

Supplemental Digital Content is available in the text.
Keywords: congenital heart defects, haploinsufficiency, induced pluripotent stem cells, proteins, proteomics, ribosomes
Abstract
Rationale:
NAA15 (N-alpha-acetyltransferase 15) is a component of the NatA (N-terminal acetyltransferase complex). The mechanism by which NAA15 haploinsufficiency causes congenital heart disease remains unknown. To better understand molecular processes by which NAA15 haploinsufficiency perturbs cardiac development, we introduced NAA15 variants into human induced pluripotent stem cells (iPSCs) and assessed the consequences of these mutations on RNA and protein expression.
Objective:
We aim to understand the role of NAA15 haploinsufficiency in cardiac development by investigating proteomic effects on NatA complex activity and identifying proteins dependent upon a full amount of NAA15.
Methods and Results:
We introduced heterozygous loss of function, compound heterozygous, and missense residues (R276W) in iPSCs using CRISPR/Cas9. Haploinsufficient NAA15 iPSCs differentiate into cardiomyocytes, unlike NAA15-null iPSCs, presumably due to altered composition of NatA. Mass spectrometry analyses reveal ≈80% of identified iPSC NatA targeted proteins displayed partial or complete N-terminal acetylation. Between null and haploinsufficient NAA15 cells, N-terminal acetylation levels of 32 and 9 NatA-specific targeted proteins were reduced, respectively. Similar acetylation loss in few proteins occurred in NAA15 R276W induced pluripotent stem cells. In addition, steady-state protein levels of 562 proteins were altered in both null and haploinsufficient NAA15 cells; 18 were ribosomal-associated proteins. At least 4 proteins were encoded by genes known to cause autosomal dominant congenital heart disease.
Conclusions:
These studies define a set of human proteins that requires a full NAA15 complement for normal synthesis and development. A 50% reduction in the amount of NAA15 alters levels of at least 562 proteins and N-terminal acetylation of only 9 proteins. One or more modulated proteins are likely responsible for NAA15-haploinsufficiency mediated congenital heart disease. Additionally, genetically engineered induced pluripotent stem cells provide a platform for evaluating the consequences of amino acid sequence variants of unknown significance on NAA15 function.
Editorial, see p 1170
In This Issue, see p 1119
Meet the First Author, see p 1120
Congenital heart disease (CHD), which affects about 1% of newborns, reflects defects in the developmental program of the heart.1,2 Defining the genetic causes of CHD will provide new insights into mechanisms of cardiac development that may eventually benefit patients with CHD and their families. Whole exome sequencingof CHD probands and their parents have enabled the identification of recurrent damaging variants in multiple genes that likely are critical for normal cardiac development.3 Functional studies are necessary to further define these gene functions and the pathogenetic mechanisms of damaging variants. While loss of function (LoF) variants infer that haploinsufficiency of the encoded protein contributes to CHD, the consequence of missense variants on protein function are less readily interpreted, often leading to classification of these as variants of unknown significance although some may contribute to the CHD.2
We previously reported 2 patients with CHD with de novo heterozygous LoF variants in NAA15 (N-alpha-acetyltransferase 15), which encodes a protein subunit of the NAT (N-terminal [Nt] acetyltransferase) complex.2–4 In addition to CHD, these patients had extra-cardiac disorders including neurodevelopmental deficits.2,3 Prior studies have reported damaging NAA15 variants in patients with other congenital malformations and neurodevelopmental abnormalities.5–7
Acetylation of the N-terminus of proteins is a prevalent modification that occurs in ≈85% of yeast and human proteins.8,9 The effect of Nt-acetylation on proteins is diverse and includes changes to protein stability, complex formation, protein folding, and aggregation.10 The NatA complex, one of 8 NAT types, is essential in most, if not all, eukaryotes and is responsible for the majority of Nt-acetylation.10,11 This complex binds the ribosome and was shown to acetylate nascent polypeptide chains at specific Nt amino acids (Ser-, Thr-, Ala-, Val-, Gly-, and Cys-) after the initiating methionine is removed.8,10,12,13 The NatA complex is formed by the catalytic subunit NAA10 and the auxiliary unit NAA15. HYPK (Huntingtin interacting protein K) is a chaperone protein that attaches to the NatA complex along with subunit NAA50.14–17 NAA50 also forms part of the NatE complex, which displays a distinct substrate specificity compared with NatA.10,18 NAA15 is a subunit of both the NatA and NatE complexes, and its role is to position the catalytic subunits in close vicinity to the nascent polypeptides; in the case of NAA10, it also modulates its substrate specificity.10,19–21 Additionally, NAA15 interaction with NAA10 and HYPK has been implicated in regulation of protein folding and Nt-acetylation fidelity.15,22 Abnormal NatA complex function has been previously associated with human cancers and neurological disorders.5,23,24 To date, the relationship between Nt-acetylation, NAA15, and CHD has not been investigated.
We studied human isogenic induced pluripotent stem cell (iPSC) lines that were engineered to contain NAA15 variants identified in patients with CHD and predicted to be damaging. We evaluated whether iPSCs with NAA15 variants differentiated into cardiomyocytes. By assessing both Nt-acetylation and protein levels by mass spectrometry (MS), we demonstrate that NAA15 haploinsufficiency perturbs normal function of undifferentiated iPSCs. We identify proteins that require the full complement of NAA15 to preserve the integrity of these stem cells for cardiac development.
Methods
Data Availability
All data and materials have been made publicly available. Further details are provided in the Major Resources Table located in the Data Supplement.
Study Cohort With CHD
CHD subjects (n=4511) were recruited to the Congenital Heart Disease Network Study of the Pediatric Cardiac Genomics Consortium (CHD GENES: URL: https://www.clinicaltrials.gov; Unique identifier: NCT01196182) or the DNA biorepository of the Single Ventricle Reconstruction trial after approval from Institutional Review Boards as previously described.1,25,26 All subjects or their parents provided informed consent. Clinical diagnoses, including cardiac and noncardiac congenital anomalies, were obtained from review of patient charts and family interview.
CRISPR Gene Editing and Mutation Confirmation
Isogenic personal genome project 1 iPSCs were modified using CRISPR/Cas9 technology to create NAA15 LoF or missense mutation cell lines.27 Further details provided in Methods in the Data Supplement.
Label-Free Quantitative Shotgun Proteomics and Data Analysis
Two independent iPSC lines of both wild type (WT) cells and NAA15 mutant cells were grow in 10 cm petri dishes until near confluency. iPSCs were detached and using Accutase (Millipore) and collected in 15 cm centrifuge tubes. Cells were centrifuged (Beckman) in a 15 mL tube at 1000 rpm for 5 minutes. Cells were washed with PBS and pelleted by centrifugation. Eight samples of each genotype was collected, and a total of 24 samples were prepared for LC-MS/MS/ analysis. Further details provided in Methods in the Data Supplement.
The MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifiers PXD017672 and PXD018013.
Bioinformatic Analysis and Gene Ontology Analysis
Gene ontology annotation proteome was derived using the R package clusterProfiler.28 Proteins were classified by Gene ontology annotation based on biological process, molecular function, and cellular component. Quantified proteins detected by MS of iPSCs were used as a background and other parameters with default. The P values were adjusted by Bonferroni correction for multiple testing.
Statistics
Single comparisons were analyzed by using the Student t test, with significance defined as P<0.05. Multiple comparisons between genotypes were analyzed using 1-way ANOVA with post hoc Tukey HSD, with significance defined as P<0.05. For proteomics, pairwise SAM t tests (or pairwise statistical testing using the SAM method29) were performed and significant hits determined using as cutoff values a permutation based false discovery rate (FDR) of 0.01 (1000 permutations) and a background variance parameter s0 of 1. For all experiments analyzed for statistical significance, only within-test corrections were made.
Results
NAA15 Variants Are Associated With CHD and Other Extra Cardiac Anomalies
Whole exome sequencing of 4511 patients with CHD 1,2,4 identified 4 subjects with a rare LoF variant (allele frequency <0.00005) in the NAA15 gene, resulting in NAA15 haploinsufficiency (Figure 1A). Parental analyses indicated that 3 of these LoF variants (p.Ser761*, p.Lys336Lys fs*6, and p.Arg470*) arose de novo in the probands.2,4 The inheritance of the p.Ala718fs variant is uncertain, as parental samples were unavailable. Among ≈125 000 subjects in the gnomAD database,12 14 NAA15 LoF variants are reported, inferring an 8.9-fold higher frequency of NAA15 LoF variants in CHD probands (P=0.002).
Figure 1.
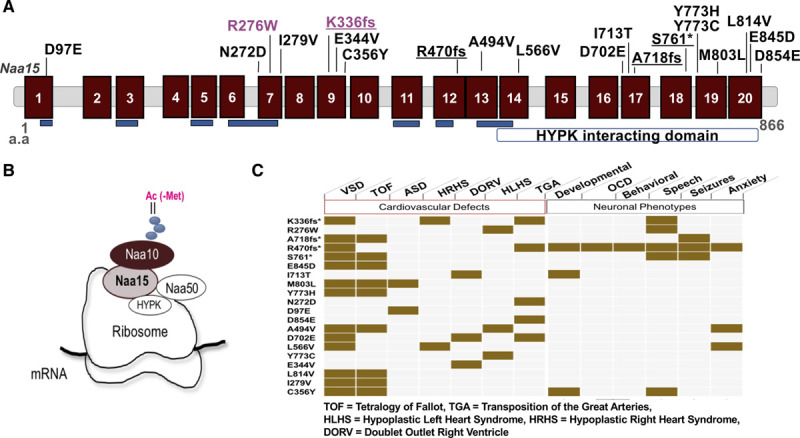
NAA15 variants discovered in patients with congenital heart disease patients. A, Schematic diagram of the NAA15 (N-alpha-acetyltransferase 15) gene. The NAA15 gene consists of tetracopeptide repeats (blue) essential for interaction with the NatA (N-terminal acetyltransferase) complex subunit NAA10 and a HYPK (Huntingtin interacting protein K) interacting domain at the C terminus. Location of variants identified in patients with congenital heart disease (CHD; loss of function [LoF] variants are underlined) and CRISPR/Cas9-derived variants in induced pluripotent stem cell (iPSCs; magenta) are shown. B, The subunit composition of NatA and NatE complexes. NAA15 is the auxiliary unit for both complexes. C, Table of cardiovascular and neuronal clinical phenotypes of patients with CHD. *indicates LoF variants.
The CHD phenotypes included tetralogy of Fallot, heterotaxy with d-looped ventricles, transposition of the great arteries, and hypoplastic left or right heart syndrome (Figure 1C and Table I in the Data Supplement). In addition to cardiac anomalies, all 4 patients with CHD with a NAA15 LoF variant had extracardiac anomalies including seizures, neurobehavioral, ophthalmologic, auditory, or orthopedic disorders (Figure 1C and Table I in the Data Supplement).5
We also identified 15 very rare (allele frequency <1.0×10−5e-5 or absent from the gnomAD database12) inherited NAA15 missense variants among these 4511 patients with CHD (Figure IA and Table I in the Data Supplement). In addition, one missense variant, R276W (Figure 1A and Figure IA and IB in the Data Supplement), could not be assessed for inheritance. The frequency of rare missense alleles in the Pediatric Cardiac Genomics Consortium (PCGC) cohort was 0.0035 (n=16 of 4511 CHD probands), significantly higher than the frequency observed among ≈115 000 Gnomad subjects (frequency, 0.002; n=198; P=0.02; odds ratio, 1.8). CHD probands with rare NAA15 missense variants had notably fewer extracardiac anomalies than CHD probands with NAA15 LoF variants (Table I in the Data Supplement). Hence, despite the observation that these very rare NAA15 missense variants were transmitted from an unaffected parent, we suspected that some of them contribute to CHD.
Genetically Engineered iPSCs Model NAA15 Haploinsufficiency
We introduced NAA15 variants into the iPSC line, personal genome project 1, using CRISPR/Cas9 gene editing (see Methods in the Data Supplement) to create human iPSCs with reduced or no NAA15 protein. Two independent cell lines were constructed with each genotype NAA15−/−, NAA15+/−, and NAA15+/R276W (Figure 1A, Figure IA through IC and Table II in the Data Supplement) and studied as detailed below. NAA15 variants in iPSCs were confirmed both by Sanger sequencing and next generation sequencing of PCR amplified products (Figure IIA through IIC in the Data Supplement).
RNA expression levels for each of the 2 independent iPSC lines in NAA15+/+ and NAA15-mutant iPSCs, were first characterized by RNAseq. NAA15 mRNA levels were reduced by 56% and 24% in the NAA15+/− and NAA15+/R276W iPSCs, respectively (Figure 2A and Table III in the Data Supplement). There was a 99% decrease in NAA15 mRNA in NAA15−/− iPSCs, suggesting that these variants triggered nonsense-mediated decay (Figure 2A). We performed MS-based shotgun proteomics in at least 3 replicates for each of the 2 independent lines. NAA15 protein was near-normal levels in NAA15+/− iPSCs but absent from NAA15−/− iPSCs (Figure 2B). The amount of NAA15 protein detected by MS in NAA15+/− iPSCs could represent a proportion of truncated NAA15 polypeptides. To confirm this hypothesis, NAA15 protein levels were measured in 2 independent biological replicates of both WT and NAA15 mutant iPSCs by Western blotting (Figure IID and IIE in the Data Supplement), which supported this conclusion. NAA15 protein was reduced in NAA15+/− iPSCs by about 50%, and full length NAA15 protein was not detectable in NAA15−/− iPSCs (Figure IID and IIE in the Data Supplement).
Figure 2.
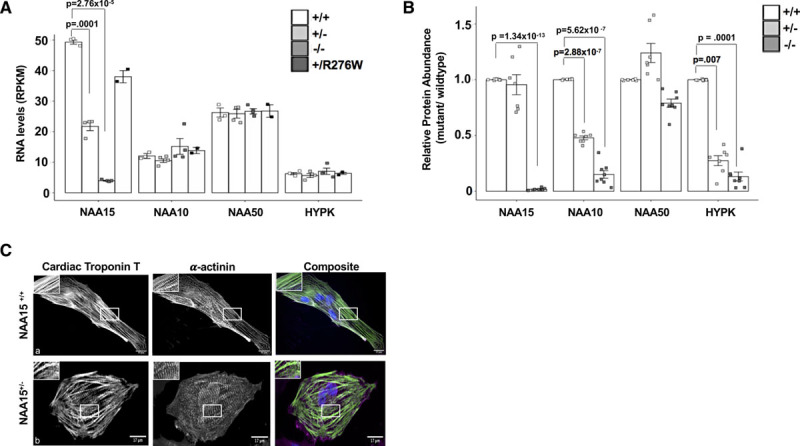
NAA15+/− and NAA15+/R276W induced pluripotent stem cells (iPSCs) develop into cardiomyocytes, while NAA15 (N-alpha-acetyltransferase 15) null iPSCs have reduced viability. A, Graph shows RNA expression of the NatA (N-terminal acetyltransferase) components in NAA15-mutant iPSCs. NAA15 RNA levels are significantly lowered in iPS cells with NAA15+/− and NAA15−/− variants compared with wildtype iPSCs. Data was collected from 2 independent cell lines for each genotype and as technical replicates for selected lines. Total cell lines analyzed: NAA15+/+ (n=3), NAA15−/− (n=4), NAA15+/− (n=4), and NAA15+/R276W (n=2). Significance of differences between NAA15 mutant iPSCs and wildtype iPSCs were evaluated by Student t tests, P<0.05 (P values were adjusted by Bonferroni correction; only significant adjusted P values are displayed). All data points are presented and plotted as mean±SEM. B, Graph represents fold change ratios of relative protein levels in NAA15 mutant iPSCs compared with wildtype iPSCs for Nat complex subunits. Relative protein abundance was quantified by MaxLFQ algorithms integrated in the MaxQuant software. Data was collected from two independent undifferentiated iPS cell lines for each NAA15 mutant genotype and as technical replicates for each cell line. Total cell lines analyzed: NAA15+/− (n=8), NAA15−/− (n=8), NAA15+/− (n=7), and NAA15+/R276W (n=8). Significant changes indicated by P values were calculated using pairwise comparison SAM t test method and a permutation based false discovery rate (FDR) of 0.01 (1000 permutations) as cutoff values with a background variance parameter s0 of 1. All data points are presented and plotted as mean±SEM. C, Representative images of NAA15+/+ iPSCs and NAA15+/− iPSCs stained with cardiac troponin T antibody (green), α-actinin (magenta), and DAPI for nuclei (blue). A representative image for each cell type is presented. Normal sarcomeres were observed in both NAA15 mutant and wildtype iPSC-CMs. Magnification, 60×; scale bar 25um.
Because NAA15 is normally associated with components NAA10, HYPK, and NAA5015,17,18 (Figure 1B), we assessed the levels of these proteins in the mutant iPSCs using MS. Both NAA15−/− and NAA15+/− iPSCs showed decreased protein levels of NAA10 (Figure 2B). NAA50, the catalytic unit of NatE and binding partner of NAA15 protein, was reduced in NAA15−/− iPSCs. Significant reduction of the NAA50 protein did not occur in NAA15+/− iPSCs (Figure 2B). Noticeably, HYPK, a NAA15 binding partner and subunit of the NatA complex was significantly reduced in both NAA15+/− and NAA15−/− iPSCs (Figure 2B).
We further explored the functional effects of NAA15+/− and NAA15+/R276W using a yeast assay in which the hNatA (human NatA complex) functionally replaced yeast NatA, as shown by complementation of growth phenotypes with partial rescue of the NatA-specific Nt-acetylome.8 HsNatA D335fs and S761* failed to rescue the temperature-sensitive growth phenotype of yNatAΔ (Figure IIIA through IIIC in the Data Supplement), suggesting that NAA15+/− results in impaired NatA functionality. However, in this yeast assay, human HsNatA R276W rescued yeast growth suggesting at least partial NatA function in yeast (Figure IIIC in the Data Supplement). Noticeably, Schizosaccharomyces pombe NAA15 has a tryptophan residue at position 276 while all mammals have an arginine residue, which suggests that this residue is functionally important in human cells. As such, we suggest that rescue of yeast growth experiments might not fully provide functional assessments of mutant human NAA15 proteins.
NAA15+/− and NAA15+/R276W iPSCs Develop Into Contractile Cardiomyocytes, While NAA15−/− iPSCs Have Reduced Viability
To test whether NAA15 variants have an effect on iPSC maturation, 2 independent iPSC lines for each genotype (NAA15−/−, NAA15+/−, and NAA15+/R276W) were differentiated into cardiomyocytes using a 13-day differentiation protocol to assess for the development and contractility of sarcomeres.30,31 Both NAA15+/− and NAA15+/R276W cells differentiated into cardiomyocytes (Figure 2C, a and b), however, NAA15−/− iPSCs grew slowly (data not shown) and failed to differentiate. Cell viability of NAA15-mutant iPSCs was assessed (see Methods in the Data Supplement). Similar to observations reported in NAA15/NatA yeast knockout and human knockdown cells,19,23,32–34 NAA15−/− iPSCs had significantly retarded growth and cell death (P=0.001; average cell death is 12% in NAA15+/+ iPSCs versus 32% in NAA15−/− iPSCs, n=2). NAA15+/− iPSC-derived cardiomyocytes (iPSC-CMs) were stained with cardiac troponin T and actinin antibodies to visualize sarcomere structures (Figure 2C, a and b). NAA15 mutant cell sarcomeres were indistinguishable from WT cell sarcomeres (Figure 2C, a and b and Figure IV and Table IV in the Data Supplement).
Contractility of NAA15+/− and NAA15+/R276W iPSC-CMs was monitored by live image analysis (Movies I and II in the Data Supplement). Two biological replicates of NAA15 mutant iPSC-CMs and one of 2 biological replicates for WT iPSC-CMs were incubated with GFP (green fluorescent protein)-actinin lentivirus to enable high fidelity tracking of sarcomere function35(Movies III through V in the Data Supplement). The lengths of GFP-labeled sarcomeres were measured during the contractile cycle. Contractile measurements of unloaded (ie, sarcomeres that were not working against resistance) demonstrated no difference in rates of sarcomere shortening or contraction between mutant and WT cells (Figure VA in the Data Supplement). To better recapitulate native cardiomyocyte architecture and mechanics, the function of loaded sarcomeres from these cells grown as 3-dimensional micro-tissue structures,36,37 where sarcomeres work against resistance, were created from WT iPSC-CMs and 2 biological replicates of NAA15 mutant iPSC-CMs (Movies VI through VIII in the Data Supplement). In contrast to unloaded 2-dimensional tissue sarcomere function, image analysis show loaded NAA15+/− iPSC-CM sarcomeres exhibited a smaller percent contraction than WT iPSC-CM sarcomeres (Figure VB in the Data Supplement).
Nt-acetylation in NAA15 Haploinsufficient, Null, and R276W Missense iPSCs
We studied Nt-acetylation of 2 biological replicates in iPSCs with NAA15 variants and WT using a MS-based Nt enrichment assay.8,38–40 NatA acetylates N-termini containing Ser-, Thr-, Ala-, Val-, Gly-, or Cys.10,12 We identified a total of 989 previously annotated N-termini41 in one or both of the 2 independent cell lines for WT and NAA15 mutant iPSCs (Table V in the Data Supplement); ≈650 N-termini peptides were identified in each cell line (Tables V and VI in the Data Supplement). NatA targeted sequences represented ≈60% of the 989 detected N-termini peptides (Tables V and VI in the Data Supplement); no Cys-starting peptides were detected. In NAA15+/+ iPSCs, we observed 17 Nt peptides that were partially acetylated (10%–95%) and 359 with complete (>95%) Nt-acetylation (Figure 3A and Tables V and VI in the Data Supplement). Of the completely or partially Nt-acetylated peptides,8,10 98% have an alanine, threonine, or serine at position 2 (Figure 3B and Tables VI and VII in the Data Supplement). However, peptides with partial acetylation have a much higher fraction of Nt glycine or valine residues (Figure 3C and Tables VI and VII in the Data Supplement). The preference for acetylation of proteins with Nt alanine and serine residues was preserved in NAA15-mutant iPSCs as observed in previous studies (Tables VI and VII in the Data Supplement).23
Figure 3.
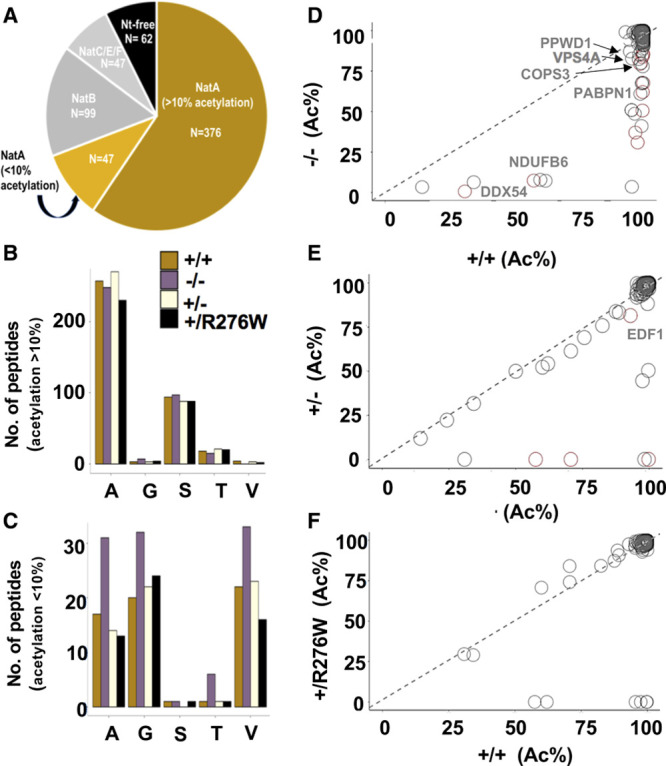
N-terminal acetylation in NAA15+/− and NAA15−/− induced pluripotent stem cells (iPSCs). A, N-terminal peptides acetylated in NAA15+/+ iPSCs are categorized by NAT-type substrate specificity. NatA (N-terminal acetyltransferase) targets are displayed according to percent Nt-acetylation; Nt-acetylation >10% highlighted in gold and Nt-acetylation <10% highlighted in light gold. B and C, The number of Nt-acetylated NatA-type N-terminal peptides represented by residues A, G, S, T, or V in NAA15 mutant and wildtype iPSCs. Data is displayed as the absolute number of peptides measured from the sum of 2 biological replicates for each genotype. Alanine and serine residues are preferentially acetylated in peptides with >10% Nt-acetylation; glycine and valine residues are representative of N-termini with <10% Nt-acetylation. Primary peptide summary provided in Table V in the Data Supplement. There were no statistically significant differences between genotypes (hypergeometric test; data not shown). D–F, Representative scatter plots display the correlation of the degrees of Nt-acetylation of all N-termini identified as NatA targets in NAA15−/−, NAA15+/−, NAA15+/R276W, and NAA15+/+ iPSCs. Each plot compares NAA15 (N-alpha-acetyltransferase 15) mutant iPSCs to wildtype iPSCs. Differentially acetylated N-termini are found below the line of identity (y intercept=0, slope=1). Peptides lose acetylation moieties in NAA15-mutant iPSCs. NAA15−/− iPSCs have the largest number of peptides (n=32) with reduced Nt-acetylation. There are N-termini with reduced N-terminal acetylation in NAA15+/− iPSCs (n=9) and NAA15+/R276W iPSCs (n=8). Decreased protein expression changes are highlighted in red. COPS3 indicates COP9 signalosome complex subunit 3; DDX54, ATP-dependent RNA helicase; NDUFB6, NADH dehydrogenase (ubiquinone) 1β subcomplex subunit 6; PABPN1, polyadenylate-binding protein 2; PPWD1, peptidyl-prolyl isomerase domain and WD repeat-containing protein 1; and VPS4A, vacuolar protein sorting-associated protein 4.
Protein Nt-acetylation in NAA15 mutant iPSCs was compared with Nt-acetylation in WT iPSCs. Only a limited number of putative NatA-type N-termini substrates had >10% difference in acetylation between wild type and mutant iPSCs (Figure 3, Table 1, and Figure VI in the Data Supplement). In NAA15−/− iPSCs 32 proteins showed these changes (Figure 3B through 3D and Table 1). Notably, all proteins that had partial acetylation in WT iPSCs lacked Nt-acetylation in NAA15−/−. Nine of these proteins were altered in NAA15+/− iPSCs and 8 had altered Nt-acetylation in NAA15+/R276W iPSCs (Figure 3E and 3F, Table 1).
Table 1.
Proteins With Altered N-Terminal Acetylation in Wildtype and NAA15 Mutant iPSCs*
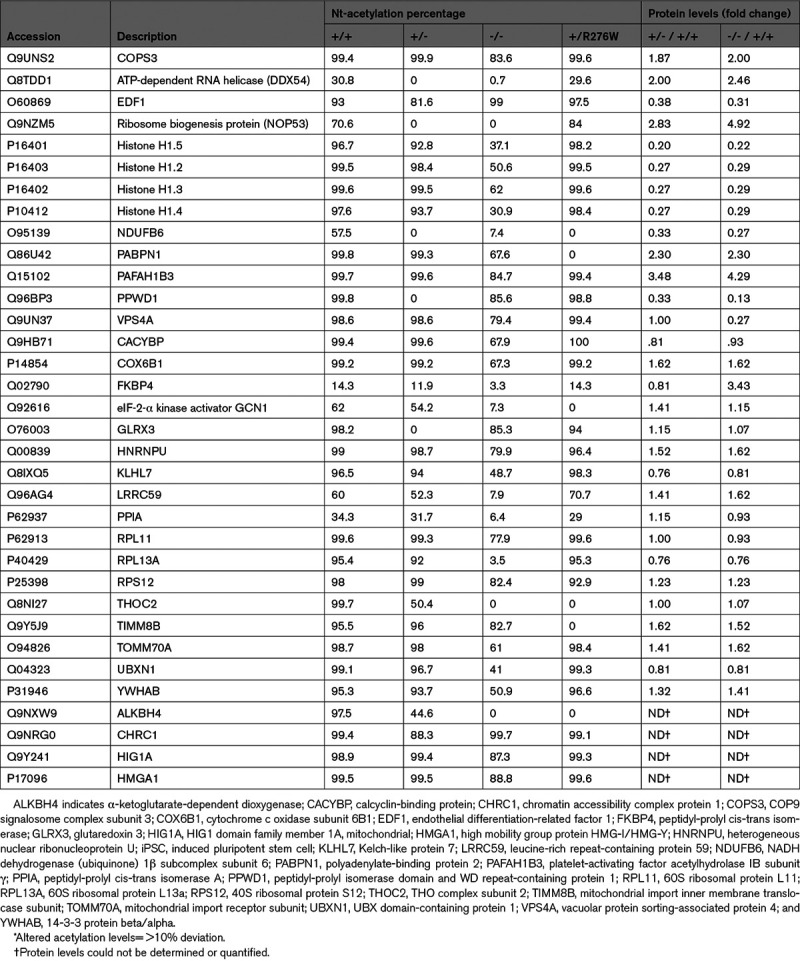
All proteins with reduced acetylation in mutant iPSCs displayed NatA-type substrate specificity (Tables 1 and 2, Table V in the Data Supplement). Greater than 50% of proteins with altered Nt-acetylation had an alanine residue at position 2 and >30% also had an alanine residue at position 3 (Table 2).23 The distribution of N-terminal residues in proteins with altered Nt-acetylation did not differ significantly from the distribution of N-terminal residues in proteins with unchanged N-terminal acetylation in these cells (not shown). We deduced that NAA15 variants or deficiency altered N-terminal acetylation in a small number of NatA substrate proteins and with no preference for an extended N-terminal amino acid substrate specificity.
Table 2.
Second and Third Amino Acid Identities of Peptides With Altered Nt-Acetylation in NAA15 Mutant iPSCs
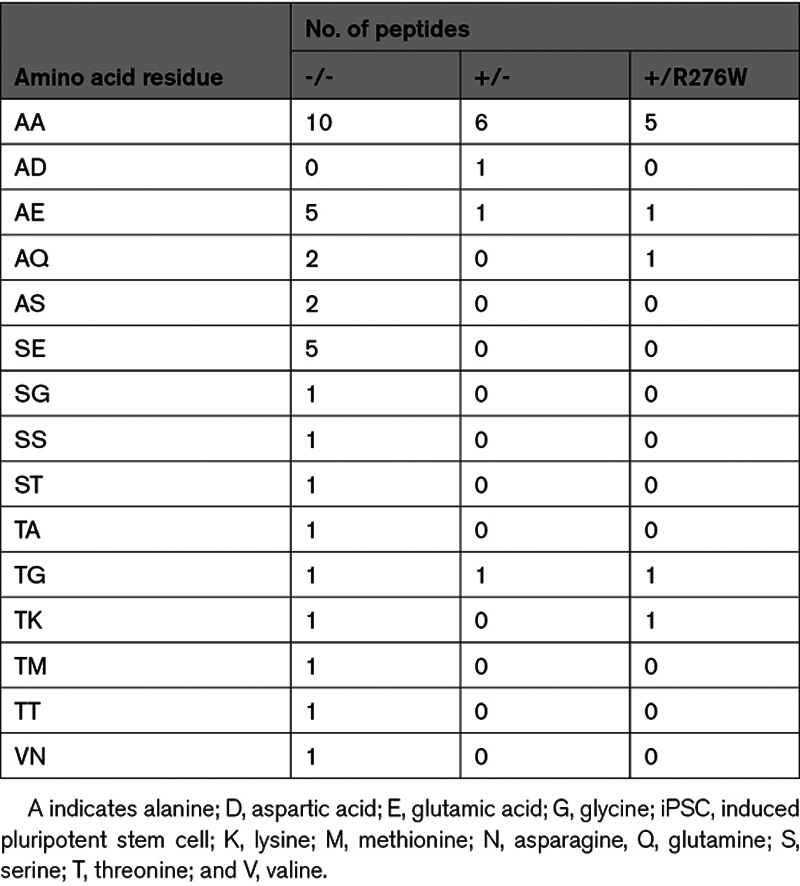
We noticed that some proteins reduced in Nt-acetylation (Figure 3D) have functions in cellular proliferation and survival,42–46 perhaps accounting for the deficits in growth and viability of NAA15−/− iPSC.41–46 To understand whether the change in Nt-acetylation status compromised the function of these proteins and contributed to the phenotype of NAA15−/− iPSCs, we used a combination of shotgun MS-based proteomics and pairwise comparison by SAM t test (FDR=0.01; Tables VIII through X in the Data Supplement). Nine proteins with reduced acetylation correlated with changes in protein expression in NAA15−/− iPSCs (Figure 3D and 4A). Among these, measurable acetylation occurred in 3 proteins in NAA15+/− and in one protein in NAA15+/R276W iPSCs (Figure 3E and 3F).
Figure 4.
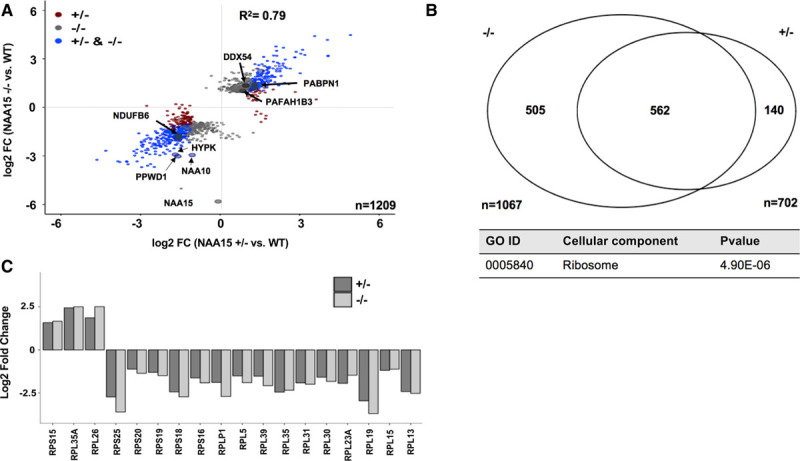
Proteins differentially expressed in NAA15+/− and NAA15−/− induced pluripotent stem cells (iPSCs). A, Scatterplot displays log2 fold changes of differentially expressed proteins in NAA15+/− and NAA15−/− iPSCs compared with wildtype (WT) iPSCs. Approximately 1200 proteins are differentially expressed, 9 and 3 proteins of which have reduced Nt-acetylation in NAA15−/− iPSCs and NAA15+/− iPSCs, respectively. Data was collected from 2 independent undifferentiated iPS cell lines for each NAA15 (N-alpha-acetyltransferase 15) mutant genotype and at least 3 technical replicates for each sample. B, Comparison of differentially expressed proteins in NAA15−/− and NAA15+/− iPSCs. Differential expression was considered for log2 fold change |1|. Identified proteins: 5196, quantified proteins: 3911 proteins detected by mass spectrometry (C) gene ontology (GO) enrichment of differentially expressed proteins in both NAA15−/− and NAA15+/− iPSCs. A total of 41 proteins are localized to the ribosome. Raw P value presented in data table. D, Bar graph represents log2 fold change of ribosomal proteins that are differentially expressed in both NAA15−/− and NAA15+/− iPSCs.
Altered Protein Expression Due to NAA15 Haploinsufficiency or Deficiency
Using shotgun proteomics, we studied 2 independent undifferentiated iPSC lines for each NAA15 mutant genotype and at least 3 technical replicates for each sample to assess protein levels (Table VIII in the Data Supplement). Among 5196 proteins identified by MS, extracts from NAA15+/- iPSCs, NAA15-/- iPSCs, or both revealed a total of 1209 proteins that were differentially expressed (pairwise comparison by t test) compared with WT iPSCs (Figure 4A and 4B, Figure VII and Tables VIII through X in the Data Supplement). Five hundred sixty-two proteins were differentially expressed in both NAA15+/− and NAA15−/− iPSCs compared with WT iPSCs and 505 proteins in NAA15−/− iPSCs but not NAA15+/− iPSCs (Figure 4B, Figure VII and Tables VIII through X in the Data Supplement). More than 60% of differentially expressed proteins had a NatA-target residue at position 2 of the mature protein (Table XI in the Data Supplement). NatA-target residues, alanine and serine, were frequently observed at position 2. Similar frequencies are seen in proteins with no expression changes (Table XI in the Data Supplement). Despite the large numbers of differentially expressed proteins, there were very few, if any, significant differences in mRNA levels, as assessed by RNAseq analysis (Tables III and IV in the Data Supplement).
Among differentially expressed proteins identified in NAA15+/− iPSCs and NAA15−/− iPSCs, the levels of HYPK, a component of the NatA complex that interacts with NAA15, were significantly reduced (Figure 1C).14–17,22 A total of 29 out of 54 known interactors of HYPK were detected in the WT iPSCs.47 Although HYPK was decreased in MS measurements, there was no enrichment of HYPK interacting proteins that were differentially expressed. Only 6 of 29 known HYPK interacting proteins were decreased in NAA15-mutant iPSCs (P=0.3; Tables IX and X in the Data Supplement).
To test the hypothesis that differentially expressed proteins shared a common function, the 562 proteins differentially expressed in both NAA15+/− and NAA15−/− iPSCs were analyzed by gene ontology analysis using clusterProfiler (implemented in R; Methods in the Data Supplement).28 Of the 3 gene ontology enrichment processes that were performed, we found no enrichment after Bonferroni correction for proteins with shared biological function or molecular processes; however, 41 ribosomal (-associated) proteins with altered expression levels were observed (P=5×10−6), Table XII in the Data Supplement, Figure 4C). These proteins included the catalytic subunit of the NatA complex, NAA10, and ribosomal proteins of the 60S large and 40S small ribosome subunits that function in protein synthesis (Figure 1B, Table XII in the Data Supplement, and Figure 4D and 4E).48–52
Ribosomal Protein Deficiency Due to NAA15 Haploinsufficiency
Twenty-nine ribosomal proteins of the 84 proteins that comprise 60S or 40S ribosomal subunit53,54 were affected in NAA15+/− and NAA15−/− iPSCs; eleven 60S large ribosomal proteins and 6 small ribosomal proteins were affected in both NAA15+/− and NAA15−/− iPSCs (Tables IX and X in the Data Supplement, Figure 4D). Twelve ribosomal proteins were differentially expressed in NAA15−/− iPSCs but not in NAA15+/− iPSCs (Tables IX and X in the Data Supplement).
NAA15 facilitates binding of the NatA complex to the ribosome.55–58The general docking site for NatA resides at the ribosomal exit tunnel near large ribosomal unit RPL25 (L23A), a general docking platform for various factors that are transiently associated with ribosomes.55,57,59–61 Dysregulated ribosomal proteins observed in both NAA15+/− and NAA15−/− iPSCs clustered near the NatA docking site (Figure 5A through 5C). Affected small ribosomal subunits (RPS18, RPS25, RPS16, RPS19, RPS20) were clustered at the head of the small ribosomal subunit53 (Figure 5C). Large ribosomal units53 (RPL5 [ribosomal protein L5], RPL13, RPL15, RPL19, RPL30, RPL31, RPL35, RPL23A, RPL39) with low protein levels were clustered at the exit tunnel of the ribosome near the NAA15/NatA docking site (Figure 5B).
Figure 5.
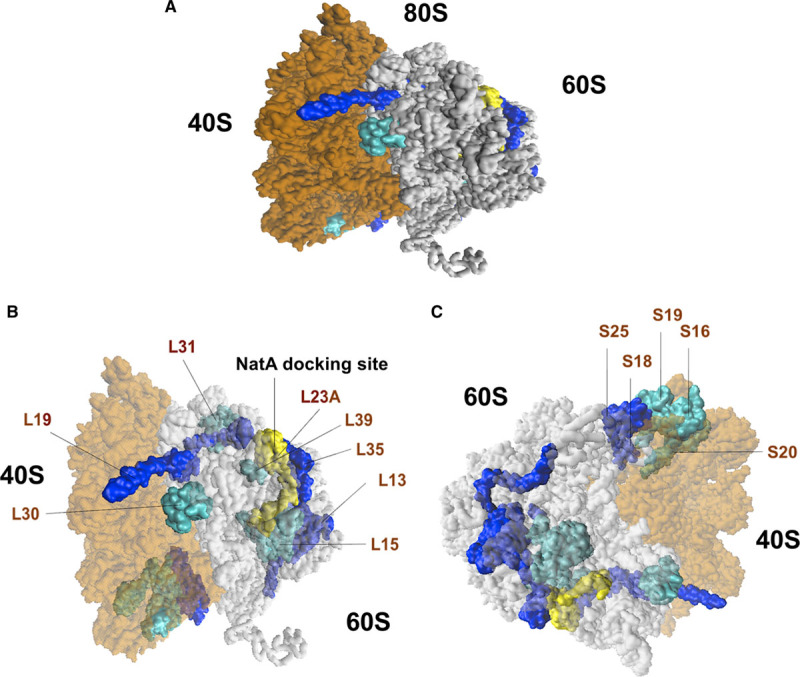
Ribosomal proteins of the 40S small and 60S large subunits affected in NAA15 (N-alpha-acetyltransferase 15)-mutant iPSCs. A, Graphical representation of the human 80S ribosomes with affected 40S small and 60S large subunits in both NAA15−/− and NAA15+/− iPSCs. Ribosomal proteins down in expression highlighted in dark blue (log2 fold change <−2) and light blue (−2< log2 fold change >−1). NatA (N-terminal acetyltransferase) Docking site is highlighted in yellow. B, Diagram showing location of affected 60S large subunits and (C) 40S small subunits. Large ribosomal proteins are clustered and located near the NatA docking site. Small ribosomal proteins affected in their expression clustered together.
Fetal Heart Expression and Association With CHD of Proteins Perturbed in NAA15-Haploinsufficient iPSCs
Because extensive protein expression studies of human and mouse fetal heart are not available, we assessed expression of the RNAs encoding these proteins in the mouse and human fetal heart62; ≈50% of the 562 proteins are expressed in the mouse and human fetal heart (unpublished). Of these 562 proteins, 4 (DHCR7 [7-dehydrocholesterol reductase], MAP2K2 [mitogen-activated protein kinase kinase 2], NSD1 [nuclear receptor binding SET domain protein 1], and RPL5; Table XIII in the Data Supplement) are encoded by genes known to cause autosomal dominant CHD (Table XIII in the Data Supplement). RNA encoding all 4 of these proteins are found in multiple cell types in the human fetal heart (Figure VIII in the Data Supplement).
Discussion
Exome sequence analyses of 4511 CHD subjects with extra-cardiac phenotypes identified 20 subjects with rare inherited or de novo variants that are predicted to perturb NAA15 (Table I and Figure I in the Data Supplement, Figure 1A). Similar NAA15 variants occur in patients with neurological abnormalites.5,7 We explored the consequences of NAA15 variants using human iPSCs by both transcriptome (RNAseq) and proteomic analyses (Figure 2; Figure II and Tables III, IV, VIII, IX, and X in the Data Supplement). NAA15+/− cells had ≈50% of normal levels but like NAA15+/R276W iPSCs, these differentiated into cardiomyocytes. NAA15+/− iPSC-CMs displayed normal unloaded contractility, but impaired contractility when loaded (ie, working against resistance; Figure V in the Data Supplement). NAA15−/− iPSCs produced no NAA15 and like yeast cells lacking NAA15, grew slowly (Figure 2D).19 NAA15-deficiency had minimal effect on the transcriptome but significantly altered the proteome consistent with its role in protein modification.
Because NAA15 is a component of the NatA complex, we assessed the level of N-terminal protein acetylation in iPSCs carrying NAA15 variants using positional proteomics. Approximately 650 proteins with N-terminal sequences that were predicted to be NatA targets could be assessed. Only 32 and 9 proteins had altered Nt-acetylation in NAA15 null and haploinsufficient cells, respectively. By contrast, levels of 562 proteins were altered in both NAA15-null and NAA15-haploinsufficient cells suggesting a possible role for NAA15 and NatA in translation efficiency and protein stability in addition to Nt-acetylation. If NAA15 was the only available ribosome anchor for NAA10 and the NatA complex in human cells as it is in yeast cells,8,55,58 NAA15 null cells should present with a complete lack of Nt-acetylation of all NatA substrates. The explanation for the partial effect observed may be due to the presence of the NAA15 paralogue NAA16 in human cells; however, the low expression of NAA16 in iPSCs suggests the need for additional studies to measure whether the paralogue to NAA15 had any effect on N-terminal acetylation. It has been reported that NAA16-NAA10 complexes may perform NatA-type Nt-acetylation in human cells and partially act as a backup system for the NAA15-NAA10 NatA complex.63
The levels of 18 of the 81 identified ribosomal proteins were altered in NAA15-haploinsufficent cells. NAA15 anchors the NatA complex to the ribosomal large subunit via RPL25 (L23A).55–58,64 Consistent with this model, some of these affected proteins clustered near the NAA15/NatA docking site. Presumably, NAA15 binding to one or more of these proteins56 protects them from subsequent degradation either during ribosome assembly or subsequent ribosomal activity. Previous studies have demonstrated that ribosomal subunit deficiency leads to a variety of pathological states.48,50,51,65 Damaging human variants in ribosomal subunits RPS19, RPL5,4 and RPL35A have been identified in patients with CHD4 (PCGC consortium data not shown) and also contribute to Diamond-Blackfan anemia,50,66,67 a disorder that is accompanied by CHD in 30% of patients.68
Among differentially expressed proteins in NAA15 haploinsufficient cells, 4 (DHCR7, MAP2K2, NSD1, and RPL5) are encoded by known CHD genes (Table XIII and Figure VIII in the Data Supplement) and are most likely responsible for NAA15-haploinsufficient mediated CHD.4,69–71 As the precise mechanisms by which each haploinsufficiency of each of these genes is defined, we anticipate the mechanism(s) will be shared by NAA15 haploinsufficient patients.
Exome analyses of CHD probands also identified 16 NAA15 missense variants, including R276W, a variant of uncertain significance. iPSCs carrying the NAA15 R276W variant had eight proteins with altered Nt-acetylation, of which 4 had altered Nt-acetylation in NAA15-haploinsufficient cells. These data suggest that the R276W variant likely impairs NAA15 function, and consequently NatA function, and like heterozygous LoF variants contributes to CHD. Future studies of protein levels in genetically engineered iPSCs carrying other NAA15 variants found in CHD probands, will help identify those variants contributing to cardiac abnormalities and those variants that are likely benign.
In this study, we use genetically engineered iPSC models to recapitulate NAA15 LoF and missense variants discovered in CHD patients. We observe that variants in NAA15 causing NAA15 haploinsufficiency or deficiency result in either a reduction or complete depletion of NAA15 and NatA protein levels. We find that aberrant protein expression of NAA15 alters Nt-acetylation of a small number of proteins. Most importantly, a reduction of the NAA15 protein interrupts its interaction with the ribosome. The failed interaction causes ribosomal deficiency and mis-expression of a large number of proteins most likely involved in heart development. Protein expression changes are possibly due to obstructed ribosomal machinery and defects in protein synthesis. One or more proteins affected in both NAA15-mutant iPSCs have been documented as a cause of congenital heart defects and are likely candidates required for normal cardiac developmental processes.
Acknowledgments
We would like to thank Paula Montero Llopis of the MicRoN (Microscopy Resources On the North Quad) core for her support and assistance, specifically for collecting images in Figure 2. We appreciate the study participants and their families, without whom this work would not have been possible.
Sources of Funding
We gratefully acknowledge the National Heart, Lung, and Blood Institute Pediatrics Cardiac Genomics Consortium (PCGC) and Cardiovascular Development Consortium (CVDC) investigators (B.D. Gelb, W.K. Chung, E. Goldmuntz, G.A. Porter, R.P. Lifton, M. Brueckner, H.J. Yost, B.G. Bruneau, C.E. Seidman, J.G. Seidman) for their support and expertise in cardiovascular development. Funding support for this study was provided by grants to the PCGC and CVDC by the US National Heart Lung and Blood Institute (UM1 HL098123 [B.D. Gelb], U01 HL098153 [EG], UM1HL098179 [B.G. Bruneau], R01 HL151257 [C.E. Seidman], 1UM1HL098166 [J.G. Seidman]), SFARI and the JPB Foundation (W.K. Chung), Harvard Medical School Diversity and Inclusion Dean’s Postdoctoral Fellowship (T. Ward), Stanley J. Sarnoff Cardiovascular Research Foundation (W. Tai), HHMI Medical Research Fellowship (M.Y. Jang), FAPESP number 19/11921-1 and 18/13706-0 (G. Venturini), K. Zhang acknowledges fellowship support from the American Heart Association, award number 17PRE33660967, John S. Ladue Memorial Fellowship at Harvard Medical School (Y. Kim), S. Varland and T.Arnesen were funded by the Research Council of Norway through a FRIPRO mobility grant 261981, which is cofounded by the European Union’s Seventh Framework Programme under Marie Curie grant agreement no 608695, the Research Council of Norway (project 249843), the Norwegian Health Authorities of Western Norway (project F-12540), and the Norwegian Cancer Society. Funding was supported by the Engineering Research Centers Program of the National Science Foundation, no.EEC-1647837(C.S. Chen). K. Gevaert acknowledges support from The Research Foundation—Flanders (FWO), project number G008018N. Funding was supported by the Howard Hughes Medical Institute (C.E. Seidman), and Fondation Leducq 16 CVD 03 (J.G. Seidman).
Disclosures
None.
Supplemental Materials
Expanded Online Materials and Methods
Online Figures I–VIII
Online Movies I–VIII
Data Set of Online Tables I–XIII
References 72–76
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- CHD
- congenital heart disease
- GFP
- green fluorescent protein
- iPSC
- induced pluripotent stem cells
- iPSC-CMs
- induced pluripotent stem cell-derived cardiomyocytes
- LoF
- loss of function
- NAT
- N-terminal acetyltransferase
- Nt
- N-terminal
K. Gevaert, C. Seidman, and J.G. Seidman contributed equally.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.316966.
For Sources of Funding and Disclosures, see page 1167.
Contributor Information
Tarsha Ward, Email: Tarsha_Ward@hms.harvard.edu.
Warren Tai, Email: warrentai1@gmail.com.
Sarah Morton, Email: Sarah.Morton@childrens.harvard.edu.
Francis Impens, Email: francis.impens@vib-ugent.be.
Petra Van Damme, Email: Petra.vandamme@ugent.be.
Delphi Van Haver, Email: delphi.vanhaver@vib-ugent.be.
Evy Timmerman, Email: evy.timmerman@vib-ugent.be.
Gabriela Venturini, Email: venturini.gabriela@gmail.com.
Kehan Zhang, Email: kehan@bu.edu.
Min Young Jang, Email: megan.jang@gmail.com.
Jon A.L. Willcox, Email: Jon_Willcox@hms.harvard.edu.
Alireza Haghighi, Email: AHAGHIGHI@BWH.HARVARD.EDU.
Bruce D. Gelb, Email: bruce.gelb@mssm.edu.
Wendy K. Chung, Email: wkc15@cumc.columbia.edu.
Elizabeth Goldmuntz, Email: goldmuntz@email.chop.edu.
George A. Porter, Jr, Email: george_porter@urmc.rochester.edu.
Richard P. Lifton, Email: rickl@mail.rockefeller.edu.
Martina Brueckner, Email: martina.brueckner@yale.edu.
H. Joseph Yost, Email: jyost@genetics.utah.edu.
Benoit G. Bruneau, Email: bbruneau@gladstone.ucsf.edu.
Joshua Gorham, Email: jgorham@genetics.med.harvard.edu.
Yuri Kim, Email: ykim@genetics.med.harvard.edu.
Alexandre Pereira, Email: alexandre.pereira@incor.usp.br.
Jason Homsy, Email: jason.homsy@gmail.com.
Craig C. Benson, Email: craigcbenson@gmail.com.
Steven R. DePalma, Email: depalma@genetics.med.harvard.edu.
Sylvia Varland, Email: Sylvia.Varland@uib.no.
Christopher S. Chen, Email: chencs@bu.edu.
Thomas Arnesen, Email: Thomas.Arnesen@uib.no.
Kris Gevaert, Email: kris.gevaert@vib-ugent.be.
Christine Seidman, Email: seidman@genetics.med.harvard.edu.
Novelty and Significance
What Is Known?
NAA15 (N-alpha-acetyltransferase 15) is an 866 amino acid subunit of the NatA (N-terminal acetyltransferase), which acetylates the N-terminal residue of many eukaryotic proteins.
Patients with congenital heart disease (CHD) have genetic variants in the NAA15 gene predicted to be harmful, yet there is limited knowledge of how NAA15 variants cause CHD.
Human induced pluripotent stem cells (iPSCs) can be used as a human model to study biological consequences of genetic variants that may cause disease.
What New Information Does This Article Contribute?
Exome sequence analysis of 4511 patients with CHD identified 20 subjects with rare inherited or de novo variants predicted to perturb NAA15, which may be responsible for their condition.
NAA15 haploinsufficiency in iPSCs has little effect on the transcriptome. However, a 50% reduction of NAA15 protein does lead to impaired cardiomyocyte contractility and proteome integrity.
Expression levels of ribosome-associated proteins and 4 proteins that encode genes known to cause autosomal dominant CHD are altered in NAA15 haploinsufficient and deficient iPSCs.
CHD, which affects about 1% of newborns, reflects defects in heart development during fetal life. Defining genetic causes of CHD provides new insights into mechanisms of cardiac development that may eventually benefit patients with CHD and their families. We have used induced pluripotent stem cells to model NAA15 haploinsufficiency. In iPSCs, NAA15 haploinsufficiency and deficiency cause loss of N-terminal acetylation of 9 and 32 proteins. In addition, we discovered 562 proteins with altered expression levels; 18 proteins are ribosome-associated. Among affected proteins, 4 proteins are known CHD genes and are likely candidates that require a full complement of NAA15. We conclude that patients with CHD with NAA15 haploinsufficiency likely have altered ribosomal activity, which effects steady-state levels of proteins essential for early cardiac development. Genetically engineered iPSCs can be used as a platform for defining the potential pathogenicity of NAA15 variants of unknown significance.
References
- 1.van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–2247. doi: 10.1016/j.jacc.2011.08.025 [DOI] [PubMed] [Google Scholar]
- 2.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. doi: 10.1038/ng.3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H, Dharmadhikari AV, Varland S, Ma N, Domingo D, Kleyner R, Rope AF, Yoon M, Stray-Pedersen A, Posey JE, et al. Truncating variants in NAA15 are associated with variable levels of intellectual disability, autism spectrum disorder, and congenital anomalies. Am J Hum Genet. 2018;102:985–994. doi: 10.1016/j.ajhg.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo H, Wang T, Wu H, Long M, Coe BP, Li H, Xun G, Ou J, Chen B, Duan G, et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol Autism. 2018;9:64. doi: 10.1186/s13229-018-0247-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao JJ, Halvardson J, Zander CS, Zaghlool A, Georgii-Hemming P, Månsson E, Brandberg G, Sävmarker HE, Frykholm C, Kuchinskaya E, et al. Exome sequencing reveals NAA15 and PUF60 as candidate genes associated with intellectual disability. Am J Med Genet B Neuropsychiatr Genet. 2018;177:10–20. doi: 10.1002/ajmg.b.32574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F, et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A. 2009;106:8157–8162. doi: 10.1073/pnas.0901931106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta. 2016;1864:1372–1401. doi: 10.1016/j.bbapap.2016.06.007 [DOI] [PubMed] [Google Scholar]
- 10.Aksnes H, Drazic A, Marie M, Arnesen T. First things first: vital protein marks by N-terminal acetyltransferases. Trends Biochem Sci. 2016;41:746–760. doi: 10.1016/j.tibs.2016.07.005 [DOI] [PubMed] [Google Scholar]
- 11.Aksnes H, Ree R, Arnesen T. Co-translational, post-translational, and non-catalytic roles of N-terminal acetyltransferases. Mol Cell. 2019;73:1097–1114. doi: 10.1016/j.molcel.2019.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frottin F, Martinez A, Peynot P, Mitra S, Holz RC, Giglione C, Meinnel T. The proteomics of N-terminal methionine cleavage. Mol Cell Proteomics. 2006;5:2336–2349. doi: 10.1074/mcp.M600225-MCP200 [DOI] [PubMed] [Google Scholar]
- 13.Arnold RJ, Polevoda B, Reilly JP, Sherman F. The action of N-terminal acetyltransferases on yeast ribosomal proteins. J Biol Chem. 1999;274:37035–37040. doi: 10.1074/jbc.274.52.37035 [DOI] [PubMed] [Google Scholar]
- 14.Choudhury KR, Raychaudhuri S, Bhattacharyya NP. Identification of HYPK-interacting proteins reveals involvement of HYPK in regulating cell growth, cell cycle, unfolded protein response and cell death. PLoS One. 2012;7:e51415. doi: 10.1371/journal.pone.0051415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottlieb L, Marmorstein R. Structure of human NatA and its regulation by the huntingtin interacting protein HYPK. Structure. 2018;26:925–935.e8. doi: 10.1016/j.str.2018.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raychaudhuri S, Sinha M, Mukhopadhyay D, Bhattacharyya NP. HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity. Hum Mol Genet. 2008;17:240–255. doi: 10.1093/hmg/ddm301 [DOI] [PubMed] [Google Scholar]
- 17.Weyer FA, Gumiero A, Lapouge K, Bange G, Kopp J, Sinning I. Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nat Commun. 2017;8:15726. doi: 10.1038/ncomms15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng S, Magin RS, Wei X, Pan B, Petersson EJ, Marmorstein R. Structure and mechanism of acetylation by the N-terminal dual enzyme NatA/Naa50 complex. Structure. 2019;27:1057–1070.e4. doi: 10.1016/j.str.2019.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullen JR, Kayne PS, Moerschell RP, Tsunasawa S, Gribskov M, Colavito-Shepanski M, Grunstein M, Sherman F, Sternglanz R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989;8:2067–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnesen T, Anderson D, Baldersheim C, Lanotte M, Varhaug JE, Lillehaug JR. Identification and characterization of the human ARD1–NATH protein acetyltransferase complex 2005;386:433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liszczak G, Goldberg JM, Foyn H, Petersson EJ, Arnesen T, Marmorstein R. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat Struct Mol Biol. 2013;20:1098–1105. doi: 10.1038/nsmb.2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnesen T, Starheim KK, Van Damme P, Evjenth R, Dinh H, Betts MJ, Ryningen A, Vandekerckhove J, Gevaert K, Anderson D. The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol Cell Biol. 2010;30:1898–1909. doi: 10.1128/MCB.01199-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myklebust LM, Van Damme P, Støve SI, Dörfel MJ, Abboud A, Kalvik TV, Grauffel C, Jonckheere V, Wu Y, Swensen J, et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum Mol Genet. 2015;24:1956–1976. doi: 10.1093/hmg/ddu611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fluge Ø, Bruland O, Akslen LA, Varhaug JE, Lillehaug JR. NATH, a novel gene overexpressed in papillary thyroid carcinomas. Oncogene. 2002;21:5056–5068. doi: 10.1038/sj.onc.1205687 [DOI] [PubMed] [Google Scholar]
- 25.Gelb B, Brueckner M, Chung W, Goldmuntz E, Kaltman J, Kaski JP, Kim R, Kline J, Mercer-Rosa L, Porter G, et al. ; Pediatric Cardiac Genomics Consortium. The congenital heart disease genetic network study. Circulation Research. 2013;112:698–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohye RG, Sleeper LA, Mahony L, Newburger JW, Pearson GD, Lu M, Goldberg CS, Tabbutt S, Frommelt PC, Ghanayem NS, et al. ; Pediatric Heart Network Investigators. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010;362:1980–1992. doi: 10.1056/NEJMoa0912461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma A, Toepfer CN, Ward T, Wasson L, Agarwal R, Conner DA, Hu JH, Seidman CE. CRISPR/Cas9-mediated fluorescent tagging of endogenous proteins in human pluripotent stem cells. Current protocols in human genetics. 2018;96:21 11 1-21 11 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. 2012;109:E1848–E1857. doi: 10.1073/pnas.1200250109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnesen T, Gromyko D, Pendino F, Ryningen A, Varhaug JE, Lillehaug JR. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-alpha-acetyltransferase complex. Oncogene. 2006;25:4350–4360. doi: 10.1038/sj.onc.1209469 [DOI] [PubMed] [Google Scholar]
- 33.Gromyko D, Arnesen T, Ryningen A, Varhaug JE, Lillehaug JR. Depletion of the human Nα-terminal acetyltransferase A induces p53-dependent apoptosis and p53-independent growth inhibition. Int J Cancer. 2010;127:2777–2789. doi: 10.1002/ijc.25275 [DOI] [PubMed] [Google Scholar]
- 34.Kalvik TV, Arnesen T. Protein N-terminal acetyltransferases in cancer. Oncogene. 2013;32:269–276. doi: 10.1038/onc.2012.82 [DOI] [PubMed] [Google Scholar]
- 35.Toepfer CN, Sharma A, Cicconet M, Garfinkel AC, Mücke M, Neyazi M, Willcox JAL, Agarwal R, Schmid M, Rao J, et al. SarcTrack. Circ Res. 2019;124:1172–1183. doi: 10.1161/CIRCRESAHA.118.314505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 2015;349:982–986. doi: 10.1126/science.aaa5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Legant WR, Pathak A, Yang MT, Deshpande VS, McMeeking RM, Chen CS. Microfabricated tissue gauges to measure and manipulate forces from 3D microtissues. Proc Natl Acad Sci USA. 2009;106:10097–10102. doi: 10.1073/pnas.0900174106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gevaert K, Goethals M, Martens L, Van Damme J, Staes A, Thomas GR, Vandekerckhove J. Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat Biotechnol. 2003;21:566–569. doi: 10.1038/nbt810 [DOI] [PubMed] [Google Scholar]
- 39.Staes A, Impens F, Van Damme P, Ruttens B, Goethals M, Demol H, Timmerman E, Vandekerckhove J, Gevaert K. Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat Protoc. 2011;6:1130–1141. doi: 10.1038/nprot.2011.355 [DOI] [PubMed] [Google Scholar]
- 40.Van Damme P, Van Damme J, Demol H, Staes A, Vandekerckhove J, Gevaert K. A review of COFRADIC techniques targeting protein N-terminal acetylation. BMC Proc. 2009;3suppl 6S6. doi: 10.1186/1753-6561-3-S6-S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan J, Walz K, Nakamura H, Carattini-Rivera S, Zhao Q, Vogel H, Wei N, Justice MJ, Bradley A, Lupski JR. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol Cell Biol. 2003;23:6798–6808. doi: 10.1128/mcb.23.19.6798-6808.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Cooke M, Panjwani S, Cao K, Krauth B, Ho PY, Medrzycki M, Berhe DT, Pan C, McDevitt TC, et al. Histone h1 depletion impairs embryonic stem cell differentiation. PLoS Genet. 2012;8:e1002691. doi: 10.1371/journal.pgen.1002691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mangé A, Coyaud E, Desmetz C, Laurent E, Béganton B, Coopman P, Raught B, Solassol J. FKBP4 connects mTORC2 and PI3K to activate the PDK1/Akt-dependent cell proliferation signaling in breast cancer. Theranostics. 2019;9:7003–7015. doi: 10.7150/thno.35561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teng T, Mercer CA, Hexley P, Thomas G, Fumagalli S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Mol Cell Biol. 2013;33:4660–4671. doi: 10.1128/MCB.01174-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhattacharjee RB, Bag J. Depletion of nuclear poly(A) binding protein PABPN1 produces a compensatory response by cytoplasmic PABP4 and PABP5 in cultured human cells. PLoS One. 2012;7:e53036. doi: 10.1371/journal.pone.0053036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choudhury KR, Bucha S, Baksi S, Mukhopadhyay D, Bhattacharyya NP. Chaperone-like protein HYPK and its interacting partners augment autophagy. Eur J Cell Biol. 2016;95:182–194. doi: 10.1016/j.ejcb.2016.03.003 [DOI] [PubMed] [Google Scholar]
- 48.Cheng Z, Mugler CF, Keskin A, Hodapp S, Chan LY, Weis K, Mertins P, Regev A, Jovanovic M, Brar GA. Small and large ribosomal subunit deficiencies lead to distinct gene expression signatures that reflect cellular growth rate. Mol Cell. 2019;73:36–47.e10. doi: 10.1016/j.molcel.2018.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregory B, Rahman N, Bommakanti A, Shamsuzzaman M, Thapa M, Lescure A, Zengel JM, Lindahl L. The small and large ribosomal subunits depend on each other for stability and accumulation. Life Sci Alliance. 2019;2:e201800150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peisker K, Braun D, Wölfle T, Hentschel J, Fünfschilling U, Fischer G, Sickmann A, Rospert S. Ribosome-associated complex binds to ribosomes in close proximity of Rpl31 at the exit of the polypeptide tunnel in yeast. Mol Biol Cell. 2008;19:5279–5288. doi: 10.1091/mbc.e08-06-0661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steffen KK, McCormick MA, Pham KM, MacKay VL, Delaney JR, Murakami CJ, Kaeberlein M, Kennedy BK. Ribosome deficiency protects against ER stress in Saccharomyces cerevisiae. Genetics. 2012;191:107–118. doi: 10.1534/genetics.111.136549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khatter H, Myasnikov AG, Natchiar SK, Klaholz BP. Structure of the human 80S ribosome. Nature. 2015;520:640–645. doi: 10.1038/nature14427 [DOI] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38, 27. doi: 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 55.Gautschi M, Just S, Mun A, Ross S, Rücknagel P, Dubaquié Y, Ehrenhofer-Murray A, Rospert S. The yeast N(alpha)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol Cell Biol. 2003;23:7403–7414. doi: 10.1128/mcb.23.20.7403-7414.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knorr AG, Schmidt C, Tesina P, Berninghausen O, Becker T, Beatrix B, Beckmann R. Ribosome-NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat Struct Mol Biol. 2019;26:35–39. doi: 10.1038/s41594-018-0165-y [DOI] [PubMed] [Google Scholar]
- 57.Polevoda B, Brown S, Cardillo TS, Rigby S, Sherman F. Yeast N(alpha)-terminal acetyltransferases are associated with ribosomes. J Cell Biochem. 2008;103:492–508. doi: 10.1002/jcb.21418 [DOI] [PubMed] [Google Scholar]
- 58.Varland S, Arnesen T. Investigating the functionality of a ribosome-binding mutant of NAA15 using Saccharomyces cerevisiae. BMC Res Notes. 2018;11:404. doi: 10.1186/s13104-018-3513-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kramer G, Boehringer D, Ban N, Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat Struct Mol Biol. 2009;16:589–597. doi: 10.1038/nsmb.1614 [DOI] [PubMed] [Google Scholar]
- 60.Kramer G, Rauch T, Rist W, Vorderwülbecke S, Patzelt H, Schulze-Specking A, Ban N, Deuerling E, Bukau B. L23 protein functions as a chaperone docking site on the ribosome. Nature. 2002;419:171–174. doi: 10.1038/nature01047 [DOI] [PubMed] [Google Scholar]
- 61.Wilson DN, Nierhaus KH. Ribosomal proteins in the spotlight. Crit Rev Biochem Mol Biol. 2005;40:243–267. doi: 10.1080/10409230500256523 [DOI] [PubMed] [Google Scholar]
- 62.Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, Hu Y, Dong J, Li Q, Wu X, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26:1934–1950.e5. doi: 10.1016/j.celrep.2019.01.079 [DOI] [PubMed] [Google Scholar]
- 63.Arnesen T, Gromyko D, Kagabo D, Betts MJ, Starheim KK, Varhaug JE, Anderson D, Lillehaug JR. A novel human NatA Nalpha-terminal acetyltransferase complex: hNaa16p-hNaa10p (hNat2-hArd1). BMC Biochem. 2009;10:15. doi: 10.1186/1471-2091-10-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magin RS, Deng S, Zhang H, Cooperman B, Marmorstein R. Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLoS One. 2017;12:e0186278. doi: 10.1371/journal.pone.0186278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warren AJ. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv Biol Regul. 2018;67:109–127. doi: 10.1016/j.jbior.2017.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, Talbot CC, Jr, Meltzer P, Esposito D, Beggs AH, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112:1582–1592. doi: 10.1182/blood-2008-02-140012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue MF, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83:769–780. doi: 10.1016/j.ajhg.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vlachos A, Osorio DS, Atsidaftos E, Kang J, Lababidi ML, Seiden HS, Gruber D, Glader BE, Onel K, Farrar JE, et al. Increased prevalence of congenital heart disease in children with Diamond Blackfan anemia suggests unrecognized Diamond Blackfan anemia as a cause of congenital heart disease in the general population: a report of the Diamond Blackfan Anemia Registry. Circ Genom Precis Med. 2018;11:e002044. doi: 10.1161/CIRCGENETICS.117.002044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Machet L. [Genetic diseases on the Internet: OMIM]. Ann Dermatol Venereol. 1998;125:645. [PubMed] [Google Scholar]
- 70.Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore. Accessed April 2020. https://omim.org/
- 71.Oyston J. Online Mendelian Inheritance in Man. Anesthesiology. 1998;89:811–812. doi: 10.1097/00000542-199809000-00060 [DOI] [PubMed] [Google Scholar]
- 72.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR; 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials have been made publicly available. Further details are provided in the Major Resources Table located in the Data Supplement.