

Abstract
Background
Sexual dimorphisms in immune responses contribute to coronavirus disease 2019 (COVID-19) outcomes, but the mechanisms governing this disparity remain incompletely understood.
Methods
We carried out sex-balanced sampling of peripheral blood mononuclear cells from hospitalized and non-hospitalized individuals with confirmed COVID-19, uninfected close contacts, and healthy control individuals for 36-color flow cytometry and single-cell RNA sequencing.
Findings
Our results revealed a pronounced reduction of circulating mucosal-associated invariant T (MAIT) cells in infected females. Integration of published COVID-19 airway tissue datasets suggests that this reduction represented a major wave of MAIT cell extravasation during early infection in females. Moreover, MAIT cells from females possessed an immunologically active gene signature, whereas cells from males were pro-apoptotic.
Conclusions
Our findings uncover a female-specific protective MAIT cell profile, potentially shedding light on reduced COVID-19 susceptibility in females.
Funding
This work was supported by NIH/NIAID (U01AI066569 and UM1AI104681), the Defense Advanced Projects Agency (DARPA; N66001-09-C-2082 and HR0011-17-2-0069), the Veterans Affairs Health System, and Virology Quality Assurance (VQA; 75N93019C00015). The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health. COVID-19 samples were processed under Biosafety level 2 (BSL-2) with aerosol management enhancement or BSL-3 in the Duke Regional Biocontainment Laboratory, which received partial support for construction from NIH/NIAID (UC6AI058607).
Keywords: SARS-CoV-2, innate immunity, monocyte, IL-7, apoptosis
Graphical abstract
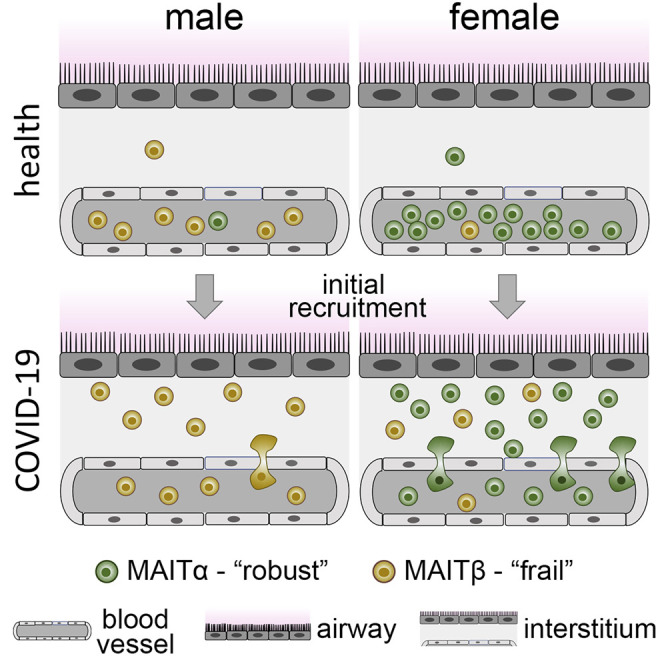
Context and significance
Why are women twice as less likely than men to experience severe COVID-19? Answering this question may help us more completely understand the immune defenses that counter SARS-CoV-2. This study from researchers at Duke University suggests that a type of white blood cells, mucosal-associated invariant T (MAIT) cells, is superior in women with COVID-19. The potential importance of the finding is that these highly specialized white blood cells have been shown to contribute critically to immune defenses in other viral and bacterial infections. These findings may shed light on the underlying reasons for reduced COVID-19 susceptibility in women and highlights the potential role of MAIT cells in countering SARS-CoV-2.
Responses in COVID-19 patients are distinct across sexes. Yu et al. show that, compared with male patients, female COVID-19 patients exhibit evidence of a major recruitment wave of circulating mucosal-associated invariant T (MAIT) cells into the airways and demonstrate that these cells display an immunologically active phenotype.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to a global pandemic of coronavirus disease 2019 (COVID-19) and a death toll of more than 2.3 million people and rising.1 According to reported sex-disaggregated data, males are affected disproportionately by SARS-CoV-2, with a higher incidence of cases, mortality, and morbidity.2 This follows a similar trend of higher case fatality rates for males in SARS-CoV and Middle East respiratory syndrome CoV (MERS-CoV) and experiments using SARS-CoV mouse models.2, 3, 4, 5, 6 Sex differences in the immune response are thought to be a key contributing factor to these CoV disease outcomes, agreeing with the current body of knowledge indicating that innate and adaptive immune responses are altered substantially according to sex.7, 8, 9, 10 Specific to SARS-CoV-2 infection, responses of lymphocytes and myeloid cells have been shown to be associated with COVID-19 outcomes.11, 12, 13, 14, 15, 16, 17, 18, 19 Correspondingly, a recent study of sex differences in COVID-19 immune responses uncovered an association between poor disease outcomes in males and weak T cell responses in CD4+ and CD8+ compartments, whereas poor outcomes in females were associated with high innate immune cytokines, tumor necrosis factor superfamily 10 (TNFSF-10), and interleukin-15 (IL-15).20 The sex differences elucidated in this seminal study further cement the need to better understand the mechanisms governing sex-specific susceptibility to SARS-CoV-2.
Results
In this study, we carried out sex-balanced sampling of peripheral blood mononuclear cells (PBMCs) from individuals with COVID-19 and control subjects for 36-color flow cytometry and single-cell RNA sequencing (scRNA-seq) analyses. A total of 88 samples were analyzed from 45 individuals. Details regarding subject demographics and sample information are summarized in Figure 1 A and Tables 1 and S1). Briefly, we analyzed samples from 28 individuals with COVID-19 as confirmed by positive SARS-CoV-2 PCR and/or immunoglobulin G (IgG) seroconversion. These included 9 hospitalized subjects (20%), 7 requiring intensive care, hereafter referred to as “hospitalized.” An additional 19 subjects were identified in non-hospital settings (42.2%), hereafter referred to as “infected.” Most of these confirmed COVID-19 cases were sampled longitudinally (range, 1–28 days), including pre- and post-anti-SARS-CoV-2 IgG seroconversion. The dates of symptom onset for all subjects with confirmed COVID-19 were recorded at enrollment, providing an illness range of 1–40 days. We also recorded symptom severity, obtained via investigator survey on 39 symptoms related to COVID-19 (STAR Methods). Additionally, we included 7 subjects (15.6%), hereafter referred to as “exposed,” who were also sampled at multiple time points. These subjects, despite being close contacts of infected individuals, remained with negligible symptom scores, were negative for SARS-CoV-2 by PCR, and did not demonstrate detectable anti-SARS-CoV-2 IgG for at least 2 months after enrollment. Last, we included a group of 10 “healthy” subjects (22.2%) who were enrolled prior to the pandemic in 2019 and did not show any symptoms associated with COVID-19 or other respiratory illness.21
Figure 1.

Loss of peripheral CD8+CD161hi T cell frequencies correlates with increased severity of COVID-19
(A) Overview of groups in this study.
(B) UMAP visualization of PBMC subsets identified by FlowSOM clustering. Samples from all participants were pooled and down-sampled to 3,000 live CD45+ cells per sample. MO, monocyte; NK, natural killer; DC, dendritic cell; PMN, polymorphonuclear neutrophil; Baso, basophil.
(C) Representative marker expression by CD4+ T cell, CD8+ T cell, MO, NK cell, and B cell subsets.
(D) Correlation analysis of immune cell subsets as shown in (C) with disease severity rank.
(E) Expression of CD161, TCR γδ and CD56 in CD8+ T cell subsets. NK cells were used as a positive control for CD56 expression.
(F) UMAP of samples grouped by disease severity rank. Samples collected within 3 days from initial symptom score recording were included.
(G) Frequencies of CD161hi T cells (mean ± standard error) in different severity groups (left) and their correlation with severity rank (right).
(H and I) Age comparisons among severity rank and between sexes.
(J) Multiple linear regression of age (x1) and severity rank (x2) with CD161hi cell frequencies. Regression models with p values are shown for age and severity rank.
Significance was determined by Kruskal-Wallis test with Dunn’s test (E) or ANOVA (H and I): ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Correlation efficiency was calculated by Spearman’s rank correlation (D and G). See also Figure S1 and Tables 1 and S1.
Table 1.
Summary of demographics and sample information
| Group | Healthy | Exposed | Infected | Hospitalized | Total | |
|---|---|---|---|---|---|---|
| Age (mean ± SD [range]) | 39.70 ± 13.68 (25–61) | 42.57 ± 13.93 (17–60) | 36.73 ± 13.88 (20–65) | 59.44 ± 15.11 (31–76) | 42.84 ± 16.06 (17–76) | |
| Number of subjects (female:male [F:M] ratio) | n = 10 (5:5) | n = 7 (3:4) | n = 19 (8:11) | n = 9 (4:5) | n = 45 (20:25) | |
| Race (n [%]) | African American | 3 (30.00%) | 0 | 1 (5.26%) | 5 (55.56%) | 9 (20.00%) |
| Asian | 0 | 1 (14.29%) | 2 (10.53%) | 0 | 3 (6.67%) | |
| White | 7 (70.00%) | 5 (71.42%) | 16 (84.21%) | 3 (33.33%) | 31 (68.89%) | |
| Others/ unknown | 0 | 1 (14.29%) | 0 | 1 (11.11%) | 2 (4.44%) | |
| Days since onset when enrolled (mean ± SD [range]) | N/A | 15.17 ± 11.25 (5–33) | 11.18 ± 4.30 (3–19) | 8.25 ± 6.14 (1–18) | 11.19 ± 6.73 (1–33) | |
| # samples of flow cytometry (F:M ratio) | n = 10 (5:5) | n = 20 (9:11) | n = 44 (21:23) | n = 9 (4:5) | n = 83 (39:44) | |
| # samples of scRNA-seq (F:M ratio) | n = 5 (3:2) | n = 8 (3:5) | n = 29 (12:17) | n = 6 (1:5) | n = 48 (19:29) | |
| Days since onset when samples collected mean ± SD (range) | N/A | 26.94 ± 14.94 (5–61) | 18.27 ± 8.44 (3–40) | 8.25 ± 6.14 (1–18) | 19.32 ± 11.44 (1–61) | |
See also Table S1.
Immune profiling of PBMCs from individuals with COVID-19 reveals the association of CD8+CD161hi T cell frequencies with disease severity
With these flow cytometry data, we generated a map of immune cell populations and their subsets by down-sampling to 3,000 viable CD45+ singlets per sample and concatenated all data for uniform manifold approximation and projection (UMAP)22 and unbiased clustering via flow self-organizing maps (FlowSOM).23 Unique marker expression of respective populations facilitated our annotation of major PBMC populations (Figures 1B and 1C), including CD4+ and CD8+ (αβ) T cells, γδ T cells, B cells, plasmablasts, natural killer (NK) cells, monocytes (MOs), and dendritic cells (DC), which were also confirmed by manual analysis (Figures S1A–S1H). Subpopulations were also annotated in this manner, such as CD45RA+ CD27+ CCR7+ naive, CD45RA– CCR7+ central memory (CM), CD45RA– CCR7– effector memory (EM), and CD45RA+ CD27− CCR7– terminally differentiated EM (terminally differentiated effector memory cells re-expressing CD45RA [EMRA]) CD8+ T cells and CD8+ CD161hi T cells and other indicated subpopulations (Figures 1B, 1C, and S1A–S1H). We noted that a minor population of basophils (Basos) and neutrophils (polymorphonuclear neutrophils [PMNs]), primarily from hospitalized individuals, were detected (Figure 1B) despite using a PBMC isolation protocol.
We next set out to screen our flow cytometry dataset for immune populations that exhibited major quantitative changes associated with COVID-19 severity. Regression analysis revealed a significant (p < 0.05) association of several major PBMC populations, including CD8+ T cells, NK cells, B cells, and MOs relative to disease severity rank (i.e., healthy, exposed, infected, and hospitalized) (Figure 1D). The highest degree of association with the most significance was found in CD8+ T cells (Figure 1D). Additional analysis of CD8+ T subpopulations also revealed significant changes in CD8+ EM T cells (Figure S1I) and in CD8+ CD161hi T cells with high significance (p = 0.0006) (Figure S1I). Other significant changes were also observed, such as in B cell subpopulations (naive, IgD+ non-class switch, and plasmablasts), NK cells (CD56lo populations), DCs (CD11c+ DCs and plasmacytoid DCs [pDCs]), MOs (classical, intermediate, and nonclassical). and CD4+ (EM) αβ T cells (Figure S1I). Our findings therefore suggest that the frequencies of certain lymphocyte and myeloid populations are affected in COVID-19, including a major effect on CD8+ CD161hi (αβ) T cells.
The robust statistical changes in total CD8+ T cells and the CD8+ CD161hi T subpopulation prompted us to look at these cells more closely. Regarding annotation of this CD161hi cluster, because the overwhelming majority of events were low to negative for CD56 and for T cell receptor γδ (TCRγδ) (Figure 1E), the phenotype was largely consistent with mucosal-associated invariant T (MAIT) cells but not NK T or γδ T cells. This designation is congruent with recent work with COVID-19 PBMCs.24, 25, 26, 27 Next we performed a more focused analysis of this cluster in COVID-19 by assessing their frequencies by severity rank using samples taken within 3 days of enrollment, the time point most proximal to the initial symptom score recordings. Results were displayed via UMAP contour plots, revealing a reduction in these cells in the SARS-CoV-2 settings (Figure 1F). Manual gating (CD3+ TCRγδ− CD8+ CD161hi) from all flow cytometry events revealed a significant reduction when comparing healthy (p = 0.0036) or exposed (p = 0.0488) subjects versus hospitalized subjects and a negative correlation (p = 0.0002) with disease severity rank (Figure 1G). Moreover, the ages of the hospitalized group are significantly higher than those of other groups (Figure 1H), although the ages of sampled subjects were similar between sexes at each severity rank (Figure 1I). To address the potential confounding factor of age, we built a multiple regression model with age and severity rank as two independent variables (Figure 1J). Our results showed that age does not significantly (p = 0.1165) affect CD161hi cells but severity rank does (p = 0.0034). Therefore, we concluded that changes of these CD161hi cells are associated with COVID-19 disease severity.
Elucidation of sex differences in CD8+ lymphocytes during SARS-CoV-2 infection
Given the previously reported sex differences in immune responses in COVID-19,2 , 20 we further grouped the data by sex and characterized the frequencies of CD8+ subsets stratified by disease severity rank (Figures 2 A, 2B, S2A, and S2B), time after symptom onset (early, ≤14 days; middle, 15–21 days; late, >21 days) (Figures 2C–2E), and seroconversion status (Figure 2F). The severity rank results showed that healthy females had greater frequencies of CD161hi cells relative to males and that females had a more dramatic loss of peripheral CD161hi cells compared with males in COVID-19 (Figures 2A–2C). We also found that, within the CD8+ compartments of healthy subjects, males had greater frequencies of CD8+ memory T cells (combined EMRA, EM, and CM) (Figures 2D and 2E). Although the memory cell predominance in males was preserved at all time points, the greater abundance of CD161hi cells in females was lost at early and middle time points but not in late disease (Figures 2D and 2E). The loss of this difference was due to a precipitous drop of CD161hi cells in females at early and middle time points (Figure 2F). No obvious changes of naive CD8+ T cells were found (Figure S2C). Last, when analyzing data from individuals with confirmed COVID-19 by seroconversion status, we found that CD161hi cells were higher in females relative to males prior to seroconversion, whereas CD8+ memory cells were higher in males in seroconverted subjects (Figure 2G). We identified a female-specific decline in circulating CD161hi cell frequencies upon exposure to/infection with SARS-CoV-2. This sex-specific reduction may be due to extravasation into airway tissue, suggesting a sex-specific role of these CD161hi cells in COVID-19.
Figure 2.

Sex-specific responses of circulating CD8+ T cells in individuals with COVID-19
(A) UMAP of samples stratified by sex and severity rank as shown in Figure 1F.
(B) Frequencies of CD161hi cells between females and males at each rank group.
(C) Linear regression of CD161hi cells with severity rank between sexes. Dashed lines indicate 95% confidence intervals. Regression models with p values are shown for each sex.
(D) UMAP of samples stratified by sex and time after symptom onset (early, ≤14 days; middle, >15 days and ≤21 days; late, >22 days).
(E) Frequencies of CD161hi and memory CD8+ T cells between sexes and time points.
(F) Sex-specific changes of CD161hi cells frequencies shown in (E).
(G) Frequencies of CD161hi and memory CD8+ T cells in samples from subjects with confirmed COVID-19 pre- and post-seroconversion.
Data were plotted as mean ± standard error (B and E–G). Significance was determined by Mann-Whitney test (B, E, and G) and Kruskal-Wallis test with Dunn’s test (F): ∗p < 0.05. See also Figure S2.
scRNA-seq uncovers the role of CD161hi T lymphocytes in SARS-CoV-2 immune responses
Next we analyzed scRNA-seq (10x Genomics) data of 48 different PBMC samples from 24 subjects across all groups (Tables 1 and S1) to further characterize these CD161hi cells. Data were processed using the Seurat 3 package,28 and subsequent transcript-based annotation was carried out (Figures 3 A, S3A, and S3B). Within T cell subsets (Figure 3B; Table S2), we were able to identify CD161hi cells in a single cluster containing high KLRB1 (i.e., CD161) expression and co-expression of CD3D, CD8A, and TRAV1-2 (Figures 3C and S3B), which encodes the Vα7.2 invariant TCR α chain on MAIT cells. Grouping these data by disease severity showed that hospitalized individuals had lower frequencies of T cells, including CD161hi cells (Figure 3D), agreeing with our flow cytometry findings and consistent with reported lymphopenia in individuals with severe COVID-19.11 , 20 , 29, 30, 31, 32 Also showing the same trend as our flow cytometry data was the higher frequency of CD161hi cells in healthy adults (Figure 3E), although it did not reach statistical significance because of the variations between females and males. Next, to address the functional role of this CD161hi cluster in COVID-19, we performed gene enrichment analysis using differentially expressed genes (DEGs) calculated by comparing the CD161hi cluster from all samples with all other T cell clusters. With several top-ranked hits consisting of immune pathways related to cytokine signaling (IL-4, IL-13, IL-10, and transforming growth factor β [TGF-β] signaling) and signal transduction and transcription (an NGF-TRKA signaling axis, nuclear events, and RAF-independent mitogen-activated protein kinase (MAPK) 1/3 activation) and an estrogen-dependent pathway (Figure 3F), our results inferred a possible sex-specific immune response of these CD161hi cells in COVID-19.
Figure 3.

Characterization of CD8+CD161hi T cells among COVID-19 PBMCs using scRNA-seq
(A) UMAP and unsupervised cluster analysis of PBMCs. RBC, red blood cell; PB, plasmablast; PLT, platelet.
(B and C) Visualization of T cell subsets with high resolution in UMAP (B) and expression of their marker genes as indicated in violin plots (C). The T∗ cluster likely represents a dropout population with low unique molecular identifier (UMI) counts. N, naive; EM, effector memory; CM, central memory; DN, double negative; rep, replicating.
(D) Changes of T cell subsets with severity rank. N, number of individuals.
(E) Frequencies of CD161hi clusters relative to all T cell subsets. Females were plotted in red and males in blue. The red dashed box delineates healthy females.
(F) Top enriched pathways of CD161hi clusters in the Reactome Pathway Database, ranked by false discovery rate (FDR; −log10 scale).
Data were plotted as mean ± standard error. Significance was determined by Kruskal-Wallis test (E). See also Figure S3 and Table S2.
Considering the critical roles of MOs in activating MAIT cells33 and the association of dysregulated MOs with severe COVID-19 outcomes,12 , 13 , 20 , 34 we applied the CellphoneDB package35 and analyzed ligand-receptor interactions between MO and T cell clusters in our data. We identified 5 MO clusters, including three conventional subsets (classical, non-classical, and intermediate) and two subsets of activated MOs (CD16 MOA and CD14 MOA) associated with COVID-19 (Figures 4A and 4B). These two activated MO subsets, which were also reported by other studies,13 , 34 express high levels of interferon-related genes (Figure S3B). Although cell-cell communications may not take place in the blood in the same way as in solid tissue, our results inferred unique interactions between CD161hi cells and MOs with the following gene pairs: KLRB1_CLEC2D, CCL5_CCR1, CXCR6_CXCL16, and IL18_IL-18R (Figures 4C and 4D). Moreover, the number of interaction counts of MOs was most abundant with the CD161hi cluster relative to all major T cell populations (Figure 4E). A recent study reported IL-18-dependent MAIT cell activation by SARS-CoV-2-infected macrophages, which supports the existence of unique interactions of MAIT cells with MOs/macrophages in COVID-19.27 These transcriptome findings further suggest a role of circulating CD161hi cells in the SARS-CoV-2 immune response and their possible interactions with MO-derived cells.
Figure 4.

Receptor-ligand interaction inferences uncover unique interactions between CD8+CD161hi T cells and MOs
(A and B) Visualization (A) and percentage distribution (B) of different MO subsets identified in Figure S3A. Three MO subsets represent resting classical (cl) CD14, non-classical (nc) CD16, and intermediate (int) MO as seen in healthy subjects. Two MO subsets involved in IFN signaling are associated with individuals with COVID-19. Based on differential expression of CD14 and CD16, they are referred to as activated CD14 and activated CD16 MO (CD14 MOA and CD16 MOA, respectively).
(C) Overview of selected ligand-receptor interactions inferenced using CellPhoneDB in the COVID-19 PBMC single-cell dataset. The red box delineates specific interactions of CD161hi T cells with MOs. p values and scales are indicated by circle size and color, respectively.
(D) Expression of representative ligand and receptor pairs between MAIT cells and MOs as indicated. Red and blue circles indicate CD161hi clusters and MOs, respectively.
(E) Heatmap of interaction counts between major T cell and MO subsets.
Sex-specific heterogeneity of circulating MAIT cells in COVID-19
To analyze our scRNA-seq dataset for potential sex differences in circulating CD161hi cells, we first sought to examine phenotypic heterogeneity in this population. To do this, we performed a focused subcluster analysis, which generated 3 distinct clusters (Figure 5 A). However, this added resolution revealed a cluster that expressed TRDC, encoding the constant region of the δ chain expressed by γδ T cells (Figure 5B), which was excluded from subsequent analyses. In contrast, the other two clusters had higher KLRB1 and TRAV1-2 expression (Figure 5B), and they are therefore referred to here as MAITα and MAITβ clusters. Of note, these 2 clusters make up approximately 80% of CD161hi PBMCs, which is consistent with a previously reported frequencies of circulating MAIT cells.24 Our results showed that the MAITα cluster possessed upregulated genes associated with cytotoxic T cells (GNLY, CD8A, and CD8B), migration/adhesion (CXCR4 and ITGB2), and cytokine signaling (IRF1, B2M, NFKBIA, JUNB, and FOS) (Figure 5C; Table S3). Although the MAITα cluster showed expression of the hemoglobin B gene (HBB), this cluster did not express other hemoglobin genes (Figures S4A and S4B), and its HBB expression level was much lower than in red blood cells (RBCs), suggesting that this cluster is not contaminated with RBCs. The MAITβ cluster was enriched for genes of ribosomal proteins, apoptosis (BAX and STUB1), and the linker histone H1 associated with apoptosis (HIST1H1C, HIST1H1D, and HIST1H1E) (Figure 5C; Table S3). Gene enrichment analysis further supported a functional dichotomy for α and β clusters. Although MAITα was enriched with several immune process pathways (e.g., interferon γ [IFN-γ], IL-4, and IL-13 signaling as well as antigen processing and presentation), MAITβ was enriched in cellular responses to external stimuli, metabolism of RNA, viral infection, and programmed cell death but not immune processes (Figures 5D and 5E; Table S4). Hence our results suggest MAIT cell heterogeneity, with the MAITα signature representing an immunologically poised/active phenotype and the MAITβ signature representing a stressed/apoptotic phenotype.
Figure 5.

Heterogeneity and distinct dynamics of circulating MAIT cells across sexes in COVID-19
(A) Subclustering of CD161hi cells (n = 21,610), showing two MAIT cell clusters and one γδ T cluster.
(B) Marker gene expression of three CD161hi clusters.
(C) Heatmap of the top 25 discriminative genes between MAITα and MAITβ clusters. Expression level was scaled by Z score distribution.
(D and E) Representative top enriched pathways of MAITα and MAITβ in the Reactome Pathway Database (ranked by FDR, −log10 scale). The top 100 DEGs ranked by fold change between MAITα and MAITβ were used for this analysis.
(F and G) UMAP visualization of MAIT cluster changes (F) and their frequencies (G) with severity rank.
(H) Frequencies of MAIT clusters, grouped by time after symptom onset.
(I and J) Sex differences of MAIT clusters as shown in (H).
Data were plotted as mean ± standard error (G–J). Significance was determined by Mann-Whitney test (I). ∗p < 0.05. See also Figure S4 and Tables S3 and S4.
Last for this series of experiments, we sought to determine the dynamics of the two phenotypically distinct clusters by sex over the COVID-19 disease course. By first grouping our data by severity and time after symptom onset, we found that MAITα was the major phenotype in healthy individuals, whereas MAITβ predominated in exposed and infected groups (Figures 5F and 5G) and at early, middle, and late time points (Figure 5H). There was a noted exception for hospitalized individuals (Figure 5F), who had very few cells, as seen in our flow cytometry data, consistent with lymphopenia, which occurs in severe COVID-19.36 When further stratified by sex, we found that MAIT cell frequencies were higher in healthy females (Figures 5I and 5J), corroborating our flow cytometry results. In healthy females, these cells were skewed toward the MAITα cluster, whereas the few cells present in healthy males consisted mostly of the MAITβ cluster (Figure 5I). However, this difference was lost in the exposed/infected setting, where the cells of individuals of both sexes were comprised mostly of the MAITβ cluster (Figure 5J). Nonetheless, MAITβ cluster percentages were statistically greater in females in late disease (Figure 5J), which reflects the increased MAIT cells during late infection in females, as shown by our flow cytometry findings. Regarding expression of CD69, a T cell activation marker, we did not observe major differences across cluster or sex but did observe elevated expression in the hospitalized group (Figures S4C–S4F). This possibly suggests an altered MAIT cell response in hospitalized individuals.24, 25, 26, 27 These results suggest sex-specific MAIT cell differences at the quantitative and phenotypic levels in health and COVID-19.
Respiratory tract MAIT cell responses differ by sex in COVID-19
To test our hypothesis that the sex-specific change of circulating MAIT cells is due to their extravasation into airway tissues, we utilized published scRNA-seq datasets of bronchoalveolar lavage fluid (BALF)37 and nasopharyngeal swabs (NPSs)38 to assess MAIT cells in airway tissues of individuals with COVID-19. Beginning our analysis with the BALF dataset, we identified the MAIT cell cluster by expression of TRAV1-2, CD3D, KLRB1, and SLC4A10 (Figures 6 A–6C). With this annotation, we found a significant increase (p = 0.0188) of MAIT cells in individuals with COVID-19 (including mild and severe cases) relative to normal control individuals and a higher MAIT cell frequency (p = 0.0332) in females relative to males among subjects with COVID-19 (Figure 6D). This detection of increased MAIT cells in BALF from females, along with the drop in these cells we observed in peripheral blood from females, suggest female-dominant extravasation of MAIT cells in COVID-19.
Figure 6.

Sex differences of MAIT cells in airway tissue samples from individuals with COVID-19
(A and B) Clustering analysis of scRNA-seq data from the COVID-19 BALF dataset with subtracted T and NK cells.37
(C) MAIT cell cluster indicated by marker genes.
(D) Frequencies of MAIT cells in BALF between normal subjects and individuals with COVID-19 (left) and across sexes within subjects with COVID-19 (right).
(E) Integrated clustering analysis of NPSs with BALF using Seurat 3. Cluster 11, MAIT cells.
(F) Referenced MAIT cell cluster in NPSs by expression of TRAV1-2 in BALF and the indicated marker genes in NPSs.
(G) Frequencies of MAIT cells in NPSs from healthy subjects and subjects with COVID-19.
(H and I) Visualization (H) and frequencies (I) of MAIT cells in NPSs, grouped by disease severity.
(J) Volcano plot showing DEGs of BALF MAIT cells between sex with log2 fold change and −log10 FDR.
(K–M) Expression of DEGs in IL-7 signaling (K), transcription factors (L), and CCL2 (M).
Data were plotted as mean ± standard error (D, G, and I), with females in red and males in black. Significance was determined by unpaired one-tailed Student’s t test (D), Mann-Whitney test (G), and Kruskal-Wallis test with Dunn’s post hoc test (I): ∗p < 0.05, ∗∗p < 0.01. See also Figure S5.
To perform the same analysis with NPS samples, we integrated the T cell data from NPSs with those from BALF to identify MAIT cells in NPSs (Figure 6E and 6F) because TCR genes were not aligned in the NPS dataset.38 With this annotation, we identified 232 and 631 MAIT cells in BALF and NPS, respectively, and quantified MAIT cell frequencies in the NPS dataset, again observing a significant increase (p = 0.0139) in individuals with COVID-19 (Figure 6G). We also analyzed the data across severity, observing a significant increase (p = 0.0038) in subjects with moderate disease relative to normal subjects and a decrease (p = 0.0366) relative to individuals with critical COVID-19 (Figures 6H and 6I). We could not perform the same analysis by sex because of insufficient numbers of samples from females (Figure 6I). Nonetheless, these results match the reduced circulating MAIT cell frequencies seen in our hospitalized individuals, suggesting that a lymphopenic state that occurs in severe COVID-19 affects MAIT cells, consistent with other reports.24, 25, 26 , 39
In a final experiment, we sought to characterize MAIT cell transcriptomes by sex in the BALF and NPS datasets and determine whether these cells resembled α and β phenotypes we identified in circulating MAIT cells. Cluster analysis was not warranted here, given the low cell numbers in these datasets. Instead, we leveraged gene modules derived from our respective α and β clusters of circulating MAIT cells. We found that MAIT cells in BALF and NPS data were largely skewed toward the β module, with minimal sex differences (Figure S5A). However, when we directly examined DEGs (DEGs between sexes), we were able to detect sex differences associated with α and β phenotypes. Specifically, in BALF, we found increased IL7R expression in females (Figure 6J) and other IL-7 signaling-associated genes (CISH and SOCS1) (Figure 6K). Given the key role of this signaling in T cell survival, we explored additional pathway genes, finding that female MAIT cells had upregulated anti-apoptotic genes (BCL2 and FOXP1) and downregulated pro-apoptotic genes (BAX and CASP3) (Figures S5B and S5C). Also observed in cells from females were upregulated anti-proliferative genes (CDKN1B and BTG2) (Figure S5D). These patterns matched MAITα gene changes in our PBMC dataset. We were able to find other sex differences, including increased expression of several transcription factors (KLF2, MYC, and CEBPD) (Figure 6L). Conversely, males had higher expression of CCL2 (Figure 6M), which has been linked to COVID-19 immunopathology.40 In short, our results infer sex differences at the qualitative level in COVID-19, with MAIT cells from females possessing a pro-survival and immunologically active phenotype.
Discussion
Despite the knowledge of sex differences in the immune response as an underlying factor in COVID-19 disease outcomes, the sexually dimorphic responses of MAIT cells, an unconventional T cell population deemed important in this disease, has remained unknown. Although MAIT cells may be known primarily for their role in host defense against bacteria, responding via their invariant TCRs to microbial vitamin B metabolites presented on major histocompatibility complex class I-related molecules,33 , 41 they are also capable of responding in a TCR-independent manner to cytokines such as IL-12 and IL-18 and can be critical players in immune responses against certain viruses.42, 43, 44 We now demonstrate that MAIT cells in females are quantitatively and qualitatively more robust in the SARS-CoV-2 setting, potentially helping us understand the immunological reasons for reduced COVID-19 susceptibility of females.
Our finding that MAIT cell recruitment to airway tissues may be more robust in females with COVID-19 was aided first by our observation of higher frequencies of circulating MAIT cells in females in the healthy setting. This difference can be explained by the rate of physiological aging-related attrition of MAIT cells, which is substantially less pronounced in the blood of females.45, 46, 47 The resultant higher frequencies in circulation enabled us to readily uncover the precipitous percentage drop we saw with MAIT cells relative to exposed/infected females. When trying to elucidate the potential cause of this drop, we considered two possible scenarios, (1) lymphopenia and (2) extravasation, which are not necessarily mutually exclusive. It is accepted that lymphopenia is associated with severe COVID-19 infection,32 , 36 , 48 , 49 which agrees with our observations in our hospitalized group (comprised of 77.8% of individuals in intensive care). Similarly, lymphopenia could partially explain the reduction in MAIT cells described by Jouan et al.,26 who studied male-dominated samples from critically ill individuals with COVID-19, although extravasation also likely occurred. In our study, however, we demonstrated that the frequencies of circulating MAIT cells dropped in our group of infected individuals. Because these subjects were not critically ill, our findings point to extravasation as a major reason for the sex-specific drop in the frequencies of circulating MAIT cells. The same pattern may also exist in several other studies.24 , 25 , 27 For example, although it has been demonstrated that circulating MAIT cells are reduced in individuals with moderate COVID-19 relative to healthy subjects in aggregated data,24 it is possible that the frequencies in healthy females contributed to reaching a statistical difference. Further supporting our conclusion, we were able to show, with publicly available scRNA-seq data from COVID-19 BALF samples,37 that females in that study had an increased MAIT cell percentage relative to males, allowing us to conclude that MAIT cell extravasation during COVID-19 may be quantitatively more robust in females.
Our results also suggest that MAIT cells may be qualitatively superior in females with respect to anti-viral immune activity in COVID-19. Leading us to this conclusion, our scRNA-seq analysis of PBMCs from affected individuals revealed two distinct clusters of MAIT cells, MAITα and MAITβ. The α cluster was enriched for various immune pathways, such as IFN-γ signaling, inferring a capacity for anti-viral immune function. In contrast, the β cluster was enriched for cell stress and apoptosis pathways, inferring a frail phenotype roughly similar to a previously described population of double-negative MAIT cells.50 , 51 We showed in the healthy setting that MAIT cells in females were skewed toward the α cluster, whereas those in males were comprised of the β cluster. Although, from these results, it could be inferred that the α cluster should be overrepresented in airways of females with COVID-19, this was not the case with BALF. However, we reasoned that such a finding would be very difficult to prove for two main reasons. First, extravasated MAIT cells with an α phenotype would be restricted to the early wave of recruitment because circulating cells are almost completely skewed to the β module in exposed/infected individuals. Second, a certain level of transcriptional reprogramming would occur upon immune cell extravasation into the tissue and potentially again upon accessing the alveolar space. Still, we were able to show in BALF that certain gene patterns remained consistent with the α signature in females versus males. In addition, our finding that BALF samples from females had quantitatively more MAIT cells gives further credence that differences revealed in the blood would likewise extend to tissue.
Limitations of study
The following limitations of this study should be considered when interpreting results. It is possible that decreased COVID-19 severity and elevated levels of MAIT cells are associated independently with the female sex. We showed the immunologically active signature of MAIT cells from females at the transcriptomic level, but functional analysis of MAIT cells in SARS-CoV-2 infection would be needed to demonstrate whether MAIT cells directly protect females with COVID-19. The same argument is relevant with respect to MAITα and MAITβ subsets in lung tissues of individuals with COVID-19. Last, although our analyses included over 80 sex-balanced samples, the current study may not be powered to distinguish other immune cell types or specific sequelae that are associated with sex differences in COVID-19, and larger cohorts with more comprehensive longitudinal sampling would be needed in the future.
We conclude that MAIT cells in females are quantitatively and qualitatively distinct from those of males, and we surmise that this distinction provides a protective advantage in COVID-19. Indeed, females in general tend to have elevated frequencies of circulating MAIT cells, as also gleaned from large independent studies with European45, South Korean,46 and Chinese populations.47 Further supporting this argument, it has now been recorded that fatality rates of adults with COVID-19 tend to be lower in females at all ages in 37 of 38 different countries or regions where MAIT cell frequencies tend to be higher in females, including North America, Europe, and Asia.2 These points also argue against the possibility that an immunologically more robust MAIT cell response has a net negative effect; for example, by immunological misfiring20 or cytokine storm-related immunopathology.40 However, one open question our findings raise is whether the males in our study, who had more circulating CD8+ memory T cells, would have an advantage in the reinfection setting or following vaccination.52 Future studies are needed to explore this question and to better understand sex differences in MAIT cells in general and in COVID-19.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV421 anti-human CCR7 (clone G043H7) | BioLegend | Cat# 353208 |
| BV711 anti-human CCR6 (clone G034E3) | BioLegend | Cat# 353436 |
| BV750 anti-huamn CXCR5 (clone RF8B2) | BD Biosicences | Cat# 747111 |
| BUV563 anti-huamn CCR5 (clone 2D7) | BD Biosicences | Cat# 741401 |
| PE-Cyanine7 anti-human CXCR3 (clone CEW33D) | ThermoFisher | Cat# 25-1839-42 |
| PerCP-eFluor710 anti-human TCR γδ (clone B1.1) | ThermoFisher | Cat# 46-9959-42 |
| APC-Fire810 anti-human CD38 (clone HIT2) | BioLegend | Cat# 303549 |
| FITC anti-human CD57 (clone HNK-1) | BioLegend | Cat# 359604 |
| PE-Cyanine5 anti-huamn CD95 (clone DX2) | ThermoFisher | Cat# 15-0959-42 |
| Spark-NIR685 anti-huamn CD19 (clone H1B19) | BioLegend | Cat# 302270 |
| Spark-NIR550 anti-human CD14 (clone 63D3) | BioLegend | Cat# 367148 |
| PerCP anti-human CD45 (clone H130) | ThermoFisher | Cat# MHCD4531 |
| PE anti-huamn CD25 (clone BC96) | ThermoFisher | Cat# 12-0259-42 |
| APC anti-human CD27 (clone O323) | ThermoFisher | Cat# 17-0279-42 |
| APC-eFluor780 anti-human HLA-DR (clone L243) | ThermoFisher | Cat# 47-9952-42 |
| PerCP-Cyanine5.5 anti-human CD11b (clone ICRF44) | BioLegend | Cat# 301328 |
| PE-eFluor610 anti-human CD24 (clone eBioSN3 (SN3 A5-2H10)) | ThermoFisher | Cat# 61-0247-41 |
| Alexa Fluor®647 anti-human CD1c (clone L161) | BioLegend | Cat# 331510 |
| APC-R700 anti-human CD127 (clone HIL-7R-M21) | BD Biosicences | Cat# 565185 |
| BUV496 anti-human CD16 (clone 3G8) | BD Biosicences | Cat# 612944 |
| BV480 anti-human IgD (clone IA6-2) | BD Biosicences | Cat# 566138 |
| BUV395 anti-human CD45RA (clone 5H9) | BD Biosicences | Cat# 740315 |
| BUV737 anti-human CD56 (clone NCAM16.2) | BD Biosicences | Cat# 612766 |
| BUV805 anti-human CD8 (clone SK1) | BD Biosicences | Cat# 612889 |
| BUV661 anti-human CD11c (clone B-ly6) | BD Biosicences | Cat# 612967 |
| SuperBright436 anti-human CD123 (clone 6H6) | ThermoFisher | Cat# 62-1239-42 |
| BV570 anti-human IgM (clone MHM-88) | BioLegend | Cat# 314517 |
| BV650 anti-human CD28 (clone CD28.2) | BioLegend | Cat# 302946 |
| BB515 anti-human CD141 (clone 1A4) | BD Biosicences | Cat# 566017 |
| eFluor450 anti-human CD161 (clone HP-3G10) | ThermoFisher | Cat# 48-1619-42 |
| BV510 anti-human CD3 (clone OKT3) | BioLegend | Cat# 317332 |
| Pacific Orange anti-human CD20 (clone HI47) | ThermoFisher | Cat# MHCD2030 |
| BV605 anti-human IgG (clone G18-145) | BD Biosicences | Cat# 563246 |
| BV785 anti-human PD-1 (clone EH12.2H7) | BioLegend | Cat# 329929 |
| BUV615 anti-human CD4 (clone SK3) | BD Biosicences | Cat# 612987 |
| Biological samples | ||
| Peripheral blood from healthy and COVID-19 related subjects | Duke University Health System | NA |
| Chemicals, peptides, and recombinant rroteins | ||
| Live/Dead UV Blue | ThermoFisher | Cat# L34962 |
| Brilliant Stain Buffer Plus | BD Biosicences | Cat# 566385 |
| Critical commercial assays | ||
| Dead Cell Removal Kit | Miltenyi Biotec | Cat# 130-090-101 |
| Chromium Next GEM Single Cell 5′ Library & Gel Bead Kit v1 | 10x Genomics | |
| Deposited data | ||
| BALF scRNA-seq data | Liao et al.37 | GSE145926 |
| NPS scRNA-seq data | Chua et al.38 | https://figshare.com/articles/COVID-19_severity_correlates_with_airway_epithelium-immune_cell_interactions_identified_by_single-cell_analysis/12436517 |
| Supplemental data | This study | https://dx.doi.org/10.17632/csc9p34d5t.1 |
| scRNA-seq data of PBMCs | This study | GSE171555 |
| Flow cytometry data of PBMCs | This study | https://flowrepository.org/id/FR-FCM-Z3WR |
| Software and algorithms | ||
| FlowJo version 10.6.1 | Tree Star | https://www.flowjo.com/ |
| Omiq | Omiq | https://www.omiq.ai/ |
| Cell Ranger version 3.1.0 | 10x Genomics | https://www.10xgenomics.com/ |
| Seurat v3 R package | https://satijalab.org/seurat/ | |
| CellPhone DB v2 | https://www.cellphonedb.org/ | |
| Reactome pathway database | https://reactome.org/ | |
| GraphPad Prism version 8 | https://www.graphpad.com/ | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel R. Saban (daniel.saban@duke.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Publicly available scRNA-seq data of BALF37 were downloaded from GEO with the accession number GSE145926, and the count data of NPS38 were downloaded from https://figshare.com/articles/COVID-19_severity_correlates_with_airway_epithelium-immune_cell_interactions_identified_by_single-cell_analysis/12436517. All clinical metadata of participants and samples in this study are included in Table S1. The fcs files of flow cytometry are available to download from https://flowrepository.org/id/FR-FCM-Z3WR. The scRNA-seq of PBMC data generated in this study are available in Gene Expression Omnibus (GEO) with the accession number GSE171555. Additional Supplemental Items are available from Mendeley Data at https://dx.doi.org/10.17632/csc9p34d5t.1. All analytic scripts will be also made available per request.
Experimental model and subject details
Ethics statement
This study and relevant protocols were approved by the Institutional Review Boards of Duke University Health System (DUHS). All procedures were performed in accordance with the Declaration of Helsinki, applicable regulations, and local policies.
Participants in this study
Inpatients (hospitalized) and outpatients (infected) with confirmed infection of SARS-CoV-2 were identified through the DUHS and enrolled into the Molecular and Epidemiological Study of Suspected Infection (MESSI, Pro00100241). The PCR testing for SARS-CoV-2 was performed at either the North Carolina State Laboratory of Public Health or at clinical laboratories of the DUHS. The exposed group, who closely contacted with COVID-19 patients, presented negative PCR test and negative serology test during longitudinally sampling from the first visit to at least 2 months after, typically 0, 7, 14, and 28 days relative to enrollment. Initial severity scores of individuals were recorded through a self-reporting survey on 38 defined symptoms related to COVID-19 plus “other” when enrolled. The exposed group (average score = 9.71) showed a lower severity scores compared with infected (outpatients) group (average score = 18.16). The hospitalized patients presented severe disease symptoms with breath difficulty, cough, fever or chest pain when enrolled, and 77.8% of them for this study required intensive care unit (ICU) care. All COVID-19 patients were also longitudinally sampled with serology test from enrollment to convalescent phase. Healthy donors were enrolled in 2019 (Duke IRB Protocol Pro00009459) with no diagnosis or symptoms consistent with COVID-19 or other respiratory illness. Written informed consent was obtained from all subjects or legally authorized representatives. Patient Demographics are summarized in Tables 1 and S1.
In keeping with inclusion criteria above, all participants with the minimum age of 17 were maintained in the analysis. Investigators were blinded during experiments in terms of sex, time post symptom onset and anti-SARS-COV-2 IgG serology condition.
Method details
Collection of peripheral blood mononuclear cells (PBMCs)
Peripheral whole blood was collected in EDTA vacutainer tubes and processed within 8 hours via Ficoll-Hypaque density gradient method to obtain cryopreserved PBMCs. Blood was diluted 1:2 in PBS, layered onto the the Ficoll-Hypaque in 50 mL conicals, and centrifuged at 420 g for 25 min. Buffy coat was used to collect PBMCs and cell pellets were washed twice with D-PBS following centrifugation at 400 g for 10 min. Counts and cell viability were obtained using Vi-Cell automated cell counter (Beckman-Coulter) before PBMCs were adjusted to 10x106 cells/ml in cryopreservation media (90% FBS, 10% DMSO) and aliquoted into cryopreservation vials on ice. Finally, cells were subjected to controlled freezing at −80°C using CoolCell LX (BioCision) for 12-24 hours, and transferred to liquid nitrogen vapor phase.
Sample processing for flow cytometry and single cell RNA-sequencing (scRNA-seq)
Counts and cell viability of thawed PBMCs were measured by Countess II after a wash with DMEM 10% FBS. The cell viability of hospitalized patients ranged from 70%–80% whereas all other samples exceeded 80% viability. An additional dead cell removal step (Miltenyi Biotec) was conducted on hospitalized PBMC samples prior to aliquot for scRNA-seq. To perform scRNA-seq, 200,000 cells per sample were aliquoted, spun down, resuspended in 30 μL PBS supplemented with 0.04% BSA and 0.2U/μl RNase inhibitor and counted using Countess II.
Panel and Staining for Flow Cytometry
Approximately 0.5-2x106 cells per cryopreserved sample were stained for flow cytometry analysis. Antibody titrations used in this study were previously established by Cytek Biosciences with slight modifications. All staining procedures were performed at room temperature. PBMCs were stained with live/dead Blue (Thermofisher) for 15 min, washed with FACS-EDTA buffer and spun down at 1500 rpm for 5 min. Samples were resuspended with Brilliant Stain Buffer Plus (BD Biosciences) and sequentially stained with anti-CCR7 for 10 min, all other chemokine receptor mix of CCR6, CXCR5, CCR5 and CXCR3 for 5 min, anti-TCR gamma/delta for 10 min and the rest surface receptor mix for 30 min. After incubation, PBMCs were washed with FACS-EDTA buffer and spun down at 1500 rpm for 5 min. Samples were fixed with 1% PFA in PBS for 20 min, spun down and resuspended in FACS-EDTA buffer.
36-color Full Spectrum Flow Cytometry
Samples were acquired using a four-laser Cytek Aurora Spectral Flow Cytometry System. Single color controls for spectral unmixing were acquired with PBMCs from healthy control blood and UltraComp eBeads (ThermoFisher). Raw data were unmixed and further analyzed using either FlowJo for manual gating or Omiq (https://www.omiq.ai) for clustering visualization and analysis.
High-dimensional data analysis of flow cytometry data
Uniform Manifold Approximation and Projection (UMAP) and FlowSOM clustering analyses were performed on Omiq (https://www.omiq.ai), using equal random sampling of 3000 live CD45+ singlets. from each FCS file. The UMAP plot was generated with the parameters of 15 neighbors and 0.4 minimum distance. All markers in flow panel were used for analysis except live/dead and CD45.
ScRNA-seq using 10x Genomics platform
10x Genomics Single Cell 5′ v1 chemistry was used to generate Gel Bead-In Emulsions (GEM), and perform post GEM-RT cleanup, cDNA amplification, as well as library construction. An agilent DNA ScreenTape assay was used for quality control. Libraries were pooled and sequenced to saturation or 20,000 unique reads per cell on average using an Illumina NovaSeq6000 with 150-bp paired-end reads.
Processing and quality control of scRNA-seq
Raw sequencing data were initially processed with 10x Genomics Cell Ranger pipelines (V3.1.0). Briefly, BCL files were demultiplexed to generate FASTQ files. FASTQ files were aligned with STAR aligner to the human genome reference GRCh38 from Ensemble database. Feature barcode processing and UMI counting were then performed according to the standard workflow. (QC summary after sequencing). The following criteria were applied as quality control of single cells from all individual samples. Cells that had fewer than 1000 UMI counts or 500 genes, as well as cell that had greater than 10% of mitochondrial genes were removed from further analysis. Genes that were expressed by fewer than 10 cells were also excluded. After filtering, a total of 424,080 cells with 18,765 gene features were kept for the downstream analysis.
Quantification and statistical analysis
Dimensionality reduction and clustering analysis
The filtered gene-barcode matrix was analyzed using Seurat 3.28 All the procedures were conducted with the default parameters unless otherwise specified. Briefly, data were first normalized using log transformation and adjusted with a scale factor of 10,000. The top 2,000 variable genes were identified, and percentages of mitochondrial genes were regressed out when scaling data. Principle component analysis (PCA) was performed using these top variable genes, and top 25 principle components (PCs) were selected for graph-based clustering with Shared Nearest Neighbor (SNN) and visualization in UMAP. The resolution was set to 0.35 to identify major immune cell subsets in PBMCs. Sub-clustering of CD161hi T cells (21,610 cells) was also performed using the analytic pipeline mentioned above with two modifications: top 10 PCs were used, and the resolution was set to 0.1 to identify MAIT cell clusters.
Differential gene expression analysis
Differentially expressed genes (DEGs) were identified using Seurat 3 (FindAllMarkers or FindMarkers Functions) with either ‘wilcox’ for all cluster markers or ‘DESeq2′.53 Randomly downsampled data with 100,000 cells were used to find all markers of PBMC clusters. A gene was considered significant with adjusted p value or false discovery rate (FDR) < 0.05. DEGs results of all PBMCs and MAIT cells are listed in Tables S2 and S3.
Pathway enrichment analysis
Top 100 DEGs of MAIT clusters were used for pathway enrichment analysis using Reactome Pathway Database (https://reactome.org). A pathway was considered significantly over-presented with FDR < 0.05. The full pathway enrichment results are summarized in Table S4.
Inference of ligand-receptor interactions between T cells and monocytes
Ligand-receptor interactions between T cells and monocytes were inferred using CellPhoneDB.35 PBMC scRNA-seq data were randomly downsampled to 50,000 cells and T and monocyte clusters were extracted based on the expression of their lineage markers. CellPhoneDB was with default parameters (https://github.com/Teichlab/cellphonedb). The inferred interactions are considered significant when p value < 0.05.
Integration of BALF and NPS dataset
Publicly available scRNA-seq data of BALF37 and of NPS38 were downloaded and processed using Seurat 3 as previously described.28 All T cell clusters, were extracted from both datasets and integrated via the Single Cell Transform (SCT) method in Seurat 3. Top 3,000 variable features were selected for the integration. Dimensionality reduction was conducted using PCA and UMAP embedding of the top 100 PCs. Clusters were visualized at a resolution of 0.8 after constructing a SNN graph using the first 50 PCs.
Calculations of the feature scores in MAIT cells
The DEGs between MAIT1 and MAIT2 were used to generate their feature scores as previously described.54 The feature scores were calculated using AddModuleScore function in Seurat 3. MAIT cells from different single cell dataset were plotted with MAIT1 feature and MAIT2 feature for visualization.
Statistical analysis
Data normality and homogeneity of variance were assessed using Kolmogorov-Smirnov test and Bartlett’s test, respectively. Due to the distribution and variance of human data, non-parametric statistical tests were favorably used throughout this study unless otherwise specified. Mann Whitney U test was used for two-group comparisons, and Kruskal-Wallis with post hoc Dunn’s test was used for comparisons of three groups and more. Spearman’s correlation efficiency was used to quantify the correlation of the ranked disease severity (from healthy as 1, to hospitalized as 4). Simple linear regression and multiple linear regression were calculated using GraphPad Prism 8. Data were transformed to meet the assumptions of linear regression when necessary. To adjust p values for multiple hypothesis testing, FDR correction was performed using the Benjamini-Hochberg procedure when appropriate. Two-tailed tests were used unless otherwise specified. A p value or FDR < 0.05 is consider statistically significant. Graphical data of quantifications presented throughout are expressed as the means ± SEMs and were plotted using Graphpad Prism 8. Other graphs in this study were generated using either the corresponding analytic packages or R package ggplot2.
Acknowledgments
This work was supported by NIH/NIAID (U01AI066569, UM1AI104681), the U.S. Defense Advanced Projects Agency (DARPA, N66001-09-C-2082 and HR0011-17-2-0069), the Veterans Affairs Health System, and Virology Quality Assurance (VQA) 75N93019C00015. COVID-19 samples were processed under Biosafety level (BSL)-2 with aerosol management enhancement or BSL-3 in the Duke Regional Biocontainment Laboratory which received partial support for construction from NIH/NIAID (UC6AI058607). The flow cytometry was performed at Duke Eye Center with the support from NIH/NEI (P30EY005722, R01EY02179) and Research to Prevent Blindness (Unrestricted, Duke Eye Center). We would like to thank Monica DeLay and Patrick Duncker (Cytek Biosciences) for help with spectral flow cytometry and Chris Ciccolella and Geoff Kraker (Omiq, Inc.). We would also like to thank Maria Miggs, Deborah Murray, Tyffany Locklear, Robert Rolfe, Jack Anderson, Allison Fullenkamp, Raul Louzuo, Thad Gurley, and Julie Steinbrink for their work and the Durham Veterans Affairs Health Care System and Duke Regional Hospital for support.
Author contributions
C.Y., S.L., X.S., and D.R.S. designed the experiments. C.Y., S.L., M.T.M., C.W.W., X.S., and D.R.S. helped conceive and implement the study. C.Y., S.L., and D.R.S. performed data analyses, interpreted the data, and wrote the manuscript, which was critically revised by all other authors. E.R.K., E.L.T., M.T.M., and C.W.W. contributed to clinical study design. M.T.M., E.P., and T.W.B. acquired clinical samples and patient metadata. C.Y., R.M., and J.K. performed flow cytometry. N.S.G., S.D., H.A.C., G.O.R., T.R., and R.X. performed sample preparation and single-cell sequencing. G.D.S. and T.N.D. performed serology and PCR assessments.
Declaration of interests
M.T.M. reports grants for biomarker diagnostics from the Defense Advanced Research Projects Agency (DARPA), National Institutes of Health (NIH), Sanofi, and the Department of Veterans Affairs. T.W.B. reports grants from DARPA and is a consultant for Predigen. M.T.M., T.W.B., E.L.T., and C.W.W. report pending patents on molecular methods to diagnose and treat respiratory infections. E.L.T. reports grants on biomarker diagnostics from DARPA and the NIH/Antibacterial Resistance Leadership Group (ARLG) and an ownership stake in Predigen. G.S.G. reports an ownership stake in Predigen. C.W.W. reports grants for biomarker diagnostics from DARPA, NIH/ARLG, Predigen, and Sanofi and has received consultancy fees from bioMerieux, Roche, Biofire, Giner, and Biomeme.
Published: April 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2021.04.008.
Supplemental information
References
- 1.Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scully E.P., Haverfield J., Ursin R.L., Tannenbaum C., Klein S.L. Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat. Rev. Immunol. 2020;20:442–447. doi: 10.1038/s41577-020-0348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alghamdi I.G., Hussain I.I., Almalki S.S., Alghamdi M.S., Alghamdi M.M., El-Sheemy M.A. The pattern of Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive epidemiological analysis of data from the Saudi Ministry of Health. Int. J. Gen. Med. 2014;7:417–423. doi: 10.2147/IJGM.S67061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlberg J., Chong D.S., Lai W.Y. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am. J. Epidemiol. 2004;159:229–231. doi: 10.1093/aje/kwh056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leong H.N., Earnest A., Lim H.H., Chin C.F., Tan C., Puhaindran M.E., Tan A., Chen M.I., Leo Y.S. SARS in Singapore--predictors of disease severity. Ann. Acad. Med. Singap. 2006;35:326–331. [PubMed] [Google Scholar]
- 6.Channappanavar R., Fett C., Mack M., Ten Eyck P.P., Meyerholz D.K., Perlman S. Sex-Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J. Immunol. 2017;198:4046–4053. doi: 10.4049/jimmunol.1601896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein S.L., Flanagan K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016;16:626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 8.Polanczyk M.J., Carson B.D., Subramanian S., Afentoulis M., Vandenbark A.A., Ziegler S.F., Offner H. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004;173:2227–2230. doi: 10.4049/jimmunol.173.4.2227. [DOI] [PubMed] [Google Scholar]
- 9.Furman D., Hejblum B.P., Simon N., Jojic V., Dekker C.L., Thiébaut R., Tibshirani R.J., Davis M.M. Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc. Natl. Acad. Sci. USA. 2014;111:869–874. doi: 10.1073/pnas.1321060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griesbeck M., Ziegler S., Laffont S., Smith N., Chauveau L., Tomezsko P., Sharei A., Kourjian G., Porichis F., Hart M., et al. Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-α Production in Women. J. Immunol. 2015;195:5327–5336. doi: 10.4049/jimmunol.1501684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silvin A., Chapuis N., Dunsmore G., Goubet A.G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mann E.R., Menon M., Knight S.B., Konkel J.E., Jagger C., Shaw T.N., Krishnan S., Rattray M., Ustianowski A., Bakerly N.D., et al. NIHR Respiratory TRC. CIRCO Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci. Immunol. 2020;5:eabd6197. doi: 10.1126/sciimmunol.abd6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Oxford Immunology Network Covid-19 Response T cell Consortium. ISARIC4C Investigators Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S., et al. Karolinska COVID-19 Study Group Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- 18.Weiskopf D., Schmitz K.S., Raadsen M.P., Grifoni A., Okba N.M.A., Endeman H., van den Akker J.P.C., Molenkamp R., Koopmans M.P.G., van Gorp E.C.M., et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020;5:eabd2071. doi: 10.1126/sciimmunol.abd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Del Valle D.M., Kim-Schulze S., Huang H.H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A., et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi T., Ellingson M.K., Wong P., Israelow B., Lucas C., Klein J., Silva J., Mao T., Oh J.E., Tokuyama M., et al. Yale IMPACT Research Team Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature. 2020;588:315–320. doi: 10.1038/s41586-020-2700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClain M.T., Constantine F.J., Henao R., Liu Y., Tsalik E.L., Burke T.W., Steinbrink J.M., Petzold E., Nicholson B.P., Rolfe R., et al. Dysregulated transcriptional responses to SARS-CoV-2 in the periphery support novel diagnostic approaches. medRxiv. 2020 doi: 10.1101/2020.07.20.20155507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018 doi: 10.1038/nbt.4314. Published online December 3, 2018. [DOI] [PubMed] [Google Scholar]
- 23.Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 24.Parrot T., Gorin J.B., Ponzetta A., Maleki K.T., Kammann T., Emgård J., Perez-Potti A., Sekine T., Rivera-Ballesteros O., Gredmark-Russ S., et al. Karolinska COVID-19 Study Group MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci. Immunol. 2020;5:eabe1670. doi: 10.1126/sciimmunol.abe1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A., et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jouan Y., Guillon A., Gonzalez L., Perez Y., Boisseau C., Ehrmann S., Ferreira M., Daix T., Jeannet R., François B., et al. Phenotypical and functional alteration of unconventional T cells in severe COVID-19 patients. J. Exp. Med. 2020;217:e20200872. doi: 10.1084/jem.20200872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flament H., Rouland M., Beaudoin L., Toubal A., Bertrand L., Lebourgeois S., Rousseau C., Soulard P., Gouda Z., Cagninacci L., et al. Outcome of SARS-CoV-2 infection is linked to MAIT cell activation and cytotoxicity. Nat. Immunol. 2021;22:322–335. doi: 10.1038/s41590-021-00870-z. [DOI] [PubMed] [Google Scholar]
- 28.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H., et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130:2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Q., Meng M., Kumar R., Wu Y., Huang J., Deng Y., Weng Z., Yang L. Lymphopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A systemic review and meta-analysis. Int. J. Infect. Dis. 2020;96:131–135. doi: 10.1016/j.ijid.2020.04.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan L., Wang Q., Zhang D., Ding J., Huang Q., Tang Y.Q., Wang Q., Miao H. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct. Target. Ther. 2020;5:33. doi: 10.1038/s41392-020-0148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Godfrey D.I., Koay H.F., McCluskey J., Gherardin N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019;20:1110–1128. doi: 10.1038/s41590-019-0444-8. [DOI] [PubMed] [Google Scholar]
- 34.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 36.Chen Z., John Wherry E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020;20:529–536. doi: 10.1038/s41577-020-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 38.Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 39.De Biasi S., Meschiari M., Gibellini L., Bellinazzi C., Borella R., Fidanza L., Gozzi L., Iannone A., Lo Tartaro D., Mattioli M., et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020;11:3434. doi: 10.1038/s41467-020-17292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merad M., Martin J.C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kjer-Nielsen L., Patel O., Corbett A.J., Le Nours J., Meehan B., Liu L., Bhati M., Chen Z., Kostenko L., Reantragoon R., et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature. 2012;491:717–723. doi: 10.1038/nature11605. [DOI] [PubMed] [Google Scholar]
- 42.van Wilgenburg B., Scherwitzl I., Hutchinson E.C., Leng T., Kurioka A., Kulicke C., de Lara C., Cole S., Vasanawathana S., Limpitikul W., et al. STOP-HCV consortium MAIT cells are activated during human viral infections. Nat. Commun. 2016;7:11653. doi: 10.1038/ncomms11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ussher J.E., Bilton M., Attwod E., Shadwell J., Richardson R., de Lara C., Mettke E., Kurioka A., Hansen T.H., Klenerman P., Willberg C.B. CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL-12+IL-18 in a TCR-independent manner. Eur. J. Immunol. 2014;44:195–203. doi: 10.1002/eji.201343509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Wilgenburg B., Loh L., Chen Z., Pediongco T.J., Wang H., Shi M., Zhao Z., Koutsakos M., Nüssing S., Sant S., et al. MAIT cells contribute to protection against lethal influenza infection in vivo. Nat. Commun. 2018;9:4706. doi: 10.1038/s41467-018-07207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Novak J., Dobrovolny J., Novakova L., Kozak T. The decrease in number and change in phenotype of mucosal-associated invariant T cells in the elderly and differences in men and women of reproductive age. Scand. J. Immunol. 2014;80:271–275. doi: 10.1111/sji.12193. [DOI] [PubMed] [Google Scholar]
- 46.Lee O.J., Cho Y.N., Kee S.J., Kim M.J., Jin H.M., Lee S.J., Park K.J., Kim T.J., Lee S.S., Kwon Y.S., et al. Circulating mucosal-associated invariant T cell levels and their cytokine levels in healthy adults. Exp. Gerontol. 2014;49:47–54. doi: 10.1016/j.exger.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Chen P., Deng W., Li D., Zeng T., Huang L., Wang Q., Wang J., Zhang W., Yu X., Duan D., et al. Circulating Mucosal-Associated Invariant T Cells in a Large Cohort of Healthy Chinese Individuals From Newborn to Elderly. Front. Immunol. 2019;10:260. doi: 10.3389/fimmu.2019.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y., et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I., et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- 50.Gérart S., Sibéril S., Martin E., Lenoir C., Aguilar C., Picard C., Lantz O., Fischer A., Latour S. Human iNKT and MAIT cells exhibit a PLZF-dependent proapoptotic propensity that is counterbalanced by XIAP. Blood. 2013;121:614–623. doi: 10.1182/blood-2012-09-456095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dias J., Leeansyah E., Sandberg J.K. Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proc. Natl. Acad. Sci. USA. 2017;114:E5434–E5443. doi: 10.1073/pnas.1705759114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Provine N.M., Amini A., Garner L.C., Spencer A.J., Dold C., Hutchings C., Silva Reyes L., FitzPatrick M.E.B., Chinnakannan S., Oguti B., et al. MAIT cell activation augments adenovirus vector vaccine immunogenicity. Science. 2021;371:521–526. doi: 10.1126/science.aax8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Publicly available scRNA-seq data of BALF37 were downloaded from GEO with the accession number GSE145926, and the count data of NPS38 were downloaded from https://figshare.com/articles/COVID-19_severity_correlates_with_airway_epithelium-immune_cell_interactions_identified_by_single-cell_analysis/12436517. All clinical metadata of participants and samples in this study are included in Table S1. The fcs files of flow cytometry are available to download from https://flowrepository.org/id/FR-FCM-Z3WR. The scRNA-seq of PBMC data generated in this study are available in Gene Expression Omnibus (GEO) with the accession number GSE171555. Additional Supplemental Items are available from Mendeley Data at https://dx.doi.org/10.17632/csc9p34d5t.1. All analytic scripts will be also made available per request.