

SUMMARY
Mitochondrial carriers (MCs) mediate the passage of small molecules across the inner mitochondrial membrane (IMM), enabling regulated crosstalk between compartmentalized reactions. Despite MCs representing the largest family of solute carriers in mammals, most have not been subjected to a comprehensive investigation, limiting our understanding of their metabolic contributions. Here, we functionally characterize SFXN1, a member of the non-canonical, sideroflexin family. We find that SFXN1, an integral IMM protein with an uneven number of transmembrane domains, is a TIM22 complex substrate. SFXN1 deficiency leads to mitochondrial respiratory chain impairments, most detrimental to complex III (CIII) biogenesis, activity, and assembly, compromising coenzyme Q levels. The CIII dysfunction is independent of one-carbon metabolism, the known primary role for SFXN1 as a mitochondrial serine transporter. Instead, SFXN1 supports CIII function by participating in heme and α-ketoglutarate metabolism. Our findings highlight the multiple ways that SFXN1-based amino acid transport impacts mitochondrial and cellular metabolic efficiency.
Graphical Abstract
In brief
Acoba et al. show that the amino acid transporter SFXN1 is a cargo of the TIM22 translocon that is important for maintaining complex III function and supports coenzyme Q, heme, and α-ketoglutarate metabolism.
INTRODUCTION
Mitochondria are classically viewed as energy-producing organelles. Lining the folds of the inner mitochondrial membrane (IMM) cristae is the respiratory chain—a group of multimeric complexes (complexes I–IV) and mobile electron carriers—that facilitates the transfer of electrons from NADH and FADH2 to O2 while pumping protons to the intermembrane space (IMS). The use of the proton gradient powers the ATP synthase (complex V), whose rotary action creates ATP by oxidative phosphorylation (OXPHOS) (Abrahams et al., 1994; Hutton and Boyer, 1979; Mitchell, 1961). Because this is the major source of cellular ATP, perturbations in OXPHOS underlie various mitochondrial pathologies, including rare syndromes and a growing number of aging-related diseases (Sun et al., 2016; Vafai and Mootha, 2012).
Beyond a role in bioenergetics, mitochondria participate in the biosynthesis of nucleotides, lipids, amino acids, and numerous metabolites. The vital position of mitochondria in cells is underscored by the emerging appreciation that they contribute to intracellular signaling and regulatory networks (Spinelli and Haigis, 2018). At the core of this are the mitochondrial carriers (MCs) that coordinate the transport of diverse solutes into and out of mitochondria. Collectively, MCs enable mitochondria to communicate with the rest of the cell to achieve homeostasis and allow this complex organelle to influence metabolic flexibility and plasticity, attributes that are paramount for survival in a constantly changing metabolic landscape (Palmieri and Monné, 2016; Taylor, 2017).
Dysfunction in MCs results in pathologies affecting different organ systems and manifesting as various severities (Ogunbona and Claypool, 2019). Many of the disorders involve OXPHOS defects, whereas others lead to specific metabolic problems. For example, deficiencies in the adenine nucleotide translocase (ANT) isoform 1 and the phosphate carrier (PiC) cause myopathy and hypertrophic cardiomyopathy (Bakker et al., 1993; Bhoj et al., 2015; Echaniz-Laguna et al., 2012; Körver-Keularts et al., 2015; Mayr et al., 2007; Palmieri et al., 2005; Thompson et al., 2016). On the other hand, mutations in SLC25A38, which code for a glycine transporter, impinge on heme production and are associated with sideroblastic anemia (Guernsey et al., 2009; Harigae and Furuyama, 2010; Horvathova et al., 2010). Numerous MCs have also been implicated in cancer progression, including the understudied amino acid transporters that support biomass production for increased cell proliferation (Lytovchenko and Kunji, 2017).
To gain further insight into the functional organization of MCs, recent work by our group used a proteomic approach to systematically map the binding partners of human ANT1 and ANT2 (Lu et al., 2017)—the archetypal members of the solute carrier 25 (SLC25) family—that mediate the exchange of ADP and ATP across the IMM (Klingenberg, 2008; Pebay-Peyroula et al., 2003). Among the interactors is another group of mitochondrial membrane proteins: the sideroflexin (SFXN) family, an enigmatic group whose function is only now emerging. SFXN1–SFXN5 are categorized under SLC56, a group separate from SLC25, because they are predicted to possess 4 to 5 transmembrane domains (TMDs) (Fleming et al., 2001), in contrast to the canonical view that MCs consist of 6 TMDs, forming a structure with 3-fold pseudosymmetry (Palmieri, 2004). SFXN1, the founding member of the SLC56 family, was identified by positional cloning of the mutation in the flexed-tail mouse model with sideroblastic anemia (Fleming et al., 2001), although its role as the causal mutation has since come into question (Hegde et al., 2007; Lenox et al., 2005). SFXN1 was recently identified as a mitochondrial serine transporter essential for one-carbon (1C) metabolism (Kory et al., 2018), a process in which folate species are generated and used in biosynthetic pathways required for cell proliferation (Ducker and Rabinowitz, 2017). Notably, although its role as a serine transporter was shown, competition assays indicate that SFXN1 can transport additional amino acids (Kory et al., 2018). Mutations in another isoform, namely, SFXN4, are associated with mitochondriopathy and macrocytic anemia (Hildick-Smith et al., 2013). However, the nature and extent of how SFXNs impact mitochondrial biology are still essentially unexplored.
Here, we investigated SFXN1 in mammalian cells. We found that in HEK293 cells, SFXN1 is the most abundant isoform among the SFXNs and demonstrate that it is an integral polytopic protein in the IMM whose topology is consistent with its 5 predicted TMDs. The steady-state protein abundance of SFXNs is reduced without acylglycerol kinase (AGK), a metazoan-specific component of the TIM22 translocon crucial for the biogenesis of 6 TMD-containing MCs (Kang et al., 2017; Vukotic et al., 2017); this finding indicates SFXNs are TIM22 complex substrates. Although SFXN1 is predicted to be involved in the mitochondrial transport of a component necessary for iron use (Fleming et al., 2001), it does not govern iron homeostasis in HEK293 cells. Rather, we find that CIII biogenesis, activity, and assembly, together with coenzyme Q (CoQ) metabolism, are most severely impaired in the absence of SFXN1. Intriguingly, these results cannot be explained by a defective 1C metabolism, which represents the only well-documented physiological role for SFXN1 (Kory et al., 2018). Instead, we show that SFXN1 is important for heme and α-ketoglutarate (αKG) metabolism, of which both support CIII function to various extents. Collectively, these data define additional roles and thus provide a richer context for how SFXN1-mediated amino acid transport maintains mitochondrial integrity and promotes metabolic flexibility.
RESULTS
SFXN1 is a multipass IMM protein
SFXNs, consisting of 5 isoforms in humans (Figure S1A), are predicted to be integral polytopic IMM proteins with 4–5 TMDs (Fleming et al., 2001). The different isoforms have wide-ranging mRNA abundance across various tissues (Figure S1B). SFXN1 is highly expressed in the kidney, small intestine, ovary, and especially the liver. SFXN3 and SFXN5 have notable expression in the brain, whereas SFXN4 is the only isoform expressed well in heart and skeletal muscle. In HEK293, SFXN1 is the most abundant isoform based on protein level, followed by SFXN3 (Figure 1A), to which it shares the highest amino acid similarity (Figure S1A). SFXN1 is highly conserved across eukaryotic species and is closely related to yeast Fsf1p, having a 75% similarity and 37% identity (Figure S1C).
Figure 1. SFXN1, an integral inner mitochondrial membrane protein, is a TIM22 complex substrate.
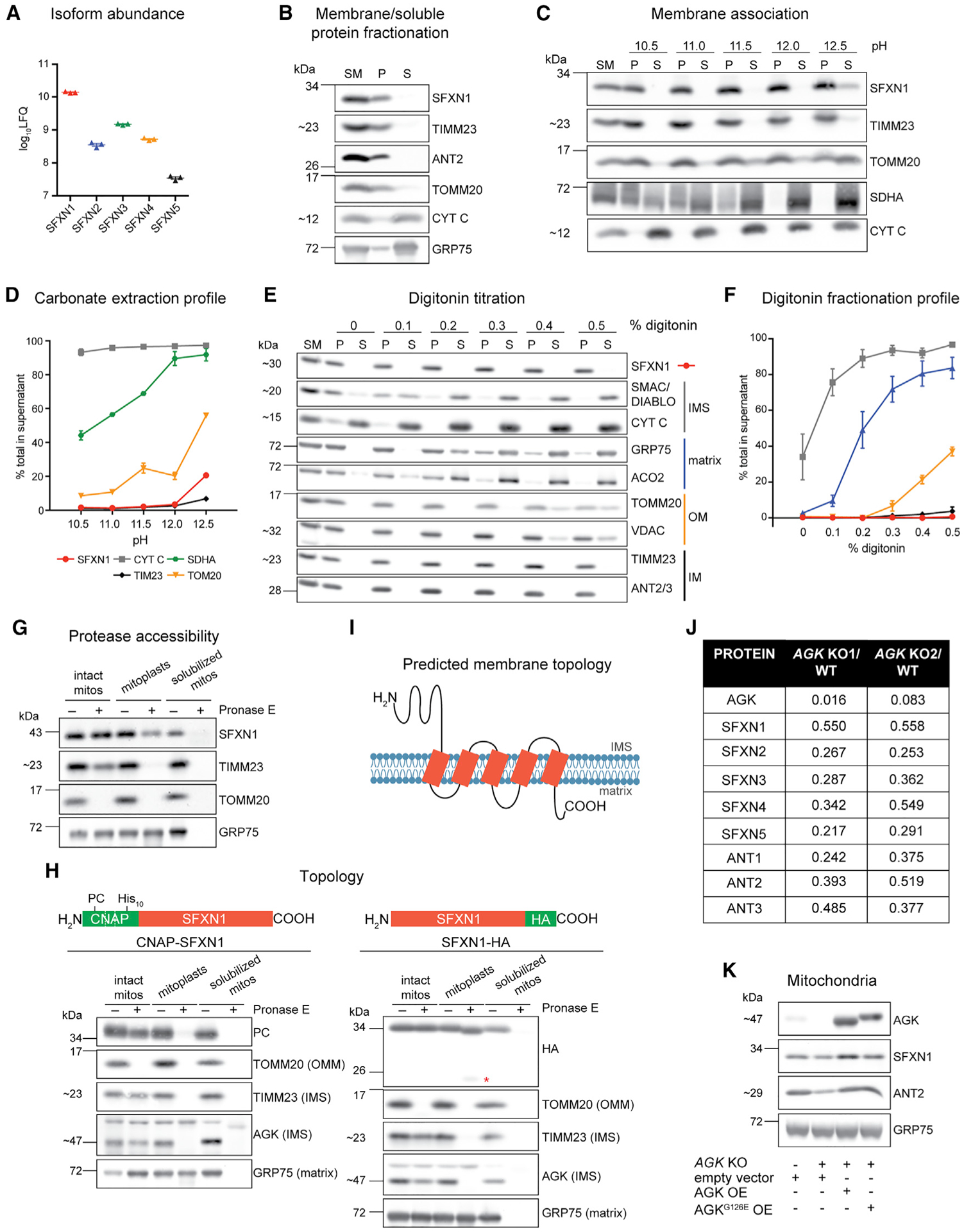
(A) Relative protein abundance of SFXN isoforms in HEK293 mitochondria as, determined by mass spectrometry and label-free quantification (LFQ) (mean ± SEM, n = 3).
(B) Sonication and centrifugation of mitochondria to separate membrane-bound from soluble proteins. SM, starting material; P, pellet; S, supernatant.
(C) Carbonate extraction of mitochondrial membrane proteins to distinguish between peripheral (appear in S) and integral (remain mostly in P) proteins.
(D) Band intensities of P and S fractions in (C) were quantified and plotted as % of protein released in the supernatant (mean ± SEM, n = 3).
(E) Digitonin titration for fractionation of mitochondrial subcompartments. Equal volumes of P and S fractions were analyzed.
(F) Band intensities of P and S fractions (E) were quantified. Average band intensity of representative mitochondrial proteins from each subcompartment was plotted as % of protein released in the supernatant (mean ± SEM, n = 3).
(G) Submitochondrial localization of endogenous SFXN1. HEK293 mitochondria were osmotically ruptured to yield mitoplasts or solubilized with sodium deoxycholate. Samples were treated with Pronase E where indicated.
(H) Submitochondrial localization of tagged SFXN1. HEK293 mitochondria lacking endogenous SFXN1 and expressing CNAP-SFXN1 or SFXN1-HA were processed as in (G). *, matrix-protected fragment.
(I) Predicted membrane topology of SFXN1 based on (H).
(J) Proteomic analysis of AGK KOs versus WT. Shown are relative protein amounts of SFXN and ANT isoforms in the presence or absence of AGK.
(K) Mitochondria from AGK KOs rescued with AGK, AGKG126E, or empty vector were resolved by SDS-PAGE and immunoblotted for the indicated proteins.
SFXNs have been annotated as mitochondrial proteins based on large-scale proteomics (Calvo et al., 2016). We sought to biochemically characterize the membrane association, submitochondrial localization, and topology of SFXN1, the most abundant isoform in HEK293 (Figure 1A; Lu et al., 2017). After sonication of mitochondria and ultracentrifugation to separate membrane bound from soluble proteins, SFXN1 was found in the pellet (Figure 1B), meaning it is a membrane protein. To determine how it is associated with the membrane, we carried out carbonate extraction at increasing pH. In contrast to SDHA and CYT C, peripheral membrane proteins attached to membrane-spanning protein partners or the IMM, respectively, SFXN1 was mostly retained in the pellet and barely released from mitochondrial membranes even at pH 12.5 (Figure 1C). Its extraction profile is most similar to that of integral membrane proteins TIMM23 and TOMM20 (Figure 1D). Thus, SFXN1 possesses membrane-spanning domains.
We then investigated the submitochondrial localization of endogenous SFXN1 by digitonin titration (Figures 1E and 1F). Incubation of mitochondria with increasing digitonin concentrations results in the sequential release into the supernatant of IMS, matrix, outer mitochondrial membrane (OMM), and IMM proteins. The fractionation profile of SFXN1 resembled that of IMM residents. This result was further corroborated by a protease accessibility assay (Figure 1G). SFXN1 was degraded only when protease treatment was combined with detergent solubilization, indicating that the epitope recognized by the SFXN1 antibody was not exposed to the IMS. This finding is in agreement with the localization of FLAG-tagged SFXN1, as determined by super-resolution microscopy (Kory et al., 2018).
Because there are conflicting results regarding the topology of SFXN1 (Lee et al., 2016, 2017), we performed a protease protection assay for N- and C-terminal epitope-tagged SFXN1 (Figure 1H). After Pronase E treatment of mitoplasts, the CNAP tag of CNAP-SFXN1 was degraded similarly to the IMS-exposed domains of TIMM23 and AGK. However, Pronase E caused SFXN1-hemagglutinin (HA) to migrate at a smaller size in mitoplasts, indicating that IMS-facing domain(s) in SFXN1 was degraded, and an even smaller band was detected that likely corresponds to an additional SFXN1-HA fragment that is matrix protected. The HA tag was degraded only upon detergent inclusion. Altogether, these findings are in agreement with the study that mapped the topological direction of endogenous IMM proteins in live cells (Lee et al., 2017) and support that the N and C termini of SFXN1 face the IMS and the matrix, accordingly (Figure 1I). A model of SFXN1 with the 2 termini at opposing sides of the IMM is consistent with the notion that it possesses 5 TMDs and is thus likely to have a unique transport mechanism compared to that of SLC25 members.
SFXNs are TIM22 complex substrates
MCs in the SLC25 family possess internal targeting elements and are integral polytopic proteins characterized by 6 TMDs. Their import and integration into the IMM are mediated by the TIM22 complex (Chacinska et al., 2009; Koehler, 2004; Neupert and Herrmann, 2007; Rehling et al., 2004). Recent studies have highlighted the substrate preference of TIM22 complex subunits and identified AGK as a metazoan-specific component essential for the import of MCs in the SLC25 family (Callegari et al., 2016; Kang et al., 2016, 2017; Vukotic et al., 2017). Because SFXN1 is a polytopic IMM protein, we asked whether SFXNs are also AGK-dependent TIM22 complex substrates. We used SILAC (stable isotope labeling by amino acids in cell culture)-based quantitative proteomics using high-resolution tandem mass spectrometry on HEK293 (herein called wild-type [WT]) and AGK knockouts (KOs) generated by CRISPR-Cas9 (Figures S2A–S2C; Mali et al., 2013; Ran et al., 2013) and found that protein levels of SFXN1–5, similar to ANT1–3, were reduced in the absence of AGK (Figure 1J). Immunoblotting confirmed that in AGK KOs, for which the abundance of the pore-forming TIMM22 subunit was significantly compromised, all SFXN isoforms were down to various extents (Figures S2E and S2F).
Other substrates of the TIM22 complex include subunits of the TIM23 complex, another import machinery that is dedicated to presequence-containing matrix and IMM proteins (Káldi et al., 1998; Stojanovski et al., 2012). To address whether the inefficient biogenesis of SFXNs is due to an indirect effect of TIM22 complex destabilization of the TIM23 translocon, we turned to patient fibroblasts harboring a mutation in TIMM50, which codes for a core component of the TIM23 complex (Reyes et al., 2018; Figures S2G and S2H). Compared to control, patient fibroblasts had reduced TIMM50 (~35%) and TIMM23 (~60%) protein levels, respectively, as previously documented (Reyes et al., 2018). Although we could not detect AGK in either control or patient fibroblasts by using our antibody, we observed a minor impact of TIMM50 mutation on TIMM22. The alterations in TIM23 subunits did not decrease levels of SFXNs, demonstrating that SFXN biogenesis does not rely on the integrity of the TIM23 complex.
Other SFXN isoforms being AGK substrates, similar to SFXN1, also indicates that they are IMM residents. This finding is in contrast to a recent report proposing that FLAG-tagged SFXN2 is in the OMM (Mon et al., 2019). As expected (Kang et al., 2017; Vukotic et al., 2017), SFXN1 protein abundance was not dependent on AGK kinase activity (Figures 1K and S2D; AGKG126E is kinase dead). In summary, SFXNs are TIM22 complex substrates whose steady-state abundance is dependent on AGK.
Absence of SFXN1 does not severely impair iron homeostasis
To probe the physiological role of SFXN1, we generated SFXN1 KOs in HEK293 by CRISPR-Cas9. We used 2 guide RNAs (gRNAs) targeting different parts of exon 2 and screened by immunoblotting (Figures S3A and S3B). Introduced genetic lesions in SFXN1 KO1 and 2, generated by gRNA 1 and 2, respectively, were analyzed by Sanger sequencing. Both contained missense mutations leading to premature stop codons (Figure S3C).
SFXN1 was identified by positional cloning as the causative gene responsible for the phenotypic anomalies of the flexed-tail mouse, characterized by axial skeletal abnormalities, temporary embryonic and neonatal anemia, and mitochondrial iron deposits (Chui et al., 1977; Fleming et al., 2001; Hunt et al., 1933; Mixter and Hunt, 1933). However, other mutations potentially responsible for the observed deficiencies were subsequently found (Hegde et al., 2007; Lenox et al., 2005). Due to this finding, the role of SFXN1 in iron homeostasis has been debated.
To test whether a loss of SFXN1 disrupts iron homeostasis, we compared cell viability of WT and SFXN1 KOs treated with increasing concentrations of deferoxamine mesylate (DFO). No difference between WT and SFXN1 KOs was observed upon DFO-mediated iron chelation (Figure 2A), indicating that SFXN1 KOs are not more sensitive to iron depletion and thus unlikely deficient in cellular iron. Indeed, cellular and mitochondrial iron levels as measured by inductively coupled mass spectroscopy (ICP-MS) were not altered in the absence of SFXN1 (Figures 2B and 2C). Although the levels of other physiologically relevant metals were unaffected, cellular Mn2+ was decreased in SFXN1 KOs. Because manganese plays a chief role as an enzyme cofactor, most notably for superoxide dismutases (Wang et al., 2018), this decrease in cellular Mn2+ could make SFXN1 KOs more susceptible to oxidative stress. Collectively, these data provide evidence that the absence of SFXN1 alone does not severely impact iron homeostasis in HEK293 cells.
Figure 2. Deletion of SFXN1 alone does not disrupt iron metabolism.
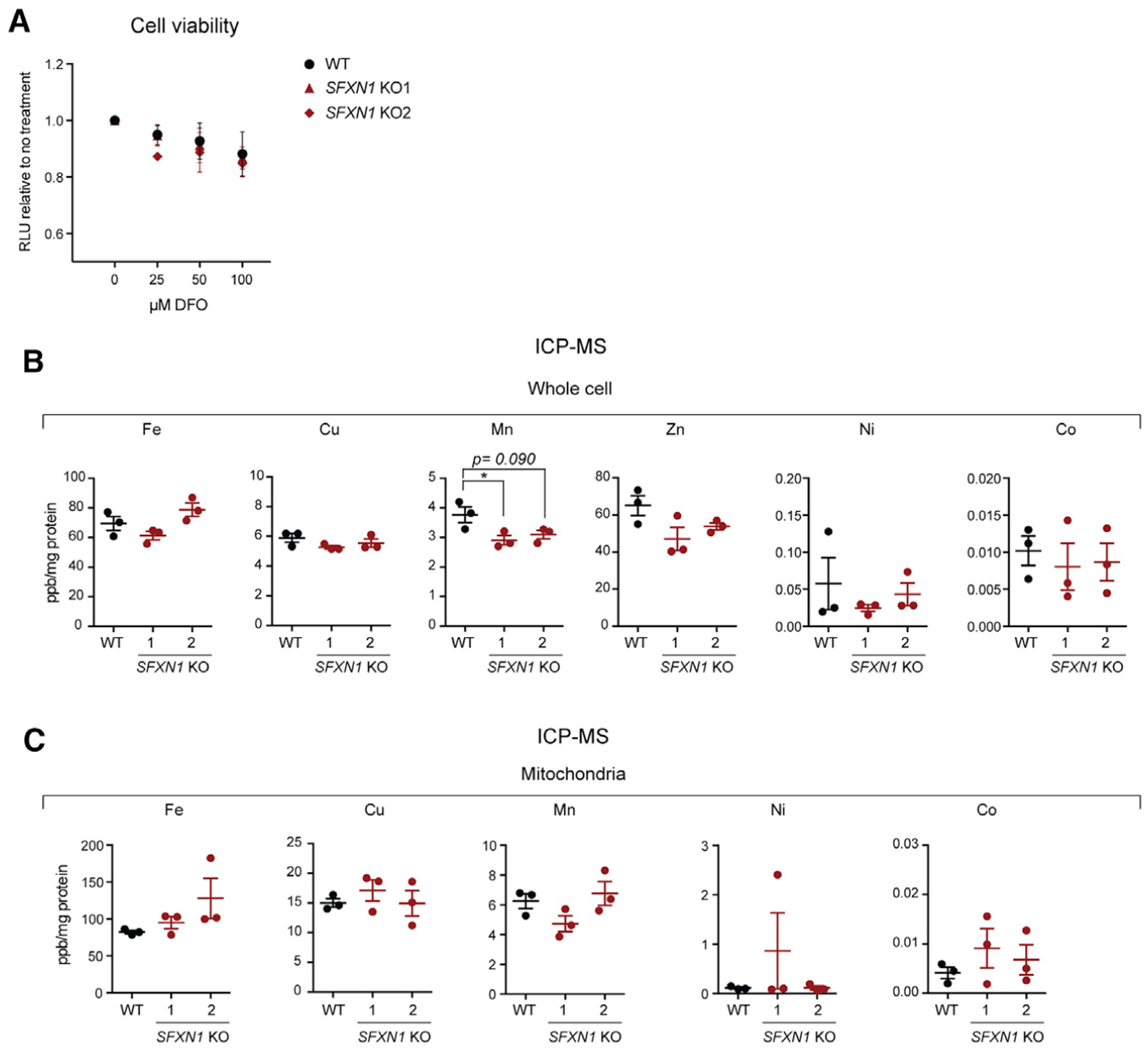
(A) Cell viability of HEK293 WT and SFXN1 KOs upon treatment with the indicated concentration of the iron chelator deferoxamine mesylate (DFO) for 24 h (mean ± SEM, n = 3).
(B and C) Relative metal concentrations in WT and SFXN1 KO cells (B) and mitochondria (C), as determined by ICP-MS (mean ± SEM, n = 3).
*p < 0.05; unpaired Student’s t test.
Loss of SFXN1 causes CIII-related defects
Because no gross alterations in metal balance were observed in SFXN1 KOs, we investigated the potential role of SFXN1 in general mitochondrial function. We analyzed the protein levels of respiratory complex subunits, import machinery components, and players important for mitochondrial morphology (Figures 3A, 3D, and S3D). Comparing SFXN1 KOs with WT, the level of UQCRC2, a CIII subunit, was significantly decreased in both KOs. Also, COX4 (COXIV) was slightly decreased and NDUFB6 (CI) was modestly increased. Other proteins involved in OXPHOS or biogenesis or that reside in IMM or matrix were unaffected or inconsistently altered in KOs relative to WT. A limited survey of electron transport chain (ETC) proteins containing iron-sulfur (Fe-S) clusters determined that in SFXN1 KOs, UQCRFS1 (CIII) was remarkably decreased, SDHB (CII) was significantly reduced, and all Fe-S-ligating subunits of CI tested were unaltered (Figures 3B, 3D, and S3D), indicating that SFXN1 absence impacts only a subset of Fe-S cluster-containing mitochondrial proteins. Because the tested CIII subunits were reduced upon SFXN1 loss, we asked whether the protein abundance of MTCYB, the sole mtDNA-encoded CIII subunit, was also altered. In SFXN1 KOs, the MTCYB protein was reduced by ~75% relative to untreated WT, whereas MTCO1 and MTCO2, mtDNA-encoded CIV subunits, were minimally affected (Figures 3C and 3D). Altogether, these data provide evidence that SFXN1 deletion is most detrimental to CIII subunit protein levels.
Figure 3. Absence of SFXN1 leads to complex-III-related defects.
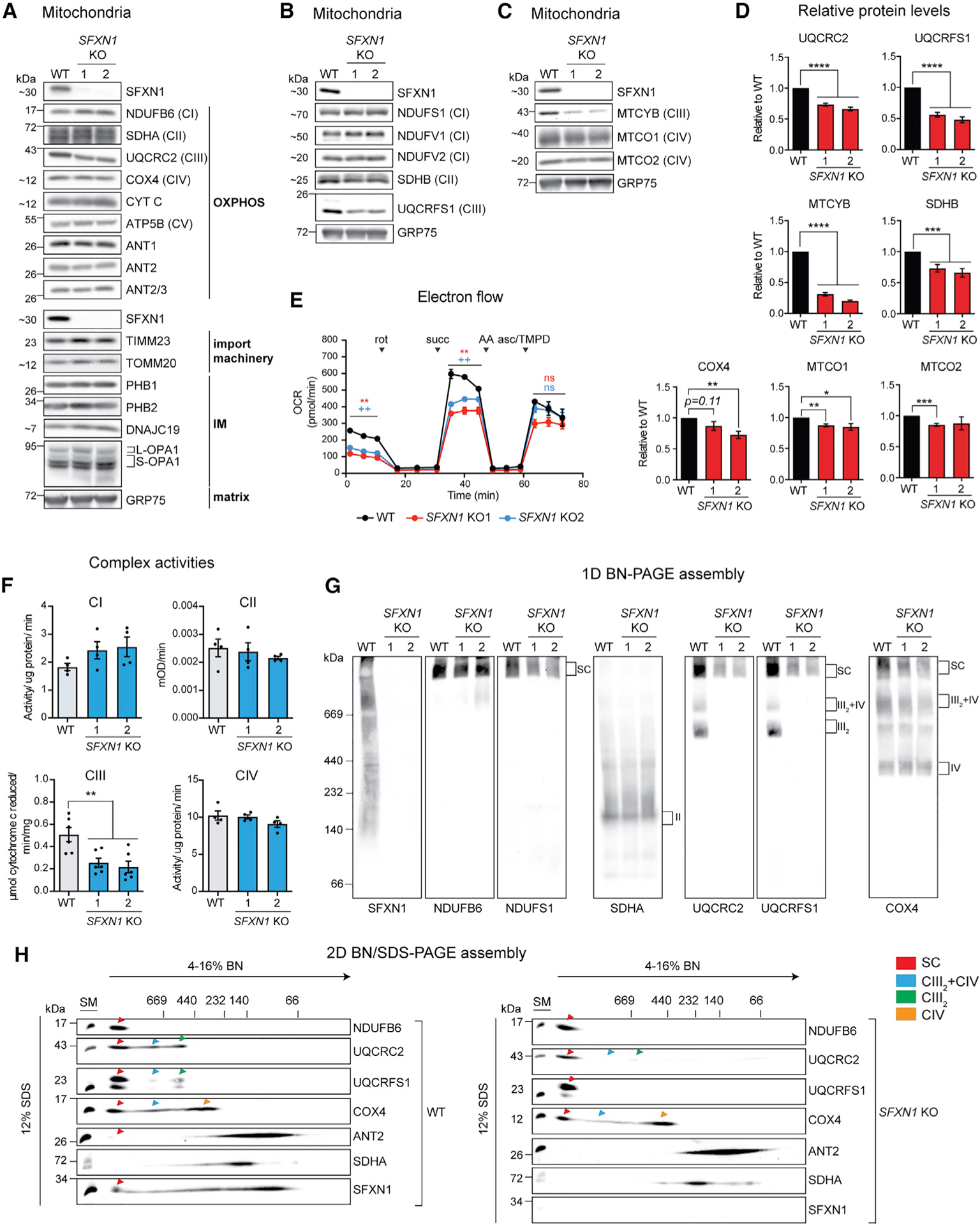
(A) Steady-state abundance of select mitochondrial proteins, including OXPHOS components, subunits of import machineries, and other proteins in the IMM and matrix.
(B) Immunoblotting for Fe-S containing subunits of respiratory complexes in mitochondrial extracts. GRP75 served as loading control.
(C) Immunoblotting for mtDNA-encoded subunits in mitochondrial extracts. GRP75 served as loading control.
(D) Densitometric analysis of bands for select proteins in (A), (B), and (C). Protein levels in WT were set to 1.0 (mean ± SEM, n ≥ 4).
(E) Electron flow through the respiratory chain in intact mitochondria. Base buffer contains 10 mM pyruvate, 2 mM malate, and 4 μM carbonyl cyanide m-chlorophenyl hydrazone (CCCP). Injections of complex inhibitors/substrates were performed as indicated. rot, rotenone; succ, succinate; AA, antimycin A; asc/TMPD, ascorbate/N,N,N′,N′-tetramethyl-p-phenylenediamine.
(F) Spectrophotometric assays using detergent-solubilized mitochondria to monitor individual complex activities. CI, oxidation of NADH to NAD+; CII, ubiquinol production; CIII, cytochrome c reduction; CIV, cytochrome c oxidation (mean ± SEM, n ≥ 4).
(G) 1D BN assembly. Mitochondria solubilized in 1% (w/v) digitonin were resolved on a 4%–16% BN gel and immunoblotted for the indicated subunits.
(H) Mitochondria solubilized in 1% (w/v) digitonin were resolved by 2D BN/SDS-PAGE and immunoblotted for the indicated subunits. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; unpaired Student’s t test.
To determine whether SFXN1 ablation leads to non-optimal ETC function, we assessed electron flow through ETC complexes by sequentially feeding specific substrates and inhibitors to intact mitochondria (Figure 3E). SFXN1 KO mitochondria registered significantly lower oxygen consumption rates (OCRs) when pyruvate (CI substrate) or succinate (CII substrate) was supplied but not after addition of ascorbate/TMPD (CIV substrate), indicating that there is a problem pre-CIV. To further isolate the pre-CIV ETC defect, the individual CI to CIV activities were measured in detergent-solubilized mitochondria that demonstrated a specific defect in CIII (Figures 3F and S3E).
Next, we interrogated the effect of SFXN1 deletion on the capacity of respiratory complexes to associate into higher order structures. Digitonin-solubilized mitochondria were subjected to 1D blue native (BN) (Figure 3G) and 2D-BN/SDS-PAGE (Figure 3H). The absence of SFXN1 did not prevent the formation of respiratory supercomplexes (SCs). Strikingly, however, the CIII2-CIV subcomplex and CIII2 were prominently decreased in SFXN1 KOs when immunoblots were probed with antibodies against 2 CIII subunits (UQCRC2 and UQCRFS1). Using the COX4 antibody, we confirmed this reduction in the CIII2-CIV subcomplex, which was more obvious in 2D BN/SDS-PAGE. We found that CI assembly, monitored by using NDUFB6 and NDUFS1 antibodies, appeared normal. Similarly, ANT2 and SDHA assemblies were unchanged in the absence of SFXN1 (Figure 3H). The reduction in CIII2-CIV was still apparent even following extraction with n-dodecyl-β-maltoside (DDM), a detergent that unlike digitonin destabilizes many of the larger SCs (Figure S3F). Moreover, there were no changes in the overall electrophoretic pattern of complexes analyzed by 1D-BN and Coomassie staining (Figure S3G). Knocking out SFXN1 in HeLa cells also rendered CIII dysfunctional (Figures S4A–S4C). Moreover, stably overexpressing SFXN1 in SFXN1 KO partially improved CIII function (Figures S4D and S4E). We speculate that reintroducing SFXN1 at a physiological level may provide a better rescue, as overexpression of MCs has been shown to induce an aggregation-based, cytosolic stress response (Liu et al., 2019).
In SFXN1 KOs, the lower CIII subunit protein levels and activity and the specific reduction of CIII2-CIV and CIII2 cannot be accounted for by a transcriptional mechanism (Figure S4F) or by changes in mtDNA content (Figure S4G). They also cannot be readily explained by a mitochondrial translation defect (Figures S4H and S4I).
CIII couples electron transfer from CoQ to CYT C to proton pumping into the IMS. The mammalian CIII monomer is composed of 11 subunits. Assembly of dimeric CIII begins with the incorporation of chaperone-bound MTCYB into the IMM followed by the attachment of nuclear-encoded structural subunits to form pre-CIII2 (Fernandez-Vizarra and Zeviani, 2018; Signes and Fernandez-Vizarra, 2018). BCS1L and LYRM7 are assembly factors required for the proper incorporation of UQCRFS1 into pre-CIII2 and thus the full maturation of active CIII2 (Fernández-Vizarra and Zeviani, 2015). Notably, the CIII assembly defect observed in SFXN1 KOs is reminiscent of that documented in BCS1L mutant and LYRM7HA-overexpressing cell lines (Bottani et al., 2017; Fernandez-Vizarra et al., 2007; Sánchez et al., 2013). However, BCS1L and LYRM7 protein levels were not altered in the absence of SFXN1 (Figure S4J). These results suggest that SFXN1 influences CIII integrity by another mechanism.
We reasoned that impaired CIII function may affect the oxidation of CoQ, a lipid that transfers electrons from CI/CII to CIII, which in turn can impact CoQ content. We therefore measured its 2 isoforms in humans, namely, CoQ9 and CoQ10. Both isoforms were markedly reduced in the absence of SFXN1, and total CoQ in SFXN1 KOs was ~50% that of WT (Figure 4A). Because a saturating amount of reduced CoQ was used in CIII activity assays (Figures 3F and S3E) and CYT C protein levels were largely unaffected (Figures 3A and S3D), SFXN1 null cells were characterized by an intrinsic CIII defect that may affect CoQ biosynthesis and/or turnover.
Figure 4. SFXN1 loss results in metabolic perturbations.
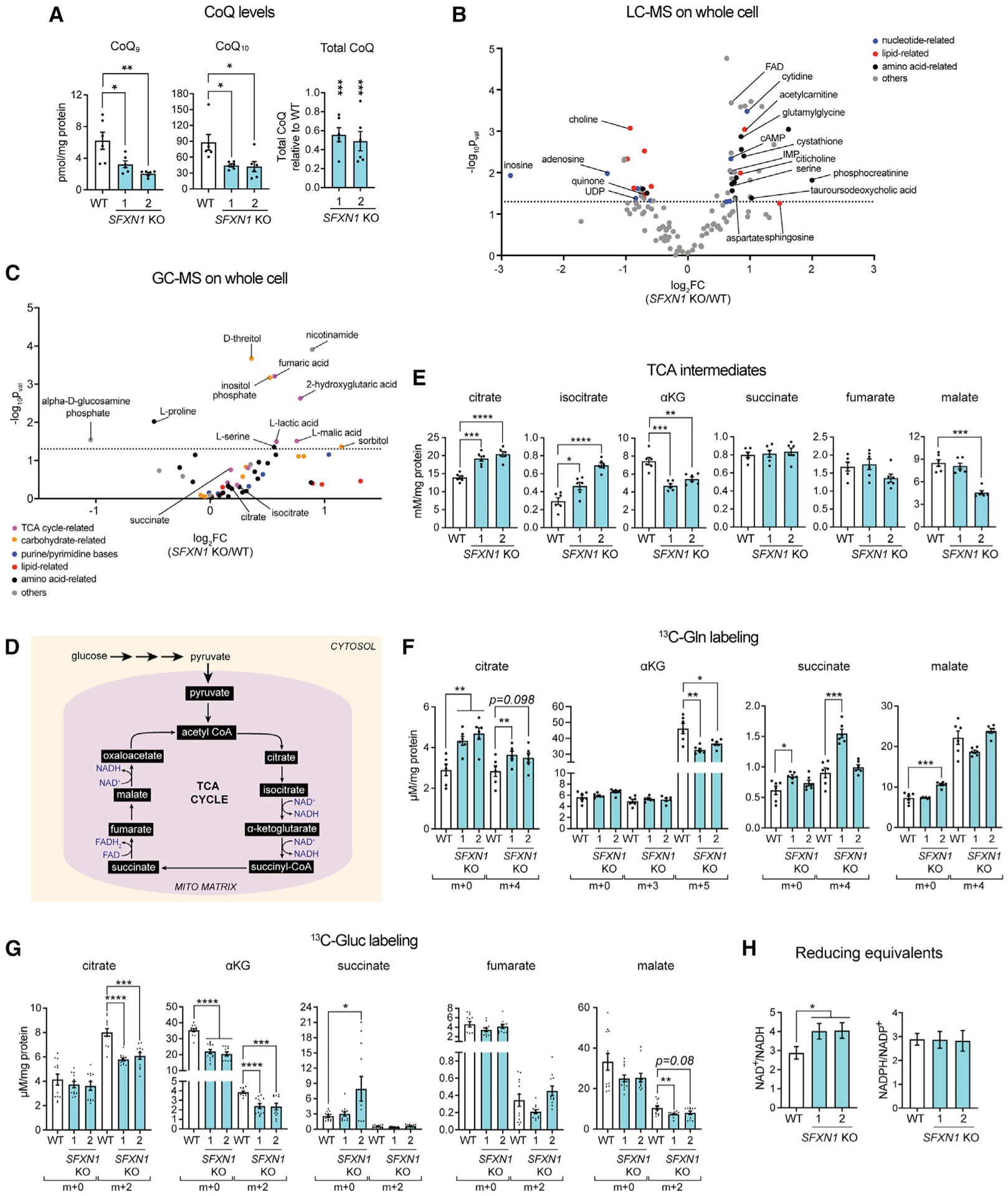
(A) CoQ measurements in cellular lipid isolates by reversed phase high-performance liquid chromatography-MS (RP-HPLC/MS) (mean ± SEM, n = 6).
(B and C) Metabolite abundance in whole cells obtained by untargeted LC-MS (B) or GC-MS (C) analysis (n = 6 per group).
(D) TCA cycle.
(E) Steady-state intracellular concentrations of TCA cycle metabolites obtained by LC-MS/MS (mean ± SEM, n = 6). αKG, α-ketoglutarate.
(F and G) Levels of m+x-labeled metabolites as determined by LC-MS/MS upon [U-13C]-glutamine labeling (mean ± SEM, n = 6) (F) and [U-13C]-glucose labeling (mean ± SEM, n = 12) (G).
(H) Quantification of NAD+/NADH (mean ± SEM, n ≥ 8) and NADPH/NADP+ ratios (mean ± SEM, n = 6). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; unpaired Student’s t test.
Metabolic perturbations upon SFXN1 deletion
SFXN1 transports serine and, based on their ability to inhibit serine flux across the IMM, additional physiologically important amino acids, including alanine, cysteine, and glycine (Kory et al., 2018). Thus, we explored other metabolic consequences upon SFXN1 loss by performing liquid chromatography-MS (LC-MS)- and gas chromatography-MS (GC-MS)-based metabolomics analysis on WT and SFXN1 KO cells (Figures 4B and 4C; Table S1). Changes in nucleotide-related metabolites were noticed (Figure 4B), which is consistent with the role of SFXN1 in 1C metabolism (Kory et al., 2018). Also observed were alterations in lipid-related metabolites, including quinone, choline, and citicoline (Figure 4B). These alterations may be linked with the role of SFXN1 in compartmentalizing serine, which in turn can be used for phospholipid, sphingosine, and ceramide biosynthesis (Gao et al., 2018). Perturbations in central carbon metabolism were also noted, as some changes were observed for tricarboxylic acid (TCA) cycle- and carbohydrate-related metabolites (Figure 4C). One of the upregulated metabolites identified upon SFXN1 loss relative to WT is 2-hydroxyglutaric acid. A closer look at TCA intermediates by targeted LC-MS revealed that in SFXN1 KOs, citrate and isocitrate were elevated and αKG was reduced (Figure 4E).
Amino acid metabolism is tightly coupled with the TCA cycle (Figure 4D). To better understand the observed central-carbon-metabolism-linked changes, WT and SFXN1 KOs were incubated with [U-13C]-glutamine or [U-13C]-glucose for the optimal duration (Figures S5A–S5D) and labeling of TCA metabolites examined (Figures 4F and 4G). SFXN1 KOs had lower m+5-labeled αKG, indicating inefficient conversion of glutamine to αKG (Figure 4F). This finding is consistent with changes in activities of enzymes involved in αKG metabolism in the absence of SFXN1 (Figures S5E–S5H). Activity of glutamate dehydrogenase (GDH), which mediates αKG formation from glutamate (Hudson and Daniel, 1993), was decreased in SFXN1 KOs. Activity of alanine aminotransferase (ALT), which facilitates the reversible addition of an amino group from L-alanine to αKG to yield pyruvate and glutamate (Groen et al., 1982; Yang et al., 2002), was also decreased in SFXN1 KOs, likely due to the lower αKG in these samples.
[U-13C]-Glucose tracing (Figure 4G) indicates that in the absence of SFXN1, glucose labeling of citrate, αKG, and malate was reduced, suggesting that incorporation of at least 2 carbon sources to TCA intermediates is altered upon SFXN1 deletion. Also, we observed a higher NAD+/NADH with normal NADPH/NADP+ ratio in SFXN1 KOs (Figure 4H), consistent with a disturbance in central carbon metabolism.
Despite these metabolic alterations, mitochondrial respiration (Figure 5A) and glycolytic parameters (Figure 5B) were not impacted in intact SFXN1 KO cells. This result is in stark contrast to isolated mitochondria, in which the loss of SFXN1 diminished basal and/or ADP-stimulated oxygen consumption driven by various fuels that donate electrons to CI, CII, and CIII, which is in agreement with a CIII defect (Figure 5C). We therefore asked how SFXN1 KOs are able to maintain cellular baseline respiration. We observed higher glucose use, with largely unaltered reliance on glutamine and fatty acid pathways (Figure 5D). It remains a possibility that SFXN1 KOs were able to switch to other fuels not tested in this study. Overall, these results highlight the metabolic flexibility at the cellular level driven by the loss of SFXN1.
Figure 5. Cells lacking SFXN1 display metabolic flexibility.
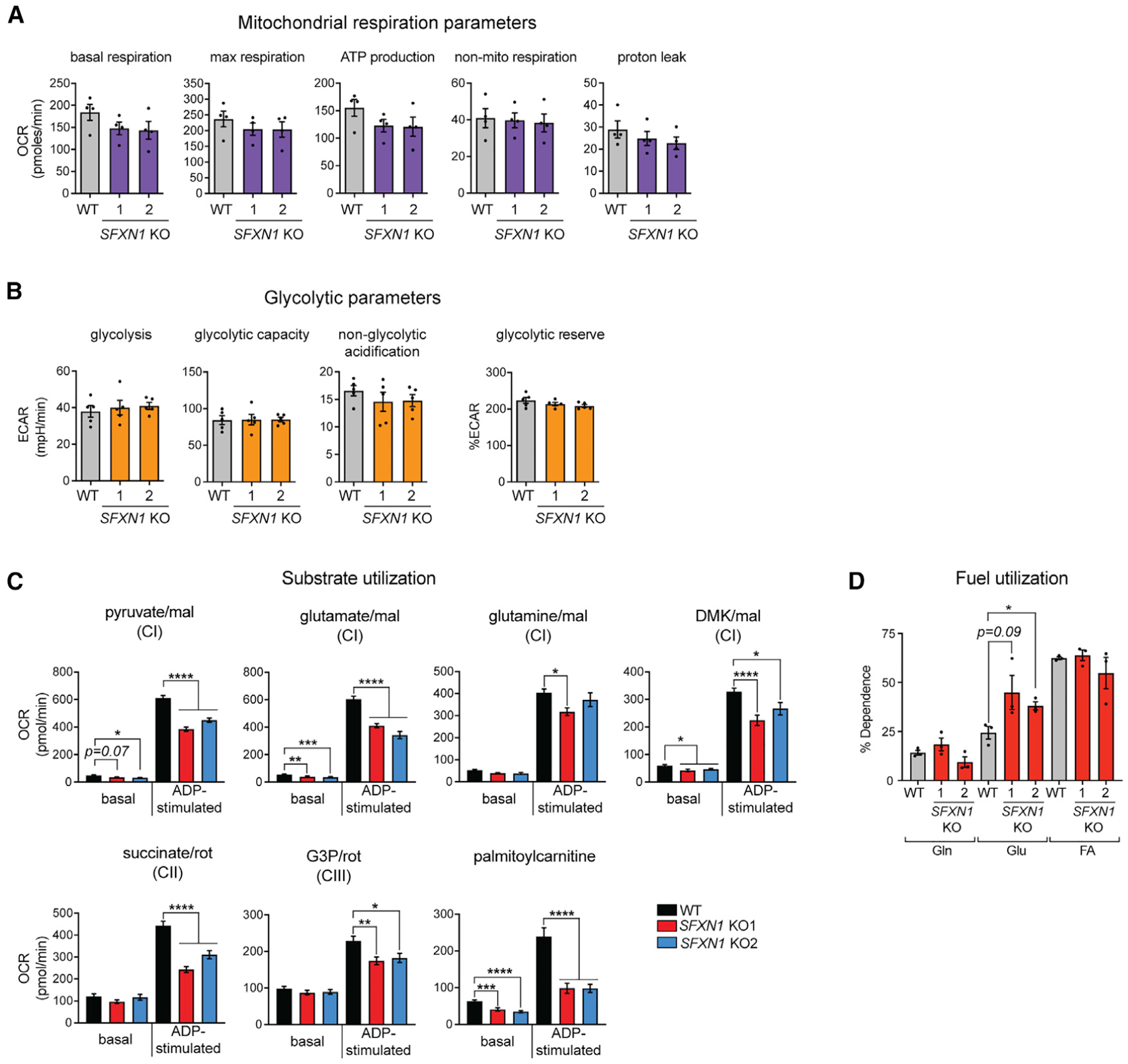
(A) Oxygen consumption rate (OCR) in intact cells. Presented are values normalized by DNA content (mean ± SEM, n = 4).
(B) Extracellular acidification rate (ECAR) in intact cells. Presented are values normalized by DNA content (mean ± SEM, n = 5).
(C) OCR in isolated mitochondria using the specified substrates (mean ± SEM, n ≥ 9 wells from at least 3 independent experiments). mal, malate; DMK, dimethyl αKG; G3P, glycerol-3-phosphate.
(D) Percent dependency on glutamine (Gln), glucose (Gluc), and fatty acid (FA) use (mean ± SEM, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; unpaired Student’s t test.
SFXN1 deletion impedes heme biosynthesis
Next, we assessed the relative amounts of 2 substrates of SFXN1 (Kory et al., 2018). Total serine in SFXN1 KOs was elevated, whereas total glycine was unmodified compared to WT (Figures S5I and S5J). Previous work reported that SFXN1 loss in Jurkat and K562 cells leads to increased serine and decreased glycine levels (Kory et al., 2018). Despite the difference in the effect on glycine, both studies identified a higher serine:glycine ratio in cells lacking SFXN1.
To analyze whether WT and SFXN1 KOs differentially consume exogenous serine, we grew cells in medium free of serine and glycine, to prevent glycine-serine interconversion, and supplemented with [U]-13C-serine. A striking amount of labeled serine (m+3) remained in the media of SFXN1 KOs, indicating they have a diminished ability to use serine (Figure 6A). Given that the medium used was initially lacking serine and glycine, the elevated unlabeled serine (m+0) and reduced unlabeled glycine (m+0) detected in the medium from SFXN1 KOs suggest that these cells robustly generate serine but fail to convert a large portion of it to glycine (Figures 6A and 6B). Thus, the absence of SFXN1 results in serine accumulation likely due to a combination of inefficient serine-to-glycine conversion as well as upregulation of serine biosynthesis.
Figure 6. Heme biosynthesis is compromised in SFXN1 Kos.
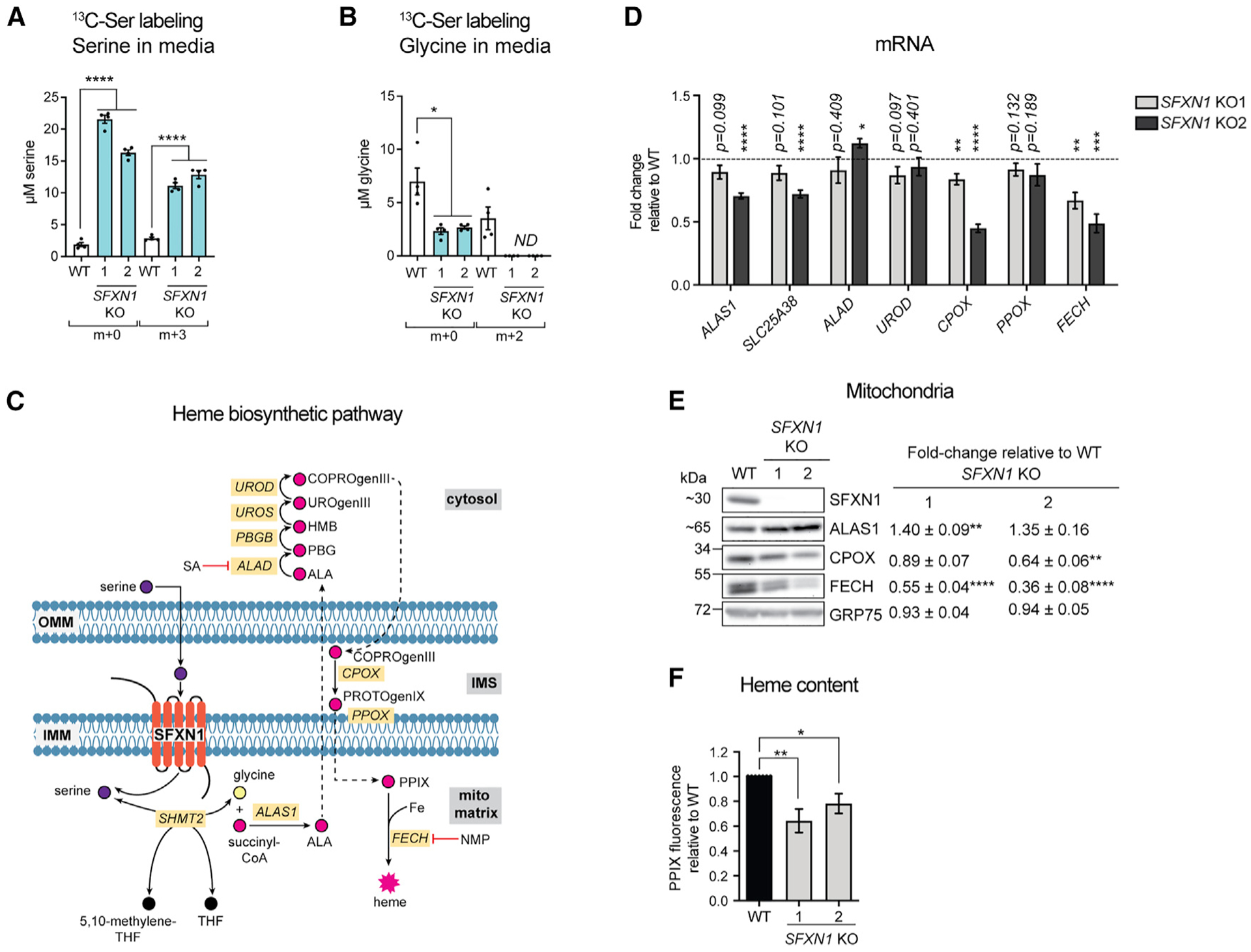
(A and B) Quantification of m+x serine (A) and m+x glycine (B) in media by LC-MS/MS. Cells were grown in serine- and glycine-free media with [U]-13C-serine for 24 h (mean ± SEM, n = 4).
(C) Heme biosynthetic pathway. SA, succinylacetone; NMP, N-methyl protoporphyrin; PPIX, protoporphyrin IX.
(D) Gene expression analysis of heme biosynthetic enzymes by qPCR (mean fold-change [FC] ± SEM, n = 4).
(E) Immunoblotting for enzymes involved in heme biosynthesis. Values shown are fold-change protein steady-state abundance in SFXN1 KOs over HEK293 WT (mean ± SEM, n = 5).
(F) Relative total heme content obtained by analyzing total heme fluorescence (mean ± SEM, n = 7). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; unpaired Student’s t test.
Serine reaching the mitochondrial matrix has several metabolic fates (Mattaini et al., 2016). Its cleavage yields 5,10-methylenetetrahydrofolate (5,10-methyl-THF) and glycine. 5,10-Methyl-THF feeds into 1C metabolism that contributes to nucleotide biosynthesis (Lewis et al., 2014), whereas glycine serves as a precursor for glutathione (Wu et al., 2004) and heme (Radin et al., 1950). SFXN1 has been shown to play a role in 1C metabolism (Kory et al., 2018), but whether it affects other branches of serine catabolism vital for mitochondrial function has not been addressed. We hypothesized that insufficient serine and/or glycine import into mitochondria upon SFXN1 loss might also impair heme generation, a tightly regulated, multistep metabolic pathway (Figure 6C). Consistent with this possibility, the mRNA levels of CPOX and FECH (Figure 6D) were reduced in the absence of SFXN1. The steady-state protein amount of FECH (Figure 6E), an Fe-S containing protein that catalyzes Fe2+ insertion into protoporphyrin IX (PPIX) (Ajioka et al., 2006), was also decreased in the absence of SFXN1 and could be partially rescued upon its re-introduction (Figure S4E). ALAS1, which performs the initial and rate-limiting step of heme production (Figure 6C), was increased in SFXN1 KOs compared to WT (Figure 6E). ALAS1 is subject to negative feedback-mediated regulation by heme (Kubota et al., 2016), suggesting that heme levels may be limiting when SFXN1 is missing. Indeed, heme was decreased in SFXN1 KOs (Figure 6F), consistent with a role for SFXN1 in promoting heme biosynthesis.
Hemin or dimethyl αKG (DMK), but not formate, supplementation improves CIII function in SFXN1 Kos
Because SFXN1 has the ability to transport several amino acids in vitro (Kory et al., 2018), we tested whether inefficient import of a particular substrate into the mitochondrion can explain the CIII defects observed in SFXN1 KOs. Exogenous serine, glycine, alanine, or cystine did not restore CIII subunit protein abundance in SFXN1 KOs to WT levels and had no obvious effect on other ETC proteins assessed (Figure S5K). It remains a possibility that an amino acid cocktail instead of an individual amino acid can account for the defects observed. It is also plausible that without SFXN1, the specific amino acid supplied cannot robustly reach the mitochondrial matrix to exert an effect. If true, this finding would imply that other MCs, including other SFXNs, may be partially, but not completely, functionally redundant with SFXN1. We therefore asked whether suboptimal generation of specific metabolites due to inefficient transport of precursors into the mitochondrial matrix could better explain the CIII-related phenotype of SFXN1 KOs. Candidate metabolites are formate and heme. Formate, one of the most studied products of serine catabolism, is involved in mitochondrial translation and thus can affect respiratory chain function (Lucas et al., 2018; Minton et al., 2018; Morscher et al., 2018). On the other hand, heme moieties are present in CIII (Kim et al., 2012); thus, it is possible that the CIII dysfunction in SFXN1 KOs stems from impaired heme metabolism (Figure 6).
In cells lacking SFXN1, formate shortage is responsible for their slower proliferation in serine-deficient media (Kory et al., 2018). We noted WT already displayed a proliferation defect upon serine withdrawal in glucose-based media, and this defect was exacerbated when SFXN1 was deleted (Figure 7A). Formate supplementation, but not hemin, a cell-permeable derivative of heme (Nakamichi et al., 2005; Wang et al., 2009), rescued this phenotype.
Figure 7. Reinforcement of heme and αKG metabolism, but not the one-carbon pathway, partially restores complex III function in the absence of SFXN1.
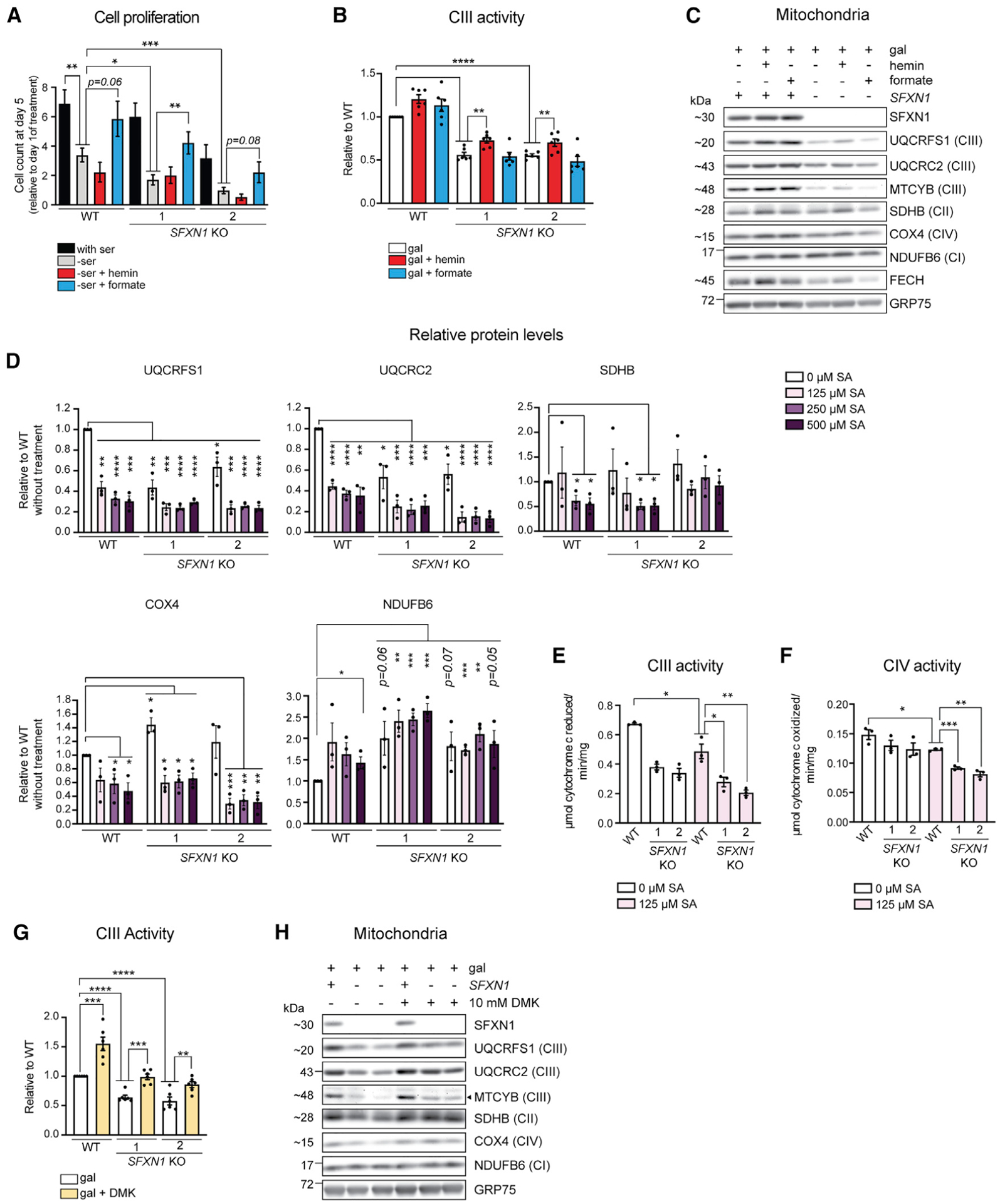
(A) Cell proliferation in full media (with serine), without serine (-ser), and upon supplementation of serine-free media with 15 μM hemin (−ser + hemin) or 1 mM formate (−ser + formate) (mean ± SEM, n ≥ 4).
(B) Complex III activity in DDM-solubilized mitochondria. At 48 h before mitochondrial isolation, cells were switched to media containing galactose only, galactose with 15 μM hemin, or galactose with 1 mM formate (mean ± SEM, n = 6).
(C) Immunoblotting for select respiratory complex subunits using mitochondrial isolates detailed in (B).
(D) Steady-state protein abundance of select respiratory complex subunits in cell lysates. Cells were grown in glucose-based media and treated with the indicated concentration of SA, an inhibitor of heme biosynthesis, for 2 days.
(E and F) Complex III (E) or IV (F) activity in DDM-solubilized mitochondria. At 48 h before mitochondrial isolation, cells were switched to media containing galactose only or galactose with 125 μM SA (mean ± SEM, n = 3).
(G) Complex III activity in DDM-solubilized mitochondria. At 48 h before mitochondrial isolation, cells were switched to media containing galactose only or galactose with 10 mM dimethyl αKG (DMK) (mean ± SEM, n = 6).
(H) Immunoblotting for select respiratory complex subunits using mitochondrial isolates detailed in (G). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; unpaired Student’s t test.
Curiously, other studies have determined that formate depletion mainly affects activities of CI, CIV, and CV and their subunit protein abundances but not CIII (Lucas et al., 2018; Minton et al., 2018; Morscher et al., 2018). Indeed, exogenous formate did not reverse the reduced CIII activity (Figure 7B) and CIII subunit protein levels (Figure 7C) in SFXN1 KOs. The inability of formate to restore CIII expression or activity is consistent with the preserved mtDNA translation in SFXN1 KOs (Figures S4H and S4I). In contrast to formate, exogenous hemin enhanced CIII activity in both WT and SFXN1 KOs (Figure 7B). These results indicate that in absence of SFXN1, the 1C pool is a limiting factor for nucleotide synthesis and therefore cell proliferation (Kory et al., 2018; Labuschagne et al., 2014), whereas deficient heme is partially responsible for the decreased CIII function.
We noted that serine withdrawal from glucose-containing media is not sufficient to impact CIII activity or CIII subunit protein levels in WT or SFXN1 KOs (Figures S6A and S6C). Still, CIII impairments manifested in SFXN1 KOs even with glucose as the carbon source instead of galactose, wherein cells were not forced to respire (Figures S6B, S6C, and S6D). In this metabolic setting, supplementation with hemin, but not formate, again improved CIII activity in cells lacking SFXN1 (Figure S6B).
An addition of hemin, however, only minimally affected CIII subunit protein abundance and assembly in SFXN1 KOs regardless of carbon source (Figures 7C, S6C, and S6D). This result suggests that the lower heme resulting from SFXN1 loss contributes to the decreased CIII activity but that there are other metabolites formed following SFXN1-mediated transport of serine or other amino acids that can influence CIII stability. It also remains a possibility that exogenous hemin is not as readily used for processes that ensure proper CIII expression and assembly.
To interrogate whether heme deficiency affects protein abundance of ETC complex subunits independent of a perturbed serine metabolism, we treated cells with increasing concentrations of succinylacetone (SA), which blocks heme biosynthesis by inhibiting ALAD (Figure 6C; Sassa and Kappas, 1983; Tschudy et al., 1981). SA treatment decreased protein levels of CII, CIII, and CIV but not CI, which does not bear heme, and upregulated ALAS1 in response to the expected aminolevulinic acid (ALA) accumulation (Maitra et al., 2019; Figures 7D and S7A). Although CII (SDHB) and CIV (COX4) protein levels were reduced by ~50% upon treatment of WT cells with 500 μM SA, CIII (UQCRC2 and UQCRFS1) was decreased by ~75% (Figures 7D and S7A). This result suggests that CIII expression and/or stability is highly sensitive to SA-mediated heme deprivation. Deleting SFXN1 furthered this drop in CIII subunit protein abundance upon SA treatment (Figures 7D and S7A). CIII activity compared to CIV activity in WT was also more responsive to SA treatment, and both were disrupted further in the absence of SFXN1 (Figures 7E and 7F). This finding is in agreement with the already reduced heme levels in these cells and is also indicative that other roles of SFXN1 contribute to the CIII-related phenotype.
On the other hand, treatment with N-methyl protoporphyrin (NMP), which represses FECH by being a PPIX transition analog (Figure 6C), affected only CIV (Figure S7B). Selective susceptibility of CIV assembly and subunit protein levels to NMP treatment has been previously documented (Atamna et al., 2001). Based on our data, this susceptibility seems to be a NMP-specific response, suggesting that this drug may preferentially affect heme a that is only present in CIV (Atamna et al., 2001; Kim et al., 2012). In summary, inadequate heme may also be a contributing factor to the lower CIII subunit protein levels in SFXN1 KOs.
Because αKG was reduced in SFXN1 KOs, we asked whether the addition of DMK, a cell-permeable version of αKG, has a positive effect on CIII function. Surprisingly, DMK supplementation restored CIII activity in SFXN1 KOs and even enhanced WT CIII activity (Figure 7G). It also reinforced CIII subunit protein abundance in SFXN1 KOs and increased the levels of SDHB and COX4, which were also slightly down in cells lacking SFXN1 (Figures 7H and S7C). In toto, our data support a model in which SFXN1 regulates CIII expression and activity by both promoting heme synthesis and maintaining an intact αKG metabolism, possibly by fine-tuning flux into the TCA cycle.
DISCUSSION
MCs represent important nodes of metabolism. In this study, we uncover additional functions for SFXN1, an unconventional MC that transports select amino acids including serine, outside its role in the generation of 1C units (Kory et al., 2018). We demonstrate that SFXN1 is important for CIII integrity, as well as CoQ, heme, and αKG metabolism.
With a series of biochemical experiments, we show that SFXN1 is an integral membrane protein in the IMM with N and C termini exposed to the IMS and mitochondrial matrix, respectively, consistent with having 5 TMDs. Also with a protease protection assay, another study has shown that the N terminus of FLAG-SFXN4 is facing the IMS (Hildick-Smith et al., 2013). Together with SFXN1, SFXN4 and SFXN2 are modeled to have 5 TMDs, whereas SFXN3 and SFXN5 are predicted to have only 4. Whether or not this feature imparts a substrate preference of each isoform is unknown. Structural information of the SFXN family, including insight into determinants of ligand selectivity that may dictate the transport mechanism, are all currently lacking.
We show that together with SLC25 family members including the prototypical ANTs (Kang et al., 2017; Vukotic et al., 2017), SFXNs are AGK-reliant TIM22 complex cargos. Consistent with this finding, a recent study showed reduced import and assembly of SFXNs in mitochondria without AGK or with mutated TIMM9 (Jackson et al., 2020), a chaperone of the TIM22 pathway (Adam et al., 1999). This reduction, however, is not observed with knockdown of TIMM29, another metazoan-specific TIM22 constituent important for the IMM integration of members of the TIM family, such as TIMM23 and TIMM17 (Callegari et al., 2016; Jackson et al., 2020; Kang et al., 2016), indicating only select components of the TIM22 machinery are essential for SFXNs. Interestingly, there seems to be a range of AGK dependency for the different ANT and SFXN isoforms (Figures 1J, S2E, and S2F). This suggests that there are perhaps unknown players required for the import/assembly of specific carriers or that the number of TMDs or some other structural element may govern their dependence on AGK. Orthodox TIM22 substrates contain 4 or 6 TMDs (Rehling et al., 2003), but a recent report expanded this catalog to include subunits of the mitochondrial pyruvate carrier, consisting of 2 or 3 TMDs (Gomkale et al., 2020). The odd number of TMDs in SFXN1 thus makes it another atypical TIM22 complex substrate.
We find that SFXN1 is vital for CIII integrity and CoQ metabolism. Being a mitochondrial serine transporter, SFXN1 is positioned to influence numerous mitochondrial functions, as there is a close relationship between ETC efficiency and serine metabolism. It has been shown that mtDNA depletion activates serine biosynthesis and that respiratory complex lesions perturb 1C metabolism by limiting formate production from serine (Bao et al., 2016; Nikkanen et al., 2016). Our study contributes to growing evidence that, conversely, disruption of serine metabolism also exerts a toll on mitochondrial function. Supplementation of formate, which maintains tRNA methylation needed for proper mitochondrial protein translation (Lucas et al., 2018; Minton et al., 2018; Morscher et al., 2018), did not restore any tested aspect of CIII function. This finding indicates that a problematic 1C metabolism is not the underlying cause of any of the SFXN1-related CIII defects as defined here. Serine deprivation has been shown to induce changes in the lipid profile that subsequently leads to mitochondrial fragmentation (Gao et al., 2018), serving as a precedent that serine-derived metabolites other than 1C units influence mitochondrial physiology. These findings also argue that SFXN1 is more than just a player in 1C metabolism.
We illustrate that in the context of CIII dysfunction, the inefficient generation of at least 2 metabolites—heme and αKG—contributes to the phenotype. SFXN1 is linked to heme biosynthesis by its ability to deliver serine, and perhaps glycine (Kory et al., 2018), into the mitochondrial matrix where the pathway is initiated. Consistently, SFXN1 is a constituent of the mitochondrial heme metabolon (Medlock et al., 2015). In endothelial cells, depletion of cellular serine by deletion of PHGDH, encoding the rate-limiting enzyme in de novo serine biosynthesis, results in inadequate heme and impedes OXPHOS capacity (Vandekeere et al., 2018). In our HEK293-based SFXN1 KO model, there is an accumulation of total serine and an elevated serine:glycine ratio, implying that it is the ability to take serine into the mitochondrion for glycine conversion and not low serine levels per se that underlies the observed heme-related defect. This finding highlights the significance of achieving threshold metabolite concentrations in specific subcellular compartments to drive biosynthetic processes.
Notably, other SFXNs have been implicated in heme metabolism, including SFXN4 that has not been demonstrated to compensate for serine-transport-based derangements in the absence of SFXN1 (Kory et al., 2018; Mon et al., 2019; Paul et al., 2019). At least in murine erythroleukemia cells, glutamine is the major source of succinyl-coenzyme A (CoA) for ALA production (Burch et al., 2018). It is thus conceivable that SFXN4 and perhaps other isoforms can impact heme generation by transporting amino acids other than serine.
Consistent with a contribution of reduced heme content to the CIII-associated phenotype, we show that hemin supplementation improved CIII activity in cells lacking SFXN1. This result is in agreement with other studies showing that heme deprivation is associated with lower activities of heme-containing RCs II–IV (Gamble et al., 2000; Lin et al., 2019; Vandekeere et al., 2018). CIII subunit protein levels and assembly proved to be more difficult to rescue with hemin. Potentially, there is a difference in the bioavailability of exogenously supplied hemin and endogenously synthesized heme for incorporation into CIII that subsequently affects its stability. Suboptimal delivery of exogenous heme to mitochondria has been previously documented (Kim et al., 2016). This may be a potential explanation for why hemin supplementation may result in only a partial rescue of RC function (Shetty et al., 2019; Vandekeere et al., 2018).
We showed that impeding heme production at 2 unique steps of the pathway differentially affects RC subunits. SA, which acts at an early point by inhibiting ALAD, reduces steady-state protein levels of CII–CIV, with CIII subunits being most sensitive. However, NMP, which blocks the last step by inhibiting FECH, only decreased CIV. Interestingly, although both are known to deplete heme, we observed that SA and NMP treatments have opposite effects on the ALAS1 protein (Figures 7D, S7A, and S7B). SA caused upregulation, possibly as a response to ALA buildup, whereas NMP led to downregulation, likely due to PPIX accumulation (Maitra et al., 2019). Hence, the impact of heme metabolism on RC subunit protein levels and assembly appears to be more complex than being just about total heme amount. Going forward, it will be important to consider how genetic or pharmacological interventions affect heme biosynthetic intermediates and specific heme types.
SFXN1 KOs displayed a substantial reduction in αKG, in part due to ineffective glutamate and glucose conversion to αKG. Supplying αKG to the TCA cycle is relevant for the production of NADH and FADH2. αKG is connected to the serine synthesis pathway—glutamate, acting as a nitrogen donor, is converted to αKG as phosphoserine is formed from PHGDH-generated 3-phosphohydroxypyruvate (Amelio et al., 2014). It will be compelling to obtain a quantitative perspective of other metabolic flux alterations in the absence of SFXN1. The reversal of CIII activity and subunit protein abundance in SFXN1 KOs upon DMK treatment emphasizes the tight coordination of flux into the TCA cycle and OXPHOS. In cell models with moderate OXPHOS decay, stimulation of αKG anaplerosis by exogenous DMK is beneficial for cell proliferation and respiration in galactose-based media (Chen et al., 2018). Here, we provide evidence that these effects may be mediated by elevation in CIII activity and protein levels of RCs. Exactly how αKG does this warrants further investigation.
Why is CIII more sensitive to SFXN1 ablation than other RCs? Although there is presently no clear answer to this enigma, it is intriguing that SFXN1 has been documented as a binding partner of TTC19, a CIII assembly factor (Bottani et al., 2017). It is becoming increasingly appreciated that different aspects of mitochondrial biology are subject to metabolic regulation, from mitochondrial biogenesis (Scarpulla, 2011) to dynamics (Mishra and Chan, 2016). An emerging form of metabolic communication is mediated by metabolite-dependent post-translational modifications of proteins that modulate ETC biogenesis, assembly, and activation (Van Vranken et al., 2018). It is an interesting possibility that MCs, by delivering substrates, work closely with these factors.
Overall, our functional investigation significantly extends our understanding of how SFXN1 influences mitochondrial and cellular metabolic efficiency. In light of the data indicating that SFXN1 likely transports other amino acids besides serine (Kory et al., 2018), together with the documented substrate promiscuity of MCs (Boulet et al., 2018; Fiermonte et al., 2009; Seifert et al., 2015; Wohlrab and Flowers, 1982), it is plausible that SFXN1 participates in other anabolic and catabolic reactions beyond those surveyed in the present study. Dissecting the mechanisms governing substrate selection and transport by SFXN1 and other MCs in specific physiological and pathological contexts and how this is coordinated with the rest of the MC system is necessary to ultimately decode the mitochondrion’s complex regulatory network.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Steven M. Claypool (sclaypo1@jhmi.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
The accession number for the SILAC-based quantitative proteomics data reported in this paper and deposited to the ProteomeXchange Consortium is PRIDE: PXD019826 (http://proteomecentral.proteomexchange.org/cgi/GetDataset).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Flp-In 293 cells (Thermo Fisher Scientific) were grown in DMEM with 4.5 g/L glucose (Corning), supplemented with 10% fetal bovine serum (Seradigm, GIBCO), 2 mM L-glutamine (GIBCO), and 100 μg/mL zeocin (Thermo Fisher Scientific), and kept at 37°C in 5% CO2. HeLa cells (ATCC) were maintained in the same media but without zeocin. For galactose-containing media, DMEM without glucose (GIBCO) was supplemented with 10 mM galactose (Acros Organics), 10% FBS and 2 mM L-glutamine. Control and immortalized patient fibroblasts were grown in DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (GIBCO) added with 100 μg/mL uridine, 10% FBS, and 1× penicillin-streptomycin (GIBCO).
To make media lacking serine, DMEM without glucose, glutamine, serine, glycine, and sodium pyruvate (US Biological) was supplemented with 25 mM glucose (RPI), 4 mM glutamine, 30 mg/L glycine (RPI), sodium bicarbonate (Acros Organics), and 10% heat-inactivated dialyzed FBS (Gemini Bio, Thermo Fisher Scientific). For full media, 42 mg/L serine (RPI) was additionally included. 15 μM hemin (stock always freshly prepared in 20 mM NaOH) or 1 mM formate was supplemented as indicated.
Bacterial strains
For cloning, Escherichia coli DH5α cells were used for transformation. Details for the generation of plasmid constructs are described below.
METHOD DETAILS
Cell proliferation
Cells were seeded in 6-well plates (2×104 cells/well) in glucose-based media and allowed to adhere overnight. Per genotype, 6 wells were counted to monitor initial cell count at day of treatment (Day 1). Cells in another 6-well plate were washed twice with 1X PBS prior to switch to treatment media. Cells were counted at Day 5 by staining with trypan blue solution (GIBCO) and using the Countess II FL automated cell counter (Thermo Fisher Scientific). Data were presented as cell count at Day 5 relative to Day 1 of treatment.
Knock-out cell line generation via CRISPR/Cas9
Two guide RNAs (gRNAs) that target exon 2 of human SFXN1 were cloned separately into PX459, a gift from Feng Zhang (Addgene) (Ran et al., 2013). Cells were seeded in 6-well plates and grown in DMEM with 4.5 g/L glucose, 10% FBS and 2 mM L-glutamine, without antibiotics, and transfected with either construct using FuGENE HD Transfection Reagent (Promega) according to manufacturer’s protocol. HEK293 cells were transfected with gRNA #1 or #2 while HeLa cells were transfected with gRNA #2. After 24 hr, the medium was replaced with one containing the same components mentioned, but further supplemented with 1 mM sodium pyruvate (GIBCO) and 50 μg/mL uridine (Sigma-Aldrich) (which were subsequently found to be not essential additions). 48 hr post-transfection, cells were selected with 2 μg/mL puromycin (GIBCO) for 2 days, then grown in puromycin-free medium until they reached confluency. They were passaged and a portion was used for genomic DNA extraction using the Puregene Core Kit A (QIAGEN) and T7 endonuclease assay (Genecopoeia) to test if indels were introduced. The remaining cells were used for seeding at low density in 10-cm dishes. Single colonies were isolated by ring cloning, expanded, and analyzed by immunoblotting with SFXN1 antibody to select KO clones.
gRNA that targets human AGK, under the U6 promoter, was synthesized as a gene block fragment (Integrated DNA technologies) and cloned by In-Fusion cloning (Clontech) into the pEF6-nls-YFP-2A-Cas9 vector, as previously detailed (Bowman et al., 2017). Blasticidin (7.5 μg/mL; InvivoGen) selection was done 48 hr after transfection and carried out for two weeks. YFP-positive clones were selected by flow cytometry and surviving clones were screened for the absence of AGK by genotyping and immunoblotting.
Genotyping of SFXN1- and AGK null clones
Genomic DNA was extracted from WT and KO clones. The region surrounding the gRNA target site was amplified by PCR, followed by digestion with HindIII/SacII (NEB), and ligation into pBSK(−). At least 10 individual transformants were analyzed by sequencing to determine the mutations introduced. Nucleotide sequence alignments were performed by Clustal Omega.
Molecular biology
Human SFXN1 cDNA was amplified from HEK293 mRNA by using GoScript™ Reverse Transcription Mix, Oligo(dT) (Promega). Human AGK was copied from HeLa cDNA. CNAP-SFXN1, SFXN1-HA, and AGKG126E were generated by overlap-extension polymerase chain reaction (PCR) using Platinum™ Pfx DNA polymerase (Thermo Fisher Scientific). All were cloned into pcDNA5/FRT (Invitrogen) and sequenced before use for transfection. To purify recombinant AGK and recombinant ANT2, human AGK and ANT2 were cloned into pET28a vector (Novagen) containing a N-terminal hexahistidine tag. Protein induction, extraction and affinity purification were carried out as described previously (Lu et al., 2017).
Whole cell extraction
Cells were washed with ice-cold 1X PBS and incubated in RIPA buffer (20 mM HEPES-KOH, pH 7.4, 1% (v/v) Triton X-100, 50 mM NaCl, 1 mM EDTA, 2.5 mM MgCl2, 0.1% (w/v) SDS) with 1 mM PMSF for 30 min at 4°C, with constant agitation. Soluble and insoluble materials were separated by centrifugation at 21,000xg for 30 min at 4°C. The supernatant was transferred to a fresh tube, and protein quantification was performed with Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific).
Mitochondrial isolation
Cells were seeded onto 150 mm × 25 mm culture dishes and expanded. At around 80% confluency, glucose-based medium was changed to galactose-based medium and the cells were further grown for 48 hr. For supplementation experiments, the galactose-containing media was supplemented with 15 μM hemin, 1 mM formate, 10 mM dimethyl α-ketoglutarate (DMK; Sigma-Aldrich), 2 mM serine, 2 mM glycine, 2 mM alanine (RPI), 2 mM cystine dihydrochloride (RPI), or 125 μM succinylacetone (SA), as indicated. For the minus (−) serine panel, cells were seeded into glucose-based medium and allowed to adhere for 48 hr before washing with 1X PBS and adding the indicated medium.
Mitochondrial isolation was performed based on a previous protocol (Frezza et al., 2007). Briefly, cells were washed with ice-cold 1X PBS and resuspended in 3 mL IBc buffer (200 mM sucrose, 10 mM Tris-MOPS, 1 mM EGTA/Tris, pH 7.4). Homogenization was performed using a Teflon Potter-Elvehjem motor-driven dounce set at 1600 rpm. Lysates were centrifuged twice at 600xg for 10 min at 4°C to remove cellular debris and the nuclear fraction. The supernatant was then spun down at 7,000xg for 10 min at 4°C. The resulting pellet was resuspended in IBc buffer and centrifuged again at 7,000xg for 10 min at 4°C. An additional centrifugation at 10,000xg for 10 min at 4°C was performed and the pellet, which represents the crude mitochondria, was resuspended in IBc buffer to a final concentration of 10mg/mL. If not used right away, mitochondrial extracts were aliquoted, snap frozen in liquid nitrogen, and stored at −80°C.
Immunoblotting
Equal protein amounts (20–50 μg depending on samples or antibodies used) were loaded onto SDS-PAGE gels (12% or 10%–16%) and immunoblotting was carried out as described previously (Lu et al., 2016). Images were captured using Fluorchem Q (Cell Biosciences, Inc.) quantitative digital imaging system. Antibodies used for this study were as follows: AGK (AB Clonal Technology; this study), ALAS1 (Abcam), ANT1 (Lu et al., 2017), ANT2 (Pacific Immunology; this study), ANT2/3 (Panneels et al., 2003), ATP5B (Peter Pedersen, Johns Hopkins University), β-actin (Sigma-Aldrich), COX4 (Abcam), CPOX (Proteintech), MTCYB (Proteintech), CYTC (BD Biosciences), DNAJC19 (Proteintech), FECH (Proteintech), GRP75 (Antibodies Inc.), HA (Sigma-Aldrich), MTCO1 (Thermo Fisher Scientific), MTCO2 (Thermo Fisher Scientific), NDUFB6 (Abcam), NDUFS1 (Abcam), NDUFV1 (Proteintech), NDUFV2 (Proteintech), OPA1 (BD Biosciences), Protein C (Genscript), PHB1 (Biolegend), PHB2 (BioLegend), SDHA (Invitrogen, Abcam), SDHB (Abcam), SFXN1 (Proteintech), SFXN2 (Sigma-Aldrich), SFXN3 (Sigma-Aldrich), SFXN4 (Cusabio), SFXN5 (Cusabio), TIMM22 (Abcam), TIMM23 (BD Biosciences), TIMM50 (Proteintech), TOMM20 (Santa Cruz Biotechnology), UQCRC2 (Abcam), and UQCRFS1 (Abcam). Goat anti-rabbit (Pierce) or mouse (Pierce) secondary antibodies conjugated to horseradish peroxidase were utilized. Images were minimally processed in Adobe Photoshop and assembled in Adobe Illustrator.
Membrane association assays
HEK293 WT mitochondria (250 μg) were resuspended in 1 mL cold 5 mM Tris pH 7.5, incubated on ice for 30 min, and centrifuged at 21,000xg for 10 min at 4°C. The pellet was resuspended in 0.5 mL Sonication Buffer (0.6 M sucrose, 3 mM MgCl2, 20 mM HEPES-KOH, pH 7.4), sonicated thrice for 10 s, with 30 s intervals on ice, and spun down at 21,000xg for 10 min at 4°C. Ultracentrifugation (Optima MAX, TLA-120.1 rotor) at 175,000xg for 30 min at 4°C was carried out for the supernatant. The resulting soluble fraction was subjected to precipitation using 20% (v/v) TCA and 0.07% (w/v) sodium deoxycholate, while the pellet was directly resuspended in 1:1 2X RSB: 0.1 N NaOH. Analysis was done by SDS-PAGE and immunoblotting.
For carbonate extraction, 100 μg HEK293 mitochondria was resuspended in 0.5 mL of 0.1 M Na2CO3 of varying pH (pH 10.5, 11.0,11.5, 12.0, 12.5), vortexed and incubated on ice for 30 min. Samples were subjected to ultracentrifugation and downstream steps as detailed above.
Digitonin-based mitochondrial fractionation
The protocol used was adapted from a previous study (Lu et al., 2016). 100 μg mitochondria were resuspended in 50 μL SEHK buffer (250 mM sucrose, 5 mM EDTA, pH 7.0, 10 mM HEPES-KOH, pH 7.4, 200 mM KCl) supplemented with 0, 0.1, 0.2, 0.3, 0.4 or 0.5% (w/v) digitonin and 1 mM PMSF. Samples were briefly vortexed on low and incubated on ice for 2 min. 450 μL ice-cold SEHK buffer was then added to halt solubilization. Samples were fractionated by ultracentrifugation at 100,000xg for 10 min at 4°C. The resulting supernatants were subjected to TCA precipitation. Pellets from membrane and soluble fractions were resuspended in equal volumes of reducing sample buffer and analyzed by SDS-PAGE.
Protease protection assay
To determine the submitochondrial localization of endogenous SFXN1, CNAP-SFXN1 (CNAP consists of a protein C and His10 tags) or SFXN1-HA, 200 μg intact mitochondria, mitoplasts, or solubilized mitochondria were treated with 125 μg/mL Pronase E (Sigma-Aldrich) for 30 min on ice, as indicated. Mitoplasting and solubilization was done by osmotic swelling in 5 mM Tris-HCl pH 7.5 or by utilizing 0.5% (v/v) sodium deoxycholate, respectively. Protease digestion was halted by adding 5 mM PMSF. Samples were TCA-precipitated and heated at 65°C for 5 min. This was followed by incubation on ice for 1 hr, and spinning at 21,000xg for 10 min at 4°C. Pellets were washed with 1 mL cold acetone, dried, resuspended in 1:1 2X RSB: 0.1 N NaOH, and boiled for 5 min. Analysis was done by SDS-PAGE and immunoblotting.
Blue native (BN)-PAGE analysis
Mitochondria (100 or 150 μg for 1D, 150 μg for 2D, 150 μg for Coomassie staining) were solubilized in lysis buffer (20 mM Tris-Cl, 10% (v/v) glycerol, 100 mM NaCl, 20 mM imidazole, 1 mM CaCl2, pH 7.4) containing either 1% (w/v) digitonin (Biosynth International) or 1% (w/v) n-dodecyl-β-maltoside (DDM) (Thermo Fisher Scientific), and supplemented with protease inhibitors (1 mM PMSF, 2 μM pepstatin A, 10 μM leupeptin). Isolates were clarified by spinning at 21,000xg for 30 min at 4°C. Supernatants with BN sample buffer added were resolved using in-house-made 4%–16% BN-PAGE gels for immunoblotting, or 6%–16% BN-PAGE gels for Coomassie staining (0.25% (w/v) Coomassie Brilliant Blue R250, 40% methanol, 10% glacial acetic acid). For 1D BN-PAGE, the steps following transfer were adapted as previously described (Maio et al., 2017). Briefly, the PVDF membrane was soaked in 8% acetic acid for 20 min, rinsed with water, and air-dried. The membrane was washed with methanol until most of the residual Coomassie Blue G-250 was removed, before proceeding to standard immunoblotting procedures. The 2D BN/SDS-PAGE analysis was performed as previously described (Claypool et al., 2008).
SILAC
HEK293 WT and two AGK KOs were subjected to three-plex SILAC. For set 1, WT was grown in “light” media, AGK KO1 in “medium” media, and AGK KO2 in “heavy” media. For set 2, the media for AGK KO1 and KO2 were swapped. DMEM without lysine, arginine, and sodium pyruvate (GIBCO) was the base for all three media types. Light media was prepared by supplementing the base media with unlabeled L-lysine:2HCl (0.8 mM) (Acros Organics) and L-arginine:HCl (0.4 mM) (Acros Organics). To the same final concentrations, medium and heavy media contained added 2H4-L-Lysine:2HCl and 13C6-L-Arginine:HCl, or 13C6 15N2-L-Lysine:2HCl and 13C6 15N4-L-Arginine:HCl (Cambridge Isotope Laboratories), accordingly. Cells were passaged in their respective medium at least five times before initiating the experiment to ensure that the isotopic amino acids were incorporated. Two days before mitochondrial isolation, cells were cultivated in medium with the same formulation except that glucose was replaced by 10 mM galactose. A preliminary MS run was carried out to make sure that isotope labeling was > 95% before proceeding to the actual experiment. Equal concentrations of mitochondria as determined by BCA assay were mixed and processed for liquid chromatography-tandem mass spectrometry (LC-MS/MS), as previously described (Wu et al., 2014). For each sample, 1 μg was injected twice in an Orbitrap Fusion Lumos mass spectrometer coupled to an Easy nanoLC 1200 ultra-high pressure liquid chromatography (UPLC) system (Thermo Scientific). Peptides were separated on an analytical column (50 cm × 75 μm, PepMap C18 1.9 μm, 100Å, EasySpray, Thermo Scientific) using a linear gradient of 5%–30% solvent B (acetonitrile in 0.1% (v/v) formic acid) over 85 min with total run time of 120 min. Both MS and MS/MS were measured using an Orbitrap mass analyzer in top speed mode. Raw data was searched using Sequest algorithm against Human protein database (NCBI RefSeq 85) in Proteome Discoverer Software Suite v2.0 (Thermo Scientific). The search parameters included a maximum of two missed cleavages; carbamidomethylation at cysteine as a fixed modification; N-terminal acetylation, oxidation at methionine, SILAC labeling 13C6,15N2-lysine, 2H4-lysine, 13C6-arginine and 13C6,15N2-arginine as variable modifications. Precursor tolerance was set to 10 ppm and MS/MS tolerance to ± 0.02 Da. False discovery rate was set to 1% at the peptide-spectrum matches (PSMs), peptide and protein level. Data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019826.
Iron chelation
Cells were depleted of iron by treatment with deferoxamine mesylate (DFO) (Sigma-Aldrich), an iron chelator. Cell viability of HEK293 WT and SFXN1 KOs in different DFO concentrations was assessed using CellTiter-Glo® 2.0 (Promega). 1×104 cells were seeded per well in a white-walled microplate and allowed to adhere overnight before DFO treatment for 24 hr. Data were presented as luminescence values relative to no treatment.
Metal level quantification
Whole cell or mitochondrial pellets were snap-frozen until analysis by inductively-coupled mass spectroscopy (ICP-MS). Samples were warmed to room temperature and centrifuged for 2 min at 14,000xg. To each sample, 200 μL of concentrated HNO3 was added, the samples vortexed and then incubated at 80°C overnight. Samples were vortexed for 30 s while still warm, and diluted with 2 mL of milliQ water, transferred to 15 mL conical tubes, and analyzed by ICP-MS (University of Maryland School of Pharmacy Mass Spectrometry Center).
Enzyme activity measurements
Activities of Complex I (NADH dehydrogenase), Complex II (succinate dehydrogenase), and Complex IV (cytochrome c oxidase) were determined using Abcam’s Complex I (Abcam), Complex II (Abcam), or Complex IV (Abcam) Human Enzyme Activity Microplate Assay Kits, accordingly. Mitochondria (50 μg for Complex I and II, 10 μg for Complex IV) were solubilized at 5 mg/mL in the provided detergents supplemented with 1 mM PMSF based on manufacturer’s recommendations. Complex I activity was measured by monitoring the increase in absorbance at 450 nm which is coupled to the oxidation of NADH to NAD+. Complex II activity was determined as the rate of decrease in absorbance at 600 nm, when ubiquinol is generated and causes the reduction of DCPIP. Complex IV activity was quantified based on cytochrome c oxidation, which leads to a decrease in absorbance at 550 nm.
Complex III activity was measured as previously documented (Tzagoloff et al., 1975). Briefly, mitochondria were solubilized in reaction buffer (50 mM KPi, 2 mM EDTA, pH 7.4) containing 0.5% (w/v) DDM, or 0.5, 1.0, or 1.5% (w/v) digitonin, and spiked with protease inhibitors. 30 μg was added to reaction buffer with 0.08% (w/v) equine heart cytochrome c (Sigma-Aldrich) and 1 mM potassium cyanide. The reaction was started by adding 100 μM decylubiquinol. The reduction of cytochrome c was recorded as the rate of absorbance increase at 550 nm.
Alanine aminotransferase (ALT) activity was analyzed using the ALT Assay Kit (Sigma-Aldrich). 1×106 cells were homogenized in 200 μL ice-cold ALT assay buffer. Samples were spun at 15,000xg for 10 min at 4°C. Per reaction, 20 μL of the supernatant was used. The initial fluorescent intensity was measured (λex/λem = 535/587 nm), then the samples were incubated at 37°C with a reading taken every 5 min for 1 hr. Glutamate dehydrogenase (GDH) activity was analyzed using the GDH Assay Kit (Sigma-Aldrich). 1×106 cells were lysed in 200 μL ice-cold GDH assay buffer. Samples were incubated on ice for 10 min prior to centrifugation at 13,000xg for 10 min at 4°C. Per reaction, 50 μL of the supernatant was used. Samples were incubated at 370°C for 3 min and initial absorbance was measured at 450 nm. Further incubation at 37°C took place for 1 hr, with readings every 5 min. Calculations were performed per manufacturer’s protocol. Enzyme activities were normalized to protein concentrations of remaining supernatants as determined by BCA assay.
Gene expression analysis by quantitative PCR (qPCR)
Total RNA extracts were prepared using PureLink® RNA Mini Kit and DNase set (Invitrogen) according to manufacturer’s protocol. 25 ng RNA was used as template in 20 μL reactions for qPCR using EXPRESS One-Step SYBR GreenER kit (Thermo Fisher Scientific), with ROX as reference dye, and 200 nM each of forward and reverse primers for genes of interest. No template and no reverse transcriptase controls were included and samples were analyzed in duplicate. Reactions were run using the QuantStudio 6 Flex Real-Time PCR System (Applied Biosciences) with the following conditions: (a) 5 min at 50°C and 2 min at 95°C (cDNA synthesis), (b) 15 s at 95°C and 1 min at 60°C (40-cycle amplification), and (c) melt curve analysis. Gene expression was expressed as dCT or fold-change (FC) relative to WT, calculated based on the 2−ΔΔCT method, with GAPDH as the reference gene.
mtDNA content
Genomic DNA was isolated using Gentra Puregene Cell Kit (QIAGEN). qPCR was carried out with FastStart Universal SYBR Green Master Rox (Roche) according to manufacturer’s guidelines, with 10 ng gDNA as template in a 20 μL reaction mixture. QuantStudio 6 Flex Real-Time PCR System was used. ND1 is a mtDNA-encoded subunit of Complex I whereas β2M serves as a nuclear-encoded reference gene.
Translation of mitochondrial-encoded proteins
In vivo labeling of mitochondrial translation products was carried out based on a previous protocol (Sasarman and Shoubridge, 2012). Cells seeded on 6-well plates were washed with warm 1X PBS, switched to labeling media (DMEM without methionine and cysteine (GIBCO), 10% dialyzed FBS (GIBCO), 1X GlutaMAX™ (Thermo Fisher Scientific), 110 mg/L sodium pyruvate (GIBCO), and 2 mM L-Glutamine (GIBCO)) and incubated at 37°C, 5% CO2 for 30 min. After washing with 1X PBS, cells were treated with 100 μg/mL anisomycin (Sigma-Aldrich) and incubated at 37°C, 5% CO2 for 5 min to inhibit mitochondrial translation. Per well, 240 μCi of EasyTag™ EXPRESS35S protein labeling mix (Perkin Elmer) was added. Labeling took place at 37°C, 5% CO2 for 30 min, after which the media was switched to media A (DMEM with 10% FBS, 2 mM L-glutamine and 1 mM sodium pyruvate) and the plates returned to the incubator for 10 min. Cells were then washed with 1X PBS thrice before collection, resuspension in 1X PBS with 1 mM PMSF, and protein quantification via BCA assay. Per sample, 50 μg were resuspended in gel loading buffer (93 mM Tris-Cl, pH 6.8, 7.5% (v/v) glycerol, 1% (w/v) SDS, 0.25 mg/mL bromophenol blue, 3% β-mercaptoethanol) and resolved on a 15%–20% SDS-PAGE gel. Gel was stained with Coomassie solution, destained, vacuum-dried, and exposed to Imaging K-screen (Bio-Rad). Radiolabeled proteins were visualized by autoradiography (Pharos FX, Quantity One 1-D Analysis Software).
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurements
The XFe96 extracellular flux analyzer (Agilent) was utilized to record OCR and ECAR. For analyzing mitochondrial respiration and glycolytic parameters in intact cells, the XF Cell Mito Stress Test (Agilent) and Glycolysis Stress Test (Agilent) kits were used. Cells were seeded at 10,000 cells/well in a Seahorse XF96 V3 PS cell culture microplate (Agilent) coated with 0.001% (w/v) poly-L-lysine (Sigma-Aldrich) for improved cell adherence. After 48 hr, cells were washed twice with XF base medium (Agilent) supplemented with 1 mM sodium pyruvate, 2 mM L-glutamine, and for the Mito Stress Test only, 10 mM glucose. Cells were then incubated in a humidified non-CO2 incubator at 37°C for a 1-hr degassing. For the Mito Stress Test, OCR values were recorded under basal conditions, and after the addition of 2 μM oligomycin, 250 nM FCCP, or 1 mM rotenone/antimycin A.
For the Glycolysis Stress Test, ECAR values were obtained under basal conditions, and after the addition of 10 mM glucose, 2 μM oligomycin or 50 mM 2-deoxyglucose. Basal ECAR prior to glucose injection is designated as non-glycolytic acidification. Glycolysis rate is the maximum rate after glucose injection minus non-glycolytic acidification. Glycolytic capacity is the maximum rate after oligomycin injection minus non-glycolytic acidification. Glycolytic reserve (%) is glycolytic capacity/glycolysis converted to percent.
To measure the dependence of cells on the utilization of glucose/pyruvate, glutamine or fatty acids, inhibitors of pathways utilizing these substrates were used: 2 μM UK-5099 (Tocris), 3 μM BPTES (Tocris) and 4 μM etomoxir (Tocris) inhibit the mitochondrial pyruvate carrier (MPC), carnitine palmitoyltransferase 1A (CPT1A), and glutaminase (GLS), respectively. Cells were seeded at 12,500 cells/well and allowed to adhere for 48 hr. After media exchange with XF base medium supplemented with 2 mM L-glutamine and 10 mM glucose, without sodium pyruvate, cells were incubated in a humidified non-CO2 incubator at 37°C for 1 hr. Baseline OCR was recorded after 24 min, then the inhibitor of the targeted pathway was injected. Target inhibitor OCR was recorded after 48 min, and then the inhibitors of the two remaining pathways were injected. OCR was monitored following an additional 48 min incubation in presence of all three inhibitors. % dependency is calculated as: (baseline OCR-target inhibitor OCR)/(baseline OCR-all inhibitors OCR).
After each experiment, media was removed and the plate was frozen at −20°C in preparation for DNA content quantification with CyQUANT Assay (Thermo Fisher Scientific). Calculations were performed per manufacturer’s instructions, and OCR and ECAR measurements were normalized to DNA content.
For OCR measurements in isolated mitochondria, fresh extracts were diluted to 1 mg/mL in IBc buffer. For the electron flow assay (Figure 3E), 5 μg mitochondria were dispensed per well. To enhance mitochondrial adherence, the microplate was spun at 2,000xg for 20 min at 4°C. 50 μL of 1X MAS, pH 7.2 (220 mM D-mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2·6H2O, 2 mM K+ HEPES, pH 7.4, 1 mM EGTA, 0.2% (w/v) fatty acid-free BSA) with 10 mM pyruvate, 2 mM malate and 4 μM CCCP was added carefully to each well. Compounds reconstituted in 1X MAS without BSA were delivered to cartridge ports so as to have the following final concentrations after sequential injections: port A, 4 μM rotenone; port B, 10 mM succinate; port C, 4 μM antimycin A; port D, 10 mM ascorbate/100 μM TMPD. For the substrate utilization experiment (Figure 5C), the following were prepared in 1X MAS, pH 7.2, with 0.2% (w/v) fatty-acid free BSA: 10 mM pyruvate/2 mM malate, 10 mM glutamate/10 mM malate, 10 mM DMK/2 mM malate, 10 mM glutamine/2 mM malate, 5 mM succinate/10 μM rotenone, 10 mM glycerol-3-phosphate/2 μM rotenone, and 80 μM palmitoylcarnitine/2 mM malate. 5 μg mitochondria/well was used except when palmitoylcarnitine/malate served as substrate, wherein 10 μg was utilized instead. After allowing the mitochondria to adhere to the plate by centrifugation, 50 μL of 1X MAS with BSA containing the substrates of interest was added per well. Injections were as follows, with the final concentrations indicated: port A, 4 mM ADP; port B, 2 μM oligomycin; port C, 4 mM CCCP; 4 μM rotenone/antimycin A.
NAD+/NADH and NADPH/NADP+ measurements
Cellular NAD+/NADH and NADPH/NADP+ were determined using the NAD/NADH-Glo™ Assay (Promega) and NADP/NADPH-Glo™ Assay (Promega), accordingly. 5 × 105 cells were seeded into 6-well plates and incubated at 37°C, 5% CO2, for 2 days. On the day of the experiment, cells were washed with 1X PBS and 400 μL of ice-cold lysis buffer (1:1 1X PBS:1% (w/v) dodecyltrimethylammonium bromide (DTAB; Sigma-Aldrich) in 0.2 N NaOH) added. Lysates were frozen at −80°C prior to the assay, which was performed as previously described (Sullivan et al., 2015).
CoQ measurements
HEK293 cellular CoQ content was measured as previously described (Fernández-Del-Río et al., 2017). In brief, cell pellets were resuspended in 1X PBS, a portion of which was used for protein quantification by BCA assay while the remaining was subjected to lipid extraction using methanol and petroleum ether containing the internal standard dipropoxy-CoQ10. CoQ9 and CoQ10 standards containing dipropoxy-CoQ10 were also prepared and lipid extracted to generate standard curves. RP-HPLC MS/MS was employed to measure CoQ9 and CoQ10 levels, with the reconstituted lipid extracts first separated on a Luna 5 μM PFP(2) 100A column (100 × 4.6 mm, 5 μm; Phenomenex) in 90% solvent A (95:5 methanol/isopropanol, 2.5 mM ammonium formate) and 10% solvent B (isopropanol, 2.5 mM ammonium formate) at a constant flow rate of 1 mL/min. Quantifications were based on total peak areas normalized against the internal standard dipropoxy-CoQ10, calculated using the CoQ9 and CoQ10 standard curves, and normalized to protein content.
Metabolite extraction
Cells (75 μL) was extracted with 400 μL of ice cold 75% ACN in H2O + 0.1% ammonium hydroxide containing 0.1 μg/mL d9-carnitine and 1 μg/mL of d4-tyrosine internal standards. A process blank (PB) was created at this time containing the extraction solvent and 75 μL of H2O. Samples were vortexed for 30 s then sonicated on ice for 2 min. Samples were chilled at −20°C for 1 hr, and then centrifuged at 20,000xg for 10 min at 4°C. The supernatant was collected and transferred to fresh tubes. To remove lipids, 750 μL of cold MTBE and 50 μL of cold H2O + 0.1% ammonium hydroxide was added to each sample. Samples were vortexed, then frozen at −80°C for 30 min. The MTBE layer was discarded and the aqueous layer dried overnight under vacuum. Samples were reconstituted with 400 μL 50% ACN in H2O prior to analysis.
LC-MS
An Agilent 6545 UPLC-QToF run in both positive and negative modes was used for analysis. Separation was achieved using a Sequant Zic-pHILIC 2.1 × 100 mm column (Millipore Sigma) with Phenomenex Krudkatcher (Phenomenex). An initial concentration of 99% ACN with 5% ddH2O (buffer B) and 5% 25 mM ammonium carbonate in ddH2O (buffer B) was held for 1 min at a flow rate of 0.2. B was decreased to 10% over 14 min and held for 5 min. B was returned to starting conditions over 0.1 min, and the system was allowed to re-equilibrate for 10 min between runs at a 0.2 mL/min flow rate.
GC-MS
GC-MS analysis was performed with an Agilent 5977b GC-MS MSD-HES and an Agilent 7693A automatic liquid sampler. Dried samples were suspended in 40 μL of a 40 mg/mL O-methoxylamine hydrochloride (MOX) (MP Biomedicals) in dry pyridine (EMD Millipore) and incubated at 37°C for 1 hr, in a sand bath. 25 μL of this solution was added to auto sampler vials. 60 μL of N-methyl-N-trimethylsilyltrifluoracetamide (MSTFA with 1%TMCS, Thermo Scientific) was added automatically via the auto sampler and incubated at 37°C for 30 minutes. After incubation, samples were vortexed and 1 μL of the prepared sample was injected into the gas chromatograph inlet in the split mode with the inlet temperature held at 250°C. A 10:1 split ratio was used for analysis for the majority of metabolites. Any metabolites that saturated the instrument at the 10:1 split were analyzed at a 50:1 split ratio. The gas chromatograph had an initial temperature of 60°C for 1 min followed by a 10°C/min ramp to 325°C and a hold time of 10 min. A 30-m Agilent Zorbax DB-5MS with 10 m Duraguard capillary column was employed for chromatographic separation. Helium was used as the carrier gas at a 1 mL/min rate.
Data analyses
Data were collected using MassHunter software (Agilent). For LC-MS, molecular features were identified using MassHunter Profinder10.0 and their peak area was recorded using MassHunter Quant 9.0. For GC-MS, metabolites were identified and their peak area was recorded using MassHunter Quant (for GC-MS). Metabolite identity was established using a combination of an in-house metabolite library developed using pure purchased standards, the NIST library and the Fiehn library.
Additional metabolite quantifications
TCA metabolite measurements were obtained via LC-MS/MS. HEK293 WT and SFXN1 KOs were seeded at a density of 2×105 cells/well in 24-well plates. After 48 hr, cells were washed twice with ice-cold 1X PBS, and extracted with ice-cold 80:20% (v/v) methanol:MS-grade water. Samples were kept on dry ice during the collection process. Samples were then vortexed vigorously and centrifuged at 12,000 rpm for 15 min at 4°C. Pellets were used for protein quantification while supernatants were dried overnight by vacuum centrifugation.
At the time of analysis, dried samples were reconstituted with water containing 0.2% (v/v) formic acid. TCA cycle intermediates were separated on Nexera UHPLC (Shimadzu) using X-Terra C18 UHPLC column (2.6um, 50mm, 2.1mm; Waters, Milford, MA) using a previously published method with a minor change in gradient (Al Kadhi et al., 2017). 0.2% (v/v) formic acid in water and 0.2% (v/v) formic acid in acetonitrile were used as mobile phases A and B, respectively. Total flow was set to 0.2 mL/min, with a linear gradient applied as follows: starting condition 0% B was linearly increased to 5% B in 4 min; 2 min at 100% B was added for column washing purposes. In between samples, the column was re-equilibrated for 3 min. Sample injection volume was 2 μL. Samples were kept at 4°C, and oven temperature was set to 21°C. Eluates were detected using AB SCIEX 4500 triple quadrupole instrument. Mass spectroscopy parameters were set for each metabolite by direct infusion of corresponding standards. Instrument was operated under following conditions: Curtain gas, 20V; Temperature, 500°C; Ion source gas 1, 50V; Ion source gas 2, 40V; Ion spray voltage, −4500V; CAD gas, Low; Entrance Potential, −10V. MultiQuant (AB SCIEX) was used to profile peaks and peak area was used for metabolite profiling. Mixture of TCA standards was utilized for standard curve generation in the μM range.
For the glutamine or glucose labeling experiments, cells were incubated with 4 mM [U-13C]-glutamine (Cambridge Isotope Laboratories) or 25 mM [U-13C]-glucose (Cambridge Isotope Laboratories) for 4 or 9 hr, respectively, prior to processing as described above. Isotopomers were quantified using MultiQuant. For the serine uptake assay, cells were incubated in DMEM without serine and glycine, supplemented with 400 μM [U]-13C-serine (Cambridge Isotope Laboratories) for 24 hr. Isotopomers of serine and glycine in the media were quantified. Metabolite abundances were normalized to protein content.
Cellular glycine and serine concentrations were measured using the Glycine and Serine Fluorimetric Assay Kits (Biovision), accordingly, following the manufacturer’s protocol. Briefly, 10×106 cells were used to generate lysates which were deproteinized using 10 kDa spin columns (Millipore Sigma) and 5 or 10 μL of the filtrate was used for the assay. Readings were normalized to protein concentration as determined by BCA assay.
Heme content
Total heme content was measured based on total heme fluorescence as previously documented (Sassa, 1976; Sinclair et al., 2001), with some modifications. 1×105 cells was resuspended in 500 μL 20 mM oxalic acid and kept in the dark at 4°C. After 24 hr of acid extraction, 500 μL of 2 M oxalic acid (Sigma-Aldrich) was added and the samples were mixed by pipetting. Half was transferred to new amber tubes, which were then incubated at 98°C for 30 min in order to remove iron from heme and liberate PPIX. The remaining half was kept at RT for the same duration. Samples were centrifuged at RT at 12,000xg for 2 min before transferring 200 uL of the heated or unheated samples in duplicate into black-walled 96-well plates. Fluorescence was measured using a CLARIOstar microplate reader (BMG Labtech), at excitation and emission wavelengths of 400 nm and 620 nm, respectively.
Treatment with heme biosynthesis inhibitors
One day post-seeding in glucose-based media, cells were treated with 0, 125, 250 or 500 μM succinylacetone (SA; Sigma-Aldrich) or 10 mM N-methyl protoporphyrin IX (NMP; Santa Cruz) for 48 hr prior to whole cell extraction and immunoblotting for proteins of interest.
Oligonucleotides used in this study
| Primer name | Primer sequence (5′−3′) |
|---|---|
| SFXN1-F | TCTGGAGAACTACCACCAAAC |
| SFXN1-R | CGGGGTACCTTACAATCCCTTATTGAAGT |
| CNAP-SFXN1-rxn1-F | CTAGCTAGCGTTTAAACTTAAGCTTATGGAAGACCAAG |
| CNAP-SFXN1-rxn1-R | GTTTGGTGGTAGTTCTCCAGAAGGCATGTGATGATGGTG |
| CNAP-SFXN1-rxn2-F | TCTGGAGAACTACCACCAAAC |
| CNAP-SFXN1-rxn2-R | CGGGGTACCTTACAATCCCTTATTGAAGT |
| SFXN1-HA-rxn1-F | TACCCTTACGATGTACCGGATTACGCATGAGGTACCCCG |
| SFXN1-HA-rxn1-R | CGGGGTACCTCATGCGTAATCCGGTACATCGTAAGGGTA |
| SFXN1-HA-rxn2-F | CTAGCTAGCATGTCTGGAGAACTACCACC |
| SFXN1-HA-rxn2-R | TACATCGTAAGGGTACAATCCCTTATTGAA |
| recomb_Nde1-AGK-F | GGAATTCCATATGACGGTGTTCTTTAAAAC |
| recomb_AGK-HindIII-R | CCCAAGCTTTCACTGGGTGGGGCTTGTGAG |
| recomb_Nde1-hANT2-F | CTAGCTAGCATGACAGATGCCGCTGTGTCCTTCGCCAAG |
| recomb_hANT2-HindIII-R | ACGCGTCGACCTTATGTGTACTTCTTGATTTCATC |
| subclone_AGK-F | CTAGCTAGCGCCATGACGGTGTTCTTTAAAACGC |
| subclone_AGK-R | GGGGTACCTCACTGGGTGGGGCTTGTGAGCATC |
| mutate_AGKG126E-rxn1-F | TAATACGACTCACTATAGGGAG |
| mutate_AGKG126E rxn1-R | CAGTGTCTCATCTCCTCCTGCAACAATGATCACATC |
| mutate_AGKG126E-rxn2-F | GAGGAGATGAGACACTGCAGGAGGTTGTTACTG |
| mutate_AGKG126E-rxn1-R | TAGGTCCCGTGTATGAGATAG |
| genotype_HindIII-sfxn1-F | CCCAAGCTTGGGACGTAGTGACATTGTAAG |
| genotype_sfxn1-SacII-R | TCCCCGCGGGGAGATAGCAGCCTAACATGA |
| genotype_HindIII-agk-F | CCCAAGCTTGGGAGGCCAGAGGAGAAGTGGTA |
| genotype_agk-SacII-R | TCCCCGCGGGGATGCAGATGCAAAGGTGGGAT |
| qpcr_mtcyb-F | TGAAACTTCGGCTCACTCCT |
| qpcr_mtcyb-R | TGGGCGATTGATGAAAAGGC |
| qpcr_nd1-F | TTCAGCAATTTAGGAACCACCC |
| qpcr_nd1-R | GGTCACACTTAATTTGCCACCAA |
| qpcr_uqcrc2-F | TTCAGCAATTTAGGAACCACCC |
| qpcr_uqcrc2-R | GGTCACACTTAATTTGCCACCAA |
| qpcr_uqcrfs1-F | CTGAATACCGCCGCCTTGAA |
| qpcr_uqcrfs1-R | ATGCGACACCCACAGTAGTTA |
| qpcr_alas1-F | GCCAGGCTGTGAGATTTACT |
| qpcr_alas1-R | CTGAGGTGGCTGACATCATT |
| qpcr_alad-F | GGGTGGAGGGAGAATACTAAAC |
| qpcr_alad-R | GCCTTGGAGTTGGACTACAT |
| qpcr_cpox-F | CAGGGCTGTAGTTCCTTCTTAC |
| qpcr_cpox-R | GGAGTGAAGAGGCCAAACTT |
| qpcr_fech-F | ATGGAGAGAGATGGCCTAGAA |
| qpcr_fech-R | GCTTCCGTCCCACTTGATTA |
| qpcr_ppox-F | GGAGGCTGACCACGTTATTA |
| qpcr_ppox-R | GGCACCAAATGTCCAAATCC |
| qpcr_slc25a38-F | CTATGGACTACGTGGCTTCTTC |
| qpcr_slc25a38-R | TCCTCTCTTGGTCAGGACTT |
| qpcr_urod-F | TTCCAAGCCATCACCCTTAC |
| qpcr_urod-R | GACTAGCCTGAGGTCTCTGATA |
| mtDNA_ND1-F | ACGGGCTACTACAACCCTTC |
| mtDNA_ND1-R | GCCTAGGTTGAGGTTGACCA |
| mtDNA_B2M-F | GGAGAGCTGTGGACTTCGTC |
| mtDNA_B2M-R | ATTCTACAAACGTCGCGTGC |
QUANTIFICATION AND STATISTICAL ANALYSIS
Densitometric band analysis for protein quantitation was performed using Quantity One 1-D Analysis Software (Bio-Rad). Results are presented as means, with error bars representing the standard error of the mean (SEM). Unpaired Student’s t test was used to evaluate statistical significance of differences between means (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Statistical analysis was performed by Prism 8. Statistical details of experiments can be found in the figure legends. n represents independent experiments or independently prepared samples, unless otherwise noted.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-AGK, mouse monoclonal | AB Clonal Technology; This study | clone 506-1-1-1 |
| anti-ALAS1 (clone EPR10247), rabbit monoclonal | Abcam | Cat#ab154860 |
| anti-ANT1, mouse monoclonal | Lu et al., 2017 | clone IF3F11 |
| anti-ANT2, rabbit polyclonal | Pacific Immunology; This study | clone 5695 |
| anti-ANT2/3 (clone 5H7), mouse monoclonal | Panneels et al., 2003 | N/A |
| anti-ATP5B, rabbit polyclonal | Peter Pedersen, JHU | N/A |
| anti-β-actin, mouse monoclonal | Sigma-Aldrich | Cat#A5441; RRID:AB_476744 |
| anti-COX4, rabbit polyclonal | Abcam | Cat#16056; RRID:AB_443304 |
| anti-CPOX, rabbit polyclonal | Proteintech | Cat# 12211-1-AP; RRID:AB_2084233 |
| anti-CYTB, rabbit polyclonal | Proteintech | Cat#55090-1-AP; RRID:AB_2881266 |
| anti-CYTC (clone 7H8.2C12), mouse monoclonal | BD Biosciences | Cat# 556433; RRID:AB_396417 |
| anti-DNAJC19, rabbit polyclonal | Proteintech | Cat#12096-1-AP; RRID:AB_2094914 |
| anti-FECH, rabbit polyclonal | Proteintech | Cat#14466-1-AP; RRID:AB_2231579 |
| anti-GRP75, mouse monoclonal | Antibodies Inc. | Cat#75-127 |
| anti-HA (clone HA-7), mouse monoclonal | Sigma-Aldrich | Cat#H9658; RRID:AB_439705 |
| anti-MTCO1 (clone 1D6E1A8), mouse monoclonal | Thermo Fisher Scientific | Cat#459600; RRID:AB_2532240 |
| anti-MTCO2 (clone 12C4F12), mouse monoclonal | Thermo Fisher Scientific | Cat#A-6404; RRID:AB_221584 |
| anti-NDUFB6 (clone 21C11BC11), mouse monoclonal | Abcam | Cat#ab110244; RRID:AB_10865349 |
| anti-NDUFS1 (clone EPR11521(B)), rabbit monoclonal | Abcam | Cat#ab169540; RRID:AB_2687932 |
| anti-NDUFV1, rabbit polyclonal | Proteintech | Cat#11238-1-AP; RRID:AB_2149040 |
| anti-NDUFV2, rabbit polyclonal | Proteintech | Cat#15301-1-AP; RRID: AB_2149048 |
| anti-OPA1 (clone 18), mouse monoclonal | BD Biosciences | Cat#612606; RRID:AB_399888 |
| anti-Protein C (clone HPC4), rabbit polyclonal | Genscript | Cat#A01774-100; RRID:AB_2744686 |
| anti-PHB1 (clone 6031), rabbit polyclonal | BioLegend | Cat#603102; RRID:AB_2164630 |
| anti-PHB2 (clone 6118), rabbit polyclonal | BioLegend | Cat#611802; RRID:AB_2164497 |
| anti-SDHA (clone 2E3GC12FB2AE2), mouse monoclonal | Invitrogen/Abcam | Cat#459200; RRID:AB_2532231/Cat# ab14715; RRID:AB_301433 |
| anti-SDHB (clone 21A11AE7), mouse monoclonal | Abcam | Cat#ab14714; RRID:AB_301432 |
| anti-SFXN1, rabbit polyclonal | Proteintech | Cat#12296-1-AP; RRID:AB_2185814 |
| anti-SFXN2, rabbit polyclonal | Sigma-Aldrich | Cat#HPA026834; RRID:AB_1856791 |
| anti-SFXN3, rabbit polyclonal | Sigma-Aldrich | Cat#HPA048105; RRID:AB_2680265 |
| anti-SFXN4, rabbit polyclonal | Cusabio | Cat#CSB-PA744046LA01HU |
| anti-SFXN5, rabbit polyclonal | Cusabio | Cat#CSB- PA819464LA01HU |
| anti-TIMM22 (clone EPR9973), rabbit monoclonal | Abcam | Cat#ab167423; RRID:AB_2802138 |
| anti-TIMM23, mouse monoclonal | BD Biosciences | Cat#611332; RRID:AB_398755 |
| anti-TIMM50, rabbit polyclonal | Proteintech | Cat#22229-1-AP; RRID:AB_2879039 |
| anti-TOMM20, rabbit polyclonal | Santa Cruz Biotechnology | Cat# sc-11415 (discontinued); Cat# sc-17764; RRID:AB_2207533 |
| anti-UQCRC2 (clone 13G12), mouse monoclonal | Abcam | Cat# ab14745; RRID:AB_2213640 |
| anti-UQCRFS1 (clone 5A5), mouse monoclonal | Abcam | Cat#ab14746; RRID:AB_301445 |
| Goat anti-rabbit IgG (H+L), secondary antibody, HRP | Thermo Fisher Scientific | Cat#31460; RRID:AB_228341 |
| Goat anti-mouse IgG (H+L), secondary antibody, HRP | Thermo Fisher Scientific | Cat#62-6520; RRID:AB_2533947 |
| Chemicals, peptides, and recombinant proteins | ||
| anisomycin | Sigma-Aldrich | Cat#A9789 |
| blasticidin | InvivoGen | Cat#ant-bl-05 |
| 1,4-benzoquinone | Sigma-Aldrich | Cat#PHR1028 |
| BPTES | Tocris | Cat#5301 |
| coenzyme Q9 | Sigma-Aldrich | Cat#27597 |
| coenzyme Q10 | Sigma-Aldrich | Cat#C9538 |
| cytochrome c | Sigma-Aldrich | Cat#C2506 |
| decylubiquinone | Enzo Life Science | Cat#BML-CM115 |
| dialyzed FBS | Gemini Bio; Thermo Fisher Scientific | Cat#100-108; Cat#A3382001 |
| digitonin | Biosynth International | Cat#D-3200 |
| n-dodecyl-β-maltoside | Thermo Fisher Scientific | Cat#D4641 |
| 4,6-Dioxoheptanoic acid (succinylacetone) | Sigma-Aldrich | Cat#D1415 |
| DMEM, with 4.5 g/L glucose and L-glutamine, without sodium pyruvate | Corning | Cat#10-013-CM |
| DMEM, with 4.5 g/L glucose, L-glutamine, and sodium pyruvate | GIBCO | Cat#11995040 |
| DMEM without glucose | GIBCO | Cat#11966025 |
| DMEM with L-glutamine, without D-glucose, without L-arginine HCl, without L-lysine HCl, without sodium pyruvate | GIBCO | Cat#ME120220 |
| DMEM with high glucose, no glutamine, no methionine, no cystine | GIBCO | Cat#21013024 |
| DMEM without glucose, glutamine, serine, glycine, and sodium pyruvate | US Biological | Cat#D9800-16 |
| EasyTag™ EXPRESS35S protein labeling mix | Perkin Elmer | Cat#NEG772002MC |
| Fetal Bovine Serum | Seradigm; GIBCO | Cat#1500; Cat#16000-044 |
| Fugene HD Transfection Reagent | Promega | Cat#E2311 |
| deferoxamine mesylate salt | Sigma-Aldrich | Cat#D9533 |
| dimethyl α-ketoglutarate | Sigma-Aldrich | Cat#349631 |
| D(+)-galactose | Acros Organics | Cat#150615000 |
| D(+)-glucose | RPI | Cat#G32040 |
| etomoxir | Tocris | Cat#4539 |
| glycine | RPI | Cat#G36050 |
| hemin | Sigma-Aldrich | Cat#51280 |
| hygromycin B | Thermo Fisher Scientific | Cat#10687010 |
| L-alanine | RPI | Cat#A20060 |
| L-arginine:HCl | Acros Organics | Cat#AC105000250 |
| L-cystine dihydrochloride | RPI | Cat#C81060 |
| L-glutamine | GIBCO | Cat#25030081 |
| 1X GlutaMAX™ | GIBCO | Cat#35050061 |
| L-lysine:2HCl | Acros Organics | Cat#AC413360250 |
| 2H4-L-Lysine:2HCl | Cambridge Isotope Laboratories | Cat#CLM-2247-H-PK |
| 13C6-L-Arginine:HCl | Cambridge Isotope Laboratories | Cat#CLM-2265-H-PK |
| 13C615N2-L-Lysine:2HCl | Cambridge Isotope Laboratories | Cat#CNLM-291-H-PK |
| 13C615N4-L-Arginine:HCl | Cambridge Isotope Laboratories | Cat#CNLM-539-H-PK |
| L-serine | RPI | Cat#S22020 |
| oxalic acid dihydrate | Sigma-Aldrich | Cat#O0376 |
| palmitoyl-L-carnitine chloride | Sigma-Aldrich | Cat#P1645 |
| potassium borohydride | Sigma-Aldrich | Cat#455571 |
| Platinum™ Pfx DNA polymerase | Thermo Fisher Scientific | Cat#11708 |
| Pronase E | Sigma-Aldrich | Cat#7433-2 |
| penicillin-streptomycin | GIBCO | Cat#15140122 |
| puromycin | GIBCO | Cat#A11138 |
| sn-glycerol 3-phosphate lithium salt | Sigma-Aldrich | Cat#94124 |
| sodium bicarbonate | Acros Organics | Cat#S233 |
| sodium formate | Sigma-Aldrich | Cat#71539 |
| sodium pyruvate | GIBCO | Cat#11360070 |
| [U-13C]D-glucose | Cambridge Isotope Laboratories | Cat#CLM-1396-5 |
| [U-13C]L-glutamine | Cambridge Isotope Laboratories | Cat#CLM-1822-H-PK |
| [U-13C]L-serine | Cambridge Isotope Laboratories | Cat#CLM-1574-H |
| UK-5099 | Tocris | Cat#4186 |
| uridine | Sigma-Aldrich | Cat#U3750 |
| zeocin | Thermo Fisher Scientific | Cat#R25001 |
| Critical commercial assays | ||
| Complex I Enzyme Activity Microplate Assay Kit | Abcam | Cat#ab109721 |
| Complex II Enzyme Activity Microplate Assay Kit | Abcam | Cat#ab109908 |
| Complex IV Human Enzyme Activity Microplate Assay Kit | Abcam | Cat#ab109909 |
| DL-Serine Assay Kit | Biovision | Cat#K743 |
| dodecyltrimethylammonium bromide | Sigma-Aldrich | Cat#D8638 |
| Glycine Assay Kit | Biovision | Cat#K589 |
| GoScript™ Reverse Transcription Mix, Oligo(dT) | Promega | Cat#A2791 |
| PureLink™ RNA Mini Kit | Thermo Fisher Scientific | Cat#12183018A |
| PureLink™ DNase Set | Thermo Fisher Scientific | Cat#12185010 |
| Gentra Puregene Cell Kit | QIAGEN | Cat#158767 |
| T7 endonuclease assay | Genecopoeia | Cat#IC005 |
| Seahorse XF Mito Stress Test Kit | Agilent | Cat#103015-100 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent | Cat#103020-100 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| Cell Titer Glo 2.0 | Promega | Cat#G9241 |
| ALT Assay Kit | Sigma-Aldrich | Cat#MAK052 |
| GDH Assay Kit | Sigma-Aldrich | Cat#MAK099 |
| EXPRESS One-Step SYBR GreenER Kit | Cat#11780200 | |
| CyQUANT Cell Proliferation Assay | Thermo Fisher Scientific | Cat#C7026 |
| NAD/NADH-Glo™ Assay | Promega | Cat#G9071 |
| NADP/NADPH-Glo™ Assay | Promega | Cat#G9081 |
| Deposited data | ||
| SILAC proteomics (HEK293 WT versus AGK KOs) | This study | PRIDE: PXD019826 |
| Experimental models: cell lines | ||
| Human: Flp-In™ 293 | Thermo Fisher Scientific | Cat#R75007 |
| Human: HeLa | ATCC | Cat#CCL-2 |
| Human: 293 SFXN1 KO | This study | N/A |
| Human: HeLa SFXN1 KO | This study | N/A |
| Human: 293 SFXN1 KO + SFXN1 | This study | N/A |
| Human: 293 SFXN1 KO + CNAP-SFXN1 | This study | N/A |
| Human: 293 SFXN1 KO + SFXN1-HA | This study | N/A |
| Human: 293 AGK KO1 | This study | N/A |
| Human: 293 AGK KO2 | This study | N/A |
| Human: 293 AGK KO + AGK | This study | N/A |
| Human: 293 AGK KO + AGKG126E | This study | N/A |
| Human: control fibroblasts | Reyes et al., 2018 | N/A |
| Human: TIMM50 mut fibroblasts | Reyes et al., 2018 | N/A |
| Recombinant DNA | ||
| pcDNA5/FRT | Thermo Fisher Scientific | Cat#V6010-20 |
| pOG44 | Thermo Fisher Scientific | Cat#V600520 |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Addgene | Cat#62988 |
| pEF6-nls-YFP-2A-Cas9 | Bowman et al., 2017 | N/A |
| pET28a | Novagen | Cat#69864 |
| PX459-SFXN1-1 | This study | N/A |
| PX459-SFXN1-2 | This study | N/A |
| pEF6-nls-YFP-2A-Cas9-agk1-1 | This study | N/A |
| pcDNA5/FRT-SFXN1 | This study | N/A |
| pcDNA5/FRT-CNAP-SFXN1 | This study | N/A |
| pcDNA5/FRT-SFXN1-HA | This study | N/A |
| pcDNA5/FRT-AGK | This study | N/A |
| pcDNA5/FRT-AGKG126E | This study | N/A |
| pET28a-AGK | This study | N/A |
| pET28a-hANT2 | This study | N/A |
| Software and algorithms | ||
| Proteome Discoverer Software Suite ver 2.0 | Thermo Scientific | N/A |
| MultiQuant ver 3 | AB SCIEX | N/A |
| Quantity One 1-D Analysis Software | Biorad | N/A |
| Prism 8 | Graphpad | N/A |
| FluoChemQ | Cell Biosciences, Inc. | N/A |
Highlights.
SFXNs are atypical TIM22 complex substrates
Loss of SFXN1 leads to respiratory chain impairments, most damaging to complex III
SFXN1 contributes to coenzyme Q, heme, α-ketoglutarate, and one-carbon metabolism
Metabolite supplementation improves complex III function in the absence of SFXN1
ACKNOWLEDGMENTS
We are grateful to Dr. Valeria Culotta and Dr. Sabrina Schatzman for suggestions regarding iron measurements. We thank Dr. Heather Neu of the Mass Spectrometry Center at the University of Maryland School of Pharmacy for processing and analyzing the samples for ICP-MS; Dr. Pete Pederson for the ATP5B antisera; Dr. Massimo Zeviani of the University of Padova and Dr. Pedro Pinheiro of the University of Cambridge for the control and TIMM50 mutation patient fibroblasts; Dr. James Cox and Dr. Jonathan Leon Catrow of the University of Utah’s Metabolomics Core Facility (supported by the NCRR Shared Instrumentation Grants 1S10OD016232-01, 1S10OD018210-01A1, and 1S10OD021505-01) for the metabolomics analysis; and Dr. Adam Cornish for technical assistance. This work was supported in part by NIH grants (R01HL108882 to S.M.C., R35GM131701-01 to O.K., R01DK120530 and R01DK116746 to M.J.W., and Biochemistry, Cellular, and Molecular Biology Program Training Grant T32GM007445 to Y.-W.L.), an NSF grant (MCB-1330803 to C.F.C.), and pre-doctoral fellowships from the American Heart Association (16PRE31140006 to M.G.A. and 12PRE11910004 to Y.-W.L.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108869.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abrahams JP, Leslie AG, Lutter R, and Walker JE (1994). Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628. [DOI] [PubMed] [Google Scholar]
- Adam A, Endres M, Sirrenberg C, Lottspeich F, Neupert W, and Brunner M (1999). Tim9, a new component of the TIM22.54 translocase in mitochondria. EMBO J 18, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajioka RS, Phillips JD, and Kushner JP (2006). Biosynthesis of heme in mammals. Biochim. Biophys. Acta 1763, 723–736. [DOI] [PubMed] [Google Scholar]
- Al Kadhi O, Melchini A, Mithen R, and Saha S (2017). Development of a LC-MS/MS Method for the Simultaneous Detection of Tricarboxylic Acid Cycle Intermediates in a Range of Biological Matrices. J. Anal. Methods Chem 2017, 5391832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio I, Cutruzzolá F, Antonov A, Agostini M, and Melino G (2014). Serine and glycine metabolism in cancer. Trends Biochem. Sci 39, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H, Liu J, and Ames BN (2001). Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: revelance to aging. J. Biol. Chem 276, 48410–48416. [DOI] [PubMed] [Google Scholar]
- Bakker HD, Scholte HR, Van den Bogert C, Ruitenbeek W, Jeneson JA, Wanders RJ, Abeling NG, Dorland B, Sengers RC, and Van Gennip AH (1993). Deficiency of the adenine nucleotide translocator in muscle of a patient with myopathy and lactic acidosis: a new mitochondrial defect. Pediatr. Res 33, 412–417. [DOI] [PubMed] [Google Scholar]
- Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, et al. (2016). Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj EJ, Li M, Ahrens-Nicklas R, Pyle LC, Wang J, Zhang VW, Clarke C, Wong LJ, Sondheimer N, Ficicioglu C, and Yudkoff M (2015). Pathologic Variants of the Mitochondrial Phosphate Carrier SLC25A3: Two New Patients and Expansion of the Cardiomyopathy/Skeletal Myopathy Phenotype With and Without Lactic Acidosis. JIMD Rep 19, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottani E, Cerutti R, Harbour ME, Ravaglia S, Dogan SA, Giordano C, Fearnley IM, D’Amati G, Viscomi C, Fernandez-Vizarra E, and Zeviani M (2017). TTC19 Plays a Husbandry Role on UQCRFS1 Turnover in the Biogenesis of Mitochondrial Respiratory Complex III. Mol. Cell 67, 96–105. e4. [DOI] [PubMed] [Google Scholar]
- Boulet A, Vest KE, Maynard MK, Gammon MG, Russell AC, Mathews AT, Cole SE, Zhu X, Phillips CB, Kwong JQ, et al. (2018). The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem 293, 1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CE, Rodriguez S, Selen Alpergin ES, Acoba MG, Zhao L, Hartung T, Claypool SM, Watkins PA, and Wolfgang MJ (2017). The Mammalian Malonyl-CoA Synthetase ACSF3 Is Required for Mitochondrial Protein Malonylation and Metabolic Efficiency. Cell Chem. Biol 24, 673–684. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch JS, Marcero JR, Maschek JA, Cox JE, Jackson LK, Medlock AE, Phillips JD, and Dailey HA Jr. (2018). Glutamine via α-ketoglutarate dehydrogenase provides succinyl-CoA for heme synthesis during erythropoiesis. Blood 132, 987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callegari S, Richter F, Chojnacka K, Jans DC, Lorenzi I, Pacheu-Grau D, Jakobs S, Lenz C, Urlaub H, Dudek J, et al. (2016). TIM29 is a subunit of the human carrier translocase required for protein transport. FEBS Lett 590, 4147–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, and Pfanner N (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, Valsecchi F, Primiano G, Calder EL, Carelli V, et al. (2018). Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells with Mitochondrial DNA Mutations. Cell Metab 27, 1007–1025. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui DH, Sweeney GD, Patterson M, and Russell ES (1977). Hemoglobin synthesis in siderocytes of flexed-tailed mutant (f/f) fetal mice. Blood 50, 165–177. [PubMed] [Google Scholar]
- Claypool SM, Oktay Y, Boontheung P, Loo JA, and Koehler CM (2008). Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol 182, 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-Carbon Metabolism in Health and Disease. Cell Metab 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Chassagne M, Ceresuela J, Rouvet I, Padet S, Acquaviva C, Nataf S, Vinzio S, Bozon D, and Mousson de Camaret B (2012). Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J. Med. Genet 49, 146–150. [DOI] [PubMed] [Google Scholar]
- Fernández-Del-Río L, Nag A, Gutiérrez Casado E, Ariza J, Awad AM, Joseph AI, Kwon O, Verdin E, de Cabo R, Schneider C, et al. (2017). Kaempferol increases levels of coenzyme Q in kidney cells and serves as a biosynthetic ring precursor. Free Radic. Biol. Med 110, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Vizarra E, and Zeviani M (2015). Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front. Genet 6, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, and Zeviani M (2018). Mitochondrial complex III Rieske Fe-S protein processing and assembly. Cell Cycle 17, 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Bugiani M, Goffrini P, Carrara F, Farina L, Procopio E, Donati A, Uziel G, Ferrero I, and Zeviani M (2007). Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum. Mol. Genet 16, 1241–1252. [DOI] [PubMed] [Google Scholar]
- Fiermonte G, Paradies E, Todisco S, Marobbio CM, and Palmieri F (2009). A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme A and adenosine 3′,5′-diphosphate in human mitochondria. J. Biol. Chem 284, 18152–18159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming MD, Campagna DR, Haslett JN, Trenor CC III, and Andrews NC (2001). A mutation in a mitochondrial transmembrane protein is responsible for the pleiotropic hematological and skeletal phenotype of flexed-tail (f/f) mice. Genes Dev 15, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Gamble JT, Dailey HA, and Marks GS (2000). N-Methylprotoporphyrin is a more potent inhibitor of recombinant human than of recombinant chicken ferrochelatase. Drug Metab. Dispos 28, 373–375. [PubMed] [Google Scholar]
- Gao X, Lee K, Reid MA, Sanderson SM, Qiu C, Li S, Liu J, and Locasale JW (2018). Serine Availability Influences Mitochondrial Dynamics and Function through Lipid Metabolism. Cell Rep 22, 3507–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomkale R, Cruz-Zaragoza LD, Suppanz I, Guiard B, Montoya J, Callegari S, Pacheu-Grau D, Warscheid B, and Rehling P (2020). Defining the Substrate Spectrum of the TIM22 Complex Identifies Pyruvate Carrier Subunits as Unconventional Cargos. Curr. Biol 30, 1119–1127. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen AK, Sips HJ, Vervoorn RC, and Tager JM (1982). Intracellular compartmentation and control of alanine metabolism in rat liver parenchymal cells. Eur. J. Biochem 122, 87–93. [DOI] [PubMed] [Google Scholar]
- Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, Lachance M, Matsuoka M, Nightingale M, Rideout A, et al. (2009). Mutations in mitochondrial carrier family gene SLC25A38 cause non-syndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet 41, 651–653. [DOI] [PubMed] [Google Scholar]
- Harigae H, and Furuyama K (2010). Hereditary sideroblastic anemia: pathophysiology and gene mutations. Int. J. Hematol 92, 425–431. [DOI] [PubMed] [Google Scholar]
- Hegde S, Lenox LE, Lariviere A, Porayette P, Perry JM, Yon M, and Paulson RF (2007). An intronic sequence mutated in flexed-tail mice regulates splicing of Smad5. Mamm. Genome 18, 852–860. [DOI] [PubMed] [Google Scholar]
- Hildick-Smith GJ, Cooney JD, Garone C, Kremer LS, Haack TB, Thon JN, Miyata N, Lieber DS, Calvo SE, Akman HO, et al. (2013). Macrocytic anemia and mitochondriopathy resulting from a defect in sideroflexin 4. Am. J. Hum. Genet 93, 906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvathova M, Ponka P, and Divoky V (2010). Molecular basis of hereditary iron homeostasis defects. Hematology 15, 96–111. [DOI] [PubMed] [Google Scholar]
- Hudson RC, and Daniel RM (1993). L-glutamate dehydrogenases: distribution, properties and mechanism. Comp. Biochem. Physiol. B 106, 767–792. [DOI] [PubMed] [Google Scholar]
- Hunt HR, Mixter R, and Permar D (1933). Flexed Tail in the Mouse, Mus Musculus. Genetics 18, 335–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton RL, and Boyer PD (1979). Subunit interaction during catalysis. Alternating site cooperativity of mitochondrial adenosine triphosphatase. J. Biol. Chem 254, 9990–9993. [PubMed] [Google Scholar]
- Jackson TD, Hock D, Palmer CS, Kang Y, Fujihara KM, Clemons NJ, Thorburn DR, Stroud DA, and Stojanovski D (2020). The TIM22 complex regulates mitochondrial one-carbon metabolism by mediating the import of Sideroflexins. bioRxiv 10.1101/2020.02.06.937920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káldi K, Bauer MF, Sirrenberg C, Neupert W, and Brunner M (1998). Biogenesis of Tim23 and Tim17, integral components of the TIM machinery for matrix-targeted preproteins. EMBO J 17, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Baker MJ, Liem M, Louber J, McKenzie M, Atukorala I, Ang CS, Keerthikumar S, Mathivanan S, and Stojanovski D (2016). Tim29 is a novel subunit of the human TIM22 translocase and is involved in complex assembly and stability. eLife 5, e17463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Stroud DA, Baker MJ, De Souza DP, Frazier AE, Liem M, Tull D, Mathivanan S, McConville MJ, Thorburn DR, et al. (2017). Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 67, 457–470. e5. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Khalimonchuk O, Smith PM, and Winge DR (2012). Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 1823, 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Jeong MY, Parnell TJ, Babst M, Phillips JD, and Winge DR (2016). The Plasma Membrane Protein Nce102 Implicated in Eisosome Formation Rescues a Heme Defect in Mitochondria. J. Biol. Chem 291, 17417–17426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M (2008). The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 1778, 1978–2021. [DOI] [PubMed] [Google Scholar]
- Koehler CM (2004). New developments in mitochondrial assembly. Annu. Rev. Cell Dev. Biol 20, 309–335. [DOI] [PubMed] [Google Scholar]
- Körver-Keularts IM, de Visser M, Bakker HD, Wanders RJ, Vansenne F, Scholte HR, Dorland L, Nicolaes GA, Spaapen LM, Smeets HJ, et al. (2015). Two Novel Mutations in the SLC25A4 Gene in a Patient with Mitochondrial Myopathy. JIMD Rep 22, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N, Wyant GA, Prakash G, Uit de Bos J, Bottanelli F, Pacold ME, Chan SH, Lewis CA, Wang T, Keys HR, et al. (2018). SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 362, eaat9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Nomura K, Katoh Y, Yamashita R, Kaneko K, and Furuyama K (2016). Novel Mechanisms for Heme-dependent Degradation of ALAS1 Protein as a Component of Negative Feedback Regulation of Heme Biosynthesis. J. Biol. Chem 291, 20516–20529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, and Maddocks OD (2014). Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep 7, 1248–1258. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kang MG, Park JS, Lee G, Ting AY, and Rhee HW (2016). APEX Fingerprinting Reveals the Subcellular Localization of Proteins of Interest. Cell Rep 15, 1837–1847. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kang MG, Shin S, Kwak C, Kwon T, Seo JK, Kim JS, and Rhee HW (2017). Architecture Mapping of the Inner Mitochondrial Membrane Proteome by Chemical Tools in Live Cells. J. Am. Chem. Soc 139, 3651–3662. [DOI] [PubMed] [Google Scholar]
- Lenox LE, Perry JM, and Paulson RF (2005). BMP4 and Madh5 regulate the erythroid response to acute anemia. Blood 105, 2741–2748. [DOI] [PubMed] [Google Scholar]
- Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, Vokes NI, Feist AM, Vander Heiden MG, and Metallo CM (2014). Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KH, Xie A, Rutter JC, Ahn YR, Lloyd-Cowden JM, Nichols AG, Soderquist RS, Koves TR, Muoio DM, MacIver NJ, et al. (2019). Systematic Dissection of the Metabolic-Apoptotic Interface in AML Reveals Heme Biosynthesis to Be a Regulator of Drug Sensitivity. Cell Metab 29, 1217–1231. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang X, Coyne LP, Yang Y, Qi Y, Middleton FA, and Chen XJ (2019). Mitochondrial carrier protein overloading and misfolding induce aggresomes and proteostatic adaptations in the cytosol. Mol. Biol. Cell 30, 1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YW, Galbraith L, Herndon JD, Lu YL, Pras-Raves M, Vervaart M, Van Kampen A, Luyf A, Koehler CM, McCaffery JM, et al. (2016). Defining functional classes of Barth syndrome mutation in humans. Hum. Mol. Genet 25, 1754–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YW, Acoba MG, Selvaraju K, Huang TC, Nirujogi RS, Sathe G, Pandey A, and Claypool SM (2017). Human adenine nucleotide translocases physically and functionally interact with respirasomes. Mol. Biol. Cell 28, 1489–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas S, Chen G, Aras S, and Wang J (2018). Serine catabolism is essential to maintain mitochondrial respiration in mammalian cells. Life Sci. Alliance 1, e201800036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytovchenko O, and Kunji ERS (2017). Expression and putative role of mitochondrial transport proteins in cancer. Biochim. Biophys. Acta Bioenerg 1858, 641–654. [DOI] [PubMed] [Google Scholar]
- Maio N, Kim KS, Singh A, and Rouault TA (2017). A Single Adaptable Co-chaperone-Scaffold Complex Delivers Nascent Iron-Sulfur Clusters to Mammalian Respiratory Chain Complexes I-III. Cell Metab 25, 945–953. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra D, Bragazzi Cunha J, Elenbaas JS, Bonkovsky HL, Shavit JA, and Omary MB (2019). Porphyrin-Induced Protein Oxidation and Aggregation as a Mechanism of Porphyria-Associated Cell Injury. Cell. Mol. Gastroenterol. Hepatol 8, 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaini KR, Sullivan MR, and Vander Heiden MG (2016). The importance of serine metabolism in cancer. J. Cell Biol 214, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr JA, Merkel O, Kohlwein SD, Gebhardt BR, Böhles H, Fötschl U, Koch J, Jaksch M, Lochmüller H, Horváth R, et al. (2007). Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am. J. Hum. Genet 80, 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlock AE, Shiferaw MT, Marcero JR, Vashisht AA, Wohlschlegel JA, Phillips JD, and Dailey HA (2015). Identification of the Mitochondrial Heme Metabolism Complex. PLoS One 10, e0135896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton DR, Nam M, McLaughlin DJ, Shin J, Bayraktar EC, Alvarez SW, Sviderskiy VO, Papagiannakopoulos T, Sabatini DM, Birsoy K, and Possemato R (2018). Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Mol. Cell 69, 610–621. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, and Chan DC (2016). Metabolic regulation of mitochondrial dynamics. J. Cell Biol 212, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. [DOI] [PubMed] [Google Scholar]
- Mixter R, and Hunt HR (1933). Anemia in the Flexed Tailed Mouse, Mus Musculus. Genetics 18, 367–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mon EE, Wei FY, Ahmad RNR, Yamamoto T, Moroishi T, and Tomizawa K (2019). Regulation of mitochondrial iron homeostasis by sideroflexin2. J. Physiol. Sci 69, 359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morscher RJ, Ducker GS, Li SH, Mayer JA, Gitai Z, Sperl W, and Rabinowitz JD (2018). Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamichi I, Habtezion A, Zhong B, Contag CH, Butcher EC, and Omary MB (2005). Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase-1 induction. J. Clin. Invest 115, 3007–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert W, and Herrmann JM (2007). Translocation of proteins into mitochondria. Annu. Rev. Biochem 76, 723–749. [DOI] [PubMed] [Google Scholar]
- Nikkanen J, Forsström S, Euro L, Paetau I, Kohnz RA, Wang L, Chilov D, Viinamäki J, Roivainen A, Marjamäki P, et al. (2016). Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab 23, 635–648. [DOI] [PubMed] [Google Scholar]
- Ogunbona OB, and Claypool SM (2019). Emerging Roles in the Biogenesis of Cytochrome c Oxidase for Members of the Mitochondrial Carrier Family. Front. Cell Dev. Biol 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F (2004). The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 447, 689–709. [DOI] [PubMed] [Google Scholar]
- Palmieri F, and Monné M (2016). Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 1863, 2362–2378. [DOI] [PubMed] [Google Scholar]
- Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, et al. (2005). Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet 14, 3079–3088. [DOI] [PubMed] [Google Scholar]
- Panneels V, Schüssler U, Costagliola S, and Sinning I (2003). Choline head groups stabilize the matrix loop regions of the ATP/ADP carrier ScAAC2. Biochem. Biophys. Res. Commun 300, 65–74. [DOI] [PubMed] [Google Scholar]
- Paul BT, Tesfay L, Winkler CR, Torti FM, and Torti SV (2019). Sideroflexin 4 affects Fe-S cluster biogenesis, iron metabolism, mitochondrial respiration and heme biosynthetic enzymes. Sci. Rep 9, 19634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, and Brandolin G (2003). Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44. [DOI] [PubMed] [Google Scholar]
- Radin NS, Rittenberg D, and Shemin D (1950). The rôle of glycine in the biosynthesis of heme. J. Biol. Chem 184, 745–753. [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehling P, Model K, Brandner K, Kovermann P, Sickmann A, Meyer HE, Kühlbrandt W, Wagner R, Truscott KN, and Pfanner N (2003). Protein insertion into the mitochondrial inner membrane by a twin-pore translocase. Science 299, 1747–1751. [DOI] [PubMed] [Google Scholar]
- Rehling P, Brandner K, and Pfanner N (2004). Mitochondrial import and the twin-pore translocase. Nat. Rev. Mol. Cell Biol 5, 519–530. [DOI] [PubMed] [Google Scholar]
- Reyes A, Melchionda L, Burlina A, Robinson AJ, Ghezzi D, and Zeviani M (2018). Mutations in TIMM50 compromise cell survival in OxPhos-dependent metabolic conditions. EMBO Mol. Med 10, e8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez E, Lobo T, Fox JL, Zeviani M, Winge DR, and Fernández-Vizarra E (2013). LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial Complex III assembly in human cells. Biochim. Biophys. Acta 1827, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F, and Shoubridge EA (2012). Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol. Biol 837, 207–217. [DOI] [PubMed] [Google Scholar]
- Sassa S (1976). Sequential induction of heme pathway enzymes during erythroid differentiation of mouse Friend leukemia virus-infected cells. J. Exp. Med 143, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa S, and Kappas A (1983). Hereditary tyrosinemia and the heme biosynthetic pathway. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J. Clin. Invest 71, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert EL, Ligeti E, Mayr JA, Sondheimer N, and Hajnóczky G (2015). The mitochondrial phosphate carrier: Role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun 464, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty T, Sishtla K, Park B, Repass MJ, and Corson TW (2019). Heme synthesis inhibition blocks angiogenesis via mitochondrial dysfunction. bioRxiv 10.1016/j.isci.2020.101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signes A, and Fernandez-Vizarra E (2018). Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem 62, 255–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair PR, Gorman N, and Jacobs JM (2001). Measurement of heme concentration. Curr. Protoc. Toxicol Chapter 8, Unit 8.3. [DOI] [PubMed] [Google Scholar]
- Spinelli JB, and Haigis MC (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol 20, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D, Bohnert M, Pfanner N, and van der Laan M (2012). Mechanisms of protein sorting in mitochondria. Cold Spring Harb. Perspect. Biol 4, a011320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, and Finkel T (2016). The Mitochondrial Basis of Aging. Mol. Cell 61, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB (2017). Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol 27, 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson K, Majd H, Dallabona C, Reinson K, King MS, Alston CL, He L, Lodi T, Jones SA, Fattal-Valevski A, et al. (2016). Recurrent De Novo Dominant Mutations in SLC25A4 Cause Severe Early-Onset Mitochondrial Disease and Loss of Mitochondrial DNA Copy Number. Am. J. Hum. Genet 99, 860–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschudy DP, Hess RA, and Frykholm BC (1981). Inhibition of delta-aminolevulinic acid dehydrase by 4,6-dioxoheptanoic acid. J. Biol. Chem 256, 9915–9923. [PubMed] [Google Scholar]
- Tzagoloff A, Akai A, and Needleman RB (1975). Assembly of the mitochondrial membrane system: isolation of nuclear and cytoplasmic mutants of Saccharomyces cerevisiae with specific defects in mitochondrial functions. Journal of Bacteriology 122, 826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, and Mootha VK (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. [DOI] [PubMed] [Google Scholar]
- Van Vranken JG, Nowinski SM, Clowers KJ, Jeong MY, Ouyang Y, Berg JA, Gygi JP, Gygi SP, Winge DR, and Rutter J (2018). ACP Acylation Is an Acetyl-CoA-Dependent Modification Required for Electron Transport Chain Assembly. Mol. Cell 71, 567–580. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekeere S, Dubois C, Kalucka J, Sullivan MR, García-Caballero M, Goveia J, Chen R, Diehl FF, Bar-Lev L, Souffreau J, et al. (2018). Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab 28, 573–587. e13. [DOI] [PubMed] [Google Scholar]
- Vukotic M, Nolte H, König T, Saita S, Ananjew M, Krüger M, Tatsuta T, and Langer T (2017). Acylglycerol Kinase Mutated in Sengers Syndrome Is a Subunit of the TIM22 Protein Translocase in Mitochondria. Mol. Cell 67, 471–483. e7. [DOI] [PubMed] [Google Scholar]
- Wang JY, Lee YT, Chang PF, and Chau LY (2009). Hemin promotes proliferation and differentiation of endothelial progenitor cells via activation of AKT and ERK. J. Cell. Physiol 219, 617–625. [DOI] [PubMed] [Google Scholar]
- Wang Y, Branicky R, Noë A, and Hekimi S (2018). Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol 217, 1915–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlrab H, and Flowers N (1982). pH gradient-dependent phosphate transport catalyzed by the purified mitochondrial phosphate transport protein. J. Biol. Chem 257, 28–31. [PubMed] [Google Scholar]
- Wu G, Fang YZ, Yang S, Lupton JR, and Turner ND (2004). Glutathione metabolism and its implications for health. J. Nutr 134, 489–492. [DOI] [PubMed] [Google Scholar]
- Wu X, Renuse S, Sahasrabuddhe NA, Zahari MS, Chaerkady R, Kim MS, Nirujogi RS, Mohseni M, Kumar P, Raju R, et al. (2014). Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat. Commun 5, 4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RZ, Blaileanu G, Hansen BC, Shuldiner AR, and Gong DW (2002). cDNA cloning, genomic structure, chromosomal mapping, and functional expression of a novel human alanine aminotransferase. Genomics 79, 445–450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the SILAC-based quantitative proteomics data reported in this paper and deposited to the ProteomeXchange Consortium is PRIDE: PXD019826 (http://proteomecentral.proteomexchange.org/cgi/GetDataset).