

Abstract
Regulatory T-cells (Tregs) are key players in the maintenance of immune homeostasis by preventing immune responses to self-antigens. Defects in Treg frequency and/or function result in overwhelming CD4 and CD8 T cell immune responses participating in the autoimmune attack. Perpetuation of autoimmune damage is also favored by Treg predisposition to acquire effector cell features upon exposure to a proinflammatory challenge.
Treg impairment plays a permissive role in the initiation and perpetuation of autoimmune liver diseases, namely autoimmune hepatitis, primary biliary cholangitis and primary sclerosing cholangitis. In this Review, we outline studies reporting the role of Treg impairment in the pathogenesis of these conditions and discuss methods to restore Treg number and function either by generation/expansion in the test tube or through in vivo expansion upon administration of low dose IL-2. Challenges and caveats of these potential therapeutic strategies are also reviewed and discussed.
Keywords: regulatory T cells, effector lymphocytes, immune tolerance, autoimmune hepatitis, immunotherapy
1. Regulatory T cells: definition, development and modulation
Regulatory T cells (Tregs) are suppressive lymphocytes central to immunoregulation and maintenance of immune homeostasis [1]. Though lymphocytes capable of suppression include also regulatory CD8 [2], B regulatory (Bregs) [3, 4], invariant NKT (iNKT) [5], and T regulatory type 1 (Tr1) cells [6], it is the subset of CD4+ T cells co-expressing the IL-2 receptor α chain (CD25) that has been the focus of major studies over the past two decades, because of their pivotal role in actively promoting and maintaining immune tolerance. A clearer understanding of Treg role was gained in the 1990s thanks to the discoveries by Sakaguchi and colleagues, showing that the 10% of CD4 T cells constitutively expressing CD25 were responsible for the maintenance of immune tolerance in mice [7, 8]. Subsequently, in 2003, the transcription factor box P3 (FOXP3) was shown to control the generation and function of murine Tregs [9, 10] and was found to be expressed by human CD4 cells with the highest expression levels of CD25 [11]. Differently from mice where Foxp3 remains a marker to identify bona fide Tregs, in human CD4 cells FOXP3 can be also upregulated – though transiently – on naïve effectors that have undergone activation; this representing a significant challenge for Tregs identification and subsequent isolation, which is also hampered by FOXP3 being an intracellular marker. Measurement of CD127, the α-chain of the IL-7 receptor that is normally present on activated Teff [12], is now usually carried out to differentiate CD4 effector cells that express CD127 at high levels from bona fide Tregs, typically CD127low/neg.
Tregs are commonly classified according to their developmental pathway. The majority of FOXP3+ Tregs present in the periphery originate in the thymus (tTregs) and their generation is favored by intermediate affinity self-peptides/MHC interactions that generate high intensity TCR signals [13, 14]. There are also peripheral Tregs (pTregs) that differentiate from T cells in certain peripheral sites like the gut mucosa [15] and acquire stable FOXP3 expression; as well as FOXP3+ and FOXP3− induced Tregs (iTregs) that derive in vitro from Tconv after exposure to suboptimal antigen stimulation in the presence of anti-inflammatory mediators [16–18].
Both tTregs and pTregs display hypomethylated regions in the genome like the so-called Treg specific demethylated region (TSDR), within the Foxp3 CNS2 region [19]. Foxp3 DNA methylation has been regarded as a typical feature of bona fide Tregs. There are also other Treg defining genes, i.e. Cd25, Ctla4, Helios and Eos that also contain DNA hypomethylated regions [20]. Although a phenotypic distinction between tTregs and pTregs/iTregs can be made in mouse based on the levels of Helios, which is highly expressed by tTregs [21], such differentiation is less clear when considering human cells.
Additional studies by Sakaguchi’s laboratory have shown that analysis of FOXP3, CD25 and CD45RA enables to identify different fractions of human FOXP3+CD4+ T cells with distinct activation and functional properties: fraction 1, i.e. CD45RA+FOXP3lowCD25low including resting or naïve Tregs that possess suppressive properties and demethylated TSDR; fraction 2, i.e. CD45RA−FOXP3highCD25high defining effector Tregs, which are highly proliferating cells with strong suppressor capabilities and demethylated TSDR; and fraction 3, i.e. CD45RA−FOXP3lowCD25low that do not have suppressive properties, secrete IL-2 and IFN-γ, mainly representing Tconv (reviewed in [22]). In some circumstances, Tregs gain effector cell properties while losing expression of Foxp3 [23, 24]. Tregs can also adapt to their environment and acquire expression of chemokines and transcription factors normally associated with effector cells [25–29]; this was suggested to enable Treg trafficking to inflammatory sites to suppress the corresponding effector cells [1].
2. Mechanisms of suppression
Tregs can suppress immune responses either by direct targeting of T cells or through the modulation of antigen presenting cell (APC) maturation and function. A wealth of in vitro studies has demonstrated that Tregs suppress via direct cell-to-cell contact [30, 31]. There is also evidence, however, that Tregs can mediate suppression upon the release of cytokines like TGF-β [32–34], IL-10 [35, 36] and IL-35 [37]. Cytokine mediated Treg suppression is predominantly present in the gut, skin and lungs [38–40].
Tregs can also suppress by killing their targets via induction of apoptosis; either by promoting a caspase cascade, resulting from the interaction of the death ligands tumor necrosis factor (TNF)-related-apoptosis-inducing ligand (TRAIL) [41] and Fas-ligand [42] on Tregs with the death receptors on their targets; release of granzyme A and B [43–47] and perforin [43, 46]; or through Galectin 9, which binds to the T-cell-immunoglobulin-and-mucin-domain-3 (Tim-3) expressed by activated effector T cells [48].
Treg suppression might also result from metabolic disruption. In this regard, Tregs deplete IL-2 from the environment, depriving effector T cells of this cytokine and leading to Bim-mediated apoptosis [49]. Moreover, Tregs express cyclic adenosine monophosphate (cAMP), which has implications for suppressive ability [50] and can be directly transferred to effector T cells via gap junctions; this leading to the upregulation of the inducible cAMP early repressor (ICER) [51]. ICER elevations were found to limit nuclear factor of activated T cells (NFAT) and IL-2 transcription [52]. Another important mechanism operated by Tregs and involving metabolic disruption is mediated by CD39, an ectonucleotidase that initiates a hydrolysis cascade that ultimately converts pro-inflammatory adenosine triphosphate and adenosine diphosphate into immunosuppressive adenosine [53]. Previous studies have shown that Tregs obtained from CD39−/− mice have impaired suppressor ability in vitro and fail to block allograft rejection in vivo [54]. In the autoimmune setting, CD39+ Tregs are defective in the peripheral blood of patients with multiple sclerosis [55] and, in rheumatoid arthritis, Tregs obtained from patients unresponsive to methotrexate express low CD39 levels and display impaired suppressive properties [56].
An additional mechanism of Treg suppression involves direct interactions between Tregs and APCs through the binding of CTLA4 on Tregs to CD80 and CD86 costimulatory molecules on APCs. By lowering CD80 and CD86 expression, Tregs render APCs less capable of initiating an immune response [57]. In both mice and humans, CTLA4 can also lead to physical removal of CD80 and CD86 from the surface of APCs via trans-endocytosis and degradation [58]. Mechanisms of Treg suppression are summarized in Figure 1.
Figure 1. Treg mechanisms of suppression.
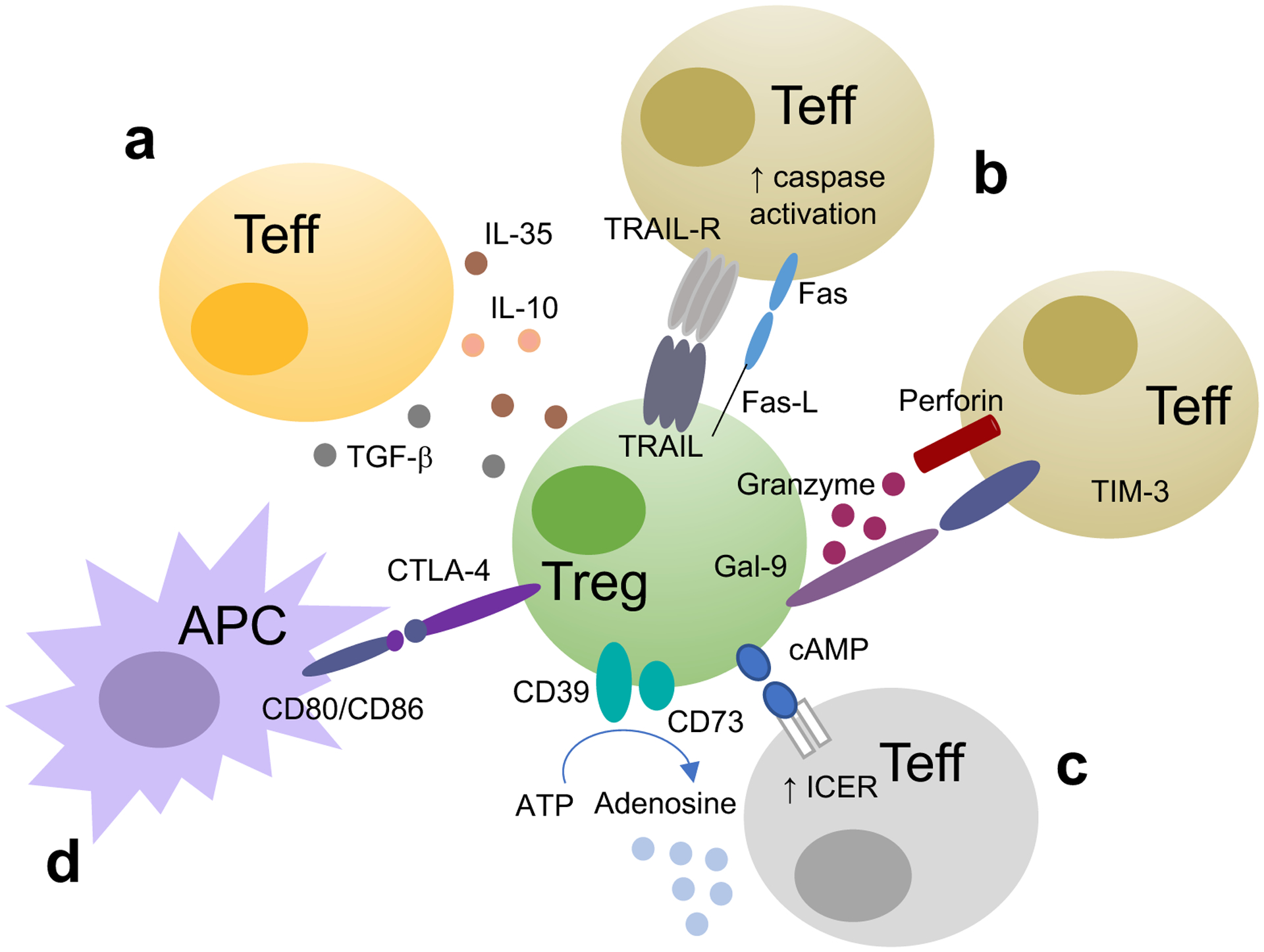
Tregs suppress effector T cell (Teff) function upon (a) secretion of cytokines like IL-10, TGF-β and IL-35; (b) by inducing apoptosis upon activation of caspase following interaction of TNF-related-apoptosis-inducing ligand (TRAIL) and Fas-ligand (Fas-L) with TRAIL receptor (TRAIL-R) and Fas, release of granzyme and perforin and upon interaction between galectin-9 in Tregs and T-cell-immunoglobulin-and-mucin-domain-3 (Tim-3) on Teff. Tregs can also suppress (c) by operating metabolic disruption, by transferring cyclic adenosine monophosphate (cAMP) to Teff. cAMP has immunoregulatory properties because it activates the inducible cAMP early repressor (ICER) to limit IL-2 transcription. Tregs can also generate immunosuppressive adenosine through the tandem action of CD39 and CD73. Tregs can also suppress (d) through modulation of antigen presenting cell (APC) function by lowering CD80/CD86 expression on APC surface.
3. Tregs and autoimmunity: the paradigm of autoimmune hepatitis
Treg defects are associated with several autoimmune conditions in mice and man, their impairment playing an important role in the initiation, progression and perpetuation of tissue damage of diseases like rheumatoid arthritis [59], multiple sclerosis [60], Graves’ disease [61] and type 1 diabetes [62]. In these autoimmune conditions, numerical and/or functional Treg impairment was reported [59–61]. This key role of Tregs has been demonstrated over the years also in autoimmune hepatitis (AIH), a severe hepatopathy characterized by hypergammaglobulinemia, interface hepatitis on histology and seropositivity for disease defining autoantibodies [63–65]. The type of serum autoantibodies differentiates two subtypes of the disease, type 1 AIH (AIH-1), characterized by anti-nuclear (ANA) and/or anti-smooth muscle (ASMA) autoantibodies; and type 2 AIH (AIH-2), with seropositivity for liver kidney microsomal antibody type 1 (LKM-1) and/or liver cytosol 1 (LC1) autoantibodies. In AIH, the autoimmune reaction leading to liver damage initiates with the presentation of a liver autoantigen by APCs to an uncommitted T lymphocyte that, following antigen encounter, becomes activated. Autoreactive immunity in AIH is amplified by the presence of HLA class II molecules on unconventional APCs, like hepatocytes [66, 67]. Following activation, Th0 cells differentiate into Th1, Th2 and Th17 cells that have been shown having a pathogenetic role in AIH liver damage. Secretion of IFN-γ and IL-2 by Th1 cells results in macrophage activation and upregulation of HLA class I and HLA class II by hepatocytes [68, 69]. B cell activation in the presence of IL-4 and IL-10 secreted by Th2 lymphocytes results in their maturation into plasma cells that produce autoantibodies, which in turn mediate cell cytotoxicity [70, 71]. Activation of Th17 cells has been associated with induction of pro-fibrotic events [72, 73]. If these events are not opposed by effective immunoregulation, the autoimmune attack continues perpetrating and favoring the progression of tissue damage.
3.1. Treg in AIH: historical overview
A wealth of studies has supported a role for Treg impairment in the initiation and progression of AIH tissue damage (Figure 2). It remains still unclear, however, whether Treg defects in AIH represent the initial causative event leading to immune tolerance breakdown; or result from the disease itself. Initial studies on suppressor cells in AIH were conducted by Hodgson et al., who reported that concanavalin-A stimulated suppressor activity was not elicited in mononuclear cells from patients with AIH – at that time called chronic active hepatitis – when compared with patients with acute hepatitis, acute inflammatory diseases and controls [74]. This was demonstrated in cells obtained at remission and during relapse, in the presence or absence of prednisone therapy, and varied substantially during the course of the disease [74]. Subsequent findings from Nouri-Aria and colleagues showed that pre-incubation of AIH-derived lymphocytes with low-dose prednisolone in vitro, resulted in significant improvement of suppressor cell activity; whereas this effect was not noted in cells obtained from HBsAg+ chronic active hepatitis [75]. Further investigations corroborated the notion of an impairment of suppressor cells in AIH by demonstrating that AIH derived lymphocytes generated T lymphocyte migration inhibitory factors (T-LIF) in the presence of liver-specific protein. Generation of T-LIF was blocked when AIH T cells were cultured at 9:1 ratio with T cells, obtained from normal subjects and HBsAg+ chronic hepatitis patients [76]. Several years later a numerical and functional defect in CD4+CD25+/high and FOXP3+ cells was reported in patients with AIH-1 and AIH-2, as well as in patients with an overlap between AIH-1 and sclerosing cholangitis (autoimmune sclerosing cholangitis, AISC) [77, 78] when compared to healthy subjects [79–81]. Tregs isolated from AIH patients are also impaired in their ability to expand [79]. When obtained from patients studied at diagnosis, before immunosuppressive treatment is instituted, Tregs are unable to regulate CD8 T cell proliferation and IL4 production [80]. Re-acquisition of this property at remission suggests a role for immunosuppressive treatment in the reconstitution of Treg function [80]. As in the case of healthy controls, Tregs isolated from AIH patients suppress via direct contact while modifying CD4 and CD8 T cell cytokine profile [81]. The extent of Treg suppression over CD4 and CD8 T cell effector function (i.e. proliferation and inflammatory cytokine release) remains lower in AIH than in health [81]. Defective suppressive function was also reported in the case of AIH T-reg co-culture with monocytes [82]. In a study of 51 patients with autoimmune liver disease (AILD, including AIH and AISC), we reported enhanced monocyte spontaneous migration in both AILD patients with active and inactive disease. Importantly, addition of Tregs restrained monocyte migration and function in healthy controls, leaving it unchanged or even exacerbating it in the case of AIH Tregs. This aberrant pattern displayed by Tregs in AIH might be linked to increased expression of the activation marker CD127 [82]. Depletion of CD127 in Tregs resulted in further ability to decrease monocyte migration, augmented IL-10 production in healthy subjects and, although to a lesser extent, also in AIH patients [82]. Impairment in peripheral blood derived Treg frequencies and function has been shown in pediatric and adult AIH along with other immune cell alterations, like increase in γδ T cells that displayed an inverted Vδ1/Vδ2 ratio and higher IFN-γ and granzyme B production [83]. Additional studies reported that in AIH patients concomitantly suffering from systemic lupus erythematosus, Tregs display a distinctive phenotype with high expression levels of IFN-γ and CD127, suggestive of skewing towards Th1 cells. These effector-like features of AIH/SLE Tregs possibly account for severe impairment of suppressive function [84]. A German group was later unable to detect abnormal frequency and function of circulating Tregs in AIH [85], a discrepancy likely related to differences in methodology, patients’ demographics, disease stage and treatment duration.
Figure 2. Treg impairment in AIH.
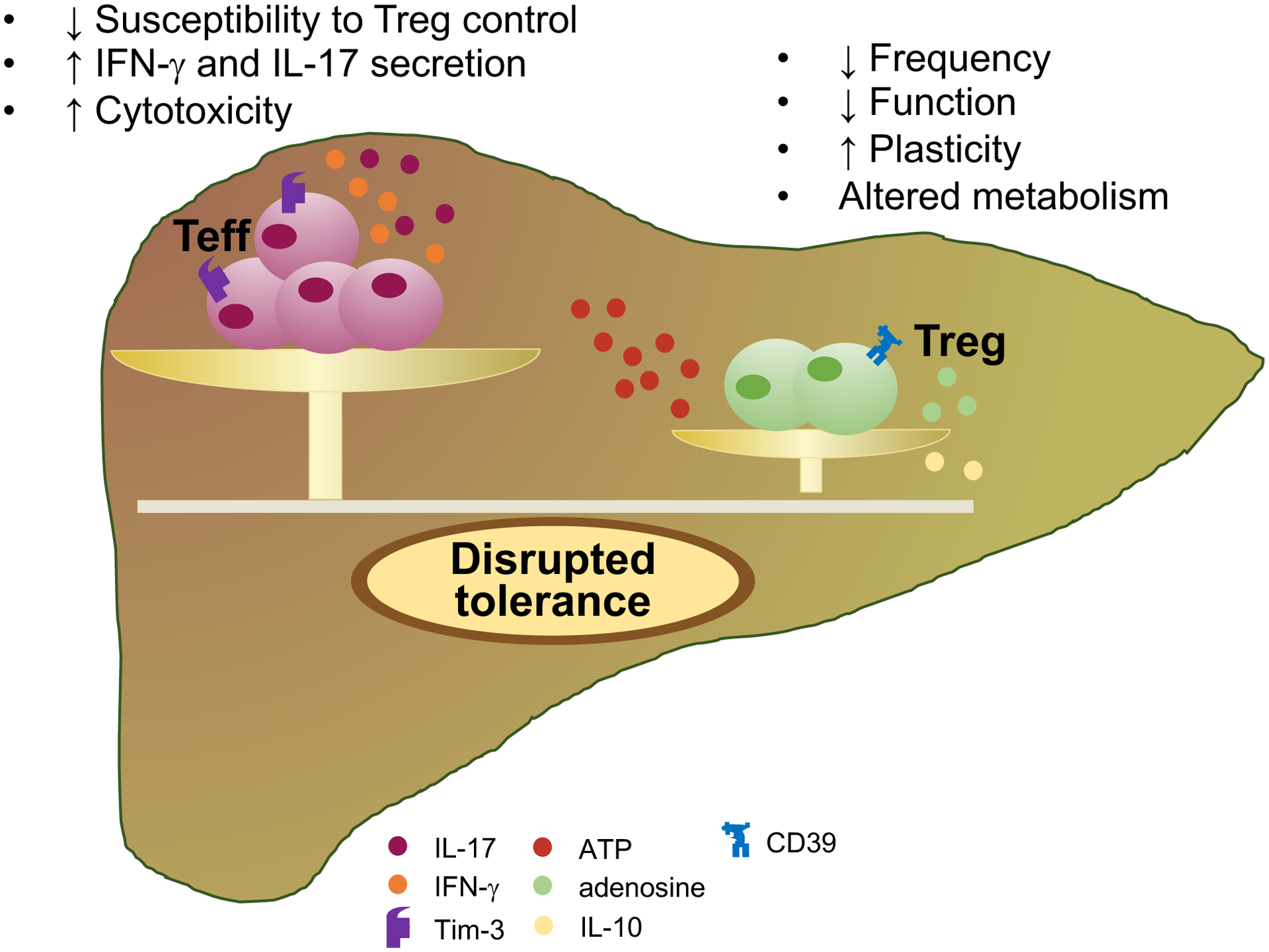
Imbalance between effector cells and Tregs contribute to the initiation and progression of tissue damage in AIH. Overwhelming T effector cell immunity that results in enhanced cytotoxicity against hepatocytes and in augmented secretion of IFN-γ and IL-17 pro-inflammatory cytokines is permitted by impaired Treg cell immune responses. Treg impairment might derive from reduced frequency, defective function, increased tendency to acquire effector cell features (plasticity) and altered metabolism, i.e. reduced ability to generate adenosine and to produce IL-10. In turn, effector cells obtained from AIH patients display impaired susceptibility to Treg control. T effector cell (Teff), regulatory T cell (Treg); T-cell-immunoglobulin-and-mucin-domain-3 (Tim-3); adenosine triphosphate (ATP); IFN-gamma (IFN-γ).
Further investigations focusing on liver-derived Tregs reported intra-hepatic Treg accumulation in untreated AIH-1 patients, followed by disproportionate decrease during immunosuppressive treatment [86]; it was postulated that this decrease during treatment could explain high relapse rate when immunosuppression is discontinued [86]. Similar results were reported by the same Authors when analyzing liver derived Tregs in a pediatric population of AIH patients [87]. Subsequent investigations conducted with the view of determining the impact of corticosteroid treatment on Treg immunophenotype and function, revealed heightened number of Tregs with activated memory phenotype (i.e. FOXP3+, CTLA4+ and CD39+) that progressively decline during corticosteroid treatment [88].
3.2. Tregs in AIH mouse models
Impairment in Treg frequencies was also found in a humanized mouse model of AIH, generated upon injection of human cytochrome P4502D6 (CYP2D6)/formiminotrasferase cyclodeaminase (FTCD) fusion protein into HLA-DR3+ or HLA-DR3− NOD recipient mice. Importantly, lower Treg numbers were associated with enhanced Th1 cell immunity and HLA-DR3+ mice had the most severe form of the disease that was accompanied by alterations in gut microbiota that included decreased bacteria diversity and total load [89]. In a mouse model of double transgenic TF-OVAxDEREG (Depletion of REGulatory T cells) mice, depletion of Tregs resulted in dramatic amplification of T cell mediated hepatitis [90]. In another model, injection of CYP2D6/FTCD into humanized HLA-DR4 transgenic mice resulted in decrease in hepatic Tregs that also displayed reduced suppressor activity [91].
3.3. Mechanisms of Treg impairment in AIH
Impaired Treg function in AIH has been linked to defective levels of Galectin-9, a β galactoside binding protein that binds to Tim-3 on CD4 effector cells. In healthy control cells, binding of Galectin-9 to Tim-3 induces apoptosis in CD4 effector cells; thus low Galectin-9 levels result in defective Treg ability to control effectors. Importantly CD4 effector cells obtained from AIH patients display impaired Tim-3 levels, suggesting that, in AIH, Tregs are functionally ineffective and effector lymphocytes are refractory to their control [92]. Notably, in vitro exposure of CD4 effectors to tacrolimus and cyclosporin resulted in ameliorated effector cell responsiveness to Treg control [93].
Subsequent studies highlighted defects in the expression of the ectonucleotidase CD39 in Tregs obtained from AIH patients. Circulating CD39+ Tregs are decreased in frequency in AIH where they also fail to keep under control production of IL-17 by CD4 effectors. CD39+ Tregs from AIH patients are more prone to plasticity than their normal counterpart and acquire features of effectors when exposed to a pro-inflammatory challenge, supporting the evidence that defective immunoregulation in this condition might also derive from increased conversion of Tregs into effector lymphocytes, this favoring perpetration of autoimmune liver damage [94].
Recent studies have demonstrated that impaired CD39 levels and activity in AIH Tregs derive from alterations of aryl hydrocarbon receptor (AhR) signaling [95]. AhR is a mediator of toxin responses and adaptive immunity, including Treg cell immunity [96, 97]. Upon binding to endogenous or exogenous ligands, AhR undergoes activation, this resulting in CD39 upregulation. Defective Treg ability to upregulate CD39 in AIH derives from aberrantly high levels of aryl hydrocarbon receptor repressor that inhibits AhR; and is also linked to increased levels of estrogen receptor alpha (Erα) that binds to AhR. Importantly, in AIH, AhR binds Erα with higher affinity than aryl hydrocarbon receptor nuclear translocator (ARNT), the classical AhR binding partner; this non-conventional binding was shown to result in impaired CD39 upregulation [95]. Defective AhR signaling in AIH was also suggested by a study by Lyttan and colleagues, reporting reduced indoleamine-2,3-dioxygenase-1 (IDO-1) activity, as reflected by lower degradation of tryptophan into kynurenine, an AhR endogenous ligand. Decreased Kynurenine/tryptophan ratio was observed in active AIH patients and postulated being important in determining Treg dysfunction in AIH [98].
In additional studies, defective Treg function in AIH was linked with reduced ability to produce IL-10; this, in turn, resulting from poor response to IL-2 as reflected by impaired ability to upregulate the phospho signal transducer and activator of transcription 5 (pSTAT-5) [87].
In the context of de novo AIH after liver transplant, heightened secretion of IL-12 and IL-6 by monocytes/macrophages was suggested to account for Treg functional impairment linked to aberrant production of IFN-gamma (IFN-γ [99]. Subsequent studies in the same disease setting reported that silencing of Toll-like-receptors (TLRs) 2 and 4, which mediate inflammasome activation in CD14++ monocytes, significantly decreased IFN-γ production by Tregs, further emphasizing the role of monocytes in conferring Treg proinflammatory properties in de novo AIH [100].
3.4. Potential for Treg immunotherapy in AIH
That Treg number and function recovers - at least to some extent - during AIH remission, suggests the potential of these cells to be restored, this having important implications for therapeutic purposes (Figure 3). In a study on 24 AIH-1 patients and 22 healthy subjects we tested the ability of Tregs to be expanded in vitro using polyclonal stimuli, namely anti-CD3/anti-CD28 and high dose IL-2. Expanded Tregs retained the conventional Treg phenotype and exhibited enhanced FOXP3 levels and suppressive function, also in AIH [101]. Exposure to anti-CD3/anti-CD28 and high dose IL-2 resulted also in Treg generation from CD4+CD25− effectors; an important finding implicating that not only Tregs could be expanded from the existing pool but also generated de novo from effector cells. Further studies from our group provided evidence that inhibition of IL-17 could favor differentiation of newly generated Tregs, derived from CD4+CD25− effectors, following polyclonal expansion; this representing another strategy to consider when aiming to generate and expand Tregs for immunotherapeutic purposes [102].
Figure 3. Treg generation and expansion.
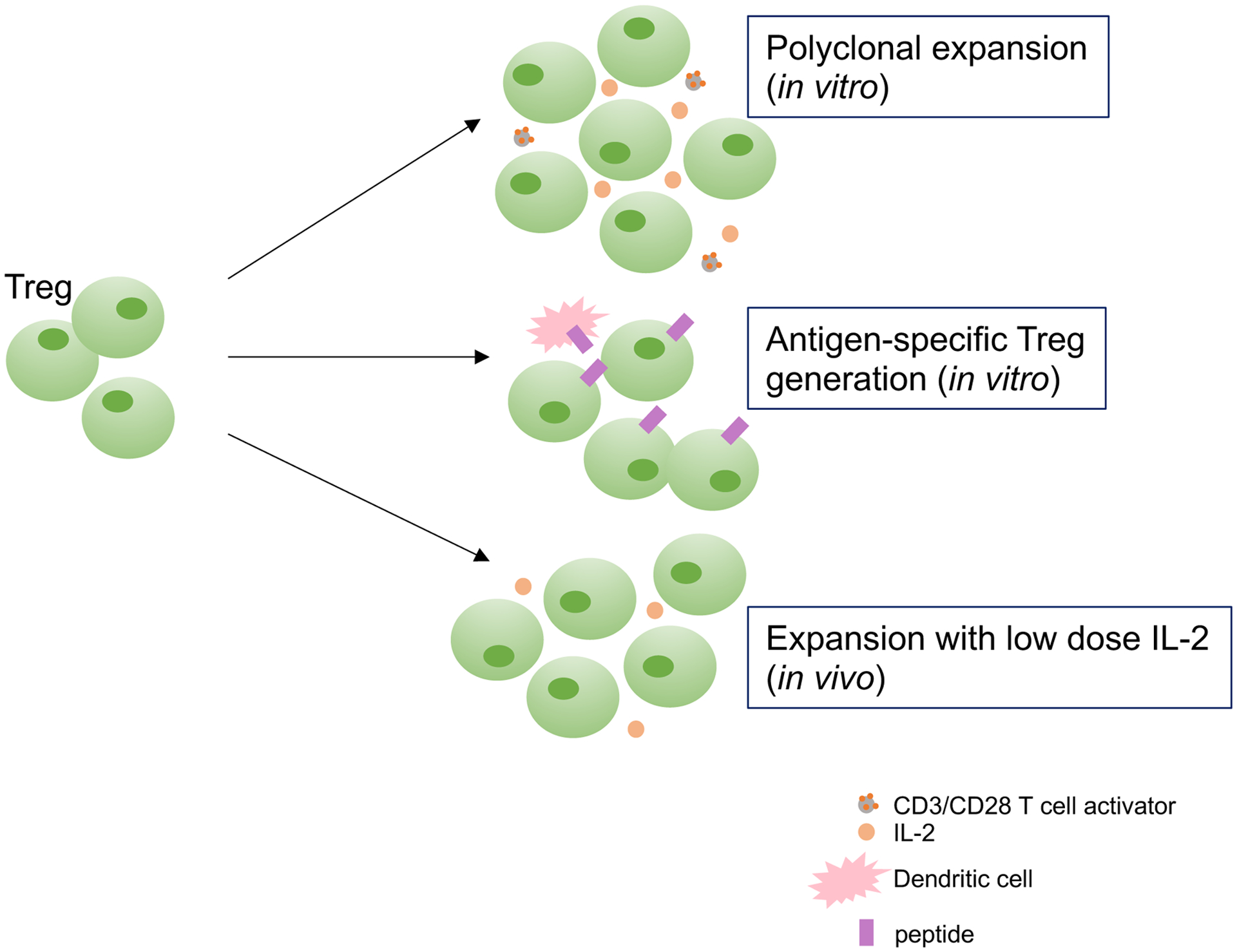
Increased Treg frequencies noted in AIH patients at remission indicate that these cells could be restored, at least to some extent. Previous studies have shown that Tregs could be expanded in vitro using polyclonal stimuli, i.e. high dose IL-2 and anti-CD3/anti-CD28 T cell expander (polyclonal expansion); generation of Tregs with liver autoantigen specificity could be obtained upon Treg co-culture with semi-mature dendritic cells presenting the autoantigenic peptide of interest. Recent work has reported the possibility to expand Tregs using low dose IL-2 in vivo. Further work is, however needed to validate all of these approaches.
In the case of AIH-2 where characterization of the key autoantigen has been performed and knowledge of the key CD4 and CD8 immunodominant epitopes acquired, generation of Tregs with autoantigen specificity would represent a valuable tool to provide a more tailored control over autoreactive T cell immunity. In this regard, we could generate Tregs with specificity for HLA class II restricted T cell epitopes in the presence of semi-mature dendritic cells (smDCs). CYP2D6-specific Tregs obtained in the presence of smDCs express heightened FOXP3 and display strong suppressor ability over CD4 T cell proliferation and cytokine production, and CD8 T cell cytotoxicity. Possession of the appropriate HLA-DR molecule and recognition of the specific CYP2D6 autoantigenic sequence are critical elements to antigen specific Treg suppressive function. Lack of either factor results in reduced control over responder cell proliferation and pro-inflammatory cytokine secretion [103]. Importantly, we found that exposure to all-trans-retinoic-acid (ATRA) could stabilize antigen-specific Tregs [104]. ATRA limited the increase in Th1 and Th17 transcription factors as well as the decrease in antigen-specific Treg function following pro-inflammatory challenge with IL-6 and IL-1β [104].
Studies by Oo and colleagues have shown that the liver represents a good target for Treg cell therapy, as Tregs isolated from leukapheresis products, labelled with indium tropolonate and re-infused i.v. into AIH patients, home to the liver where they could be retained for up to 72 hours [105]. Importantly, Umeshappa et al. reported that peptide-major histocompatibility class II (pMHC-II) based nanomedicines displaying tissue specific autoantigenic epitopes like CYP2D6398–412 could limit autoantigen specific T cell immunity in a mouse model of AIH, obtained upon infection of NOD mice with a replication-defective adenovirus encoding human FTCD [106, 107]. The beneficial effects of pMHC-II-based nanomedicine therapeutic activity was associated with the formation and expansion of Tr1 cells, a regulatory subset predominant in the chronic phase of inflammation [6, 106].
3.4. IL-2 and expansion of Tregs ex vivo
As autoantigen specific Tregs cannot be always generated because the autoantigen remains unknown in a large proportion of patients with AIH-1, treatment with low dose IL-2 has been considered as additional strategy to restore the defective Treg pool in AIH. In a clinical trial on 46 patients with various autoimmune diseases, including AIH and sclerosing cholangitis, participants were administered low dose IL-2 (1 million IU/day) for 5 days, followed by fortnightly injections for 6 months. The Authors reported Treg expansion and activation in all cases, although specific data on AIH patients (n=2) are not presented [108]. Of note, in all other cases, increase in Treg percentage (expressed as fold change compared to baseline level) was mainly evident on day 8 and was then contained thereafter, despite levels remained slightly higher when compared to baseline. Further, effects on clinical disease score is provided for patients affected by rheumatological disorders, ulcerative colitis and psoriasis but not for the two AIH patients enrolled in the study; therefore, based on these reported observations, the effects of low dose IL-2 in AIH patients remains unclear. In another study on two AIH patients with persistent disease activity, as assessed by serum biochemistry and liver biopsy, IL-2 was administered at 1 million IU for five days monthly for a total of six months [109]. In one of the two patients, AST and IgG decreased to normal levels by the end of the treatment; whereas the proportion of circulating Tregs increased in both cases with a peak observed at day 9 and returning to baseline levels at day 28. This suggests that the effect of low dose IL-2 on Treg frequency is transient. Further studies in larger numbers of subjects should be performed to assess low dose IL-2 efficacy as well as the long-term effects of this treatment on Tregs, particularly on their suppressive function, expansion and plasticity.
4. Tregs in other autoimmune liver diseases
4.1. Primary biliary cholangitis
The role of Tregs in the maintenance of immune tolerance has been studied also in other autoimmune liver diseases, namely primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). Defects in suppressor cells in PBC were reported in early studies in the 1980s [110]. Later studies reported impaired frequencies of CD4+CD25high [111, 112] or CD4+FOXP3+ [113], but normal suppressive function [111, 114]. Other immunoregulatory cell defects have been reported in PBC, these mainly involving the CD8+CD28− subset, a subpopulation of regulatory lymphocytes that suppress via IL-10 and TGF-β [114, 115]. The findings of increased FOXP3+ cells in the liver and of a direct correlation between the expression of FOXP3 and portal inflammation indicate either a compartmentalization of Tregs in the areas of tissue damage or upregulation of FOXP3 subsequent to activation in non-regulatory lymphocytes. Importantly IL-2Rα/CD25 deficient mice were found to develop portal inflammation and biliary ductular damage similar to what observed in PBC patients [116]. Notably, in these mice, lack of Tregs resulted in CD8 T cell mediated damage [117].
The importance of Treg mediated immune homeostasis in PBC was further supported by a subsequent study in scurfy mice that have a mutation in the gene encoding the Foxp3 transcription factor leading to complete deletion of Foxp3+ Tregs. These investigations showed that between 3 to 4 weeks of age all animals develop AMA autoantibodies along with a moderate to severe lymphocytic infiltrate in the portal areas and bile duct damage [118]. Further evidence of the key role of Treg immune responses in protecting from liver damage in murine autoimmune cholangitis has been provided by the work of Huang et al [119] who emphasized the importance of the permissive role played by defective Tregs in favoring CD8 T cell cytotoxicity in this model. In another murine model of PBC, the dominant negative transforming growth factor β receptor II (dnTGFβRII) mice, there was a substantial decrease in the expression of Treg related transcription factors, including Eos, Ahr, Klf2 and Foxp1 [120] that was associated with a pro-inflammatory phenotype of regulatory cells [120]. Recent findings from Liaskou and colleagues showed that Tregs obtained from PBC patients have increased susceptibility to low dose IL-12 that favors their differentiation into IFN-γ secreting cells. The acquisition of proinflammatory properties by Tregs might be pivotal in the perpetuation of tissue and bile duct damage in PBC [121].
As for AIH, administration of pMHC-based nanomedicines that display autoantigenic epitopes, specifically the pyruvate dehydrogenase complex II (PDC-E2)166–188 and PDC-E282–96 resulted in Tr1 cell expansion and recruitment to the liver in NOD.c3c4 mice that spontaneously develop a form of autoimmune biliary disease resembling human PBC [122]. Migration of PDC-E2 specific Tregs to the liver enabled control of liver autoimmunity without impacting systemic or local autoimmunity against infections or tumors [106].
4.2. Primary sclerosing cholangitis
In PSC, another autoimmune liver disease characterized by inflammation and fibrosis of intra and extrahepatic bile ducts that eventually progresses to biliary cirrhosis and hepatic decompensation, mucosal and functional impairment of peripheral blood derived CD4+CD25highFOXP3+CD127low cells has been reported [123]. In a similar way, liver derived Tregs from PSC patients are also fewer in numbers when compared to other conditions like PBC [123]. Akin to PSC, pediatric patients with AISC display numerically and functionally impaired Tregs [79, 81, 92]. Administration of low dose IL-2 has been considered to achieve in vivo Treg expansion in MDR2−/− mice, a model of PSC [124]. Low dose IL-2 administration resulted in upregulation of CD39 on Tregs that enabled control over CD8 T cell effector function [124].
Collectively, these studies in murine models and humans support the key role of Tregs mediated immune homeostasis while preventing tissue damage progression.
5. Future perspectives
Restoring Treg pool, either through adoptive transfer of autologous Tregs - freshly isolated or generated in vitro - or through administration of low dose IL-2, would not only enable to control autoreactive immune responses and inflammation, but would also favor re-establishing immune tolerance. This might have strong, important implications for curing, rather than only treating, AILD, provided Treg restoration is stable and long-lasting. Because of the potential of reconstituting immune tolerance, Treg adoptive transfer is preferable to any form of drug-based immunosuppression that is non-specific and does not always prevent progression of liver tissue damage. Although low dose IL-2 has been shown having some beneficial effects by expanding the already existing Treg pool, it remains a non-specific approach that could also target other non-regulatory cell populations, including effector CD4 and CD8 lymphocytes and NK cells. As indicated above, the applications of this approach in the AILD context have been so far limited to few cases only, therefore highlighting the need for larger and more comprehensive clinical investigations.
When considering adoptive Treg transfer, numerous pre-clinical studies in the autoimmune and transplantation settings have provided evidence about antigen-specific Treg superior efficacy, when compared to polyclonal Tregs [125–128]. In addition to enhanced efficacy, antigen-specific Tregs provide a more tailored form of immunosuppression by localizing at the site of antigen presentation and targeting effectors of the same antigen specificity. There is also evidence that antigen-specific Tregs suppress effectors of different antigen specificity through bystander suppression and could also promote the emergence of other suppressor cells, like Tr1 cells.
Since the use of antigen-specific Tregs could be hampered by their low expansion capabilities, recent studies have reported the possibility of engineering antigen-specific Tregs, by deriving them from polyclonal Tregs through transduction of effector cell, or preferably, Treg TCR, or chimeric antigen receptor (CAR) (reviewed in [129]). CAR consists of the antigenic binding site of a monoclonal antibody in the extracellular domain and of T cell stimulatory and costimulatory intracellular domains. Because of the limitations related to the use of recombinant viral vectors inserting the transgene and the potential that the transgene could randomly integrate in the genome, CRISPR Cas9 mediated approaches, where endogenous TCRs could be replaced by a recombinant one, would represent a better option. Additional approaches based on induced FOXP3 expression in antigen-specific conventional CD4 cells would require further evidence before being applied, due to the risk of obtaining cells that, although overexpressing FOXP3, still maintain the function and epigenetic make-up of effectors.
Additional issues that should be addressed include plasticity, which is associated with loss of FOXP3 by Tregs and conversion to antigen-specific effectors, a risk that should be taken into consideration especially when obtaining Tregs from conventional CD4+ T cells; and stability that could be addressed by either inducing FOXP3 overexpression or upon knockout of selective pro-inflammatory genes. Another challenge might be represented by Treg persistence in the bloodstream. Since Tregs depend on IL-2 for their survival, expansion and function, several approaches have been developed to obtain IL-2 mutants that, by prolonging the half-life of IL-2, promote Treg expansion in vivo [130, 131]. Another strategy that has been proposed consists of the administration of monoclonal antibody/IL-2 complex to selectively expand Tregs in a mouse model of colitis [132] and AIH [133]. In this regard, Karakus et al. recently reported that a complex of a newly identified anti-human IL-2 antibody, UFKA-20, and IL-2 could stimulate Tregs among freshly isolated human T cells ex vivo and in rhesus macaques in vivo [134].
Treg cell homing would be another aspect to consider when implementing cell therapy-based approaches. In a study on autoantigen-specific Tregs in the AIH-2 setting, we found that antigen-specific Tregs obtained after co-culture with smDCs express CXCR3, a chemokine receptor normally present on lymphocytes trafficking to the liver [103].
An additional challenge is represented by the high costs required to generate and expand Tregs under GMP conditions; this represents a major limiting factor for a wide application of this form of immunotherapy. Further, it is still unclear whether antigen-specific Tregs should be administered once inflammation has been controlled, given previous studies demonstrating that even potent antigen-specific Tregs fail to suppress in the presence of a pro-inflammatory environment [135].
Although these challenges might limit the clinical use of adoptively transferred antigen-specific Tregs in AILD and other autoimmune conditions, continuous efforts and resources should be allocated to optimize or identify novel strategies enabling to attain immune homeostasis reconstitution, rather than aiming at containing inflammatory responses only.
6. Conclusions
Treg defects play a key permissive role in autoimmune disease pathogenesis by favoring overwhelming effector T cell immunity and, consequently, tissue damage exacerbation and progression. Numerical and functional impairment of Tregs and alterations of the associated signaling pathways is pivotal in determining CD4 and CD8 T cell autoreactivity in AIH and other autoimmune liver diseases. Treg impairment can be also associated with phenotypic and functional plasticity that results in Treg skewing to effector cells; this being regarded as a mechanism enabling the maintenance of proinflammatory T cell pools. Potential strategies aimed at restoring polyclonal and/or antigen specific Tregs, either through the generation and expansion of these cells in the test tube or in vivo, upon administration of low dose IL-2, have been reviewed and discussed. Future studies are warranted to determine the efficacy and the feasibility of these approaches in large patients’ cohorts.
Highlights.
Treg impairment plays a permissive role in AILD immunopathogenesis
Treg impairment in AILD results in overwhelming effector cell immunity
In AIH Treg defects are associated with increased functional plasticity
Challenges remain when implementing strategies for Treg generation/expansion
Funding
MSL is supported by the National Institute of Health (R01 DK108894 and R01 DK124408) and by the Seed Grant Award (Department of Anesthesia, Critical Care and Pain Medicine, BIDMC).
Abbreviations:
- Tregs
regulatory T cells
- AIH
autoimmune hepatitis
- AIH-1
type 1 AIH
- AIH-2
type 2 AIH
- AISC
autoimmune sclerosing cholangitis
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- FOXP3
transcription factor box P3
- Tconv
conventional T cells
- APC
antigen presenting cell
- AhR
aryl hydrocarbon receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interest statement: The Authors declare that no competing interests exist
References
- [1].Wing JB, Sakaguchi S. Multiple treg suppressive modules and their adaptability. Front Immunol, 2012;3:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang YM, Alexander SI. CD8 regulatory T cells: what’s old is now new. Immunol Cell Biol, 2009;87:192–3. [DOI] [PubMed] [Google Scholar]
- [3].Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol, 2010;10:236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity, 2015;42:607–12. [DOI] [PubMed] [Google Scholar]
- [5].Subleski JJ, Ortaldo JR. Editorial: NKT cells: to suppress or not to suppress, that is the question. J Leukoc Biol, 2009;86:751–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol, 2014;380:39–68. [DOI] [PubMed] [Google Scholar]
- [7].Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med, 1996;184:387–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol, 1995;155:1151–64. [PubMed] [Google Scholar]
- [9].Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol, 2003;4:330–6. [DOI] [PubMed] [Google Scholar]
- [10].Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol, 2003;4:337–42. [DOI] [PubMed] [Google Scholar]
- [11].Roncador G, Brown PJ, Maestre L, Hue S, Martinez-Torrecuadrada JL, Ling KL et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol, 2005;35:1681–91. [DOI] [PubMed] [Google Scholar]
- [12].Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med, 2006;203:1701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol, 2001;2:301–6. [DOI] [PubMed] [Google Scholar]
- [14].Wirnsberger G, Hinterberger M, Klein L. Regulatory T-cell differentiation versus clonal deletion of autoreactive thymocytes. Immunol Cell Biol, 2011;89:45–53. [DOI] [PubMed] [Google Scholar]
- [15].Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol, 2013;14:307–8. [DOI] [PubMed] [Google Scholar]
- [16].Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med, 2004;199:1401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol, 2005;6:1219–27. [DOI] [PubMed] [Google Scholar]
- [18].Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol, 2007;8:931–41. [DOI] [PubMed] [Google Scholar]
- [19].Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol, 2008;38:1654–63. [DOI] [PubMed] [Google Scholar]
- [20].Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity, 2012;37:785–99. [DOI] [PubMed] [Google Scholar]
- [21].Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev, 2014;259:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol, 2020;38:541–66. [DOI] [PubMed] [Google Scholar]
- [23].Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol, 2009;21:281–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity, 2013;39:949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol, 2009;10:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest, 2011;121:4503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol, 2011;12:304–11. [DOI] [PubMed] [Google Scholar]
- [28].Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science, 2015;349:993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science, 2015;349:989–93. [DOI] [PubMed] [Google Scholar]
- [30].Kullberg MC, Hay V, Cheever AW, Mamura M, Sher A, Letterio JJ et al. TGF-beta1 production by CD4+ CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur J Immunol, 2005;35:2886–95. [DOI] [PubMed] [Google Scholar]
- [31].von Boehmer H Mechanisms of suppression by suppressor T cells. Nat Immunol, 2005;6:338–44. [DOI] [PubMed] [Google Scholar]
- [32].Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med, 2005;201:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med, 2001;194:629–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med, 1996;183:2669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med, 1999;190:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol, 2001;166:3789–96. [DOI] [PubMed] [Google Scholar]
- [37].Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature, 2007;450:566–9. [DOI] [PubMed] [Google Scholar]
- [38].Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity, 2008;28:546–58. [DOI] [PubMed] [Google Scholar]
- [39].Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol, 2007;8:942–9. [DOI] [PubMed] [Google Scholar]
- [40].Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell, 2006;126:1121–33. [DOI] [PubMed] [Google Scholar]
- [41].Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+)CD25(+) regulatory T cells. Cell Death Differ, 2007;14:2076–84. [DOI] [PubMed] [Google Scholar]
- [42].Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol, 2009;182:1469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity, 2007;27:635–46. [DOI] [PubMed] [Google Scholar]
- [44].Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol, 2005;174:1783–6. [DOI] [PubMed] [Google Scholar]
- [45].Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood, 2006;107:3925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity, 2004;21:589–601. [DOI] [PubMed] [Google Scholar]
- [47].Askenasy N Enhanced killing activity of regulatory T cells ameliorates inflammation and autoimmunity. Autoimmun Rev, 2013;12:972–5. [DOI] [PubMed] [Google Scholar]
- [48].Kashio Y, Nakamura K, Abedin MJ, Seki M, Nishi N, Yoshida N et al. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J Immunol, 2003;170:3631–6. [DOI] [PubMed] [Google Scholar]
- [49].Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol, 2007;8:1353–62. [DOI] [PubMed] [Google Scholar]
- [50].Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med, 2007;204:1303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vaeth M, Gogishvili T, Bopp T, Klein M, Berberich-Siebelt F, Gattenloehner S et al. Regulatory T cells facilitate the nuclear accumulation of inducible cAMP early repressor (ICER) and suppress nuclear factor of activated T cell c1 (NFATc1). Proc Natl Acad Sci U S A, 2011;108:2480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bodor J, Bopp T, Vaeth M, Klein M, Serfling E, Hunig T et al. Cyclic AMP underpins suppression by regulatory T cells. Eur J Immunol, 2012;42:1375–84. [DOI] [PubMed] [Google Scholar]
- [53].Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med, 2012;367:2322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med, 2007;204:1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol, 2009;183:7602–10. [DOI] [PubMed] [Google Scholar]
- [56].Peres RS, Liew FY, Talbot J, Carregaro V, Oliveira RD, Almeida SL et al. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc Natl Acad Sci U S A, 2015;112:2509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate costimulatory molecules on antigen-presenting cells. Eur J Immunol, 2000;30:1538–43. [DOI] [PubMed] [Google Scholar]
- [58].Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science, 2011;332:600–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med, 2004;200:277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med, 2004;199:971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mao C, Wang S, Xiao Y, Xu J, Jiang Q, Jin M et al. Impairment of regulatory capacity of CD4+CD25+ regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves’ disease. J Immunol, 2011;186:4734–43. [DOI] [PubMed] [Google Scholar]
- [62].Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes, 2005;54:92–9. [DOI] [PubMed] [Google Scholar]
- [63].Liberal R, Grant CR, Longhi MS, Mieli-Vergani G, Vergani D. Diagnostic criteria of autoimmune hepatitis. Autoimmun Rev, 2014;13:435–40. [DOI] [PubMed] [Google Scholar]
- [64].Heneghan MA, Yeoman AD, Verma S, Smith AD, Longhi MS. Autoimmune hepatitis. Lancet, 2013;382:1433–44. [DOI] [PubMed] [Google Scholar]
- [65].Grant CR, Liberal R, Mieli-Vergani G, Vergani D, Longhi MS. Regulatory T-cells in autoimmune diseases: challenges, controversies and--yet--unanswered questions. Autoimmun Rev, 2015;14:105–16. [DOI] [PubMed] [Google Scholar]
- [66].Herkel J, Jagemann B, Wiegard C, Lazaro JF, Lueth S, Kanzler S et al. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocyutes. Hepatology, 2003;37:1079–85. [DOI] [PubMed] [Google Scholar]
- [67].Crispe IN. Liver antigen-presenting cells. J Hepatol, 2011;54:357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lobo-Yeo A, Senaldi G, Portmann B, Mowat AP, Mieli-Vergani G, Vergani D. Class I and class II major histocompatibility complex antigen expression on hepatocytes: a study in children with liver disease. Hepatology, 1990;12:224–32. [DOI] [PubMed] [Google Scholar]
- [69].Franco A, Barnaba V, Ruberti G, Benvenuto R, Balsano C, Musca A. Liver-derived T cell clones in autoimmune chronic active hepatitis: accessory cell function of hepatocytes expressing class II major histocompatibility complex molecules. Clin Immunol Immunopathol, 1990;54:382–94. [DOI] [PubMed] [Google Scholar]
- [70].Cochrane AM, Moussouros A, Smith A, Portmann B, Eddleston AL, Williams R. Lymphocyte cytotoxicity in chronic active hepatitis: effect of therapy and correlations with clinical and histological changes. Gut, 1978;19:308–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cochrane AM, Moussouros A, Thomsom AD, Eddleston AL, Wiiliams R. Antibody-dependent cell-mediated (K cell) cytotoxicity against isolated hepatocytes in chronic active hepatitis. Lancet, 1976;1:441–4. [DOI] [PubMed] [Google Scholar]
- [72].Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One, 2011;6:e18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Beringer A, Miossec P. IL-17 and IL-17-producing cells and liver diseases, with focus on autoimmune liver diseases. Autoimmun Rev, 2018;17:1176–85. [DOI] [PubMed] [Google Scholar]
- [74].Hodgson HJ, Wands JR, Isselbacher KJ. Alteration in suppressor cell activity in chronic active hepatitis. Proc Natl Acad Sci U S A, 1978;75:1549–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Nouri-Aria KT, Hegarty JE, Alexander GJ, Eddleston AL, Williams R. Effect of corticosteroids on suppressor-cell activity in “autoimmune” and viral chronic active hepatitis. N Engl J Med, 1982;307:1301–4. [DOI] [PubMed] [Google Scholar]
- [76].Vento S, Hegarty JE, Bottazzo G, Macchia E, Williams R, Eddleston AL. Antigen specific suppressor cell function in autoimmune chronic active hepatitis. Lancet, 1984;1:1200–4. [DOI] [PubMed] [Google Scholar]
- [77].Gregorio GV, Portmann B, Karani J, Harrison P, Donaldson PT, Vergani D et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology, 2001;33:544–53. [DOI] [PubMed] [Google Scholar]
- [78].Terziroli Beretta-Piccoli B, Vergani D, Mieli-Vergani G. Autoimmune sclerosing cholangitis: Evidence and open questions. J Autoimmun, 2018;95:15–25. [DOI] [PubMed] [Google Scholar]
- [79].Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol, 2004;41:31–7. [DOI] [PubMed] [Google Scholar]
- [80].Longhi MS, Ma Y, Mitry RR, Bogdanos DP, Heneghan M, Cheeseman P et al. Effect of CD4+ CD25+ regulatory T-cells on CD8 T-cell function in patients with autoimmune hepatitis. J Autoimmun, 2005;25:63–71. [DOI] [PubMed] [Google Scholar]
- [81].Longhi MS, Hussain MJ, Mitry RR, Arora SK, Mieli-Vergani G, Vergani D et al. Functional study of CD4+CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol, 2006;176:4484–91. [DOI] [PubMed] [Google Scholar]
- [82].Longhi MS, Mitry RR, Samyn M, Scalori A, Hussain MJ, Quaglia A et al. Vigorous activation of monocytes in juvenile autoimmune liver disease escapes the control of regulatory T-cells. Hepatology, 2009;50:130–42. [DOI] [PubMed] [Google Scholar]
- [83].Ferri S, Longhi MS, De Molo C, Lalanne C, Muratori P, Granito A et al. A multifaceted imbalance of T cells with regulatory function characterizes type 1 autoimmune hepatitis. Hepatology, 2010;52:999–1007. [DOI] [PubMed] [Google Scholar]
- [84].Longhi MS, Ma Y, Grant CR, Samyn M, Gordon P, Mieli-Vergani G et al. T-regs in autoimmune hepatitis-systemic lupus erythematosus/mixed connective tissue disease overlap syndrome are functionally defective and display a Th1 cytokine profile. J Autoimmun, 2013;41:146–51. [DOI] [PubMed] [Google Scholar]
- [85].Peiseler M, Sebode M, Franke B, Wortmann F, Schwinge D, Quaas A et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol, 2012;57:125–32. [DOI] [PubMed] [Google Scholar]
- [86].Taubert R, Hardtke-Wolenski M, Noyan F, Wilms A, Baumann AK, Schlue J et al. Intrahepatic regulatory T cells in autoimmune hepatitis are associated with treatment response and depleted with current therapies. J Hepatol, 2014;61:1106–14. [DOI] [PubMed] [Google Scholar]
- [87].Diestelhorst J, Junge N, Schlue J, Falk CS, Manns MP, Baumann U et al. Pediatric autoimmune hepatitis shows a disproportionate decline of regulatory T cells in the liver and of IL-2 in the blood of patients undergoing therapy. PLoS One, 2017;12:e0181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Jeffery HC, Braitch MK, Bagnall C, Hodson J, Jeffery LE, Wawman RE et al. Changes in natural killer cells and exhausted memory regulatory T Cells with corticosteroid therapy in acute autoimmune hepatitis. Hepatol Commun, 2018;2:421–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Yuksel M, Wang Y, Tai N, Peng J, Guo J, Beland K et al. A novel “humanized mouse” model for autoimmune hepatitis and the association of gut microbiota with liver inflammation. Hepatology, 2015;62:1536–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].An Haack I, Derkow K, Riehn M, Rentinck MN, Kuhl AA, Lehnardt S et al. The Role of Regulatory CD4 T Cells in Maintaining Tolerance in a Mouse Model of Autoimmune Hepatitis. PLoS One, 2015;10:e0143715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yuksel M, Xiao X, Tai N, Vijay M, Gulden E, Beland K et al. The induction of autoimmune hepatitis in the human leucocyte antigen-DR4 non-obese diabetic mice autoimmune hepatitis mouse model. Clin Exp Immunol, 2016;186:164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Liberal R, Grant CR, Holder BS, Ma Y, Mieli-Vergani G, Vergani D et al. The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin-9/tim-3 pathway. Hepatology, 2012;56:677–86. [DOI] [PubMed] [Google Scholar]
- [93].Grant CR, Holder BS, Liberal R, Heneghan MA, Ma Y, Mieli-Vergani G et al. Immunosuppressive drugs affect interferon (IFN)-gamma and programmed cell death 1 (PD-1) kinetics in patients with newly diagnosed autoimmune hepatitis. Clin Exp Immunol, 2017;189:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Grant CR, Liberal R, Holder BS, Cardone J, Ma Y, Robson SC et al. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology, 2014;59:1007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Vuerich M, Harshe R, Frank LA, Mukherjee S, Gromova B, Csizmadia E et al. Altered aryl-hydrocarbon-receptor signalling affects regulatory and effector cell immunity in autoimmune hepatitis. J Hepatol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature, 2008;453:65–71. [DOI] [PubMed] [Google Scholar]
- [97].Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature, 2008;453:106–9. [DOI] [PubMed] [Google Scholar]
- [98].Lytton SD, Osiecki M, MalgorzataWozniak, Cukrowska B, Wierzbicka A, Goliszek M et al. Tryptophan-kynurenine profile in pediatric autoimmune hepatitis. Immunol Res, 2019;67:39–47. [DOI] [PubMed] [Google Scholar]
- [99].Arterbery AS, Osafo-Addo A, Avitzur Y, Ciarleglio M, Deng Y, Lobritto SJ et al. Production of Proinflammatory Cytokines by Monocytes in Liver-Transplanted Recipients with De Novo Autoimmune Hepatitis Is Enhanced and Induces TH1-like Regulatory T Cells. J Immunol, 2016;196:4040–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Arterbery AS, Yao J, Ling A, Avitzur Y, Martinez M, Lobritto S et al. Inflammasome Priming Mediated via Toll-Like Receptors 2 and 4, Induces Th1-Like Regulatory T Cells in De Novo Autoimmune Hepatitis. Front Immunol, 2018;9:1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Longhi MS, Meda F, Wang P, Samyn M, Mieli-Vergani G, Vergani D et al. Expansion and de novo generation of potentially therapeutic regulatory T cells in patients with autoimmune hepatitis. Hepatology, 2008;47:581–91. [DOI] [PubMed] [Google Scholar]
- [102].Longhi MS, Liberal R, Holder B, Robson SC, Ma Y, Mieli-Vergani G et al. Inhibition of interleukin-17 promotes differentiation of CD25(−) cells into stable T regulatory cells in patients with autoimmune hepatitis. Gastroenterology, 2012;142:1526–35 e6. [DOI] [PubMed] [Google Scholar]
- [103].Longhi MS, Hussain MJ, Kwok WW, Mieli-Vergani G, Ma Y, Vergani D. Autoantigen-specific regulatory T cells, a potential tool for immune-tolerance reconstitution in type-2 autoimmune hepatitis. Hepatology, 2011;53:536–47. [DOI] [PubMed] [Google Scholar]
- [104].Holder BS, Grant CR, Liberal R, Ma Y, Heneghan MA, Mieli-Vergani G et al. Retinoic acid stabilizes antigen-specific regulatory T-cell function in autoimmune hepatitis type 2. J Autoimmun, 2014;53:26–32. [DOI] [PubMed] [Google Scholar]
- [105].Oo YH, Ackrill S, Cole R, Jenkins L, Anderson P, Jeffery HC et al. Liver homing of clinical grade Tregs after therapeutic infusion in patients with autoimmune hepatitis. JHEP Rep, 2019;1:286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J et al. Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat Commun, 2019;10:2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hardtke-Wolenski M, Fischer K, Noyan F, Schlue J, Falk CS, Stahlhut M et al. Genetic predisposition and environmental danger signals initiate chronic autoimmune hepatitis driven by CD4+ T cells. Hepatology, 2013;58:718–28. [DOI] [PubMed] [Google Scholar]
- [108].Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis, 2019;78:209–17. [DOI] [PubMed] [Google Scholar]
- [109].Lim TY, Martinez-Llordella M, Kodela E, Gray E, Heneghan MA, Sanchez-Fueyo A. Low-Dose Interleukin-2 for Refractory Autoimmune Hepatitis. Hepatology, 2018;68:1649–52. [DOI] [PubMed] [Google Scholar]
- [110].Vento S, O’Brien CJ, McFarlane BM, McFarlane IG, Eddleston AL, Williams R. T-lymphocyte sensitization to hepatocyte antigens in autoimmune chronic active hepatitis and primary biliary cirrhosis. Evidence for different underlying mechanisms and different antigenic determinants as targets. Gastroenterology, 1986;91:810–7. [DOI] [PubMed] [Google Scholar]
- [111].Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology, 2006;43:729–37. [DOI] [PubMed] [Google Scholar]
- [112].Wang D, Zhang H, Liang J, Gu Z, Zhou Q, Fan X et al. CD4+ CD25+ but not CD4+ Foxp3+ T cells as a regulatory subset in primary biliary cirrhosis. Cell Mol Immunol, 2010;7:485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Rong G, Zhou Y, Xiong Y, Zhou L, Geng H, Jiang T et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol, 2009;156:217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Bernuzzi F, Fenoglio D, Battaglia F, Fravega M, Gershwin ME, Indiveri F et al. Phenotypical and functional alterations of CD8 regulatory T cells in primary biliary cirrhosis. J Autoimmun, 2010;35:176–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Vuddamalay Y, van Meerwijk JP. CD28(−) and CD28(low)CD8(+) Regulatory T Cells: Of Mice and Men. Front Immunol, 2017;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH et al. IL-2 receptor alpha(−/−) mice and the development of primary biliary cirrhosis. Hepatology, 2006;44:1240–9. [DOI] [PubMed] [Google Scholar]
- [117].Hsu W, Zhang W, Tsuneyama K, Moritoki Y, Ridgway WM, Ansari AA et al. Differential mechanisms in the pathogenesis of autoimmune cholangitis versus inflammatory bowel disease in interleukin-2Ralpha(−/−) mice. Hepatology, 2009;49:133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhang W, Sharma R, Ju ST, He XS, Tao Y, Tsuneyama K et al. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology, 2009;49:545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Huang W, Kachapati K, Adams D, Wu Y, Leung PS, Yang GX et al. Murine autoimmune cholangitis requires two hits: cytotoxic KLRG1(+) CD8 effector cells and defective T regulatory cells. J Autoimmun, 2014;50:123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wang YH, Yang W, Yang JB, Jia YJ, Tang W, Gershwin ME et al. Systems biologic analysis of T regulatory cells genetic pathways in murine primary biliary cirrhosis. J Autoimmun, 2015;59:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Liaskou E, Patel SR, Webb G, Bagkou Dimakou D, Akiror S, Krishna M et al. Increased sensitivity of Treg cells from patients with PBC to low dose IL-12 drives their differentiation into IFN-gamma secreting cells. J Autoimmun, 2018;94:143–55. [DOI] [PubMed] [Google Scholar]
- [122].Irie J, Wu Y, Wicker LS, Rainbow D, Nalesnik MA, Hirsch R et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med, 2006;203:1209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Sebode M, Peiseler M, Franke B, Schwinge D, Schoknecht T, Wortmann F et al. Reduced FOXP3(+) regulatory T cells in patients with primary sclerosing cholangitis are associated with IL2RA gene polymorphisms. J Hepatol, 2014;60:1010–6. [DOI] [PubMed] [Google Scholar]
- [124].Taylor AE, Carey AN, Kudira R, Lages CS, Shi T, Lam S et al. Interleukin 2 Promotes Hepatic Regulatory T Cell Responses and Protects From Biliary Fibrosis in Murine Sclerosing Cholangitis. Hepatology, 2018;68:1905–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med, 2004;199:1455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med, 2004;199:1467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol, 2009;39:1108–17. [DOI] [PubMed] [Google Scholar]
- [128].Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest, 2003;112:1688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol, 2020;20:158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature, 2012;484:529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity, 2015;42:826–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Spangler JB, Trotta E, Tomala J, Peck A, Young TA, Savvides CS et al. Engineering a Single-Agent Cytokine/Antibody Fusion That Selectively Expands Regulatory T Cells for Autoimmune Disease Therapy. J Immunol, 2018;201:2094–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Buitrago-Molina LE, Pietrek J, Noyan F, Schlue J, Manns MP, Wedemeyer H et al. Treg-specific IL-2 therapy can reestablish intrahepatic immune regulation in autoimmune hepatitis. J Autoimmun, 2020;117:102591. [DOI] [PubMed] [Google Scholar]
- [134].Murar CE, Ninomiya M, Shimura S, Karakus U, Boyman O, Bode JW. Chemical Synthesis of Interleukin-2 and Disulfide Stabilizing Analogues. Angew Chem Int Ed Engl, 2020;59:8425–9. [DOI] [PubMed] [Google Scholar]
- [135].Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med, 2007;13:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]