

SUMMARY
Repetitive elements (REs) compose ~50% of the human genome and are normally transcriptionally silenced, although the mechanism has remained elusive. Through an RNAi screen, we identified FBXO44 as an essential repressor of REs in cancer cells. FBXO44 bound H3K9me3-modified nucleosomes at the replication fork and recruited SUV39H1, CRL4, and Mi-2/NuRD to transcriptionally silence REs post-DNA replication. FBXO44/SUV39H1 inhibition reactivated REs, leading to DNA replication stress and stimulation of MAVS/STING antiviral pathways and interferon (IFN) signaling in cancer cells to promote decreased tumorigenicity, increased immunogenicity, and enhanced immunotherapy response. FBXO44 expression inversely correlated with replication stress, antiviral pathways, IFN signaling, and cytotoxic T cell infiltration in human cancers, while a FBXO44-immune gene signature correlated with improved immunotherapy response in cancer patients. FBXO44/SUV39H1 were dispensable in normal cells. Collectively, FBXO44/SUV39H1 are crucial repressors of RE transcription, and their inhibition selectively induces DNA replication stress and viral mimicry in cancer cells.
In Brief
Targeting FBXO44, which regulates the transcriptional silencing of REs in the human cancer genome, promotes antiviral signaling and decreases tumorigenesis as well as overcomes resistance to immune checkpoint blockade.
Graphical Abstract
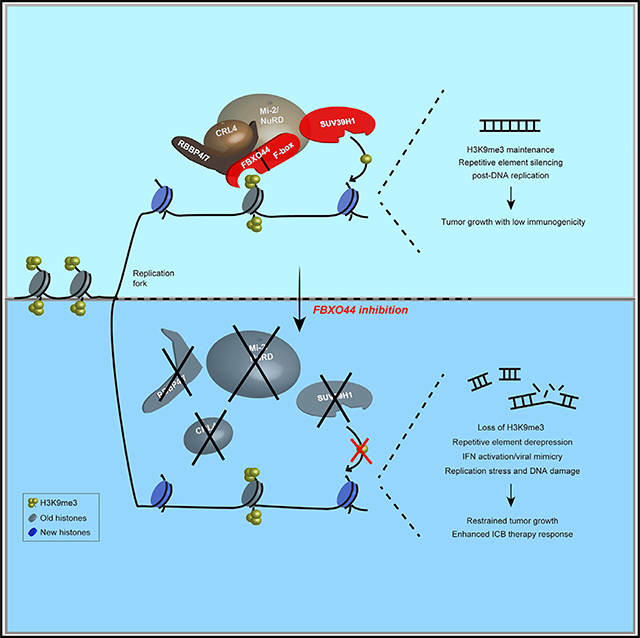
INTRODUCTION
Repetitive elements (REs) maintain genomic stability and chromosome architecture and drive human genome evolution and diversity (Slotkin and Martienssen, 2007). Major types of REs in humans include interspersed LTR-based endogenous retroviruses (ERVs; 8% of genome) and non-LTR-based short- and long-interspersed nuclear elements (SINEs and LINEs, respectively; 35% of genome). Other REs include DNA transposons (3% of genome) and tandemly arrayed simple repeats, such as satellite and telomeric repeats (3% of genome) that result from improper replication (Ishak et al., 2016; Padeken et al., 2015). The regulatory sequences of several REs, such as LTRs, have been co-opted through evolution as cis-regulatory promoter/enhancer elements to control gene regulatory networks involved in development, inflammatory response, and fertility, among other processes (Chuong et al., 2017; Thompson et al., 2016).
Transposable elements and tandem repeats promote replication fork stalling and non-allelic recombination events between homologous repeats, thus posing a major threat to genome integrity (Konkel and Batzer, 2010; Padeken et al., 2015). To counteract these adverse effects, REs are normally transcriptionally silenced in somatic cells through repressive epigenetic modifications that are thought to include DNA methylation (5-methyl-cytosine) and nucleosome modifications, including deacetylation, trimethylation of histone H3 lysine 9 (H3K9me3), and trimethylation of histone H4 lysine 20 (H4K20me3) (Slotkin and Martienssen, 2007). These repressive modifications are facilitated by multiple, functionally redundant enzymes. For example, H3K9me3 modifications can be mediated by histone methyltransferases (HMTs) SETDB1, G9a (EHMT2), SUV39H1, and SUV39H2. However, mechanistic details regarding how REs are silenced in human cells, especially in relation to the maintenance of repressive modifications through DNA replication cycles, have not been fully elucidated.
Compromised RE silencing can activate their transcription, potentially leading to the accumulation of cytosolic double-stranded (ds)RNA and dsDNA replication intermediates that are recognized by pattern recognition receptors (PRRs) RIG-I/MDA5 and cGAS, respectively, which normally function to detect exogenous pathogens (Jones et al., 2019; Kassiotis and Stoye, 2016). RIG-I and MDA5 activate mitochondrial antiviral-signaling protein (MAVS), whereas cGAS stimulates a signaling cascade that activates stimulator of interferon (IFN) genes (STING). Activation of either pathway stimulates IFN regulatory factor (IRF)3/7-dependent transcription of IFN stimulatory genes (ISGs) that initiate IFN-driven antiviral immune responses. Robust stimulation of these pathways can also trigger regulated cell death (RCD) (Vanpouille-Box et al., 2018). Recent studies have shown that induction of RE transcription in cancer cells beyond a threshold level of tolerance through treatment with DNA demethylating agents or histone deacetylase (HDAC) inhibitors augments the effectiveness of antitumor therapies, including immunotherapy (Ishak et al., 2018). To date, tumor-specific RE silencing mechanisms that can be effectively targeted for treatment have not been identified.
Based on this background, we undertook a discovery effort to identify novel regulators of REs in cancer cells through a genetic screen for potential modifiers of the key histone modification, H3K9me3.
RESULTS
FBXO44 is essential for H3K9me3-mediated transcriptional silencing of REs in cancer cells
To identify H3K9me3 regulators in cancer cells, we performed a cell image-based RNA interference (RNAi) screen that detected changes in levels of H3K9me3 and phosphorylated (p)-RPA32T21 (DNA replication stress marker) in a panel of cancer cell lines (Figure 1A) (Rantala et al., 2011). Among the top hits, short interfering RNA (siRNA)-mediated knockdown (KD) of FBXO44 decreased chromatin-associated H3K9me3 modifications and increased p-RPA32T21 levels (Figures 1B–1D). FBXO44 is a member of the F-box protein family that shares a function as substrate recognition factors for the SKP1-CUL1-F-box (SCF)-type ubiquitin ligase (Skaar et al., 2013). Fractionation experiments detected FBXO44 in the cytoplasm, nucleus, and bound to chromatin (Figure 1E). Immunoprecipitation (IP) verified FBXO44 interacted with histone H3 in chromatin fractions (Figure 1F). Immunoblotting confirmed that FBXO44 KD reduced chromatin-associated H3K9me3, and to a lesser extent H3K9me1 and H3K9me2 modifications (Figure 1G). H3K27me3, H3K36me3, and H3K79me2 modifications were unaffected. Chromatin immunoprecipitation sequencing (ChIP-seq) revealed that FBXO44 was highly enriched at repetitive DNA (94.6%) and heterochromatin (2.4%), with little binding at promoters, enhancers, or transcribed regions (<3% total) (Figure 1H; Table S1). Specifically, FBXO44 binding localized to various RE subtypes (Figure 1I). FBXO44 chromatin binding sites strongly co-localized with H3K9me3, and much less with H3K27me3 or H3K4me3 modifications, across the genome and specifically at REs (Figures 1J–1M and S1A; Table S2). ChIP experiments confirmed enrichment of FBXO44 bound at various subfamilies of satellite repeats (MajSAT, mcBox, and SATIII), SINE/LINEs (Alu and L1), and ERV (HERV-K) (Figure 1N). FBXO44 targeting diminished H3K9me3 modifications at these REs (Figures 1M, 1O, and S1A). qRT-PCR analysis showed that FBXO44 KD activated the transcription of various RE subfamilies (Figures 1P and S1B), including REs that displayed decreased H3K9me3 level in FBXO44 KD cells, indicating they were direct targets of FBXO44. RNA sequencing (RNA-seq) confirmed the upregulated expression of various REs in FBXO44 KD cells (Figure S1C; Table S3). Of note, the RE subtypes activated by FBXO44 KD partially overlapped with those activated by targeting histone H3K4 demethylase, LSD1 (Figure S1D) (Sheng et al., 2018). Together, these findings demonstrate that FBXO44 co-localized with H3K9me3 modifications and was essential for RE transcriptional silencing in cancer cells.
Figure 1. FBXO44 regulates H3K9me3-mediated transcriptional silencing of REs in cancer cells.

(A and B) Schematic (A) and result (B) of the H3K9me3 regulator screen.
(C) IF images of chromatin-associated H3K9me3 in MDA-MB-231 cells. Scale bar, 20 μm.
(D) Quantification of H3K9me3 relative intensity (n = 15) (top panel) and immunoblots (bottom panel) for cells in (C).
(E) Immunoblot of FBXO44 in cytoplasmic, nuclear, and chromatin fractions.
(F) CoIP of endogenous FBXO44 with histone H3 in chromatin fractions.
(G) Immunoblots of the indicated chromatin modifications in chromatin fractions.
(H) ChIP-seq results for FBXO44 chromatin binding peaks categorized by chromosome feature.
(I) RE annotation of FBXO44 chromatin binding peaks.
(J) Heatmaps of FBXO44 and H3K9me3 ChIP-seq signals.
(K) ChIP-seq enrichment profiles of FBXO44 and H3K9me3 peaks.
(L) Venn diagram plots of ChIP-seq peaks for FBXO44 and H3K9me3, H3K27me3, and H3K4me3.
(M) Visualization of FBXO44 chromatin binding sites and H3K9me3 modifications for a segment of chromosome 8 harboring satellite repeats (SAR). H3K27me3 and H3K4me3 modifications are shown.
(N) ChIP analysis of FBXO44 binding to the indicated REs (n = 3).
(O) ChIP analysis of H3K9me3 levels at the indicated REs (n = 3).
(P) qRT-PCR analysis of the indicated REs (n = 3).
Data represent mean ± SEM. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test (D), and two-way ANOVA followed by Tukey’s multiple comparisons test (N), Tukey’s multiple comparisons test (O), Sidak’s multiple comparisons test (P).
FBXO44 recruits SUV39H1, CRL4, and Mi-2/NuRD to REs
Protein mass spectrometry analysis revealed that FBXO44 interacted with several chromatin modifiers/remodelers previously implicated in heterochromatin assembly (Figure 2A; Table S4), including components of Mi-2/NuRD (GATAD2A/B, CHD4, MBD2, MTA1–3, and HDAC1–2), CTBP transcriptional corepressor (PRMT5, MEP50, and WIZ), and polycomb repressor complex (PRC2)/EED-EZH2 (EED) (Kadoch et al., 2016; Simon et al., 2015; Sims and Wade, 2011). FBXO44 also interacted with the CRL4 ubiquitin ligase (DDB1 and CUL4B), as well as DDB1 and CUL4-associated factors (DCAFs) RBBP4 and RBBP7, which was implicated in regulation of H3K9me3 and H3K27me3 in human cells (Higa et al., 2006; Hu et al., 2012). Coimmunoprecipitation (coIP) confirmed FBXO44 interacted with components of Mi-2/NuRD and CRL4, as well as RBBP4/7 (Figures 2B and S2A). Moreover, FBXO44 interacted with H3K9me3 methyltransferase SUV39H1 (Figure 2B). To determine if FBXO44 cooperated with SUV39H1, CRL4RBBP4/7, and Mi-2/NuRD in RE silencing, we targeted each enzyme/complex component and analyzed H3K9me3 levels and RE transcription. Targeting SUV39H1 or CUL4B, or co-KD of Mi-2/NuRD components GATAD2A+B, decreased total chromatin-associated H3K9me3 modifications, comparable to FBXO44 KD (Figure 2C). ChIP experiments showed targeting SUV39H1, CUL4B, GATAD2A+B, or RBBP4+7 decreased H3K9me3 modifications at various REs, similar to FBXO44 KD (Figure 2D). These KD cells also activated RE transcription comparable to FBXO44 KD cells (Figure 2E). Of note, although IP experiments demonstrated that FBXO44 interacted with CUL1 in chromatin fractions (Figure 1F), ChIP experiments showed CUL4B, but not CUL1, was enriched at REs (Figure S2B). Further, CUL1 KD did not activate RE transcription (Figure S2C). Collectively, these results suggest FBXO44 serves a CUL1-independent function by cooperating with SUV39H1, CRL4RBBP4/7, and Mi-2/NuRD to transcriptionally silence REs in cancer cells.
Figure 2. FBXO44 recruits SUV39H1, CRL4RBBP4/7, and Mi-2/NuRD to REs.

(A) STRING network plot for interactions among FBXO44-interacting proteins.
B) CoIP of endogenous FBXO44 with SUV39H1 and components of CRL4 and Mi-2/NuRD.
(C) IF images of chromatin associated H3K9me3 in MDA-MB-231 cells (top panel). Scale bar, 10 μm. Quantification is shown (n = 15) (bottom panel).
(D) ChIP analysis of H3K9me3 levels at indicated REs (n = 3).
(E) qRT-PCR analysis of RE transcripts (n = 3).
(F) CoIP of endogenous SUV39H1 with CUL4B and DDB1.
(G–I) CoIP of endogenous CUL4B with DDB1 (G) and endogenous FBXO44 with SUV39H1 (H) and GATAD2A (I).
(J) ChIP analysis of H3K9me3 levels at the indicated REs (n = 3).
(K) Immunoblots of the indicated proteins in chromatin fractions.
(L and M) ChIP analysis of binding of the indicated Flag-tagged proteins to various REs (n = 3).
Data represent mean ± SEM. ns, not significant; **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test (C), and two-way ANOVA followed by Sidak’s multiple comparisons test (D and L), Tukey’s multiple comparisons test (E, J, and M).
Next, we characterized FBXO44’s molecular interactions with SUV39H1, CRL4RBBP4/7, and Mi-2/NuRD. CoIP experiments showed that endogenous SUV39H1 interacted with CRL4 components, CUL4B and DDB1, and these interactions were FBXO44-dependent (Figure 2F). However, FBXO44 KD did not affect CRL4 complex assembly (Figure 2G). Interaction of FBXO44 and SUV39H1 depended on Mi-2/NuRD components GATAD2A+B, although interaction between FBXO44 and GATAD2B was SUV39H1-independent (Figures 2H and 2I). FBXO44 consists of an N-terminal F-box domain and C-terminal F-box-associated domain (FBA), which presumably binds substrates. ChIP experiments showed that expression of an siRNA-resistant cDNA encoding F-box deleted FBXO44 (ΔF-FBXO44) that did not interact with CUL1 but appropriately localized in cells (Figures S2D and S2E), could not compensate for FBXO44 in mediating H3K9me3 modifications at REs in FBXO44 KD cells (Figure 2J). CoIP experiments revealed that ΔF-FBXO44 interacted with histone H3.1, CUL4B/DDB1, and RBBP4/7, but not GATAD2B or SUV39H1 (Figures S2F–S2J). Together, these data suggest that FBXO44 functions as an adaptor protein, with its FBA domain mediating interactions with chromatin and CRL4RBBP4/7 and N-terminal F-box-containing region required for interaction with SUV39H1 and Mi-2/NuRD.
As FBXO44 targeting decreased the interaction of SUV39H1, CUL4B, and Mi-2/NuRD with chromatin, a phenotype rescued by expression of an siRNA-resistant FBXO44 cDNA (Figure 2K), we hypothesized that FBXO44 might recruit these enzymes to REs to initiate transcriptional silencing. FBXO44 KD prevented recruitment of SUV39H1, CUL4B, RBBP4/7, and Mi-2/NuRD (GATAD2A/B and CHD4) to REs in ChIP experiments (Figure 2L). Targeting RBBP4+7 inhibited recruitment of SUV39H1, CUL4B, and GATAD2A/B, but not FBXO44 (Figure S2K). CUL4B reduction prevented SUV39H1 and GATAD2A/B recruitment, although FBXO44 was unaffected (Figure S2L). Further, targeting GATAD2A+B inhibited SUV39H1 recruitment, but not FBXO44 or CUL4B (Figure S2M). In contrast, SUV39H1 KD had no effect on FBXO44, CUL4B, RBBP4/7, or GATAD2A/B recruitment to REs (Figure S2N). These data suggest that FBXO44 functions upstream of SUV39H1, CRL4RBBP4/7, and Mi-2/NuRD in RE silencing. In support of this, introduction of ΔF-FBXO44 into FBXO44 KD cells promoted recruitment of RBBP4/7, but not SUV39H1, to REs (Figure 2M). ChIP experiments also demonstrated that FBXO44 KD diminished mono-ubiquitylation of H2AK119 at REs (Figure S2O), a repressive chromatin modification mediated, in part, by CRL4 (Hu et al., 2012).
FBXO44 binds H3K9me3-modified nucleosomes at the replication fork and initiates RE silencing post-DNA replication
Mass spectrometry revealed that FBXO44 interacted with several DNA replication proteins, including PCNA, CHAF1A/B, and MCM7 (Figure 2A; Table S4). In addition, FBXO44 accumulated in the nucleus in S phase (Figure 3A). CoIP experiments also showed that FBXO44 preferentially interacted with DNA replication-associated histone H3.1, relative to histone H3.3 (Figure 3B), which is deposited by a DNA synthesis-independent pathway (Tagami et al., 2004). Therefore, we hypothesized that FBXO44 could coordinate RE silencing with DNA replication and investigated FBXO44’s interaction with chromatin as a function of DNA replication by aniPOND analysis (Leung et al., 2013). FBXO44 specifically interacted with newly replicated chromatin and dissociated within 60 min following DNA replication (Figure 3C). To identify the potential trigger for FBXO44 chromatin binding, we performed in vitro binding assays using recombinant FBXO44 and synthetic H3K9me3−, H3K9me1−, or un-modified nucleosomes revealing that FBXO44 selectively bound to H3K9me3-modified nucleosomes (Figure 3D), consistent with ChIP-seq data that showed FBXO44 co-localized with H3K9me3 modifications in cells (Figures 1J–1M and S1A). A modified aniPOND experiment demonstrated that FBXO44 bound H3K9me3-modified nucleosomes of newly replicated chromatin (Figure 3E). Together, these results demonstrate that FBXO44 bound H3K9me3-modified nucleosomes at the replication fork to initiate RE silencing post-DNA replication (Figure 3F).
Figure 3. FBXO44 promotes RE silencing post-DNA replication.

(A) IF images of FBXO44 (middle panel) in HeLa cells synchronized at the indicated cell cycle phases (left panel). Scale bar, 10 μm. Quantification of cells with FBXO44 nuclear localization is shown (n = 5) (right panel).
(B) CoIP of endogenous FBXO44 with Flag-histone H3.1 or H3.3.
(C) aniPOND analysis of FBXO44 chromatin binding. Schematic of protocol (left panel) and immunoblots of FBXO44, DNA replication fork protein PCNA, and histone H3 (control) are shown (right panel).
(D) In vitro binding assay using recombinant FBXO44 and H3K9me1−, H3K9me3−, or un-modified nucleosomes.
(E) aniPOND analysis of FBXO44 binding to H3K9me3-modified nucleosomes. Schematic of protocol (left panel) and immunoblots are shown (right panel).
(F) Model of FBXO44 regulation of H3K9me3-mediated RE silencing post-DNA replication.
(G) Flow cytometry analysis of cell cycle (left panel) and quantification (n = 3) (right panel).
(H) IF images of EdU incorporation in DNA of MDA-MB-231 cells (left panel) and quantification (n = 5) (right panel). Scale bar, 20 μm.
(I) Immunoblots of DNA replication checkpoint and DNA damage response (DDR) proteins. *p-RPA32T21.
(J) ChIP analysis of γH2AX levels at the indicated REs (n = 3).
Data represent mean ± SEM. ns, not significant; *p < 0.05, ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test (A), Dunnett’s multiple comparisons test (H), and two-way ANOVA followed by Dunnett’s multiple comparisons test (G), Tukey’s multiple comparisons test (J).
See also Figure S3.
FBXO44/SUV39H1 inhibition promotes DNA replication stress and DSBs in cancer cells
FBXO44/SUV39H1 KD increased the level of p-RPA32T21 in cancer cells, indicating DNA replication stress (Figure S3A). FBXO44 KD cells also accumulated in S phase and displayed reduced EdU incorporation (Figures 3G and 3H). Furthermore, FBXO44 KD activated the DNA replication checkpoint, as indicated by increased p-ATRS428 and p-CHK1S345 (Figure 3I). Targeting FBXO44/SUV39H1, GATAD2A+B, CUL4B, or RBBP4+7 increased γH2AX, indicating DNA double-strand breaks (DSBs) (Figures 3I and S3B) and activated p53 expression (Figure 3I). FBXO44 KD-induced γH2AX was enriched at REs compared to randomly selected control genes, on ChIP analysis (Figure 3J). Thus, FBXO44/SUV39H1 inhibition promotes DNA replication stress and DSBs in cancer cells.
FBXO44/SUV39H1 inhibition activates antiviral pathways and IFN signaling and enhances cancer cell immunogenicity
FBXO44/SUV39H1 targeting induced the accumulation of cytosolic dsRNA and dsDNA in cancer cells (Figures 4A and 4B). IP of the cytosolic dsRNA and RNase A protection experiments revealed that FBXO44 KD cells contained dsRNA generated from various REs subtypes (Figures 4C and S4A). FBXO44 KD cells also contained cytosolic dsDNA generated from these RE subtypes (Figure 4D). Accordingly, RIG-I/MDA5-MAVS and cGAS-STING antiviral pathways were activated in FBXO44/SUV39H1 KD cells (Figures 4E, S1B, and S4B). FBXO44 KD cells also displayed increased p-IRF3S386, a modification that stimulates IRF3 nuclear translocation and activity (Li and Chen, 2018), and increased IRF7 expression (Figure 4E) (Honda et al., 2006). Transfection of cytosolic dsRNA or dsDNA isolated from FBXO44 KD cells activated MAVS or STING transcription, respectively, in recipient cells (Figure 4F). FBXO44 KD cells also exhibited increased micronuclei that stained positive for cGAS and γH2AX compared to control cells on immunofluorescence (IF) analysis (Figure 4G), consistent with their generation via genomic instability (Mackenzie et al., 2017; Bakhoum et al., 2018). Together, these data suggest that cytosolic dsRNA and dsDNA generated from REs, as well as DNA replication stress, contribute to activate antiviral pathways in FBXO44 KD cells.
Figure 4. FBXO44 inhibition activates RIG-I/MDA5-MAVS and cGAS-STING antiviral pathways and IFN signaling and enhances cancer cell immunogenicity.

(A and B) IF images of MDA-MB-231 cells (left panels) and quantification of relative intensity (right panels) of dsRNA (A) and dsDNA (B, arrows) (n = 15). Scale bar, 10 μm.
(C and D) qRT-PCR analysis of the indicated REs (n = 3) (right panels). Protocols are shown (left panels).
(E) qRT-PCR analysis (n = 3) (left panel). Immunoblots of the indicated proteins (right panel).
(F) qRT-PCR analysis (n = 3) (middle and right panels). Schematic of protocol (left panel).
(G) IF images of cGAS and γH2AX positive micronuclei (left panel). DNA stained with DAPI. Scale bar, 5 μm. Quantification is shown (n = 5) (right panel).
(H) Pathway enrichment map for significantly enriched gene sets in GSEA of FBXO44 KD RNA-seq. p < 0.01; false discovery rate (FDR) <0.1.
(I) GSEA enrichment plots for selected gene sets in FBXO44 KD RNA-seq.
(J) ELISA quantification of IFN-β, CCL5, and CXCL10 (n = 3).
(K) qRT-PCR analysis of IFN-β (n = 3). Day is time post-KD.
(L) GSEA analysis of immune-stimulatory pathways.
(M) Heatmap of representative genes from RNA-seq data.
Data represent mean ± SEM. ns, not significant; nd, not detected; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test (A, B, and F), Dunnett’s multiple comparisons test (G), two-way ANOVA followed by Tukey’s multiple comparisons test (C and K), Dunnett’s multiple comparisons test (D), Sidak’s multiple comparisons test (E), and unpaired Student’s t test (J).
Consistently, gene set enrichment analysis (GSEA) of FBXO44 KD RNA-seq data revealed positive regulation of defense response, activation of innate immune response, and inflammatory response among the most upregulated gene expression pathways in FBXO44 KD cells (Figures 4H and 4I; Table S5). FBXO44 KD increased the expression of many ISGs in RNA-seq (Figure S4C; Table S5). We confirmed that FBXO44/SUV39H1 KD activated the expression of IFN-α/β and several ISGs, including chemokines CCL5, CXCL9, and CXCL10, by qRT-PCR (Figures 4E, S1B, and S4B). FBXO44/SUV39H1 KD enhanced the secretion of IFN-β, CCL5, and CXCL10 (Figure 4J), which promote intratumoral infiltration of effector T cells (Jones et al., 2019; Topper et al., 2017). None of the protein-coding genes bound by FBXO44 and harboring H3K9me3 modifications were antiviral genes or ISGs on ChIP-seq (Table S2), indicating the induced antiviral pathways and ISGs were indirect effects of FBXO44 targeting. In support of these data, transfection of cytosolic dsRNA or dsDNA isolated from FBXO44 KD cells activated IFN-β expression in recipient cells (Figure 4F). Next, we investigated the importance of antiviral pathways in inducing IFN in FBXO44 KD cells. FBXO44 KD induced the expression of IFN-α/β in cancer cell lines that failed to activate either MAVS or STING, indicating stimulation of either dsRNA or dsDNA sensing pathway was sufficient to trigger the antiviral response (Figure S1B). Co-targeting of MAVS+STING in FBXO44 KD cells rescued the induced IFN-β (Figure 4K). Similarly, simultaneous targeting of IRF3+IRF7 rescued the induced IFN-β in FBXO44 KD cells more efficiently than KD of IRF3 or IRF7 alone (Figure S4D). Taken together, these findings demonstrate that FBXO44/SUV39H1 inhibition promotes cytosolic accumulation of dsRNA and dsDNA that trigger RIG-I/MDA5-MAVS and cGAS-STING antiviral pathways and IFN signaling in cancer cells.
GSEA analysis of FBXO44 KD cellular RNA-seq data identified several upregulated immune-related gene expression pathways, including cytokine-cytokine receptor interaction and antigen processing cross presentation (Figure 4L). FBXO44 KD increased expression of IFNGR1 and IFNGR2, which encode the IFN-γ receptor, and decreased IFN-γ signaling inhibitor PTPN2, indicating potentially augmented responses to immune cell-derived IFN-γ (Figure 4E). Given that FBXO44 KD associated with enhanced antigen processing and presentation, we analyzed the effects on cancer cell immunogenicity. FBXO44 KD induced ~40 cancer/testis antigens (CTAs) on RNA-seq data (Figure 4M; Table S5), including MAGE-A and SSX family members that are immunotherapy targets in human cancers (Gjerstorff et al., 2015). FBXO44 KD increased SSX1 protein expression on IF analysis (Figure S4E). In orthogonal experiments, FBXO44 KD enhanced SSX1 surface expression on flow cytometry (Figure S4F). Similarly, FBXO44 KD cells increased expression of several natural killer group 2D (NKG2D) ligands, including ULBP2, which was enhanced on the cell surface (Figures 4M, S4E, and S4F). Thus, FBXO44 inhibition promotes cancer cell immunogenicity in vitro.
FBXO44/SUV39H1 inhibition selectively decreases cancer cell proliferation and survival in vitro
Activation of IFN signaling and genomic instability can promote cellular proliferation arrest and apoptosis (Sheng et al., 2018; Topper et al., 2017; Vanpouille-Box et al., 2018; Recolin et al., 2014). In preclinical studies, efficient FBXO44 KD diminished the proliferation of all cancer cell lines tested (Figure 5A), including patient-derived glioblastoma cultures (GSC1517 and GSC1552) enriched in cancer stem cells (CSCs), accompanied by increased apoptosis (Figure 5B). Consistent with reduced proliferation, FBXO44 KD downregulated cancer-associated gene expression pathways, including cell cycle, cell division, and DNA replication, on RNA-seq (Figures 4H and S5A). FBXO44 KD also decreased the fraction of breast cancer cells that expressed CSC markers (CD24− CD44+) and inhibited tumorsphere formation of breast cancer and glioblastoma cells (Figures 5C and S5B). FBXO44 KD decreased cancer cell migration and invasion in vitro (Figure 5D). Co-targeting of MAVS+STING partially rescued the decreased proliferation of FBXO44 KD cells in vitro (Figure 5E). In addition, the S phase accumulation and decreased viability of FBXO44 KD cells were partially rescued by co-KD of MAVS+STING (Figures 5F and S5C). In contrast, co-KD of MAVS+STING failed to rescue the induced γH2AX in FBXO44 KD cells (Figure S5D).
Figure 5. FBXO44/SUV39H1 inhibition selectively decreases cancer cell proliferation and viability in vitro.

(A) Growth curves of the indicated cancer cell lines and patient-derived glioblastoma cultures (n = 3).
(B) Representative flow cytometry analysis of apoptotic (annexin V+) cells (left panel). Quantification is shown (n = 3) (right panel).
(C) Representative images (left panel) and quantification (right panel) of tumorspheres (n = 3) at day 14. Scale bar, 100 μm.
(D) Representative images (left panel) and quantification (n = 3) (right panel) of migration and invasion analyses of MDA-MB-231 cells. Scale bar, 50 μm.
(E) Growth curves (n = 3) (left panel) and immunoblots of FBXO44, MAVS, and STING (right panel).
(F) Flow cytometry analysis of cell cycle (left panel) and quantification (n = 3) (right panel).
(G) Viabilities of the indicated cells after treatment with F5446 for 48 h (n = 3).
(H) Growth curves of HMECs and astrocytes (n = 3) (left panels) and immunoblots of the indicated proteins (right panels).
Data represent mean ± SEM. ns, not significant; ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Dunnett’s multiple comparisons test (B and D), and two-way ANOVA followed by Dunnett’s multiple comparisons test (A), Sidak’s multiple comparisons test (C), Tukey’s multiple comparisons test (E, F, G, and H).
See also Figure S5.
To assess whether this epigenetic regulatory pathway could potentially be targeted for cancer treatment, we focused on SUV39H1 and its chemical inhibitor F5446 (Lu et al., 2019). F5446 treatment reduced chromatin-associated H3K9me3 (Figure S5E) and activated the expression of various REs and IFN signaling in cancer cells (Figures S5F and S5G), similar to SUV39H1 KD (Figure S4B). F5446 treatment also reduced the survival of cancer cell lines and patient-derived glioblastoma cultures (Figure 5G).
To establish the potential of a therapeutic index for FBXO44/SUV39H1, we examined the role of FBXO44/SUV39H1 in RE silencing in normal cells. FBXO44 KD did not affect the proliferation of human mammary epithelial cells (HMECs) or primary astrocytes (Figure 5H), nor increased the expression of REs, MAVS/STING, or IFN-β (Figure S5H), or increased γH2AX (Figure S5I). In addition, F5446 treatment minimally affected HMEC and astrocyte viability (Figure 5G). HMECs and astrocytes displayed decreased H3K9me3 levels and FBXO44/SUV39H1 binding at REs compared to cancer cell lines on ChIP (Figures S5J–S5L), supporting cancer cell-specific effects of FBXO44/SUV39H1 inhibition on RE transcription.
FBXO44/SUV39H1 inhibition decreases tumor growth, enhances antitumor immune response, and overcomes resistance to immune checkpoint blockade therapy
FBXO44 KD decreased metastatic seeding of cancer cells to the lung, brain, and bone in immunodeficient mice (Figures S6A–S6C). FBXO44 KD also decreased orthotopic mammary tumor growth (Figures S6D–S6F). Co-targeting of MAVS+STING only partially rescued the decreased tumor growth induced by FBXO44 KD, consistent with in vitro data showing it could not fully rescue the anti-proliferation effect of FBXO44 KD cells (Figure 5E). Thus, FBXO44/SUV39H1 inhibition decreases cell-autonomous tumor growth in mice only partially dependent on MAVS/STING signaling.
Considering the activation of IFN signaling, we next examined whether inhibition of tumor cell-intrinsic FBXO44/SUV39H1 influenced the tumor immune microenvironment. Short hairpin RNAs (shRNAs) were applied to diminish FBXO44 or SUV39H1 expression in mouse 4T1 breast cancer cells to levels that activated REs, IFN-α/β, and ISGs, but permitted tumor growth, thus mimicking pharmacological inhibition in cancer treatment (Figures 6A and S6G). FBXO44/SUV39H1 KD increased the intratumoral abundance of CD8+ T and natural killer (NK) cells (Figure 6B). The CD8+/Treg cell ratio and number of IFN-γ+CD8+ T cells also increased in FBXO44/SUV39H1 KD tumors, indicating a functional immune response (Figures 6B and 6C). Immunohistochemical (IHC) analysis confirmed enhanced infiltration of CD8+ T and NK cells in FBXO44/SUV39H1 KD tumors (Figure 6D). FBXO44/SUV39H1 KD also enhanced PD-L1 and H2Kd MHC class I alloantigen surface expression on 4T1 tumor cells in vivo (Figure 6E), corroborating in vitro qRT-PCR data (Figures 4E, S1B, S4B, S5F, S5G, and S6G), demonstrating enhanced immunogenicity in vivo and potentially circumventing a major pathway of cancer cell immune evasion, consistent with other studies using DNA methylation or LSD1 inhibitors (Chiappinelli et al., 2015; Sheng et al., 2018). Co-targeting of MAVS+STING rescued the increased intratumoral infiltration of CD8+, NK, and IFN-γ+ CD8+ cells and enhanced PD-L1 and H-2Kd MHC class I alloantigen surface expression on tumor cells induced by FBXO44 KD (Figures S6H–S6J), consistent with in vitro data showing the induced IFN-β expression in FBXO44 KD cells was dependent on MAVS+STING (Figure 4K).
Figure 6. FBXO44/SUV39H1 inhibition decreases tumor growth, enhances antitumor immune response, and overcomes resistance to ICB therapy.

(A) Representative images of syngeneic immunocompetent mice at day 22 post-transplantation (top panel) and tumor growth curves (n = 6) (bottom panel).
(B and C) Flow cytometry quantification of the indicated infiltrating immune cells in tumors in (A) (n = 6).
(D) Representative IHC images of indicated infiltrating immune cells in tumors in (A) (left panel) and quantification (n = 6) (right panels). Scale bar, 50 μm.
(E) Flow cytometry analysis of PD-L1 and MHC-I (H-2Kd) surface expression on 4T1 tumor cells in (A). shCtrl (n = 4), shFBXO44 (n = 6), and shSUV39H1 (n = 4).
(F) Growth curves of tumors in syngeneic immunocompetent mice (n = 5).
(G) Survival curves for mice in (F).
(H) Growth curves of tumors in immunodeficient mice (n = 10 for days 12, 17, and 21; n = 7 for day 25).
(I) Representative bioluminescent images of mice in (H) following treatment with vehicle or F5446 at day 25.
(J) Images of mammary tumors dissected from mice in (H) following treatment with vehicle or F5446 at day 25.
(K) Representative IHC images of tumors stained with anti-γH2AX or anti-cleaved caspase 3 antibody (top panel) and quantification (n = 6) (bottom panel). Scale bar, 50 μm.
(L) qRT-PCR analysis for tumors dissected from mice in (H) (n = 3).
(M) Growth curves of tumors in syngeneic immunocompetent mice (n = 6).
(N) Representative bioluminescent images of tumors in (M) at day 28 (left panel) and quantification of total body radiance (n = 6) (right panel).
(O) Survival curves for mice in (M).
Data represent mean ± SEM. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Dunnett’s multiple comparisons test (B, C, D [for CD45+ cells], and E), Tukey’s multiple comparisons test (N), two-way ANOVA followed by Tukey’s multiple comparisons test (A, D [for CD8+ T and NK cells], F, H, L, and M), Sidak’s multiple comparisons test (K), and log-rank test (G and O).
See also Figure S6.
FBXO44 inhibition induced IFN signaling, enhanced cancer cell immunogenicity, and increased intratumoral infiltration of CD8+ T cells; phenotypes associated with a favorable response to immune checkpoint blockade (ICB) therapy (Jones et al., 2019; Keenan et al., 2019). To investigate whether targeting tumor cell-intrinsic FBXO44/SUV39H1 could overcome resistance to anti-PD-1 therapy, we treated immunocompetent mice bearing 4T1 cell-derived control, FBXO44 KD, or SUV39H1 KD mammary tumors with either anti-PD-1 or isotype immunoglobulin G (IgG) control antibodies (Figure 6F). As expected, 4T1 control tumors were refractory to anti-PD-1 treatment (Bertrand et al., 2017), whereas FBXO44/SUV39H1 KD tumors displayed enhanced sensitivity. Mice bearing FBXO44/SUV39H1 KD tumors treated with anti-PD-1 therapy exhibited increased survival compared to control mice (Figure 6G). Co-targeting of MAVS+STING rescued the enhanced antitumor response to anti-PD-1 therapy and increased survival induced by FBXO44 KD (Figures S6K and S6L). Thus, targeting tumor cell-intrinsic FBXO44/SUV39H1 enhanced immunogenicity and ICB therapy response in a MAVS/STING-dependent manner, in addition to anti-proliferative and anti-tumorigenic effects.
Next, we evaluated the effects of pharmacologically targeting SUV39H1 on tumor growth and immunotherapy response. F5446 treatment inhibited mammary tumor growth in immunocompromised mice in a dose-dependent manner (Figures 6H–6J). F5446-treated tumors exhibited a dose-dependent increase in γH2AX and cleaved caspase 3, a marker of apoptosis (Figure 6K). Consistent with the observed effects in vitro (Figures S5F and S5G), F5446-treated tumors upregulated expression of REs, IFN-α/β, and ISGs (Figure 6L). In addition, F5446 treatment enhanced the sensitivity of 4T1 tumors to anti-PD-1 therapy, resulting in increased mouse survival (Figures 6M–6O).
FBXO44 is associated with poor clinical outcomes in cancer patient datasets
To investigate whether FBXO44 associated with human tumorigenesis and therapeutic responses, we interrogated public cancer transcriptomic data, revealing FBXO44 overexpression in many human cancer types compared to normal adjacent tissues (Figure 7A). FBXO44 was expressed at low levels in normal breast tissues and increased with tumor stage (Figure 7B). High FBXO44 expression correlated with poor patient outcome in several major cancer types (Figure 7C).
Figure 7. FBXO44 is associated with poor clinical outcomes in cancer patient datasets.

(A) Analysis of FBXO44 expression in indicated cancer types relative to normal adjacent tissue (Oncomine, Compendia Bioscience, Ann Arbor, MI).
(B) Representative IHC images (left panels) and quantification of FBXO44 expression in normal breast tissue and breast tumors (n = 8 normal, n = 24 stage II, and n = 16 stage III) (right panel). Scale bar, 0.5 mm; inset scale bar, 50 μm.
(C) Survival plots for patients with FBXO44 high- versus low-expressing tumors (http://www.kmplot.com).
(D) Pan-cancer analysis of TCGA dataset for FBXO44 expression with indicated gene expression signatures.
(E) Pathway enrichment map for GSEA for gene sets enriched among significantly upregulated (red) or downregulated (blue) genes in FBXO44 high- versus low-expressing tumors in pan-cancer analysis of the TCGA dataset. FDR <0.1.
(F) GSEA analysis of various immune-stimulatory pathways in FBXO44 high- versus low-expressing tumors in pan-cancer analysis of the TCGA dataset.
(G) Correlation analysis between FBXO44 expression level and z-scores of the indicated gene sets in different cancer types from the TCGA dataset.
(H) Boxplots of FBXO44-immune gene signature Z scores in non-responder and responder groups of patients with anti-PD-1 or TIL therapy in the indicated datasets.
(I) Heatmap of the FBXO44-immune gene signature differentially enriched in responder versus non-responder patients in Harel anti-PD-1 therapy dataset.
(J) Model of FBXO44/SUV39H1 inhibition-induced antitumor effects and enhancement of immunotherapy response.
Data represent mean ± SEM. *p < 0.05, ****p < 0.0001 by two-way ANOVA followed by Tukey’s multiple comparisons test (B) and unpaired Student’s t test (H).
Next, we interrogated The Cancer Genome Atlas (TCGA) to determine if FBXO44 expression associated with the tumor immune microenvironment in cancer patients. Pan-cancer analysis showed that FBXO44 expression inversely correlated with gene expression signatures of CD8+ T and NK cell infiltration and antigen processing and presentation (Figures 7D and S7A; Table S6). FBXO44 expression inversely correlated with innate immune system, cytosolic DNA sensing pathway, RIG-I/MDA5-mediated induction of IFN-α/β pathways, and regulation of IFN-γ signaling, among other immune-related processes (Figures 7E, 7F, and S7B), confirming our results from in vitro and mouse models. Specifically, FBXO44 expression strongly anti-correlated with antiviral mechanism by IFN stimulated genes and interferon signaling in various human cancers (Figure 7G; Table S7). FBXO44 expression also strongly anti-correlated with activation of ATR in response to replication stress (Figure S7C; Table S7), consistent with activation of the DNA replication checkpoint in FBXO44 KD cells in vitro (Figure 3I).
To investigate the clinical relevance of FBXO44/SUV39H1 inhibition for cancer immunotherapy, we created a FBXO44-immune gene signature and evaluated its predictive value across multiple public cancer immunotherapy datasets, including anti-PD-1 and tumor-infiltrating T cell (TIL) therapies (Figure S7D). Responders to immune therapies expressed higher levels of the FBXO44-immune gene signature than non-responders across most of the datasets (Figures 7H, 7I, and S7E), suggesting that FBXO44/SUV39H1 inhibition sensitizes cancers to immunotherapy.
DISCUSSION
During replication of heterochromatin, parental H3K9me3-modified H3-H4 tetramers are evicted ahead of the replication fork and randomly re-deposited along with newly synthesized tetramers onto sister chromatids, resulting in a half reduction in H3K9me3 modifications and potential transcriptional activation of these regions (Allshire and Madhani, 2018; Chen et al., 2008; Kloc et al., 2008; Lu and Gilbert, 2007). Here, we demonstrated that FBXO44 functions as an epigenetic “reader” that binds H3K9me3-modified nucleosomes at the replication fork and recruits an enzymatic complex that includes SUV39H1 to rapidly reassemble heterochromatin and transcriptionally silence REs to avoid potentially adverse effects on genome integrity. SUV39H1’s interaction with chromatin and REs in cancer cells depended on FBXO44 and, indirectly, through its interaction with Mi-2/NuRD, as co-targeting of GATAD2A+B inhibited SUV39H1 recruitment. This epigenetic regulatory complex is similar to the DIM-5/7/9-CUL4-DDB1 complex (DCDC) in Neurospora crassa, in which the scaffold protein DIM-7 targets H3K9me3 methyltransferase DIM-5 and CUL4-DDB1-DIM-9(DCAF) to heterochromatic domains (Gessaman and Selker, 2017; Lewis et al., 2010). Consistent with the proposed role for FBXO44 in DNA replication-coupled RE silencing, H3K9me3 modifications primarily occur in S phase (Alabert et al., 2015; Di Micco et al., 2011; Xu et al., 2012), concomitant with FBXO44’s translocation to the nucleus. In addition, SUV39H1 was found enriched at nascent chromatin using a quantitative proteomics approach (Alabert et al., 2014). Moreover, CRL4 is intricately involved in DNA replication and repair (Abbas and Dutta, 2011) and cooperates with SUV39H1 in H3K9me3-mediated repression of Igfbp3 in cancer cells (Yang et al., 2015). FBXO44 and CRL4 functionally cooperate in proteasomal degradation of regulator of G protein signaling 2 (RGS2) (Sjogren et al., 2015).
Although FBXO44/SUV39H1 were essential for RE silencing in cancer cells, FBXO44/SUV39H1 levels were decreased at REs and FBXO44/SUV39H1 targeting did not activate RE transcription in normal cells. The cancer cell-specific role of FBXO44/SUV39H1 in RE silencing could be attributed to the decreased levels of DNA methylation at RE in human cancers (Anwar et al., 2017; Ehrlich, 2002) and/or absence of compensatory H3K9me3 modification pathways. Alternatively, FBXO44/SUV39H1-mediated RE silencing could be uniquely required for rapidly proliferating cells to prevent DNA replication stress and genome instability that could result from the progressive loss of H3K9me3 with each DNA replication cycle. Consistent with this, SUV39H1/2 promotes H3K9me3 modifications at intact LINE and ERV sequences in rapidly dividing mouse embryonic stem cells (mESCs), although LINE silencing is largely governed by DNA methylation in somatic cells (Bulut-Karslioglu et al., 2014; Martens et al., 2005).
Our data demonstrate that FBXO44/SUV39H1 are crucial suppressors of DNA replication stress and IFN signaling in cancer cells (Figure 7J). FBXO44/SUV39H1 targeting restricted cancer cell autonomous growth and stimulated antitumor T cell immunity. The anti-proliferative and apoptotic effects of FBXO44/SUV39H1 targeting on cancer cells were partially rescued by MAVS+STING KD, suggesting the induced DNA replication stress and DSBs in these cells likely contribute to these phenotypes. However, the increased IFN-β expression and enhanced CD8+ and NK cell intratumoral infiltration induced by FBXO44 KD were dependent on these antiviral pathways. The triggers that activated antiviral pathways in FBXO44 KD cancer cells were cytosolic dsRNA and dsDNA, at least partially generated from various RE subtypes, and genomic instability. In contrast, the anti-proliferative effects on cancer cells treated with low-dose DNA methylation inhibitor or LSD1 inhibitor are thought to be mediated through reactivation of ERVs and stimulation of RIG-I/MDA5-MAVS (Chiappinelli et al., 2015; Sheng et al., 2018), as well as IRF7 (Roulois et al., 2015), suggesting that FBXO44/SUV39H1 inhibition could promote a more robust viral mimicry in cancer cells compared to these agents.
The majority of patients with solid tumors do not receive long-term benefit from ICB therapy. Mechanisms of resistance include decreased CD8+ T cell abundance and defective IFN signaling or antigen presentation (Tumeh et al., 2014; Gao et al., 2016; Zaretsky et al., 2016; Sade-Feldman et al., 2017). FBXO44/SUV39H1 targeting sensitized normally refractory cancer cells to anti-PD-1 therapy. Therefore, FBXO44/SUV39H1 inhibitors could potentially convert “cold” tumors, which are poorly immunogenic and non-responsive to immunotherapy, to “hot” (Sharma et al., 2017). Of note, SUV39H1 targeting reactivated expression of effector genes in tumor-infiltrating cytotoxic T cells in a pre-clinical mouse cancer model indicating reprogramming of T cell exhaustion (Lu et al., 2019), a phenotype also observed with DNA methylation inhibitor treatment (Ghoneim et al., 2017).
In summary, our study demonstrates that FBXO44 promotes DNA replication-coupled transcriptional silencing of RE in cancer cells. Moreover, FBXO44/SUV39H1 are crucial suppressors of DNA replication stress and IFN signaling in cancer cells. FBXO44/SUV39H1 inhibitors could have widespread application in cancer treatment, as stand-alone anti-cancer therapies and enhancers of immunotherapy response.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles Spruck (cspruck@sbpdiscovery.org).
Materials availability
All unique reagents generated in this study will be made available upon request. An agreement with our institute’s Materials Transfer Agreement (MTA) may be required.
Data and code availability
The ChIP-seq and RNA-seq datasets generated during this study are available at GEO: GSE139973 and GSE139974.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cancer cell lines were purchased from the American Tissue Culture Collection (ATCC) and grown in recommended culture media. Specifically, MDA-MB-231, BT-549, MCF7, A549, and H446 cells were cultured in RPMI 1640 (Corning), HEK293T, HEK293FT, and HeLa cells were cultured in DMEM (Corning), MDA-MB-231-luc cells were cultured in MEM (Corning), U2OS and HCT116 cells were cultured in McCoy’s 5A (Corning). The above media were supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 100 U/mL penicillin and streptomycin. Patient-derived glioma stem cells (GSCs) were obtained (Xie et al., 2018) and cultured in Neurobasal medium (Thermo Fisher Scientific, #11360070) supplemented with B27 without vitamin A (Thermo Fisher Scientific, #A3353501), EGF, and bFGF (20 ng/mL each; R&D Systems), sodium pyruvate (Thermo Fisher Scientific, #11360070), and GlutaMAX (Thermo Fisher Scientific, # 35050061). HMECs (Lonza, #CC-2551) were cultured in MEGM BulletKit Medium (Lonza, #CC-3150). Primary astrocytes were cultured in Astrocyte Medium (ScienCell, #1801). All cells were incubated at 37°C in 5% CO2. The cell cultures were authenticated by short tandem repeat (STR) analysis.
Animal studies
Mice were maintained under pathogen free conditions in 14 hr light / 10 hr dark cycle with ad libitum access to food and water. All animal handling and procedures used were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of SBP or UCSD. For intracardiac injections, 105 MDA-MB-231-luc cells transduced with lentivirus expressing non-targeting or FBXO44 shRNA were resuspended in 100 μL DPBS and injected into the left ventricle of 6-week old female athymic nude mice (Envigo no. 069) anesthetized with isoflurane. Metastatic seeding was monitored weekly by intraperitoneal (i.p.) injection of 100 μL of 30 mg/mL D-Luciferin (Xenogen no. XR-1001) 10 min prior to imaging using the IVIS Spectrum Xenogen Imaging System (Caliper Life Sciences). Images were analyzed using Living Image 3.0 software (Caliper Life Sciences). For ex vivo imaging of tissues, mice were injected with D-Luciferin prior to euthanasia, tissues harvested and placed in 24-well tissue culture plates containing 1 mL of 0.3 mg/mL D-Luciferin and imaged. Organs/tissues were fixed in 10% formalin and subjected to hematoxylin and eosin staining. For orthotopic injections of NSG mice, 3 × 106 MDA-MB-231-luc cells were suspended in 100 μL of PBS and injected into the mammary fat pads of 4- to 6-week-old female mice. For F5446 drug treatments, mice were randomly divided into 3 groups at day 12. The mice were administered vehicle (10% Cremophor EL in PBS) or F5446 at doses of 10 and 20 mg/kg body weight via i.p. injection on days 12, 14, 17, 19, 21 and 24 post-inoculation of cells. Tumor size was measured every 4–5 days (long diameter and short diameter) with a caliper and tumor volume calculated as 0.5 × length × width × width. Mice were imaged weekly to monitor tumor growth and metastasis in vivo using an IVIS Spectrum Xenogen Imaging System (Caliper Life Sciences). All mice were euthanized at day 25 except 3 mice were taken out from each group for tumor IHC staining of γH2AX and cleaved caspase 3 and quantitative RT-PCR analysis at day 21. For 4T1 tumor experiments, 105 4T1 cells were inoculated into the mammary fat pads of 6- to 8-week-old female BALB/c mice (The Jackson Laboratory). Tumor size was measured twice weekly. After 22 days, mice were euthanized and portions of each tumor processed for flow cytometry and histological analysis. For antibody treatments, mice were administered 200 μg rat IgG2a isotype control (BioXCell no. BP0089) or anti-PD-1 (BioXCell no. BP0273) antibody via i.p. injection on days 15, 17, 19, 21 and 23 post-inoculation of 4T1 cells. For combination treatments, mice were administered vehicle (10% Cremophor EL in PBS) or F5446 at 10 mg/kg body weight via i.p. injection on days 12, 14, 16, 18, 20, and 22 post-inoculation of cells and administered 200 μg rat IgG2a isotype (control) or anti-PD-1 antibody via i.p. injection on days 15, 17, 19, 21, and 23 post-inoculation of 4T1 cells. Mouse survival was monitored with tumor volume exceeding 2000 mm3, weight loss >20%, and decreasing behavioral conditions considered as endpoints.
METHOD DETAILS
Cell transfection and viral transductions
Plasmid transfections were performed using jetPRIME transfection reagent (Polyplus) for HEK293T and HEK293FT cells and Lipofectamine 2000 (Thermo Fisher Scientific) for all other cell lines, following the manufacturer’s instructions. Lentiviruses were generated using HEK293FT cells (Thermo Fisher Scientific) and vectors pLenti CMV Puro DEST (Addgene plasmid no. 17452), pLiX_402 (Addgene plasmid no. 41394), or pLKO.1 (Addgene plasmid no. 10878) using standard techniques. Single and multiple viral transductions were performed in the presence of 8 μg/ml polybrene (Sigma-Aldrich).
Gene silencing
The quantitative image-based H3K9me3 regulator screen was performed using a cell-spot microarray approach (Rantala et al., 2011), modified for detection of DAPI, p-RPA32T21, and H3K9me3. FBXO44 siRNAs (no. SI00145663, SI00145670, SI03078551) were purchased from QIAGEN, with SI00145663 and SI00145670 used in most experiments. Other siRNAs used included SUV39H1 (QIAGEN no. SI00048685 and SI02665019), GATAD2A (QIAGEN no. SI04318636), GATAD2B (Dharmacon no. J-013892–06-0005), CHD4 (QIAGEN no. SI00024563), CUL4B (QIAGEN no. SI04215015 and SI04292540), CUL1 (QIAGEN no. SI02225657 and SI02225664), RBBP4 (Dharmacon no. L-012137–00-0005), RBBP7 (Dharmacon no. L-011375–00-0005), IRF3 (QIAGEN no. SI02626526), and IRF7 (QIAGEN no. SI00448672). siRNA transfections were performed using Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific). Unless otherwise noted, cells were harvested 72 hr post-transfection. Lentiviral shRNA vectors for human cells included non-targeting control (Dharmacon no. RHS6848 and Sigma no. SCH002), FBXO44 #1 (Dharmacon no. RHS3979–201785935), FBXO44 #2 (Dharmacon no. RHS3979–201789150), LSD1 #1 (Sigma no. TRCN0000046071), LSD1 #2 (Sigma no. TRCN0000382249), MAVS (Sigma no. TRCN0000236031), and STING (Sigma no. TRCN0000163296). Lentiviral shRNA vectors for mouse cells included non-targeting control (Dharmacon no. RHS6848 and Sigma no. SCH002), FBXO44 (Dharmacon no. RMM3981–201914287), SUV39H1 (Dharmacon no. RMM3981–201816562), MAVS (Sigma no. TRCN0000124769), and STING (Sigma no. TRCN0000346320).
Immunoblotting and immunoprecipitation
For immunoblotting, cells were lysed in cold RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.01% SDS) supplemented with protease and phosphatase inhibitors (50 mM NaF, 1 mM PMSF, 1 mM Na3VO4, 1 μg/mL Aprotinin, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin). Lysates were briefly sonicated, clarified, then subjected to SDS-PAGE and transferred to PVDF membranes using a Bio-Rad transfer apparatus. Membranes were blocked with 5% non-fat milk or BSA in TBS containing 0.1% Tween-20 (TBST) at 25°C for 1 hr, followed by incubation with primary antibody overnight at 4°C. Membranes were washed 3 × 10 min in TBST and incubated with species-specific HRP-conjugated secondary antibodies (Pierce) for 1 hr at 25°C. After 3 × 10 min washes in TBST, membranes were developed using an enhanced chemiluminescence (ECL) reagent before being exposed to film or ChemiDoc Imaging System (Bio-Rad). For IP, cells were lysed in IP buffer (50 mM Tris-HCl [pH 7.4], 125 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) with inhibitors (50 mM NaF, 1 mM PMSF, 1 mM Na3VO4, 1 μg/mL Aprotinin, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin) followed by sonication and centrifugation to clear insoluble debris. Lysates were then incubated with protein G agarose beads and IgG antibody of the same species as the IP antibody for 2 hr at 4°C to reduce non-specific binding. The cleared lysates were then incubated with IP antibody overnight at 4°C. Specifically, for Flag IP, 5 μg of anti-Flag antibody (Sigma-Aldrich no. F7425) was used for 2500 μg of protein lysate. For Myc IP, anti-Myc antibody (Cell Signaling Technology no. 2276) was used at 1:1000 dilution. For FBXO44 IP, 5 μL of anti-FBXO44 antibody (Sigma-Aldrich no. HPA003363) was used for 106 cells. For CUL4B IP, 2 μg of anti-CUL4B antibody (Proteintech no. 12916–1-AP) was used for 2500 μg of protein lysate. For SUV39H1 IP, anti-SUV39H1 antibody (Millipore no. 05–615) was used at 1:100 dilution. Beads were washed with IP buffer, boiled in 1× SDS gel loading buffer, and subjected to electrophoresis as described above.
qPCR analyses
Total RNA was isolated using the RNeasy Plus Mini Kit (QIAGEN no. 74134). cDNA was synthesized using qScript cDNA SuperMix (Quantabio no. 95048) following the manufacturer’s instructions. qPCR reactions were performed using SYBR Select Master Mix (Thermo Fisher Scientific no. 4472908) and a Stratagene Mx3000P instrument (Agilent Technologies). Thermal cycling conditions included an initial denaturation step of 95°C for 10 min and 40 cycles at 95°C for 15 s and 60°C for 60 s. Analyses were carried out in triplicate for each data point. The qPCR primers used for gene expression analysis of human REs and genes included
MajSAT- GGCGAGAAAACTGAAAATCACG and CTTGCCATATTCCACGTCCT, mcBox- AGGGAATGTCTTCCCATAAAAACT and GTCTACCTTTTATTTGAATTCCCG, SATIII- AATCAACCCGAGTGCAATCGAATGGAATCG and TCCATTCCATTCCTGTACTCGG, Alu- AATGGTACGATCTCGGCTCA and TAGCCAGGTGTGGTGACTTG, L1 ORF1- TGGCCCCCACTCTCTTCT and TCAAAGG AAAGCCCATCAGACTA, L1 ORF2- GCCATTGCTTTTGGTGTTTT and AAATGGTGCTGGGAAAACTG, L1 5′-UTR- AAGCAAGC CTGGGCAATG and ACGGAATCTCGCTGATTGCTA, HERV-E- GGTGTCACTACTCAATACAC and GCAGCCTAGGTCTCTGG, HERV-F- CCTCCAGTCACAACAACTC and TATTGAAGAAGGCGGCTGG, HERV-K- CAGTCAAAATATGGACGGATGGT and ATTGGCAACACCGTATTCTGCT, ERVL- ATATCCTGCCTGGATGGGGT and GAGCTTCTTAGTCCTCCTGTGT, GAPDH- CGACCA CTTTGTCAAGCTCA and AGGGGTCTACATGGCAACTG, FBXO44- GCCCAGTAATGAGTGTCCACG and AGATTGCGGGTCCAAG TACC, SUV39H1- CCTGCCCTCGGTATCTCTAAG and ATATCCACGCCATTTCACCAG, IFN-α- AATGACAGAATTCATGAAAGCGT and GGAGGTTGTCAGAGCAGA, IFN-β- GCCATCAGTCACTTAAACAGC and GAAACTGAAGATCTCCTAGCCT, MX1- CTGCACAG GTTGTTCTCAGC and GTTTCCGAAGTGGACATCGCA, IRF3- CATGTCCTCCACCAAGTCCT and GGCTTGTGATGGTCAAGGTT, IRF7- TCAACACCTGTGACTTCATGT and GTGGACTGAGGGCTTGTA, CCL5- CCAGCAGTCGTCTTTGTCAC and CTCTGGGTTGG CACACACTT, CXCL9- CCAGTAGTGAGAAAGGGTCGC and AGGGCTTGGGGCAAATTGTT, CXCL10- GCCTTCGATTCTGGATT CAG and GTGGCATTCAAGGAGTACCTC, cGAS- GCCGCCGTGGAGATATCAT and GGCGGTTTTGGAGAAGTTGA, STING-ATATCTGCGGCTGATCCTGC and TTGTAAGTTCGAATCCGGGC, RIG-I, CCAGCATTACTAGTCAGAAGGAA and CACAGTGCA ATCTTGTCATCC, MAVS- CAGAACTGGGCAGTACCC and AGGAGACAGATGGAGACACA, IFNGR1- TTCCATCTCGGCATACAG CAA and TCTTTGGGTCAGAGTTAAAGCCA, IFNGR2- CTCCTCAGCACCCGAAGATTC and GCCGTGAACCATTTACTGTCG, PTPN2- TGCAGTTTAACACGACTGTGA and GAAGAGTTGGATACTCAGCGTC, PD-L1- GTGGCATCCAAGATACAAACTCAA and TCCTTCCTCTTGTCACGCTCA, GAS6- GGTAGCTGAGTTTGACTTCCG and GACAGCATCCCTGTTGACCTT, EYA2- CAGCGATT GTCTGGATAAACTGA and GGAGGTGGGTAAGCTGTATAGG, HES1- CGTGCGAGGGCGTTAATA and GGGTAGGTCATGGCATT GAT, and LSD1- GTGGACGAGTTGCCACATTTC and TGACCACAGCCATAGGATTCC. qPCR primers used for analysis of mouse REs and genes included FBXO44- TACCTTCCATTCATCGCCTCC and CATTGACCTGGTTACACTCTGG, SUV39H1- TGTCAACCA TAGTTGTGATCC and GCATGTTGTAATCAAAGGTGAG, MajSAT II- CTTGCCATATTCCACGTCCT and GGCGAGAAAACTGAAAAT CACG, MinSAT II- TTGGAAACGGGATTTGTAGA and CGGTTTCCAACATATGTGTTTT, LINE1- CTGCCGTCTACTCCTCTTGG and TTTGGGACACAATGAAAGCA, MERV-L- GACACCTTTTTTAACTATGCGAGCT and TTTCTCAAGGCCCACCAATAGT, STINGGGTCACCGCTCCAAATATGTAG and CAGTAGTCCAAGTTCGTGCGA, MAVS- CTGCCTCACAGCTAGTGACC and CCGGCGCTG GAGATTATTG, IFN-α- CGGTGCTGAGCTACTGGC and TTTGTACCAGGAGTGTCAAGG, IFN-β- GGTGGAATGAGACTATTGTTG and AGGACATCTCCCACGTC, IFN-γ- AAAGAGATAATCTGGCTCTGC and GCTCTGAGACAATGAACGCT, CCL5- TCCTTCGAGT GACAAACACG and CCCTCACCATCATCCTCACT, CXCL9- TGAGGTCTTTGAGGGATTTGTAGTG and GGAACCCTAGTGATAAG GAATGCA, CXCL10- GACGGTCCGCTGCAACTG and CTTCCCTATGGCCCTCATTCT, PD-L1- GACCAGCTTTTGAAGGGAAATG and CTGGTTGATTTTGCGGTATGG, and GAPDH- TGACCTCAACTACATGGTCTACA and CTTCCCATTCTCGGCCTTG.
RNase protection assay
RNase protection assay was performed as described previously (Roulois et al., 2015) with modifications. Total RNA from MDA-MB-231 cells (2 × 106) transfected with control or 2 different FBXO44 siRNAs was isolated using the RNeasy Plus Mini Kit (QIAGEN no. 74134) and 4 μg of total RNA dissolved in 49.5 μL RNase protection buffer (10 mM Tris-HCl [pH 7.5], 350 mM NaCl). Then 0.5 μL RNase A (10 mg/ml, Sigma-Aldrich no. R6513) was added and the mixture incubated for 30 min at 37°C. dH2O was added to the control sample. dsRNA was isolated and denatured at 95°C for 5 min. Reverse transcription was carried out using qScript cDNA SuperMix (Quantabio no. 95048) and qPCR reactions performed using primers listed in the “qPCR analyses” section. dsRNA enrichment for selected REs was calculated by (RE/GAPDH) RNase A / (RE/GAPDH) control.
RNA-seq
PolyA+ RNA was isolated using the NEBNext Poly(A) mRNA Magnetic Isolation Module and barcoded libraries made using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB). Libraries were pooled and single end sequenced (1 × 75) on an Illumina NextSeq 500 using the High Output V2 Kit (Illumina). Read data was processed in BaseSpace (https://basespace.illumina.com:443/). Reads were then aligned to the Homo sapiens genome (hg19) using STAR aligner (https://code.google.com/archive/p/rna-star) with default settings. Differential transcript expression was determined using the Cufflinks Cuffdiff package (https://github.com/cole-trapnell-lab/cufflinks). GSEA was performed on pre-ranked gene lists upon FBXO44 KD using gene set permutation for statistical testing. GSEA results were visualized using Enrichment Map in Cytoscape. For analysis of RE expression from RNA-seq data, reads were mapped to the Homo sapiens genome (hg19) using Bowtie 2 and assigned to REs using RepEnrich2 with the recommended parameters (https://github.com/nerettilab/RepEnrich2) (Criscione et al., 2014). The RE annotation file (hg19_repeatmasker_clean.txt) was provided by RepEnrich2. The resulting counts for REs were analyzed by the edgeR package to obtain CPM (counts per million reads) values.
ChIP and ChIP-seq
For Flag and FBXO44 ChIP experiments, cells were cross-linked with 1% formaldehyde for 10 min at 25°C and reactions stopped by adding glycine to a final concentration of 0.125 M for 5 min at 25°C. Cells were rinsed 3× with PBS and re-suspended in ChIP lysis buffer (50 mM Tris-HCl [pH 8.1], 1% SDS, 10 mM EDTA, 0.2 mM PMSF, 1 μg/mL Aprotinin, 1 μg/mL Leupeptin). Specifically, lysates prepared from 106 cells in 130 μL lysis buffer were introduced into microsonication tubes (Covaris no. 520045). For H3K9me3 ChIP experiments, native ChIP was performed. Cells were harvested and washed with cold PBS and native chromatin extracted using a commercial chromatin extraction kit (Abcam no. ab117152). For H2AK119ub native ChIP, 1 μL of anti-H2AK119ub antibody (Cell Signaling Technology no. 8240) was used for 106 cells. For all ChIP experiments, DNA was fragmented by sonication in a S220 Focused-Ultrasonicator (Covaris) for 7 min (Duty cycle- 5%, Intensity- 4, Cycles/Burst- 200). After centrifugation, the supernatant was diluted 1:10 by adding ChIP dilution buffer (Tris-HCl [pH 8.1], 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM NaCl, 0.2 mM PMSF, 1 μg/mL Aprotinin, 1 μg/mL Leupeptin), 5% saved for input, and the remaining lysate pre-cleared for 2 hr with 15 μl protein G agarose before overnight incubation with the indicated antibody. Specifically, for Flag ChIP, 2.5 μL of anti-Flag antibody (Cell Signaling Technology no. 14793) was used for 106 cells. For FBXO44 ChIP, 5 μL of anti-FBXO44 antibody (Sigma-Aldrich no. HPA003363) was used for 106 cells. For SUV39H1 ChIP, 6 μL of anti-SUV39H1 antibody (Millipore no. 05–615) was used for 106 cells. For γH2AX ChIP, 4 μL of anti-γH2AX antibody (Abcam no. 2893) was used for 106 cells. For H3K9me3 ChIP, the ChIPAb+ Tri-methyl-Histone H3 (Lys9) kit (Millipore no. 17–10242) was used, typically with 1 μL of anti-H3K9me3 antibody for 106 cells. Bound material was recovered after incubation with 15 μL of protein G beads at 4°C. Beads were then washed sequentially with low salt buffer (20 mM Tris-HCl [pH 8.1], 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl), high salt buffer (20 mM Tris-HCl [pH 8.1], 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl), LiCl buffer (10 mM Tris-HCl [pH 8.1], 0.25 M LiCl, 1% IGEPAL-CH630, 1% Na deoxycholate, 1 mM EDTA) and TE (10 mM Tris-HCl [pH 8.1], 1 mM EDTA). Elution was performed by adding 200 μL of freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3) to 15 μL protein G beads and rotating the sample for 15 min at 25°C. After centrifugation, the chromatin IP and input were reverse-cross-linked by adding 5M NaCl (4 μL for input and 8 μL for IP sample) and incubated overnight at 65°C, and then DNA extracted using the Gel Extraction Kit (QIAGEN no. 28704). Primers used included
MajSAT- GGCGAGAAAACTGAAAATCACG and CTTGCCATATTCCACGTCCT, mcBox- AGGGAATGTCTTCCCATAAAAACT and GTCTACCTTTTATTTGAATTCCCG, SATIII- AATCAACCCGAGTGCAATCNGAATGGAATCG and TCCATTCCATTCCTGTACTCGG, ALU- AATGGTACGATCTCGGCTCA and TAGCCAGGTGTGGTGACTTG, L1 5′UTR- GATGATGGTGATGTACAGATGGG and AGCC TAACTGGGAGGCACCC, L1 3′UTR- TGTATACATGTGCCATGCTGGTGC and AATGAGATCACATGGACACAGGAAG, HERV-K-AGAGGAAGGAATGCCTCTTGCAGT and TTACAAAGCAGTATTGCTGCCCGC, and Gapdh- TCGAACAGGAGGAGCAGAGAG and TACTAGCGGTTTTACGGGCG.
For ChIP-seq, libraries were made using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB). Libraries were pooled and single end sequenced (1 × 75) on an Illumina NextSeq 500 using the High Output V2 kit (Illumina). Read data was processed in Base-Space (https://basespace.illumina.com:443/). ChIP-seq reads were then aligned to the Homo sapiens genome (hg19) using Bowtie 2 with default parameters, which allows multiple alignment. Peaks were called using Partek software with default parameters. The midpoint of each estimated fragment by Partek and its location on the genome was calculated. The genome was divided into non-overlapping windows of the default 100 bp. An aligned read was considered to be located in a window if the midpoint of its estimated fragment was within the window. The number of midpoints in each window was counted and an empirical distribution of window counts was created. A zero-truncated negative binomial model was fit to the distribution, and a peak was determined based on the FDR (0.001, default) calculated from the model. Overlapping enriched windows were merged into regions and reported. For analysis of REs from ChIP-seq data, a list of FBXO44 peaks associated with REs was created by intersecting FBXO44 binding peaks with RE loci obtained from RepeatMasker, which was used in the downstream analysis. Reads were mapped and assigned to the FBXO44-associated REs using RepEnrich2 with the recommended parameters (https://github.com/nerettilab/RepEnrich2). The resulting counts for REs were analyzed by the edgeR package to obtain CPM values. The H3K27me3 and H3K4me3 ChIP-seq data in MDA-MB-231 cells were obtained from GEO: GSE87169 (Lee et al., 2016). The BigWig files were obtained using deeptools bamCoverage command with–normalizeUsing CPM. The heatmap and profile plots for ChIP-seq data were performed by deeptools plotHeatmap and plotProfile in the scale-regions mode. Venn diagrams were obtained using VennDiagram package. The ChIP-seq binding signal from BAM files was visualized in The Integrative Genomics Viewer (IGV).
Cytoplasmic DNA isolation and analyses
Equal amounts of MDA-MB-231 cells (5 × 106) transfected with control or 2 different FBXO44 siRNAs were harvested and washed with cold PBS. Cytoplasmic fractions were extracted from the cells after the nuclear fraction was removed through centrifugation using the Nuclear Extract Kit (Active Motif no. 40010) according to manufacturer’s instructions. Cytoplasmic fraction (390 μL solution) was combined with 25 μL 5M NaCl, then treated with 8 μL RNase A/T1 Mix (Thermo Fisher Scientific no. EN0551) for 30 min at 37°C prior to DNA extraction using the Gel Extraction Kit (QIAGEN no. 28704). The amount of indicated RE DNA in cytosol were determined using qPCR primers in “ChIP and ChIP-seq” section and normalized based on gapdh level.
Cytoplasmic dsRNA isolation and analyses
Control or FBXO44 KD MDA-MB-231 cells (4.5 × 106) were harvested and cytoplasmic fractions extracted using the Nuclear Extract Kit (Active Motif no. 40010) according to the manufacturer’s instructions. An equal volume of 70% ethanol (350 μL) was added to the cytoplasmic fractions and then RNA isolated using the RNeasy Plus Mini Kit (QIAGEN no. 74134). The total RNA was dissolved with 38 μL RNase-free H2O. Then 2 μL total RNA was used as input and the remainder divided into 2 tubes (18 μL in each). J2 antibody (Scicons no. 10010200) and normal mouse IgG (Santa Cruz no. sc-2025) were conjugated (2 μg per pulldown) to 20 μL protein G agarose (Millipore no. 16–266) by rotation for 2 hr at 4°C. To each tube containing total RNA, 1 μL of RNase A (10 mg/ml, Sigma-Aldrich no. R6513) was added and then mixed with 1 mL IP buffer (50 mM Tris-HCl [pH 7.4], 125 mM NaCl, 1 mM EDTA, 0.1% Triton X-100), followed by incubation with conjugated protein G agarose beads for 2 hr at 4°C. Beads were washed 3× with IP buffer and incubated in 50 μL proteinase K digestion solution (1× TE, 100 mM NaCl, 1% SDS, and 1 μL of 20 mg/mL Proteinase K solution (Thermo Fisher Scientific no. AM2546)) for 20 min at 45°C. After centrifugation, the eluate (50 μL) was added to 300 μL Buffer RLT Plus from the RNeasy Plus Mini Kit (QIAGEN no. 74134) and RNA isolated. The final product containing dsRNA was denatured at 95°C for 5 min, followed by reverse transcription and qPCR reactions using the primers in “qPCR analyses” section.
Cytoplasmic dsRNA/DNA re-transfection
Preparation of cytoplasmic fractions of cultured cells was performed and cytoplasmic DNA extracted using the Gel Extraction Kit (QIAGEN no. 28704) after incubation with RNase A/T1 Mix (Thermo Fisher Scientific no. EN0551) as detailed in the “Cytoplasmic DNA isolation and qPCR analyses” section. For dsRNA extraction, the cytoplasmic fraction was used for total RNA isolation using the RNeasy Plus Mini Kit (QIAGEN no. 74134) and total RNA digested with RNase A solution to preserve dsRNA as detailed in the “RNase protection assay” section. The concentrations of cytoplasmic dsRNA and DNA were measured using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific). For the cytoplasmic DNA re-transfection assay, 1 μg cytoplasmic DNA was digested with 1 μL DNase I (Thermo Fisher Scientific no. EN0521) or 1 μL H2O (mock digestion) in 10 μL reaction mixture for 30 min at 37°C prior to re-transfection into MDA-MB-231 cells with 1.5 μL Lipofectamine 2000 (Thermo Fisher Scientific). Transfection of 0.8 μg poly (dA:dT) (InvivoGen no. tlrl-patn) was used as a positive control. For cytoplasmic dsRNA re-transfection, 1 μg of dsRNA was digested with 1 μL RNase III (Thermo Fisher Scientific no. AM2290) or 1 μL H2O (mock digestion) in 5 μL reaction mixture for 60 min at 37°C prior to re-transfection into MDA-MB-231 cells with 1.5 μL Lipofectamine 2000. Transfection of 0.8 μg poly (I:C) (InvivoGen no. tlrl-pic) was used as positive control. After 72 hr, RNA was isolated and qPCR reactions performed using primers in “qPCR analysis” section.
IF and IHC
For IF analyses, cells were grown on laminin-coated coverslips (neuVitro no. GG-12-laminin), washed twice with cold PBS, and then fixed with 4% paraformaldehyde for 20 min at 25°C followed by permeabilization with 0.5% Triton X-100 in PBS for 5 min. To detect chromatin associated H3K9me3, cells were fixed with chilled methanol for 5 min at −20°C. After 3 × 10 min washes in PBS, cells were incubated in blocking buffer (10 mM Tris-Cl [pH 7.5], 120 mM KCl, 20 mM NaCl, 0.5 mM EDTA, 0.1% Triton X-100, 10% milk, 2% BSA) overnight at 4°C. The relevant primary antibodies diluted in blocking buffer were then added. Specifically, 1:500 dilution for anti-H3K9me3 (Abcam no. ab8898), 1:2000 dilution for anti-p-RPA32T21 (GeneTex no. GTX62664), 1:60 dilution for anti-dsRNA (Millipore no. MABE1134), 1:1000 dilution for anti-dsDNA (Abcam no. ab27156), 1:50 dilution for anti-ULBP2 (Santa Cruz no. sc-53135), and 1:100 dilution for anti-SSX1 (Novus Biologicals no. NBP2–00614). Cells were washed 3 × 10 min with KCMT buffer (10 mM Tris-Cl [pH 7.5], 120 mM KCl, 20 mM NaCl, 0.5 mM EDTA, 0.1% Triton X-100) and incubated with Alexa Fluor 488-labeled anti-rabbit (Thermo Fisher Scientific no. A-21441) or Alexa Fluor 594-labeled anti-mouse (Cell Signaling Technologies no. 8890) antibodies at 1:500 dilution for 1 hr at 25°C. After 3 × 10 min washes with KCMT buffer, cells were stained with DAPI (Thermo Fisher Scientific no. D1306), coverslips mounted with anti-fade mounting medium (Vector Laboratories no. H-1000), and slides examined by fluorescence microscopy (Nikon Inverted TE300) or confocal microscopy (Zeiss LSM 710 NLO). IHC analysis of normal breast and breast carcinoma specimens (Novus Biologicals no. NBP2–30212) were performed by antigen retrieval and blocking endogenous peroxidase activity using the BOND RX automated IHC system (Leica) followed by staining with anti-FBXO44 antibody (Sigma-Aldrich no. HPA003363) at 1:200 dilution. IHC staining was performed using the Bond Polymer Refine Detection Kit (Leica no. DS9800) and images scanned using Aperio ScanScope AT2 (Aperio Technologies). FBXO44 staining intensity was quantified using Aperio software. For analysis of mouse lung specimens, lungs were removed and fixed in 10% formalin for 24 hr and paraffin embedded. Serial sections cut at 5 μm thickness were stained with hematoxylin (Leica no. 3801560) for 4 min followed by eosin (Leica no. 3801600) for 30 s. Slides were then dehydrated, cleaned, and mounted with Cytoseal 60 mounting media (Thermo Fisher Scientific no. 8310–4). Tissue sections were scanned using Aperio ScanScope AT2. For MDA-MB-231-luc and 4T1 tumors, staining was performed as above using the BOND RX automated IHC system (Leica) with anti-cleaved caspase 3 (Cell Signaling Technology no. 9664; 1:300 dilution), anti-γH2AX (Cell Signaling Technology no. 80312; 1:200 dilution), anti-CD45 (BD no. 550539; 1:50 dilution), anti-CD8a (Thermo Fisher Scientific no. 14–0808-82; 1:50 dilution), and NK Cell Marker (ANK61) (Santa Cruz no. sc-59340; 1:50 dilution) antibodies.
Chromatin extraction
For direct western blotting, chromatin was extracted using a commercial kit (Abcam no. ab117152), sonicated in RIPA buffer, and dissolved in 4× SDS loading buffer. For chromatin fractionation experiments, cells were resuspended in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 50 mM NaF, 1 mM PMSF, 1 mM Na3VO4, 1 μg/mL Aprotinin, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin) and incubated on ice for 5 min, followed by centrifugation at 1,300 × g for 5 min. The cytoplasmic supernatant was then collected. The nuclei pellet was washed once in buffer A and lysed in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, with protease/phosphatase inhibitors described above) on ice for 30 min. After centrifugation at 12,000 × g for 20 min, the nucleoplasmic supernatant was collected. The insoluble chromatin pellet was then washed once with buffer B and centrifuged as described above. The chromatin pellet was then resuspended in RIPA buffer for 20 min at 4°C and sonicated extensively to release chromatin-bound proteins. After centrifugation at 12,000 × g for 20 min, chromatin-associated proteins were collected for electrophoresis.
Mass spectrometry
For analysis of FBXO44 in whole cell extract (WCE), Flag-FBXO44 was introduced into Flp-In TREx-HeLa cells (ThermoFisher Scientific no. R78007) and FBXO44 protein complexes affinity purified. For analysis of FBXO44 in chromatin fractions, MDA-MB-231 (control) or MDA-MB-231-Flag-FBXO44 cells were enriched in S phase by double thymidine block (two sequential 2 mM overnight incubations) and released into normal medium for 3 hr and harvested, washed 2× with cold PBS, and chromatin isolated. Chromatin was dissolved in 2 mL IP buffer and sonicated until the solution cleared. After centrifugation at 12,000 × g for 20 min at 4°C, the supernatants were collected and adjusted to 3 mL with IP buffer and pre-cleared by incubation with 10 μL of normal rabbit IgG and 60 μL of protein G agarose beads for 2 hr at 4°C. The supernatants were then incubated with 12.5 μL of anti-Flag antibody (Sigma-Aldrich no. F7425) overnight at 4°C. After 3 × 10 min washes with IP buffer, the immunoprecipitates were eluted 3× with 150 μL of Flag peptide elution buffer (Sigma-Aldrich no. FLAGIPT1). Trichloroacetic acid (Sigma-Aldrich no. T0699) was then added to the eluents to a final concentration of 20% and incubated on ice for 1 hr. After centrifugation at 14,000 × g for 25 min at 4°C, pellets were washed with 500 μl of ice-cold acetone. After another centrifugation at 14,000 × g for 25 min at 4°C, the pellets were air-dried in a fume hood for 30 min and stored at −20°C. Mass spectrometry analysis was performed using duplicate samples as described (Vashisht et al., 2015).
In vitro binding assays
Synthetic biotinylated nucleosomes (EpiCypher no. 16–0006, 16–0315, 16–0325; 25 μg) were incubated with 10 μL of streptavidin beads (Cell Signaling Technology no. 3419) in binding buffer (50 mM Tris-HCl [pH 7.4], 125 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 50 mM NaF, 1 mM PMSF, 1 mM Na3VO4, 1 μg/mL Aprotinin, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin) on a rotator for 30 min at 4°C. Then, 8 μg of recombinant FBXO44 protein (OriGene no. TP760409) was added and incubated for 2 hr at 4°C in 250 μL total volume. Freeze-thaw cycles were avoided with recombinant proteins. After centrifugation, the beads were washed 3 × 10 min with binding buffer and boiled in 1× SDS gel loading buffer prior to electrophoresis.
aniPOND analysis
aniPOND was performed as described (Leung et al., 2013), with modifications. Briefly, MDA-MB-231 cells (~8.5 × 107) were cultured in medium containing 10 μM EdU for 30 min and then harvested or chased with medium containing 10 μM thymidine for 60 min and harvested. Nuclei were isolated by addition of nuclear extraction buffer. Biotin-azide click reactions were performed on a rotator for 1 hr at 4°C, and 50 μL of streptavidin beads (Cell Signaling Technology no. 3419) were used for streptavidin capture for each sample. Beads were washed 3 × 10 min and boiled in 1× SDS gel loading buffer prior to electrophoresis. For aniPOND analysis of FBXO44 binding to H3K9me3-modified nucleosomes, MDA-MB-231-Flag-FBXO44 cells (~1.7 × 108) were incubated with 10 μM EdU for 30 min, harvested, subjected to biotin-azide click reactions, and chromatin sonicated prior to IP with IgG or anti-Flag antibody (Sigma-Aldrich no. F3165). Chromatin was eluted 3× with Flag peptide solution (150 ng/μL) and subjected to affinity purification with streptavidin beads.
Cell migration/invasion assays
For migration assays, 4 × 104 MDA-MB-231 cells in 40 μl of medium containing 0.5% FBS were plated into the upper chambers of 24-well inserts with 8 μm pores (Trevigen no. 3484–024-01). Bottom wells contained 360 μL of medium supplemented with 10% PBS. After 24 hr, cells were fixed with cold methanol for 20 min. Non-migrating cells that remained in the upper chamber were gently removed using a cotton swab and cells that migrated to the bottom chambers were stained with 1% crystal violet. Invasion assays were performed using well inserts pre-coated with basement membrane (medium density). For both experiments, cells that migrated to the bottom chambers were quantified by imaging 4 randomly selected fields.
Tumorsphere assays
For MCF7 and MDA-MB-231 tumorsphere assays, 2.5 × 103 cells expressing the indicated shRNAs were plated into 6-well ultra-low attachment plates (Corning no. 3471) containing 500 μL of complete MammoCult Human Medium (STEMCELL Technologies no. 05620). Tumorspheres were counted after 10 days using a light microscope. For GSC tumorsphere assays, 1 × 105 cells expressing the indicated shRNAs were plated into regular 6-well plates containing Neurobasal medium supplemented with B27 without vitamin A, EGF, bFGF, sodium pyruvate, and GlutaMAX. Tumorspheres were counted 6 days after plating for Extreme Limiting Dilution Analysis (http://bioinf.wehi.edu.au/software/elda/). All experiments were performed in triplicate.
Flow cytometry
Apoptotic cells were detected using the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences no. 556547) according to the manufacturer’s protocol. Cells were analyzed on an LSR Fortessa instrument (BD PharMingen). For breast CSC quantifications, cells were stained with anti-CD24-APC and anti-CD44-PE antibodies using the Breast Cancer Stem Cell Isolation Kit (MagCellect no. MAGH111). Cells were analyzed on a NovoCyte 3000 flow cytometer (ACEA Biosciences). For cell surface expression of ULBP2 and SSX1, MDA-MB-231 cells were transfected with non-targeting or FBXO44 siRNAs and stained with anti-ULBP2 antibody (GeneTex no. GTX53048) at 1:50 dilution or anti-SSX1 antibody (Novus Biologicals no. NBP2–00614) at 1:50 dilution and analyzed on a LSR Fortessa instrument (BD PharMingen). For 4T1 experiments, tumors were dissociated into single cells using the gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec) as described in the protocol for the Tumor Dissociation Kit (Miltenyi Biotec). Cells were passed through a 70 μm filter to remove clumps and maintain single cell suspensions. Cell surface staining was performed with the indicated antibodies before fixation and permeabilization of cells for intracellular staining. For IFN-γ detection, Cell Activation Cocktail (BioLegend) was used for stimulation (5 hr) prior to cell surface staining. All antibodies (anti-CD45.2, anti-CD3ε, anti-CD4, anti-CD8, anti-CD25, anti-CD335, anti-FOXP3, anti-IFN-γ, anti-H-2Kd, anti-PD-L1) were purchased from BioLegend. Samples were analyzed using an LSR Fortessa instrument (BD PharMingen) and data analyzed with FlowJo Software (Treestar).
Pan-cancer analysis of TCGA dataset
The pan-cancer gene expression and patient annotation datasets of TCGA were retrieved from the Genomic Data Commons (GDC) of the National Cancer Institute (https://gdc.cancer.gov/) (Thorsson et al., 2018). Sample-wise gene set activities of different pathways were calculated in GSVA using the “ssGSEA” method, and signature scores of different types of immune cells were calculated in GSVA using the “z-score” method. Spearman correlation and multiple testing corrections were done in R 3.5. GSEA analysis was performed using the GSEA v3.0 desktop application.
FBXO44-immune gene signature analysis in immunotherapy datasets
The FBXO44-immune gene signature was defined as genes whose expression were upregulated at least 1.5 fold in FBXO44 knockdown RNA-seq, and also belonged to 6 sets of immune response related gene sets enriched in GSEA analysis, including “GO_INFLAMMATORY_RESPONSE,” “GO_ACTIVATION_OF_INNATE_IMMUNE_RESPONSE,” “GO_POSITIVE_REGULATION_OF_DEF ENSE_RESPONSE,” “GO_POSITIVE_REGULATION_OF_LEUKOCYTE_MIGRATION,” “KEGG_CYTOKINE_CYTOKINE_RECEPTOR_INTERACTION” and “REACTOME_ANTIGEN_PROCESSING_CROSS_PRESENTATION.” The immunotherapy datasets for anti-PD1 therapy and adoptive cell transfer of TILs were analyzed from these studies (Harel et al., 2019; Lauss et al., 2017; Riaz et al., 2017). The processed protein or RNA expression data were retrieved from the publication or Gene Expression Omnibus (GSE91061 and GSE100797). The FBXO44-immune gene signature score was calculated in each dataset using the “z-score” method in GSVA. The difference of signature score between responders and non-responders were tested using unpaired Student’s t test.
QUANTIFICATION AND STATISTICAL ANALYSIS
The information about statistical details and methods is indicated in the figure legends, text, or methods. Statistical significance tests, including unpaired Student’s t test, one-way ANOVA, two-way ANOVA, Spearman correlation test and normality test, were performed using R or Graphpad Prism software, as denoted in each analysis. Data are presented as mean ± SEM for a minimum of three independent experiments unless otherwise indicated. For box-and-whisker plots, the box indicates interquartile range (IQR), the line in the box indicates the median, the whiskers indicate points within Q3+1.53IQR and Q1−1.53IQR and the points beyond whiskers indicate outliers. Q1 and Q3, the first and third quartiles, respectively. All statistical tests were two-tailed. p < 0.05 of the two tail was taken to indicate statistical significance unless otherwise specified.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Rabbit anti-human FBXO44 | Sigma-Aldrich | Cat# HPA003363; RRID:AB_1078831 |
| Mouse anti-human FBXO44 | Santa Cruz | Cat# sc-398020 |
| Rabbit anti-human H3K9me3 | Abcam | Cat# ab8898; RRID:AB_306848 |
| Rabbit anti-human H3K9me3 | GeneTex | Cat# GTX121677; RRID:AB_10721938 |
| Rabbit anti-human H3K9me2 | Cell Signaling | Cat# 4658; RRID:AB_10544405 |
| Rabbit anti-human H3K9me1 | Abcam | Cat# ab176880; RRID:AB_2751009 |
| Rabbit anti-human p-RPA32T21 | GeneTex | Cat# GTX62664; RRID:AB_10620643 |
| Rabbit anti-human p-Histone H2A.XS139 | Cell Signaling | Cat# 9718; RRID:AB_2118009 |
| Rabbit anti-human p-Histone H2A.XS139 | Abcam | Cat# ab2893; RRID:AB_303388 |
| Mouse anti-human p-Histone H2A.XS139 | Millipore | Cat# 05–636-I; RRID:AB_2755003 |
| Rabbit anti-human SUV39H1 | Cell Signaling | Cat# 8729; RRID:AB_10829612 |
| Mouse anti-human SUV39H1 | Millipore | Cat# 05–615; RRID:AB_2196724 |
| Rabbit anti-human H3K27me3 | Millipore | Cat# ABE44; RRID:AB_10563660 |
| Rabbit anti-human H3K36me3 | Millipore | Cat# ABE435 |
| Rabbit anti-human H3K79me2 | Millipore | Cat# 04–835; RRID:AB_1587126 |
| Rabbit anti-human H2AK119ub | Cell Signaling | Cat# 8240; RRID:AB_10891618 |
| Rabbit anti-human histone H3 | Abcam | Cat# ab18521; RRID:AB_732917 |
| Mouse anti-HA tag | BioLegend | Cat# 901513; RRID:AB_2565335 |
| Rabbit anti-human CUL4B | Bethyl Laboratories | Cat# A303–863A-T; RRID:AB_2620214 |
| Rabbit anti-human CUL4B | Proteintech | Cat# 12916–1-AP; RRID:AB_2086699 |
| Rabbit anti-human DDB1 | Cell Signaling | Cat# 6998; RRID:AB_10829458 |
| Rabbit anti-human CUL1 | Thermo Fisher Scientific | Cat# 71–8700; RRID:AB_2534002 |
| Mouse anti-Flag tag | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| Rabbit anti-Flag tag | Cell Signaling | Cat# 14793; RRID:AB_2572291 |
| Mouse anti-Myc tag | Cell Signaling | Cat# 2276; RRID:AB_331783 |
| Rabbit anti-GFP | Cell Signaling | Cat# 2555; RRID:AB_10692764 |
| Mouse anti-human GATAD2A | Santa Cruz | Cat# sc-514987 |
| Mouse anti-human GATAD2B | Abnova | Cat# H00057459-A01; RRID:AB_463225 |
| Rabbit anti-human RBBP4/7 | Cell Signaling | Cat# 9067; RRID:AB_11178523 |
| Mouse anti-human a-Tubulin | Sigma-Aldrich | Cat# T5168; RRID:AB_477579 |
| Mouse anti-human b-Actin | Sigma-Aldrich | Cat# A5441; RRID:AB_476744 |
| Rat anti-human ORC2 | Cell Signaling | Cat# 4736; RRID:AB_2157716 |
| Rabbit anti-human GAPDH | Santa Cruz | Cat# sc-25778; RRID:AB_10167668 |
| Rabbit anti-human cleaved caspase 3 | Cell Signaling | Cat# 9664; RRID:AB_2070042 |
| Rabbit anti-human PARP | Cell Signaling | Cat# 9542; RRID:AB_2160739 |
| Rabbit anti-human ATM | Cell Signaling | Cat# 2873; RRID:AB_2062659 |
| Mouse anti-human p-ATMS1981 | Cell Signaling | Cat# 4526; RRID:AB_2062663 |
| Rabbit anti-human ATR | Cell Signaling | Cat# 2790; RRID:AB_2227860 |
| Rabbit anti-human p-ATRS428 | Cell Signaling | Cat# 2853; RRID:AB_2290281 |
| Mouse anti-human CHK1 | Cell Signaling | Cat# 2360; RRID:AB_2080320 |
| Rabbit anti-human p-CHK1S345 | Cell Signaling | Cat# 2348; RRID:AB_331212 |
| Mouse anti-human CHK2 | Cell Signaling | Cat# 3440; RRID:AB_2229490 |
| Rabbit anti-human p-CHK2T68 | Cell Signaling | Cat# 2197; RRID:AB_2080501 |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Mouse anti-dsDNA | Abcam | Cat# ab27156; RRID:AB_470907 |
| Mouse J2 | Scicons | Cat# 10010200; RRID:AB_2651015 |
| Rabbit anti-human cGAS | Cell Signaling | Cat# 15102; RRID:AB_2732795 |
| Rabbit anti-human STING | Cell Signaling | Cat# 13647; RRID:AB_2732796 |
| Rabbit anti-human MAVS | Cell Signaling | Cat# 3993; RRID:AB_823565 |
| Rabbit anti-human IRF3 | Cell Signaling | Cat# 4302; RRID:AB_1904036 |
| Rabbit anti-human phosphorylated-IRF3S386 | GeneTex | Cat# GTX130422 |
| Rabbit anti-human IRF7 | Cell Signaling | Cat# 4920; RRID:AB_2127551 |
| Mouse anti-human ULBP2 | Santa Cruz | Cat# sc-53135; RRID:AB_630415 |
| Mouse anti-human ULBP2 | GeneTex | Cat# GTX53048 |
| Mouse anti-human SSX1 | Novus Biologicals | Cat# NBP2–00614 |
| Rat anti-mouse PD-1 | BioXCell | Cat# BP0273 |
| Rat IgG2a isotype control | BioXCell | Cat# BP0089 |
| Rat anti-mouse CD45 | BD | Cat# 550539; RRID:AB_2174426 |
| Rat anti-mouse CD8a | Thermo Fisher Scientific | Cat# 14–0808-82; RRID:AB_2572861 |
| Rat anti-mouse NK Cell Marker (ANK61) | Santa Cruz | Cat# sc-59340; RRID:AB_784846 |
| Mouse anti-mouse BV605-CD45.2 | BioLegend | Cat# 109841; RRID:AB_2563485 |
| Rat anti-mouse PerCP/Cyanine5.5-CD4 | BioLegend | Cat# 100539; RRID:AB_893332 |
| Rat anti-mouse FITC-CD8 | BioLegend | Cat# 100706; RRID:AB_312745 |
| Rat anti-mouse BV711-CD25 | BioLegend | Cat# 102049; RRID:AB_2564130 |
| Hamster anti-mouse APC-CD3ε | BioLegend | Cat# 152306; RRID:AB_2632669 |
| Rat anti-mouse PE-FOXP3 | BioLegend | Cat# 126403; RRID:AB_1089118 |
| Rat anti-mouse BV421-CD335 | BioLegend | Cat# 137611; RRID:AB_10915472 |
| Rat anti-mouse PE-IFN-γ | BioLegend | Cat# 505807; RRID:AB_315401 |
| Mouse anti-mouse FITC-H-2Kd | BioLegend | Cat# 116605; RRID:AB_313740 |
| Rat anti-mouse PE-PD-L1 | BioLegend | Cat# 124307; RRID:AB_2073557 |
| Normal rabbit IgG | Santa Cruz | Cat# sc-2027; RRID:AB_737197 |
| Normal mouse IgG | Santa Cruz | Cat# sc-2025; RRID:AB_737182 |
| Rabbit anti-Flag tag | Sigma-Aldrich | Cat# F7425; RRID:AB_439687 |
| Biological samples | ||
| Human Breast Tissue MicroArray (Cancer) | Novus Biologicals | Cat# NBP2–30212 |
| Chemicals, peptides, and recombinant proteins | ||
| Proteinase K | Thermo Fisher Scientific | Cat# AM2546 |
| Streptavidin beads | Cell Signaling | Cat# 3419 |
| Protein G agarose | Millipore | Cat# 16–266 |
| 3x FLAG Peptide | Sigma-Aldrich | Cat# F4799 |
| Recombinant FBXO44 protein | OriGene | Cat# TP760409 |
| Synthetic biotinylated nucleosomes | EpiCypher | Cat# 16–0006; 16–0325; 16–0325 |
| Sodium L-ascorbate | TCI | Cat# A0539 |
| MammoCult Medium | STEMCELL | Cat# 05620 |
| Biotin-azide | Lumiprobe | Cat# C3730 |
| Copper (II) sulfate | Acros Organics | Cat# AC197715000 |
| Anti-fade Mounting Medium | Vector Laboratories | Cat# H-1000 |
| D-Luciferin | Xenogen | Cat# XR-1001 |
| Trichloroacetic acid | Sigma-Aldrich | Cat# T0699 |
| DAPI | Thermo Fisher Scientific | Cat# D1306 |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Cremophor EL | Millipore | Cat# 238470 |
| RNase A | Sigma-Aldrich | Cat# R6513 |
| F5446 | Kebin Liu (Augusta University, USA) |
N/A |
| RNase A/T1 Mix | Thermo Fisher Scientific | Cat# EN0551 |
| Critical commercial assays | ||
| Click-iT EdU Alexa Fluor 488 Imaging Kit | Thermo Fisher Scientific | Cat# C10337 |
| EZ Nucleosomal DNA Prep Kit | Zymo | Cat# D5220 |
| Tumor Dissociation Kit | Miltenyi | Cat# 130–096-730 |
| FITC Annexin V Apoptosis Detection Kit | BD Biosciences | Cat# 556547 |
| 7-AAD Viability Staining Solution | BioLegend | Cat# 420403 |
| Chromatin Extraction Kit | Abcam | Cat# ab117152 |
| Nuclear Extract Kit | Active Motif | Cat# 40010 |
| H3K9me3 ChIP Kit | Millipore | Cat# 17–10242 |
| FLAG Immunoprecipitation Kit | Sigma-Aldrich | Cat# FLAGIPT1 |
| Zombie UV Fixable Viability Kit | BioLegend | Cat# 423107 |
| Breast Cancer Stem Cell Isolation Kit | MagCellect | Cat# MAGH111 |
| Deposited data | ||
| RNA-seq of FBXO44 knockdown | This paper | GEO: GSE139974 |
| ChIP-seq of FBXO44 | This paper | GEO: GSE139973 |
| TCGA pan-cancer datasets | The Genomic Data Commons (GDC) |
https://gdc.cancer.gov/about-data/publications/panimmune |
| Experimental models: cell lines | ||
| Human: BT-549 (female, 72 years old) | ATCC | Cat# HTB-122 |
| Human: MDA-MB-231 (female, 51 years old) | ATCC | Cat# HTB-26 |
| Human: MCF7 (female, 69 years old) | ATCC | Cat# HTB-22 |
| Human: A549 (male, 58 years old) | ATCC | Cat# CCL-185 |
| Human: H446 (male, 61 years old) | ATCC | Cat# HTB-171 |
| Human: HEK293T (female, fetus) | ATCC | Cat# CRL-3216 |
| Human: HeLa (female, 31 years old) | ATCC | Cat# CCL-2 |
| Human: U2OS (female, 15 years old) | ATCC | Cat# HTB-96 |
| Human: HCT116 (male, 48 years old) | ATCC | Cat# CCL-247 |
| Mouse: 4T1 (female) | ATCC | Cat# CRL-2539-LUC2 |
| Human: HMEC (female, adult) | Lonza | Cat# CC-2551 |
| Human: GSC1517 (female, 54 years old) | Mack et al., 2019 | N/A |
| Human: GSC1552 (male, 56 years old) | Mack et al., 2019 | N/A |
| Human: primary astrocytes | ScienCell | Cat# 1800 |
| Experimental models: organisms/strains | ||
| Mouse: NOD.Cg-Prkdcscid
Il2rgtm1Wjl/SzJ (female, 4 to 6 weeks old) |
Jackson Laboratory | Cat# 005557 |
| Mouse: Athymic Nude-Foxn1nu (female, 6 weeks old) |
Envigo | Cat# 069 |
| Mouse: BALB/c (female, 6 to 8 weeks old) | Jackson Laboratory | Cat# 000651 |
| Oligonucleotides | ||
| Sequences provided in STAR methods | This paper | N/A |
| Recombinant DNA | ||
| pCMV-Myc-FBXO44 | This paper | N/A |
| pCMV-Myc-DF-FBXO44 | This paper | N/A |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| pFlag-CMV2-FBXO44 | This paper | N/A |
| pFlag-CMV2-SUV39H1 | This paper | N/A |
| pFlag-CMV2-GATAD2A | This paper | N/A |
| pFlag-CMV2-GATAD2B | This paper | N/A |
| pFlag-CMV2-CHD4 | This paper | N/A |
| pFlag-CMV2-CUL4B | This paper | N/A |
| pFlag-CMV2-RBBP4 | This paper | N/A |
| pFlag-CMV2-RBBP7 | This paper | N/A |
| pFlag-CMV2-H3.1 | This paper | N/A |
| pFlag-CMV2-H3.3 | This paper | N/A |
| pcDNA3.1-HA-CUL1 | This paper | N/A |
| Software and algorithms | ||
| Cufflinks v2.2.1 | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| GSEA v4.0.3 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Cytoscape v3.8 | Shannon et al., 2003 | https://cytoscape.org/ |
| Bowtie 2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| RepEnrich2 | Criscione et al., 2014 | https://github.com/nerettilab/RepEnrich2 |
| Partek Genomics Suite | Partek | https://www.partek.com/ |
| Deeptools v3.1.2 | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/3.1.2/ |
| VennDiagram R package | Chen and Boutros, 2011 | https://cran.r-project.org/web/packages/VennDiagram/index.html |
| GSVA R package | Hanzelmann et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| Graphpad Prism v7 | GraphPad | N/A |
| R v3.6.1 | R | https://www.r-project.org/ |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| FlowJo v10 | Treestar | https://www.flowjo.com/ |
Highlights.
FBXO44 promotes H3K9me3-mediated repetitive element silencing in cancer cells
FBXO44 recruits SUV39H1, CRL4, and Mi-2/NuRD at the replication fork
Targeting FBXO44 stimulates antiviral pathways and replication stress in cancer cells
FBXO44/SUV39H1 inhibition overcomes resistance to immune checkpoint blockade therapy
ACKNOWLEDGMENTS
The authors thank Peter Adams (Sanford Burnham Prebys Medical Discovery Institute [SBP], USA) for critically reading the manuscript, Kebin Liu (Augusta University, USA) for F5446, BioLegend (San Diego, CA, USA) for help with ELISA assays, and Sumit Chanda and Ani Deshpande labs (SBP, USA) for reagents and helpful comments. Buddy Charboro, Alexey Eroshkin, and Andrew Hodges provided technical support. SBP’s Animal Facility, Bioinformatics, Cell Imaging, Tumor Analysis, and Flow Cytometry Shared Resources are supported by an NCI Cancer Center support grant (P30 CA030199). This study was funded by grants from the DoD BCRP (W81XWH-15-1-0383) and CBCRP (21IB-0113). J.N.R. is funded by NIH (NS103434, CA238662, and CA197718). O.S. is funded by grants from the Swedish Childhood Cancer Foundation, Swedish Cancer Society, and Radiumhemmets Research Fund. J.W. is funded by NIH (GM089778).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.11.042.
REFERENCES
- Abbas T, and Dutta A (2011). CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle 10, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Barth TK, Reveron-Gomez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, and Groth A (2015). Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 29, 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, and Madhani HD (2018). Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol 19, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar SL, Wulaningsih W, and Lehmann U (2017). Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. Int. J. Mol. Sci 18, e974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, Rochaix P, Andrieu-Abadie N, Levade T, Meyer N, et al. (2017). TNF alpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun 8, 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut-Karslioglu A, De La Rosa-Velazquez IA, Ramirez F, Barenboim M, Onishi-Seebacher M, Arand J, Galan C, Winter GE, Engist B, Gerle B, et al. (2014). Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 55, 277–290. [DOI] [PubMed] [Google Scholar]
- Chen H, and Boutros PC (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, and Grewal SIS (2008). Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451, 734–737. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, and Feschotte C (2017). Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet 18, 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione SW, Zhang Y, Thompson W, Sedivy JM, and Neretti N (2014). Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d’Ario G, Montani E, et al. (2011). Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol 13, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M (2002). DNA methylation in cancer: too much, but also too little. Oncogene 21, 5400–5413. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessaman JD, and Selker EU (2017). Induction of H3K9me3 and DNA methylation by tethered heterochromatin factors in Neurospora crassa. Proc. Natl. Acad. Sci. USA 114, E9598–E9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG, and Youngblood B (2017). De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 170, 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjerstorff MF, Andersen MH, and Ditzel HJ (2015). Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget 6, 15772–15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, Baruch EN, Tripple V, Arama-Chayoth M, Greenberg E, et al. (2019). Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 179, 236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa LA, Wu M, Ye T, Kobayashi R, Sun H, and Zhang H (2006). CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol 8, 1277–1283. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, and Taniguchi T (2006). Type I inteferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360. [DOI] [PubMed] [Google Scholar]
- Hu HL, Yang Y, Ji QH, Zhao W, Jiang BC, Liu RQ, Yuan JP, Liu Q, Li X, Zou YX, et al. (2012). CRL4B Catalyzes H2AK119 Monoubiquitination and Coordinates with PRC2 to Promote Tumorigenesis. Cancer Cell 22, 781–795. [DOI] [PubMed] [Google Scholar]
- Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch ID, et al. (2016). An RBEZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 64, 1074–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak CA, Classon M, and De Carvalho DD (2018). Deregulation of Retroelements as an Emerging Therapeutic Opportunity in Cancer. Trends Cancer 4, 583–597. [DOI] [PubMed] [Google Scholar]
- Jones PA, Ohtani H, Chakravarthy A, and De Carvalho DD (2019). Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 19, 151–161. [DOI] [PubMed] [Google Scholar]
- Kadoch C, Copeland RA, and Keilhack H (2016). PRC2 and SWI/SNF Chromatin Remodeling Complexes in Health and Disease. Biochemistry 55, 1600–1614. [DOI] [PubMed] [Google Scholar]
- Kassiotis G, and Stoye JP (2016). Immune responses to endogenous retroelements: taking the bad with the good. Nat. Rev. Immunol 16, 207–219. [DOI] [PubMed] [Google Scholar]
- Keenan TE, Burke KP, and Van Allen EM (2019). Genomic correlates of response to immune checkpoint blockade. Nat. Med 25, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloc A, Zaratiegui M, Nora E, and Martienssen R (2008). RNA interference guides histone modification during the S phase of chromosomal replication. Curr. Biol 18, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel MK, and Batzer MA (2010). A mobile threat to genome stability: The impact of non-LTR retrotransposons upon the human genome. Semin. Cancer Biol 20, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, Salim M, Vallon-Christersson J, Torngren T, Kvist A, et al. (2017). Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun 8, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Kim M, and Kim HP (2016). Epigenetic regulation of long noncoding RNA UCA1 by SATB1 in breast cancer. BMB Rep. 49, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KHT, Abou El Hassan M, and Bremner R (2013). A rapid and efficient method to purify proteins at replication forks under native conditions. Biotechniques 55, 204–206. [DOI] [PubMed] [Google Scholar]
- Lewis ZA, Adhvaryu KK, Honda S, Shiver AL, Knip M, Sack R, and Selker EU (2010). DNA Methylation and Normal Chromosome Behavior in Neurospora Depend on Five Components of a Histone Methyltransferase Complex, DCDC. PLoS Genet. 6, e1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, and Gilbert DM (2007). Proliferation-dependent and cell cycle-regulated transcription of mouse pericentric heterochromatin. J. Cell Biol 179, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Yang D, Klement JD, Oh IK, Savage NM, Waller JL, Colby AH, Grinstaff MW, Oberlies NH, Pearce CJ, et al. (2019). SUV39H1 Represses the Expression of Cytotoxic T-Lymphocyte Effector Genes to Promote Colon Tumor Immune Evasion. Cancer Immunol. Res 7, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, Bernatchez JA, Zhu Z, Gimple RC, Kim LJY, et al. (2019). Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J. Exp. Med 216, 1071–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JH, O’Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, and Jenuwein T (2005). The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 24, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padeken J, Zeller P, and Gasser SM (2015). Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev 31, 12–19. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantala JK, Makela R, Aaltola AR, Laasola P, Mpindi JP, Nees M, Saviranta P, and Kallioniemi O (2011). A cell spot microarray method for production of high density siRNA transfection microarrays. BMC Genomics 12, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recolin B, van der Laan S, Tsanov N, and Maiorano D (2014). Molecular mechanisms of DNA replication checkpoint activation. Genes (Basel) 5, 147–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. (2015). DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 162, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH, et al. (2018). LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JM, Parker JS, Liu F, Rothbart SB, Ait-Si-Ali S, Strahl BD, Jin J, Davis IJ, Mosley AL, and Pattenden SG (2015). A Role for Widely Interspaced Zinc Finger (WIZ) in Retention of the G9a Methyltransferase on Chromatin. J. Biol. Chem 290, 26088–26102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JK, and Wade PA (2011). Mi-2/NuRD complex function is required for normal S phase progression and assembly of pericentric heterochromatin. Mol. Biol. Cell 22, 3094–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren B, Swaney S, and Neubig RR (2015). FBXO44-Mediated Degradation of RGS2 Protein Uniquely Depends on a Cullin 4B/DDB1 Complex. PLoS ONE 10, e0123581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, and Pagano M (2013). Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol 14, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, and Martienssen R (2007). Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet 8, 272–285. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, and Nakatani Y (2004). Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51–61. [DOI] [PubMed] [Google Scholar]
- Thompson PJ, Macfarlan TS, and Lorincz MC (2016). Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 62, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M, et al. (2017). Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 171, 1284–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Demaria S, Formenti SC, and Galluzzi L (2018). Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 34, 361–378. [DOI] [PubMed] [Google Scholar]
- Vashisht AA, Yu CC, Sharma T, Ro K, and Wohlschlegel JA (2015). The Association of the Xeroderma Pigmentosum Group D DNA Helicase (XPD) with Transcription Factor IIH Is Regulated by the Cytosolic Iron-Sulfur Cluster Assembly Pathway. J. Biol. Chem 290, 14218–14225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Wu TP, Gimple RC, Li Z, Prager BC, Wu Q, Yu Y, Wang P, Wang Y, Gorkin DU, et al. (2018). N(6)-methyladenine DNA Modification in Glioblastoma. Cell 175, 1228–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Wang WX, Chen S, and Zhu B (2012). A model for mitotic inheritance of histone lysine methylation. EMBO Rep. 13, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu R, Qiu R, Zheng Y, Huang W, Hu H, Ji Q, He H, Shang Y, Gong Y, and Wang Y (2015). CRL4B promotes tumorigenesis by coordinating with SUV39H1/HP1/DNMT3A in DNA methylation-based epigenetic silencing. Oncogene 34, 104–118. [DOI] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP-seq and RNA-seq datasets generated during this study are available at GEO: GSE139973 and GSE139974.