

Abstract
The evolution of gene order rearrangements within bacterial chromosomes is a fast process. Closely related species can have almost no conservation in long-range gene order. A prominent exception to this rule is a >40 kb long cluster of five core operons (secE-rpoBC-str-S10-spc-alpha) and three variable adjacent operons (cysS, tufB, and ecf) that together contain 57 genes of the transcriptional and translational machinery. Previous studies have indicated that at least part of this operon cluster might have been present in the last common ancestor of bacteria and archaea. Using 204 whole genome sequences, ∼2 Gy of evolution of the operon cluster were reconstructed back to the last common ancestors of the Gammaproteobacteria and of the Bacilli. A total of 163 independent evolutionary events were identified in which the operon cluster was altered. Further examination showed that the process of disconnecting two operons generally follows the same pattern. Initially, a small number of genes is inserted between the operons breaking the concatenation followed by a second event that fully disconnects the operons. While there is a general trend for loss of gene synteny over time, there are examples of increased alteration rates at specific branch points or within specific bacterial orders. This indicates the recurrence of relaxed selection on the gene order within bacterial chromosomes. The analysis of the alternation events indicates that segmental genome duplications and/or transposon-directed recombination play a crucial role in rearrangements of the operon cluster.
Keywords: bacterial evolution, gene order, proteobacteria, firmicutes, SNAP, transposon
Significance
The linear order of genes within the bacterial chromosome is fluid, everchanging as life evolves, but little is known about the underlying mechanisms that drive chromosomal rearrangements on an evolutionary time scale. Here, the evolutionary history of a cluster of 57 genes was reconstructed for >2 billion years and 163 events were identified in which the linear order of the genes was altered. The analysis of these events indicates that gene duplications and transposon-directed recombination are potential forces that drive chromosome fluidity.
Introduction
It is generally accepted that all life on earth has evolved from a universal common ancestor which would entail that all life forms share a single ancestral gene order (Woese 2000; Koonin 2003, 2014; Forterre 2015; Booth et al. 2016; Weiss et al. 2018). As life evolved, the order of genes on the chromosome changed over time until selection, genetic drift and horizontal gene transfer (HGT) removed almost all traces of the last common gene order from modern chromosomes (Koonin et al. 1996; Tatusov et al. 1996). Despite this general trend there are a few genes that display a significant degree of synteny across the bacterial domain of life which suggests that this gene order was present in at least the last common ancestor of all bacteria. This higher degree of synteny has been used to identify functional groups of genes, to supplement traditional phylogenetic analysis methods, and to reconstruct the organization of ancestral genomes (Overbeek et al. 1999; Snel et al. 2002; Wang et al. 2012; Anselmetti et al. 2015; Rajaraman and Ma 2016). These genes are generally organized within operons and gene synteny could be driven by selection for coregulation and/or the ability of horizontal transfer of fully functional units (Moreno-Hagelsieb et al. 2001; Price et al. 2005; Rocha 2006). Conservation on a higher order, operon synteny, is virtually absent in bacteria (Tamames 2001).
A prominent exception to this rule is the secE-rpoBC-str-S10-spc-alpha operon cluster (Watanabe et al. 1997; Wachtershauser 1998; Barloy-Hubler et al. 2001; Coenye and Vandamme 2005; Brandis et al. 2019). In the Proteobacteria, this cluster is ∼33 kb long and contains seven operons (tufB, secE, rpoBC, str, S10, spc, and alpha) that encode up to 50 genes of the transcriptional and translational machinery (precise numbers vary between species) and operon concatenation is maintained due to operon coregulation (Brandis et al. 2019). At least part of this operon cluster is similarly organized in archaea indicating that it might have been present in the last common ancestor of bacteria and archaea (Coenye and Vandamme 2005). The potential ancestral operon cluster included the genes encoding the main subunits of the RNA polymerase (RpoA, RpoB, and RpoC) (Zhang et al. 1999), up to 31 out of the 33 universal ribosomal proteins (Ban et al. 2014), three of the five translation initiation and elongation factors (IF-1, EF-Tu, and EF-G) (Ramakrishnan 2002), and two subunits of the Sec translocase (du Plessis et al. 2011). This indicates that the operon cluster could be a remnant of a primordial operon cluster encoding the full transcriptional and translational machinery. The synteny between genes in the operon cluster is among the most highly conserved in bacteria but even this cluster can be altered (Watanabe et al. 1997; Itoh et al. 1999; Tamames 2001; Coenye and Vandamme 2005). The exceptional degree of operon synteny conservation makes the secE-rpoBC-str-S10-spc-alpha operon cluster an ideal tool to study chromosomal reorganization on an evolutionary time scale.
Here, the organization of the secE-rpoBC-str-S10-spc-alpha operon cluster was compared in 204 modern species belonging to the Proteobacteria, Acidobacteria, Firmicutes, and Tenericutes. Using gene synteny and maximum parsimony, the ancestral operon cluster was reconstructed for the last common ancestor of the Gammaproteobacteria and the last common ancestor of the Bacilli. The evolutionary history from the ancestral secE-rpoBC-str-S10-spc-alpha operon clusters to the organization within the modern species was reconstructed. It was previously estimated that this corresponds to 2.5 Gy of evolution for the families of Gammaproteobacteria and 2.0 Gy of evolution for the families of Bacilli included in this study (Marin et al. 2017). A total of 163 independent evolutionary events (115 rearrangements and 48 deletions) were identified in which the operon cluster was altered. Ten events were further analyzed and molecular mechanisms that could be responsible for the operon alterations are discussed.
Materials and Methods
Software
Sequence alignments and tree constructions were performed using CLC Main Workbench version 8.1 (QIAGEN, Aarhus) and PhyML version 3.3.20190321 (Guindon et al. 2009).
Phylogenetic Analysis
A total of 204 annotated genomes were downloaded from the NCBI database for the analysis (supplementary table S1, Supplementary Material online). Genes that were not annotated at their expected genetic locations within the operon cluster were manually checked to rule out annotation errors which were common for tRNAs and the small rpmJ gene. The genomes were split into two groups: 1) Proteobacteria and Acidobacteria (115 species) to reconstruct the evolution to the last common ancestor of the Gammaproteobacteria, and 2) Firmicutes and Tenericutes (89 species) for the reconstruction to the last common ancestor of the Bacilli. Protein alignments were performed with CLC using the CLC progressive alignment algorithm using the standard settings (Gap open cost: 10.0; Gap extension cost: 1.0; End gap cost: As any other; Alignment: Very accurate) and the MUSCLE alignment algorithm (Edgar 2004). No masking or local realignments were performed and all relevant protein alignments were concatenated. The tufB gene (duplicate of tufA) within the Gammaproteobacteria and Acidobacteria and genes that were absent in at least one modern genome were excluded from the analyses. For the Proteobacteria and Acidobacteria the concatenated alignment included 39 proteins with a total length of 9,429 amino acids (based on the mean length of each protein) and the concatenated alignment for the Firmicutes and Tenericutes included 44 proteins corresponding to 10,199 amino acids (supplementary tables S2 and S3, Supplementary Material online). Maximum likelihood phylogenetic trees based on the concatenated protein alignments were constructed using the CLC maximum likelihood phylogeny algorithm with standard settings (Tree construction method: maximum likelihood [Felsenstein 1981]; Protein substitution model: WAG [Whelan and Goldman 2001]; estimate topology [Felsenstein 1981]; Bootstrapping: 100 replicates [Efron 1982]) and the PhyML software with standard settings (Model of amino-acids substitution: LG [Le and Gascuel 2008] or WAG [Whelan and Goldman 2001]; Amino acid frequencies: model; Proportion of invariable sites: fixed [P-invar = 0.00]; One category of substitution rate: no; Number of substitution rate categories: 4; Gamma distribution rates across sites: yes; Gamma distribution parameter: estimated; Optimize tree topology: yes; Starting tree: BioNJ; Tree topology search operations: SPR moves; Add random starting trees: no; Nonparametric bootstrap analysis: yes [100 replicates]; Approximate likelihood ratio test: no). Bootstrapping was performed to provide support values and nodes with a bootstrap value below 80% were collapsed. Each tree was rooted between the two phyla that were part of the respective analysis and trees that did not properly separate the outgroups were removed from the further analysis. In total, three trees were produced to reconstruct the evolution to the last common ancestor of the Gammaproteobacteria and two for the reconstruction to the last common ancestor of the Bacilli (figs. 1 and 2, supplementary figs. S8–S12, Supplementary Material online).
Fig. 1.

Phylogeny of the Gammaproteobacteria. A maximum likelihood phylogeny tree was produced using the PhyML algorithm (WAG substitution model) based on the concatenated CLC alignments of 39 proteins within the secE-rpoBC-str-S10-spc-alpha operon cluster (supplementary table S2, Supplementary Material online). Support for each node was evaluated by bootstrapping and nodes with a bootstrap value below 80% were collapsed. Support values for nodes are shown when these are below 100%. Two additional nodes that were identified based on the gene synteny analysis are indicated with dashed lines. Branch lengths for the outgroup species was reduced by a factor of two (see scale value in parenthesis). Taxonomic orders are designated according to NCBI and evolutionary events are indicated in the tree. See supplementary figure S9, Supplementary Material online and supplementary table S6, Supplementary Material online for event details.
Reconstruction of Ancestral Operon Cluster Organization
Reconstruction of the ancestral operon cluster was performed using gene synteny and maximum parsimony. Each gene pair could take one of three states: 1) connected, 2) disconnected, and 3) one of the two genes is deleted. No distinction was made for the distance between two disconnected gene pairs (supplementary fig. S13, Supplementary Material online). For each potentially connected gene pair the number of minimal state changes was determined for the case that the gene pair is connected in the ancestral operon cluster (Nconnected) and for the case that the gene pair is not connected within the ancestral operon cluster (Ndisconnected). A gene pair was defined to be likely connected within the ancestral operon cluster if 1) Nconnected ≤ Ndisconnected and 2) the organization of the gene pair within the respective outgroup species agrees with the connection. The decision trees for all gene pairs that are not fully conserved throughout the Gammaproteobacteria or Bacilli are shown in supplementary figures S14 and S15, Supplementary Material online.
Operon Assignments
Operons in the ancestral operon clusters were defined according to the E. coli nomenclature. For three genes (rpmG, tRNA-Trp and rpl7ae) this assignment was ambiguous because they are not part of the operon cluster in E. coli and are located between two operons. The rpmG and tRNA-Trp genes were assigned to the secE operon and rpl7ae to the str operon according to the operon structure of the Bacilli. The ancestral operon cluster in the Bacilli contained five additional genes upstream (cysS-rnc-rlmB-orf1-sigH) and six additional genes downstream (ecfA1-ecfA2-ecfT-truA-rplM-rpsI) (fig. 3, supplementary table S5, Supplementary Material online). The additional genes identified upstream and downstream of the operon cluster in the Bacilli were grouped into two operons and named after their respective first gene: cysS and ecf (fig. 3).
Fig. 3.

Reconstructed ancestral secE-rpoBC-str-S10-spc-alpha operon cluster in the last common ancestor of the Gammaproteobacteria and the last common ancestor of the Bacilli. (A) Overview over the operon concatenation. (B) Overview over the operon content. Operons present in the last common ancestor of the Gammaproteobacteria are indicated by a “γ” and operons present in the last common ancestor of the Bacilli are indicated by a “B.”
Identification of Evolutionary Events
Evolutionary events that alter the organization of the operon cluster (deletions or rearrangements) were identified by comparing the organization within modern species to the reconstructed ancestral organization. Initially, a gene synteny analysis was performed for all genomic regions that deviated from the proposed ancestral operon cluster organization. For regions with insertions (<10 kb sequence between two genes of the operon cluster), all inserted genes were included in the analysis. For gene pairs that were fully disconnected within a modern species, only the five first genes adjacent to the two respective disconnected genes were included (supplementary fig. S16A and supplementary tables S4, S5, Supplementary Material online). Novel gene neighborhoods were initially identified based on the annotated protein function and comparison of sequence lengths. Uncertain gene neighborhoods (based on a single proteins or common protein functions) were further confirmed by protein sequence alignments using the CLC alignment algorithm as described above. Evolutionary events were then identified with all five constructed trees (supplementary figs. S8–S12, Supplementary Material online) using maximum parsimony and the results of the gene synteny analysis. Novel gene neighborhoods that were identified were set to be dominant so that not all descendent species were required to contain the specific rearrangement (e.g., due to loss of the inserted genes). A layout for the identification process is shown in supplementary figure S16B, Supplementary Material online. The trees that described the evolutionary events with the least alterations and highest bootstrap values were chosen for further analysis (figs. 1 and 2).
Fig. 2.

Phylogeny of the Bacilli. A maximum likelihood phylogeny tree was produced using the CLC algorithm (WAG substitution model) based on the concatenated CLC alignments of 44 proteins within the secE-rpoBC-str-S10-spc-alpha operon cluster (supplementary table S3, Supplementary Material online). Support for each node was evaluated by bootstrapping and nodes with a bootstrap value below 80% were collapsed. Support values for nodes are shown when these are below 100%. Branch lengths for the outgroup species was reduced by a factor of two (see scale value in parenthesis). Taxonomic orders and families are designated according to NCBI and evolutionary events are indicated in the tree. See supplementary figure S11 and supplementary table S6, Supplementary Material online for event details.
Test for Long Distance HGT
Each of the deletions and rearrangements that were part of the in-depth analysis were tested for the contribution of HGT from distantly related species. Long distance HGT was tested by comparison of the gene trees of the potentially transferred genes with their associated species trees (figs. 1 and 2). For deletion events, potentially transferred genes were defined as the corresponding copy of the gene located outside the operon cluster. For the rearrangement events, potentially transferred genes were defined as the genes included in the minimal inserted segment (fig. 6). Species were included in the analysis based on two parameters: 1) species must include the full set of genes to be analyzed, and 2) the chosen set must include closely related species before and after the potential HGT event. Maximum likelihood phylogenetic trees based on the concatenated alignments of the proteins to be tested were constructed using the identical settings that were used for constructing the respective species trees. The generated gene trees were then compared with their associated species trees and the RF distance was calculated based on an 80% bootstrap threshold (Robinson and Foulds 1981).
Fig. 6.
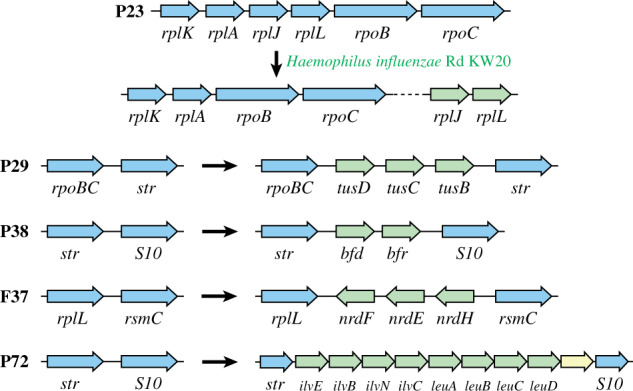
Overview over the four rearrangement events (P29, P38, F37, and F72) and one of the deletion events (P23) selected for further analysis. Genes/operons that are colinear at the time of the insertion are shown in blue and genes that represent the minimal inserted segment are green. The last gene of the insertion event F72 (yellow) is a putative amino acid transporter and was not part of the further analysis.
Results and Discussion
Reconstruction of the Ancestral Operon Clusters
A total of 204 bacterial genomes within the Proteobacteria, Acidobacteria, Firmicutes, and Tenericutes were chosen for the analysis of the operon cluster (supplementary table S1, Supplementary Material online). The majority of genomes chosen belong to the Gammaproteobacteria (103 genomes) and the Bacilli (80 genomes) to support a detailed analysis within these two important classes of bacteria and to include both Gram-negative and Gram-positive bacteria in the study. Based on a phylogenetic analysis (figs. 1 and 2), the most likely ancestral secE-rpoBC-str-S10-spc-alpha operon cluster was reconstructed for the last common ancestor of the Gammaproteobacteria and the last common ancestor of the Bacilli (fig. 3, supplementary tables S4 and S5, Supplementary Material online). Operons in the ancestral operon clusters were defined according to the Escherichia coli nomenclature to be consistent with previous studies. The tufB operon was absent from the operon cluster in all Bacilli but the ancestral operon cluster in the Bacilli contained two additional operons, the cysS operon upstream (cysS-rnc-rlmB-orf1-sigH) and the ecf operon downstream (ecfA1-ecfA2-ecfT-truA-rplM-rpsI) (fig. 3, supplementary table S5, Supplementary Material online). Overall, the two reconstructed ancestral operon clusters can be divided into a core cluster (secE-rpoBC-str-S10-spc-alpha) that is almost identical in both ancestors (40 out of 47 genes, 85% are present in both) and the variable tail operons (tufB, cysS, and ecf) that differ between the two ancestors (fig. 3). Interestingly, the outgroup species in the phylogenetic analysis of the Gammaproteobacteria strongly suggest that the rpmG, adk, map and infA genes were present in the secE and spc operons of the last common ancestor of the Proteobacteria (supplementary table S4, Supplementary Material online). Furthermore, the outgroup species in the phylogenetic analysis of the Bacilli suggest that the tufB operon was present in the last common ancestor of the Firmicutes (supplementary table S5, Supplementary Material online) which agrees with a previous study that suggests that the tuf duplication precedes the evolution of the Firmicutes (Lathe and Bork 2001). The potential ancestral operon cluster that was present in the last common ancestor of the Proteobacteria and the Firmicutes is shown in supplementary figure S1, Supplementary Material online.
Identification of Evolutionary Events
The organization of the genes within the operon cluster of the 204 modern genomes included in this study was compared with their respective reconstructed ancestral organizations. All alterations within the organization were classified into two classes: deletions and rearrangement. “Deletions” were defined as alterations that remove a single gene from an operon but leave the operon–operon concatenation intact (the gene might be lost from the chromosome or effectively translocated), and “rearrangements” as changes that affect the operon structure and/or the operon–operon concatenation (inversions, gene insertions and loss of concatenation). Two species within the Gammaproteobacteria (Legionella pneumophila and Halorhodospira halophila) contained the full ancestral operon cluster and a further 17 species that belong to nine different orders only contained changes within the tRNA genes of the tufB operon. Overall, the seven operons of the ancestral operon cluster remained connected (<10 kb sequence between any two genes) in 38 out of 103 (37%) Gammaproteobacteria within thirteen orders (supplementary table S4, Supplementary Material online). Among the Bacilli, no modern species contained the full ancestral operon cluster but thirteen species among the Bacillaceae and Thermoactinomycetaceae differ only by a rpsD deletion and one species of the Alicyclobacillaceae differs only by an ecfA1A2T deletion from the ancestral operon cluster. The eight operons of the ancestral operon cluster remained connected in 34 out of 80 (43%) Bacilli within seven bacterial families (supplementary table S5, Supplementary Material online).
Next, the phylogenetic analysis was combined with the identified operon alterations to pinpoint evolutionary events that led to the modern operon organization. The four tRNA genes encoded in the tufB operon (tRNAThr-tRNATyr-tRNAGly-tRNAThr) displayed a high degree of variability (e.g., frequent deletions of one or both tRNAThr genes) and were therefore disregarded for this analysis. Overall, a total of 163 evolutionary events were identified, consisting of 48 deletions and 115 rearrangements (figs. 1 and 2, supplementary table S6, Supplementary Material online). The majority of the rearrangement events (75 out of 115, 65%) disrupt operon–operon concatenation but leave the operons intact. All operon pairs are affected except for the spc-alpha operon pair which remains concatenated in every genome that was analyzed. Two deletions and two rearrangements each were chosen from the Gammaproteobacteria and the Bacilli for further analysis (representing 10 independent events). For the deletions the focus was on deletions that are present in a small number of modern species and where the remaining operon organization is intact. Rearrangement events were selected based on the formation of conserved novel gene neighborhoods. Each of the eight deletions and rearrangements were tested for the influence of HGT from distantly related species that might transfer a rearranged operon structure into the chromosome (closely related species share the operon organization) and potential alternative mechanisms of operon cluster alterations are discussed.
Deletions of rplJL (P23) and rpmJ (P50, P51, P52) within the Gammaproteobacteria
A single species within the Gammaproteobacteria (Haemophilus influenzae Rd KW20) carries a deletion (event P23) of the rplJ and rplL genes, encoding ribosomal proteins RplJ (L10) and RplL (L7), from the rpoBC operon (figs. 1 and 4). Both proteins are essential for bacterial growth (Baba et al. 2006) and the H. influenzae Rd KW20 genome contains a rplJL gene pair at a different genomic location. A test for HGT based on the Robinson–Foulds distance (Robinson and Foulds 1981) indicates that no HGT occurred from a distantly related species (RF = 0 based on an 80% bootstrap threshold, supplementary fig. S2, Supplementary Material online) leaving HGT from closely related species and intrachromosomal rearrangements as possible explanations for the rplJL deletion event.
The ribosomal protein RpmJ (L36) was deleted from the operon cluster in three independent events (P50, P51, and P52) resulting in six modern Gammaproteobacteria that lack the rpmJ gene within the operon cluster (fig. 1). RpmJ is not essential for bacterial growth in E. coli K-12 (Baba et al. 2006) indicating that the Gammaproteobacteria that lack the rpmJ gene within the operon cluster might produce RpmJ-free ribosomes. However, each of the six genomes contains a copy of the rpmJ gene outside the operon cluster. A screening of the genomes of all Proteobacteria and Acidobacteria included in this study revealed that 35% (40/115) of all genomes contain two copies of rpmJ (supplementary table S7, Supplementary Material online), suggesting a duplication or HGT event early in the evolution of the Proteobacteria. The rpmJ genes of the 115 Proteobacteria and Acidobacteria were classified as type 1 (rpmJ1, located within the spc operon) and type 2 (rpmJ2, located outside the spc operon) and a phylogenetic analysis was conducted to test if the rpmJ genes within the six modern species that lack the gene within the operon cluster are related to a novel duplication event (rpmJ1) or to the pre-existing duplication (rpmJ2). Only a single node that separates the rpmJ1 and rpmJ2 genes into two distinct clades displayed a bootstrap value >80% (with the exception of one rpmJ2 gene which is discussed below, fig. 4A). The amino acid consensus sequence of the two rpmJ types shows 40% sequence identity and a three amino acid insertion that all rpmJ2 genes share (fig. 4B, C). Two of the rpmJ2 genes were identified in Helicobacter pylori J99 and Campylobacter jejuni 269.97, suggesting that the rpmJ2 gene was present outside the operon cluster in the last common ancestor of the Epsilonproteobacteria and Gammaproteobacteria. Although this data do not reveal the origin of the rpmJ2 gene (duplication or HGT), it shows that the three deletion events of the rpmJ1 genes from the spc operon in the Gammaproteobacteria represent the deletion of a duplicate gene resulting in a genome with a single copy rpmJ.
Fig. 4.

Analysis of rpmJ deletion events within the Gammaproteobacteria (events P50, P51, and P52). (A) Circular maximum likelihood phylogram (PhyML algorithm with WAG substitution model based on CLC alignments, nodes with bootstrap value below 80% were collapsed) of RpmJ proteins encoded within (RpmJ1, black) and outside (RpmJ2, red) the spc operon. (B) Logo of alignments of all RpmJ1 and all RpmJ2 proteins within the Proteobacteria and Acidobacteria. (C) Alignment of consensus sequences of RpmJ1 and RpmJ2 proteins within the Proteobacteria and Acidobacteria. Identical amino acids are indicated by a dot. (D) Organization of duplicate spc-alpha operons within Psychromonas ingrahamii. (E, F) Maximum likelihood phylogeny trees were produced using the PhyML algorithm (WAG substitution model) based on the concatenated CLC alignments of (E) 39 proteins within the secE-rpoBC-str-S10-spc-alpha operon cluster (supplementary table S2, Supplementary Material online) and (F) RpmJ, RpsM, RpsK, RpsD, RpoA, and RpsQ. Support for each node was evaluated by bootstrapping. The duplication event is indicated in the phylogenetic trees by a yellow star. RF value between the trees was calculated based on an 80% bootstrap threshold.
A single rpmJ2 gene (found in Psychromonas ingrahamii) was closely related to the rpmJ1 genes (fig. 4A). Further analysis of the genomic location of the gene revealed that it was located together with a second copy of the alpha operon (rpmJ2-rpsM2-rpsK2-rpsD2-rpoA2-rplQ2) (fig. 4D). A phylogenetic analysis based on the six proteins indicates that the duplication is most likely a result of an intrachromosomal duplication event within the P. ingrahamii chromosome or was acquired by horizontal transfer from a closely related species (RF = 0 based on an 80% bootstrap threshold, fig. 4E, F).
Deletions of rpsN (F81) and secY (F84) within the Bacilli
The species within the Leuconostocaceae clade are the only Bacilli that lack the rpsN gene within the spc operon of the operon cluster (event F81). The rpsN gene encodes ribosomal protein RpsN (S14) that is essential for growth in E. coli (Baba et al. 2006). As expected, each of the four species within the Leuconostocaceae clade carry a copy of rpsN in the chromosome outside the spc operon. Screening the Firmicutes and Tenericutes included in this study for second copies of the rpsN gene showed that 46 out of the 89 genomes (52%) carry two copies of rpsN (supplementary table S8, Supplementary Material online). The rpsN genes within the Firmicutes and Tenericutes were classified as type 1 (rpsN1, located within the spc operon) and type 2 (rpsN2, located outside the spc operon) and a phylogenetic analysis based on the amino acid alignments was conducted (fig. 5). The results show two distinct versions of RpsN, a short isoform that is located within the spc operon (RpsN1, 31 aa) and a long isoform that is found outside the spc operon (RpsN2, 89 aa). There are six exceptions to this classification. Two species (Bacillus coagulans and Streptococcus mutans) carry a second copy of rpsN1 outside the spc operon and four species carry the rpsN2 isoform within the spc operon (fig. 5A). Interestingly, although three of the four species with the rpsN2 isoform within the spc operon are members of the outgroup species (Acholeplasma laidlawii, Erysipelothrix rhusiopathiae, and Veillonella parvula) there is also a single species within the Bacilli (Streptococcus pneumoniae) that carries the longer rpsN2 isoform within the spc operon. An alignment of the rplE-rpsN-rpsH segment of the spc operon within S. pneumoniae and the two closest related species (Streptococcus parasanguinis and Streptococcus intermedius) indicates that only the rpsN coding sequence was changed within S. pneumoniae whereas the neighboring genes remained unchanged (fig. 5D, E). The two RpsN isoforms display a high degree of protein sequence identity at the beginning (10 out of the first 11 amino acids) and the end (20 out of the last 21 amino acids) of the coding sequence (fig. 5B, C). Since the last common ancestor of the Streptococcaceae most likely carried both rpsN isoforms in its chromosome (supplementary table S8, Supplementary Material online) it is possible that the changed rpsN sequence in S. pneumoniae is the result of an intrachromosomal gene conversion event as seen for the duplicate tuf genes (Abdulkarim and Hughes 1996; Brandis et al. 2018). Alternatively, the novel rpsN2 sequence within the spc operon of S. pneumoniae could be the result of an HGT event in which only the rpsN coding sequence was exchanged. Independent of the precise mechanism, these results show that the coding sequences of a single gene within the operon cluster can be exchanged to a significantly different isoform without affecting the neighboring genes or the intergenic sequences.
Fig. 5.

Analysis of rpsN deletion events within the Bacilli (events F81). (A) Circular maximum likelihood phylogram (CLC algorithm with WAG substitution model based on CLC alignments, nodes with bootstrap value below 80% were collapsed) of RpsN proteins encoded within (RpsN1, black) and outside (RpsN2, red) the spc operon. (B) Logo of alignments of all RpmJ1 and all RpmJ2 proteins within the Firmicutes and Tenericutes. (C) Alignment of consensus sequences of RpsN1 and RpsN2 proteins within the Firmicutes and Tenericutes. Identical amino acids are indicated by a dot. (D, E) Nucleotide sequence alignment of the rplE-rpsN-rpsH segment in the spc operon of Streptococcus pneumoniae with the corresponding segments in (D) Streptococcus parasanguinis and (E) Streptococcus intermedius. The black line indicates the DNA and coding sequences are shown above. The nucleotide sequence identity (averaged over 50 nt) is shown below. The red crosses indicate the likely location of a recombination event that inserted the rpsN2 gene into the spc operon of S. pneumoniae.
Bacillus selenitireducens is the only species within this study that has the secY gene, encoding the essential Sec translocon subunit SecY (Baba et al. 2006), located outside the spc operon (event F84). A phylogenetic analysis of the SecY proteins in the Listeriaceae, Bacillaceae, and Sporolactobacillaceae was conducted to test if the deletion of the secY gene from the spc operon in B. selenitireducens is the result of a long-distance HGT event. The resulting tree is fully compatible with the species tree of the respective species based on the full operon cluster (RF = 0 based on an 80% bootstrap threshold, supplementary fig. S3, Supplementary Material online). This indicates that no long-distance HGT event seemed to complement the deletion of the gene from the spc operon.
Rearrangements of rpoBC-Str (P29) and str-S10 (P38) within the Gammaproteobacteria
Two rearrangement events were chosen for the Gammaproteobacteria that most likely consisted of the insertion of a small number of genes between the operon pairs (fig. 6). The tusDCB genes were inserted between the rpoBC and str operon pair (event P29), and bfd-bfr between the str and S10 operons (event P38). The bfr gene encodes a bacterioferritin that is responsible for iron storage within the bacterial cell (Andrews et al. 1989; Sevcenco et al. 2011) and bfd encodes a ferredoxin that binds Bfr and helps regulating iron homeostasis (Garg et al. 1996; Yao et al. 2012). The TusBCD sulfurtransferase complex is an important part of the 2-thiouridine synthesis of the modified wobble base mnm5s2U in tRNA (Ikeuchi et al. 2006; Numata et al. 2006). Based on the novel gene neighborhood present within the modern species, both insertion events most likely occurred within the last common ancestor of Psychromonas, Moritella, and all Aeromondales, Vibrionales, Pateurellales, Orbales, and Enterobacterales (fig. 1, supplementary figs. S4 and S5, Supplementary Material online). In many modern species, this novel gene order is not maintained but the operon pairs (rpoBC-str and str-S10) are fully disconnected (separated by >10 kb) suggesting that the gene insertions relaxed the selection for operon synteny (supplementary figs. S4A and S5A, Supplementary Material online). Phylogenetic analyses indicate that neither event is likely to be the result of a gene transfer event from a distantly related species (supplementary figs. S4 and S5, Supplementary Material online). The only node within the phylogenetic tree based on the TusDCB proteins that significantly differs from the species tree places Moritella yayanosii within the neighboring Vibrionales and Aeromondales clade but the node is only supported by a bootstrap value of 80% and the tree is still fully consistent with the insertion event (supplementary fig. S4, Supplementary Material online).
Rearrangements of rplL-rsmC (F37) and str-S10 (F72) within the Bacilli
The rearrangement events within the Bacilli that were chosen for further analysis occurred within the Lactobacillaceae (event F37) and within the Sporolactobacillaceae (event F72). Both events consist of insertions of a small number of genes (fig. 6). The nrdHEF genes were inserted between the rplL and rsmC genes within the rpoBC operon (event F37) and a set of amino acid biosynthesis genes, ilvEBNC-leuABCD between the str and S10 operons (event F72). NrdH is a glutaredoxin-like protein that acts as an electron donor for the class Ib ribonucleotide reductase NrdEF which is involved in the biosynthesis of dNTPs (Jordan et al. 1994), and the IlvBCEN and LeuABCD proteins are part of the biosynthetic synthesis pathway of branched chain amino acids (Salmon et al. 2006). As seen for the insertion events within the Gammaproteobacteria, the novel gene order created by the insertion of the nrdHEF genes is not maintained in all modern species and the rplL and rsmC gene pair is fully disconnected in two out of the five modern species (supplementary fig. S6A, Supplementary Material online). The phylogenetic analysis based on the NrdHEF proteins shows no indications of HGT from a distantly related species (RF = 0 based on an 80% bootstrap threshold, supplementary fig. S6, Supplementary Material online) but the tree based on the IlvBCEN and LeuABCD proteins shows some deviations from the species tree (RF = 5 based on an 80% bootstrap threshold). However, all discrepancies are located within the Bacillaceae and Sporolactobacillaceae clades. The Planococcaceae clade in which the insertion event occurred is identical to the species tree (supplementary fig. S7, Supplementary Material online). It is therefore unlikely that either of the two events is the result of a long-distance HGT event.
Potential Mechanisms That Could Drive Operon Cluster Alterations
The four deletions that were part of the further analysis were either 1) deletions of genes with a homolog in a different chromosomal location that has most likely been present in the last common ancestors of the Gammaproteobacteria (rpmJ, events P50, P51, and P52) or the Bacilli (rpsN, event F81), or 2) the result of a novel duplication (intrachromosomal or by HGT from a closely related species) followed by a deletion of the gene within the operon cluster (rplJL, event P23 and secY, event F84). For the rearrangement events the analysis indicates that the disconnection of two concatenated operons often follows the same pattern: initially, a small number of genes invade the interoperon region of the operon pair thus breaking the concatenation. Due to the relaxed selection for operon colocalization further rearrangement events can fully separate the operons. This also agrees with the observation that 67% of the events (77 out of 115 events) can be traced back to gene insertions (supplementary table S6, Supplementary Material online).
A potential mechanism that could explain each of the events that did not involve a pre-existing gene duplication is the recently suggested SNAP (Selection during Niche Adaptation) model. This model proposes that a segmental chromosomal duplication is selected and stabilized within a bacterial population during adaptation to a novel environment. Subsequently, the asymmetrical loss of duplicate genes can lead to alterations in the gene order on the chromosome (fig. 7) (Brandis and Hughes 2020). Segmental duplications are probably the most common type of mutations in the bacterial genome as they appear at very high frequencies even in the absence of selection (Anderson and Roth 1977; Haack and Roth 1995; Reams et al. 2010), are often found during laboratory selection conditions (Riehle et al. 2001; Knöppel et al. 2016), and play a role in shaping genome diversification and the evolution of new genes (Bergthorsson et al. 2007; Näsvall et al. 2012; Zhou et al. 2012). Additionally, a duplication of a chromosomal region would not disrupt operon integrity or operon concatenations. The subsequent loss of duplicate genes/operons is a slow process enabling the bacteria to adapt to potential negative consequences caused by the novel gene order. Thus, the SNAP model could explain the observed operon cluster alterations by combining high-frequency events and overcoming counter-selective barriers (Brandis and Hughes 2020). This model could also explain the duplicate section of the operon cluster (rpmJ-rpsM-rpsK-rpsD-rpoA-rplQ) found in P. ingrahamii (supplementary fig. S3D, Supplementary Material online). Alternatively, the gene insertion events could be facilitated by direct transposition, be the net outcome of multiple consecutive inversion events, or the result of the integration of a horizontally acquired segment from a closely related species (fig. 7B). A limitation with these three mechanisms is that they would be based on recombination between very short or nonhomologous sequences and are therefore expected to occur, if at all, at very low frequencies (Watt et al. 1985; Shen and Huang 1986; Brandis et al. 2018). Additionally, each of them leads to the sudden disruption of the operon integrity or operon concatenations and will most likely cause a reduction of cellular fitness (Campo et al. 2004; Brandis et al. 2019). Thus, these mechanisms couple a very infrequent event with a counter-selected fitness cost. A potential way to overcome these limitations would be “hijacking” of transposable elements. The integration of a transposon within or between operons could provide adequate sequence homologies for recombination that could ultimately lead to the insertion of genes between the operons. Transposon-directed integration is for example frequently observed with the integration of the F plasmid into the chromosome (Chumley et al. 1979). Interestingly, six of the events observed in the Gammaproteobacteria (P46) and Bacilli (F23, F33, F47, F76, and F103) consisted of integrations of transposable elements within the operon cluster (supplementary table S6, Supplementary Material online). This indicates that transposable elements could play a crucial role in the rearrangements of the operon cluster.
Fig. 7.

Potential mechanisms for operon cluster alteration events by intrachromosomal recombination (gray boxes) or horizontal gene transfer. (A) Genes can be deleted from the operon cluster if the chromosome contains a pre-existing homolog (a). Alternatively, the acquisition of a duplicate gene by intrachromosomal duplication (b) or horizontal gene transfer (d) allows for the deletion of the duplicate gene within the operon cluster (c). (B) Insertion of genes into the operon cluster could be facilitated by an intrachromosomal duplication event (a) followed by asymmetrical loss of duplicate genes (b), by two consecutive transposon-directed inversions (c, d), by transposon-directed transposition (e, f), or horizontal gene transfer (g) followed by transposon-directed insertion (f).
It is not possible to determine the precise mechanisms by which the analyzed operon cluster alterations occurred but the data suggest that duplications and transposable elements could be involved. Thus, deletions are most likely the result of a pre-existing ancient duplication or a novel duplication event that occurred intrachromosomally or by HGT from a closely related species (fig. 7A). Insertion events are most likely the result of a segmental duplication followed by asymmetrical loss of duplicate genes, transposon-directed intrachromosomal rearrangements, or transposon-directed integration of a horizontally acquired segment from a closely related species (fig. 7B).
Operon Cluster Alteration Events Are Not Equally Distributed
The selection to maintain the linear gene order within bacterial chromosomes is weak on an evolutionary time scale (Tamames 2001). The secE-rpoBC-str-S10-spc-alpha operon cluster is a remarkable example of an exception to this rule as can be seen in species that maintained the full operon cluster organization over 2 Gy. This is further highlighted by the observation that changes to the operon cluster occur infrequently. Almost all branches within the two trees display no (70%) or single (20%) alteration events (supplementary table S9, Supplementary Material online). Interestingly, there are three branches within the Bacilli with seven or eight “simultaneous” events that might represent time points with unusually many operon cluster alteration events. To test this hypothesis, the ratio of alteration events per amino acid substitution (based on branch length) was calculated for every branch with at least one alteration in the Gammaproteobacteria and Bacilli trees. On average, there were 3.7 ± 3.8 alteration events per 1,000 amino acid substitutions with no significant difference (P = 0.37, t-test) between the Gammaproteobacteria (3.3 ± 3.6 alterations per 1,000 substitutions) and the Bacilli (4.0 ± 3.9 alterations per 1,000 substitutions). Two of the three branches with a large number of alteration events were indistinguishable from this average rate (3.4 and 4.9 alterations per 1,000 substitutions) but the branch that represents the last common ancestor of the Streptococcaceae displayed a 3-fold increased operon alteration rate of 12 alterations per 1,000 amino acid substitutions. This indicates that the last common ancestor of the Streptococcaceae encountered a period with relaxed selection on the gene order within operon cluster. Another noteworthy observation is that a third of the events identified within the Gammaproteobacteria (33%, 17 out of 51 events) are located within the Pasteurellales whereas these only represent 12% of the analyzed species (fig. 1). An order-wide operon cluster alteration rate (including all branches and alterations unique to each order) was determined within the Gammaproteobacteria (supplementary table S10, Supplementary Material online). The alteration rate within the Pasteurellales was 3.24 alterations per 1,000 substitutions. This rate was 14-fold higher that the average rate of all other orders (0.24 ± 0.24 alterations per 1,000 substitutions). A possible explanation for this observation might be that the Pasteurellales are naturally competent. Natural competence per se is not uncommon among the Gammaproteobacteria (Johnston et al. 2014) but the Pasteurellales have a strong preference in the uptake of conspecific DNA (Bakkali et al. 2004; Redfield et al. 2006; Mell and Redfield 2014). It has been shown that natural transformation of conspecific DNA frequently generates duplications within bacterial genomes (Johnston et al. 2013). It is therefore possible that the natural competence coupled with preferential uptake of conspecific DNA could provide an increase of duplication formations that is ultimately responsible for the observed increase in operon rearrangements (fig. 7A). Alternatively, direct integration of the conspecific DNA into the chromosome could result in operon cluster alterations that would be indistinguishable from intrachromosomal translocations (fig. 7B).
Conclusions
Here, the evolutionary history of the secE-rpoBC-str-S10-spc-alpha operon cluster was reconstructed for a period of 2 Gy back to the last common ancestors of the Gammaproteobacteria and the Bacilli. A total of 163 independent evolutionary events were identified in which the operon cluster was altered (figs. 1 and 2) and a subset of gene deletion and operon rearrangement events were further analyzed. The main findings were:
The operon cluster remains stable in many Gammaproteobacteria and Bacilli over billions of years with no or little change.
Single genes within the operon cluster can be exchanged with distantly related homologs without affecting the neighboring genes.
Segmental duplications of the operon cluster can be found within the chromosomes.
Deletions of genes within the operon cluster are the result of novel duplications or pre-excising homologs.
Operons concatenation is broken by an invasion-separation mechanism.
Intrachromosomal duplications and/or transposon-directed recombination play a crucial role in the rearrangement of the operon cluster.
Long-distance HGT does not play a major role in the events that were further investigated.
There are examples of accumulation of operon cluster alterations at specific branch points or within specific bacterial orders.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Data Availability
All genome sequences included in this study are publicly available in the NCBI database. Accession numbers are provided in supplementary table S1, Supplementary Material online.
Supplementary Material
Acknowledgments
This study was supported by grants to Diarmaid Hughes from the Swedish Science Research Council (Vetenskapsrådet, grant number 2017-03953) and the Carl Trygger Foundation (grant number CTS17:204).
Literature Cited
- Abdulkarim F, Hughes D.. 1996. Homologous recombination between the tuf genes of Salmonella typhimurium. J Mol Biol. 260(4):506–522. [DOI] [PubMed] [Google Scholar]
- Anderson RP, Roth JR.. 1977. Tandem genetic duplications in phage and bacteria. Annu Rev Microbiol. 31:473–505. [DOI] [PubMed] [Google Scholar]
- Andrews SC, Harrison PM, Guest JR.. 1989. Cloning, sequencing, and mapping of the bacterioferritin gene (bfr) of Escherichia coli K-12. J Bacteriol. 171(7):3940–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmetti Y, et al. 2015. Ancestral gene synteny reconstruction improves extant species scaffolding. BMC Genomics 16(Suppl 10):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkali M, Chen TY, Lee HC, Redfield RJ.. 2004. Evolutionary stability of DNA uptake signal sequences in the Pasteurellaceae. Proc Natl Acad Sci USA. 101(13):4513–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, et al. 2014. A new system for naming ribosomal proteins. Curr Opin Struct Biol. 24:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barloy-Hubler F, Lelaure V, Galibert F.. 2001. Ribosomal protein gene cluster analysis in eubacterium genomics: homology between Sinorhizobium meliloti strain 1021 and Bacillus subtilis. Nucleic Acids Res. 29(13):2747–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergthorsson U, Andersson DI, Roth JR.. 2007. Ohno's dilemma: evolution of new genes under continuous selection. Proc Natl Acad Sci USA. 104(43):17004–17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth A, Mariscal C, Doolittle WF.. 2016. The modern synthesis in the light of microbial genomics. Annu Rev Microbiol. 70:279–297. [DOI] [PubMed] [Google Scholar]
- Brandis G, Cao S, Hughes D.. 2018. Co-evolution with recombination affects the stability of mobile genetic element insertions within gene families of Salmonella. Mol Microbiol. 108(6):697–710. [DOI] [PubMed] [Google Scholar]
- Brandis G, Cao S, Hughes D.. 2019. Operon concatenation is an ancient feature that restricts the potential to rearrange bacterial chromosomes. Mol Biol Evol. 36:1900–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandis G, Hughes D.. 2020. The SNAP hypothesis: chromosomal rearrangements could emerge from positive Selection during Niche Adaptation. PLoS Genet. 16(3):e1008615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo N, Dias MJ, Daveran-Mingot ML, Ritzenthaler P, Le Bourgeois P.. 2004. Chromosomal constraints in Gram-positive bacteria revealed by artificial inversions. Mol Microbiol. 51(2):511–522. [DOI] [PubMed] [Google Scholar]
- Chumley FG, Menzel R, Roth JR.. 1979. Hfr formation directed by Tn10. Genetics 91(4):639–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenye T, Vandamme P.. 2005. Organisation of the S10, spc and alpha ribosomal protein gene clusters in prokaryotic genomes. FEMS Microbiol Lett. 242(1):117–126. [DOI] [PubMed] [Google Scholar]
- du Plessis DJ, Nouwen N, Driessen AJ.. 2011. The Sec translocase. Biochim Biophys Acta. 1808(3):851–865. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B. 1982. The jackknife, the bootstrap, and other resampling plans. Philadelphia (PA: ): Society for Industrial and Applied Mathematics. [Google Scholar]
- Felsenstein J. 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 17(6):368–376. [DOI] [PubMed] [Google Scholar]
- Forterre P. 2015. The universal tree of life: an update. Front Microbiol. 6:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg RP, Vargo CJ, Cui X, Kurtz DM Jr.. 1996. A [2Fe-2S] protein encoded by an open reading frame upstream of the Escherichia coli bacterioferritin gene. Biochemistry 35(20):6297–6301. [DOI] [PubMed] [Google Scholar]
- Guindon S, Delsuc F, Dufayard JF, Gascuel O.. 2009. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 537:113–137. [DOI] [PubMed] [Google Scholar]
- Haack KR, Roth JR.. 1995. Recombination between chromosomal IS200 elements supports frequent duplication formation in Salmonella typhimurium. Genetics 141(4):1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi Y, Shigi N, Kato J, Nishimura A, Suzuki T.. 2006. Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol Cell. 21(1):97–108. [DOI] [PubMed] [Google Scholar]
- Itoh T, Takemoto K, Mori H, Gojobori T.. 1999. Evolutionary instability of operon structures disclosed by sequence comparisons of complete microbial genomes. Mol Biol Evol. 16(3):332–346. [DOI] [PubMed] [Google Scholar]
- Johnston C, et al. 2013. Natural genetic transformation generates a population of merodiploids in Streptococcus pneumoniae. PLoS Genet. 9(9):e1003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston C, Martin B, Fichant G, Polard P, Claverys JP.. 2014. Bacterial transformation: distribution, shared mechanisms and divergent control. Nat Rev Microbiol. 12(3):181–196. [DOI] [PubMed] [Google Scholar]
- Jordan A, et al. 1994. A second class I ribonucleotide reductase in Enterobacteriaceae: characterization of the Salmonella typhimurium enzyme. Proc Natl Acad Sci USA. 91(26):12892–12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöppel A, Näsvall J, Andersson DI.. 2016. Compensating the fitness costs of synonymous mutations. Mol Biol Evol. 33(6):1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 2003. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol. 1(2):127–136. [DOI] [PubMed] [Google Scholar]
- Koonin EV. 2014. Carl Woese's vision of cellular evolution and the domains of life. RNA Biol. 11(3):197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Mushegian AR, Rudd KE.. 1996. Sequencing and analysis of bacterial genomes. Curr Biol. 6(4):404–416. [DOI] [PubMed] [Google Scholar]
- Lathe WC 3rd, Bork P.. 2001. Evolution of tuf genes: ancient duplication, differential loss and gene conversion. FEBS Lett. 502(3):113–116. [DOI] [PubMed] [Google Scholar]
- Le SQ, Gascuel O.. 2008. An improved general amino acid replacement matrix. Mol Biol Evol. 25(7):1307–1320. [DOI] [PubMed] [Google Scholar]
- Marin J, Battistuzzi FU, Brown AC, Hedges SB.. 2017. The Timetree of prokaryotes: new insights into their evolution and speciation. Mol Biol Evol. 34(2):437–446. [DOI] [PubMed] [Google Scholar]
- Mell JC, Redfield RJ.. 2014. Natural competence and the evolution of DNA uptake specificity. J Bacteriol. 196(8):1471–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Hagelsieb G, Treviño V, Pérez-Rueda E, Smith TF, Collado-Vides J.. 2001. Transcription unit conservation in the three domains of life: a perspective from Escherichia coli. Trends Genet. 17(4):175–177. [DOI] [PubMed] [Google Scholar]
- Näsvall J, Sun L, Roth JR, Andersson DI.. 2012. Real-time evolution of new genes by innovation, amplification, and divergence. Science 338(6105):384–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata T, Fukai S, Ikeuchi Y, Suzuki T, Nureki O.. 2006. Structural basis for sulfur relay to RNA mediated by heterohexameric TusBCD complex. Structure 14(2):357–366. [DOI] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N.. 1999. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA. 96(6):2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Huang KH, Arkin AP, Alm EJ.. 2005. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Res. 15(6):809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaraman A, Ma J.. 2016. Reconstructing ancestral gene orders with duplications guided by synteny level genome reconstruction. BMC Bioinformatics 17(Suppl 14):414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan V. 2002. Ribosome structure and the mechanism of translation. Cell 108(4):557–572. [DOI] [PubMed] [Google Scholar]
- Reams AB, Kofoid E, Savageau M, Roth JR.. 2010. Duplication frequency in a population of Salmonella enterica rapidly approaches steady state with or without recombination. Genetics 184(4):1077–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfield RJ, et al. 2006. Evolution of competence and DNA uptake specificity in the Pasteurellaceae. BMC Evol Biol. 6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle MM, Bennett AF, Long AD.. 2001. Genetic architecture of thermal adaptation in Escherichia coli. Proc Natl Acad Sci USA. 98(2):525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DF, Foulds LR.. 1981. Comparison of phylogenetic trees. Math Biosci. 53(1–2):131–147. [Google Scholar]
- Rocha EP. 2006. Inference and analysis of the relative stability of bacterial chromosomes. Mol Biol Evol. 23(3):513–522. [DOI] [PubMed] [Google Scholar]
- Salmon KA, Yang CR, Hatfield GW.. 2006. Biosynthesis and regulation of the branched-chain amino acids. EcoSal Plus. 2(1). doi: 10.1128/ecosalplus.3.6.1.5. [DOI] [PubMed] [Google Scholar]
- Sevcenco AM, et al. 2011. Exploring the microbial metalloproteome using MIRAGE. Metallomics 3(12):1324–1330. [DOI] [PubMed] [Google Scholar]
- Shen P, Huang HV.. 1986. Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics 112(3):441–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA.. 2002. The identification of functional modules from the genomic association of genes. Proc Natl Acad Sci USA. 99(9):5890–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamames J. 2001. Evolution of gene order conservation in prokaryotes. Genome Biol. 2(6):RESEARCH0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, et al. 1996. Metabolism and evolution of Haemophilus influenzae deduced from a whole-genome comparison with Escherichia coli. Curr Biol. 6(3):279–291. [DOI] [PubMed] [Google Scholar]
- Wachtershauser G. 1998. Towards a reconstruction of ancestral genomes by gene cluster alignment. Syst Appl Microbiol. 21:473–477. [Google Scholar]
- Wang Y, et al. 2012. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40(7):e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Mori H, Itoh T, Gojobori T.. 1997. Genome plasticity as a paradigm of eubacteria evolution. J Mol Evol. 44(Suppl 1):S57–64. [DOI] [PubMed] [Google Scholar]
- Watt VM, Ingles CJ, Urdea MS, Rutter WJ.. 1985. Homology requirements for recombination in Escherichia coli. Proc Natl Acad Sci USA. 82(14):4768–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MC, Preiner M, Xavier JC, Zimorski V, Martin WF.. 2018. The last universal common ancestor between ancient Earth chemistry and the onset of genetics. PLoS Genet. 14(8):e1007518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S, Goldman N.. 2001. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 18(5):691–699. [DOI] [PubMed] [Google Scholar]
- Woese CR. 2000. Interpreting the universal phylogenetic tree. Proc Natl Acad Sci USA. 97(15):8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, et al. 2012. The structure of the BfrB-Bfd complex reveals protein–protein interactions enabling iron release from bacterioferritin. J Am Chem Soc. 134(32):13470–13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, et al. 1999. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell 98(6):811–824. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gu J, Li YQ, Wang Y.. 2012. Genome plasticity and systems evolution in Streptomyces. BMC Bioinformatics 13(Suppl 10):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genome sequences included in this study are publicly available in the NCBI database. Accession numbers are provided in supplementary table S1, Supplementary Material online.