

Abstract
Dollo’s law posits that evolutionary losses are irreversible, thereby narrowing the potential paths of evolutionary change. While phenotypic reversals to ancestral states have been observed, little is known about their underlying genetic causes. The genomes of budding yeasts have been shaped by extensive reductive evolution, such as reduced genome sizes and the losses of metabolic capabilities. However, the extent and mechanisms of trait reacquisition after gene loss in yeasts have not been thoroughly studied. Here, through phylogenomic analyses, we reconstructed the evolutionary history of the yeast galactose utilization pathway and observed widespread and repeated losses of the ability to utilize galactose, which occurred concurrently with the losses of GALactose (GAL) utilization genes. Unexpectedly, we detected multiple galactose-utilizing lineages that were deeply embedded within clades that underwent ancient losses of galactose utilization. We show that at least two, and possibly three, lineages reacquired the GAL pathway via yeast-to-yeast horizontal gene transfer. Our results show how trait reacquisition can occur tens of millions of years after an initial loss via horizontal gene transfer from distant relatives. These findings demonstrate that the losses of complex traits and even whole pathways are not always evolutionary dead-ends, highlighting how reversals to ancestral states can occur.
Keywords: gene loss, evolution, GAL cluster, Dollo’s law, horizontal gene transfer, yeasts, lateral gene transfer
Introduction
Understanding the interactions between a species’ phenotype, genotype, and environment is a central goal of evolutionary biology. Of particular interest are the mechanisms by which the environment selects for changes in phenotype and subsequently genome content. Budding yeast are present in an extraordinary range of environments, and accordingly, display remarkable physiological diversity (Hittinger et al. 2015). Alongside robustly characterized physiologies (Kurtzman et al. 2011) and the availability of an unrivaled set of genome sequences (Hittinger et al. 2015; Shen et al. 2016, 2018), budding yeasts provide a unique subphylum-level eukaryotic model for studying the interplay between the genome, phenotype, and the environment.
Trait reversal is an intriguing phenomenon whereby the character state of a particular evolutionary lineage returns to its ancestral state. For more than a century, trait reversal after a loss event has been thought to be highly unlikely; Dollo’s law of irreversibility states that, once a trait is lost, it is unlikely for the same trait to be found in a descendant lineage, thereby excluding certain evolutionary paths (Dollo 1893; Simpson 1953). Despite this purist interpretation, many examples of apparent violations to Dollo’s law have been documented (Collin and Cipriani 2003; Whiting et al. 2003; Kohlsdorf and Wagner 2006; Brandley et al. 2008; Kohlsdorf et al. 2010; Lynch and Wagner 2010; Wiens 2011; Xu et al. 2016; Recknagel et al. 2018), and it is clear that evolutionary processes sometimes break Dollo’s law (Collin and Miglietta 2008; Seher et al. 2012; Esfeld et al. 2018). Nonetheless, the molecular and genetic mechanisms leading to trait reversal have only been determined in a few cases (Seher et al. 2012; Esfeld et al. 2018). For example, it was recently shown that flower color reversal in a Petunia species was facilitated by the resurrection of a pseudogene (Esfeld et al. 2018). In this case, the reversal was temporally rapid, which is in agreement with the hypothesis that traits flicker on and off during speciation (Collin and Miglietta 2008). These results underscore that complex traits do indeed undergo reversal and help identify one possible genetic mechanism for doing so. In other cases, traits have been reversed long after the speciation process and long after pseudogenes are undetectable (Collin and Cipriani 2003; Chippindale et al. 2004), raising the question of how trait reversal can occur millions of years after the initial loss.
The Leloir pathway of galactose utilization in the model budding yeast Saccharomyces cerevisiae (subphylum Saccharomycotina) is one of the most intensely studied and well-understood genetic, regulatory, and metabolic pathways of any eukaryote (Johnston 1987; Jayadeva Bhat and Murthy 2001; Hittinger et al. 2004; Hittinger and Carroll 2007; Martchenko et al. 2007; Hittinger et al. 2010; Slot and Rokas 2010; Wolfe et al. 2015; Kuang et al. 2016, 2018). Although its regulatory genes are unlinked, the GAL genes encoding the three key catabolic enzymes (GAL1, GAL7, and GAL10) are present in a localized gene cluster (Slot and Rokas 2010). A critical consequence of clustering genes in fungi is a marked increase in the rate of gene loss (Hittinger et al. 2004; Slot and Rokas 2010; Campbell et al. 2013; Wisecaver et al. 2014; Wisecaver and Rokas 2015) and a striking increase in the incidence of horizontal gene transfer (HGT) of those genes (Wisecaver and Rokas 2015; Slot 2017). The principal mode of evolution for the GAL gene cluster has been differential gene loss from an ancestral species that possessed the GAL genes in a cluster (Hittinger et al. 2004; Slot and Rokas 2010; Riley et al. 2016; Shen et al. 2018). In one case, the budding yeast GAL enzymatic gene cluster was horizontally transferred into the fission yeast Schizosaccharomyces pombe (subphylum Taphrinomycotina) (Slot and Rokas 2010). Nonetheless, this transferred cluster is not functional in typical growth assays, suggesting Sc. pombe GAL cluster may not be deployed catabolically or may respond to induction signals other than galactose (Matsuzawa et al. 2011). Dairy and some other strains of S. cerevisiae may have horizontally acquired a more active, transcriptionally rewired GAL pathway from an unknown outgroup of the genus Saccharomyces (Legras et al. 2018; Duan et al. 2019), or they may have preserved these two versions of the pathway through balancing selection (Boocock et al. 2019); nonetheless, trait reversal is highly unlikely under either interpretation because S. cerevisiae and its closest relatives are generally able to consume galactose. Collectively, these prior observations suggest that both cis-regulatory features and unlinked regulators play crucial roles in determining the function of horizontally transferred genes. Due to the widespread loss of GAL genes and the apparent ability for the GAL enzymatic gene cluster to be horizontally transferred intact, we hypothesized that budding yeast GAL clusters might break Dollo’s law under some conditions.
To address this hypothesis, we explored the genetic content and phenotypic capabilities of a diverse set of budding yeast genomes. Despite being deeply embedded within clades that underwent ancient losses of galactose metabolism, the genera Brettanomyces and Wickerhamomyces both contain representatives that could utilize galactose. Analyses of their genome sequences revealed GAL gene clusters that exhibited an unusually high degree of synteny with gene clusters in distantly related species. Further analysis of the genome of Nadsonia fulvescens showed that it also contains a GAL gene cluster that is remarkably similar to a distantly related species. Through rigorous phylogenetic hypothesis testing, we found strong evidence for the complete losses of the genes encoding the enzymes necessary for galactose catabolism, followed by their reacquisitions via independent yeast-to-yeast HGT events in at least two, and possibly three, cases. Genes lost in fungi have been regained via HGT from bacterial donors in several cases (Hall and Dietrich 2007; Keeling and Palmer 2008; Marcet-Houben and Gabaldón 2010; Fitzpatrick 2012; Alexander et al. 2016; Gonçalves et al. 2018; Kominek et al. 2019), but here we demonstrate an exceptionally clear example of a complex trait and its corresponding genes being lost and then regained to a eukaryotic form similar to its ancestral one. We conclude that multiple distantly related lineages of yeasts have circumvented evolutionary irreversibility, both at the molecular and phenotypic level, via eukaryotic HGT and that evolutionary paths are not absolutely constrained after trait loss.
Methods
GAL gene identification
We analyzed 96 publicly available genome sequences used in a recent study of the Saccharomycotina phylogeny (Shen et al. 2016) (86 Saccharomycotina, 10 outgroups), as well 10 additional species belonging to clades where we identified potentially deep losses of the GAL gene cluster. Of the latter 10 species, five genome sequences, including N. fulvescens var. fulvescens, were published recently (Shen et al. 2018), while genome sequences for five new species are published here. Due to their importance to this study and since previously published genome sequences may have been from different strains that were unavailable for phenotyping, eight additional genome sequences were generated for taxonomic type strains. In total, 104 genome sequences were analyzed. All genome sequences generated after a backbone phylogeny was compiled from data published before 2016 (Shen et al. 2016) are denoted Y1000+ in Supplementary Figures S7–S10. The presence of GAL genes in the genome assemblies was inferred with TBLASTN (Altschul et al. 1990) v2.7.1 using the Candida albicans Gal1, Gal7, and Gal10 sequences as queries, followed by extraction of the open reading frame centered on the location of the best hit. The structure and synteny of the clusters were manually curated and documented. For Saccharomyces kudriavzevii, where balanced variation is segregating for the GAL pathway (Hittinger et al. 2010), phylogenetic analyses were performed with the taxonomic type strain (cannot grow on galactose), whereas summary figures (Supplementary Figure S3 and Figures 1, 3, and 6) show a reference strain (ZP591) that can grow on galactose.
Figure 1.

Evolutionary history of galactose metabolism in budding yeasts. Left, species-level presence or absence of galactose utilization is mapped onto the relative divergence timetree (Supplementary Figures S2 and S3) with some clades collapsed (triangles). Branch color denotes the ability to metabolize galactose: blue (+) and red (–). The labeled black bars mark the branches of three key events in the evolution of the GAL cluster (A-cluster formation, B-relocation of ORF-Y into the cluster, and C- relocation of ORF-X into the cluster), which were manually inferred from the phylogeny and GAL cluster structures. Bolded species names indicate the three clades with unexpected GAL clusters. The dashed branch of the Nadsonia lineage indicates the ambiguity of the ancestral character state due to its extremely long branch. Right, GAL cluster structures are shown. For collapsed clades, a consensus cluster is shown (see Supplementary Figure S3 for all cluster structures).
Sequencing and assembly of genomes
For the new genomes sequenced here, genomic DNA was sonicated and ligated to Illumina sequencing adaptors as previously described (Hittinger et al. 2010). The paired-end library was sequenced on an Illumina HiSeq 2500 instrument, conducting a rapid 2 × 250 run. To generate whole-genome assemblies, paired-end Illumina reads were used as input to a meta-assembler pipeline iWGS (Zhou et al. 2016). The quality of the assemblies was assessed using QUAST (Gurevich et al. 2013) v3.1, and the best assembly for the newly described species was chosen based on N50 statistics.
GAL gene similarity analysis
To calculate the percent identities between Gal proteins, we first aligned the protein sequences for each species (see Supplementary Table S1 for species used) of Gal1, Gal7, and Gal10 and generated percent identity matrices using Clustal Omega (Sievers et al. 2011). These results were then subdivided into four groups: (A) the percent identities between species within the potential HGT recipient clade, (B) the percent identities between species of the recipient clade and their closest relative with GAL genes, (C) the percent identities between species of the recipient clade and species in the donor lineage, and (D) the percent identities between species of the recipient clade and an outgroup lineage (i.e., S. cerevisiae). Next, a similarity score was calculated by normalizing the percent identity values of each group to the average value of the fourth group:
When interpreting these results, the critical comparison is between group B and group C. If the difference in the mean similarity score of group B versus group C is negative, the score is consistent with a violation of the assumption of vertical inheritance. This interpretation is because one would expect proteins in group B to be more similar based on sharing a more recent common ancestor than the species compared in group C. However, further phylogentic hypothesis testing is required to formally test HGT.
Codon analysis
Codon content analysis, frequency, and relative synonymous codon usage (RSCU) of the GAL genes were carried out in DAMBE [v7.0.28; (Xia 2018)] using the appropriate species codon table (standard and yeast alternative nuclear). Genome-wide RSCU and codon frequency data were obtained from table S14 of LaBella et al. (2019).
Phylogenetic analyses
Sequence alignments were conducted using MAFFT (Katoh and Standley 2014) v 7.409 run in the “–auto” mode. Alignments were subjected to maximum-likelihood (ML) phylogenetic reconstruction using RAxML (Stamatakis 2014) v8.1.0 with 100 rapid bootstrap replicates. Constrained phylogenetic trees were generated with RAxML using the “-g” option. The constraint tree that served as our null hypothesis included all taxa, except those that are part of the three HGT recipient candidate species/lineages (Supplementary Figure S11A). Note that during ML inference, RAxML allows all taxa omitted from the constraint tree to be placed anywhere on the tree. Thus, the placement of the taxa associated with the three HGT recipient candidate species/lineages did not follow the species phylogeny. To test whether the placement of each of the three recipient candidate species/lineages was consistent with the species phylogeny (note that our null hypothesis is HGT and our alternative hypothesis is vertical descent), we also generated three constraint trees that were identical to the null constraint tree but also constrained each of the individual HGT recipient candidate species’/lineages’ placement to that expected according to the species phylogeny (Supplementary Figure S11, B–D). By testing the null hypothesis against the three alternative hypotheses (each of which constrained an HGT recipient candidate species/lineage to conform to the species phylogeny), we were able to independently test whether each HGT placement was statistically better supported than its species-phylogeny placement. Statistical support for the HGT events involving GAL genes was determined using the Approximately Unbiased (AU) test, by comparing 1-on-1, the ML tree for the null hypothesis with each of the ML trees corresponding to the alternative hypotheses. The AU test was performed with IQ-TREE (Nguyen et al. 2015) v1.6.8 (-au option), which was run with the General Time Reversible model, substitution rate heterogeneity approximated with the gamma distribution (-m GTR+G), and with 10,000 replicates (-zb 10000).
Regulatory motif enrichment
Sequences of 800-bp upstream of the start codon of all identified GAL genes were extracted and subjected to a regulatory motif identification analysis using MEME (Bailey et al. 2009) v5.0.2, with the following constraints: maximum number of motifs = 20 (-nmotifs 20), maximum length of motif = 25 bases (-maxw 25), any number of motif repetitions (-anr), active search of reverse complement of the used sequence (-revcomp), and the log-likelihood ratio method (-use_llr). Selective enrichment of motifs was determined by splitting the sequences into Saccharomycetaceae and non-Saccharomycetaceae groups and running AME (McLeay and Bailey 2010) v5.0.2, with each group being the control group in one analysis and the test group in a second analysis.
Species tree reconstruction
Our data matrix was composed of 104 budding yeasts and 10 outgroups, comprising 1219 BUSCO genes (601,996 amino acid sites); each gene had a minimum sequence occupancy ≥57 taxa and sequence length ≥167 amino acid residues. For the concatenation-base analysis, we used RAxML version 8.2.3 and IQ-TREE (Nguyen et al. 2015) version 1.5.1 to perform ML estimations under an unpartitioned scheme (an LG + GAMMA model) and a gene-based partition scheme (1219 partitions; each gene has its own model), respectively. As a result, four ML trees produced by two different phylogenetic programs and two different partition strategies were topologically identical. Branch support for each internode was evaluated with 100 rapid bootstrap replicates using RAxML (Stamatakis et al. 2008). For the coalescence-based analysis, we first estimated individual gene trees with their best-fitting amino acid models, which were determined by IQ-TREE (Nguyen et al. 2015) (the “–m TESTONLY” option); we then used those individual gene trees to infer the species tree implemented in the ASTRAL program (Mirarab and Warnow 2015), v4.10.2. The reliability for each internode was evaluated using the local posterior probability measure (Sayyari and Mirarab 2016). Finally, internode certainty (IC) was used to quantify the incongruence by considering the most prevalent conflicting bipartitions for each individual internode among individual gene trees (Salichos and Rokas 2013; Salichos et al. 2014; Kobert et al. 2016), implemented in RAxML (Stamatakis 2014) v8.2.3. The relative divergence times were estimated using the RelTime (Tamura et al. 2012) in MEGA7 (Kumar et al. 2016). The ML topology was used as the input tree (Supplementary Figures S2 and S3).
Growth assays
We previously published galactose growth data for the majority of species (Opulente et al. 2018; Shen et al. 2018). Growth experiments were performed for an additional nine species separately (Supplementary Table S3). All species were struck onto yeast extract peptone dextrose (YPD) plates from freezer stocks and grown for single colonies. Single colonies were struck onto three types of plates minimal media base (5 g/L ammonium sulfate, 1.71 g/L Yeast Nitrogen Base (w/o amino acids, ammonium sulfate, or carbon), 20 g/L agar) treatments with either: 2% galactose, 1% galactose, or 2% glucose (to test for auxotrophies). We also re-struck the specific colony onto YPD plates as a positive control. All growth experiments were performed at room temperature. After initial growth on treatment plates, growth was recorded for the first round, and we struck colonies from each treatment plate onto a second round of the respected treatment to ensure there was no nutrient carryover from the YPD plate. For example, a single colony from 2% galactose minimal media plate was struck for a second round of growth on a 2% galactose minimal media plate. We inspected plates every 3 days for growth for up to a month. Yeasts were recorded as having no growth on galactose if they did not grow on either the first or second round of growth on galactose.
Data availability
The authors state that all data necessary for confirming the conclusions presented in the manuscript are represented fully within the manuscript. Raw DNA sequencing data were deposited in GenBank under Bioproject ID PRJNA647756. Whole-genome shotgun assemblies have been deposited at DDBJ/ENA/GenBank under the accessions JADIOP000000000—JADIOY000000000 and JADLIC000000000—JADLIE000000000 (Supplementary Table S1). The versions described in this paper are version JADIOP010000000–JADIOY010000000 and JADLIC010000000–JADLIE010000000.
Supplementary material is available at figshare DOI: https://doi.org/10.25386/genetics.13224920.
Results
Genome selection and sequencing
To reconstruct the evolution of galactose metabolism in the budding yeast subphylum Saccharomycotina, we first selected a set of genomes to analyze that spanned the backbone of the subphylum (Shen et al. 2016, 2018). Next, we sequenced the genomes of five additional species at strategically positioned branches: Brettanomyces naardenensis; a yet-to-be described Wickerhamomyces species, Wickerhamomyces sp. UFMG-CM-Y6624; Candida chilensis; Candida cylindracea; and Candida silvatica. All strains used in this study can be found in Supplementary Table S1. Finally, we reconstructed a species-level phylogeny, analyzing the genome sequences of 96 Saccharomycotina and 10 outgroup species (Supplementary Figures S1 and S2).
Recurrent loss of yeast GAL clusters
This dataset suggests that the GAL enzymatic gene cluster (hereafter GAL cluster) of budding yeasts formed prior to the last common ancestor of the CUG-Ser1, CUG-Ser2, CUG-Ala, Phaffomycetaceae, Saccharomycodaceae, and Saccharomycetaceae major clades (Figure 1 and Supplementary Figure S3) (Slot and Rokas 2010). This inference is supported by the presence of the fused bifunctional GAL10 gene in these lineages and the absence of the fused protein in species outside these lineages (Figure 1 and Supplementary Figure S3) (Slot and Rokas 2010). Since galactose metabolism has been repeatedly lost over the course of budding yeast evolution and the enzymatic genes are present in a gene cluster, we next asked whether the trait of galactose utilization had undergone trait reversal. We reasoned that species or lineages that utilize galactose, but who are deeply embedded in clades that predominantly cannot utilize galactose, would represent prime candidates for possible trait reversal events. When we mapped both GAL gene presence and galactose utilization onto our phylogeny (Figure 1 and Supplementary Figure S3), we inferred repeated loss of the GAL gene clusters (Figure 1 and Supplementary Figure S3) and a strong association between genotype and phenotype (Supplementary Table S2). However, we identified two genera, Brettanomyces and Wickerhamomyces, as containing candidates for trait reversal (Figure 1). This unusual trait distribution led us to consider the possibility that the GAL clusters of these two lineages were not inherited vertically.
Unusual synteny patterns of GAL clusters
If the observed distribution of galactose metabolism was to be explained by only vertical reductive evolution, then GAL cluster losses have occurred even more frequently than currently appreciated. Interestingly, we noted that the structures of Brettanomyces and Wickerhamomyces GAL clusters are strikingly syntenic to the GAL clusters belonging to distantly related yeasts, specifically those belonging to the CUG-Ser1 clade, which includes C. albicans (Figure 2 and Supplementary Figure S4).
Figure 2.
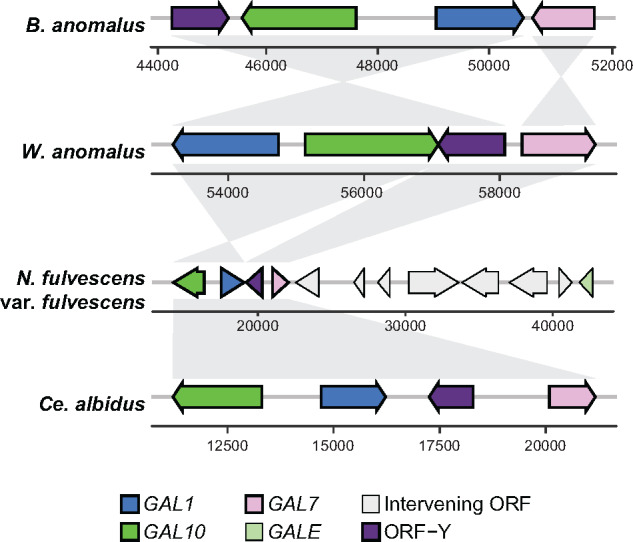
Syntenic GAL clusters between distantly related groups of yeasts. The GAL clusters of four representative species are shown. Numbers correspond to positions in each scaffold or contig. Shaded regions indicate regions of synteny between clusters. Further details and examples are provided in Supplementary Figures S3 and S4.
Several lines of evidence suggest that these GAL clusters did not evolve independently from previously unclustered genes. First, previously documented cases of de novo GAL cluster formation illustrate that gene relocation resulted in completely different structures of the cluster (Slot and Rokas 2010). Second, the GAL clusters in question here all contain ORF-Y, a gene associated with the CUG-Ser1 GAL cluster. Third, these GAL clusters all contain the fused GAL10 gene encoding a bifunctional protein (a fusion of galactose mutarotase domain encoded by GALM and the UDP-galactose 4-epimerase domain encoded by GALE), which is present in similar CUG-Ser1 GAL clusters. These observations suggest a model wherein the Brettanomyces and Wickerhamomyces GAL clusters share ancestry with GAL clusters from the CUG-Ser1 clade, rather than with those from their much closer organismal relatives.
Unexpectedly, we observed distinct GAL clusters in Lipomyces starkeyi and N. fulvescens (Figure 2 and Supplementary Figure S3), two species that diverged from the rest of the Saccharomycotina prior to the formation of the canonical GAL cluster. L. starkeyi, a species belonging to a lineage that is sister to the rest of the budding yeasts, contains a large gene cluster consisting of two copies of GAL1, a single copy of GAL7, GALE, and a gene encoding a zinc-finger domain (Supplementary Figure S3). Based on the phylogenetic positioning of L. starkeyi (Figure 1 and Supplementary Figure S1) and the novel content and configuration of this cluster (Figure 2 and Supplementary Figure S3), we propose that its GAL gene cluster may have formed independently of the canonical budding yeast GAL cluster.
Remarkably, the structure of the GAL cluster of N. fulvescens is nearly identical to that of the CUG-Ser1 species Cephaloascus albidus (Figure 2 and Supplementary Figures S3 and S4), despite the fact that these two lineages are separated by hundreds of millions of years of evolution (Shen et al. 2018). This synteny suggests that the GAL cluster of N. fulvescens was either horizontally acquired or that it independently evolved the bifunctional GAL10 gene and a GAL cluster with the same gene arrangement. Interestingly, N. fulvescens var. elongata has a pseudogenized GAL10 gene (indicated by multiple inactivating mutations along the gene; Supplementary Figure S5), while N. fulvescens var. fulvescens has an intact GAL10 gene, and the varieties’ phenotypes were consistent with their inferred GAL10 functionality (Supplementary Figure S1and Table S3). Both varieties also contain a linked GALE gene, which resides ∼20 kb downstream of GAL7, suggesting the ongoing replacement of an ancestral GALE-containing GAL cluster by a CUG-Ser1-like GAL cluster containing GAL10. A similar fusion of GAL clusters has also been reported in the genus Torulaspora (Wolfe et al. 2015; Venkatesh et al. 2020). Notably, GALE or GAL10 genes are present in some budding yeast species that do not utilize galactose (Riley et al. 2016), and N. fulvescens var. fulvescens has only CUG-Ser1-like copies of the GAL7 and GAL1 genes required for galactose utilization. While parsimony suggests that the last common ancestor of N. fulvescens and its relative Yarrowia lipolytica was able to utilize galactose, N. fulvescens rests on an unusually long branch with no other known closely related species. Thus, in this case, we cannot infer whether partial cluster loss and trait loss (i.e., to the state of possessing only GALE and not utilizing galactose) preceded acquisition of the new functional cluster.
Allowing reacquisition is more parsimonious than enforcing loss
These synteny observations suggest three independent reacquisitions of the GAL cluster and at least two independent reacquisitions of the galactose utilization trait. To test the hypothesis of trait reversal, we next investigated whether, in some cases, reacquisition of the GAL cluster offered a more parsimonious explanation than reductive evolution. To reconcile the observed topologies of the gene and species phylogenies, we manually reconstructed the evolutionary events using a parsimony framework, either assuming Dollo’s law of irreversibility to be true (only gene loss was possible) or false (both gene loss and reacquisition were possible). When there was variation segregating below the species level [e.g., N. fulvescens and S. kudriavzevii (Hittinger et al. 2010)], we treated the species as positive for galactose utilization. When Dollo’s law was enforced, we inferred 15 distinct loss events for galactose metabolism (Figure 3A). When we allowed for the violation of Dollo’s law, assuming an equal weight to the probability of loss and reacquisition, we replaced a portion of the loss events with two reacquisition events, arriving at a more parsimonious inference of 11 distinct events: 9 losses and 2 reacquisitions (Figure 3A). The most parsimonious scenario did not infer trait loss for Nadsonia, but even adding one loss and one gain of galactose metabolism, instead of the cluster replacement scenario, still yielded a more parsimonious solution of 13 distinct events. While it is likely gene loss and reacquisition do not occur with equal probability during evolution, the weight of reacquisition must exceed three times that of loss for a loss-only scenario to be more parsimonious. Moreover, any weightings given to evolutionary losses and reacquisitions completely ignore the power of selection, which is critical to the likelihood that a rare mutational event, such as HGT, would rise to fixation.
Figure 3.
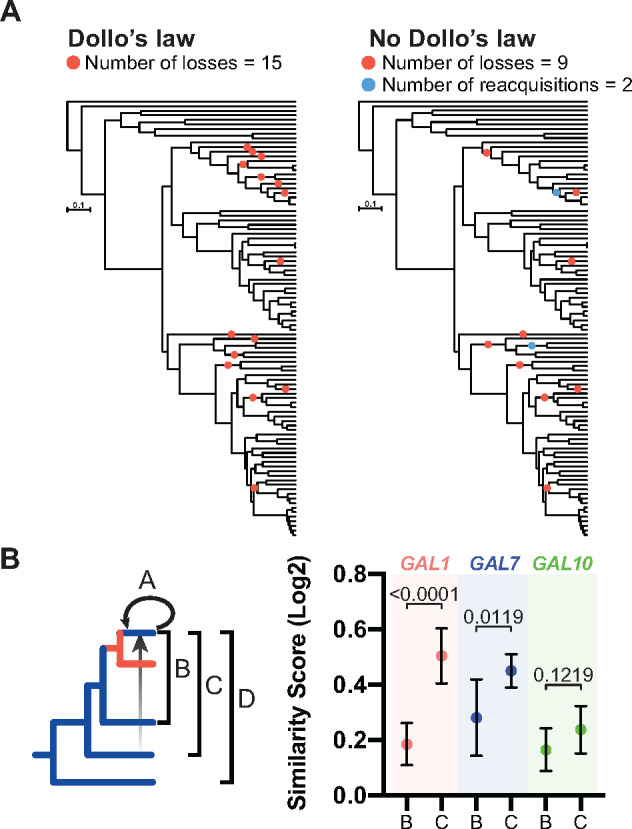
Comparison of Dollo’s law versus reacquisition of the GAL genes from the CUG-Ser1 clade. (A) Evolutionary trait reconstruction based on a parsimony framework either assuming that traits cannot be regained (left) or that traits can be regained (right). (B) Similarity scores of the Gal1, Gal7, and Gal10 proteins as calculated by protein sequence similarity and the comparisons shown on the left; means with standard deviations are depicted on the right. Raw percent identity values and all other comparisons are shown in Supplementary Figure S6. Comparisons used to calculate similarity scores: A, between species within the clade with potentially transferred GAL genes (recipient clade); B, between the recipient clade and their closest relative with GAL genes; C, between the recipient clade and the potential donor lineage (CUG-Ser1 clade); and D, between the recipient clade and an outgroup lineage. Student’s t-tests of the mean differences in similarity scores between groups are shown (n = 7).
Yeast GAL gene clusters have been horizontally transferred multiple times
From these synteny and trait reconstructions, we hypothesized that the GAL clusters of Brettanomyces, Wickerhamomyces, and Nadsonia were horizontally transferred from the CUG-Ser1 clade. This hypothesis predicts that the coding sequences of their GAL genes should be more similar to species in the CUG-Ser1 clade than to their closest relative possessing GAL genes. Thus, we calculated the percent identities of Gal1, Gal7, and Gal10 proteins between four groups of species; (A) between species in the candidate HGT recipient clade, (B) between the candidate HGT recipient clade and their closest relative with GAL genes, (C) between the candidate HGT recipient clade and the candidate donor clade, and (D) between the candidate HGT recipient clade and an outgroup lineage (Figure 3, B and C). If the genes were vertically acquired, one would expect the percent identities to be highest in group A and then decrease in the order of group B to C to D. If the genes were acquired horizontally, then the percent identities would be higher in group C than in group B. Indeed, we found that the mean similarity score of the Gal1 and Gal7 proteins of group C were significantly greater than group B (Figure 3B and Supplementary Figure S6). While these results are congruent with the model where the GAL clusters of Brettanomyces, Wickerhamomyces, and Nadsonia were acquired horizontally from the CUG-Ser1 clade, we note that sequence similarity is not always a good measure of relatedness. Therefore, we next sought to explicitly test this model using phylogenetic hypothesis testing.
We next reconstructed ML phylogenies for each of the GAL genes, as well as for the concatenation of all three (Supplementary Figures S7–S10). Interestingly, we observed a consistent pattern of phylogenetic placement of Brettanomyces, Wickerhamomyces, and Nadsonia GAL genes, which grouped to different lineages than would be expected based on their species taxonomy or phylogeny (Supplementary Figure S1). The Wickerhamomyces GAL genes formed a clade with Hyphopichia; the Brettanomyces GAL genes formed a clade with several genera from the families Debaryomycetaceae and Metschnikowiaceae; and the Nadsonia GAL genes formed a clade with those from the family Cephaloascaceae. These observations are consistent with three independent HGTs of GAL clusters into these lineages from the CUG-Ser1 clade.
To formally test the hypothesis of GAL HGT, we used AU tests (Supplementary Figure 4A). Specifically, we generated multiple ML phylogenetic trees using alignments of GAL genes with constraints on the placements of various taxa: (1) constrained to follow the species tree, except for the three HGT candidate lineages (Brettanomyces, Wickerhamomyces, and Nadsonia); (2) the same constraint as in (1) but with the Brettanomyces lineage additionally constrained to follow the species tree; (3) the same constraint as in (1) but with the Wickerhamomyces lineage additionally constrained to follow the species tree; and (4) the same constraint as in (1) but with the Nadsonia lineage additionally constrained to follow the species tree (Supplementary Figure S11). We then used the AU test to conduct 1-on-1 comparisons of the trees with the unconstrained placement of all three candidate lineages (null hypothesis) against trees with constraints placed on individual lineages (alternative hypotheses). In each case, we found that the alternative hypothesis (i.e., that an HGT candidate lineage placed consistently with the species tree and was inherited vertically) was rejected with strong statistical support (Figure 4B). These results were consistent across individual alignments of the GAL genes, as well as when all three genes were examined together (Figure 4B). From these results, we conclude that the GAL clusters of the Brettanomyces, Wickerhamomyces, and Nadsonia lineages were likely acquired via HGT from ancient CUG-Ser1 yeasts.
Figure 4.
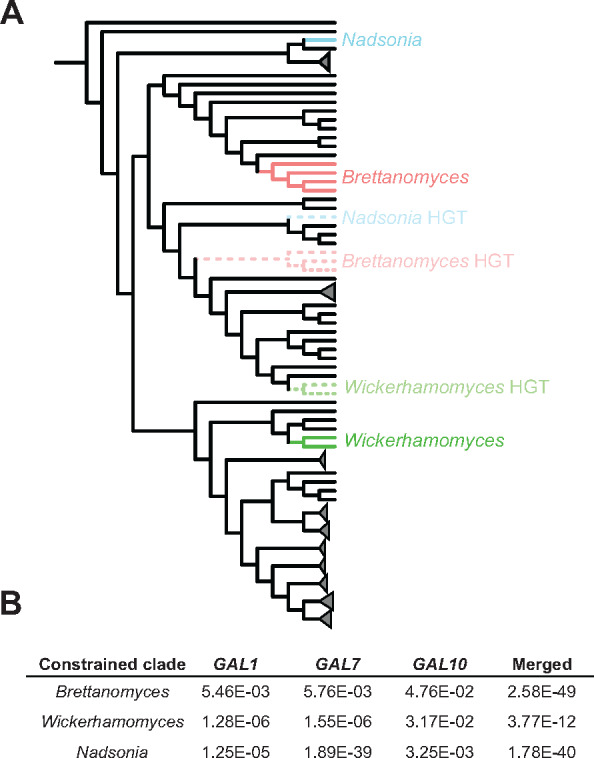
The GAL clusters of three lineages were acquired by HGT. (A) Diagrammatic representation of AU tests performed. The null tree was constrained to follow the species tree, except for the three HGT candidate lineages, which were placed by ML (dashed, lightly colored; Supplementary Figures S7–S10). Three alternative hypotheses were tested that independently constrained a single HGT candidate to its placement in the species tree (solid, darkly colored). Constraint trees used in the AU tests are provided in Supplementary Figure S11. (B) P-values of the AU tests are shown. All tests significantly reject their alternative constrained topologies and vertical inheritance, which is consistent with HGT as the mechanism of reacquisition.
CUG codon reassignment was likely not a barrier to HGT from the CUG-Ser1 clade
Codon reassignments, such as those seen in the CUG-Ser1 clade, have the potential to act as a barrier to HGT. Indeed, we recently showed CUG-Ser1 clade species have significantly fewer genes horizontally transferred from bacteria than other yeast clades (Shen et al. 2018). But, do the same constraints that apply to recipient lineages also apply to donor lineages? To address this question, we first examined the genome-wide frequency of the CUG codon across the Saccharomycotina. In the 94 CUG-Ser1 clade species from Shen et al. (2018), the average RSCU of CUG was 0.532, where a value below 1 indicates that it is used less often than other serine codons. Among extant CUG-Ser1 clade species, many GAL genes have zero CUG codons, and nearly all have very few (Supplementary Table S5). In contrast, within the recipient clades, the CUG codon (which encodes leucine) is used at a more normal frequency with RSCU values of 0.990, 0.960, and 1.259 for Phaffomycetaceae (e.g., Wickerhamomyces), Pichiaceae (e.g., Brettanomyces), and Dipodascaceae/Trichomonascacea (e.g., Nadsonia) clades, respectively. It is therefore likely that the GAL genes that underwent HGT originally contained so few CUG codons that they did not significantly conflict with the recipient species’ codon usages. We conclude that, although codon reassignment may serve as a barrier to HGT into the CUG-Ser1 clade as a recipient, codon reassignment was unlikely to pose much of a barrier to HGT from the CUG-Ser1 clade as a donor.
Regulatory mode correlates with the HGTs
Gal4 is the key transcriptional activator of the GAL cluster in S. cerevisiae and responds to galactose through the co-activator Gal3 and co-repressor Gal80. This mode of regulation is thought to be restricted to the family Saccharomycetaceae and is absent in other yeasts and fungi (Choudhury and Whiteway 2018). In other budding yeasts (including C. albicans, the most thoroughly studied CUG-Ser1 species, as well as Y. lipolytica, an outgroup to S. cerevisiae and C. albicans), regulation of the GAL cluster is thought to be under the control of the activators Rtg1 and Rtg3 (Dalal et al. 2016). These two regulatory mechanisms respond to different signals and have dramatically different dynamic ranges. In Gal4-regulated species, the GAL cluster is nearly transcriptionally silent in the presence of glucose and is rapidly induced to high transcriptional activity when only galactose is present. In contrast, Rtg1/Rtg3-regulated species have high basal levels of transcription and are weakly induced in the presence of galactose (Dalal et al. 2016).
Intriguingly, all putative donor lineages of the GAL genes were from the CUG-Ser1 clade of yeasts, and no transfers occurred from or into the family Saccharomycetaceae. To examine whether the relaxed Rtg1/Rtg3 regulatory regimen of the CUG-Ser1 yeasts might have facilitated their role as an HGT donor, as opposed to the Gal4-mediated regulation of the Saccharomycetaceae, we identified sequence motifs that were enriched 800-bp upstream from the coding regions of the GAL1, GAL7, and GAL10 genes (Supplementary Table S4). Then, based on the existing experimental evidence on the regulation of the GAL genes (Hittinger and Carroll 2007; Martchenko et al. 2007; Dalal et al. 2016), we divided the yeast species into Saccharomycetaceae and non-Saccharomycetaceae species. We then ran a selective motif enrichment analysis to determine if any regulatory motifs were enriched in one group, but not the other. We found that the top enriched motifs corresponded to the known Gal4-binding site in the Saccharomycetaceae (Johnston 1987) and the known Rtg1-binding site in the non-Saccharomycetaceae species (Dalal et al. 2016) (Figure 5, A and B and Supplementary Table S4), consistent with the previously documented regulatory rewiring of the GAL genes that occurred at the base of the family Saccharomycetaceae (Dalal et al. 2016). In general, the enrichment of Rgt1-binding sites was patchier and did not include the HGT recipient lineages, the previously characterized Rtg1-regulated GAL cluster of Y. lipolytica (Dalal et al. 2016), or several CUG-Ser1 clade species (e.g., Ce. albidus).
Figure 5.

Enrichment of transcription factor-binding sites in the promoters of GAL enzymatic genes. (A) ML phylogeny of Saccharomycotina. Colors indicate highlighted clades: light blue—Nadsonia, red—Brettanomyces, yellow—CUG-Ser1 clade, green—Wickerhamomyces, and blue—Saccharomycetaceae. (B) Heatmap of enrichment for either Rtg1- or Gal4-binding sites in the promoters of the GAL genes (GAL1, GAL10, and GAL7). White-shaded boxes indicate lineages lacking the GAL gene cluster.
Taken together, our new results suggest that the switch to the Gal4-mode of regulation, which is tighter and involves multiple unlinked and dedicated regulatory genes, reduced the likelihood of horizontal transfer into naïve genomes or genomes that had lost their GAL pathways. Specifically, any GAL cluster regulated by Gal4 would not be able to be transcribed or properly regulated if it were horizontally transferred into a species lacking GAL4 and other regulatory genes. In contrast, Rtg1 and Rtg3 are more broadly conserved, and any horizontally transferred GAL cluster regulated by them would likely be sufficiently transcriptionally active, providing an initial benefit to the organism.
Discussion
Budding yeasts have diversified from their metabolically complex most recent common ancestor over the last 400 million years (Kurtzman et al. 2011; Shen et al. 2018). While they have evolved specialized metabolic capabilities, their evolutionary trajectories have been prominently shaped by reductive evolution (Dujon et al. 2004; Kurtzman et al. 2011; Shen et al. 2018; Steenwyk et al. 2019). Here, we present evidence that losses of the GAL genes and galactose metabolism in some lineages were offset, tens of millions of years after their initial losses, by eukaryote-to-eukaryote HGT (Figure 6). While reacquired ancestral traits have been documented in several eukaryotic lineages, our observation of galactose metabolism reacquisition differs in a few regards. First, the majority of reported events did not identify the molecular mechanism or the genes involved in the reacquired traits. Second, few studies have comprehensively sampled taxa and constructed robust genome-scale phylogenies onto which the examined traits were mapped, a requirement for robustly inferring trait evolution. Remarkably, we observed trait reversal in at least two independent lineages, with a third possible lineage, suggesting that the recovery of lost eukaryotic metabolic genes may be an important and underappreciated driver in trait evolution in budding yeasts, and perhaps more generally in fungi and other eukaryotes. In line with our study, budding yeasts also have reacquired lost metabolic traits from bacteria, supporting the hypothesis that regains via HGT offset reductive evolution (Gonçalves et al. 2018).
Figure 6.
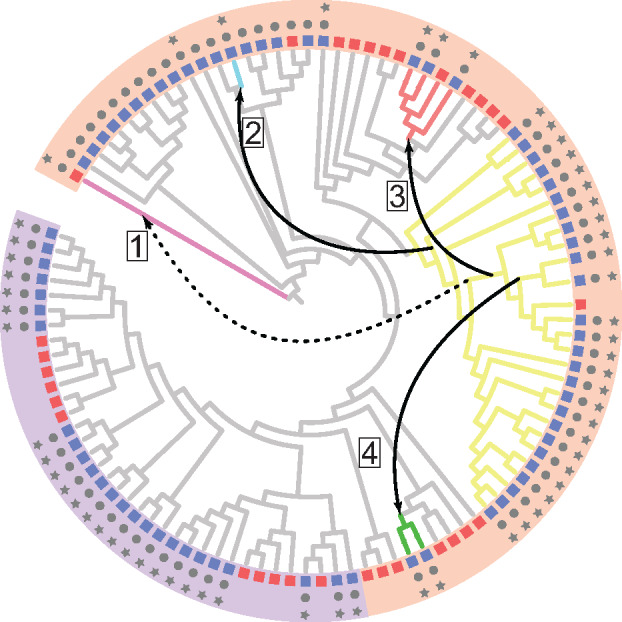
The CUG-Ser1 clade serves as a common donor of the GAL gene cluster to other yeasts. A cladogram of the ML phylogeny is presented with the leaf labels removed for simplicity. The colored boxes represent the species’ ability to utilize galactose (blue = positive/variable, red = negative), gray circles indicate the presence of a full set of GAL enzymatic genes, and gray stars indicate that those GAL genes are clustered. Five lineages on the cladogram are colored: pink—Sc. pombe (a member of the subphylum Taphrinomycotina with a transferred GAL cluster that does not confer utilization), light blue—Nadsonia, red—Brettanomyces, yellow—CUG-Ser1 clade, and green—Wickerhamomyces. Numbered boxes and arrows depict the four horizontal transfer events of the GAL cluster, one previously documented case into Sc. pombe (dashed arrow) (Slot and Rokas 2010) and the three described here (solid arrows). The colored arcs encompassing the cladogram represent the predicted regulatory mode of the GAL genes: orange—Rtg1/Rtg3 (non-Gal4) and purple—Gal4.
The absence of GAL HGTs from Saccharomycetaceae into other major clades provides clues into the potential limits on ancestral trait reacquisition via HGT. We propose the transcriptional rewiring to Gal4-mediated regulation imposed a restriction on the potential for benefit of transferred GAL clusters. Since Gal4-mediated gene activation is tightly coordinated and the off-state is less leaky (Choudhury and Whiteway 2018), any transferred GAL cluster lacking Gal4-binding sites into a species with exclusively Gal4-mediated activation in response to galactose would not be able to activate the transferred genes. Similarly, transfer of a Gal4-regulated gene cluster into a species lacking GAL4 and other upstream regulators would have limited potential for activation. For the case of transfer between two species whose regulation does not rely on Gal4, the transferred GAL cluster would be transcriptionally active because the broadly conserved transcription factors Rtg1 and Rtg3 could further enhance moderate basal transcriptional activity (Dalal et al. 2016). Thus, even leaky levels of transcription would provide a benefit in the presence of galactose that could further be refined, possibly to become regulated by lineage-specific networks. Under this model, the likelihood of HGT is partly determined by the potential activity of the transferred genes and by the recipient’s ancestral regulatory mode.
More generally, our findings demonstrate that reductive evolution is not always a dead end, and gene loss can be circumvented by HGT from distantly related taxa. However, the scope of genes that can be regained in this fashion is likely limited. In particular, the GAL genes of the CUG-Ser1 clade of budding yeasts represent something of a best-case scenario. First, all enzymatic genes needed for phenotypic output are encoded in a cluster, facilitating the likelihood that all necessary genes for function are transferred together (Wisecaver and Rokas 2015; Rokas et al. 2018). Second, the regulatory mode of these GAL genes is conducive to function in the recipient species, as they are loosely regulated by conserved factors with moderate basal activity. Third, the genes would provide a clear competitive advantage in environments with galactose.
The modern interpretation of Dollo’s law is that species cannot return to a previous character state after loss. Alongside recently reported character state reversals in petunias after pseudogene reactivation (Esfeld et al. 2018), our results of reacquisition of galactose metabolism and GAL genes by HGT can be considered a case of character state reversal. However, the previous example fits into the model that, for groups undergoing adaptive radiations, lost traits seem to “flicker” on and off, resulting in an unusual distribution of character states on the phylogeny. Here, and in the recently described reacquisition of alcoholic fermentation genes from bacteria in fructophilic yeasts (Gonçalves et al. 2018), the ancestral genes were completely lost from the genome, and they were restored far later than could be explained by the flickering of traits during adaptive radiations. The reacquisition of galactose metabolism in budding yeasts represents a striking example of trait reversal by eukaryote-to-eukaryote HGT and provides insight into the mechanisms by which Dollo’s law can be broken.
Acknowledgments
We are grateful to Carlos A. Rosa for providing the strain Wickerhamomyces sp. UFMG-CM-Y6624. We thank the Rokas and Hittinger labs for comments and discussions and the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing Illumina sequencing facilities and services.
M.A.B.H. (study design, preliminary phylogenetic analyses, sequence analyses, cluster analyses, and text); J.K. (study design, genome assemblies, phylogenetic analyses, motif enrichment analyses, and text); D.A.O. (genomic DNA isolation, library preparation, and yeast growth assays); X.-X.S. (phylogenomic analyses); A.L.L. (cluster analyses and CUG codon analysis); X.Z. (preliminary genome annotations and analyses); J.DeV. and A.B.H. (genomic DNA isolation and library preparation); C.P.K. (support and supervision, study design); and A.R. and C.T.H. (support and supervision, study design, and text).
Funding
This material is based upon work supported by the National Science Foundation under Grant Nos. DEB-1442113 (to A.R.) and DEB-1442148 (to C.T.H. and C.P.K.), in part by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-SC0018409 and DE-FC02-07ER64494 to Timothy J. Donohue), USDA National Institute of Food and Agriculture (Hatch Project 1020204 to C.T.H.), and National Institutes of Health (NIAID AI105619 to A.R.), and a Guggenheim fellowship (to A.R). C.T.H. is a Pew Scholar in the Biomedical Sciences, a Vilas Early Career Investigator, and an H. I. Romnes Faculty Fellow, supported by the Pew Charitable Trusts, Vilas Trust Estate, and Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation, respectively. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture (USDA). The USDA is an equal opportunity provider and employer.
Conflicts of interest
None declared.
Literature cited
- Alexander WG, Wisecaver JH, Rokas A, Hittinger CT. 2016. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proc Natl Acad Sci USA. 113:4116–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, et al. 2009. MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boocock J, Sadhu MJ, Bloom JS, Kruglyak L. 2019. Ancient balancing selection maintains incompatible versions of a conserved metabolic pathway in yeast. bioRxiv 829325. 10.1101/829325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandley MC, Huelsenbeck JP, Wiens JJ. 2008. Rates and patterns in the evolution of snake-like body form in squamate reptiles: evidence for repeated re-evolution of lost digits and long-term persistence of intermediate body forms. Evolution. 62:2042–2064. [DOI] [PubMed] [Google Scholar]
- Campbell MA, Staats M, van Kan JAL, Rokas A, Slot JC. 2013. Repeated loss of an anciently horizontally transferred gene cluster in Botrytis. Mycologia. 105:1126–1134. [DOI] [PubMed] [Google Scholar]
- Chippindale PT, Bonett RM, Baldwin AS, Wiens JJ. 2004. Phylogenetic evidence for a major reversal of life-history evolution in Plethodontid salamanders. Evolution. 58:2809–2822. [DOI] [PubMed] [Google Scholar]
- Choudhury BI, Whiteway M. 2018. Evolutionary transition of GAL regulatory circuit from generalist to specialist function in ascomycetes. Trends Microbiol. 26:692–702. [DOI] [PubMed] [Google Scholar]
- Collin R, Cipriani R. 2003. Dollo’s law and the re-evolution of shell coiling. Proc R Soc Lond B. 270:2551–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin R, Miglietta MP. 2008. Reversing opinions on Dollo’s law. Trends Ecol Evol. 23:602–609. [DOI] [PubMed] [Google Scholar]
- Dalal CK, Zuleta IA, Mitchell KF, Andes DR, El-Samad H, et al. 2016. Transcriptional rewiring over evolutionary timescales changes quantitative and qualitative properties of gene expression. eLife. 5:e18981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollo L. 1893. Les lois de l’évolution. Bull Soc Belge Géol. VII: 164–166. [Google Scholar]
- Duan S-F, Shi J-Y, Yin Q, Zhang R-P, Han P-J, et al. 2019. Reverse evolution of a classic gene network in yeast offers a competitive advantage. Curr Biol. 29:1126–1136.e5. [DOI] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, et al. 2004. Genome evolution in yeasts. Nature. 430:35–44. [DOI] [PubMed] [Google Scholar]
- Esfeld K, Berardi AE, Moser M, Bossolini E, Freitas L, et al. 2018. Pseudogenization and resurrection of a speciation gene. Curr Biol. 28:3776–3786.e7. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DA. 2012. Horizontal gene transfer in fungi. FEMS Microbiol Lett. 329:1–8. [DOI] [PubMed] [Google Scholar]
- Gonçalves C, Wisecaver JH, Kominek J, Salema-Oom M, Leandro MJ, et al. 2018. Evidence for loss and adaptive reacquisition of alcoholic fermentation in an early-derived fructophilic yeast lineage. eLife. 7: 10.7554/eLife.33034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29:1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, Dietrich FS. 2007. The reacquisition of biotin prototrophy in Saccharomyces cerevisiae involved horizontal gene transfer, gene duplication and gene clustering. Genetics. 177:2293–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Carroll SB. 2007. Gene duplication and the adaptive evolution of a classic genetic switch. Nature. 449:677–681. [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Gonçalves P, Sampaio JP, Dover J, Johnston M, et al. 2010. Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature. 464:54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, Bai FY, Boekhout T, Gonçalves P, et al. 2015. Genomics and the making of yeast biodiversity. Curr Opin Genet Dev. 35:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, Carroll SB. 2004. Parallel inactivation of multiple GAL pathway genes and ecological diversification in yeasts. Proc Natl Acad Sci USA. 101:14144–14149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayadeva Bhat P, Murthy TVS. 2001. Transcriptional control of the GAL/MEL regulon of yeast Saccharomyces cerevisiae: mechanism of galactose-mediated signal transduction. Mol Microbiol. 40:1059–1066. [DOI] [PubMed] [Google Scholar]
- Johnston M. 1987. A model fungal gene regulatory mechanism: the GAL genes of Saccharomyces cerevisiae. Microbiol Rev. 51:458–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2014. MAFFT: iterative refinement and additional methods. Methods Mol Biol. 1079:131–146. [DOI] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. 2008. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 9:605–618. [DOI] [PubMed] [Google Scholar]
- Kobert K, Salichos L, Rokas A, Stamatakis A. 2016. Computing the internode certainty and related measures from partial gene trees. Mol Biol Evol. 33:1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlsdorf T, Lynch VJ, Rodrigues MT, Brandley MC, Wagner GP. 2010. Data and data interpretation in the study of limb evolution: a reply to Galis et al. on the reevolution of digits in the lizard genus Bachia. Evolution. 64:2477–2485. [Google Scholar]
- Kohlsdorf T, Wagner GP. 2006. Evidence for the reversibility of digit loss: a phylogenetic study of limb evolution in Bachia (Gymnophthalmidae: Squamata). Evolution. 60:1896–1912. [PubMed] [Google Scholar]
- Kominek J, Doering DT, Opulente DA, Shen X-X, Zhou X, et al. 2019. Eukaryotic acquisition of a bacterial operon. Cell. 176:1356–1366.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang MC, Hutchins PD, Russell JD, Coon JJ, Hittinger CT. 2016. Ongoing resolution of duplicate gene functions shapes the diversification of a metabolic network. eLife. 5: 10.7554/eLife.19027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang MC, Kominek J, Alexander WG, Cheng J-F, Wrobel RL, et al. 2018. Repeated cis-regulatory tuning of a metabolic bottleneck gene during evolution. Mol Biol Evol. 35:1968–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman CP, Fell JW, Boekhout T. 2011. The Yeasts: A Taxonomic Study, 5th ed. Elsevier, Amsterdam.
- LaBella AL, Opulente DA, Steenwyk JL, Hittinger CT, Rokas A. 2019. Variation and selection on codon usage bias across an entire subphylum. PLoS Genet. 15:e1008304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legras J-L, Galeote V, Bigey F, Camarasa C, Marsit S, et al. 2018. Adaptation of S. cerevisiae to fermented food environments reveals remarkable genome plasticity and the footprints of domestication. Mol Biol Evol. 35:1712–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch VJ, Wagner GP. 2010. Did egg-laying boas break Dollo’s law? Phylogenetic evidence for reversal to oviparity in sand boas (Eryx: Boidae). Evolution. 64:207–216. [DOI] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldón T. 2010. Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 26:5–8. [DOI] [PubMed] [Google Scholar]
- Martchenko M, Levitin A, Hogues H, Nantel A, Whiteway M. 2007. Transcriptional rewiring of fungal galactose-metabolism circuitry. Curr Biol. 17:1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa T, Fujita Y, Tanaka N, Tohda H, Itadani A, et al. 2011. New insights into galactose metabolism by Schizosaccharomyces pombe: isolation and characterization of a galactose-assimilating mutant. J Biosci Bioeng. 111:158–166. [DOI] [PubMed] [Google Scholar]
- McLeay RC, Bailey TL. 2010. Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics. 11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, Warnow T. 2015. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics. 31:i44–i52 [DOI] [PMC free article] [PubMed]
- Nguyen LT, Schmidt HA, Haeseler AV, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opulente DA, Rollinson EJ, Bernick-Roehr C, Hulfachor AB, Rokas A, et al. 2018. Factors driving metabolic diversity in the budding yeast subphylum. BMC Biol. 16:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recknagel H, Kamenos NA, Elmer KR. 2018. Common lizards break Dollo’s law of irreversibility: genome-wide phylogenomics support a single origin of viviparity and re-evolution of oviparity. Mol Phylogenet Evol. 127:579–588. [DOI] [PubMed] [Google Scholar]
- Riley R, Haridas S, Wolfe KH, Lopes MR, Hittinger CT, et al. 2016. Comparative genomics of biotechnologically important yeasts. Proc Natl Acad Sci USA. 113:9882–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Wisecaver JH, Lind AL. 2018. The birth, evolution and death of metabolic gene clusters in fungi. Nat Rev Microbiol. 16:731–744. [DOI] [PubMed] [Google Scholar]
- Salichos L, Rokas A. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature. 497:327–331. [DOI] [PubMed] [Google Scholar]
- Salichos L, Stamatakis A, Rokas A. 2014. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol Biol Evol. 31:1261–1271. [DOI] [PubMed] [Google Scholar]
- Sayyari E, Mirarab S. 2016. Fast coalescent-based computation of local branch support from quartet frequencies. Mol Biol Evol. 33:1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seher TD, Ng CS, Signor SA, Podlaha O, Barmina O, et al. 2012. Genetic basis of a violation of Dollo’s law: re-evolution of rotating sex combs in Drosophila bipectinata. Genetics. 192:1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X-X, Opulente DA, Kominek J, Zhou X, Steenwyk J, et al. 2018. The tempo and mode of genome evolution in the budding yeast subphylum. Cell. 175:1533–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X-X, Zhou X, Kominek J, Kurtzman CP, Hittinger CT, et al. 2016. Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3 (Bethesda). 6:3927–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, et al. 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson GG. 1953. The Major Features of Evolution. Columbia University Press, New York. [Google Scholar]
- Slot JC. 2017. Fungal gene cluster diversity and evolution. Adv Genet. 100:141–178. [DOI] [PubMed] [Google Scholar]
- Slot JC, Rokas A. 2010. Multiple GAL pathway gene clusters evolved independently and by different mechanisms in fungi. Proc Natl Acad Sci USA. 107:10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J, Renner S. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 57:758–771. [DOI] [PubMed] [Google Scholar]
- Steenwyk JL, Opulente DA, Kominek J, Shen X-X, Zhou X, et al. 2019. Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol. 17:e3000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, et al. 2012. Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci USA. 109:19333–19338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh A, Murray AL, Coughlan AY, Wolfe KH. 2020. Giant GAL gene clusters for the melibiose-galactose pathway in Torulaspora. Yeast epub doi:10.1002/yea.3532. [DOI] [PMC free article] [PubMed]
- Whiting MF, Bradler S, Maxwell T. 2003. Loss and recovery of wings in stick insects. Nature. 421:264–267. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. 2011. Re-evolution of lost mandibular teeth in frogs after more than 200 million years, and re-evaluating Dollo’s law. Evolution. 65:1283–1296. [DOI] [PubMed] [Google Scholar]
- Wisecaver JH, Rokas A. 2015. Fungal metabolic gene clusters-caravans traveling across genomes and environments. Front Microbiol. 6:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisecaver JH, Slot JC, Rokas A. 2014. The evolution of fungal metabolic pathways. PLoS Genet. 10:e1004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Armisén D, Proux-Wera E, ÓhÉigeartaigh SS, Azam H, et al. 2015. Clade- and species-specific features of genome evolution in the Saccharomycetaceae. FEMS Yeast Res. 15:fov035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. 2018. DAMBE7: new and improved tools for data analysis in molecular biology and evolution, (S. Kumar, Ed.). Mol Biol Evol. 35:1550–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Jerlström-Hultqvist J, Kolisko M, Simpson AGB, Roger AJ, et al. 2016. On the reversibility of parasitism: adaptation to a free-living lifestyle via gene acquisitions in the diplomonad Trepomonas sp. PC1. BMC Biol. 14:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Peris D, Kominek J, Kurtzman CP, Hittinger CT, et al. 2016. In silico Whole Genome Sequencer & Analyzer (iWGS): a computational pipeline to guide the design and analysis of de novo Genome Sequencing Studies. G3 (Bethesda). 6:3655–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors state that all data necessary for confirming the conclusions presented in the manuscript are represented fully within the manuscript. Raw DNA sequencing data were deposited in GenBank under Bioproject ID PRJNA647756. Whole-genome shotgun assemblies have been deposited at DDBJ/ENA/GenBank under the accessions JADIOP000000000—JADIOY000000000 and JADLIC000000000—JADLIE000000000 (Supplementary Table S1). The versions described in this paper are version JADIOP010000000–JADIOY010000000 and JADLIC010000000–JADLIE010000000.
Supplementary material is available at figshare DOI: https://doi.org/10.25386/genetics.13224920.