

Summary
Single-cell sequencing of environmental microorganisms is an essential component of the microbial ecology toolkit. However, large-scale targeted single-cell sequencing for the whole-genome recovery of uncultivated eukaryotes is lagging. The key challenges are low abundance in environmental communities, large complex genomes, and cell walls that are difficult to break. We describe a pipeline composed of state-of-the art single-cell genomics tools and protocols optimized for poorly studied and uncultivated eukaryotic microorganisms that are found at low abundance. This pipeline consists of seven distinct steps, beginning with sample collection and ending with genome annotation, each equipped with quality review steps to ensure high genome quality at low cost. We tested and evaluated each step on environmental samples and cultures of early-diverging lineages of fungi and Chromista/SAR. We show that genomes produced using this pipeline are almost as good as complete reference genomes for functional and comparative genomics for environmental microbial eukaryotes.
Subject areas: Genomics, Geomicrobiology, Microbiology
Graphical abstract
Highlights
-
•
We optimized single-cell methodology using a broad sample range, for EME
-
•
We combined bioinformatic and bench protocols into a concise workflow
-
•
We benchmarked the pipeline and used it on environmental samples
-
•
We selected a set of QC criteria for best genome quality prediction
Genomics ; Geomicrobiology ; Microbiology
Introduction
Single-cell genomics has significantly advanced mammalian and prokaryote studies related to human health, revisions of the tree of life, and biotechnology, as well as enhanced our understanding of the roles that microbes play in ecosystems (Gawad et al., 2016 and references inside; Linnarsson and Teichmann, 2016; Macaulay and Voet, 2014; Neu et al., 2017; Rinke et al., 2013; Stepanauskas, 2012). In some microbial eukaryotes, such as fungi and protozoa, single-cell genomics has been used to reveal the ecological and biological functions of some uncultured species (Berbee et al., 2017; Lin et al., 2014; Roy et al., 2014; Yoon et al., 2011 and based on this pipeline, Ahrendt et al., 2018). Nevertheless, to date, the majority of microbial eukaryote species are not considered feasible targets for genomic environmental studies (metagenomics and single-cell studies). In particular, genomes of uncultivated environmental microbial eukaryotes (EMEs) remain largely unavailable for the currently available genomic technology (Hyde, 2001; Lazarus and James, 2015; Sibbald and Archibald, 2017). Most studies that have recovered single-cell eukaryote genomes were performed on organisms that are abundant, have been successfully cultivated, or have a well-described molecular physiology (Gawryluk et al., 2016; Lin et al., 2014; Roy et al., 2014; Troell et al., 2016; Yoon et al., 2011; Zhang et al., 2017). The genomes of a diversity of low-abundance (at a concentration below 5% to as low as 0.01% in environments) EMEs (Tkacz et al., 2018; Wurzbacher et al., 2017 and present study target enrichment estimates) are therefore unexplored.
A quick look at fungi and Chromista shows the extent of skewed representation of annotated genomes and the knowledge gap in current genome biology. For example, in the fungal genome database MycoCosm (Grigoriev et al., 2014), seven of nine fungal phyla are early diverging lineages with more than 2,000 species described (Blackwell, 2011; Stajich et al., 2009), whereas the 173 sequenced and annotated genomes of early diverging fungi (EDF) represent only ≤10% of the total number of fungal genomes and are heavily skewed to the derived group Dikarya (1,626). Likewise, in Chromista (Ruggiero et al., 2015) or supergroup SAR (Burki et al., 2019), the phylum Ciliophora has sequenced genomes for only 4 of 11 classes. Furthermore, 20 of 23 sequenced and annotated genomes of Ciliophora species available in NCBI belong to two classes, and the majority of these genomes are incompletely annotated. The full list of unrepresented phyla and classes from these two kingdoms is much longer. However, rDNA operational taxonomic unit (OTU) screening, environmental observations, and biochemical studies suggest that many of the understudied lineages are ubiquitous, extremely diverse, and highly adaptable (Alexander et al., 2016; Berbee et al., 2017; Blackwell, 2011; Foissner, 1999, 2009; Hyde, 2001; Lazarus and James, 2015; Sibbald and Archibald, 2017; Stajich et al., 2009). Consequently, there is a need for broader sampling of eukaryotic genome diversity (Sibbald and Archibald, 2017).
In addition to developing affordable tools for deeper and broader phylogenetic sampling targeting low-abundance EMEs in their native habitats, assessing the level of genome completeness is critical for understanding EME function and evolution. We explored the latter question in our recent publication (Ahrendt et al., 2018) using the pipeline that we describe in this study. Among the large number of publications dedicated to single-cell genomics (Arriola et al., 2007; Chen et al., 2017; Clingenpeel et al., 2014, 2015; Ellegaard et al., 2013; Garvin et al., 2015; Gawad et al., 2016; Gawryluk et al., 2016; Lan et al., 2017; Lin et al., 2014; Linnarsson and Teichmann, 2016; Macaulay and Voet, 2014; Neu et al., 2017; Rinke et al., 2013, 2014; Roy et al., 2014; Spits et al., 2006; Stepanauskas, 2012; Troell et al., 2016; Zhang et al., 2017), none has addressed the specific challenges associated with obtaining high-quality annotated de novo assembled genomes of poorly studied, low-abundance EMEs from a wide range of environmental samples.
Here, we explore the possibilities and current limitations of eliminating bias in genome representation by adapting current single-cell genomics methods into a pipeline for studying and annotating the genomes of EMEs with low and ultralow abundance to produce a broader representation of eukaryotic organisms in genomic studies. We explore, step by step, the potential critical impact of each challenge on both the quality of the recovered genomes and the cost of the study and the characteristics that set the genomics of single-cell EMEs apart from those of bacteria, archaea, and abundant or cultivated eukaryotes. The analyzed set of target species spans five fungal clades: four EDF (7 species with approximately 70 genomes), one Dikarya (1 species with 6 single-cell genomes), and one Chromista (Ciliophora with 7 single-cell genomes belonging to one unknown species). To date, this is the widest set of single-cell genomics methods, benchmarked against isolate genomes and tested on a wide range of sample types (isogenic cultivated, heterogenic co-cultivated, various environmental) spanning a wide phylogenetic range of target species.
Multiple displacement amplification (MDA) is the most widely used single-cell genome amplification method. MDA-associated genome amplification bias (GAB) has been shown to affect the quantification of copy number variation, SNP, and chimera formation (Garvin et al., 2015; Hou et al., 2015; Lasken and Stockwell, 2007; Zong et al., 2012). For mammalian cells, it has been shown that whole-genome amplification (WGA) methods other than MDA have lower GAB (Foissner, 2009; Gawad et al., 2016; Hou et al., 2015; Ning et al., 2015). Nevertheless, when appropriate genome assembly algorithms are applied, MDA-generated high-molecular-weight DNA fragments are of paramount value for de novo assemblies compared with linear amplification methods involving PCR (Spits et al., 2006).
Cell wall lysis (CWL) is another common challenge for single-cell genomics of the environmental microbiome (Brown and Audet, 2008; Tighe et al., 2017), but it is not a problem for mammalian species or species lacking cell walls, for which most single-cell genomics methods have been developed. Coupling CWL with downstream genome amplification has been explored for bacterial single-cell genomics methods (Clingenpeel et al., 2014, 2015; Rinke et al., 2014). Inadequate CWL and DNA contamination have been found to affect genome completeness in bacteria and perhaps lead to amplification bias in bacterial genomes (Clingenpeel et al., 2014, 2015). The same authors observed that an early start of genome amplification (SGA) correlated with larger genome assemblies, presumably due to less biased amplification, suggesting that early SGA occurs when lysis is complete (Clingenpeel et al., 2014, 2015). Only one study has explored a number of protist species using the same lysis method (Yoon et al., 2011). In other studies of single-cell fungal, plant, or protist genomes, CWL has been tailored for a specific target (Gawryluk et al., 2016; Lin et al., 2014; Roy et al., 2014; Troell et al., 2016). To date, a universal CWL, compatible with same reaction MDA was explored only for single-cell prokaryote and mammalian genomes (reviewed in Gawad et al., 2016 and citations). The use of either low-volume same-tube reactions or microfluidics has been shown to significantly reduce both amplification bias and contamination levels and to result in more complete genomes (Rinke et al., 2014, reviewed in Gawad et al., 2016). However, existing microfluidics devices (Gawad et al., 2016; Lan et al., 2017) do not accommodate the diverse size and shape of environmental microorganisms and/or cell lysis requirements. There is a need for methods that can accommodate lysis conditions that work for a wide range of unknown organisms without having to purify nucleic acids and without inhibiting subsequent molecular reactions.
Specifically, we explored (1) the accurate single-cell isolation of the target species at a one-in-millions representation in the environmental sample by testing fluorescent-activated cell sorting (FACS) and microfluidic methods; (2) the suitability of various new and known (Brown and Audet, 2008; Clingenpeel et al., 2014, 2015; Rinke et al., 2014; Tighe et al., 2017; Yoon et al., 2011) CWL methods for a broad range of organisms and their compatibility with downstream high-throughput semiautomated processes; (3) all factors known to affect genome completeness, such as GAB due to MDA (Hou et al., 2015) and genome size, previously reported in mammalian cells and prokaryotes (Garvin et al., 2015; Hou et al., 2015; Lasken and Stockwell, 2007; Zong et al., 2012) but never explored in EMEs; (4) GC%, cell wall complexity, and genome structure as factors that have not been shown to cause MDA-GAB or genome incompleteness; (5) the costs associated with a high sequencing capacity, which has not been previously discussed in attempts for a broader and deeper phylogenomic mining of EMEs (unlike environmental prokaryotes with genomes that are 100-fold smaller than those of eukaryotes on average, mass sequencing of poor-quality or nontarget EME genomes can become prohibitively costly, even when using NovaSeq); (6) methods using shallow sequencing to evaluate genome quality before deep sequencing (Daley and Smith, 2014), which is particularly important for high throughput; (7) genome assembly tools (Bankevich et al., 2012; Butler et al., 2008; Peng et al., 2012) for accurate de novo reconstruction of single-cell EME genomes from novel lineages that suffered partial GAB and chimerization during amplification; (8) bioinformatics tools (Auch et al., 2010; Han et al., 2016; Meier-Kolthoff et al., 2013) to accurately predict single-cell intra- and interspecific genome distance for EMEs—a prerogative for assembling a genome by coassembling or selecting single-cell or multiple-cell genomes with similar and low genome distances that are characteristic of intraspecific phylogeny (coassemblies provide a higher level of genome completeness [Kogawa et al., 2018]); (9) genome completeness assessment tools (Parra et al., 2007, supplementary reference 4); and (10) correct gene structure prediction and annotation from fragmented and/or incomplete genomes.
Results
Pipeline synopsis
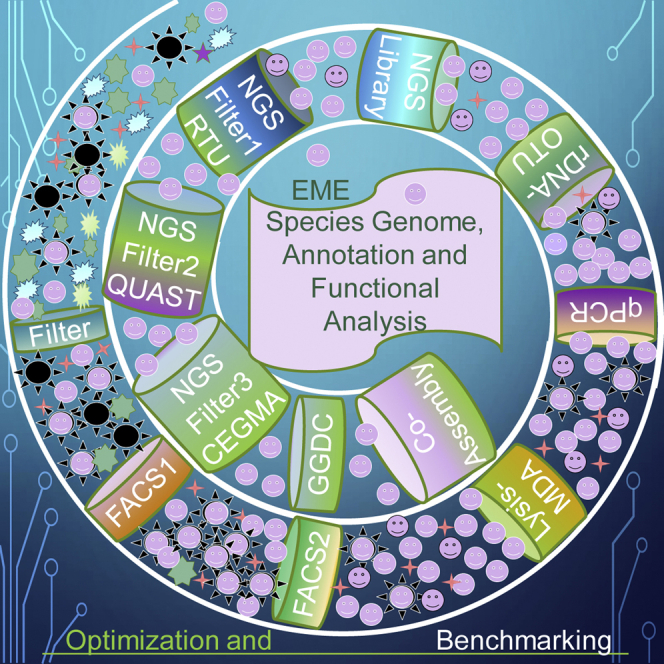
We developed and benchmarked a 7-step pipeline for EME genome recovery, as illustrated in Figure 1. Brief description of our pipeline:
-
1.
Environmental sample collection (Step 1), shipping, storage, and target species enrichment evaluation (QC1): Step 1 included sample collection, storage, and shipping. When possible and necessary, filtration steps were used for target population enrichment. Enriched filters in suitable media were used to suspend cells before storage and shipment. QC1 included target organism enrichment evaluation (TgE) via rDNA profiling, microscopic identification and counting, or both. Optimizations involved enrichment and storage methods.
-
2.
Single-cell isolation (Step 2) and target species enrichment evaluation (QC2): During step 2, samples were visualized using a microscope and a FACS instrument; target populations were identified and sorted in bulk into large tubes in 0.02 μm filtered original media. After the first round of FACS enrichment of the target population, a second round of sorting (as needed to reduce carryover contaminants) was used before single-set sorting. FACS-enriched target populations were used for FACS of single cells or batches of 10–100 cells into 384-well plates and immediately frozen on dry ice. QC2 included TgE via FACS with or without microscopic validation before single-cell isolation. Optimizations involved cell staining protocols and target cell isolation methods.
-
3.
Single-cell lysis and genome amplification (Step 3) and lysis-MDA efficiency evaluation (QC3). During step 3, isolated single cells (or batches of 10–100 cells) were lysed and amplified via MDA in the same well. Single-cell amplified genomes or multiple-cell amplified genomes were quantified in real time and at the endpoint. QC3 criteria included real-time-monitored SGA, percent positive MDA, fold genome amplification (FGA), and other criteria described in transparent methods step 3 and later. Optimizations involved the efficiency and compatibility of a number of lysis-MDA protocols.
-
4.
Confirmation of species identity, contaminant, or symbiont occurrences in single and multiple amplified genomes (Step 4) and OTU screening (QC4). During step 4, an aliquot of single and multiple amplified genomes was subjected to qPCR for rDNA genes followed by Sanger sequencing and BLAST against NCBI and AFTOL (Celio et al., 2006) databases. QC4 included OTU identification, evaluation of Step 1 through Step 3 process efficiency, and selection of target genomes for sequencing. Optimizations for step 4: a range of 18S, 16S, 28S, and ITS rDNA primers and regions were tested for suitability with a broad phylogenetic range. Optimizations for merging step 4 with step 5: various rDNA assembly methods from shallow short-read sequencing and various next-generation sequencing (NGS) library protocols for high-throughput shallow sequencing were tested for suitability for rDNA assembly methods.
-
5.
NGS library construction and shallow sequencing for single and multiple amplified genomes (Step 5) and library (QC5) and genome (QC6) quality predictions. In step 5, genomic DNA was fragmented, and NGS libraries were constructed and pooled for shallow sequencing following Illumina guidelines. QC5 included library quality evaluation following Illumina recommendations. QC6 included genome quality prediction based on criteria such as percent contaminant, random 20-mer uniqueness (RTU) and other NGS-read-QC metrics described in supplemental section step 5. Optimizations: We tested various fragmentation protocols, read length and library construction protocols, and the use of various NGS-read-QC metrics to identify libraries with the following: (1) high GAB, (2) high chimerization rate, and (3) contamination and incomplete prediction of the genome assembly outcome by eliminating poor-quality libraries.
-
6.
Deep sequencing for single and multiple amplified genome assembly, single amplified genome phylogenomic distance estimation, and coassembly of multiple single amplified genomes into a single-species genome (Step 6), single amplified genome assembly quality evaluation, and annotation quality prediction (QC7). During step 6, we assembled genomes from deep-sequenced libraries that passed QC6 evaluation. We found that single and multiple amplified genome distances and their coassembled genomes had the same intraspecific distance. For genome distance calculation for species coassembled from single and multiple amplified genome, we used Genome-to-Genome Distance Calculator (GGDC) formula 2 described in Meier-Kolthoff et al. (2013). For QC7, we used quality assessment tool for genome assemblies (QUAST) (Lazarus et al., 2017) criteria and tested the core eukaryotic gene mapping approach (CEGMA) values (Parra et al., 2007). Optimizations: We tested all 20 QUAST criteria for predictability of assembly quality for high-quality annotations and further functional predictions. We reduced the number of necessary QUAST criteria to a few essential criteria. We tested CEGMA values as a predictor of genome completeness before annotation. We tested various genome distance estimation methods for suitability with large eukaryotic genomes that were amplified, fragmented, and incomplete.
-
7.
Annotation and functional predictions (Step 7) and genome completeness evaluation (QC8). In step 7, assembled genomes were annotated using the MycoCosm pipeline toolsets (Grigoriev et al., 2014). QC8 included CEGMA values (Parra et al., 2007) to assess genome completeness (using the presence of conserved core eukaryotic genes) before functional and phylogenomic analysis (Ahrendt et al., 2018). Optimizations: We tested both CEGMA and BUSCO values for genome completeness prediction; manual curation was necessary for species that had no close genome annotations.
Figure 1.

Pipeline schematics for environmental microbial eukaryotic single-cell whole-genome recovery, de novo assembly, and annotation
Square boxes show the pipeline steps and components. QC1 through 7 and green ovals show quality check steps and the main criteria used in this study, described in the brief pipeline overview. TgE, target organism enrichment; rDNA, ribosomal DNA; FACS, fluorescent-activated cell sorting; TgBm, target organism biometrics; sc, single cell; MDA, multiple displacement amplification; SGA, start of genome amplification; SAG, single amplified genome; CAG, composite amplified genome; OTU, operational taxonomic unit; BLAST, basic local alignment search tool (Altschul et al., 1990); NGS, next-generation sequencing; LQC, NGS library quality check; RQC, Illumina sequencing read quality check; RTU, random 20-mer uniqueness; cont, contaminant identified by BLAST on NGS read; QUAST, quality assessment tool for genome assemblies (Lazarus et al., 2017); CEGMA, core eukaryotic genes mapping approach (Parra et al., 2007); BUSCO, (Simão et al., 2015). See also Data S2 and Table S10.
Sample selection for EME single-cell genomics pipeline benchmarking
We tested this pipeline on a range of samples shown and described in Figure 2, Tables 1 and 2, and Data S1. Samples ranged from pure environmental (Data S1RR–S1U) to dual cultures mimicking natural conditions (Data S1A–S1P). Samples had various levels of complexity (Table 2 and Figure 2). The complexity level was estimated based on a range of factors described in Table 2. Environmental samples ranged from high complexity (ultralow abundance of target species among high-abundance nontarget organisms in the original environment, see Data S1T and S1U) to medium complexity (see Data S1RS and S1S and Table 2). Dual cultures ranged from low (Data S1G–LM) and medium (Data S1E, S1F, S1M–S1P) to highly enriched for target species (see Data S1A–S1D). We examined the effect of organisms' shapes, sizes, and motility on single-cell isolation success and the range of enrichment levels relative to other taxa. The latter was <2%–85%; shapes were spherical, oval, cylindrical, trapezoidal, and needle-shaped; sizes were from 2 to 100 μm; motility modes were sessile or flagellated (Table 1, Figure 2, and Data S1).
Figure 2.

Main target single-cell diversity used for pipeline evaluation
Shown here are nine of the eleven samples used. For the other two samples, see Table 1. Pictures for the first through eighth species are sized relative the 5μM scale bar. Heatmap colors reflect the spectrum of values: red, highest; yellow, lowest; and green, average. TgE is the FACS target enrichment estimated in step 2 of the pipeline (for step 1 TgE, shape and other details, see Table 1 and Video S1). SCL is the sample complexity level (for details see Table 2). The genome size ($) was not known for any of the target organisms before assembly and was therefore estimated based on the assembly size and genome completeness. The genome average GC% (#) was not known before genome assembly and was therefore predicted based on the GC% of the existing genes or by estimation using the nearest phylogenetic group. ∗The phylum Zoopagomycota was established by (Spatafora et al., 2016) in part using data obtained from these four single-cell genomes. Before this study, the phylogenetic data available for this group were limited. See also Data S1.
Table 1.
Combination of critical factors affecting FACS single-cell isolation success of microbial eukaryotes from diverse samples
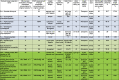 |
Color coding of the samples: green, environmental; light green, laboratory rec-reated environmental sample without artificial substrate; blue, non-axenic heterogeneous co-cultures; white, highly enriched non-axenic co-cultures.
∗ This is the concentration of an enriched FACS population containing target species.
Table 2.
Range of criteria useful for sample complexity level prediction for EME single-cell genome recovery.
| Tested Samples in this study and their level of complexity. | R.a | C.p | P.c. | No sample in this study | D.c,S.p | No sample in this study | T.s | B.h | M.b. | Compost ciliate, Gosling and Third Sister lakes Cryptomycota and Chytridiomycota unknown species |
|---|---|---|---|---|---|---|---|---|---|---|
| Complexity Level | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| target cell abundance (concentration) | 80–100% | 30–100% | 50–90% | 30–80% | 30–70% | 30–50% | 10–20% | 1–15% | 1–5% | ≤1 |
| target cell abundance (amount) | ≥102 | ≥103 | 104 – 105 | 104 – 105 | 104 – 106 | 104 – 106 | 104 – 106 | 104 – 106 | 103 – 106 | ≤1000 |
| diversity of organisms | ≤2 | ≤2 | ≤5 | 10≥5 | 50≥5 | 50≥10 | 50≥5 | 10≥2 | 100≥2 | 1000≥5 |
| presence of ‘competing’ cells (with similar biometric characteristics) | 0 | 0 | 0 | ≤2% | ≤3% | ≤4% | ≤5% | ≥10% | ≥10% | ≥10% |
| shape of the target cell | Spheroid, oval, cylindrical | Spheroid, oval, cylindrical | Spheroid, oval, cylindrical | Spheroid, oval, cylindrical | Spheroid, oval, trapezoidal, cylindrical | Needle-like, trapezoidal, Spheroid, oval, cylindrical | Needle-like, trapezoidal, Spheroid, oval, cylindrical | Needle-like, trapezoidal, Spheroid, oval, cylindrical | Needle-like, trapezoidal, Spheroid, oval, cylindrical | Needle-like, trapezoidal, Spheroid, oval, cylindrical |
| Size of the target cell | 1–20uM | 1–20uM | 1–20uM | 1–25uM | 1–25uM | 1–25uM | 1–30uM | 0.5 –50uM | 0.5 –60uM | 0.5 –80uM |
| target cell wall complexity (layers) | ≤1 | ≤1 | ≤1 | 2 | 2 | 0–2 | 0–2 | 0–2 | 0–2 | 0–3 |
Rozella zoospores after thawing from −80°C frozen original sample.
For each sample and each target species, multiple genomes were isolated and analyzed. However, only the highest quality genomes were assembled and annotated. Pipeline optimizations are described in the transparent methods and supplemental information and were performed using separate sorting of 1, 10, 30, 50, and 100 cells per reaction and the set of QC criteria described in Data S2. The results of the pipeline QC evaluation are illustrated in Figure 3 and Data S2, and they are explored in greater depth further in the article and in the transparent methods in supplemental information. Specifically, single-cell isolation methods were tested, further developed, and optimized in steps 1 and 2 on 11 samples (see Table 1, Data S1 and transparent methods steps 1, 2). Steps 3 through 7 were tested, developed, and optimized on the 9 species described in Figure 2 and transparent methods steps 3–7). We tested and optimized the merging of steps 4 and 5 using all 11 samples. We benchmarked the methods used in these steps, against the unamplified genomes of two fungal species (Rozella allomycis [Cryptomycota] and Caulochytrium protostelioides [Chytridiomycota]). Their unamplified genomes were isolated from the same highly enriched dual cultures of parasitic target fungi with their fungal host and microbiome used for single-cell isolation. Genome size and GC% variations of the known target species were used to test amplification efficiency and bias. The presence or absence of the cell wall in the target species was used to test lysis efficiency.
Figure 3.

Predictive value and applicability of the used QC criteria for a wide phylogenetic group
Twenty QC criteria examined (see Data S2) can be reduced to six shown here. Axes color: black, pre-assembly criteria; gray, assembly metrics; red, pre-annotation criteria. Gower & Hand PCA biplot represents similarity between data points, with smaller distance higher similarity. Shown plot explains 80.2% of the variability for fungi plus ciliate protists group. Any point on the plot projected orthogonally onto the axes will show the approximate value of the variable. Percent at the end of the axes labels indicates predictability value of the axes. Species full names are given in Figures 2 and S1. ED, early diverging. See also Table S10.
The EME pipeline single-cell genome recovery lowest efficiency threshold was identified as 1 target in 500 amplified genomes or 0%–2% rDNA OTU during step 1 screening, using three environmental samples: two aimed at fungal phyla (Cryptomycota and Chytridiomycota) with ultralow abundance of unknown target species and one aimed at one SAR phyla (Ciliophora) with unknown OTU (see details in Data S1R–S1U and Table 1). To refine the target enrichment and single-cell isolation methods, we used eight more samples with one known target species per sample (Figure 2 and Table 2). Six of these samples harbored different mycoparasyte target fungal species with different hosts, all dual non-axenic cultures with different microbiome diversity and target concentration levels (for details see Data S1A–S1D and S1G–S1P). Two other samples re-created the natural environment of the target species and were available in very low amount, one a symbiont of pollen (Data S1E and S1F) and the other a parasite of crustacean (Data S1O and S1P). Both had distinct target species cell shapes albeit high content of contaminating organisms. All these samples were useful for studying the impact of different sample features on single-cell isolation efficiency (Table 1).
EME single-cell genomics pipeline step-by-step bottlenecks and optimizations
We found that the following combination of factors prevented successful target single-cell FACS isolation: very low concentration of target coupled with one or two of the following: size above 30 μM, presence of non-target organisms with highly similar biometric properties (e.g., flagella, cilia, shape, size, and light refraction index), and very low volume of sample (Tables 1 and 2). On the other hand, a two-step FACS isolation, whenever possible, improved target single-cell recovery, shown in Figure S1 and described in Optimization in step 2 in transparent methods.
This finding was further supported by the lack of strong correlation between target concentration in original sample and total number of positive MDA reactions, as well as target single-cell OTU (rDNA-PCR confirmed) (see Figure S4). However, there was a strong correlation between MDA-amplified single-cell and rDNA identified target OTU, shown in Figure S4, indicating that FACS enrichment step before single-cell sorting, and cell lysis, had a significant impact on the number of recovered target genomes.
We performed an extensive optimization of the single-cell lysis methods compatible with the same-well MDA, described in step 3 of the transparent methods and Data S3. We found one lysis method that allowed for the shortest protocol and reduction of the reagent carry-over contaminating DNA (transparent methods Step 3 optimization). Although this method worked well across all species, we observe some heterogeneity between species (Figure S2) and some, but less, difference between single-cell level and multiple-cell level within one species, when start of MDA was used as a proxy for lysis-MDA efficiency.
Further optimization of the OTU screening (see Table S1 for best rDNA primers) of the amplified EME genomes led to improved quality of the single-cell genome recovery, as well as reduction of costs, as described in detail in transparent methods Optimization step 4. Merging of Steps 4 and 5 of the pipeline offered an additional benefit of cost reduction and identification of EMEs with symbionts, which otherwise are screened out in Step 4. The merging of these steps relies on rDNA assembly tool capable of correctly assembling it from MDA-NGS reads. We optimized this step, described in transparent methods Optimization step 5 and the results of the rDNA assembly tools testing are shown in Figure S5.
Another improvement in single-cell genome quality during step 5 was the use of RTU measure. We found that libraries with high RTU (above 60%) produced low-biased genomes used later for co-assembly and annotation here, as opposed to the highly biased genomes with low 20-mer uniqueness, most screened out during optimization step and some shown in Figure 5. The correlation between RTU and genome completeness as CEGMA is very high, as illustrated in Figure 3 and Data S2c. Similarly, de novo assembly tools and type of NGS had a significant impact on the genome quality (Tables S2–S4 and Figure S7), described in transparent methods optimization step 6.
Figure 5.

Intra- and interspecific genome coverage variability
Each species Circos map is scaled relative to the largest genome (Blyttiomyces helicus) true to size. Reference genome (coassembly) is shown as the outer gray circle. Coassembly GC% plotted as a red line over the gray circle. 1,000-bp bins were plotted against reference co-assembly genome and scaled proportionally to the co-assembly genome size. Five representative libraries from single-cell (black); 10-cell, 30-cell, or 50-cell depending on the species (blue); and 100-cell sorts were chosen for each species. For each cell sort category one worst case, one average case, and one best case were picked when available. In the middle of the plot numbers are coassembly genome in gray, GC% in red, and CEGMA completeness in purple. See also Figures S6 and S8.
Measuring genome completeness for de novo assemblies is an imperative requirement for quality evaluation. However, only approximate estimates could be obtained using mathematical algorithms. Two tools developed for eukaryotic genomes looked most promising: CEGMA (Parra et al., 2007) and BUSCO (Simão et al., 2015). We used CEGMA for our pipeline evaluation and later tested the newly developed BUSCO. Unexpectedly BUSCO did perform worse (detected less genes) than CEGMA for the EDF. BUSCO's inaccurate performance for EDF could be due to lower availability of a statistically significant number of EDF of a specific phylum and high diversity within phylum. We decided not to use this engine until a larger database of EDF annotated genomes is acquired and suggest periodic evaluation of BUSCO versus CEGMA for future EMEs studies.
Using CEGMA as a proxy for genome completeness, we evaluated the minimum number of single-cell amplified genomes necessary for each species to achieve a near-complete coassembly with quality comparable to isolated genomes obtained from unamplified DNA obtained from thousands of cells. To coassemble one species genome from multiple single-cell libraries, we tested various approaches for determining genome-to-genome distance (Figures 4 and 6 and Table 3 and Table S8). Once again, we found that only one of the three known approaches, GGDC tool, formula 2, performed the best for the widest range of species, described in the next section and technical details provided in transparent methods steps 4, 5, and 6: Phylogenetic and phylogenomic calculations. Owing to the random nature of the amplification bias in all but one species (C. protostelioides) with high GC%, to produce an improved species genome coassembly from single-cell assemblies, on average 3 single cells are necessary (Table S5); however, co-assemblies from best single- or multiple-cells genomes led to significantly improved results (Figures 4 and 6). Overall, we recovered a range of 5%–95% CEGMA from a single cell (median = 60%) and 60%–98% for coassemblies. The obtained single-cell and 10- to 100-cell genomes and coassemblies are examined for functional predictability in the final section.
Figure 4.

Intra- and interspecific variabilities
(A) Cryptomycota and Chytridiomycota 18S rDNA (region v6 to v9) ML tree based on the HKY85 nucleotide substitution model with bootstrap values shown above 60%.
(B) Assembled: Genome distance was calculated using GGDC formula 2, designed for incomplete isolated genomes (Auch et al., 2010; Meier-Kolthoff et al., 2013); the genome size shows the degree of variation in genome recovery between single-cell and multiple-cell sorts, and the core eukaryotic gene mapping approach (CEGMA) value reflects genome completeness. ∗Assemblies used for the genome coverage Circos plots. For all the other species, see Figure 5.
(C) Genome coverage shows mapping in 1,000-bp bins from individual select single-cell or multiple-cell libraries to the reference coassembled species genome.
See also Figure S6.
Figure 6.

Intraspecific single-cell genome variability and phylogenetic placement of single- and multi-cell genomes
Phylogenetic distance was estimated based on the 18S rDNA region v6 through v9 using PhyML package (Guindon et al., 2010). Genome distance was estimated using Genome-to-Genome Distance Calculator (GGDC), formula 2 (Meier-Kolthoff et al., 2013). See Table 3 for Genome-to-Genome Distance between genera.
(A) Zoopagomycota phylogenetic tree and GGDC, genome size, and completeness. Best nucleotide substitution model estimated HKY85, random starting tree, estimated best tree with bootstrap analysis, bootstrap shown values above 60%. Tree: Branches are shown as: Dc, Dimargaris cristalligena RSA 468; Ts, Thamnocephalis sphaerospora, Sp, Syncephalis pseudoplumigaleata; Pc, Piptocephalis cylindrospora RSA2659.
(B) Ascomycota single-cell phylogenetic tree and GGDC, genome size, and completeness. Best nucleotide substitution model estimated GTR+G+I, random starting tree, estimated best tree with bootstrap analysis, bootstrap shown values above 60%. Tree: Branches are shown as Mb, Metschnikowia bicuspidata in red with closest species in dark red.
(C) Compost ciliate single-cell phylogenetic tree and GGDC, genome size and completeness. Best nucleotide substitution model estimated HKY85+G, with bootstrap analysis, shown values above 60%. Branches for the single-cells are shown with: aqua, ciliate Protist (CiPr). Species with closest 18S are shown in dark teal. Non-Alveolata branches are shown in black.
Table 3.
Genome-to-genome distance estimates for eukaryotic genera
| Fungi | Distance: Scale: 0 to 1 | Distance: Scale: 0 to 1 |
|---|---|---|
| Ascomycota interspecific distance | Metschnikowia bicuspidata (crab parasite) | Metschnikowia fruticola |
| Metschnikowia bicuspidata yeast cell (Mby) (Daphnia pulex parasite used in this study) | 0.191 | 0.168 |
| Zoopagomycota intergeneric distance | Piptocephalis cylindrospora | Dimargaris cristalligena |
| Dimargaris cristalligena | 0.163 | |
| Syncephalis pseudoplumigaleata | 0.140 | 0.047 |
| Thamnocephalis sphaerospora | 0.160 | 0.167 |
| Piptocephalis cylindrospora | 0.163 | |
| Chytridiomycota Intergeneric distance | Cpi | Batrachochytrium dendrobatidis |
| Caulochytrium protostelioides isolate (Cpi) | 0.172 | |
| Blyttiomyces helices | 0.159 | 0.120 |
| Interphylum distance | Cpi (Chytridiomycota) | Batrachochytrium dendrobatidis (Chytridiomycota) |
| Rozella allomycis (Cryptomycota) | 1.000 | 0.145 |
| Mby (Daphnia pulex parasite used in this study) (Ascomycota) | 0.317 | 0.139 |
| Dimargaris cristalligena (Zoopagomycota) | 0.217 | 0.131 |
| Thamnocephalis sphaerospora (Zoopagomycota) | 0.200 | 0.160 |
| Piptocephalis cylindrospora (Zoopagomycota) | 0.200 | 0.141 |
| Syncephalis pseudoplumigaleata (Zoopagomycota) | 0.199 | 0.127 |
| Cilliate protists | Distance: scale: 0 to 1 | |
| CiPr_NSBU | 0.013 | |
| CiPr_NSBW | 0.013 | |
| CiPr_NSBX | 0.012 | |
| CiPr_NSBY | 0.012 | |
| CiPr_NSCG | 0.012 | |
| CiPr_NSCA | 0.012 | |
| CiPr_Co-assembly | 0.038 | |
| Astramina rara GCA_000211355.2_ASM21135v2_genomic | 0.086 | |
| Param_tetraurelia GCF_000165425.1_ASM16542v1_genomic | 0.134 | |
| Param_biaurelia GCA_000733385.1_ASM73338v1_genomic | 0.136 | |
| Param_sexaurelia GCA_000733375.1_ASM73337v1_genomic | 0.136 | |
| Sterkiella GCA_001273305.2_ASM127330v2_genomic | 0.142 | |
| Sphaeroforma GCF_001186125.1_Spha_arctica_JP610_V1_genomic | 0.158 | |
| Reticulomyxa GCA_000512085.1_Reti_assembly1.0_genomic | 0.161 | |
| Ichthyophthirius multifiliis GCF_000220395.1_JCVI-IMG1-V.1_genomic | 0.162 | |
| Naegleria_fowleri_1.0 GCA_000499105.1_genomic | 0.168 | |
| Tetra_borealis_V1 GCA_000260095.1_genomic | 0.178 | |
| Param_caudatum GCA_000715435.1_43c3d_assembly_v1_genomic | 0.179 | |
| Euglena gracilis GCA_001638955.1_Euglena_mito_Newbler_genomic | 0.180 | |
| Tetrahymena_thermoph GCF_000189635.1_JCVI-TTA1-2.2_genomic | 0.181 | |
| Trypanosoma GCA_000227375.1_ASM22737v1_genomic | 0.190 |
Whole-genome distance was estimated using genome-to-genome distance calculator (GGDC), formula 2 (Meier-Kolthoff et al., 2013). For the intraspecific distance estimates see Figure 6. Species names: CiPr, ciliate protist from compost followed by unique single-cell genome identifier. Ciliate genomes retrieved from NCBI have their NCBI genome identifier after species name.
QC criteria as predictors of genome quality and EME single-cell genomics pipeline efficiency
We examined all QC criteria described in Data S2 for the potential to predict genome quality (fragmentation, completeness, functional prediction power) and process efficiency (time plus reagent and sequencing cost). We divided the pipeline QC process into three parts: preassembly, assembly, and postassembly. For each part, we eliminated redundant and non-predictive criteria and obtained a nonredundant predictive criteria list (Figure 3).
For the preassembly, QC1 and QC2 criteria were assessed for their prediction power in target single-cell recovery (number of cells versus efficiency). QC3 criteria were examined as predictors for single-cell lysis efficiency and GAB, later found to be better predicted by QC6 criteria. QC4 criteria were used for examining contamination and target single-cell OTU confirmation. We found that the QC1 and QC2 TgE criteria were critical for the prediction of target-species single-cell recovery, but these criteria were not as critical for genome quality prediction. Based on the genome recovery rate correlation with TgE, we established three success categories. In the first category, TgE1 (target concentration in original sample) or TgE2 (post FACS enrichment) estimated at ≥50%, guaranteed a successful and cost-efficient recovery of multiple single-cell genomes. In the second group, with TgE between 2.5% and 50%, genome recovery was reliant on morphological differences between target and nontarget cells as well as lysis-amplification efficiency. In the third group, with TgE below 2%, a wider range of factors alone or in combination (including sample volume, target cell size, shape, refractive index, viability rate, and morphological difference with nontarget cells) proved to be important for the genome recovery rate. The QC3, SGA, criterion was predictive of lysis efficiency but not amplification bias (Data S3, Figures S2 and S6). Thus the correlation between SGA and CEGMA was found to be weak, ranging from 0.1 to 0.3 depending on the species (Figure S6 and transparent methods step 3). rDNA OTU screening at QC4 or QC6 proved to be indispensable for reducing the sequencing costs by excluding nontarget genomes and genomes with contamination levels affecting genome assembly and genome completeness. See transparent methods step 4 for more details.
For the prediction of the successful outcome of the assembly phase, criteria of the QC5 (NGS library quality) and QC6 (shallow NGS read quality) steps were examined as predictors of quality of the de novo assembly of the amplified single-cell genomes. In addition, the QC6 criteria were explored for OTU identification of targets and of contaminants or symbionts (transparent methods step 5). The QC5 and QC6 criteria were found to be good predictors of genome quality. The QC5 library insert size reflected sequencing success with an impact on assembly quality, with reads in the range of 300–500 bp providing the best results. The RTU value of QC6 was found to be the best predictor of amplification bias, % read contaminant for process contamination, and a number of Illumina read QC metrics for genome fragmentation and CEGMA value (transparent methods step 5 and Data S2). For fungal species with genome sizes from 10 to 30 Mb, the RTU correlation with CEGMA was positive and negative with FGA (Data S2), supporting its predictive value. Conversely, protist single-cell genomes with sizes 80–110 Mb and a very high RTU value had a strong negative correlation between RTU and CEGMA, due to the low variation and high values of CEGMA for each genome. In addition, we examined the correlation between shallow NGS read GC% and CEGMA and assembled genome size (AGS). For the whole set of species, no correlation between GC% and CEGMA or between GC% and AGS was found (Data S2). Nevertheless, in five fungal species, correlation between read GC% and CEGMA was positive, and the correlation between GC% and AGS was positive in four fungal species and the ciliate. Similarly, the correlation between GC% and FGA or between GC% and SGA in QC3 was low for the whole species set (Data S2).
Based on the observed heterogeneity between species, we investigated the role of the genome GC% in amplification bias, as well as the impact of amplification bias on the completeness of genome recovery. We compared genome assembly quality from bulk isolate unamplified DNA and single- and multiple-cell-sort amplified DNA from C. protostelioides and R. allomycis. These species had similar genome sizes and lysis-MDA efficiencies but the highest (68%) and lowest (35%) GC%, respectively. Genome assemblies for the isolate unamplified DNA from C. protostelioides had 55%–65% higher RTU than single- and multiple-cell amplified DNA (Table S9); similarly, the isolate genome size was 50% higher and CEGMA completeness was 20% higher. For the R. allomycis, RTU was 2.13% lower for the unamplified isolate genome and genome size was 26% percent larger than the MDA-amplified genome coassembly (11 Mb versus 9 Mb), whereas CEGMA was similar between amplified and unamplified genomes (Table S9). In C. protostelioides, amplification bias correlated with genome GC% (Figure S6 and Table S6). The average genome assembly GC% was 10%–15% higher for the unamplified than MDA-amplified genomes. A close-up examination of the areas that did not amplify revealed an average GC% of 68%, a number similar to the average GC% of the unamplified DNA genome assembly and 3% higher than the amplified coassembled genome (Figure S6 and Table S9). In R. allomycis, the GC% for the unamplified genome and MDA-amplified genome assembly was similar (34.5%–35%) (Table S9) and very low amplification bias (Figure 5). Both C. protostelioides and R. allomycis had a similar genome size, lysis-MDA efficiency, and final amount of amplified DNA, which suggests that FGA was not the cause of bias. Genome bias in C. protostelioides occurred during MDA amplification of the high GC regions (see the detailed description in transparent methods step 5 optimization and Figure S6 and Table S6).
For the other species, we could not isolate enough pure material to make libraries from unamplified DNA. Instead, we compared single-cell genome assemblies against the coassemblies with with the largest genome completeness scores. These species had a moderate GC% (38%–55%), and the difference between coassemblies and single-cell assemblies was insignificant. We observed in five of nine species that the single-cell amplified genomes coassembly had a much higher CEGMA than the best assemblies from either single- or multiple-cell amplified genomes (30–100 cells) (Figures 4, 6, 7 and S3). The average increase in CEGMA was 12% (range 0.2%–33%). In the case of Dimargaris cristalligena, CEGMA from a 50-cell amplified genome was already 98.7%, only 0.2% lower than that of the coassembly (Figure 7). Based on CEGMA analysis, completeness of the coassemblies or best individual assemblies for fungal genomes was estimated at an average of 91%, with a minimum of 75.5% and maximum of 98.9%; for the ciliate, the average was 94.3%. For genomes with a moderate GC% (38%–55%), FGA had the largest negative impact on genome coverage bias (Figure 7).
Figure 7.

Amplification bias for coassemblies and largest assembled genomes (from 1 or 100 cells)
(A) Genome completeness as CEGMA across species for best genomes.
(B) Correlation between fold amplification and CEGMA for coassembly and for largest genome.
(C–E) Correlation between assembly size and CEGMA. Correlation is Pierson value (R) for genome size and CEGMA. (C) For coassembly. (D) For 100-cells genomes where available. (E) For single-cell genomes.
See also Figure S4.
For the prediction of the postassembly outcome, we explored each of the QUAST (QC7) criteria and fold amplification and assembled genome size (Data S2). Genome quality was measured by CEGMA (Figures 4, 6 and 7) and the continuity of the assembled genomes by mapping each individual assembly to the reference genome shown in Figure 5. We found that the QUAST criteria MGS_N50 and MGS_L50 directly correlate with the CEGMA value (Data S2 and Tables S2–S4). The correlation between AGS and CEGMA within and between each species was positive, supported by the negative correlation between FGA and CEGMA (Data S2). However, we found a weak correlation between these values in the best single-cell or coassembled genomes across all species (Data S2). The highest genome fragmentation bias was observed in genomes with the highest amplification bias and did not correlate directly with any criterion. In summary, the pass and fail value of the QC criteria for de novo single-species genome assembly is shown in Table S10.
Intra- and interspecific variabilities in single-cell genomics and the impact on genome distance estimates for species coassembly
Comparative analysis of single-cell genome variability within and between studied species revealed that variability between species was higher than within species (Figures 4, 5, and 6). Among all impact factors, we found that high GC% (>63%) (Figure S6), poor lysis of cell walls, and small genome size each provided a basis for amplification bias and variability in genome coverage between single-cells (Figures 4, 5, and 6).
For example, species with similar, good lysis efficiency and GC%, but smaller genome size experienced higher GAB (Figure 5). Similarly, higher GC% in species with the same size and lysis efficiency led to higher genome fragmentation and poor recovery (Figure 4). Species with medium GC% and genome size, but poor lysis efficiency (M. bicuspidata) had fewer genomes recovered, but less GAB (Figures 5 and 6). Intraspecific variability was observed mostly between assemblies from different cell sorts (1 versus 10 versus 100) and supported a random intraspecific variation in genome coverage. Respectively, assemblies from multiple-cell libraries had higher CEGMA and lower fragmentation than did those from single-cell libraries. The observed intraspecific random variability had a beneficial impact on coassembly quality, resulting in a much higher species genome quality (Figures 4, 5, 6, and 8). However, for C. protostelioides, the variability between multiple-cell (100-cell) assemblies was as high as the observed variability between single-cell assemblies in other species (Figures 4, 5, and 6 and Table S9).
Figure 8.

Single-cell genome coassembly quality assessment for functional genomics studies
(A) Comparative analysis of annotated genomes. See also Table S7.
(B) Functional prediction value assessment. Izo, isolate unamplified genome assembly; COA, coassembly of several single- and/or multiple-cell assemblies, and 100-100 cell-sort genome assembly. For the fungi, the scale for the KEGG metabolic pathway signature was the same (0–678). For the CiPr (ciliate protist), the scale was 0–1252. FPi – functional genomics prediction index, where i = geomean % complete genes and % CEGMA coverage; dIA, absent entries in the amplified genome compared with the isolate genome. Abbreviations for species names are explained in Table 1. Detailed information for each KEGG entry is available in Table S9. KEGG peak numbers: 1 through 8 are category total, 9 through 18 are specific enrichments in subgroups of the respective category. 1. Amino acid metabolism, 2. Biosynthesis of secondary metabolites, 3. Carbohydrate metabolism, 4. Glycan biosynthesis and metabolism, 5. Lipid metabolism, 6. Metabolism of cofactors and vitamins, 7. Nucleotide metabolism and overview of biosynthesis of alkaloids and hormones, 8. Xenobiotic biodegradation and metabolism, 9. Tryptophan metabolism, 10. Biosynthesis of polyketides and nonribosomal peptides, 11. Biosynthesis of siderophore group nonribosomal peptides, 12. Starch and sucrose metabolism, 13. Lipopolysaccharide biosynthesis, 14. Pentose phosphate pathway, 15. Energy metabolism, 16. Nicotinate and nicotinamide metabolism (cyt p450), 17. Benzoate degradation via CoA ligation, drug metabolism cytochrome p450, gamma-hexachlorocyclohexane degradation, metabolism of xenobiotics by cytochrome p450, 18. Metabolism of other amino acids.
A requirement for creating species-level genome assemblies from multiple individual genome assemblies is determining the phylogenetic identity of individual genomes before coassembly. We compared the use of rDNA, ANI, and GGDC for intraspecific distance estimation for the purpose of creating species coassemblies (Figures 4 and 6 and Tables S3 and S8; for detailed description see transparent methods step 6 Optimization and steps 4, 5, and 6: Phylogenetic and phylogenomic calculations). Our tests found that GGDC formula 2 (Auch et al., 2010; Meier-Kolthoff et al., 2013) was the most reliable tool for establishing intraspecific distance for incomplete genomes obtained from EME single-cell amplified DNA, even though it was originally designed and tested only on unamplified DNA bulk isolate genomes. The most striking proof of formula 2's ability to correctly predict the genome distance for this group of eukaryotes is correctly placed C. protostelioides single-cell and 10-cell genomes, which had the same intraspecific distance as other species' genomes despite having much lower CEGMA values (Figures 4 and S8). Our results showed that the minimum CEGMA value necessary for accurate genome-to-genome distance prediction was lower by 10%, from the original publication (Auch et al., 2010; Meier-Kolthoff et al., 2013). Our GGDC formula 2 estimates were supported by phylogenomic placement of the C. protostelioides single-cell genome Figure S8 and discussed further in Ahrendt et al. (2018). We observed no negative impact of single-cell genome quality variability on genome distance estimates.
Genomes obtained via single-cell amplification are suitable for functional analyses
To determine if the assemblies produced by this pipeline were suitable for functional analysis, we annotated them and performed a comparative structural analysis and a functional prediction analysis (for technical details see transparent methods step 7). We annotated the best single- and multiple-cell assemblies and coassembled fungal and protist genomes using the MycoCosm annotation pipeline with manual curation when necessary (Grigoriev et al., 2014).
A comparative analysis of the genome structure, e.g., number of genes, gene density and intron/exon structure, and transcript and protein length, for each species revealed an average 372 genes/Mbp density, with small variability for the analyzed set of genomes (Table S7 and Figure 8). Highest gene density was observed in R. allomycis (497 and 535 gwnws/Mbp for coassembly and isolate, respectively). The lowest gene density was seen in D. cristalligena (242 genes/Mbp), the second largest fungal genome with the greatest intron length (Figure 8). Both are mycoparasites. The highest number of genes (12,167) was in the largest fungal genome Blyttiomyces helicus, whereas the average among fungi was 6,422 genes. The seven ciliate protist single-cell genomes were 2.5×–10× larger than the spread of fungal genomes used for the study (Figure 8 and Table S7). As expected, for this ciliate the number of genes was much larger adding up to 40,072 gene models (Tables S7 and S9). Nevertheless, gene density (331.5) was below the fungal average with slightly smaller transcript (880 bp), exon (144 bp), intron (49 bp), and protein (282aa) median length (Table S7). For fungi, average transcript length was 1,101 bp with little fluctuation between species. Average of the exon and intron length were 297 and 93 bp, respectively, ranging between 675 and 141 bp for exon median; intron median varied between 29 and 197 bp (Table S7). The longest exon median was observed in the smallest fungal genome, the yeast Metschnikowia bicuspidata. In the second and third largest fungal genomes (D. cristalligena, C. protostelioides) the intron length was the first and second highest and exon was the third and second highest. Most of the EDF had, on average, 75.5% spliced transcripts, and the median per spliced gene for introns and exons was 2.7% and 2.8%, respectively. Caulochytrium made an exception and was more alike to the yeast Metschnikowia with only 38% and 24% spliced genes for each species with median one intron and exon per spliced genes. The ciliate and five of the fungi had equal and the highest numbers of introns per spliced gene, and four of these fungi and the ciliate had equal but fewer exons per gene. The percent of the spliced genes was similar between the ciliate and the five fungi, whereas the number of spliced genes was significantly smaller in fungi, somewhat correlating with their genome size. In spite of the aforementioned variability, protein length median average was 305 aa, with little variation between species.
We used the two fungal species unamplified “isolate” genomes (Cpi: C. protostelioides isolate, and Rai: R. allomycis isolate) to benchmark and evaluate the amplified single- and multiple-cell genomes, lacking isolate references, for suitability for comparative genomic analysis. We observed that both C. protostelioides and R. allomycis had a significant percentage of incomplete genes in the coassembly and single- or multiple-cell assemblies relative to their isolate genome (Tables S7 and S9). However, coassembly and multiple- or single-cell assemblies had fewer genes and exon, transcript, and protein lengths for C. protostelioides but not for R. allomycis (Tables S7 and S9). On the other hand, the gene density was the same in the isolated genome and the coassembly genome for C. protostelioides and differed between those for R. allomycis (Figure 8 and Table S7). This variation perhaps influenced the estimation of the % spliced genes between isolates and coassemblies.
Gene model support showed that for the fungi and ciliate protists, on average 60.2% and 65% of models had homology to KEGG database proteins and 68% and 61% to Swiss-Prot proteins, respectively (Figure 8 and Table S9). The highest number of supported models was observed in Metschnikowia (95% KEGG and 86% Swiss-Prot) and the lowest in Blyttiomyces helicus (28% KEGGs and 42% Swiss-Prot) (Figure 8). In fungi the number of complete genes (from start codon to stop codon) was higher than in ciliate protists (average 74% and 56%, respectively). However, Caulochytrium had the lowest number of complete genes for the amplified genome (55%) despite similar number of hidden Markov models-supported Pfam with Rozella, Blyttiomyces, and the ciliate protists (Table S9 and Figure 8).
We used the KEGG database, gene model support and completeness, and CEGMA values (Table S9) to create a functional genomics prediction index (FPi) for this purpose (Figure 8 and Table S9). Furthermore, we assessed the quality of the coassemblies relative to the isolate annotation for the benchmarked genomes using the presence/absence score (dIA) for each KEGG category. The dIA was calculated by subtracting the number of models for each KEGG -EC (enzyme commission) pathway map of the amplified genome from the isolated genome. An average and a mean value were obtained for the entire set of KEGG ECs for this calculation (Table S9).
Our analysis of the functional genomics prediction power, using the FPi and dIA, showed (Figure 8B) that a conservative 80% cutoff for FPi value characterizes a genome nearly identical to the isolated genome in terms of KEGG values (profile and dIA). We observed a significant alteration of the KEGG profile and dIA values when FPi dropped to 66% or less. As shown in Figure 8, Rozella coassembly and multiple-cell assemblies resulted in annotations that produced a significantly lower dIA score compared with the Caulochytrium coassembly, supporting our FPi score cutoff. We observed that when the FPi value was between 70% and 80%, KEGG gene counts were reduced without changing the pattern qualitatively (e.g., presence-absence) (Figure 8 and Table S9). For example, for the most biased amplified genome (C. protostelioides), the KEGG number of genes from the coassembly was significantly lower than in the isolate, making KEGG inaccurate for gene expansion-reduction analysis. In contrast, for Rozella, the KEGG pattern was nearly identical between the coassembly and isolate. In Rozella, nearly identical numbers between the coassembly and isolate for KEGG were accompanied by similar CEGMA completeness and low GC% (34%), as well as the number of complete genes in Rozella was higher than that in the Caulochytrium coassembly (67% versus 54%).
We found that the combination of <60% complete genes, lower than 90% CEGMA, and high average GC%, as observed for the coassembly of Caulochytrium, led to lower-than-acceptable scores for reliable quantitative functional predictions (at least for KEGG-based gene counts).
Four fungal single-cell genome coassemblies without an isolate (e.g., Metschnikowia, Dimargaris, Thamnocephalis, and Syncephalis) had an FPi score above 80%. They had nearly complete CEGMA values and average GC%, further supporting FPi-inferred predictions for highly accurate KEGG profiles. In the three other species, Piptocephalis, Blyttiomyces, and the ciliate protist, a lower FPi (70%–80%) and either a lower CEGMA value (fungi) or lower percent of complete genes (protist) predicted the identified KEGG numbers to be underrepresented. However, given that the FPi for these three species was within 10% of the reliable interval (70%–80%), qualitative but not quantitative analysis would reflect the same functional predictions as their respective isolated genomes.
As an example of the types of functional analyses possible with our genomes, we investigated expansions of KEGG groups. In two fungal species (Dimargaris and Caulochytrium) and the ciliate, we found expansions in tryptophan metabolism, biosynthesis of polyketides and nonribosomal peptides, and biosynthesis of siderophore group nonribosomal peptides (Figure 8B, peaks 9, 10, and 11, respectively). In addition, Caulochytrium showed an expansion in the starch and sucrose metabolism category, whereas several other fungi had an increased count of genes from the energy metabolism category. An in-depth examination of the gene expansion and losses and functional implications for the lifestyles of these single-cell-derived eight fungal genomes was given in our recent article (Ahrendt et al., 2018).
Unlike fungi, ciliate protist genome displayed an expansion in the number of genes involved in xenobiotic biodegradation and metabolism, specifically benzoate degradation via CoA ligation, drug metabolism-cytochrome P450, gamma-hexachlorocyclohexane degradation, and metabolism of xenobiotics by cytochrome P450 (Figures 8B and Table S9). The ciliate displays a unique expansion in the pentose phosphate pathway and the lipopolysaccharide biosynthesis categories. An in-depth functional analysis of the ciliate protist comparative genomics in the context of a group of 180 species was developed in a seprate manuscript by Ciobanu et.al., (in preparation).
Discussion
In this article we review, test, and optimize single-cell genomics approaches for studying microbial eukaryotes in their natural environment. As a result, we developed a single-cell pipeline for mining the genomes of EMEs by combining known, optimized, and novel wet-bench approaches with bioinformatics tools. Our results empowered large-scale functional genomics studies that shed light on the ecological niches of EMEs and uncovered their functional plasticity (Ahrendt et al., 2018 and submission ciliate).
We benchmarked the pipeline against two fungal isolate genomes (R. allomycis and C. protostelioides) using a set of eight known fungal species, three environmental samples with unknown EME species, and a set of 20 quality check criteria. We evaluated (1) the steps that had the largest impact on genome completeness, (2) the criteria that were most predictive of the best genome quality and completeness, and (3) the correlation between genome completeness and each evaluation criterion used. As a proof of concept, we used this pipeline for genome recovery of unknown EME Chromista/SAR (Ciliophora) and fungi (Cryptomycota and Chytridiomycota) species. Neither the rDNA nor the genome of the ciliate could be recovered using standard metagenome sequencing and assembly methods in previous studies (Eichorst et al., 2013; Luo et al., 2012). The fungal EME samples had an rDNA phyla profile but were microscopically undistinguishable from other non-target organisms and were at an ultra-low concentration. Complex genomes from novel lineages that are subjected to partial chimerization during amplification require correct bioinformatics tools for genome assembly, genome-to-genome similarity estimation, genome completeness assessment, correct gene structure prediction, and annotation from fragmented and/or incomplete genomes. We tested a number of bioinformatics tools (Auch et al., 2010; Bankevich et al., 2012; Butler et al., 2008; Han et al., 2016; Meier-Kolthoff et al., 2013; Peng et al., 2012) for this purpose and, when necessary, adjusted the most suitable to achieve the aforementioned results.
The pipeline allowed recovery of high-quality genomes from individual cells with a CEGMA median genome completeness of 60% (range 5%–90%). We found that the EME target single-cell recovery rate and genome quality increased when an enrichment FACS step was performed before single-cell isolation, during presequencing part of the pipeline. For sequencing, one simple but critical improvement was the implementation of a shallow sequencing step before the deep sequencing step required for de novo assembly. Shallow sequencing for amplification bias estimation has been proposed previously (Daley and Smith, 2014). Our pipeline, unlike the suggested method in Daley and Smith (2014), used the read-QC-pipeline RTU metric to estimate amplification bias as well as the contamination level and a number of other parameters indicative of the sequence quality. We found that the best predictor for amplification bias and the highest measure of genome completeness out of all criteria used during this step was RTU. In addition, we tested the use of shallow sequencing reads for rDNA assembly followed by OTU screening. This eliminated issues related to PCR primer bias (Lazarus et al., 2017) or Sanger sequencing and allowed us to identify cases where symbiotic organisms were present despite any issues of rDNA divergence or contaminating OTUs. Several of the commonly used criteria (Gurevich et al., 2013) describing genome assembly quality (number of scaffolds in the range of 2–10 kb, 10–25 kb, and 25–50 kb, main genome scaffold _N50) correlated well with CEGMA and genome size.
We identified critical factors affecting the EME recovery rate using a range of QC and optimization steps. The main bottleneck for successful enrichment was a combination of extreme cell size and cell shape (narrow elliptical, 2 × 50 μM) along with a low sample volume (1–2 mL) or when nontarget organisms with similar morphology to the target were present at a higher concentration in the sample (e.g., more Cryptophyta (flagellate algae) than Cryptomycota (flagellate fungi), along with other organisms with the same size). Poor lysis efficiency was found to affect the number but not the quality of amplified target genomes (e.g., M. bicuspidata, B. helicus, T. sphaerospora, and S. pseudoplumigaleata). The most striking supporting example was zoospores of C. protostelioides, where despite the high number of successfully lysed and amplified cells, we observed the highest amplification bias, likely due to the higher-than-average GC (68%). In contrast, M. bicuspidata with yeast cell walls was lysed at a significantly lower percentage but had 90% higher CEGMA values for single-cell amplified genomes and 30% higher values for 100-cell sorting than C. protostelioides zoospores. Both species had a small genome, 13 and 11 Mbp, but M. bicuspidata had 10%–15% lower GC% than C. protostelioides, contributing toward resulting genome quality. Although a universally efficient method for opening cell walls in a single-cell reaction remains to be found for maximizing the number of EMEs recovered from environmental samples, our work found that the main culprit for lower genome quality was amplification bias. Amplification bias in smaller genomes has been reported previously (Dean et al., 2002; Gawad et al., 2016 and references), but the mildly high (68%) GC of the genome causing extreme MDA bias was unexpected. Several articles have indicated a mild bias for the MDA reactions caused by higher GC% (Garvin et al., 2015; Xu et al., 2014), whereas others did not find a similar correlation (Ellegaard et al., 2013). Given the strand displacement ability of the phi29 DNA polymerase that allows unwinding of DNA without nucleotide bias, it is possible that structural features (e.g., the presence of more regulatory or chromatin organization protein complexes in the higher GC% regions) were the true cause of the amplification bias observed in C. protostelioides. The genomes with an average GC% of 35%–55% and larger genomes had low amplification bias and the highest completeness.
Overall, we looked at 20 potentially predictive criteria for the pipeline outcome. Majority of criteria were highly predictive, and we reduced the redundant ones. Several were weakly predictive: The SGA and FGA (reflecting the duration of amplification and DNA amount) were weakly inversely correlated with genome completeness (assessed by CEGMA); the strength of predictability of the latter two was lower than that of the RTU. Although RTU was a good predictor of genome quality for most species, we observed that for genome size >60 Mb with RTU >80%, the RTU did not correlate with genome completeness, thus setting up the highest threshold for the RTU predictability. The largest amplification bias was observed for genomes ranging in size between 10 and 30 Mb. These genomes would benefit the most from significantly reduced amplification times. For larger genomes (e.g., 30–50 Mb), this trend was also observed but was not as strong. Supporting this observation, for the seven >100-Mb protist genomes, the RTU was very high and not correlated with CEGMA. For a generalized prediction, we speculate that the RTU reflects amplification bias for genome sizes smaller than 60 Mb using our MDA protocol (3–4 h amplification), whereas for genomes larger than 60 Mb, this is not the case. Based on this study, we suggest that for unicellular protists and genomes with size >60 Mb, a larger study is necessary to include a broader spectrum of genome average GC% to understand the impact of GC% on amplification bias.
To streamline our QC process, we reduced the set of 20 criteria to six that span the preassembly, assembly, and post-assembly pipeline steps. Based on the set of criteria with the highest predictive value, we found that EMEs with genome sizes ranging from 10 to 30 Mb clustered around 40% genome completeness. This group was the largest of the dataset and had the most statistical support (70%) for genome completeness prediction power. Genomes from 30 to 60 Mb clustered at approximately 70% genome completeness, and genomes larger than 100 Mb clustered at approximately 90% completeness. However, the proportion of organisms with larger genomes was smaller in our dataset, and therefore, the statistical support for genome completeness prediction power was lower (25%) for them (Figure 3). Several outliers supported the observation that specific combinations of factors could affect genome completeness. (1) The yeast M. bicuspidata had an extremely high CEGMA value despite its small genome size and poor lysis, perhaps due to low GC%. (2) B. helicus and (3) D. cristalligena are partial outlier species from the other EDF. Both of them, regardless of the poor start of amplification, produced the largest fungal genome sizes of the dataset with very high CEGMA values. We conclude that for smaller genomes, reducing genome amplification time paired with amplified Illumina libraries would improve genome recovery.
It has been shown (Kogawa et al., 2018) and we confirm here that completion of a species genome could be achieved via coassembly of individual genomes of the same species. A prerogative for species-specific coassembly for environmental single-cell genomics is establishing the correct genome-to-genome taxonomic distance. No tools for EME genome-to-genome similarity have been evaluated to date. We found that the best tool for intraspecific and intragenic genome-to-genome distance calculation was the GGDC from DSMZ (Auch et al., 2010; Meier-Kolthoff et al., 2013). Originally developed for unamplified prokaryotic genomes, this tool set provides several formulas suitable for various levels of genome completeness. Our tests showed that formula 2 was highly suitable for amplified eukaryotic genomes. Several authors (Auch et al., 2010; Meier-Kolthoff et al., 2013) have reported that this formula performed well with prokaryotic genomes with as low as 20% completeness, and in our study, we found that formula 2 performed accurately with amplified single-cell fungal genomes with as low as 5.9% completeness. We found that, for the most part, the quality of the coassemblies and, in some cases, single-cell as well as 10- to 100-cell genomes was comparable to that of unamplified genomes (derived from millions of cells) and could be used for comparative genomics.
Reaching genome completeness for single-cell genomes whose quality is close to that of isolated DNA genomes empowers comparative genomics studies to make meaningful functional predictions. Using our benchmarking species with isolated genomes, we proposed a new criterion called FPi to be able to estimate functional prediction value. This criterion is based on three separate genome quality standards for de novo assembled single-cell genomes. We conclude that for EME single-cell genomes that suffered serious amplification bias, it was necessary to coassemble multiple partial genomes to achieve a meaningful functional prediction score. We found that for genomes with high amplification bias, coassembling three 100-cell sorts was necessary; for moderate amplification bias, three to five single-cell sorts were sufficient; and for those with low amplification bias, single-cell assemblies were sufficient. The example of a broad and in-depth analysis of the functional value of single-cell genomics based on this fungal set of genomes was presented in our complementary article (Ahrendt et al., 2018).
A challenge not discussed so far anywhere is the costs associated with the high sequencing capacity necessary for EMEs. Given that EMEs are significantly underrepresented in the environment compared with their prokaryotic neighbors (Wurzbacher et al., 2017), mining EMEs with existing methods would require significantly more resources, starting with sample volumes and ending with computational resources, including all the steps in between discussed in this article. For example, unlike environmental prokaryotes with genomes 100-fold smaller than those of eukaryotes on average, mass sequencing of poor-quality genomes or nontarget genomes for EMEs can become prohibitively costly, even on NovaSeq when attempting broader and deeper phylogenomic mining. Therefore, establishing methods for evaluating the quality of the genome before deep sequencing is critical for affordable high-throughput EME genomics. We provide several optimizations that allow affordable exploration of EME genomics in the context of their ecological niche. We provide analytical methods to explore factors and predictors of successful genome recovery across broad-spectrum taxa of diverse genome size, GC%, cell wall composition, and phylogenetic origin. Following the general idea of using shallow sequencing as a prediction tool at an earlier step, we found a new highly predictive metric for GAB and made several highly effective changes to the amplification and screening process of single-cell genomes before the deep sequencing step, which allowed us to reduce costs and significantly improve the genome quality of the EMEs. For example, just implementing the screening after the shallow sequencing step, using HighSeq Illumina technology for deep sequencing, savings are 10-fold for smaller genomes or more for larger genomes in this study; for NovaSeq Illumina technology savings are less, but still over 10-fold for larger genomes. Considering that for a good genome de novo assembly from amplified reads we needed 50 million reads or more for 60-Mb genomes, blind sequencing on NovaSeq a full MDA plate (288 cells, or 60% of this, if taking only positive MDAs) is still costly, whereas implementing rDNA-OTU, RTU, and contaminant screening step reduced the high-quality targets to 4–6 per plate and on average 30 lower quality targets per plate. Although in this study we did not analyze the non-target EME genomes from the three environmental samples, in a broader targeted study the outcome of the pipeline would be higher. Another application of this pipeline, especially the use of shallow sequencing step rDNA or other marker genes assembly, would be larger phylogenetic studies of the tightly associated community of the target EMEs. Thus, our study offers an avenue to increase the resolution of microbial communities and allow for functional prediction of the role of EMEs in their environment at the next level of depth and breadth.
Limitations of the study
We established the target single-cell EME genome recovery limits for this pipeline. The most critical limitation is the ultra-low concentration of the target EME and the same size of the most abundant non-target prokaryote and eukaryote organisms in the same sample. The second most impactful limitation is caused by amplification bias of the high GC% genomes (here above 65%). The third less impactful limitation is amplification bias caused by fold amplification of the small genomes, which can be reduced by overall reduction of the amplification time in step 3 for all EME genomes, regardless of the knowledge about their genome size. The fourth limitation is the organisms that have a size exceeding 100 μM diameter due to the FACS limitation. This can be managed by replacing FACS with LCM or micromanipulation, but reduces the high-throughput aspect.
Resource availability
Lead contact
Doina Ciobanu, dgciobanu@lbl.gov.
Materials availability
No new unique reagents were generated in this study. Reagents sources are listed in transparent methods.
Data and code availability
The coassembled genomes and annotations of the target species reported in this paper are available through "MycoCosm:https://genome.jgi.doe.gov/fungi" using the following "MycoCosm:URLs" and "GenBank: accession numbers" reported in this paper are respectively: R. allomycis CSF55 single-cell "https://genome.jgi.doe.gov/Rozal_SC1; "GenBank: QUVT00000000", B. helicus Perch Fen single-cell "https://genome.jgi.doe.gov/Blyhe1; "GenBank:QPFV00000000", C. protostelioides ATCC 52028 single-cell "https://genome.jgi.doe.gov/Caupr_SCcomb; "GenBank:QUVS00000000", D. cristalligena RSA 468 single-cell "https://genome.jgi.doe.gov/DimcrSC1; "GenBank:QRFA00000000", P. cylindrospora RSA 2659 single-cell "https://genome.jgi.doe.gov/Pipcy3_1; "GenBank:QPFT00000000", T. sphaerospora RSA 1356 single-cell "https://genome.jgi.doe.gov/Thasp1; "GenBank:QUVU00000000", S. pseudoplumigaleata Benny S71-1 single-cell "https://genome.jgi.doe.gov/Synps1; "GenBank:QUVV00000000" and M. bicuspidata single-cell "https://genome.jgi.doe.gov/Metbi_SCcomb; "GenBank:QUVR00000000". The whole-genome sequence for the non-single-cell isolate C. protostelioides ATCC 52028 is available through "MycoCosm:https://genome.jgi.doe.gov/Caupr1" and "GenBank: QAJV00000000"". The whole-genome sequences for the non-single-cell isolate of R. allomycis CSF55 was not determined in this study and is available through "MycoCosm:https://genome.jgi.doe.gov/Rozal1_1") and "GenBank:ATJD00000000".
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
We would like to thank Meghan Dufy for the Metschnikowia sample; Joyce E. Longcore for the Blyttiomyces helicus sample; Christopher Daum and Mathew Zane for providing valuable information for sequencing optimization; Eugene Goltsman for valuable discussion and suggestions for data analysis and visualization; Catherine Adam and Yi Peng Lee for library qPCR; Laura Sandor, Jenifer Kaplan, and Jennifer Chiniquy for running Illumina and PacBio sequencing instruments at JGI during this project; Bryce Foster for running the JGI RQC pipeline; and the NERSC facility for providing excellent service.
The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. S.R.A., I.V.G., T.Y.J., and C.A.Q. are supported by NSF grant DEB-1354625. G.L.B., M.E.S., and T.Y.J. were supported by NSF grant DEB-1441677.
Author contributions
D.C. wrote the manuscript with valuable input from T.Y.J., T.W., I.V.G., A. Copeland, S.A., and C.A.Q.
T.Y.J. and I.V.G. initiated this study.
J.-F.C., D.C., T.Y.J., and I.V.G. designed the study.
J.-F.C., D.C., I.V.G., T.Y.J., A. Copeland, and A. Clum coordinated the implementation of pipeline segments into one process.
D.C. and S.C. designed and performed wet-bench protocols and pipeline evaluation.
A. Clum, A. Copeland, W.B.A., S.A., and B.F. designed and performed genome assembly tool testing, optimizations, final assemblies, and assembly QC.
W.A. and C.A.Q. performed rDNA assembly tool testing and optimization.
J.P.M.-K. and D.C. performed GGDC tool testing for protists and fungi.
P.S. and W.A. performed ANI tool testing.
A.S. and S.A. performed annotation tool testing, annotations and QC.
Y.T.T., G.L.B., M.E.S., T.Y.J., S.W.S., and C.A.Q. performed sample collection and preservation.
K.B. and S.D. performed data tracking.
Declarations of interests
The authors declare no competing interests.
Published: April 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102290.
Contributor Information
Doina Ciobanu, Email: dgciobanu@lbl.gov.
Jan-Fang Cheng, Email: jfcheng@lbl.gov.
Supplemental information
Highlighted in red are the best results.
Nucleotide identity analysis for seven single-cell amplified genomes from ciliate protist. Note that on average only 67% of the assembled genome had enough similarity for the ANI formula. Coverage heatmap shows the percentage of the genome that was used for the ANI calculation, based on the cutoff >70% identity over >70% of the fragment, fragment size 1,020 bp. ANI for genome part that has >70% identity over >70% of the fragment size 1,020 bp.
A through D and G through M. Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target concentration.
(A) Rozella allomycis (Cryptomycota) sample. Microscopic examination shows a monoculture with highly motile spores. Upper inset picture: received photo ID before shipping on ice, phase microscopy under cover glass. Lower inset picture shows two magnified zoospores. For a movie showing zoospore motility see file: Video S1.
(B) Rozella allomycis (Cryptomycota) Top panel and table: sample FACS examination reveals the presence of other organisms (gate P7). Middle panel: the gate with target organism (P9) is chosen and sorted into a tube with 1x PBS. Microscopic verification of the Syto9 DNA-labeled FACS-isolated single cell sort of target flagellate spore from gate P9 is shown in the picture above the panel. Lower panel: Purified population of the target organism (gates P6 and P9) is used for isolation of individual cells into 384-well plates.
(C) Caulochytrium protostelioides ATCC52028 (Chytridiomycota) sample microscope examination shows a monoculture with motile spores. Upper right inset: Phase transmitted light image of the zoospore shows 3 × 5 μm body size with 20-μm-long flagella; received photo ID before shipping on ice. Upper left inset: Magnified zoospore transmitted direct light image after receiving and thawing the sample. Lower left and right pictures: Sorted single cells after FACS purification (see next page) maintain motility.
(D) Caulochytrium protostelioides ATCC52028 (Chytridiomycota). Upper panel and table: FACS examination of the sample reveals the presence of a significant number of other organisms. The gate (P1) with target organism was chosen and sorted into a tube with 1× PBS. Second panel and table: Purified population (gate P1) of target organism was used for isolation of individual cells into 384-well plates. Image of Syto9 DNA-labeled FACS-isolated target zoospore from GateP1 is shown in Figure S1c.
(E) Blyttiomyces helicus sample microscopic examination shows a heterogeneity of microorganisms, likely bacterial cells of various size visible under phase transmitted light with occasional target fungal cells. Phase microscopy and Tubulin Tracker fluorescent DNA label was used for primary assessment of the sample complexity and target concentration. Tubulin Tracker fluorescent DNA label microscopic examination allows to assess organisms that have flagella among prokaryote enriched sample. Lower left inset: Phase transmitted light image of the zoospore shows 5 μm body size with 30-μm-long flagella, received photo ID prior shipping on ice.
(F) Blyttiomyces helicus sample FACS examination confirms microscopic assessment, showing high enrichment in organisms smaller than 2 μm (gate P6). Population enrichment FACS assessment shows a very low enrichment for target zoospore gate P7 and P8. Direct sorting without enrichment purification step was performed due to low concentration (table P8) and small volume (2 mL) of available sample.
(G) Dimargaris cristalligena RSA 468 sample microscope examination shows an almost pure sample, highly enriched with spores. Upper left inset shows received photo ID before shipping on ice; phase contrast image of a pear-shaped spore with 5 × 8 μm size. Lower right inset: transmitted light bright field with target pear-shaped spores magnified.
(H) Dimargaris cristalligena RSA 468 FACS examination. Upper panel and table: sample shows a distinct population of target sample (gates P4, P5) and the presence of smaller organisms and small unlabeled particles. Lower panel, table and picture: FACS image of the cleaned P5 population used for single-cell sorting into 384-well plates. Transmitted light image of sorted spores from cleaned population from gate P5.
(I) Piptocephalis cylindrospora RSA2659 sample. Microscope examination of the sample under fluorescent light reveals a large number of non-target cells of varied morphology. Upper right inset: shows received photo ID before shipping on ice. Lower picture and lower right inset: Sample examined under transmitted light looks homogeneous.
(J) Piptocephalis cylindrospora RSA2659 FACS examined sample. Top panel and table: Syto9 fluorescent DNA label used for primary assessment of the sample complexity and target enrichment shows that the target population is dominant with a large portion that does label and two smaller populations with lower fluorescent intensity and size. Middle panel: Clean target P4 gate-sorted population into tubes was further assessed sorting three gates (P2, P3, P6). Lower panel and microscope transmitted and fluorescent light images: Clean target was sorted under best combination of settings for pure single-cell sort and used for sorting into 384-well plates.
(K) Syncephalis pseudoplumigaleata Benny S71-1 sample. Upper Left and middle insets: received photo ID before shipping on ice. Upper right: Transmitted light microscopy revealed 50-μm large sporangia. Magnified sporangia head bursting open, containing 6-μm oval-shaped static single-cell spores; the sample shows some other small cells consistent with FACS assessment; see Figure S1m. Lower transmitted light microscopy picture with three insets shows a clump of sporangia surrounded by a dominant population of uniform spores. Sample was filtered through 35-μm mesh filter before sorting to eliminate sporangia.
(L) Syncephalis pseudoplumigaleata Benny S71-1. Upper panel and table: FACS sample after filtration of sporangia shows a dominant population (P5 and P4) of an organism of the targeted size and shape. Middle microscope picture and lower FACS panel show enriched purified population used for single-cell isolation. Right-side insets: Magnified 3 × 6-μm spore with distinctive morphological features, transmitted light top and phase1 bottom.
(M) Microscope examination of the Thamnocephalis sphaerospora RSA 1356 sample under transmitted light shows 20-μm sporangia (shown magnified in the inset image) containing 6-μm round single-cell spores among hyphae. Right side insets: received photo ID from the collaborator, phase microscopy shows hyphae with sporangia, isolate sporangia, and a spore. Lower panel: sorted single-cell spores from FACS-cleaned sample (see next page). Magnified single-cell sort isolated spore with visible dense chromatic regions.
(N) FACS examination of the filtered Thamnocephalis sphaerospora RSA 1356 sample shows a large population of smaller than target cells and unstained debris. Upper panel and table: Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target enrichment. Lower panel: Target population (gates P7 and P8) sorted into a tube is shown on the transmitted and fluorescent light microscopic images.
(O) Metschnikowia bicuspidata sample was available in limited amount (2 mL). Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target concentration. Microscope fluorescent (upper left image) and transmitted light (lower right image) examination of the sample showed a mixture of the parasite yeast cell, ascospores and other organisms and particles. Upper right inset: received photo ID before shipping on ice, Phase transmitted light shows 2.5 × 13-μm yeast and 2 × 60-μm ascospore cells.
(P) Metschnikowia bicuspidata sample. Upper panel: FACS examination showed a large population of small organisms and two distinct small populations: P1 and P2 of larger-size organisms. Lower panel: Transmitted light and fluorescent light images of sorted cells from P1 (yeast), P2 (ascospore), and unknown P4 population. Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target enrichment. (R) Compost samples with cysts that appear only after mc-cellulose addition. Unknown cysts were designated as target (20-μM cysts later shown to be a ciliate, see Figure 6C, whereas the 5-μM cysts were identified as fungi Cladosporium during the rDNA-OTU screening step). Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target enrichment. Left side pictures: compost sample enriched with microcrystalline cellulose displays a wide range of organisms with a large population of 15–20 μm round cysts. Right-side pictures: same sample after a number of filtration steps (see transparent methods step 1 optimization) displays mostly identified target cysts and prokaryote size microorganisms. This sample was used for the first round of FACS cleaning and target enrichment.
(S) Clockwise: FACS of the filtered compost sample identified two distinct populations: shown in green and red in the left-side panels and microscope pictures below. Upper right panel shows FACS population after first round of sorting, and the panel below with the microscope picture with yellow frame shows the sample used for single-cell sorting into the 384-well plates.
(T) Top down: Gosling and Third Sister lakes sample screening for rDNA-OTU target phyla presence and concentration. Target phyla were unknown Cryptomycota and Chytridiomycota. Microscopic evaluation of the sample. Syto9 stains DNA and shows the ensemble of the organisms. Tubulin will stain the target phyla and other eukaryotes with flagella or tubulin. FACS + itag target sample concentration estimate. Bar graph on the left shows the results of the populations sorted on the right: One 384-well plate (288 sample single-cells) was sorted per each color (population). Only P8 (yellow) sorted on fluorescence and side scatter and P4 (dark blue) sorted on fluorescence and forward scatter yielded one target genome. Each population was sorted on Syto9, WGA, and Tubulin. Only Tubulin harvested target cells.
(U) FACS enrichment of the populations of the Gosling and Third Sister lakes samples: P6 population shown is the P8 in the previous panel. Gosling lake 100-cell/well and 500-cell/well, Tubulin sorted for rDNA i-tag estimate of the target species recovery rate. This rate of genome recovery is below the efficiency threshold of the pipeline. To improve pipeline efficiency for ultra-low abundance targets, several optimizations are described in steps 4 and 5 of the transparent methods. They allowed the recovery of 7 new target genomes studied in a separate paper focused on further optimization for ultra-low abundance EMEs.
(A) Principal-component analysis monoplots for the full set of quality check criteria. A 90° angle shows no correlation, a 180-degree angle shows inverted correlation, and an angle less than 45° shows strong correlation. The total dataset of 20 quality check criteria used for the study was split into 2 groups for comprehensive visualization: Upper row shows set 1a. Lower row shows set 1b. Set 1a and set1b have common QC criteria for reference purpose: MGS_L50, MGS_N50, SCFF_2.5kb. Set 1a upper monoplots were generated based on scores from 12 quality check criteria, and set 1b lower monoplots were generated using additional 8 criteria plus three from the above dataset. Shown QC criteria: AGS, assembled genome size; EGS, estimated genome size; SGA, start of the genome amplification; FGA, fold genome amplification to 0.5–1 μg; GC%, average GC percent of the assembled genome; RTU, random 20-mer uniqueness; CEGMA, core eukaryotic genes mapping approach; %GS > 50 kb, percent genome in scaffolds over 50 kb; gap%, assembled genome scaffold gap percent; MGS_N50, main genome scaffold N50; MGS_L50, main genome scaffold L50; SCFF_N(10-500kb), number of scaffolds between preceding bin and shown bin number, e.g., SCFF_2.5kb, number of scaffolds between 2 and 2.5 kb and SCFF_5kb, number of scaffolds between 2.5 and 5 kb, etc.
(B–E) Clockwise: scatterplot correlation matrix with density ellipse and plot. Color-coded genomes with species names abbreviation and 1, 10, 50, 100 cell sorts on the right of the abbreviation. Full species names are given in Figure 3 and Data S2F. Covariance scores from set1a 12 quality check criteria. Ranked correlation matrix, shown are Pearson (r), Spearman (rs), and Kendall (tau) correlation values. Darker color in the table indicates a stronger correlation with red showing negative and blue positive correlation values. A positive correlation on matrix will correspond to the diagonal ellipse orientation from zero to infinite, and a negatively correlated data will cluster in an ellipse oriented perpendicularly to the positive correlation ellipse. B. Set1a of the 20 QC criteria for all species genomes. C. Set1b of the 20 QC criteria for all species genomes. D. Reduced set of QC criteria used in Figure 3, for fungi-only species genomes. E. Reduced set of QC criteria used in Figure 3, for all species genomes.
(F and G) Gower & Hand PCA biplot represents similarity between data points, with smaller distance higher similarity. Shown plot explains 80.2% of the variability for fungi only. Any point on the plot projected orthogonally onto the axes will show the approximate value of that variable. Percent at the end of the axes labels indicates predictability value of the axes. Axes with predictability values below 60% are masked in gray.
(F) Predictability value of the QC criteria used, for fungal species only.
(G) Predictability value of the criteria used, for all species.
X axis shows time of MDA amplification and has same scale for all images in one panel. Reference black bar shows earliest start of amplification time point in the same panel. Y axis shows fluorescence level that represents a combination of optical refraction dependent on reaction volume, DNA amount, air bubbles, and other factors that can affect the optical intensity of signal. Stock alkaline solution was 500 mM with 10 mM DTT. All other alkaline solutions were diluted with ultrapure H2O. Lysis solution conditions: Alkaline∗ was 133 mM KOH +133 mM HCl, 10 mM final concentration in MDA mix; Alkaline 1 was 30 mM KOH final in MDA mix; Alkaline 2 was 6 mM KOH final; Heat in H2O: 98°C 1 min, 0°C 2 min, room t°C 3 min, 60°C 2 min, 0°C 2 min; Alkaline with heat: 98°C 1 min, 0°C 2 min, KOH at 10 mM final in MDA mix, 60°C 2 min, 0°C 2 min; Alkaline with heat: KOH at 10 mM final in MDA, 60°C 2 min, 0°C 2 min. Detergent was 0.3% Tween with 1mM EDTA.
(A) NEB MDA—lysis solution efficiency assessment using purified DNA. In all the tests negative controls that amplified are from the reaction mixes that contain the detergent (Tween 0.3%) used in the lysis buffer composition in this test.
(B) NEB MDA—lysis solution efficiency assessment using soil prokaryote single cells.
(C–F) Right side shows quantitative assessment of the lysis buffer efficiency for single-cells 10 cells, 100cells. (C) Gram-positive bacteria (E. coli) and Gram-negative bacteria (B. subtillis). (D) Rozella allomycis NEB MDA top three left tests and Qiagen MDA, bottom left test. (E) Caulochytrium protostelioides ATCC52028 Qiagen MDA, Alkaline lysis top is Qiagen kit protocol (see methods), Alkaline∗ was the same alkaline as used for the NEB-MDA tests in (D).
(F)Caulochytrium protostelioides ATCC52028 Qiagen MDA.
References
- Ahrendt S.R., Quandt C.A., Ciobanu D., Clum A., Salamov A., Andreopoulos B., Cheng J.F., Woyke T., Pelin A., Henrissat B. Leveraging single-cell genomics to expand the fungal tree of life. Nat. Microbiol. 2018;3:1417–1428. doi: 10.1038/s41564-018-0261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander W.G., Wisecaver J.H., Rokas A., Hittinger C.T. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proc. Natl. Acad. Sci. U S A. 2016;113:4116–4121. doi: 10.1073/pnas.1517242113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arriola E., Lambros M.B., Jones C., Dexter T., Mackay A., Tan D.S., Tamber N., Fenwick K., Ashworth A., Dowsett M. Evaluation of Phi29-based whole-genome amplification for microarray-based comparative genomic hybridisation. Lab. Invest. 2007;87:75–83. doi: 10.1038/labinvest.3700495. [DOI] [PubMed] [Google Scholar]
- Auch A.F., von Jan M., Klenk H.P., Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2010;2:117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbee M.L., James T.Y., Strullu-Derrien C. Early diverging fungi: diversity and impact at the dawn of terrestrial life. Annu. Rev. Microbiol. 2017;71:41–60. doi: 10.1146/annurev-micro-030117-020324. [DOI] [PubMed] [Google Scholar]
- Blackwell M. The fungi: 1, 2, 3 ... 5.1 million species? Am. J. Bot. 2011;98:426–438. doi: 10.3732/ajb.1000298. [DOI] [PubMed] [Google Scholar]
- Brown R.B., Audet J. Current techniques for single-cell lysis. J. R. Soc. Interfaces. 2008;5:S131–S138. doi: 10.1098/rsif.2008.0009.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki F., Roger A., Brown M., Simpson A. The new tree of eukaryotes. Trends Ecol. Evol. 2019;35:43–55. doi: 10.1016/j.tree.2019.08.008. [DOI] [PubMed] [Google Scholar]
- Butler J., MacCallum I., Kleber M., Shlyakhter I.A., Belmonte M.K., Lander E.S., Nusbaum C., Jaffe D.B. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio G.J., Padamsee M., Dentinger B.T.M., Bauer R., McLaughlin D.J. Assembling the fungal tree of life: constructing the structural and biochemical database. Mycologia. 2006;98:850–859. doi: 10.3852/mycologia.98.6.850. [DOI] [PubMed] [Google Scholar]
- Chen C., Xing D., Tan L., Li H., Zhou G., Huang L., Xie X.S. Single-cell whole-genome analyses by linear amplification via transposon insertion (LIANTI) Science. 2017;356:189–194. doi: 10.1126/science.aak9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clingenpeel S., Schwientek P., Hugenholtz P., Woyke T. Effects of sample treatments on genome recovery via single-cell genomics. ISME J. 2014;8:2546–2549. doi: 10.1038/ismej.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clingenpeel S., Clum A., Schwientek P., Rinke C., Woyke T. Reconstructing each cell's genome within complex microbial communities-dream or reality? Front. Microbiol. 2015;5:771. doi: 10.3389/fmicb.2014.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley T., Smith A.D. Modeling genome coverage in single-cell sequencing. Bioinformatics. 2014;30:3159–3165. doi: 10.1093/bioinformatics/btu540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean F.B., Hosono S., Fang L., Wu X., Faruqi A.F., Bray-Ward P., Sun Z., Zong Q., Du Y., Du J. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. U S A. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichorst S.A., Varanasi P., Stavila V., Zemla M., Auer M., Singh S., Simmons B.A., Singer S.W. Community dynamics of cellulose-adapted thermophilic bacterial consortia. Environ. Microbiol. 2013;15:2573–2587. doi: 10.1111/1462-2920.12159. [DOI] [PubMed] [Google Scholar]
- Ellegaard K.M., Klasson L., Andersson S.G.E. Testing the reproducibility of multiple displacement amplification on genomes of clonal endosymbiont populations. PLoS One. 2013;8:e82319. doi: 10.1371/journal.pone.0082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foissner W. Protist diversity: estimates of the near-imponderable. Protist. 1999;150:363–368. doi: 10.1016/S1434-4610(99)70037-4. [DOI] [PubMed] [Google Scholar]
- Foissner W. Protist diversity and distribution: some basic considerations. In: Foissner W., Hawksworth D.L., editors. Protist Diversity and Geographical Distribution. Springer Netherlands; 2009. pp. 1–8. [Google Scholar]
- Garvin T., Aboukhalil R., Kendall J., Baslan T., Atwal G.S., Hicks J., Wigler M., Schatz M.C. Interactive analysis and assessment of single-cell copy-number variations. Nat. Methods. 2015;12:1058–1060. doi: 10.1038/nmeth.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawad C., Koh W., Quake S.R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 2016;17:175–188. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- Gawryluk R.M.R., Del Campo J., Okamoto N., Strassert J.F.H., Lukeš J., Richards T.A., Worden A.Z., Santoro A.E., Keeling P.J. Morphological identification and single-cell genomics of marine diplonemids. Curr. Biol. 2016;26:3053–3059. doi: 10.1016/j.cub.2016.09.013. [DOI] [PubMed] [Google Scholar]
- Grigoriev I.V., Nikitin R., Haridas S., Kuo A., Ohm R., Otillar R., Riley R., Salamov A., Zhao X., Korzeniewski F. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42:D699–D704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Gurevich A., Saveliev V., Vyahhi N., Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han N., Qiang Y., Zhang W. ANItools web: a web tool for fast genome comparison within multiple bacterial strains. Database (Oxford) 2016;2016:baw084. doi: 10.1093/database/baw084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y., Wu K., Shi X., Li F., Song L., Wu H., Dean M., Li G., Tsang S., Jiang R. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. GigaScience. 2015;4:37. doi: 10.1186/s13742-015-0068-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde K.D. Where are the missing fungi? Mycol. Res. 2001;105:1409–1412. [Google Scholar]
- Kogawa M., Hosokawa M., Nishikawa Y., Mori K., Takeyama H. Obtaining high-quality draft genomes from uncultured microbes by cleaning and co-assembly of single-cell amplified genomes. Sci. Rep. 2018;8:2059. doi: 10.1038/s41598-018-20384-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F., Demaree B., Ahmed N., Abate A.R. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol. 2017;35:640–646. doi: 10.1038/nbt.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasken R.S., Stockwell T.B. Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol. 2007;7:19. doi: 10.1186/1472-6750-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus K.L., James T.Y. Surveying the biodiversity of the Cryptomycota using a targeted PCR approach. Fungal Ecol. 2015;14:62–70. [Google Scholar]
- Lazarus K.L., Benny G.L., Ho H.M., Smith M.E. Phylogenetic systematics of Syncephalis (Zoopagales, Zoopagomycotina), a genus of ubiquitous mycoparasites. Mycologia. 2017;109:333–349. doi: 10.1080/00275514.2017.1307005. [DOI] [PubMed] [Google Scholar]
- Linnarsson S., Teichmann S.A. Single-cell genomics: coming of age. Genome Biol. 2016;17:97. doi: 10.1186/s13059-016-0960-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay I.C., Voet T. Single cell genomics: advances and future perspectives. PLoS Genet. 2014;10:e1004126. doi: 10.1371/journal.pgen.1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff J.P., Auch A.F., Klenk H.P., Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu K.E., Tang Q., Wilson P.C., Khan A.A. Single-cell genomics: approaches and utility in immunology. Trends Immunol. 2017;38:140–149. doi: 10.1016/j.it.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning L., Li Z., Wang G., Hu W., Hou Q., Tong Y., Zhang M., Chen Y., Qin L., Chen X. Quantitative assessment of single-cell whole genome amplification methods for detecting copy number variation using hippocampal neurons. Sci. Rep. 2015;5:11415. doi: 10.1038/srep11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G., Bradnam K., Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- Peng Y., Leung H.C., Yiu S.M., Chin F.Y. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- Rinke C., Schwientek P., Sczyrba A., Ivanova N.N., Anderson I.J., Cheng J.-F., Darling A., Malfatti S., Swan B.K., Gies E.A. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- Rinke C., Lee J., Nath N., Goudeau D., Thompson B., Poulton N., Dmitrieff E., Malmstrom R., Stepanauskas R., Woyke T. Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nat. Protoc. 2014;9:1038–1048. [Google Scholar]
- Roy R.S., Price D.C., Schliep A., Cai G., Korobeynikov A., Yoon H.S., Yang E.C., Bhattacharya D. Single cell genome analysis of an uncultured heterotrophic stramenopile. Sci. Rep. 2014;4:4780. doi: 10.1038/srep04780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero M.A., Gordon D.P., Orrel T.M., Bailly N., Bourgoin T., Brusca R.C., Cavalier-Smith T., Guiry M.D., Kirk P.M. A higher level classification of all living organisms. PLoS One. 2015;10:e0119248. doi: 10.1371/journal.pone.0119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibbald S.J., Archibald J.M. More protist genomes needed. Nat. Ecol. Evol. 2017;1:145. doi: 10.1038/s41559-017-0145. [DOI] [PubMed] [Google Scholar]
- Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Spatafora J.W., Chang Y., Benny G.L., Lazarus K., Smith M.E., Berbee M.L., Bonito G., Corradi N., Grigoriev I., Gryganskyi A. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia. 2016;108:1028–1046. doi: 10.3852/16-042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits C., Le Caignec C., De Rycke M., Van Haute L., Van Steirteghem A., Liebaers I., Sermon K. Whole-genome multiple displacement amplification from single cells. Nat. Protoc. 2006;1:1965–1970. doi: 10.1038/nprot.2006.326. [DOI] [PubMed] [Google Scholar]
- Stajich J.E., Berbee M.L., Blackwell M., Hibbett D.S., James T.Y., Spatafora J.W., Taylor J.W. The fungi. Cuur. Biol. 2009;19:PR840–PR845. doi: 10.1016/j.cub.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanauskas R. Single cell genomics: an individual look at microbes. Curr. Opin. Microbiol. 2012;15:613–620. doi: 10.1016/j.mib.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Tighe S., Afshinnekoo E., Rock T.M., McGrath K., Alexander N., McIntyre A., Ahsanuddin S., Bezdan D., Green S.J., Joye S. Genomic methods and microbiological technologies for profiling novel and extreme environments for the extreme microbiome project (XMP) J. Biomol. Tech. 2017;28:31–39. doi: 10.7171/jbt.17-2801-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkacz A., Hortala M., Poole P.S. Absolute quantitation of microbiota abundance in environmental samples. Microbiome. 2018;6:110. doi: 10.1186/s40168-018-0491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troell K., Hallström B., Divne A.-M., Alsmark C., Arrighi R., Huss M., Beser J., Bertilsson S. Cryptosporidium as a testbed for single cell genome characterization of unicellular eukaryotes. BMC Genomics. 2016;17:471. doi: 10.1186/s12864-016-2815-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzbacher C., Nilsson R.H., Rautio M., Peura S. Poorly known microbial taxa dominate the microbiome of permafrost thaw ponds. ISME J. 2017;11:1938–1941. doi: 10.1038/ismej.2017.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B., Li T., Luo Y., Xu R., Cai H. An empirical algorithm for bias correction based on GC estimation for single cell sequencing. In: Peng W.-C., Wang H., Bailey J., Tseng V.S., Ho T.B., Zhou Z.-H., Chen A.L.P., editors. Trends and Applications in Knowledge Discovery and Data Mining. Springer International Publishing; 2014. pp. 15–21. [Google Scholar]
- Yoon H.S., Price D.C., Stepanauskas R., Rajah V.D., Sieracki M.E., Wilson W.H., Yang E.C., Duffy S., Bhattacharya D. Single-cell genomics reveals organismal interactions in uncultivated marine protists. Science. 2011;332:714–717. [Google Scholar]
- Zhang Q., Wang T., Zhou Q., Zhang P., Gong Y., Gou H., Xu J., Ma B. Development of a facile droplet-based single-cell isolation platform for cultivation and genomic analysis in microorganisms. Sci. Rep. 2017;7:41192. doi: 10.1038/srep41192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C., Lu S., Chapman A.R., Xie X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K., Limpens E., Zhang Z., Ivanov S., Saunders D.G., Mu D., Pang E., Cao H., Cha H., Lin T. Single nucleus genome sequencing reveals high similarity among nuclei of an endomycorrhizal fungus. PLoS Genet. 2014;10:e1004078. doi: 10.1371/journal.pgen.1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rozella zoospores after thawing from −80°C frozen original sample.
Highlighted in red are the best results.
Nucleotide identity analysis for seven single-cell amplified genomes from ciliate protist. Note that on average only 67% of the assembled genome had enough similarity for the ANI formula. Coverage heatmap shows the percentage of the genome that was used for the ANI calculation, based on the cutoff >70% identity over >70% of the fragment, fragment size 1,020 bp. ANI for genome part that has >70% identity over >70% of the fragment size 1,020 bp.
A through D and G through M. Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target concentration.
(A) Rozella allomycis (Cryptomycota) sample. Microscopic examination shows a monoculture with highly motile spores. Upper inset picture: received photo ID before shipping on ice, phase microscopy under cover glass. Lower inset picture shows two magnified zoospores. For a movie showing zoospore motility see file: Video S1.
(B) Rozella allomycis (Cryptomycota) Top panel and table: sample FACS examination reveals the presence of other organisms (gate P7). Middle panel: the gate with target organism (P9) is chosen and sorted into a tube with 1x PBS. Microscopic verification of the Syto9 DNA-labeled FACS-isolated single cell sort of target flagellate spore from gate P9 is shown in the picture above the panel. Lower panel: Purified population of the target organism (gates P6 and P9) is used for isolation of individual cells into 384-well plates.
(C) Caulochytrium protostelioides ATCC52028 (Chytridiomycota) sample microscope examination shows a monoculture with motile spores. Upper right inset: Phase transmitted light image of the zoospore shows 3 × 5 μm body size with 20-μm-long flagella; received photo ID before shipping on ice. Upper left inset: Magnified zoospore transmitted direct light image after receiving and thawing the sample. Lower left and right pictures: Sorted single cells after FACS purification (see next page) maintain motility.
(D) Caulochytrium protostelioides ATCC52028 (Chytridiomycota). Upper panel and table: FACS examination of the sample reveals the presence of a significant number of other organisms. The gate (P1) with target organism was chosen and sorted into a tube with 1× PBS. Second panel and table: Purified population (gate P1) of target organism was used for isolation of individual cells into 384-well plates. Image of Syto9 DNA-labeled FACS-isolated target zoospore from GateP1 is shown in Figure S1c.
(E) Blyttiomyces helicus sample microscopic examination shows a heterogeneity of microorganisms, likely bacterial cells of various size visible under phase transmitted light with occasional target fungal cells. Phase microscopy and Tubulin Tracker fluorescent DNA label was used for primary assessment of the sample complexity and target concentration. Tubulin Tracker fluorescent DNA label microscopic examination allows to assess organisms that have flagella among prokaryote enriched sample. Lower left inset: Phase transmitted light image of the zoospore shows 5 μm body size with 30-μm-long flagella, received photo ID prior shipping on ice.
(F) Blyttiomyces helicus sample FACS examination confirms microscopic assessment, showing high enrichment in organisms smaller than 2 μm (gate P6). Population enrichment FACS assessment shows a very low enrichment for target zoospore gate P7 and P8. Direct sorting without enrichment purification step was performed due to low concentration (table P8) and small volume (2 mL) of available sample.
(G) Dimargaris cristalligena RSA 468 sample microscope examination shows an almost pure sample, highly enriched with spores. Upper left inset shows received photo ID before shipping on ice; phase contrast image of a pear-shaped spore with 5 × 8 μm size. Lower right inset: transmitted light bright field with target pear-shaped spores magnified.
(H) Dimargaris cristalligena RSA 468 FACS examination. Upper panel and table: sample shows a distinct population of target sample (gates P4, P5) and the presence of smaller organisms and small unlabeled particles. Lower panel, table and picture: FACS image of the cleaned P5 population used for single-cell sorting into 384-well plates. Transmitted light image of sorted spores from cleaned population from gate P5.
(I) Piptocephalis cylindrospora RSA2659 sample. Microscope examination of the sample under fluorescent light reveals a large number of non-target cells of varied morphology. Upper right inset: shows received photo ID before shipping on ice. Lower picture and lower right inset: Sample examined under transmitted light looks homogeneous.
(J) Piptocephalis cylindrospora RSA2659 FACS examined sample. Top panel and table: Syto9 fluorescent DNA label used for primary assessment of the sample complexity and target enrichment shows that the target population is dominant with a large portion that does label and two smaller populations with lower fluorescent intensity and size. Middle panel: Clean target P4 gate-sorted population into tubes was further assessed sorting three gates (P2, P3, P6). Lower panel and microscope transmitted and fluorescent light images: Clean target was sorted under best combination of settings for pure single-cell sort and used for sorting into 384-well plates.
(K) Syncephalis pseudoplumigaleata Benny S71-1 sample. Upper Left and middle insets: received photo ID before shipping on ice. Upper right: Transmitted light microscopy revealed 50-μm large sporangia. Magnified sporangia head bursting open, containing 6-μm oval-shaped static single-cell spores; the sample shows some other small cells consistent with FACS assessment; see Figure S1m. Lower transmitted light microscopy picture with three insets shows a clump of sporangia surrounded by a dominant population of uniform spores. Sample was filtered through 35-μm mesh filter before sorting to eliminate sporangia.
(L) Syncephalis pseudoplumigaleata Benny S71-1. Upper panel and table: FACS sample after filtration of sporangia shows a dominant population (P5 and P4) of an organism of the targeted size and shape. Middle microscope picture and lower FACS panel show enriched purified population used for single-cell isolation. Right-side insets: Magnified 3 × 6-μm spore with distinctive morphological features, transmitted light top and phase1 bottom.
(M) Microscope examination of the Thamnocephalis sphaerospora RSA 1356 sample under transmitted light shows 20-μm sporangia (shown magnified in the inset image) containing 6-μm round single-cell spores among hyphae. Right side insets: received photo ID from the collaborator, phase microscopy shows hyphae with sporangia, isolate sporangia, and a spore. Lower panel: sorted single-cell spores from FACS-cleaned sample (see next page). Magnified single-cell sort isolated spore with visible dense chromatic regions.
(N) FACS examination of the filtered Thamnocephalis sphaerospora RSA 1356 sample shows a large population of smaller than target cells and unstained debris. Upper panel and table: Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target enrichment. Lower panel: Target population (gates P7 and P8) sorted into a tube is shown on the transmitted and fluorescent light microscopic images.
(O) Metschnikowia bicuspidata sample was available in limited amount (2 mL). Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target concentration. Microscope fluorescent (upper left image) and transmitted light (lower right image) examination of the sample showed a mixture of the parasite yeast cell, ascospores and other organisms and particles. Upper right inset: received photo ID before shipping on ice, Phase transmitted light shows 2.5 × 13-μm yeast and 2 × 60-μm ascospore cells.
(P) Metschnikowia bicuspidata sample. Upper panel: FACS examination showed a large population of small organisms and two distinct small populations: P1 and P2 of larger-size organisms. Lower panel: Transmitted light and fluorescent light images of sorted cells from P1 (yeast), P2 (ascospore), and unknown P4 population. Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target enrichment. (R) Compost samples with cysts that appear only after mc-cellulose addition. Unknown cysts were designated as target (20-μM cysts later shown to be a ciliate, see Figure 6C, whereas the 5-μM cysts were identified as fungi Cladosporium during the rDNA-OTU screening step). Syto9 fluorescent DNA label was used for primary assessment of the sample complexity and target enrichment. Left side pictures: compost sample enriched with microcrystalline cellulose displays a wide range of organisms with a large population of 15–20 μm round cysts. Right-side pictures: same sample after a number of filtration steps (see transparent methods step 1 optimization) displays mostly identified target cysts and prokaryote size microorganisms. This sample was used for the first round of FACS cleaning and target enrichment.
(S) Clockwise: FACS of the filtered compost sample identified two distinct populations: shown in green and red in the left-side panels and microscope pictures below. Upper right panel shows FACS population after first round of sorting, and the panel below with the microscope picture with yellow frame shows the sample used for single-cell sorting into the 384-well plates.
(T) Top down: Gosling and Third Sister lakes sample screening for rDNA-OTU target phyla presence and concentration. Target phyla were unknown Cryptomycota and Chytridiomycota. Microscopic evaluation of the sample. Syto9 stains DNA and shows the ensemble of the organisms. Tubulin will stain the target phyla and other eukaryotes with flagella or tubulin. FACS + itag target sample concentration estimate. Bar graph on the left shows the results of the populations sorted on the right: One 384-well plate (288 sample single-cells) was sorted per each color (population). Only P8 (yellow) sorted on fluorescence and side scatter and P4 (dark blue) sorted on fluorescence and forward scatter yielded one target genome. Each population was sorted on Syto9, WGA, and Tubulin. Only Tubulin harvested target cells.
(U) FACS enrichment of the populations of the Gosling and Third Sister lakes samples: P6 population shown is the P8 in the previous panel. Gosling lake 100-cell/well and 500-cell/well, Tubulin sorted for rDNA i-tag estimate of the target species recovery rate. This rate of genome recovery is below the efficiency threshold of the pipeline. To improve pipeline efficiency for ultra-low abundance targets, several optimizations are described in steps 4 and 5 of the transparent methods. They allowed the recovery of 7 new target genomes studied in a separate paper focused on further optimization for ultra-low abundance EMEs.
(A) Principal-component analysis monoplots for the full set of quality check criteria. A 90° angle shows no correlation, a 180-degree angle shows inverted correlation, and an angle less than 45° shows strong correlation. The total dataset of 20 quality check criteria used for the study was split into 2 groups for comprehensive visualization: Upper row shows set 1a. Lower row shows set 1b. Set 1a and set1b have common QC criteria for reference purpose: MGS_L50, MGS_N50, SCFF_2.5kb. Set 1a upper monoplots were generated based on scores from 12 quality check criteria, and set 1b lower monoplots were generated using additional 8 criteria plus three from the above dataset. Shown QC criteria: AGS, assembled genome size; EGS, estimated genome size; SGA, start of the genome amplification; FGA, fold genome amplification to 0.5–1 μg; GC%, average GC percent of the assembled genome; RTU, random 20-mer uniqueness; CEGMA, core eukaryotic genes mapping approach; %GS > 50 kb, percent genome in scaffolds over 50 kb; gap%, assembled genome scaffold gap percent; MGS_N50, main genome scaffold N50; MGS_L50, main genome scaffold L50; SCFF_N(10-500kb), number of scaffolds between preceding bin and shown bin number, e.g., SCFF_2.5kb, number of scaffolds between 2 and 2.5 kb and SCFF_5kb, number of scaffolds between 2.5 and 5 kb, etc.
(B–E) Clockwise: scatterplot correlation matrix with density ellipse and plot. Color-coded genomes with species names abbreviation and 1, 10, 50, 100 cell sorts on the right of the abbreviation. Full species names are given in Figure 3 and Data S2F. Covariance scores from set1a 12 quality check criteria. Ranked correlation matrix, shown are Pearson (r), Spearman (rs), and Kendall (tau) correlation values. Darker color in the table indicates a stronger correlation with red showing negative and blue positive correlation values. A positive correlation on matrix will correspond to the diagonal ellipse orientation from zero to infinite, and a negatively correlated data will cluster in an ellipse oriented perpendicularly to the positive correlation ellipse. B. Set1a of the 20 QC criteria for all species genomes. C. Set1b of the 20 QC criteria for all species genomes. D. Reduced set of QC criteria used in Figure 3, for fungi-only species genomes. E. Reduced set of QC criteria used in Figure 3, for all species genomes.
(F and G) Gower & Hand PCA biplot represents similarity between data points, with smaller distance higher similarity. Shown plot explains 80.2% of the variability for fungi only. Any point on the plot projected orthogonally onto the axes will show the approximate value of that variable. Percent at the end of the axes labels indicates predictability value of the axes. Axes with predictability values below 60% are masked in gray.
(F) Predictability value of the QC criteria used, for fungal species only.
(G) Predictability value of the criteria used, for all species.
X axis shows time of MDA amplification and has same scale for all images in one panel. Reference black bar shows earliest start of amplification time point in the same panel. Y axis shows fluorescence level that represents a combination of optical refraction dependent on reaction volume, DNA amount, air bubbles, and other factors that can affect the optical intensity of signal. Stock alkaline solution was 500 mM with 10 mM DTT. All other alkaline solutions were diluted with ultrapure H2O. Lysis solution conditions: Alkaline∗ was 133 mM KOH +133 mM HCl, 10 mM final concentration in MDA mix; Alkaline 1 was 30 mM KOH final in MDA mix; Alkaline 2 was 6 mM KOH final; Heat in H2O: 98°C 1 min, 0°C 2 min, room t°C 3 min, 60°C 2 min, 0°C 2 min; Alkaline with heat: 98°C 1 min, 0°C 2 min, KOH at 10 mM final in MDA mix, 60°C 2 min, 0°C 2 min; Alkaline with heat: KOH at 10 mM final in MDA, 60°C 2 min, 0°C 2 min. Detergent was 0.3% Tween with 1mM EDTA.
(A) NEB MDA—lysis solution efficiency assessment using purified DNA. In all the tests negative controls that amplified are from the reaction mixes that contain the detergent (Tween 0.3%) used in the lysis buffer composition in this test.
(B) NEB MDA—lysis solution efficiency assessment using soil prokaryote single cells.
(C–F) Right side shows quantitative assessment of the lysis buffer efficiency for single-cells 10 cells, 100cells. (C) Gram-positive bacteria (E. coli) and Gram-negative bacteria (B. subtillis). (D) Rozella allomycis NEB MDA top three left tests and Qiagen MDA, bottom left test. (E) Caulochytrium protostelioides ATCC52028 Qiagen MDA, Alkaline lysis top is Qiagen kit protocol (see methods), Alkaline∗ was the same alkaline as used for the NEB-MDA tests in (D).
(F)Caulochytrium protostelioides ATCC52028 Qiagen MDA.
Data Availability Statement
The coassembled genomes and annotations of the target species reported in this paper are available through "MycoCosm:https://genome.jgi.doe.gov/fungi" using the following "MycoCosm:URLs" and "GenBank: accession numbers" reported in this paper are respectively: R. allomycis CSF55 single-cell "https://genome.jgi.doe.gov/Rozal_SC1; "GenBank: QUVT00000000", B. helicus Perch Fen single-cell "https://genome.jgi.doe.gov/Blyhe1; "GenBank:QPFV00000000", C. protostelioides ATCC 52028 single-cell "https://genome.jgi.doe.gov/Caupr_SCcomb; "GenBank:QUVS00000000", D. cristalligena RSA 468 single-cell "https://genome.jgi.doe.gov/DimcrSC1; "GenBank:QRFA00000000", P. cylindrospora RSA 2659 single-cell "https://genome.jgi.doe.gov/Pipcy3_1; "GenBank:QPFT00000000", T. sphaerospora RSA 1356 single-cell "https://genome.jgi.doe.gov/Thasp1; "GenBank:QUVU00000000", S. pseudoplumigaleata Benny S71-1 single-cell "https://genome.jgi.doe.gov/Synps1; "GenBank:QUVV00000000" and M. bicuspidata single-cell "https://genome.jgi.doe.gov/Metbi_SCcomb; "GenBank:QUVR00000000". The whole-genome sequence for the non-single-cell isolate C. protostelioides ATCC 52028 is available through "MycoCosm:https://genome.jgi.doe.gov/Caupr1" and "GenBank: QAJV00000000"". The whole-genome sequences for the non-single-cell isolate of R. allomycis CSF55 was not determined in this study and is available through "MycoCosm:https://genome.jgi.doe.gov/Rozal1_1") and "GenBank:ATJD00000000".