

Abstract
Multisystem inflammatory syndrome in children (MIS-C) is a life-threatening post-infectious complication occurring unpredictably weeks after mild or asymptomatic SARS-CoV-2 infection. We profiled MIS-C, adult COVID-19, and healthy pediatric and adult individuals using single-cell RNA sequencing, flow cytometry, antigen receptor repertoire analysis, and unbiased serum proteomics, which collectively identified a signature in MIS-C patients that correlated with disease severity. Despite having no evidence of active infection, MIS-C patients had elevated S100A-family alarmins and decreased antigen presentation signatures, indicative of myeloid dysfunction. MIS-C patients showed elevated expression of cytotoxicity genes in NK and CD8+ T cells and expansion of specific IgG-expressing plasmablasts. Clinically severe MIS-C patients displayed skewed memory T cell TCR repertoires and autoimmunity characterized by endothelium-reactive IgG. The alarmin, cytotoxicity, TCR repertoire, and plasmablast signatures we defined have potential for application in the clinic to better diagnose and potentially predict disease severity early in the course of MIS-C.
Keywords: SARS-CoV-2, MIS-C, pediatric, inflammation, alarmins, cytotoxicity, TRBV11-2, plasmablasts
Graphical abstract
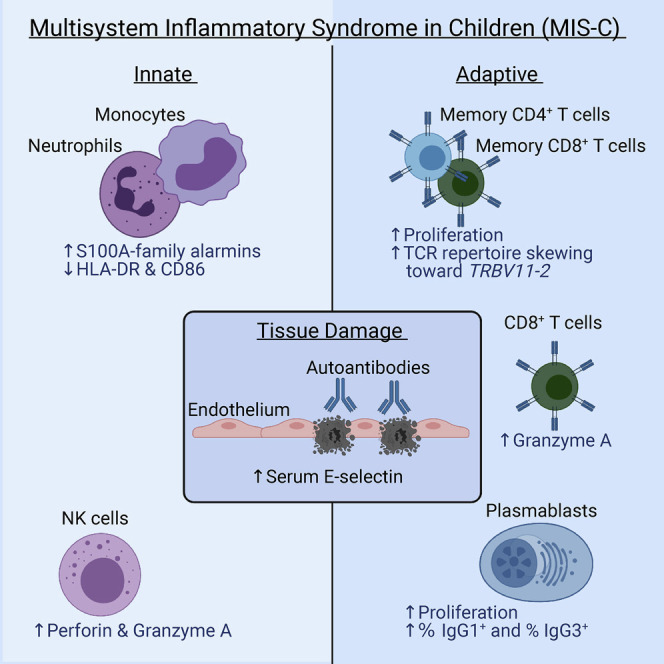
Multisystem inflammatory syndrome in children (MIS-C) is a life-threatening and unpredictable condition of unknown etiology. Ramaswamy et al. use peripheral blood single-cell transcriptomic profiling along with other techniques to define key innate and adaptive signatures that characterize MIS-C.
Introduction
Pediatric patients are largely spared of severe respiratory pathology associated with SARS-CoV-2 infection. However, a severe and delayed post-SARS-CoV-2 inflammatory response in children has been recognized around the world. This “multisystem inflammatory syndrome in children” (MIS-C) presents in youth who had a mild or asymptomatic SARS-CoV2 infection roughly 4–6 weeks prior (Feldstein et al., 2020, Whittaker et al., 2020, Jones et al., 2020, Riphagen et al., 2020, Rauf et al., 2020, Cheung et al., 2020, Verdoni et al., 2020, Toubiana et al., 2020, Klocperk et al., 2020). Symptoms in MIS-C patients vary and involve a systemic cytokine storm with fever, gastrointestinal, cardiac, vascular, hematologic, mucocutaneous, neurologic, and/or respiratory pathology. MIS-C often leads to critical illness with distributive/cardiogenic shock in up to 80% of patients and a 2% mortality rate (Feldstein et al., 2020). Most patients with this syndrome are previously healthy with no co-morbidities and recover with supportive care and immune suppressive therapy. Further understanding the pathophysiology of this disease is imperative to predict, prevent, and optimally treat MIS-C in children exposed to SARS-CoV-2.
Initial reports compared MIS-C with Kawasaki Disease (KD) because of the common presentation with fever, rash, and coronary aneurysms (Whittaker et al., 2020, Jones et al., 2020, Rauf et al., 2020, Verdoni et al., 2020, Toubiana et al., 2020). However, MIS-C predominantly affects older children with an increased prevalence among Black and Hispanic/Latino populations, whereas KD affects very young children with higher occurrence in East Asian populations. Moreover, MIS-C has distinct gastrointestinal symptoms, leukopenia, and high B-type natriuretic peptide, troponin, ferritin, and C-reactive protein, and it more frequently leads to shock (Whittaker et al., 2020, Rowley, 2020). Acute MIS-C was further characterized by high systemic inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, IL-8, IL-10, IL-17, and IFN-γ (Carter et al., 2020). Also reported was a cytokine profile indicative of NK, T cell, monocyte and neutrophil recruitment, mucosal immunity, and immune cell negative feedback (Gruber et al., 2020). Analysis of peripheral blood mononuclear cells (PBMCs) from MIS-C patients revealed CD4+, CD8+, γδT cell and B cell lymphopenia, with high HLA-DR expression on γδ and CCR7+ CD4+ T cells, elevated CD64 expression on neutrophils and monocytes, and low HLA-DR and CD86 on monocytes and dendritic cells (Carter et al., 2020). Neutralizing anti-SARS-CoV-2 antibody responses in MIS-C closely resemble convalescent COVID-19. Elevated complement C5b9 in serum and misshapen red blood cells have also been reported, which are consistent with endothelial cell activation and clinical findings of distributive and cardiogenic shock (Gruber et al., 2020, Consiglio et al., 2020, Diorio et al., 2020). Using panels of human antigens to screen for autoantibodies, recent studies found that acute MIS-C patients had increased antibody binding to antigens associated with endothelium and heart development and other common autoimmunity targets as compared to healthy controls (Gruber et al., 2020, Consiglio et al., 2020). As such, one of the dominant hypotheses to explain the immunopathology of MIS-C is autoimmunity triggered by self-reactive antibodies produced in response to SARS-CoV-2. An analogous mechanism was reported in KD where the presumed infectious trigger is often unknown (Leung et al., 1986a, Leung et al., 1989b, Leung et al., 1989a, Leung et al., 1986b). This hypothesis, however, has not yet been directly tested.
Here, we reported 23 cases of MIS-C and elucidate correlates of immunopathology using single-cell RNA sequencing with antigen receptor repertoire analysis, serum proteomics, flow cytometry, and functional studies in a subset of acute and recovered MIS-C patients compared to healthy pediatric donors, adult COVID-19 patients, and healthy adults. We found innate and adaptive immune triggering during acute MIS-C that featured elevated innate alarmins, acute inflammatory serum proteins, and heightened cytotoxicity signatures. Moreover, in severe MIS-C, we observed increased TRBV11-2 usage among CD4+ and CD8+ memory T cells and expansion of IgG plasmablasts that correlated with serum antibody binding to cultured activated human cardiac microvascular endothelial cells. Collectively, we identified myeloid, NK, plasmablast, and TCR repertoire signatures that could enable improved diagnosis and treatment of MIS-C in children after exposure to SARS-CoV-2.
Results
Clinical characteristics distinguish moderate and severe MIS-C
Our clinical cohort includes 23 MIS-C patients, divided into severe (MIS-C-S: n = 14) and moderate (MIS-C-M: n = 9) groups based on clinical criteria (Table S1). Severe patients were critically ill, with cardiac and/or pulmonary failure (requiring vasoactive medication and/or significant respiratory support with positive pressure or mechanical ventilation), although not due to primary hypoxia. Most patients presented to care 4-6 weeks after peak adult COVID-19 hospitalizations (Figure 1 A). Although a minority (43%; 10/23) of patients tested positive for SARS-CoV-2 virus near the limit of detection during hospitalization, all of the patients had positive SARS-CoV-2 serology. A majority of subjects presented with fever, gastrointestinal symptoms, conjunctivitis, rash, and distributive shock (Figure S1A). Four patients developed coronary aneurysms and 71% of the severe patients had depressed left ventricular heart function (Figure S1A). All children received steroids and aspirin, with a majority also receiving intravenous immunoglobulin (IVIG). Most severe patients also received vasoactive medications (epinephrine, norepinephrine, vasopressin, dopamine, and/or milrinone). Severely affected patients more frequently received Anakinra, an IL-1 receptor antagonist, along with heparin (enoxaparin) for anticoagulation and a course of antibiotics prior to negative culture results (Figure 1B). Principal component analysis (PCA) of clinical lab values separated MIS-C-S and MIS-C-M patients (Figures 1C, and S1B-S1C). In keeping with prior studies, clinical labs for MIS-C patients showed high ferritin, B-type natriuretic peptide (BNP), troponin, C-reactive protein (CRP), soluble CD25 (sCD25), IL-6, and IL-10 (Figure 1D and Table S2). Severe patients also had high lactate, aspartate/alanine aminotransferases (AST/ALT), and creatinine, signifying the multi-organ involvement and shock state of their presentation (Figures 1D and S1B; Table S2). One critically ill patient (P1) had a throat culture that was positive for group A streptococcus (Table S3). All of the patients improved significantly, and all but one patient (P6 with previously undiagnosed chronic kidney disease) have been discharged home after an average of 6.5 days in the hospital.
Figure 1.

Clinical features of moderate and severe MIS-C
(A) Yale New Haven Hospital (YNHH) timeline of total daily adult COVID-19 hospitalizations (blue) and MIS-C cumulative cases (red).
(B) Clinical time course of moderate and severe patients showing symptom onset and treatments relative to hospital admission (Day 0).
(C) PCA biplot for clinical parameters, where available for MIS-C patients. P13, P7, and P21 were excluded due to unavailable measurements for troponin, BNP, and ALC, respectively.
(D) Clinical laboratory data for the indicated analyte. Normal range represented by gray shading. BNP, B-type natriuretic peptide; CRP, C-reactive protein; ALC, absolute lymphocyte count; AST, aspartate aminotransferase; ALT, alanine aminotransferase; WBC, white blood cells; CKD, chronic kidney disease; CHF, chronic heart failure.
MIS-C alters immune cell subsets with no evidence of active viral or bacterial infection
We surveyed the peripheral blood immune cell landscape of MIS-C by performing single-cell RNA sequencing (scRNA-seq) on samples from six pediatric/child healthy donors (C.HD), seven MIS-C patients, and two recovered patients (MIS-C-R). We also incorporated samples from thirteen adult healthy donors (A.HD) and adult COVID-19 patients from early (COVID19-A: median of 7 days after symptom onset; n = 4) and late time points (COVID19-B: median of 16 days after symptom onset; n = 6) from our recent study (Figure S1D and Table S4; STAR Methods) (Unterman et al., 2020).
We performed integrative analysis to harmonize all 38 single-cell gene expression (GEX) datasets, followed by graph-based clustering and non-linear dimensionality reduction using uniform manifold approximation and projection (UMAP) to visualize communities of similar cells. We resolved 30 distinct PBMC cell types (Figures 2 A, 2B, S2A, and S2B). Additionally, we performed Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) on fresh PBMCs isolated from two MIS-C patients and three A.HD, allowing 189 surface antibody phenotypes to be resolved at a single-cell level together with GEX (Table S5) (Mimitou et al., 2019). To annotate memory and naive T cell GEX-based clusters, we exploited CITE-seq antibody-derived tag (ADT) signals for CD45RO and CD45RA. We also confirmed annotations of low-density neutrophils (retained after PBMC isolation) and mature NK cells using CD66b and CD57 markers, respectively (Figure S2C). We subsequently determined differences in cell type percentages among the pediatric cohorts (Figures 2C and S2D). Naive CD4+ T cells were decreased in the peripheral blood of MIS-C patients compared to C.HD. Naive B cells and plasmablasts were increased, and conventional dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs) were decreased in MIS-C compared to C.HD. We further validated this finding by flow cytometry (Figure S2E). We additionally leveraged scRNA-seq GEX data to map significant changes in ligand-receptor connectivity in MIS-C compared to C.HD (Figure S2F), and found that ligands and receptors involved in diapedesis and inflammation were coordinately up in MIS-C, including SELPG-ITGAM and MMP9-ITGB2.
Figure 2.

Altered MIS-C immune cell subsets with no evidence of active viral or bacterial infection
(A) Peripheral blood mononuclear cell (PBMC) UMAP of integrated samples from pediatric healthy donors, adult healthy donors, MIS-C patients, and COVID-19 patients.
(B) Dot plots of key PBMC cell lineage markers.
(C) Distributions of peripheral blood cell frequencies across pediatric cohorts, based on cell types inferred from scRNA-seq. A non-parametric two-sided Wilcoxon test was used to assess statistical significance between the C.HD and MIS-C groups.
(D) Donor distributions of viral and bacterial scores in monocytes and neutrophils. Module scores are calculated for each cell and averaged per donor.
To understand the possible viral or bacterial triggers for acute MIS-C onset, we evaluated well-defined signatures of respiratory viral and bacterial infections (Lydon et al., 2019, Hadjadj et al., 2020). We detected a robust anti-viral signature in monocytes and neutrophils in COVID19-A but not in the MIS-C cohort (Figures 2D and S2G). Similarly, there was no evident bacterial signature in the MIS-C cohort compared to C.HD (Figure 2D and Table S3). To determine whether an active herpesvirus could be found in patients with MIS-C, we created viral reference transcriptomes for Epstein-Barr virus (EBV) and Cytomegalovirus (CMV) for alignment of sequencing reads from our pediatric cohort. We did not identify counts aligning to these transcriptomes, with the few cells that appear positive for counts likely the result of misalignment (Figure S2H). Thus, peripheral blood cells in MIS-C patients showed significant alterations, and lack of EBV/CMV reads suggests that there may not be active herpesvirus replication, consistent with our lack of direct evidence for an active viral or bacterial infection during acute illness.
Myeloid cells undergo post-inflammatory phenotypic changes, including an elevated alarmin signature
To investigate innate immune contributions to MIS-C, we sub-clustered monocytes, neutrophils, and dendritic cells (Figures 3A and 3B). Supporting initial findings in PBMCs, cDC and pDC were decreased among myeloid cells by scRNA-seq (Figure S3A). We additionally evaluated differentially expressed genes (Table S6) in neutrophils and monocytes, and found a shared upregulation of the alarmin-related S100A genes (Bertheloot and Latz, 2017)—S100A8, S100A9, and S100A12—in MIS-C versus C.HD (Figures 3C, 3D, and S3B–S3D). Consistent with previous scRNA-seq studies, COVID19-A samples also showed an elevated S100A score compared to A.HD (Figure 3D) (Schulte-Schrepping et al., 2020). Pathway enrichment on genes with shared decreased gene expression in monocytes and neutrophils revealed a significant reduction in antigen-presentation and processing (Figure S3E). Indeed, MIS-C patient monocytes had significantly reduced expression of HLA class II (HLA-DP, DQ, and DR) antigen presenting molecules and CD86, a key molecule for providing costimulatory signals to induce T cell activation (Figures 3E and S3F). This is consistent with findings from multi-parameter flow and single-cell studies of COVID-19 (Lucas et al., 2020, Laing et al., 2020, Schulte-Schrepping et al., 2020). In order to validate the functional relevance of these observations at the protein level, we performed flow cytometry-based validation with a larger cohort, and confirmed higher protein levels of S100A9 and lower HLA-DR and CD86 (Figure 3F and Figure S3G). MIS-C classical monocytes demonstrated an elevated sepsis signature, which is linked to dysregulated monocyte activation and observed in COVID-19 patient monocytes (Figure 3G) (Reyes et al., 2020, Schulte-Schrepping et al., 2020). To comprehensively define the serum proteome landscape in MIS-C, we profiled nearly 5,000 serum proteins in three MIS-C patients (P1.1, P2.1, P3.1) and four pediatric healthy donors using SomaScan technology (Figures 3H and S3H). Overall, there was a significant enrichment in myeloid and DC-derived proteins among the differentially expressed proteins in serum (p = 1.7 × 10−12) (Figure 3H). Moreover, pathway analysis of differential proteins in the serum revealed enrichment of terms associated with “Cytokine-cytokine receptor interaction,” “Fluid shear stress and atherosclerosis,” and “Complement and coagulation cascades,” which were consistent with the inflammatory phenotype in the patients (Figure 3I). To understand the impact of these serum proteins on immune cells, we performed connectivity analysis to link elevated serum ligands with receptors expressed in PBMCs of MIS-C patients (Figure S3I). This analysis highlighted CXCL10-CXCR3, which is known to be involved in leukocyte trafficking to inflamed tissues, as a potentially relevant axis in MIS-C (Coperchini et al., 2020, Lee et al., 2009). Thus, gene expression programs in myeloid cells from MIS-C patients are characterized by increased S100A alarmin expression and decreased antigen presentation gene expression. Moreover, the serum proteome in MIS-C patients is consistent with inflammatory myeloid responses and potential endothelial cell activation.
Figure 3.

Innate inflammation in MIS-C with elevated myeloid alarmins in the S100A family
(A) Myeloid cell sub-clustering UMAP.
(B) Key markers delineating myeloid clusters.
(C) Heatmap representing top 20 up- and downregulated differentially expressed genes in monocytes between MIS-C and C.HD. Scale bar represents the scaled average expression of markers.
(D) A module score for S100A8, S100A9, and S100A12 is computed across pediatric donors and adult healthy donors in all monocytes and neutrophils depicted in UMAP. Statistical significance between cohorts is computed using a two-sided non-parametric Wilcoxon test.
(E) HLA class II score including HLA-DP, DQ, and DR molecules is computed across adult and pediatric donors as above, and CD86 expression depicted across pediatric and adult donors in monocytes. Statistical significance was assessed as in (D).
(F) Flow cytometric validation of key scRNA-seq data in gated CD14+ monocytes stained for S100A9, HLA-DR, and CD86 in C.HD (n = 6) and MIS-C (n = 10). Representative plots depict donor closest to the mean of the cohort. Statistical significance was assessed using a two-sided unpaired t-test.
(G) Sepsis-associated monocyte module score computed in classical monocytes across pediatric and adult cohorts. Statistical significance was assessed as in (D).
(H) Volcano plot showing differentially up- and downregulated serum proteins between MIS-C (n = 3) and pediatric healthy donors (n = 4). Molecule annotations are color-coded and genes of interest are labeled in black text. IL-1RN, an upregulated protein in MIS-C, likely corresponds to anakinra treatment. Significance of enrichment is calculated using Fisher’s exact test.
(I) Pathway analysis of differential proteins in serum analysis between MIS-C (n = 3) and C.HD (n = 4).
NK cells show increased expression of cytotoxicity genes in MIS-C patients
To further define the T and NK cell states in MIS-C, we sub-clustered T and NK cells (Figures 4 A, 4B, S4A, and S4B). Increased frequencies were observed in regulatory T cells and proliferating (Ki67+) T and NK cells from MIS-C compared to C.HD (Figure 4C). We confirmed elevation of proliferating CD4+ T cells (Ki67+) cells in MIS-C by flow cytometry (Figure S4C). To assess the potential for a superantigen response among total T cells, we scored a defined signature of superantigen genes, which was not altered in MIS-C compared to C.HD (Figure S4E) (Grumann et al., 2008). However, differential gene expression analysis in NK cell subsets revealed a significant increase in PRF1, GZMA, and GZMH in MIS-C compared to C.HD, with high gene expression levels retained in recovered patients (Figures 4D–F and S4D). To validate the scRNA-seq findings, we assessed Granzyme A protein levels by intracellular flow cytometry staining and confirmed elevated levels in CD56bright NK cells from MIS-C patients compared to C.HD (Figure 4G). CCL4, produced by activated NK cells, was also increased in MIS-C (Figure S4G). Additionally, ITGB7, an integrin subunit supporting lymphocyte infiltration of the gut through MAdCAM-1 binding (Habtezion et al., 2016), was increased in memory CD8+ T cells (Figure S4H). TCR diversity analysis identified a subset of MIS-C patients with decreased non-naive CD4+ T cell clonal diversity, possibly indicating clonal expansion. However, no evidence of clonal expansion was seen in MIS-C CD8+ T cells. By contrast, COVID-19 patients exhibited lower TCR richness in CD4+ T cells (Figure S4I). Thus, NK cells, and to a lesser extent CD8+ T cells, exhibit elevated cytotoxicity features with potential relevance for tissue damage.
Figure 4.

Increased cytotoxicity signatures in NK cells from MIS-C patients
(A) T cell sub-clustering UMAP.
(B) Dot plot depicting key T and NK cell markers for cluster delineation.
(C) T and NK compositions across pediatric cohorts. A two-sided Wilcoxon test was calculated for statistical significance between cohorts.
(D) Heatmap representing top 20 up- and downregulated differentially expressed genes in NK cells between MIS-C and C.HD. Highlighted are genes associated with cytotoxicity. Scale bar represents the scaled average expression of markers.
(E and F) PRF1, GZMA, and GZMH expression in NK cell subsets in MIS-C compared to C.HD and MIS-C-R donors. Scaled average expression was calculated for each donor. Statistical significance was computed as in (C).
(G) Flow cytometric confirmation of key scRNA-seq data. Quantification of granzyme A in CD56bright NK cells stained for Granzyme A in MIS-C (n = 11) and C.HD (n = 6). Statistical significance was assessed using a two-sided unpaired t-test.
IgG plasmablasts are elevated in MIS-C
To investigate whether an ongoing humoral response could underpin acute MIS-C immunopathology, we sub-clustered annotated B cells (Figures 5A and 5B). In MIS-C, proliferating (Ki67+) plasmablasts were increased compared to C.HD, and expressed apoptosis genes consistent with a short-lived phenotype (Figures 5C, S5A, and S5B). An increase in proliferating plasmablasts has also been observed in severe COVID-19 (Bernardes et al., 2020). Naive B cells were also increased, and memory B cells reduced in MIS-C versus C.HD, which was confirmed by flow cytometric analysis of CD19+CD20+ B cells (Figure S5C). Differential expression analysis between naive B cells of MIS-C and C.HD showed an enrichment of the KEGG B cell signaling pathway among differentially upregulated genes (Figures S5D and S5E). Next, we assessed antibody isotype, clonotypic diversity, and somatic hypermutation (SHM) of B cell receptors (BCRs). In memory B cells, the proportion of IgM B cells was increased in MIS-C (Figure S5F). The proportion of plasmablasts expressing IgG1 or IgG3 was elevated in MIS-C (Figures 5D and S5G), and a smaller proportion of plasmablast IgG clones in MIS-C and COVID-19 patients harbored mutated BCR variable regions (defined as >1% nucleotides mutated relative to germline) compared to age-matched controls (Figure 5E) (Consiglio et al., 2020, Kaneko et al., 2020). Moreover, a subset of MIS-C patients exhibited lower BCR clonal diversity – consistent with clonal expansion – when compared to C.HD, though this relationship was not as consistent as that between COVID-19 patients and A.HD (Figure 5F).
Figure 5.

MIS-C patients have increased proliferating plasmablasts harboring IgG1 and IgG3 and a coordinated CD4+ T cell response
(A) B cell sub-clustering UMAP.
(B) Dot plots for key B cell markers delineating naive, memory, and plasmablast subsets.
(C) Distributions of B cell frequencies within total B cells across donors. Two-sided Wilcoxon rank sum tests were used to calculate significance.
(D) IGHG1 and IGHG3 isotype frequencies as a proportion of plasmablasts (proliferating and non-proliferating) are depicted across donors. Statistical significance was calculated as in (C).
(E) Proportion mutated IGHG clones in plasmablasts. Statistical significance was calculated as in (C).
(F) Simpson’s diversity in all B cells computed across cohorts in pediatric cohorts (top) and adult cohorts (bottom). Significance calculated as above. Statistical significance was calculated as in (C).
(G) (Top): Percentage dividing plasmablasts/total B cells versus percentage Ki67+CD4+ cells/total T cells within the MIS-C cohort using scRNA-seq. Ki67+ CD4+ cells defined as CD4+ cells within the Ki67+ NK and T cell cluster (see Figure 4A). (Bottom): Correlation of dividing plasmablasts (CD272+CD382+Ki67+) among CD19+ B cells and Ki67+ CD4+ T cells among CD3+ T cells assessed by flow cytometry. Statistical significance calculated by linear regression. 95% confidence interval is shown in gray shading.
(H) Heatmap showing differential gene expression across four subsets of CD4+ T cells, comprising samples from the MIS-C scRNA-seq cohort. Scale bar represents the scaled average expression of markers.
To examine potential drivers of this plasmablast response, we looked at correlates in the CD4+ T cell response. We found a possible association between proliferating Ki67+ CD4+ T cells and Ki67+ plasmablasts by scRNA-seq, which we confirmed as a significant correlation in our expanded flow cytometry cohort (Figure 5G). Gene expression analysis of Ki67+ CD4+ T cells revealed low CXCR5, but high ICOS, PDCD1, MAF, and IL21 as well as chemokine receptors for homing to inflamed tissue including CCR2, CX3CR1, and CCR5 (Figures 5H, S5H, and S5I). These cells appear to be phenotypically similar to T peripheral helper cells seen in some autoimmune conditions (Rao et al., 2017). Together, these data indicate that plasmablasts in MIS-C patients are expanded, correlate with proliferating CD4+ T cells with putative B cell-helper function, and more frequently harbor IgG1 and IgG3 antibody isotypes compared to C.HD.
Evidence for autoreactivity in severe MIS-C patients
Severe and moderate MIS-C patients are clinically distinct (Table S1). Thus, we hypothesized that some immunopathology features in MIS-C-S patients would be more pronounced than in MIS-C-M patients and stratified the subjects for further analysis. To begin to assess TCR repertoire skewing from prior exposures, possibly in response to previous SARS-CoV2 infection, we assessed memory T cell compartments using PCA of TRBV gene usage. Both CD4+ and CD8+ memory T cells exhibited significant skewing of the V-beta repertoire, with TRBV11-2 significantly enriched in MIS-C-S (n = 4) compared to C.HD (n = 6) or MIS-C-M (n = 2) in both compartments (Figures 6 A, S6A, and S6B). COVID19-A and COVID19-B memory T cells did not exhibit a separation from A.HD (Figures S6C and S6D), possibly indicating a specific skewing event in the memory compartment in MIS-C. Moreover, effector CD8+ T cells exhibited increased gene expression of PRF1 and GZMA when comparing MIS-C-S with C.HD (Figure 6B). As confirmation, we observed a significant increase of Granzyme A by flow cytometry in the CD45RA+ CD8+ T-effector memory (TEMRA) compartment of MIS-C-S patients (Figure 6C). LAG3, an inhibitory receptor that shows enhanced expression upon T cell activation, was also increased in effector CD8+ T cells (Figure S6E). Based on data in Figure 5G demonstrating a coordinated CD4+ T cell and plasmablast response in some MIS-C patients, we assessed these associations further in MIS-C-S. We found that patients with low B cell clonal diversity also had low combined Ki67+ and CD4+ memory T cell diversity, suggestive of coordinated clonal expansion (Figure 6D). Furthermore, in MIS-C-S, we found increased plasmablast frequencies, decreased total B cell clonal diversity, and an increased proportion of mutated IgG clones, consistent with a more robust B cell response in these patients (Figure 6E). In addition, the proportion of IGHG1- and IGHG3-plasmablasts was higher in MIS-C-S patients compared to controls (Figure S6F).
Figure 6.

Distinct features of severe versus moderate MIS-C
(A) PCA of TRBV usage in CD4+ and CD8+ memory cells, along with frequency of TRBV11-2 usage, in the pediatric cohort. Statistical significance for PCA calculated by permutation test, and by one-sided Wilcoxon test for TRBV11-2 frequency comparisons.
(B) PRF1 and GZMA expression in effector memory CD8+ T cells along with dot plot depicting relative average expression and percent expression for four cytotoxicity genes (right). Two-sided Wilcoxon rank sum tests were used to calculate significance.
(C) Flow cytometric evaluation of Granzyme A in TEMRA CD8+ compartment in C.HD (n = 6), MIS-C-S (n = 7), and MIS-C-M (n = 5) patients. Statistical significance was assessed using an ordinary one-way ANOVA test.
(D) Correlation between BCR diversity and TCR diversity relating to combined Ki67+ and memory CD4+ T cells. P7.1 was excluded from TCR analysis due to low cell numbers (see STAR Methods).
(E) B cell diversity, plasmablast frequency, and proportion of mutated IGHG within MIS-C cohort.
(F) Serum E-selectin in pediatric healthy and MIS-C donors. Statistical significance was calculated as in (B).
(G) Median fluorescence intensity (normalized to average HD) of serum IgG binding to cultured human cardiac microvascular endothelial cells (HCMEC) by flow cytometry (left). A non-parametric two-sided Wilcoxon rank sum test was used to calculate significance. Error bars represent mean with SD. MIS-C-S (acute n = 3; P1-3); MIS-C-M (acute n = 2; P4-5 and recovered n = 1; P11); and HD (n = 6; 1 C.HD and 5 A.HD). Representative histogram of a patient representing the median of the MIS-C-S cohort and sampled prior to IVIG treatment (right).
Given the distributive and cardiogenic shock in MIS-C, we aimed to investigate endothelial cell involvement. Further analysis of serum proteomics data from Figure 3H showed that endothelial E-selectin, a molecule known to be expressed on inflamed endothelial cells, was also elevated in MIS-C-S serum (Figure 6F) (Ley and Kansas, 2004). Next, we assessed a possible autoantibody response directed at endothelial cells. We examined binding of MIS-C serum antibodies to activated human cardiac microvascular endothelial cells (HCMEC) in culture. Indeed, IgG from acutely ill severe (P1.1, P2.1, P3.1, the latter being pre-IVIG) but not moderate (acutely ill P4.1 and and P5.1 or recovered P11) MIS-C patients bound activated endothelial cells (Figure 6G), consistent with a potential autoimmune process. Thus, T and B cell clonal expansion, as well as cytotoxic gene expression signatures in CD8+ T cells, appear to be associated with severe MIS-C. Additionally, we provide functional evidence for MIS-C autoantibody binding to activated endothelial cells relevant for severe disease pathology.
Discussion
Our comprehensive analysis of MIS-C patients using single-cell RNA sequencing, antigen receptor repertoire analysis, serum proteomics, and in vitro assays provided in-depth data on disease immunopathology. Separation of MIS-C into moderate and severe groups based on clinical criteria uncovered signals of disease pathogenesis that otherwise would not have emerged. In innate cells, reduced HLA class II and CD86 expression, molecules involved in antigen presentation to T cells, point to a compensatory post-inflammatory feedback response or a dysregulated innate response to inflammation, though underlying altered antigen presenting function cannot be excluded. Elevated alarmin genes—including subunits of calprotectin (S100A8 and S100A9) and EN-RAGE (S100A12)—together with increased acute-phase and myeloid-derived inflammatory proteins and high endothelial E-selectin, suggest inflammatory amplification and damage as important disease correlates.
What drives the cytokine storm and multi-organ damage in MIS-C? In addition to possible innate drivers described above, our analysis of lymphocytes from MIS-C patients points to three main findings. First, NK cells and, to a lesser extent, CD8+ T cells expressed elevated perforin, granzyme A, and granzyme H. These cytotoxic molecules are relevant for tissue damage. In contrast to granzyme B, granzyme A is known to cleave pro-IL-1 and may directly contribute to inflammation beyond its cytotoxic function (Hildebrand et al., 2014, Zhou et al., 2020). Second, B cells had an expansion of proliferating plasmablasts that fits with a potential humoral response often weeks after clearance of SARS-CoV-2, raising the possibility that these are autoreactive expansions of antibody-secreting cells. Third, evaluation of severe MIS-C patients showed evidence of clonal expansion and somatic hypermutation within B cell populations, and measurable binding of serum IgG to activated cardiac endothelial cells in culture. The plasmablasts expanded in MIS-C showed evidence of being short-lived with upregulated pro-apoptotic genes, which may help explain the self-resolving nature of pathology. Collectively, our data support a model in which prior SARS-CoV-2 infection causes lasting immune alterations that set the stage for development of an acute and life-threatening post-infectious inflammatory episode in a fraction of children and adolescents.
The relative rarity of MIS-C suggests three main possibilities to explain the onset of disease. First, a rare genetic predisposition could underlie disease, and future genomics investigations will be revealing on this front. Second, similarly to rheumatic heart disease, the infectious trigger could elicit adaptive immune responses that, on rare occasion, cross-react with self-antigens. The rapid resolution of inflammation in MIS-C is evidence that, if autoimmunity drives pathology, it is transient and perhaps mediated by short-lived immune cell populations. Third, a rare combination of SARS-CoV-2 infection followed by a second microbial trigger could drive the acute MIS-C inflammatory episode. We did not find evidence of herpesvirus reactivation or peripheral blood signatures of ongoing viral or bacterial infections. We did, however, find evidence of a skewed TCR repertoire with significantly increased representation of TRBV11-2 in both CD4+ and CD8+ memory T cells in severe MIS-C patients. This skewing in the memory population may suggest relevance of exposure to a superantigen motif (Cheng et al., 2020). Alternatively, TRBV11-2 has been identified among autoreactive T cells with non-classical MHC restriction, raising the possibility that non-peptide antigens could be relevant for MIS-C pathology (Guo et al., 2018). Nonetheless, a tissue-specific response to an infectious trigger remains plausible, and the common feature of abdominal pain early in the course of MIS-C is suggestive of potential gut involvement and consistent with elevated ITGB7 in T cell subsets. Further work is required to define contributions of each of these potential triggers.
The determinants of whether a child with MIS-C develops moderate or severe disease are also unknown and may relate to prior SARS-CoV-2 viral load and immune repertoire shaping and/or differences in the putative secondary MIS-C-triggering event. Although patients with severe disease have a potential autoantibody response as measured by IgG binding to cultured endothelial cells, whether this is causative of severe disease or a result of increased tissue destruction and autoantigen exposure cannot currently be determined. Our findings in MIS-C have important implications for diagnostic and prognostic testing. Specifically, the alarmin, cytotoxicity, TCR repertoire, and plasmablast signatures we define have potential for application in the clinic to better diagnose and potentially predict severity early in the course of MIS-C. With new waves of SARS-CoV-2 outbreaks on the horizon and eventual vaccination to protect children from SARS-CoV-2-related disease as a critical goal, a better understanding of MIS-C drivers and immunopathology is urgently needed. Our data implicate innate and adaptive immune triggering with direct relevance for tissue destruction during acute MIS-C.
Limitations of the study
Our study has some limitations, and further investigations are required to definitively delineate factors that are causative and not a consequence of MIS-C pathology. Patients included in this study were severely ill and treated with immunomodulators, both of which could impact peripheral gene expression signatures despite our approaches to mitigate their impact (Franco et al., 2019). The single-cell RNA sequencing and serum proteomics techniques employed here are powerful and illuminating. Nonetheless, important next steps are to increase the number of analyzed MIS-C patients, determine functional consequences of the signatures we discovered in MIS-C using in vitro functional assays (e.g., measuring cytotoxicity and proliferation), and define the antigens recognized by expanded TRBV11-2-expressing T cells and IgG1/IgG3-expressing plasmablasts.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD38 APC-Cy7 | BioLegend | Cat. 303534; RRID:AB_2561605 |
| Anti-human CD20 BV650 | BioLegend | Cat. 302336; RRID:AB_2563806 |
| Anti-human CD27 PE-Cy7 | BioLegend | Cat. 356412; RRID:AB_2562258 |
| Anti-human CCR7 BV421 | BioLegend | Cat. 353208; RRID:AB_11203894 |
| Anti-human CD45RA FITC | BioLegend | Cat. 304106; RRID:AB_314410 |
| Anti-human Ki-67 BV510 | BioLegend | Cat. 350518; RRID:AB_2563862 |
| Anti-human Granzyme A PE | BioLegend | Cat. 507206; RRID:AB_315472 |
| Anti-human S100A9 PE | BioLegend | Cat. 350706; RRID:AB_2564008 |
| Anti-human CD138 BB700 | BD Biosciences | Cat. 745787; RRID:AB_2743244 |
| Anti-human CD8 BV510 | BD Biosciences | Cat. 563256; RRID:AB_2738101 |
| Anti-human CD11c PE-Cy7 | BD Biosciences | Cat. 561356; RRID:AB_10611859 |
| Anti-human CD14 BV650 | BioLegend | Cat. 301836; RRID:AB_2563799 |
| Anti-human CD16 AF700 | BD Biosciences | Cat. 557920; RRID:AB_396941 |
| Anti-human CD1c BB515 | BD Biosciences | Cat. 565054; RRID:AB_2716870 |
| Anti-human CD123 PerCP-Cy5.5 | BD Biosciences | Cat. 558714; RRID:AB_1645547 |
| Anti-human CD141 PE | Miltenyi Biotec | Cat. 130-113-318; RRID:AB_2726095 |
| Anti-human CD15 FITC | BD Biosciences | Cat. 347423; RRID:AB_400299 |
| Anti-human HLA-DR APC | BioLegend | Cat. 307610; RRID:AB_314688 |
| Anti-human CD86 BV421 | BD Biosciences | Cat. 562432; RRID:AB_11153866 |
| Total-seq C human cocktail | BioLegend | Custom cocktail, see Table S2 |
| Human BD Fc Block | BD Biosciences | Cat. 564219; RRID:AB_2728082 |
| F(ab’)2-Goat anti-human IgG: Alexa Fluor 549 | Invitrogen | Cat. A-11014; RRID:AB_2534081 |
| Experimental models: cell lines | ||
| HCMEC | Lonza | Cat. CC-7030 |
| Critical commercial assays | ||
| Chromium Next GEM SingleCell V(D)J Reagent Kits (v1.1 Chemistry) | 10x Genomics | PN-1000165 |
| Chemicals, peptides and recombinant proteins | ||
| Ficoll-Paque PLUS | GE Healthcare | Cat. GE17-1440-02 |
| Lymphoprep | STEMCELL Technologies | Cat. 07851 |
| Complete RPMI 1640 (cRPMI) medium | Lonza | Cat. BE12-752F |
| 2 mM Glutamine | Invitrogen | Cat. 25030149 |
| Penicillin and streptomycin | Invitrogen | Cat. 15070063 |
| EGM2 MV Microvascular Endothelial Cell Growth Medium-2 | Lonza | Cat. CC-4147 |
| Hank’s Balanced Salt Solution | GIBCO | Cat. BE10-508F |
| 4% Formaldehyde solution | BioLegend | Cat. 420801 |
| Gelatin veronal buffer | Sigma | Cat. G6514 |
| True-Stain Monocyte Blocker | BioLegend | Cat. 426101 |
| Dulbecco’s Phosphate-Buffered Saline | Corning | Cat. 20-031-CV |
| LIVE/DEAD™ Fixable Red Dead Cell Stain Kit | ThermoFisher Scientific | Cat. L34973 |
| Brilliant Stain Buffer | BD Biosciences | Cat. 563794 |
| Fixation Buffer (BD Cytofix) | BD Biosciences | Cat. BDB554655 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat. 00-5523-00 |
| Recombinant IFNg | PeproTech | Cat. 300-02 |
| Recombinant TNFa | Invitrogen | PHC3013 |
| Software and algorithms | ||
| CellRanger | Zheng et al., 2017 | v3.1.0. |
| Flowjo | Flowjo | v10 |
| Seurat | Stuart et al., 2019 | v3.2.1 |
| IgBlast | Ye et al., 2013 | v1.13.0 |
| Change-O | Gupta et al., 2015 | v1.0.0 |
| Shazam | Gupta et al., 2015 | v1.0.2 |
| Alakazam | Gupta et al., 2015 | v1.0.2 |
| AUCell | Aibar et al., 2017 | v1.12.0 |
| SingleR | Aran et al., 2019 | v1.4.0 |
| Connectome | Raredon et al., 2019; Raredon et al., 2021 | v0.2.2 |
| Deposited data | ||
| Raw and processed data for pediatric samples and select adult controls | This paper | GEO: GSE166489; FASTgenomics. See Table S4 for specific samples used |
| Raw and processed data for COVID-19 samples and select adult controls | Unterman et al., 2020 | GEO: GSE155223. See Table S4 for specific samples used. |
| Raw and processed data for select adult controls | Pappalardo et al., 2020 | dbGAP: phs002222. See Table S4 for specific samples used. |
| Other | ||
| Fortessa Flow Cytometer | BD Biosciences | Cat. 649225 |
| Illumina Novaseq 6000 platform | Illumina | Cat. 20012850 |
Resource availability
Lead Contact
Further information and requests can be directed to Carrie L. Lucas at Carrie.Lucas@yale.edu.
Materials availability
This study did not generate new and unique reagents.
Data and code availability
Raw and processed data pertaining to pediatric and select adult healthy control samples (A.HD1-3) in this paper are available at GEO: GSE166489 and FASTgenomics. Data pertaining to adult COVID-19 and additional adult healthy control samples are available from previous studies: Pappalardo et al. dbGAP: phs002222 and Unterman et al. GEO: GSE155223 (Pappalardo et al., 2020, Unterman et al., 2020). Code is made available in https://github.com/LucasiteLab/MIS-C_scRNAseq and FASTgenomics. Additional supplemental items are available from Mendeley Data at https://doi.org/10.17632/v9dkw64s6c.3.
Experimental model and subject details
All human subjects in this study provided informed consent in accordance with Helsinki principles for enrollment in research protocols that were approved by the Institutional Review Board of Yale University. Patients were enrolled from Yale New Haven Children’s Hospital (New Haven, CT) and Loma Linda Children’s Hospital (Loma Linda, CA). Blood from healthy donors was obtained at Yale under approved protocols. Select adult healthy control samples (A.HD8-13) were obtained from a previous study (Pappalardo et al., 2020). Further information about subjects can be found in Table S4.
COVID-19 samples
COVID19-A, COVID19-B, and select adult healthy control (A.HD4-7) samples were provided by Unterman et al., 2020 (Unterman et al., 2020). In our study, we included samples from six adult COVID-19 patients (patient characteristics summarized in Table S4). Blood was drawn at two time points during hospitalization (time point A and B), with a median time elapsed between time points of 4 days. Two samples, A.COV5, and A.COV6, only correspond to the latter time point B.
Method details
Blood sample processing
Human PBMCs were isolated by Ficoll-Paque PLUS (GE Healthcare) or Lymphoprep (STEMCELL Technologies) density gradient centrifugation, washed twice in PBS, and resuspended at 106 cells/mL in complete RPMI 1640 (cRPMI) medium (Lonza) containing 10% FBS, 2 mM glutamine, and 100 U/mL each of penicillin and streptomycin (Invitrogen). PBMCs were used fresh or cryopreserved in 10% DMSO in FBS and thawed prior to use. Serum was isolated by centrifugation of serum tubes and saving the supernatant in aliquots which were flash frozen in liquid nitrogen prior to cryopreservation in −80C.
Single-cell RNA-sequencing
Cryopreserved PBMCs were thawed in a water bath at 37°C for ∼2 min without agitation, and removed from the water bath when a tiny ice crystal still remains. Cells were transferred to a 15 mL conical tube and the cryovial was rinsed with growth medium (10% FBS in DMEM) to recover leftover cells, and the rinse medium was added dropwise to the 15 mL conical tube while gently shaking the tube. Next, growth medium was added at a speed of 3-5 mL/sec, achieving a final volume of 13 mL.
Fresh or thawed PBMCs were centrifuged at 400 g for 8 min at RT, and the supernatant was removed without disrupting the cell pellet. The pellet was resuspended in 1X PBS with 0.04% BSA, and cells were filtered with a 30 μM cell strainer. Cellular concentration was adjusted to 1,000 cells/μL based on the cell count and cells were immediately loaded onto the 10x Chromium Next GEM Chip G, according to the manufacturer’s user guide (Chromium Next GEM SingleCell V(D)J Reagent Kits v1.1). We aimed to obtain a yield of ∼10,000 cells per lane.
For CITE-seq staining, lyophilized Total-seq C human cocktail (BioLegend) (Table S2) was resuspended with 35 μL of 2% FBS in PBS vortexed for 10 s and incubated for 5 min at RT. To pellet the aggregated antibodies, rehydrated antibody cocktail was centrifuged at 20,000 g for 10 min just before adding to the cells. PBMCs were resuspended with wash buffer at the concentration of 10-20 × 106 cells/mL, and 0.5 × 106 cells were used for further staining. Cells were incubated on ice for 10 min with 5ul of Human Fc block and 5 μL of TrueStain Monocyte Blocker (Biolegend). Next, 10-20 μL (0.1-0.2 × 106 cells) were aliquoted into a new tube and incubated on ice for 30 min with 5 μL of Total-seq C antibody cocktail prepared as above. Cells were washed twice with wash buffer and third wash was with 2% FBS in PBS, then resuspended in 1X PBS with 0.04% BSA at 1,000 cells/μL and loaded onto the 10x Chromium Chip G, as described above.
cDNA libraries for gene expression, CITE-seq, and TCR/BCR sequencing were generated according to manufacturer’s instructions (Chromium Next GEM SingleCell V(D)J Reagent Kits v1.1). Each library was then sequenced on an Illumina Novaseq 6000 platform. The sequencing data was processed using CellRanger v3.1.0 (Zheng et al., 2017).
PBMC single-cell RNA sequencing analysis
Pediatric healthy donor, MIS-C, longitudinal recovered MIS-C, adult healthy donor, and adult COVID-19 PBMC CellRanger outputs were analyzed using the Seurat v3.2.1 package (Stuart et al., 2019). These data were filtered, log-normalized, integrated, and scaled prior to dimensionality reduction and cluster identification. For each dataset, we filtered out genes that were expressed in fewer than 5 cells, and we removed low quality cells which have over 10% mitochondrial gene content and contain fewer than 200 features. To remove batch- and single-donor effects, we integrated all 38 samples into one dataset using Seurat’s reference-based anchor finding and integration workflow, which is recommended by Seurat for integrating large numbers of datasets. We chose an adult healthy donor sample (A.HD3) and a MIS-C patient sample (P1.1) as references for anchor finding and integration, and used 2000 anchors and the first 30 principal components (PCs) for the integration steps. To reduce dependence of clustering on cell-cycle heterogeneity, we scored cells for cell-cycle phase based on a defined set of phase-specific genes and regressed out these genes during the scaling step.
Principal component analysis was performed on the scaled dataset. To define the number of principal components (PCs) to use we applied the elbow plot method and we also tested different numbers of PCs to evaluate the effects on the separation of distinct cell lineages. Based on these determinations, we chose the first 30 PCs for nearest neighbor identification and a clustering resolution of 1.0 for cluster finding. Finally, we chose UMAP as a non-linear dimensionality reduction approach to visualize clusters. To define clusters, we calculated differentially expressed genes specific to each cluster using Wilcoxon rank sum test. Cluster specific markers were found that had an absolute logFC of at least 0.25, an adjusted p value of less than 0.05, and were expressed in a minimum of 25% of cells in either cluster being compared.
Dead and dying cell clusters were identified as those with high mitochondrial gene content, low number of unique genes, and mitochondrial genes as the top cluster-specific differentially expressed genes. After removing cells belonging to these clusters, the data was re-processed using the same parameters above. Clusters were annotated using cluster specific differential expression and SingleR (v1.4.0) (Aran et al., 2019). All scRNA-seq analysis was done using R version 4.0.2 (R Core Team, 2020). All scRNA-seq plots were done using ggplot2 v3.3.2 (Wickham, 2016).
CITE-seq analysis
Of the 38 samples, 5 included CITE-seq data. After integrating all of the datasets in both the PBMC and sub-clustering analyses, we used a subset of our Seurat object corresponding to these 5 donors, and overlaid ADT information onto the GEX-based UMAP for cluster validation. ADT data was log-normalized prior to plotting feature counts.
Connectivity mapping
The Connectome v0.2.2 package was used to generate a network analysis of ligand-receptor interactions predicted to be up- or downregulated in MIS-C compared to C.HD (Raredon et al., 2019, Raredon et al., 2021). PBMC clusters were included that were represented in both MIS-C and C.HD groups. We excluded clusters containing doublets and clusters where the sum of cells was fewer than 75 cells in either MIS-C or C.HD. To minimize differences in connectivity due to cell compositions between cohorts, we down-sampled our dataset. Specifically, for each cluster, we computed the sum of cells belonging to MIS-C patients or C.HD, and used the minimum of the two values to randomly sample cells within MIS-C or C.HD in the relevant cluster.
To annotate ligands and receptors, we used a list of annotated human ligand-receptor pairs sourced from the FANTOM5 database appended with immunological ligands and receptors created in a recent scRNA-seq study (Unterman et al., 2020). Connectomes were created for each down-sampled cohort. An edge is determined as a ligand-receptor pair that is expressed in respective clusters at a level greater than 10% of cells, and edge-weights are determined as the product of normalized expression values of the markers (Raredon et al., 2019, Raredon et al., 2021). The two connectomes were then compared to create a fold-change connectome, and this was then filtered to only differentially expressed genes between MIS-C and C.HD. An absolute logFC cutoff > 0.1 was employed for differential expression testing. Finally, to visualize ligand-receptor interactions that are upregulated in MIS-C, ligand and receptor interactions were plotted where both ligand and receptor connectome logFCs > 1.
EBV/CMV analysis
To evaluate EBV or CMV infection of individuals in our cohort, we created combined human-viral genome references to align transcriptomic reads and counted the number of detected viral transcripts (Young et al., 2007, Balázs et al., 2017). To be as permissive as possible, we used CellRanger to map reads to entire viral genomes, to capture counts originating from ORFs, intergenic regions, or initial infection.
Sub-clustering analysis
Sub-clustering was done on myeloid cells, T and NK cells, and B cells. For T and NK sub-clustering the following clusters were selected: CD4+ memory, CD4+ naive I, CD56dimCD16bright NK, CD8+ naive, CD4+ naive II, CD8+ memory, γδT cells, MAIT and NKT cells, Regulatory T cells, CD4+ and CD8+ mixed naive T cells, CD56brightCD16dim NK, Activated memory T cell, Proliferating T and NK cell, NK-T doublets. For myeloid sub-clustering the following clusters were selected: Classical monocytes, Neutrophils, Non-classical monocytes, Platelets, Platelet-T cell doublets, Conventional DC, Platelet-bound monocytes, Plasmacytoid DC, NK-monocyte doublets. For B cell sub-clustering the following clusters were selected: Naive B, Memory B, Plasma cell, T-NK-B cell doublets. After selecting relevant clusters from the PBMC annotations, we performed the analysis as described above. The same references used to generate PBMC UMAP were applied in the reference-based integration for T and NK cell sub-clustering. For B cell sub-clustering integration, we added an additional reference (C.HD4) due to the unequal donor representation in the activated memory B cell cluster.
For B cell sub-clustering, the first 15 PCs were used for data integration and downstream steps, along with a clustering resolution of 0.3. A.COV5.2 was unable to be integrated into the B cell sub-clustering analysis due to low cell numbers (< 200 B cells) and was removed from this analysis. For T and NK cell sub-clustering, 30 PCs were used for data integration, and 8 PCs for downstream steps, and a clustering resolution of 0.9 was used. For myeloid sub-clustering, 30 PCs were used for data integration, and 15 PCs were used for downstream steps with a clustering resolution of 0.5.
Clusters were annotated as above. Doublet clusters were determined by co-expression of heterogeneous lineage markers (e.g., MS4A1 and CD3D) and nFeature and nCount distribution. Clusters of dead and dying cells were identified as above. Both of these classes of clusters were removed prior to finalizing the UMAPs.
Cell-type proportion plots
To calculate cell frequencies based on single-cell data, we tabulated donor cells in each cluster, and divided these by the total donor representation in the UMAP. Because of the inherent heterogeneity in our cohorts, a non-parametric two-sided Wilcoxon rank-sum test was used to calculate statistical significance between MIS-C and C.HD.
DEG analysis and heatmaps
Differentially expressed genes were computed between cohorts using the FindMarkers function in Seurat, using the same test and parameters as described above for clusters-specific marker delineation. We used a broader categories of cells to compute differential expression as follows: Monocytes (Classical monocytes I, Classical monocytes II, Classical monocytes III, Intermediate monocytes, Non classical monocytes), Neutrophils (Neutrophils I, Neutrophils II), NK cells (CD56dimS100A4+ NK cells, CD56dimCD38+ NK cells, CD56bright NK cells), CD8+ memory (Effector memory CD8+ T cells, Central memory CD8+ T cells, Terminal effector memory CD8+ T cells), Naive CD4+ (Naive CD4+ T cells I, Naive CD4+ T cells II, Naive CD4+ T cells III), Memory CD4+ T cells (Memory CD4+ T cells, CCR6+ memory CD4+ T cells, CXCR3+ memory CD4+ T cells), Naive B cells (Naive B, Activated naive B), Memory B cells (Intermediate memory, Activated memory B, Memory B cell), Plasmablast (Non-dividing plasmablasts, Dividing plasmablasts).
To prioritize genes for analysis, we chose an absolute average log fold-change (logFC) cutoff of an absolute value > 0.5, and a p-adjusted value < 0.05. The top 20 up- or downregulated genes, sorted by average logFC, were chosen to plot onto heatmaps. Heatmaps were visualized using the ComplexHeatmap v2.5.5 package. Correlation heatmaps were created using Hmisc v4.4-1 and corrplot v0.84.
Module scores
Module scores were calculated using the AddModuleScore function using the default parameters (Tirosh et al., 2016), and where indicated, enrichment was calculated using AUCell (v1.12.0) (Aibar et al., 2017). The S100A score consists of S100A8, S100A9, and S100A12. The HLA class II score consists of HLA-DRB1, HLA-DRB5, HLA-DRA, HLA-DQA1, HLA-DQA2, and HLA-DQB1. The super-antigen score includes the following genes: IL2, CXCL9, UBD, IFNG, CXCL11, IL22, ANKRD22, IL17A, IL31RA, FAM26F, CXCL1, IL3, SLAMF8, LOC729936, XCL1, XCL2, SERPING1, SUCNR1, IL27, APOL4, FCGR1A, FCGR1B, SECTM1, CCL8, IL17F, BATF2, GBP4, and ETV7 (Grumann et al., 2008). The viral score shown in Figure 2D consists of SIGLEC1, RABGAP1L, IFI27, CADM1, RSAD2, MX1, and SERPING1 (Lydon et al., 2019). The viral score shown in Figure S2G consists of IFI44L, IFI27, RSAD2, SIGLEC1, IFIT1, and ISG15 (Hadjadj et al., 2020). The bacterial score consists of SMPD1, CD44, SERPING1, SPI1, HERC1, MCTP1, FOLR3, CFAP45, PRF1, CTBP1, HLA-DRB1, ARL1, OAS3, ZER1, CHI3L1, IFIT2, and IFITM1 (Lydon et al., 2019). The sepsis score consists of PLAC8, CLU, RETN, CD63, ALOX5AP, SEC61G, TXN, and MT1X (Reyes et al., 2020).
Pathway analysis
The MIS-C patients were acutely ill and treated with high-dose steroids, which are known to affect immune gene expression programs. Prior to performing pathway analysis, we sought to remove genes that were clearly affected by steroid effects. As a reference, we used a publicly available bulk RNA-seq dataset of PBMC cell-types treated with methylprednisolone, the primary steroid administered to the MIS-C patients (Franco et al., 2019). We removed steroid related genes relevant to each cell type from our gene lists, defined as having an absolute logFC cutoff above 2 and p value less than 0.05 in the steroid dataset and regulated in the same direction as genes in our dataset. Pathway analysis was then done on differentially expressed genes using the above criteria and filtering for steroid-related genes. These differentially expressed genes were inputted to Enrichr (Chen et al., 2013, Kuleshov et al., 2016) to calculate enrichment of pathway-associated terms.
BCR analysis
B cell receptor (BCR) repertoire sequence data were analyzed using the Immcantation (www.immcantation.org) framework. Starting with CellRanger output, V(D)J genes for each sequence were aligned to the IMGT reference database v3.1.30 (Giudicelli et al., 2005) using IgBlast v1.13.0 (Ye et al., 2013). Nonproductive sequences were removed. Within each sample, sequences were grouped into clonal clusters, which contain B cells that relate to each other by somatic hypermutations from a common V(D)J ancestor. Sequences were first grouped by common IGHV gene annotations, IGHJ gene annotations, and junction lengths. Using the DefineClones.py function of Change-O v1.0.0 (Gupta et al., 2015), sequences within these groups differing by less than a length normalized Hamming distance of 0.15 within the junction region were defined as clones using single-linkage hierarchical clustering (Gupta et al., 2017). This threshold was determined by manual inspection of the distance to nearest sequence neighbor distribution for each sample using Shazam v1.0.2. These heavy-chain-defined clonal clusters were further split if their constituent cells contained light chains that differed by V and J genes. Within each clone, germline sequences were reconstructed with D segment and N/P regions masked (replaced with “N” nucleotides) using the CreateGermlines.py function within Change-O v1.0.0. All BCR analyses used R v3.6.1.
For analysis of B cell clonal diversity, we calculated Simpson’s diversity for each sample using the alphaDiversity function of Alakazam v1.0.2 (Gupta et al., 2015). To account for differences in sequence depth, samples within each comparison were down-sampled to the same number of sequences, and the mean of 100 such re-sampling repetitions was reported. Only samples with at least 100 B cells were included.
To identify mutated B cell clones of different cell types and isotypes, B cell clones were further separated by cell type and/or isotype. For all BCR analysis, unless otherwise indicated, “plasmablasts” indicate pooled dividing- and non-dividing annotated plasmablasts defined by sub-clustering annotation and filtered on cells containing BCRs. These B cell clones were considered “mutated” if the median somatic hypermutation frequency of their constituent sequences was ≥1%. This threshold is consistent with recent analyses of COVID-19 B cell repertoires (Unterman et al., 2020, Nielsen et al., 2020).
TCR analysis
The raw sequencing reads were preprocessed using the Cell Ranger V(D)J pipeline by 10X Genomics, which assembled read-pairs into V(D)J contigs for each cell, identified cell barcodes from targeted cells, annotated the assembled contigs with V(D)J segment labels and located the CDR3 regions. Only V(D)J contigs with high confidence defined by cell ranger were included for downstream analysis. These V(D)J contigs were re-annotated by aligning them to the IMGT reference database v3.1.3041 using IgBlast v1.13.0 in the Change-O V1.0.0 (Gupta et al., 2015) pipeline. Cells with ambiguous alpha chain or beta chain and cells with no beta chains were removed. For cells with multiple alpha and/or beta chains, if any chain could be captured, we retained the chain with the largest nUMI that provided a unique chain. No sample to sample contamination was identified based on across-cell overlap of cell barcodes and contig sequences.
For analysis of T cell clonal diversity, in each sample, cells with identical alpha chain and beta chain sequences in the repertoire were grouped as one TCR clone. To quantify the clonal diversity, we calculated both Simpson and Shannon diversity for each sample using R package Alakazam 1.0.2 (Gupta et al., 2015) which describes the richness and evenness of the repertoire, respectively. When comparing the diversity between samples, to account for the differences in sequencing depth across different samples, down-sampling was conducted to make different samples have the same number of sequences. For each diversity comparison, samples with less than 50 sequences were excluded and all samples were randomly down sampled for 100 times to the smallest number of sequences of the remaining samples. The mean diversity across the 100 times were reported and compared.
We selected memory CD4+ and CD8+ T cells; naive CD4+ and CD8+ T cells and NK cells for downstream analysis. We separated Ki67 + cells into CD4+ and CD8+ categories and combined these with the memory CD4+ and CD8+ cells, respectively.
The principal component analysis (PCA) was performed on TRBV gene frequency within each individual using R package stats v4.0.2. Statistical significance of PCA clustering was determined by permutation test, where the statistic was the ratio of mean intra/inter cluster Euclidean distances among points. Mean values of the fraction of each TRBV gene per sample with error bars were used to demonstrate the differential gene usage. TRBV genes were sorted according to the difference between MIS-C-S and C.HD in descending order.
Serum antibody binding to cultured endothelial cells
De-identified and discarded high-titer panel reactive antigen (PRA) sera showing > 80% reactivity to HLA Class-I and II antigens were collected from transplant patients at Yale New Haven Hospital’s tissue typing laboratory. Healthy donor and moderate and severe MIS-C patient serum was isolated by centrifugation of serum collection tubes at 840 g for 10 min. Supernatant was flash frozen in liquid nitrogen and stored in −80C prior to being thawed for experiments. To induce in situ levels of HLA antigens expression, 60% confluent human cardiac microvascular endothelial cells (HCMECs, Lonza) were pre-treated with human recombinant IFN-γ (final concentration of 100 units/mL, 48 h, Invitrogen) and TNF-α (final concentration of 10 ng/mL for 6-8 h, Invitrogen) in EGM2 MV Microvascular Endothelial Cell Growth Medium-2 (Lonza CC-4147). Cells were then washed with HBSS (GIBCO) and treated with PRA, HD or MIS-C serum in a 1:1 ratio with gelatin veronal buffer (GVB, Sigma) for 2 h. Untreated cells were washed with HBSS (GIBCO) followed by addition of GVB for 2 h. To assess antibody binding, cells were suspended in trypsin, washed in 1% BSA PBS (FACS buffer) and pelleted by centrifugation at 1000 rpm. The cells were then incubated with goat anti-human IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 594 (ThermoFisher, Catalog # A-11014) at 1:400 dilution with FACS buffer. Unbound antibodies were removed by washing three times with 1xPBS (GIBCO) followed by fixing with 4% paraformaldehyde solution (BioLegend Cat. 420801) at room temperature. Cells were washed twice in FACS buffer and resuspended in 200-300 μL FACS buffer for flow cytometric analysis.
Serum protein analysis
Relative serum protein levels were quantified by the SOMAscan v4 platform (Somalogic Inc., Boulder CO), which measures the binding of 5,284 modified single-stranded DNA aptamers (SOMAmers) to specific analytes in each sample (Gold et al., 2010). The assay and characteristics of the reagents have been described before (Emilsson et al., 2018). Briefly, 120 μLl aliquots of serum samples from three MIS-C patients were placed across two 96-well plates with four age- and gender-matched C.HD, independent controls, and other samples not analyzed in this study. C.HD used in serum analysis were distinct from C.HD used in scRNA-seq analysis. All samples were sent together on dry ice and assayed by Somalogic. Measurements were standardized using the default method performed by the manufacturer to first account for hybridization and assay bias within plates, followed by plate scaling and calibration to remove plate effects, and median normalization to a reference at the end. Based on a subset of 4,706 human protein analytes (representing 4,478 distinct proteins) that passed quality control, the median intra-subject coefficient of variation (CV) of 3.30% from eight blinded technical replicates across two plates suggests low technical variability.
Serum connectivity networks were created by selecting the top 40 differentially upregulated proteins in MIS-C compared to C.HD, and mapping them to receptors expressed in PBMC using the ligand-receptor reference used previously. We defined a receptor as being expressed in PBMCs if it is expressed in at least 25% of cells in any cluster.
Flow cytometry
Fluorochrome-conjugated antibodies against the following antigens were used for PBMC staining: CD38, IgD, CD3, CD4 and CD56 (BioLegend), CD138, CD8, CD11c, CD14, CD16, CD1c, CD123, CD141, CD15, HLA-DR and CD86 (Becton Dickinson), CD19, CD20, CD27, CCR7 and CD45RA (BioLegend). Human BD Fc Block (Becton Dickinson) and True-Stain Monocyte Blocker (BioLegend) were used in all preparations to avoid nonspecific staining.
In brief, after thawing cells were left 30 min in the incubator, then centrifuged and resuspended in Dulbecco’s Phosphate-Buffered Saline (DPBS, Corning), containing Fc Block, True-Stain Monocyte Blocker and LIVE/DEAD Fixable Red Dead Cell Stain Kit (ThermoFisher Scientific). After 10-min incubation, antibody mix in Brilliant Stain Buffer (Becton Dickinson) was added to the cells. Cells were incubated at room temperature for 15 minutes, then centrifuged and fixed with Fixation Buffer (BD Cytofix). For intracellular staining, cells were permeabilized after fixation, and stained with KI67, Granzyme A and S100A9 (BioLegend) using Foxp3/Transcription Factor Staining Buffer Set (eBioscience). Samples were acquired using the Fortessa Flow Cytometer (Becton Dickinson), and data were analyzed with FlowJo software (FlowJo). In figures, representative flow plots always depict the patient closest to the mean. Statistical analysis of flow data was done using GraphPad Prism version 9.0.
Quantification and statistical analyses
The number of samples per group and experiment repeats, as well as the statistical test used is indicated in each figure legend. Where indicated, p values were adjusted for multiple comparisons using the Benjamini Hochberg procedure (Benjamini and Hochberg, 1995). The absence of a p value where applicable indicates that the statistical comparison is not significantly different.
Acknowledgments
The authors thank the patients and their families for participation. We are also grateful to the physicians, nurses, and hospital staff who helped care for the patients and obtain samples. The authors thank J. Pober and D. Jane-Wit for critical input, R. Montgomery for support, and G. Wang and C. Castaldi at Yale Center for Genome Analysis for 10x Genomics library preparation and sequencing services; K. Raddassi for processing scRNA-seq samples; J.L. Pappalardo for providing us the healthy adult scRNA-seq data; R. Sparks and L. Failla for assistance providing healthy pediatric samples for SomaLogic. We also thank Ms. M. Bucklin, Mr. D. Murdock, and Dr. JM. Carpier for feedback and discussions. This research was supported by grants to C.L.L. from NIAID 3R21AI144315-01A1S1 and Yale University. R.W.P is supported by NHLBI 1K08HL136898-01A1. A.R. was supported by NIAID 5T32AI007019 and NSF Graduate Research Fellowship. S.H.K. and K.B.H. were funded in part by NIAID R01AI104739. P.G., W.L., B.S, N.B., J.S.T. are supported by the Division of Intramural Research, NIH.
Author contributions
A.R., N.N.B., and C.L.L. conceptualized the study. N.N.B., V.H., A.J.R., R.S., and J.C. consented patients and healthy donors. N.N.B., V.H., H.C., and M.L. collected blood samples and clinical information from all patients. N.N.B. and A.J.R. performed serum, plasma, and PBMC isolation and cryopreservation. T.S.S., and H.A. performed scRNA-seq and CITE-seq sample preparation and cDNA generation; A.R., T.S.S., M.C., H.A., A.U., A.S., S.K., D.V.D., and C.L.L. analyzed scRNA-seq gene expression data and CITE-seq data with the help of N.K. and D.A.H.; K.H. and S.K. analyzed single cell BCR data; N.L. and X.Y. analyzed single cell TCR data; W.L., B.S., N.B., J.C. and J.T. analyzed Somascan serum proteome data; M.C. performed flow cytometry analysis for patients’ PBMCs; N.N.B., A.K., and R.P. performed endothelial cell experiments; A.R., N.N.B., and C.L.L. wrote the manuscript with input from all authors; C.L.L. supervised the overall study.
Declaration of interests
D.A.H. has received research funding from Bristol-Myers Squibb, Novartis, Sanofi, and Genentech. He has been a consultant for Bayer Pharmaceuticals, Bristol Myers Squibb, Compass Therapeutics, EMD Serono, Genentech, Juno Therapeutics, Novartis Pharmaceuticals, Proclara Biosciences, Sage Therapeutics, and Sanofi Genzyme. Further information regarding funding is available on: https://openpaymentsdata.cms.gov/physician/166753/general-payments. N.K. reports personal fees from Boehringer Ingelheim, Third Rock, Pliant, Samumed, NuMedii, Indalo, Theravance, LifeMax, Three Lake Partners, RohBar in the last 36 months, and Equity in Pliant. N.K. is also a recipient of a grant from Veracyte and non-financial support from Miragen. All outside the submitted work; In addition, N.K. has patents on New Therapies in Pulmonary Fibrosis and ARDS (unlicensed) and Peripheral Blood Gene Expression as biomarkers in IPF (licensed to biotech). S.H.K. receives consulting fees from Northrop Grumman. K.B.H. receives consulting fees from Prellis Biologics. B.S. is a former SomaLogic, Inc. (Boulder, CO, USA) employee and a company shareholder. All other authors declared that they have no competing interests.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: April 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.04.003.
Supplemental information
189 markers used for CITE-seq. Listed are the surface markers, corresponding gene names, and associated sequences.
Differential expression markers between MIS-C and C.HD, in clusters corresponding to broad immune cell subsets. (see STAR Methods). Markers gated on abs(logFC) > 0.05 and p.adj < 0.05.
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balázs Z., Tombácz D., Szűcs A., Csabai Z., Megyeri K., Petrov A.N., Snyder M., Boldogkői Z. Long-Read Sequencing of Human Cytomegalovirus Transcriptome Reveals RNA Isoforms Carrying Distinct Coding Potentials. Sci. Rep. 2017;7:15989. doi: 10.1038/s41598-017-16262-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995;57(1):289–300. [Google Scholar]
- Bernardes J.P., Mishra N., Tran F., Bahmer T., Best L., Blasé J.I., Bordoni D., Franzenburg J., Geisen U., Josephs-Spaulding J., et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity. 2020;53(6):1296–1314 e9. doi: 10.1016/j.immuni.2020.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertheloot D., Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell. Mol. Immunol. 2017;14:43–64. doi: 10.1038/cmi.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter M.J., Fish M., Jennings A., Doores K.J., Wellman P., Seow J., Acors S., Graham C., Timms E., Kenny J., et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 2020;26:1701–1707. doi: 10.1038/s41591-020-1054-6. [DOI] [PubMed] [Google Scholar]
- Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M.H., Zhang S., Porritt R.A., Noval Rivas M., Paschold L., Willscher E., Binder M., Arditi M., Bahar I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proceedings of the National Academy of Sciences. 2020;117(41):25254–25262. doi: 10.1073/pnas.2010722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung E., Zachariah P., Gorelik M., Boneparth A., Kernie S., Orange J., Milner J. Multisystem Inflammatory Syndrome Related to COVID-19 in Previously Healthy Children and Adolescents in New York City. JAMA. 2020;324(3):294–296. doi: 10.1001/jama.2020.10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consiglio C., Cotugno N., Sardh F., Pou C., Amodio D., Zicari S., Ruggiero A., Pascucci G., Rodriguez L., Santilli V., Tan Z., Eriksson D., Wang J., Marchesi A., Lakshmikanth T., Campana A., Villani A., Rossi P., Team T.C.S., Landegren N., Palma P., Brodin P. The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell. 2020;183(4):968–981. doi: 10.1016/j.cell.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coperchini F., Chiovato L., Croce L., Magri F., Rotondi M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020;53:25–32. doi: 10.1016/j.cytogfr.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diorio C., Henrickson S., Vella L., McNerney K., Chase J., Burudpakdee C., Lee J., Jasen C., Balamuth F., Barrett D., Banwell B., Bernt K., Blatz A., Chiotos K., Fisher B., Fitzgerald J., Gerber J., Gollomp K., Gray C., Grupp S., Harris R., Kilbaugh T., Odom John A., Lambert M., Liebling E., Paessler M., Petrosa W., Phillips C., Reilly A., Romberg N., Seif A., Sesok-Pizzini D., Sullivan K., Vardaro J., Behrens E., Teachey D., Bassiri H. Multisystem Inflammatory Syndrome in Children and COVID-19 are distinct presentations of SARS-CoV-2 11th ed. J. Clin. Invest. 2020;130:5967–5975. doi: 10.1172/JCI140970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldstein L.R., Rose E.B., Horwitz S.M., Collins J.P., Newhams M.M., Son M.B.F., Newburger J.W., Kleinman L.C., Heidemann S.M., Martin A.A., et al. Overcoming COVID-19 Investigators. CDC COVID-19 Response Team Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N. Engl. J. Med. 2020;383:334–346. doi: 10.1056/NEJMoa2021680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco L.M., Gadkari M., Howe K.N., Sun J., Kardava L., Kumar P., Kumari S., Hu Z., Fraser I.D.C., Moir S., et al. Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J. Exp. Med. 2019;216(2):384–406. doi: 10.1084/jem.20180595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V., Chaume D., Lefranc M.P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005;33:D256–D261. doi: 10.1093/nar/gki010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber C.N., Patel R.S., Trachtman R., Lepow L., Amanat F., Krammer F., Wilson K.M., Onel K., Geanon D., Tuballes K., et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C) Cell. 2020;183(4):982–995.e14. doi: 10.1016/j.cell.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumann D., Scharf S.S., Holtfreter S., Kohler C., Steil L., Engelmann S., Hecker M., Völker U., Bröker B.M. Immune cell activation by enterotoxin gene cluster (egc)-encoded and non-egc superantigens from Staphylococcus aureus. J. Immunol. 2008;181(7):5054–5061. doi: 10.4049/jimmunol.181.7.5054. [DOI] [PubMed] [Google Scholar]
- Gupta N.T., Adams K.D., Briggs A.W., Timberlake S.C., Vigneault F., Kleinstein S.H. Hierarchical Clustering Can Identify B Cell Clones with High Confidence in Ig Repertoire Sequencing Data. J. Immunol. 2017;198(6):2489–2499. doi: 10.4049/jimmunol.1601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T., Koo M.Y., Kagoya Y., Anczurowski M., Wang C.H., Saso K., Butler M.O., Hirano N. A Subset of Human Autoreactive CD1c-Restricted T Cells Preferentially Expresses TRBV4-1+ TCRs. J. Immunol. 2018;200(2):500–511. doi: 10.4049/jimmunol.1700677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N.T., Vander Heiden J.A., Uduman M., Gadala-Maria D., Yaari G., Kleinstein S.H. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics. 2015;31(20):3356–3358. doi: 10.1093/bioinformatics/btv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habtezion A., Nguyen L.P., Hadeiba H., Butcher E.C. Leukocyte Trafficking to the Small Intestine and Colon. Gastroenterology. 2016;150(2):340–354. doi: 10.1053/j.gastro.2015.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand D., Bode K.A., Rieß D., Cerny D., Waldhuber A., Römmler F., Strack J., Korten S., Orth J.H., Miethke T., et al. Granzyme A produces bioactive IL-1β through a nonapoptotic inflammasome-independent pathway. Cell Rep. 2014;9(3):910–917. doi: 10.1016/j.celrep.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Jones V.G., Mills M., Suarez D., Hogan C.A., Yeh D., Segal J.B., Nguyen E.L., Barsh G.R., Maskatia S., Mathew R. COVID-19 and Kawasaki Disease: Novel Virus and Novel Case. Hosp. Pediatr. 2020;10(6):537–540. doi: 10.1542/hpeds.2020-0123. [DOI] [PubMed] [Google Scholar]
- Kaneko N., Kuo H.H., Boucau J., Farmer J.R., Allard-Charmand H., Mahajan V.S., Piechocka-Trocha A., Lefteri K., Osborn M., et al. Massachusetts Consortium on pathogen readiness specimen working Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell. 2020;183(1):143–157 e13. doi: 10.1016/j.cell.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocperk A., ParackovaDissou Z.J., Malcova H., Pavlicek P., Vymazal T., Dolezalova P., Sediva A. Case Report: Systemic Inflammatory Response and Fast Recovery in a Pediatric Patient With COVID-19. Front. Immunol. 2020;11:1665. doi: 10.3389/fimmu.2020.01665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90-7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I., et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- Lee E.Y., Lee Z.H., Song Y.W. CXCL10 and autoimmune diseases. Autoimmun. Rev. 2009;8(5):379–383. doi: 10.1016/j.autrev.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Leung D.Y., Collins T., Lapierre L.A., Geha R.S., Pober J.S. Immunoglobulin M antibodies present in the acute phase of Kawasaki syndrome lyse cultured vascular endothelial cells stimulated by gamma interferon. J. Clin. Invest. 1986;77:1428–1435. doi: 10.1172/JCI112454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.Y., Geha R.S., Newburger J.W., Burns J.C., Fiers W., Lapierre L.A., Pober J.S. Two monokines, interleukin 1 and tumor necrosis factor, render cultured vascular endothelial cells susceptible to lysis by antibodies circulating during Kawasaki syndrome. J. Exp. Med. 1986;164:1958–1972. doi: 10.1084/jem.164.6.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.Y., Cotran R.S., Kurt-Jones E., Burns J.C., Newburger J.W., Pober J.S. Endothelial activation in the pathogenesis of Kawasaki disease. Trans. Assoc. Am. Physicians. 1989;102:131–138. [PubMed] [Google Scholar]
- Leung D.Y., Cotran R.S., Kurt-Jones E., Burns J.C., Newburger J.W., Pober J.S. Endothelial cell activation and high interleukin-1 secretion in the pathogenesis of acute Kawasaki disease. Lancet. 1989;2:1298–1302. doi: 10.1016/s0140-6736(89)91910-7. [DOI] [PubMed] [Google Scholar]
- Ley K., Kansas G.S. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 2004;4:325–335. doi: 10.1038/nri1351. [DOI] [PubMed] [Google Scholar]
- Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Yale IMPACT Team Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydon E.C., Henao R., Burke T.W., Aydin M., Nicholson B.P., Glickman S.W., Fowler V.G., Quackenbush E.B., Cairns C.B., Kingsmore S.F., et al. Validation of a host response test to distinguish bacterial and viral respiratory infection. EBioMedicine. 2019;48:453–461. doi: 10.1016/j.ebiom.2019.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou E.P., Cheng A., Montalbano A., Hao S., Stoeckius M., Legut M., Roush T., Herrera A., Papalexi E., Ouyang Z., et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods. 2019;16:409–412. doi: 10.1038/s41592-019-0392-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S.C.A., Yang F., Jackson K.J.L., Hoh R.A., Roltgen K., Jean G.H., Stevens B.A., Lee J.Y., Rustagi A., Rogers A.J., Powell A.E., Hunter M., Najeeb J., Otrelo-Cardoso A.R., Yost K.E., Daniel B., Nadeau K.C., Chang H.Y., Satpathy A.T., Jardetzky T.S., Kim P.S., Wang T.T., Pinsky B.A., Blish C.A., Boyd S.D. Human B Cell Clonal Expansion and Convergent Antibody Responses to SARS-CoV-2. Cell Host Microbe. 2020;28(4):516–525.e5. doi: 10.1016/j.chom.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappalardo J.L., Zhang L., Pecsok M.K., Perlman K., Zografou C., Raddassi K., Abulaban A., Krishnaswamy S., Antel J., van Dijk D., Hafler D.A. Transcriptomic and clonal characterization of T cells in the human central nervous system. Sci. Immunol. 2020;5(51):eabb8786. doi: 10.1126/sciimmunol.abb8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2020. R: A language and environment for statistical computing. [Google Scholar]
- Rao D.A., Gurish M.F., Marshall J.L., Slowikowski K., Fonseka C.Y., Liu Y., Donlin L.T., Henderson L.A., Wei K., Mizoguchi F., et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–114. doi: 10.1038/nature20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raredon M.S.B., Adams T.S., Suhail Y., Schupp J.C., Poli S., Neumark N., Leiby K.L., Greaney A.M., Yuan Y., Horien C., et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2019;5(12):w3851. doi: 10.1126/sciadv.aaw3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raredon M.S.B., Yang J., Garritano J., Wang M., Kushnir D., Schupp J.C., Adams T.S., Greaney A.M., Leiby K.L., Kaminski N., et al. Connectome: computation and visualization of cell-cell signaling topologies in single-cell systems data. bioRxiv. 2021 doi: 10.1038/s41598-022-07959-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauf A., Vijayan A., John S.T., Krishnan R., Latheef A. Multisystem Inflammatory Syndrome with Features of Atypical Kawasaki Disease during COVID-19 Pandemic. Indian J. Pediatr. 2020;87(9):745–747. doi: 10.1007/s12098-020-03357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M., Filbin M.R., Bhattacharyya R.P., Billman K., Eisenhaure T., Hung D.T., Levy B.D., Baron R.M., Blainey P.C., Goldberg M.B., Hacohen N. An immune-cell signature of bacterial sepsis. Nat. Med. 2020;26:333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riphagen S., Gomez X., Gonzalez-Martinez C., Wilkinson N., Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395(10237):1607–1608. doi: 10.1016/S0140-6736(20)31094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley A.H. Understanding SARS-CoV-2-related multisystem inflammatory syndrome in children. Nat. Rev. Immunol. 2020;20:453–454. doi: 10.1038/s41577-020-0367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Bassler K., Schlickeiser S., Zhang B., Kramer B., Krammer T., Brumhard S., Bonaguro L., et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182(6):1419–1440 e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902 e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubiana J., Poirault C., Corsia A., Bajolle F., Fourgeaud J., Angoulvant F., Debray A., Basmaci R., Salvador E., Biscardi S., et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. BMJ. 2020;369:m2094. doi: 10.1136/bmj.m2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterman A., Sumida T.S., Nouri N., Yan X., Zhao A.Y., Gasque V., Schupp J.C., Asashima H., Liu Y., Cosme C., Jr., et al. The Yale IMPACT Research Team Single-Cell Omics Reveals Dyssynchrony of the Innate and Adaptive Immune System in Progressive COVID-19. medRxiv. 2020 doi: 10.1038/s41467-021-27716-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoni L., Mazza A., Gervasoni A., Martelli L., Ruggeri M., Ciuffreda M., Bonanomi E., D’Antiga L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. 2020;395(10239):1771–1778. doi: 10.1016/S0140-6736(20)31103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker E., Bamford A., Kenny J., Kaforou M., Jones C.E., Shah P., Ramnarayan P., Fraisse A., Miller O., Davies P., et al. PIMS-TS Study Group and EUCLIDS and PERFORM Consortia Clinical Characteristics of 58 Children With a Pediatric Inflammatory Multisystem Syndrome Temporally Associated With SARS-CoV-2. JAMA. 2020;324(3):259–269. doi: 10.1001/jama.2020.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Ye J., Ma N., Madden T.L., Ostell J.M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34-40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L.S., Arrand J.R., Murray P.G. In: Human Herpesviruses: Biology. Arvin A., Campadelli-Fiume G., Mocarski E., Moore P.S., Roizman B., Whitley R., Yamanishi K., editors. Therapy, and Immunoprophylaxis; 2007. EBV gene expression and regulation. [PubMed] [Google Scholar]
- Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., He H., Wang K., Shi X., Wang Y., Su Y., Wang Y., Li D., Liu W., Zhang Y., et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368(6494):eaaz7548. doi: 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
189 markers used for CITE-seq. Listed are the surface markers, corresponding gene names, and associated sequences.
Differential expression markers between MIS-C and C.HD, in clusters corresponding to broad immune cell subsets. (see STAR Methods). Markers gated on abs(logFC) > 0.05 and p.adj < 0.05.
Data Availability Statement
Raw and processed data pertaining to pediatric and select adult healthy control samples (A.HD1-3) in this paper are available at GEO: GSE166489 and FASTgenomics. Data pertaining to adult COVID-19 and additional adult healthy control samples are available from previous studies: Pappalardo et al. dbGAP: phs002222 and Unterman et al. GEO: GSE155223 (Pappalardo et al., 2020, Unterman et al., 2020). Code is made available in https://github.com/LucasiteLab/MIS-C_scRNAseq and FASTgenomics. Additional supplemental items are available from Mendeley Data at https://doi.org/10.17632/v9dkw64s6c.3.