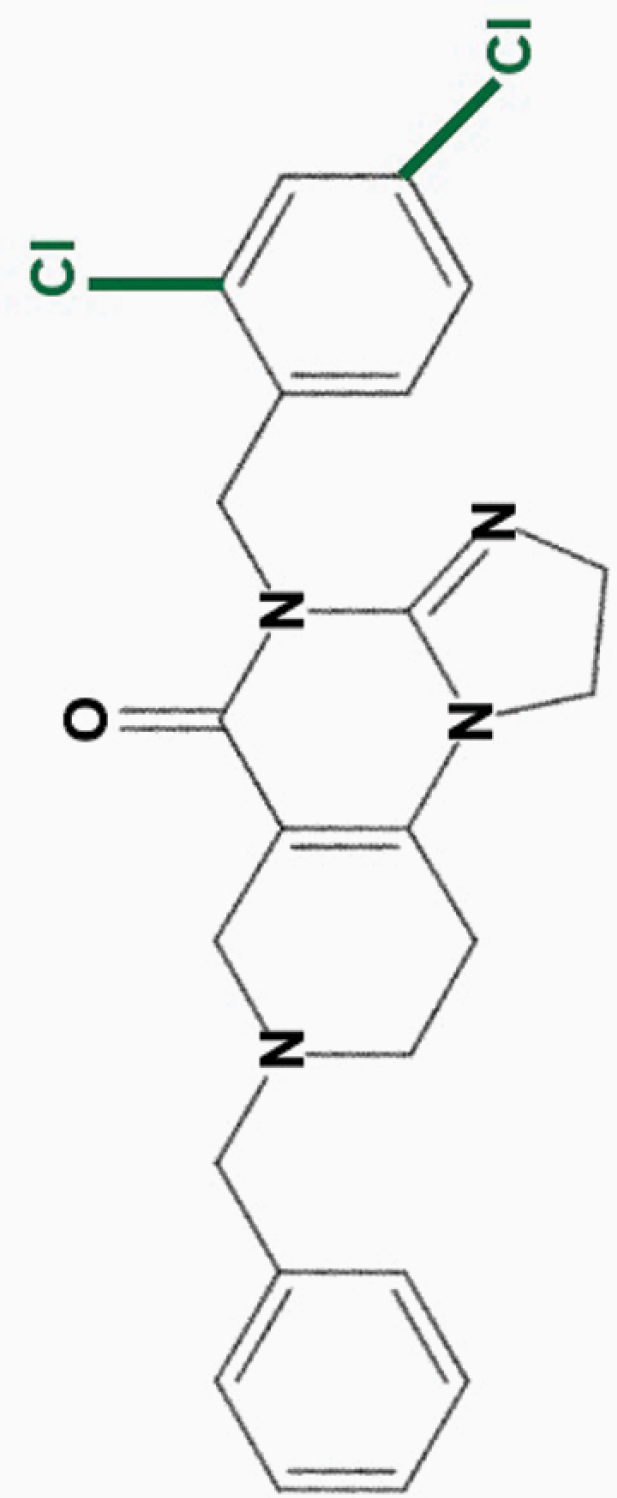
|
• Large therapeutic index in vitro | 8 | |
| • Potent efficacy against colorectal cancer cells | ||||
Abbreviation: Akt, protein kinase B; ERK, extracellular signal regulated kinase; TRAIL, tumor related apoptosis-inducing ligand; ClpP, caseinolytic protease P; ATP, adenosine triphosphate; AML, acute myeloid leukemia; OXPHOS, oxidative phosphorylation.
aONC201 is the parent compound of the imipridone family, and its modified derivatives share its core structure but harbor modifications to their peripheral benzyl moieties.8 Wagner and colleagues identified imipridone compounds harboring halogen (e.g. ONC212) or halide (e.g. ONC206, ONC209 [not shown]) substituents in the R1 position to be more potent than ONC201, and imipridones without a group at the benzyl substituent’s 2-position (e.g. ONC212, ONC213) to exhibit the widest separation between toxicity in tumor and normal cells.8 This table summarizes the properties and candidate biological targets of each imipridone. ONC201 derivatives are thought to share ONC201’s main biological targets, and as such, only additional targets specific to each compound are listed as their putative targets.
Table 2.
Clinical Trials Involving Imipridonesa
| Clinical trials involving ONC201 | |||||
|---|---|---|---|---|---|
| Trial name | Phase, Status | Start – Est. completion date | Patient criteria | Publication of results (doi) | |
| CNS | Oral ONC201 in Recurrent GBM, H3 K27M Glioma, and Midline Glioma (NCT02525692) | 2, R | 1/2016-12/2020 | GBM, DMG | 10.1007/s11060-019-03271-3 |
| Young adult, adult (16+ yrs) | |||||
| Expanded Access to ONC201 for Patients with H3 K27M-mutant and/or Midline High Grade Gliomas (NCT03134131) | TNA | 04/2017 - | H3K27M-mutant glioma, grade III/IV midline glioma, DIPG | ||
| Child, adult, older adult (3+ yrs) | |||||
| ONC201 in Pediatric H3 K27M Gliomas (NCT03416530) | 1, R | 1/2018-1/2023 | DIPG, malignant glioma | ||
| Pediatric (2–18 years) | |||||
| ONC201 in Adults With Recurrent H3 K27M-mutant Glioma (NCT03295396) | 2, R | 10/2017-12/2022 | HGG, H3K27M-mutant | ||
| Adult (18+ yrs) | |||||
| A Phase II, Open Label Study of ONC201 in Adults with EGFR-low Glioblastoma (NCT04629209) | 2, NYR | 12/2020-12/2023 | EGFR-low glioblastoma | ||
| Adult (18+ yrs) | |||||
| A First-in-human Phase I Single-agent Dose-escalation, Food Effect and Dose Expansion Study of Oral ONC206 in Recurrent and Rare Primary Central Nervous System Neoplasms (NCT04541082) | 1, R | 9/2020-02/2025 | CNS neoplasms | ||
| Adult (18+ yrs) | |||||
| Solid | Oral ONC201 in Treating Patients With Advanced Solid Tumors (NCT02250781) | 1, C | 1/2015-10/2018 | Solid tumors | 10.1186/s40425-019-0599-8 |
| Adult (18+ yrs) | |||||
| Continuation of Oral ONC201 in Treating Patients With Advanced Solid Tumors (NCT02324621) | 1, C | 2/2015-10/2018 | Solid tumors | 10.1186/s40425-019-0599-8 | |
| Adult (18+ yrs) | |||||
| ONC201 in Recurrent or Metastatic Type II Endometrial Cancer Endometrial Cancer (NCT03485729) | 2, R | 3/2018-3/2021 | Recurrent endometrial cancer (female), Adult (18+ yrs) | ||
| Single Agent ONC201 in Recurrent or Metastatic Endometrial Cancer (NCT03099499) | 2, R | 6/2017-10/2022 | Endometrial cancer (female) Adult (18+ yrs) | ||
| BrUOG 379 Phase Ib/II Trial ONC201 + Nivolumab in MSS mCRC (379) (NCT03791398) | 1/2, R | 11/2019-12/2021 | Metastatic colorectal cancer Adult (18+ yrs) | ||
| Phase 2 Study of ONC201 in Neuroendocrine Tumors (NCT03034200) | 2, R | 8/2017–9/2020 | Recurrent or metastatic neuroendocrine tumor | ||
| Adult (18+ yrs) | |||||
| ONC201 in Recurrent/Refractory Metastatic Breast Cancer and Advanced Endometrial Carcinoma (NCT03394027) | 2, R | 1/2018-12/2027 | Hormone receptor positive breast cancer (Cohort 1), TNBC (Cohort 2), Endometrial cancer (Cohort 3) | ||
| Adult (18+ yrs) | |||||
| ONC201 and Paclitaxel in Treating Patients with Platinum-Resistant Refractory or Recurrent Epithial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer (NCT04055649) | 2, NYR | 9/2019-6/2020 | Epithelial ovarian, fallopian tube, or primary peritoneal cancer, refractory disease; female; Adult (18+ yrs) | ||
| ONC201 With and Without Methionine-Restricted Diet in Patients With Metastatic Triple Negative Breast Cancer (NCT03733119) | 2, R | 11/2018-10/2021 | TNBC | ||
| Female, adult (17–64 yrs) | |||||
| Mixed | A Dose-Escalation Study of Onc201 Administered Every One or Three Weeks in Advanced Solid Tumors and Multiple Myeloma (NCT02609230) | 1, R | 11/2015-1/2022 | Advanced solid tumors, multiple myeloma | |
| Adult (18+ yrs) | |||||
| Hematologic | Akt/ERK Inhibitor ONC201 in Treating Patients With Relapsed or Refractory Non-Hodgkin’s Lymphoma (NCT02420795) | 1/2, R | 11/2015-11/2021 | Lymphoma | |
| Adult (18+ yrs) | |||||
| ONC 201 Maintenance Therapy in Acute Myeloid Leukemia and Myelodysplastic Syndrome After Stem Cell Transplant (NCT03932643) | 1, R | 7/2019-7/2024 | AML, myelodysplastic syndromes; | ||
| Adult (19+ yrs) | |||||
| ONC201 in Relapsed/Refractory Acute Leukemias and High-Risk Myelodysplastic Syndromes (HR-MDS) (NCT02392572) | 1/2, ANR | 11/2018-11/2023 | Leukemia | ||
| Adult (18+ yrs) | |||||
| Oral ONC201 in Relapsed/Refractory Multiple Myeloma (NCT02863991) | 1/2, ANR | 1/2016-12/2021 | Multiple myeloma, relapsed/refractory | ||
| Adult (18+ yrs) | |||||
Abbreviations: DMG, midline glioma; DIPG, diffuse intrinsic pontine glioma; EGFR, epidermal growth factor receptor; TNBC, triple-negative breast cancer; AML, acute myeloid leukemia; HGG, high grade glioma; GBM, glioblastoma multiforme; CNS, central nervous system.
aThe table summarizes the current state of clinical trials involving the lead imipridones ONC201 (n = 19 clinical trials) and ONC206 (n = 1). Included are trials that are recruiting (R, n = 13), temporarily not available (TNA) (n = 1), completed (C, n = 2), not yet recruiting (NYR, n = 2), or active and not recruiting (ANR, n = 2), as registered on Clinicaltrials.gov. Trials are arranged by cancer type, and the trial phase, status, study start/estimated completion dates, and patient criteria are listed.
Imipridones and cancer
ONC201 was discovered as an anticancer agent in 2013 in a bioluminescence reporter screen for inducers of TRAIL,1 and was thus named “TRAIL-inducing compound 10” (TIC10).1 ONC201 achieves sustained upregulation of TRAIL across a multitude of human cancer cell lines and induces apoptosis in malignant cells without affecting healthy cells.1,2 ONC201’s structure was first reported incorrectly as imidazo[1,2-a]pyrido[4,3-d]pyrimidine derivative based on mass spectrometry1 but was soon corrected to be an angular [3,4-e], rather than linear [4,3-d], isomer,18 confirmed by NMR and X-ray structural analysis.19 The angular structure seems to be highly essential for ONC201’s activity.18,19
Antagonism of G-protein coupled receptors
Imipridones selectively antagonize the D(2) dopamine receptor (D2R, encoded by DRD2),13,15 a GPCR that is widely expressed across cancers.13,20,21DRD2 expression correlates with ONC201 response in cancers including glioblastomas.5,13,20 However, this receptor is not essential for ONC201 sensitivity: CRISPR/Cas9-mediated knockout of DRD2 in colorectal and breast cancer cell lines does not abrogate ONC201 response.13 Modified derivatives of ONC201 may have additional GPCR targets beyond D2R; for example, ONC212 selectively activates the GPCR GPR132, which is highly expressed in leukemias and lymphomas, and in turn activates Gαq signaling in acute myeloid leukemia (AML) preclinical models.12 A list of imipridones and their putative GPCR targets is provided in Table 1.
Induction of TRAIL-mediated apoptosis
ONC201 is thought to induce TRAIL signaling by reducing levels of phosphorylated protein kinase B (Akt) and extracellular signal regulated kinase (ERK) and, in turn, phosphorylation of their target transcription factor Foxo3a.1 Through the proposed pathway, ONC201 treatment leads to dephosphorylation and subsequent translocation of Foxo3a to the nucleus, where Foxo3a binds the TRAIL promoter to induce p53-independent TRAIL expression.1 TRAIL protein then localizes to the cell membrane and binds to death receptor 4 (DR4) or 5 (DR5), activating the extrinsic pathway of apoptosis involving the initiator caspase-8.22 Indeed, ONC201 induces TRAIL upregulation in vitro across cancer cell lines including CNS,1 lung,1,23 breast, ovarian, prostate,1,2 colorectal,24 and lymphoma25 cells, without affecting healthy fibroblasts.1 In vivo, ONC201 upregulates TRAIL and induces tumor regression in colon and triple-negative breast cancer (TNBC) models, and crosses the intact blood-brain barrier (BBB) of CNS tumor-bearing mice to significantly prolong survival.1 In support of TRAIL signaling as ONC201’s target, Foxo3a knockdown inhibits TRAIL upregulation and apoptosis in ONC201-treated colon cancer cells, and TRAIL and DR5 are required for ONC201 sensitivity in breast adenocarcinoma1 and lung cancer cells.1,23 In addition, TRAIL and caspase-8 expression increase in uterine serous carcinoma cell lines treated with ONC201.26 However, in vitro kinase activity assays revealed no direct interaction between ONC201 and Akt or ERK,1 and the precise mechanism by which imipridones activate TRAIL signaling is uncertain.
Recent studies have cast doubt on TRAIL signaling as ONC201’s primary targeted pathway. These studies revealed that ONC201 elicits apoptosis in the absence of Akt/ERK inhibition27 and TRAIL or DR5 upregulation in breast,27 pancreatic,9 prostate,16 hematologic cancer,11,28,29 and glioblastoma30 cells. In fact, in contrast to the observed upregulation of TRAIL in solid tumors,1 TRAIL upregulation is limited or absent following imipridone treatment in most hematologic cancer cells.25,28 Indeed, TRAIL-resistant, caspase-8-deficient leukemic T cells are equally sensitive to ONC201-induced apoptosis as their wild-type counterparts.28 ONC201 treatment does not affect Akt/ERK/Foxo3a or TRAIL levels in hematologic cancer cell lines, and Foxo3a knockdown fails to diminish drug sensitivity in AML cells.28 In pediatric non-Hodgkin’s lymphoma, although imipridone treatment upregulated TRAIL and DR5, antibody-mediated sequestration of TRAIL only partially inhibited apoptosis,25 indicating a limited role for TRAIL.
Similar observations have been reported in solid cancers. In prostate cancer cells, 4 of 5 cell lines tested did not show significant TRAIL upregulation, and there was no correlation between TRAIL mRNA levels and ONC201 sensitivity.16 TRAIL-resistant TBNC cells remain sensitive to ONC201,31 and only modest and insignificant changes to TRAIL, DR4 and DR5 levels occur in ONC201-treated breast adenocarcinoma cells.27 Breast and endometrial cancer cell lines treated with ONC201 undergo apoptosis in the presence of the pan-caspase inhibitor Z-VAD-FMK, indicating caspase-independent cell death.27 Knockdown of DR5 or caspase-8, or treatment with caspase-8 inhibitor (Z-IETD-FMK), does not abrogate glioblastoma cell death,30 further suggesting the absence of extrinsic, caspase-8-mediated apoptosis. Moreover, only half of the patients with advanced solid tumors treated with ONC201 exhibited a modest (20%) increase in serum TRAIL.3 Together, these data indicate that while imipridones may induce TRAIL in certain cancers, their anticancer activity is not entirely dependent on TRAIL activation. Below, we expand on alternative mechanisms of drug action.
Activation of the mitochondrial Clp protease
Imipridones are potent agonists of the mitochondrial Clp protease proteolytic subunit (ClpP)11,14 (Fig. 1). ClpP localizes to the mitochondrial matrix and is essential for the normal turnover of mitochondrial proteins, including mitochondrial ribosomal subunits (e.g. Era like 12S mitochondrial rRNA chaperone 1 [ERAL1]) and metabolic enzymes such as electron transport chain (ETC) components.32,33 ClpP activity is tightly regulated by its chaperone protein ClpX, which recognizes and unfolds specific proteins, then feeds them into ClpP’s proteolytic chamber for degradation.34 Several cancers overexpress ClpP, including AML (45% of cases).35 ClpP over-activation using natural acyldepsipeptides (ADEPs) results in impaired mitochondrial oxidative phosphorylation (OXPHOS) and intrinsic cell death, and has been proposed as an anticancer strategy.36,37
Fig. 1.

Anticancer mechanism of imipridones. Data from the existing literature indicates that imipridones exert their anticancer effects primarily by binding to and potently activating the mitochondrial Clp protease proteolytic subunit ClpP, causing ClpP to lose its dependence on the chaperone protein ClpX (Step 1). Hyper-active Clp protease then depletes its target substrates including components of the respiratory complex chain, most strongly complex I and II proteins (Step 2). In turn, OXPHOS is impaired and cellular ATP is depleted (Step 2). Mitochondrial structural damage and distress occurs, concomitant with the state of energy deprivation leading to integrated stress response (ISR) activation (Step 3). The ISR is relayed to the nucleus through an undefined mechanism, involving the typical (phospho-eIF2α-dependent) or atypical (phospho-eIF2α-independent) pathway. ISR activation causes global translational attenuation, including reduced levels of cyclin D1 leading to cell cycle arrest (Step 4A). In conditions of prolonged stress, ATF4 and CHOP are upregulated, and together these transcription factors increase the expression of their target genes including GADD34, which promotes further protein synthesis and stress (thus further activating the ISR); the TRAIL receptor DR5, which can promote TRAIL-mediated extrinsic cell death; and pro-apoptotic Bcl-2 family proteins, which promote the intrinsic, mitochondrial cell death program (Step 4B). Created with BioRender.com.
A screen for ClpP activators identified imipridones (ONC201 and ONC212) as potent activators of ClpP.11 Imipridones non-covalently bind to ClpP at the interface with ClpX, as demonstrated by isothermal titration calorimetry and co-crystallization studies.11 ONC201 molecules occupy the hydrophobic pockets between adjacent ClpP subunits, involving extensive hydrophobic contact and hydrogen bonding.11 The precise interactions between ONC201 and ClpP induce the opening of ClpP’s axial entrance pore, which basally is opened only under the regulation of ClpX.11 ONC201 causes ClpP’s entrance pore radius to enlarge (from 12 to 17 Å), its N-terminal residues to exhibit increased dynamics, and its active site to change conformation, altering the placement of its catalytic triad residues.11 As a result, imipridones activate ClpP in the absence of ClpX.11 Compared to ONC201, ONC212 interacts with even higher affinity and structural complementarity to ClpP.11 ONC212’s highly electronegative p-CF3-benzyl substituent extends into ClpP’s apolar pocket and may form additional multipolar bonds with the protease.11 The precise binding properties between ClpP and other imipridones have yet to be characterized.
In imipridone-treated glioblastoma cells, ClpP significantly depletes nearly half of its target mitochondrial proteins, including strong depletion of ETC components11 explaining the resulting OXPHOS impairment38 and decrease in enzymatic activity of respiratory chain complexes I, II, and IV.11 In breast cancer cells, treatment reduces levels of mitochondrial transcription factor A, mitochondrial (TFAM) and Tu translation elongation factor, mitochondrial (TUFM), an effect that is abolished by ClpP knockdown.14 Imipridone treatment also depletes mitochondrial proteins including ClpX (which regulates several other proteins in addition to ClpP), the PAM complex component GRPEL1, ribosomal subunits MRPS7/MRPS22, and metabolic and detoxifying enzymes.17 While mitochondrial proteins are the hardest hit by ClpP hyper-activation, non-mitochondrial proteins are also targeted, including regulators of cell division and cytokinesis (e.g., aurora kinase A [AURKA], cyclin D3 [CCND3], cell division cycle 20 [CDC20]),17 indicating broad and destructive effects of ClpP agonism both within and beyond the mitochondria.
Consistent with the hyper-degradation of mitochondrial proteins, imipridones induce mitochondrial structural damage in AML11 and breast cancer cells by 3 h post treatment.27 Severe swelling, matrix lysis, cristae membrane disruption, and disintegration are evident within 6 h,27 and by 24 h, mitochondrial fragmentation and fission occur in breast cancer cells.27 Structural damage to the mitochondria is accompanied by decreased oxygen consumption rate (OCR), ATP, mtDNA, and levels of mitochondrial-encoded genes and nuclear-encoded mitochondrial genes involved in OXPHOS; as well as increased mitochondrial ROS.11,27
Targeting of mitochondrial bioenergetics
Metabolic reprogramming is a hallmark of cancer cells, and targeting of metabolic abnormalities presents a therapeutic opportunity. While cancer cells have long been thought to depend primarily on glycolysis for energy production even in the presence of oxygen (known as the Warburg effect39), it is now clear that many cancer cells upregulate mitochondrial OXPHOS for adenosine triphosphate (ATP) production.40 By hyper-activating ClpP, imipridones disrupt OXPHOS and induce a state of energy deprivation in cancer cells. Some studies have indicated that imipridones suppress both OXPHOS and glycolysis, given an observed reduction of extracellular acidification rate (ECAR, a marker of glycolysis), reduction of glycolysis-related proteins (e.g., HK2, LDH1, GLUT1), and accumulation of glycolytic metabolites in imipridone-treated glioblastoma cells.10 Importantly, treatment with ONC201, ONC206 or ONC212 significantly reduces levels of OXPHOS complexes, basal OCR, ATP, and maximal respiration in glioblastoma cells, indicating strong suppression of OXPHOS.10 Mitochondrial DNA (mtDNA) copy number levels and mitochondrial membrane potential are also depleted.10 Other findings indicate that imipridones specifically target mitochondrial OXPHOS:
First, breast cancer cells grown in glucose-containing medium and treated with ONC201 show a reduction in OCR and ATP levels, indicating suppression of OXPHOS; but also show a slight increase in ECAR, suggesting a compensatory shift towards glycolysis27 rather than suppression of both pathways. However, when cells are grown in galactose-containing medium, a stronger inhibition of OCR is observed, without a compensatory increase in ECAR,27 given that the cells cannot metabolize galactose without functioning OXPHOS machinery. Pruss and colleagues showed that imipridone-treated cancer cells strongly downregulate respiratory chain complex I and II proteins, concomitant with OCR downregulation and increased ECAR,38 again demonstrating a shift from OXPHOS to glycolysis. Importantly, cells that do not depend on mitochondrial respiration (e.g. cancer cells with reduced mtDNA quantities and those with fumarate hydratase deficiency) are ONC201-resistant.27 Together, these findings support a mechanism by which imipridones specifically target OXPHOS.
Second, the transition from OXPHOS to glycolysis renders cells vulnerable to glucose depletion: when glucose levels are decreased, imipridone treatment significantly reduces glioblastoma cell viability.38 This effect is further exacerbated when combining ONC201 with the glucose analog and glycolysis inhibitor 2-deoxyglucose (2-DG), a combination that reduces cell viability, depletes ATP and induces G2/M arrest.38 With the exception of a few protein kinases (e.g., AMPKα1, which is phosphorylated upon ATP depletion), ONC201 plus 2-DG treatment results in a state of hypo-phosphorylation across major kinases including mTOR, EGFR and PDGFRβ, as well as Akt and ERK38—the first kinases implicated in imipridone response1—indicating global downregulation of energy metabolism rather than targeting of a specific pathway (e.g., Akt/ERK-Foxo3a-TRAIL signaling).
Third, in response to imipridone treatment, cells upregulate a compensatory energy production pathway, the serine one-carbon cycle, glycine synthesis (SOG) pathway,10,38 including the enzymes phosphoglycerate dehydrogenase (PHGDH) and phosphoserine aminotransferase 1 (PSAT1). Moreover, ONC212 synergizes with the PDGDH inhibitor (NCT-503 and CBR-5884) to enhance apoptosis in glioblastoma and colon carcinoma cells in vitro, and reduce tumor size in in vivo models.10 These findings provide support for coupling imipridones with drugs that inhibit alternate metabolic pathways, to effectively target tumor cell metabolism.
ISR pathway activation
Imipridone treatment induces gene expression profiles consistent with the unfolded protein response (UPR) and integrated stress response (ISR) activation, mainly by upregulating the expression of the activating transcription factor-4 (ATF4).41 ISR is a protective mechanism in response to stressors including nutrient deprivation and misfolded protein aggregation.42,43 ATF4, the main mediator of the ISR, plays a protective role by upregulating the expression of genes that restore cellular homeostasis.44 ISR activation may follow a typical (phospho-eIF2α-dependent) or atypical (phospho-eIF2α-independent) pathway. In typical ISR, the serine/threonine kinases GCN2, PKR, HRI, or PERK are activated by specific stressors and phosphorylate the eukaryotic translation initiation factor-2-alpha (eIF2α) protein.42 eIF2α phosphorylation induces global translational attenuation, including reduced cyclin D1 levels causing G1/S cell cycle arrest.45 Only certain mRNAs, including ATF4, ATF5, CCAAT-enhancer-binding protein homologous protein (CHOP), and growth arrest and DNA-damaging inducible (GADD34), are preferentially translated to protein.45,46
Cancer cells may leverage the protective effects of the ISR to facilitate survival during conditions of stress associated with rapid growth, proliferation and hypoxia, and to evade programmed cell death.45,46 However, during prolonged or severe stress, ATF4 induces apoptosis by upregulating the transcription factor CHOP. ATF4 and CHOP bind to promoters of genes involved in protein synthesis (e.g., tRNA synthetases, translation initiation factors) and the UPR.47 In addition, CHOP upregulates GADD34 thereby promoting eIF2α dephosphorylation. Together, these changes increase protein synthesis, exacerbate cell stress, and ultimately trigger apoptosis.36,38 CHOP also downregulates antiapoptotic B-cell lymphoma 2 (Bcl-2) family members and upregulates pro-apoptotic proteins, as well as the TRAIL receptor DR5, to promote apoptosis.45
Indeed, imipridone treatment upregulates ATF4 and CHOP across cancer cell lines.8,28,41 CHOP upregulation increases the expression of target genes including GADD34 and DR5.28 Interestingly, imipridones can activate either the typical or atypical ISR in a cell type-specific manner. Typical ISR pathway activation is observed in preclinical models of AML,28 colorectal,41 and breast31 cancers. In contrast, in MCL41 and cutaneous T-cell lymphoma (CTCL) cells,48 imipridone treatment activates ATF4 via the atypical, phospho-eIF2α-independent ISR. Ishizawa and colleagues speculated that imipridones may do so by inhibiting eIF2B, which can induce ATF4 upregulation, or by inhibiting proteasomal degradation of ATF4 through a yet-undefined mechanism.28 Notably, ATF4 activation can be triggered by mitochondrial stress,49,50 as described in the next section.
Downstream targets of ISR activation
Imipridones agonize ClpP, leading to hyper-degradation of mitochondrial proteins, OXPHOS impairment, and ISR induction mediated by ATF4/CHOP (Fig. 1). However, the exact process by which mitochondrial distress relays to the nucleus to trigger the ISR is uncertain. Mitochondrial distress may trigger the atypical ISR independent of eIF2α phosphorylation,49 though this process is not fully understood. Another plausible pathway involves activation of the stress-induced mitochondrial metalloendopeptidase OMA1, which cleaves the inner mitochondrial membrane protein DAP3-binding cell death enhancer 1 (DELE1) leading to cytosolic accumulation of DELE1.50 DELE1 interacts with the eIF2α kinase HRI, which phosphorylates eIF2α triggering the ISR and ATF4 upregulation.50 Alternatively, inhibition of ATP synthases leading to mitochondrial inner-membrane hyperpolarization can induce the ISR, through an incompletely defined mechanism.51 The ability of mitochondrial distress to evoke the typical or atypical ISR may explain why both pathways have been observed in imipridone-treated cells.
Imipridone treatment culminates in the intrinsic, mitochondrial pathway of apoptosis in many cancer types. This process is regulated by Bcl-2 proteins that control mitochondrial permeability and the release of cytochrome c into the cytosol. The intrinsic apoptotic pathway involves the initiator caspase-9 and executioner caspases-3 and -7.52 In some pancreatic cancer cells, imipridone treatment induces both the extrinsic, TRAIL/caspase-8-dependent, and the intrinsic, caspase-9-dependent apoptotic pathways.53 In MCL cells, treatment upregulates pro-apoptotic Bcl-2 proteins Bax, Bak, Puma and Bim,28 and knockdown of Bax and Bak inhibits apoptosis in glioblastoma cells,30 supporting an intrinsic apoptotic mechanism. In myeloma cells, imipridone treatment upregulates Bim, whereas Bim deficiency reduces drug sensitivity.29 ONC212-treated AML cells downregulate the anti-apoptotic Bcl-2 protein MCL-1, and undergo apoptosis involving a reduction of mitochondrial membrane potential.12 TRAIL/DR5 activation is not observed, but rather cell death is caused by the intrinsic mitochondrial pathway, as evidenced by upregulation of cleaved caspase-9, -3, and PARP.29
Putative biomarkers of response to imipridones
We have listed several possible predictive biomarkers of imipridone response in Table 3. To further investigate the role of candidate biological targets in modulating sensitivity to imipridone treatment, we reanalyzed a newly generated large-scale cell viability drug screening dataset.54 This study conducted a screen of 4,518 drugs across 578 genetically characterized human cancer cell lines representing 34 different cancer types from the Cancer Cell Line Encyclopedia. Our re-analysis of 539 cell lines ranked CLPP as the primary predictor of ONC201 sensitivity at pan-cancer level (Fig. 2A). More specifically, cell lines with high CLPP expression levels responded best to ONC201, whereas cancer cell lines with low CLPP expression levels responded weakly to ONC201 (Fig. 2B). The association between ONC201 sensitivity and CLPP expression remained fully consistent within cancer types (Fig. 2D). In contrast, DRD2 expression levels demonstrated limited correlation with ONC201 sensitivity across and within cancer types (Fig. 2C, D). Notably, cancer cell lines without any detectable DRD2 expression retained the same ONC201 sensitivity profile as compared to cell lines with detectable DRD2 expression (Supplementary Figure 1), further diminishing the role of D2R antagonism in imipridones’ anticancer mechanism.
Table 3.
Candidate Biomarkers of Imipridone Sensitivitya
| Candidate biomarkers of imipridone sensitivity | ||
|---|---|---|
| Biomarker | Effect on response | Ref. |
| Bim | Bim depletion impaired ONC201-induced apoptosis in myeloma cells | 29 |
| BIP | High expression of the ER chaperone BIP was associated with ONC201 and ONC212 resistance in pancreatic cancer cells | 9 |
| CLPP | • CLPP gene was essential for ONC201 and ONC212 sensitivity in leukemia cells | 11,17 |
| • CLPP D190A mutation abrogated response to ONC201 and ONC212 in AML and colorectal cancer cells; the mutation causes loss of the negative charge at ClpP’s active site and diminishes ONC201-ClpP binding affinity | ||
| • Pretreatment CLPP expression levels correlated to ONC201 sensitivity in AML primary samples | ||
| c-Myc | Higher c-Myc levels correlated to stronger apoptosis induction in ONC201, ONC206, and ONC212-treated glioblastoma cells | 10 |
| D(2) dopamine receptor (DRD2) | • High DRD2 expression was associated with ONC201 sensitivity in hepatocellular carcinoma cell lines | 3,13 |
| • Serum prolactin induction (indicative of D2R antagonism) was reported in most (n=22/25) patients receiving ONC201 | ||
| Dopamine receptor D5 (DRD5) | • DRD5 Knockdown increased ONC201 sensitivity in colorectal cancer cells, and pharmacological inhibition increased ONC201 sensitivity in neuroblastoma and glioblastoma (but not colorectal cancer) cells | 5,13,20 |
| • DRD5 overexpression or gain-of-function (Q366R mutation) reduced ONC201 sensitivity in glioblastoma cells | ||
| • DRD5 expression levels inversely correlated to ONC201 sensitivity, and low DRD5 expression correlated to improved clinical outcome in patients with glioblastoma | ||
| EGFR | • EGFR levels inversely correlated to ONC201 response, and ectopic expression of EGFR (constitutively active EGFRvIII) conferred ONC201 resistance in glioblastoma cell lines | 55 |
| • Low EGFR expression in pre-treatment clinical specimens correlated to improved progression free and overall survival outcomes among glioblastoma patients treated with ONC201 | ||
| GPR132 | High GPR132 levels correlated to increased ONC212 sensitivity in AML cells, independent of ISR induction | 12 |
| IGF1-R | High IGF1-R levels correlated to increased ONC201 and ONC212 sensitivity in pancreatic cancer cells | 9 |
| MIPEP | MIPEP gene was essential for ONC201 and ONC212 sensitivity in leukemia cells | 17 |
| mtDNA | • Reduced mitochondrial DNA (mtDNA) quantity was associated with ONC201 resistance in breast cancer cells | 10,27 |
| • mtDNA copy number decreased in ONC201-treated glioblastoma cells | ||
| mTOR | Knockdown or inactivating mutations were associated with increased ONC201 sensitivity in colorectal cancer cells | 56 |
| XIAP | Persistent levels of X-linked inhibitor of apoptosis (XIAP), which inhibits TRAIL, was associated with ONC201 resistance in colon cancer cells | 41 |
Abbreviations: ER AML, acute myeloid leukemia; EGFR, epidermal growth factor receptor MIPEP, mitochondrial intermediate peptidase; XIAP, X-linked inhibitor of apoptosis; mTOR, Mammalian Target of Rapamycin; BIP, binding immunoglobulin protein; CLPP, caseinolytic mitochondrial matrix peptidase proteolytic subunit.
aThe table lists examples of biomarkers that may be associated with imipridone sensitivity in various cancer types.
Fig. 2.

Comprehensive cell viability screen of human cancer cell lines identified ClpP as the primary predictor of ONC201 response. Correlation between cell viability at 2.5 μM ONC201 and gene expression levels (log2 TPM) across 539 human cancer lines from the Cancer Cell Line Encyclopedia (A). Scatter plots and boxplots show ONC201 sensitivity compared to gene expression levels for CLPP (B) and DRD2 (C). Box plots show Spearman’s ρ between ONC201 sensitivity and gene expression levels for 16 individual cancer types. Calculations are based on 11 (rhabdoid) to 106 (lung) cancer cell lines (D). All gliomas among brain cancer cell lines are IDH-wild type. Primary drug screening data for human cancer cell lines was derived from the PRISM Repurposing Screen (19Q3)54 and gene expression data from the Cancer Dependency Map (20Q3).57 Spearman’s ρ and -log10P values are shown. Boxplots show the first quartile, median value, third quartile, and minimum/maximum (whiskers) of the data. Blue lines show linear regression results between ONC201 sensitivity and gene expression levels. Classification of gene expression levels into low/high is based on median. DMSO, dimethylsulfoxide.
These findings are consistent with recently published work by Jacques and colleagues, wherein a genome-wide CRISPR/Cas9 gene knockout screen in human pre-B cell lymphocytic leukemia cells revealed that CLPP and the mitochondrial intermediate peptidase MIPEP, which regulates ClpP maturation, are essential genes for ONC201 and ONC212 sensitivity.17 Again, DRD2 was not an essential gene for response, nor were other candidate targets including genes encoding TRAIL, DR5, and GPR132.17 Other resistance-associated genes encode primarily mitochondrial proteins (e.g. ETC components, mitochondrial transcription and translation factors),17 reinforcing the key role for mitochondrial function in defining cancer cell sensitivity to imipridones. Additional mitochondrial biomarkers, including mtDNA copy number, could also serve as predictors of response, under the assumption that the ISR is conditional on the availability of mitochondria in the tumor cell.
Effect on immune signaling
The role of imipridones in immune signaling is another area of active investigation. In CTCL cell lines, imipridones downregulate the pro-inflammatory cytokine IL-32.48 In colorectal cancer mouse models, imipridone treatment induces activation and accumulation of CD3+, CD4+, and CD8+ T cells and NK cells in tumors, and CD3+ and NK cells in the blood and spleen.58 Treatment also increases NK and CD3+ T cells in the blood of healthy, non-tumor-bearing mice, and cells that are resistant to ONC201 in vitro become more sensitive in in vivo models.58 These findings suggest that ONC201 treatment may induce an immune response, perhaps independent of the other actions described (e.g. OXPHOS impairment). Depletion of NK cells, but not T cells, affects imipridone response.58 NK cells may act by secreting TRAIL, and indeed treatment with the TRAIL-sequestering antibody RIK-2 reduces, but does not abolish, the cytotoxic effect of ONC201,58 indicating that the drug’s effect on NK activity expands beyond TRAIL secretion.
In clinical trials, granzyme B+ and CD56+ cells, suggestive of infiltrating NK cells, were detected in an on-treatment lymph node biopsy from a metastatic prostate cancer patient receiving ONC201.59 Serum immune cytokine and effector molecule profiling revealed a strong immune cytokine induction during the first 2 treatment cycles, followed by a strong effector induction after the second cycle.59 Stronger immune responses, defined as >50% serum perforin induction (a component of CTLs and NK cells), correlated to prolonged progression free survival (PFS).59 The first patient with MCL to receive ONC201 underwent a rectal biopsy after 6 months of treatment, revealing an increase in CHOP, CD45+ lymphocytes, and CD8+ T cells, but not NK or granzyme B+ cells.60 More studies are needed to understand the immune responses evoked by imipridones across cancer types, and clinical trials (Table 2) are incorporating the assessment of immune responses, including evaluation of NK cell and cytokine profiles, into their outcome measures.
Drug interactions
Imipridones have been tested in combination with numerous FDA-approved small molecule drugs in preclinical studies (Table 4), and several promising drug combinations have emerged, some of which have entered clinical trials (Table 2). For example, ONC201 shows increased anticancer efficacy when coupled with Bcl-2 inhibitors in glioblastoma models.30 Bcl-2 overexpression may protect against ISR signaling and imipridone-induced cell death, as shown in leukemia and lymphoma cells.28 Combining ONC201 with the Bcl-2 antagonist ABT-199 increases apoptosis in AML cells, including in high Bcl-2 expressing cells and in cells that are resistant to both ONC201 and ABT-199 as single agents.28
Table 4.
Combinatorial Treatment with Imipridones
| Imipridone | Drug | Results | Ref. |
|---|---|---|---|
| ONC201 | Everolimus (mTOR inhibitor) | • Strongly synergized to induce apoptosis in prostate cancer cells in vitro, and potently reduced tumor growth in vivo | 16 |
| • Reduced phosphorylation of mTOR downstream targets S6 and 4EBP1 | |||
| ONC201 | AZD-8055 (mTOR inhibitor) | • AZD-8066 sensitized colorectal cancer cells to ONC201 cytotoxicity | 56 |
| • Combination treatment increased caspase-8 activation and apoptosis | |||
| ONC201 | Sorafenib (multi-kinase inhibitor) | • Increased apoptosis induction in certain hepatocellular carcinoma cell lines (HepG2) | 61 |
| • Increased tumor regression in HepG2 mouse models without toxicity or weight loss | |||
| ONC201 | Gemcitabine (DNA synthesis inhibitor) | • ONC201 sensitized pancreatic cancer cells to gemcitabine-induced growth inhibition and apoptosis induction in vitro | 53 |
| • ONC201 and gemcitabine synergized in vivo to inhibit tumor growth and improve survival in pancreatic cancer mouse models | |||
| ONC201 | OTX015 (bromodomain BRD2-4 inhibitor, c-myc antagonist) | • Combination treatment increased apoptosis in glioblastoma cells | 10 |
| ONC201 | Cytarabine (anti-metabolic agent) | • Synergized to increase cytotoxicity in pediatric non-Hodgkin’s lymphoma cells | 24 |
| • Combination is in phase 1/2 clinical trial (NCT02392572) for leukemia (Table 2) | |||
| ONC201 | ABT263 (BH-3 mimetic) | • Combination decreased anti-apoptotic Bcl-2 protein MCL-1 | 30 |
| • Increased tumor regression in in vivo glioblastoma models without toxicity | |||
| ONC201 | 2-Deoxyglucose (2-DG, glucose analog and glycolysis inhibitor) | • ONC201 and 2-DG synergized to reduce cell viability and deplete ATP in glioblastoma cell lines | 38 |
| • Slightly enhanced the fraction of cells arrested at G2/M compared to single agent treatment | |||
| • Reduced tumor growth and migration | |||
| ONC212 | NCT-503 or CBR-5884 (PDGDH inhibitors) | • Dual inhibition of mitochondrial OXPHOS (via ONC212) and the SOG pathway (via PDGDH inhibition) enhanced apoptosis induction in vitro, and reduced tumor size in vivo, in glioblastoma and colon carcinoma models | 10 |
| ONC212 | ABT-199 (Bcl-2 inhibitor) | • ONC212 reduced levels of MCL-1, a resistance factor for Bcl-2 inhibition in AML, thereby sensitizing cells to Bcl-2 inhibition by ABT-199 | 12 |
| • ONC212 and ABT-199 increased apoptosis in in vitro and in vivo AML models, and prolonged survival in in vivo MCL models | |||
| ONC201/ONC212 | AG1024 (IGF1-R inhibitor) | • Synergized to reduce cell viability in 4 out of 4 pancreatic cancer in vitro | 9 |
| • Reduced tumor size in pancreatic cancer mouse model more effectively than either agent alone |
Abbreviations: mTOR, Mammalian Target of Rapamycin; AML, acute myeloid leukemia.
aThe table lists promising drug combinations with imipridones in preclinical cancer models, which may hold potential for future clinical translation.
Imipridones for CNS cancers
CNS tumors are molecularly subtyped based on genomic mutations and copy number variations,62 which may activate distinct signaling pathways and shape the metabolic dependencies of the tumor – emphasizing the need to assess imipridone response in the context of CNS tumor molecular subtype. For example, the WHO 2016 integrated classification system defines 5 adult diffuse glioma subtypes incorporating isocitrate dehydrogenase (IDH) mutation and 1p19q co-deletion status.63IDH mutation status influences the metabolic profile of the tumor, with IDH1 mutations causing increased reliance on OXPHOS,64 whereas IDH-wild-type glioma cells exhibit higher glucose uptake rates.65IDH-wild-type glioblastoma is the most common primary malignant brain tumor in adults, and a subset of these tumors harbor EGFR amplification,62–66 which may promote increased glycolysis.67,68 Importantly, recent data suggests that EGFR overexpressing adult glioblastoma cell lines are resistant to ONC201, whereas low EGFR expressing cell lines are sensitive to ONC201 treatment.55 Given that imipridones’ primary mechanism is to agonize ClpP and impair mitochondrial function, the extent to which the tumor is reliant on OXPHOS is likely to influence tumor response to treatment, potentially explaining the insensitivity of EGFR overexpressing glioblastomas to ONC201.55 These findings provide support for a recently announced phase 2 clinical trial of ONC201 for adults with EGFR-low glioblastoma (Table 2). Notably, primary glioblastomas can acquire EGFR mutations or gene amplification during therapy, or exhibit heterogeneity at diagnosis,69–71 underscoring the need to consider evolving responses to imipridones over the course of disease. More research is needed to assess imipridone response across clinical subtypes of adult and pediatric gliomas.
Although CNS tumors frequently exhibit a shift towards aerobic glycolysis to meet their high energy demands, as reviewed elsewhere,72 it is becoming increasingly clear that mitochondrial OXPHOS plays a key role in sustaining CNS tumors. For example, glioblastomas consist of heterogeneous cell populations with different metabolic demands, and OXPHOS is essential to maintain the population of glioblastoma cancer stem cells.73,74 Proteomic and phosphoproteomic analyses of IDH-wild-type glioblastoma indicate 2 distinct subtypes marked by expression of OXPHOS-related proteins.75 One group shows a protein expression signature consistent with aerobic glycolysis and overexpression of neural stem cell markers, while the second group shows an OXPHOS-dependent protein signature and overexpression of oligodendrocyte and astrocyte markers.75 Thus, protein expression-based subtyping may be helpful for identifying good candidates for imipridone treatment. Indeed, glioblastomas76 and pediatric diffuse intrinsic pontine gliomas (DIPGs)77 overexpress OXPHOS-related proteins, including respiratory complex I components—which are among the strongest downregulated proteins in imipridone-treated cells.38 Moreover, DIPG cells are sensitive to mitochondrial targeting agents, particularly when combined with PI3K/AKT/mTOR pathway inhibitors.77 The importance of both aerobic glycolysis and OXPHOS in sustaining CNS tumors highlights the need for dual targeting of multiple bioenergetics pathways to effectively shut down CNS tumor cell metabolism.
Clinical implications
Clinical trials using imipridones were first launched in 2014, shortly after ONC201’s emergence as an anticancer agent. ONC201 reached micromolar therapeutic plasma concentrations and caused only mild (grade I) adverse treatment-related events including fever, nausea, and emesis, each of which were reversed.3 The strongest clinical responses occurred in advanced-stage prostate and endometrial cancer patients with involvement of lymph nodes, bone, and lung.3 Following the demonstrated benign safety profile of ONC201 in its pilot trial,3 an extended phase 1 study was launched, continuing ONC201 treatment for 20 patients with heavily pretreated solid tumors (prostate, colon, endometrial cancers, and glioblastoma) resistant to standard-of-care treatment.59 Five patients with metastatic prostate or endometrial cancers experienced prolonged stable disease for over 6 months.59
ONC201 has since expanded into numerous phase 1 and 2 clinical trials, and ONC206 recently entered a phase 1 trial for adults with recurrent and rare primary CNS neoplasms (Table 2). In total, 5 trials are centered on patients with CNS tumors, 2 of which are open to pediatric patients. In adults with recurrent glioblastoma, ONC201 is well tolerated and achieves CNS penetration with intratumoral concentrations and pharmacodynamics responses, including elevated ATF4 and DR5, and apoptosis induction relative to archival specimens.4 The positive response in one patient diagnosed with H3K27M-mutant DMG5 resulted in an expansion of access program enrolling patients harboring H3K27M-mutant DMG, including DIPG.7 Two children with DIPG experienced PFS of 13 and 20 months,7 markedly exceeding the median PFS (7 months) and overall survival (11 months) of this devastating disease.78 These children showed radiographic evidence of regression and improvement in disease-related neurological symptoms.7 In a case report of an H3K27M-mutant DIPG patient receiving ONC201, the patient showed a remarkable response with reduced tumor size, a 22-month survival from diagnosis (censored at the time of publication), and no adverse ONC201-related events.6
Conclusions
Imipridones, and in particular ONC201 and ONC206, have emerged as promising therapeutic agents for clinical use in a wide range of cancers. The observed low toxicity profile and BBB penetration of these drugs provide an ideal opportunity for single and combination use for the treatment of malignant CNS cancers. Several questions remain to be clarified as to the anticancer activity of these drugs, including the role of the tumor microenvironment and immune signaling in modulating response across cancer types. Additionally, strategies for overcoming acquired resistance to imipridones should be explored, given the relative ease of developing resistance to small molecule anticancer drugs. Further studies are also needed to address how imipridones trigger the ISR, and how this response plays out across different cancer types. Our collective review of existing data suggests that the sensitivity of cancer cells to imipridones is likely contingent on their relative dependencies on mitochondrial OXPHOS and ability to shift to glycolysis. As described, combinatorial strategies coupling imipridones with other agents that inhibit alternate metabolic pathways may be of interest for targeting tumor cell metabolism, particularly in the context of CNS tumors. Together, the existing preclinical and clinical data supports the promise of these drugs for treating a diverse range of pediatric and adult cancers by targeting common metabolic vulnerabilities.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the generosity of all patients and their families.
Funding
This work was supported by funding from Charlie Kerr and Isabella Kerr Molina Foundation, the Kortney Rose Foundation (Oceanport, NJ), Swifty Foundation (Woodridge, IL), Michael Mosier Defeat DIPG Foundation (Bethesda, MD), ChadTough Foundation (Woodridge, IL), Smashing Walnuts Foundation (Middleburg, VA), Gabriella Miller Kids First Data Resource Center (Philadelphia, PA), the Matthew Larson Foundation (Franklin Lake, NJ), the Lilabean Foundation for Pediatric Brain Cancer Research (Silver Spring, MD), the Research Council of Norway (187615), the South-Eastern Norway Regional Health Authority, and the University of Oslo.
Conflict of interest statement. The authors declare no conflicts of interest.
Authorship statement. Reviewed articles, prepared manuscript text, created figures, and tables: ERB. Edited and revised manuscript text, performed data re-analysis, and contributed to preparation of figures: SMW. Edited and revised manuscript text: SM and MAG. Edited and revised manuscript text, figures and tables, and served as corresponding author: JN.
References
- 1. Allen JE, Krigsfeld G, Mayes PA, et al. . Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5(171):171ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allen JE, Crowder RN, Crowder R, El-Deiry WS. First-in-class small molecule ONC201 induces DR5 and cell death in tumor but not normal cells to provide a wide therapeutic index as an anti-cancer agent. PLoS One. 2015;10(11):e0143082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stein MN, Bertino JR, Kaufman HL, et al. . First-in-human clinical trial of oral ONC201 in patients with refractory solid tumors. Clin Cancer Res. 2017;23(15):4163–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arrillaga-Romany I, Chi AS, Allen JE, Oster W, Wen PY, Batchelor TT. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget. 2017;8(45):79298–79304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arrillaga-Romany I, Odia Y, Prabhu VV, et al. . Biological activity of weekly ONC201 in adult recurrent glioblastoma patients. Neuro Oncol. 2020;22(1):94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hall MD, Odia Y, Allen JE, et al. . First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: a case report. J Neurosurg Pediatr. 2019:1–7. [DOI] [PubMed] [Google Scholar]
- 7. Chi AS, Tarapore RS, Hall MD, et al. . Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J Neurooncol. 2019;145(1):97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wagner J, Kline CL, Ralff MD, et al. . Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle. 2017;16(19):1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lev A, Lulla AR, Wagner J, et al. . Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF1-R and GRP78/BIP. Oncotarget. 2017;8(47):81776–81793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishida CT, Zhang Y, Bianchetti E, et al. . Metabolic reprogramming by dual AKT/ERK inhibition through imipridones elicits unique vulnerabilities in glioblastoma. Clin Cancer Res. 2018;24(21):5392–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishizawa J, Zarabi SF, Davis RE, et al. . Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell. 2019;35(5):721–737.e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nii T, Prabhu VV, Ruvolo V, et al. . Imipridone ONC212 activates orphan G protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia. Leukemia. 2019;33(12):2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kline CLB, Ralff MD, Lulla AR, et al. . Role of dopamine receptors in the anticancer activity of ONC201. Neoplasia. 2018;20(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Graves PR, Aponte-Collazo LJ, Fennell EMJ, et al. . Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem Biol. 2019;14(5):1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Madhukar NS, Khade PK, Huang L, et al. . A Bayesian machine learning approach for drug target identification using diverse data types. Nat Commun. 2019;10(1):5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lev A, Lulla AR, Ross BC, et al. . ONC201 targets AR and AR-V7 signaling, reduces PSA, and synergizes with everolimus in prostate cancer. Mol Cancer Res. 2018;16(5):754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacques S, van der Sloot AM, C Huard C, et al. . Imipridone anticancer compounds ectopically activate the ClpP protease and represent a new scaffold for antibiotic development. Genetics. 2020;214(4):1103–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacob NT, Lockner JW, Kravchenko VV, Janda KD. Pharmacophore reassignment for induction of the immunosurveillance cytokine TRAIL. Angew Chem Int Ed Engl. 2014;53(26):6628–6631. [DOI] [PubMed] [Google Scholar]
- 19. Wagner J, Kline CL, Pottorf RS, et al. . The angular structure of ONC201, a TRAIL pathway-inducing compound, determines its potent anti-cancer activity. Oncotarget. 2014;5(24):12728–12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prabhu VV, Madhukar NS, Gilvary C, et al. . Dopamine receptor D5 is a modulator of tumor response to dopamine receptor D2 antagonism. Clin Cancer Res. 2019;25(7):2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peters MAM, Meijer C, Fehrmann RSN, et al. . Serotonin and dopamine receptor expression in solid tumours including rare cancers. Pathol Oncol Res. 2020;26(3):1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018;37(4):733–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng Y, Zhou J, Li Z, Jiang Y, Zhou Y. Small molecular TRAIL inducer ONC201 induces death in lung cancer cells: a preclinical study. PLoS One. 2016;11(9):e0162133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prabhu VV, Allen JE, Dicker DT, El-Deiry WS. Small-molecule ONC201/TIC10 targets chemotherapy-resistant colorectal cancer stem-like cells in an Akt/Foxo3a/TRAIL-dependent manner. Cancer Res. 2015;75(7):1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Talekar MK, Allen JE, Dicker DT, El-Deiry WS. ONC201 induces cell death in pediatric non-Hodgkin’s lymphoma cells. Cell Cycle. 2015;14(15):2422–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fang Z, Wang J, Clark LH, et al. . ONC201 demonstrates anti-tumorigenic and anti-metastatic activity in uterine serous carcinoma in vitro. Am J Cancer Res. 2018;8(8):1551–1563. [PMC free article] [PubMed] [Google Scholar]
- 27. Greer YE, Porat-Shliom N, Nagashima K, et al. . ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget. 2018;9(26):18454–18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishizawa J, Kojima K, Chachad D, et al. . ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9( 415):ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tu YS, He J, Liu H, et al. . The imipridone ONC201 induces apoptosis and overcomes chemotherapy resistance by up-regulation of bim in multiple myeloma. Neoplasia. 2017;19(10):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karpel-Massler G, Bâ M, Shu C, et al. . TIC10/ONC201 synergizes with Bcl-2/Bcl-xL inhibition in glioblastoma by suppression of Mcl-1 and its binding partners in vitro and in vivo. Oncotarget. 2015;6(34):36456–36471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan X, Kho D, Xu J, Gajan A, Wu K, Wu GS. ONC201 activates ER stress to inhibit the growth of triple-negative breast cancer cells. Oncotarget. 2017;8(13):21626–21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szczepanowska K, Maiti P, Kukat A, et al. . CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016;35(23):2566–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer F, Langer JD, Osiewacz HD. Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci Rep. 2015;5:18375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta. 2012;1823(1):15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cole A, Wang Z, Coyaud E, et al. . Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2015;27(6):864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wong KS, Mabanglo MF, Seraphim TV, et al. . Acyldepsipeptide analogs dysregulate human mitochondrial ClpP protease activity and cause apoptotic cell death. Cell Chem Biol. 2018;25(8):1017–1030.e1019. [DOI] [PubMed] [Google Scholar]
- 37. Dougan DA, Hantke I, Turgay K. Dysregulating ClpP: from antibiotics to anticancer? Cell Chem Biol. 2018;25(8):929–930. [DOI] [PubMed] [Google Scholar]
- 38. Pruss M, Dwucet A, Tanriover M, et al. . Dual metabolic reprogramming by ONC201/TIC10 and 2-Deoxyglucose induces energy depletion and synergistic anti-cancer activity in glioblastoma. Br J Cancer. 2020;122(8):1146–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24(11):2482–2490. [DOI] [PubMed] [Google Scholar]
- 41. Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci Signal. 2016;9(415):ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Melber A, Haynes CM. UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018;28(3):281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Quirós PM, Prado MA, Zamboni N, et al. . Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. 2017;216(7):2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16(6):533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramirez MU, Hernandez SR, Soto-Pantoja DR, Cook KL. Endoplasmic reticulum stress pathway, the unfolded protein response, modulates immune function in the tumor microenvironment to impact tumor progression and therapeutic response. Int J Mol Sci. 2019;21(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han J, Back SH, Hur J, et al. . ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ni X, Zhang X, Hu CH, et al. . ONC201 selectively induces apoptosis in cutaneous T-cell lymphoma cells via activating pro-apoptotic integrated stress response and inactivating JAK/STAT and NF-κB pathways. Oncotarget. 2017;8(37):61761–61776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Münch C, Harper JW. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature. 2016;534(7609):710–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo X, Aviles G, Liu Y, et al. . Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. 2020;579(7799): 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mick E, Titov DV, Skinner OS, Sharma R, Jourdain AA, Mootha VK. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife. 2020;9:e49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet. 2009;43:95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Q, Wang H, Ran L, Zhang Z, Jiang R. The preclinical evaluation of TIC10/ONC201 as an anti-pancreatic cancer agent. Biochem Biophys Res Commun. 2016;476(4):260–266. [DOI] [PubMed] [Google Scholar]
- 54. Corsello SM, Nagari RT, Spangler RD, et al. . Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer. 2020;1(2):235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. He Y, Li J, Koga T, et al. . Epidermal growth factor receptor (EGFR) as a molecular determinant of glioblastoma response to dopamine receptor 2 (DRD2) inhibitors. Neuro Oncol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jin ZZ, Wang W, Fang DL, Jin YJ. mTOR inhibition sensitizes ONC201-induced anti-colorectal cancer cell activity. Biochem Biophys Res Commun. 2016;478(4):1515–1520. [DOI] [PubMed] [Google Scholar]
- 57. DepMap: The Cancer Dependency Map Project at Broad Institute. DepMap 20Q3 Public. Dataset. 2020. doi: 10.6084/m9.figshare.12931238.v1. [DOI] [Google Scholar]
- 58. Wagner J, Kline CL, Zhou L, et al. . Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J Clin Invest. 2018;128(6):2325–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stein MN, Malhotra J, Tarapore RS, et al. . Safety and enhanced immunostimulatory activity of the DRD2 antagonist ONC201 in advanced solid tumor patients with weekly oral administration. J Immunother Cancer. 2019;7(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Romaguera JE, Lee HJ, Tarapore R, et al. . Integrated stress response and immune cell infiltration in an ibrutinib-refractory mantle cell lymphoma patient following ONC201 treatment. Br J Haematol. 2019;185(1):133–136. [DOI] [PubMed] [Google Scholar]
- 61. Allen JE, Prabhu VV, Talekar M, et al. . Genetic and pharmacological screens converge in identifying FLIP, BCL2, and IAP proteins as key regulators of sensitivity to the TRAIL-inducing anticancer agent ONC201/TIC10. Cancer Res. 2015;75(8):1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Louis DN, Perry A, Reifenberger G, et al. . The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 63. Molinaro AM, Taylor JW, Wiencke JK, Wrensch MR. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol. 2019;15(7):405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Grassian AR, Parker SJ, Davidson SM, et al. . IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74(12):3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Garrett M, Sperry J, Braas D, et al. . Metabolic characterization of isocitrate dehydrogenase (IDH) mutant and IDH wildtype gliomaspheres uncovers cell type-specific vulnerabilities. Cancer Metab. 2018;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network . Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Makinoshima H, Takita M, Saruwatari K, et al. . Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in epidermal growth factor receptor (EGFR)-mutated lung adenocarcinoma. J Biol Chem. 2015;290(28):17495–17504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang W, Xia Y, Cao Y, et al. . EGFR-induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell. 2012;48(5):771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Francis JM, Zhang CZ, Maire CL, et al. . EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014;4(8):956–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang J, Cazzato E, Ladewig E, et al. . Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Eskilsson E, Røsland GV, Solecki G, et al. . EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2018;20(6):743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Agnihotri S, Zadeh G. Metabolic reprogramming in glioblastoma: the influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. 2016;18(2):160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Janiszewska M, Suvà ML, Riggi N, et al. . Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26(17):1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hoang-Minh LB, Siebzehnrubl FA, Yang C, et al. . Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018;37(23):e98772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Oh S, Yeom J, Cho HJ, et al. . Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat Commun. 2020;11(1):3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Park J, Shim JK, Kang JH, et al. . Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro Oncol. 2018;20(7):954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tsoli M, Liu J, Franshaw L, et al. . Dual targeting of mitochondrial function and mTOR pathway as a therapeutic strategy for diffuse intrinsic pontine glioma. Oncotarget. 2018;9(7):7541–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cooney T, Lane A, Bartels U, et al. . Contemporary survival endpoints: an International Diffuse Intrinsic Pontine Glioma Registry study. Neuro Oncol. 2017;19(9):1279–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.