

Abstract
T lymphocytes undergo carefully orchestrated programming during development in the thymus and subsequently during differentiation in the periphery. This intricate specification allows for cell-type and context-specific transcriptional programs that regulate immune responses to infection and malignancy. Epigenetic changes, including histone modifications and covalent modification of DNA itself through DNA methylation, are now recognized to play a critical role in these cell-fate decisions. DNA methylation is mediated primarily by the actions of the DNA methyltransferase (DNMT) and ten-eleven-translocation (TET) families of epigenetic enzymes. In this review, we discuss the role of DNA methylation and its enzymatic regulators in directing the development and differentiation of CD4+ and CD8+ T-cells.
Keywords: epigenetics, lymphocyte, thymocyte development, DNMT1, DNMT3, TET
I. INTRODUCTION
T lymphocytes are a critical arm of the adaptive immune system that requires complex, tightly controlled orchestration of gene expression during developmental processes and immune responses. As T-cells develop in the thymus, they undergo a series of coordinated cellular decisions that ultimately result in commitment to a stable lineage. Then, once mature and upon egress from the thymus, naïve (antigen-inexperienced) T-cells patrol the periphery until they are activated by cognate antigen and undergo differentiation into discrete functional subsets. Activated T-cells have the potential to acquire many cell fates as they differentiate. At all stages, they must integrate a multitude of extracellular signals, including cytokines, chemokines, and metabolites that promote cellular programming. These developmental and cell-fate choices are regulated not only by transcription factor expression but also by epigenetic regulators that alter the epigenomic landscape. Epigenetic alterations include posttranslational modifications of histones and covalent modification of DNA through DNA methylation. These changes can alter the chromatin structure and accessibility at cis-regulatory elements, such as promoter or enhancer regions, to promote or repress gene transcription. Although DNA methylation has long been considered relatively stable, recent discovery of the ten-eleven translocation (TET) family of proteins identified the formation of oxidized methylcytosine derivatives and an active process of DNA demethylation. These processes are critical in the development, differentiation, and cell-fate stability of T lymphocytes. In this review, we specifically focus on how DNA methylation and its enzymatic regulators direct T-cell development and differentiation.
II. DNA METHYLATION
DNA methylation covalently modifies DNA through the methylation of the fifth carbon of a cytosine base (5-methylcytosine; 5mC), which in mammals occurs primarily in the context of CpG dinucleotides.1,2 This process can occur through de novo DNA methylation or maintenance of DNA methylation during replication. In mammals, the two major catalytically active de novo DNA methyltransferases are DNMT3A and DNMT3B.3,4 These enzymes transfer the methyl group of the coenzyme S-adenosyl-L-Methionine (SAM) to the cytosine residue of DNA5,6 and bind equally unmethylated cytosines or hemimethylated cytosines.3,7 DNMT1 is responsible for the maintenance of DNA methylation during DNA replication. In contrast to DNMT3A/B, DNMT1 preferentially binds hemimethylated DNA.8–10 In resting cells, DNMT1 is maintained in an autoinhibitory conformation.11–13 During replication, the E3 ubiquitin ligase UHRF1 recruits DNMT1 to hemimethylated CpG dinucleotides at replication forks,14,15 allowing DNMT1 to methylate the daughter DNA strand13 and thereby maintaining DNA methylation during cellular replication.
The DNMT proteins contain an N-terminal regulatory domain and a C-terminal catalytic domain (Fig. 1A). DNMT1 is by far the largest protein of the DNMT family and contains numerous domains including the N-terminal independently folded domain (NTD), replication foci-targeting sequence domain (RFTS), a CXXC motif, two bromo-adjacent homology domains (BAHs), and the catalytic domain. The NTD binds multiple proteins that can serve to regulate DNMT1 function. One of these is PCNA, a required factor for cellular replication, which binds in the NTD of DNMT1 and helps maintain the methylation of daughter DNA.16 The RFTS domain mediates DNMT1 localization to replication forks in late S-phase17 in a UHRF1-dependent manner and is required for replication-dependent maintenance of DNA methylation.14,15 The CXXC domain is a conserved zinc-finger domain that can bind unmethylated CpG-containing DNA; however, two reports10,18 have opposing findings regarding the requirement of the CXXC domain for DNMT1 enzymatic activity; thus, further work is needed to clarify its role in DNMT1 function. The function of the two BAH domains remain to be elucidated. All DNMT enzymes have a C-terminal catalytic domain that contains 10 motifs characteristic of DNA-(cytosine C5)-methyltransferases. The DNMT family uses SAM as a methyl group donor and a base-flipping mechanism that rotates the target base into the catalytic pocket.19
FIG. 1:
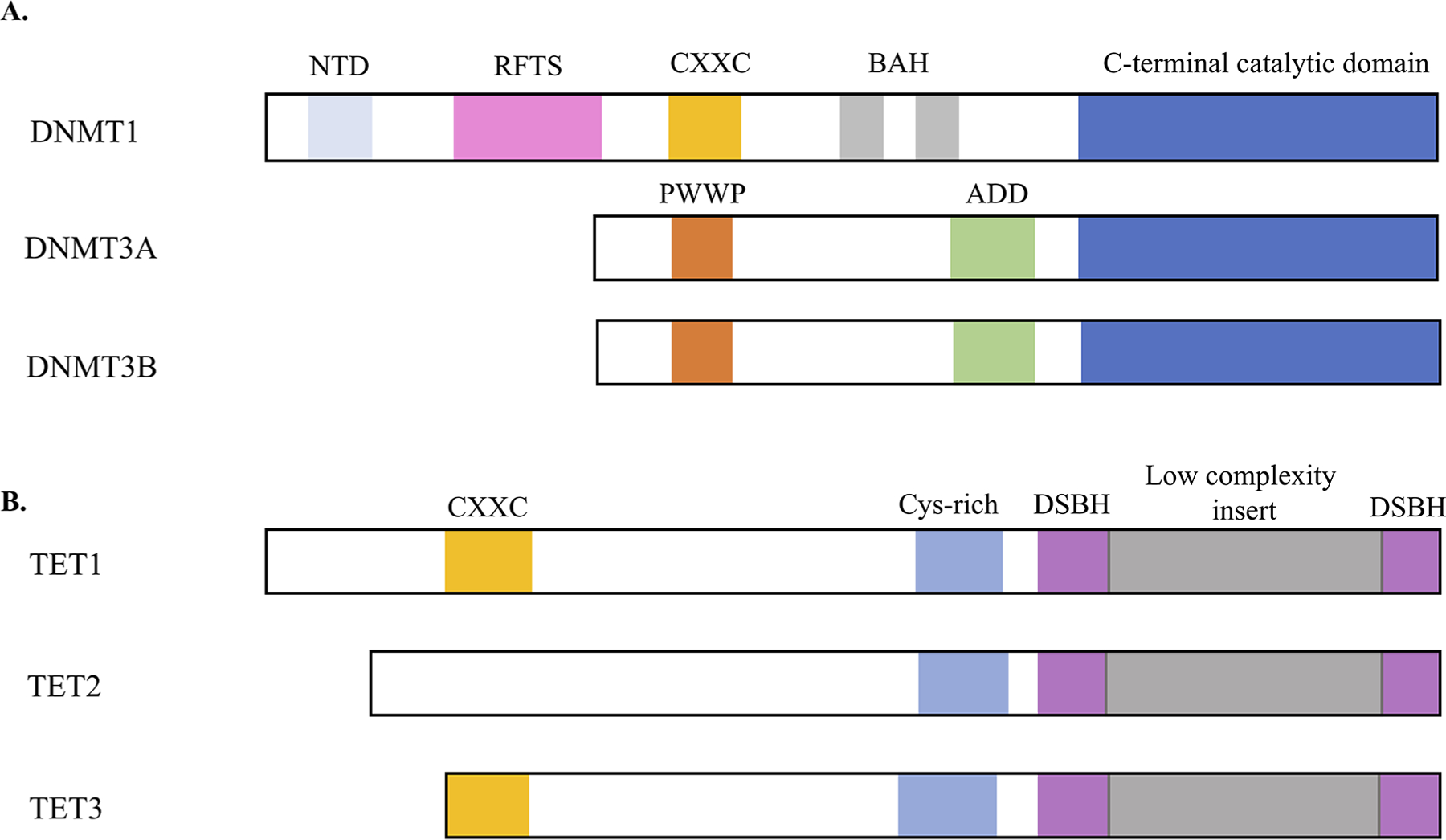
Schematic representation of structural domains of DNMT family (A) and TET family (B) members
DNMT3A and DNMT3B have similar domain structures: each contains a PWWP, an ADD, (Atrx-DNMT3-DNMT3L), and a C-terminal catalytic domain. The PWWP domain binds DNA20,21 and histone tails22 to tether DNMT3A/B to chromatin to allow for DNA methylation.23,24 Interestingly, a point mutation in the PWWP domain of DNMT3B was identified as the cause of the immunodeficiency, centrometric instabilities and facial anomalies (ICF) syndrome in humans.25 The ADD domain also interacts with histone tails,26–28 as well as multiple other reported proteins (reviewed by Tajima et al.29). The ADD domain of DNMT3A is located such that its positioning inhibits access of DNA to the catalytic domain.30 Binding of the N-terminal tail of histone H3 to the ADD domain alters the ADD domain positioning to allow DNA access to the catalytic domain, potentially enhancing DNMT3A enzymatic activity.
For many years, it was assumed that inhibition of DNMT1 activity was the primary mechanism through which DNA demethylation occurred. However, identification of the TET family of enzymes shed light on the process of active DNA demethylation. TET1 was originally identified as a fusion partner with the MLL protein in a t(10;11) translocation in acute myeloid leukemia.31,32 Three TET family members were subsequently identified (TET1–3) through homology to the trypanosome base J-binding proteins (JBP1 and JBP2), which hydrolyze the methyl group of thymine.33,34
All TET family members contain two conserved domains in the C-terminus, which together form the catalytic domain of TET proteins: a cysteine-rich region and a double-stranded β-helix (DSβH) fold domain interrupted by a low complexity nonconserved region (Fig. 1B). Contained within the DSβH domain are key residues that mediate α-ketoglutarate and Fe (II) binding, cofactors required for TET enzymatic function.33,34 TET1 and TET3 contain an N-terminal CXXC domain, which can bind unmethylated CpG dinucleotides; however, an ancestral chromosomal inversion led to the loss of the CXXC domain in the TET2 locus, and in mammals it is encoded by a distinct gene IDAX (CXXC4).35 How the structural differences in TET family members may influence their function is currently unknown. IDAX downregulated TET2 protein expression through caspase-dependent degradation in murine embryonic stem cells and a human monocytic cell line35; however, IDAX is not expressed in naïve or recently activated murine T lymphocytes36 and whether such a mechanism exists in T-cells to regulate TET2 expression is currently unclear.
TET proteins mediate DNA demethylation through the sequential oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC)33,37–39 (Fig. 2). These oxi-methylcytosine bases can promote passive DNA demethylation during replication as the DNMT1/UHRF1 complex prefers a hemimethylated substrate over a hemihydroxymethylated or unmethylated DNA base40–42; thus, their presence will inhibit maintenance of DNA methylation. Additionally, both 5fC and 5caC can be converted to an unmodified cytosine through the action of thymine DNA glycosylase (TDG) and the base excision repair pathway.37,43–45
FIG. 2:
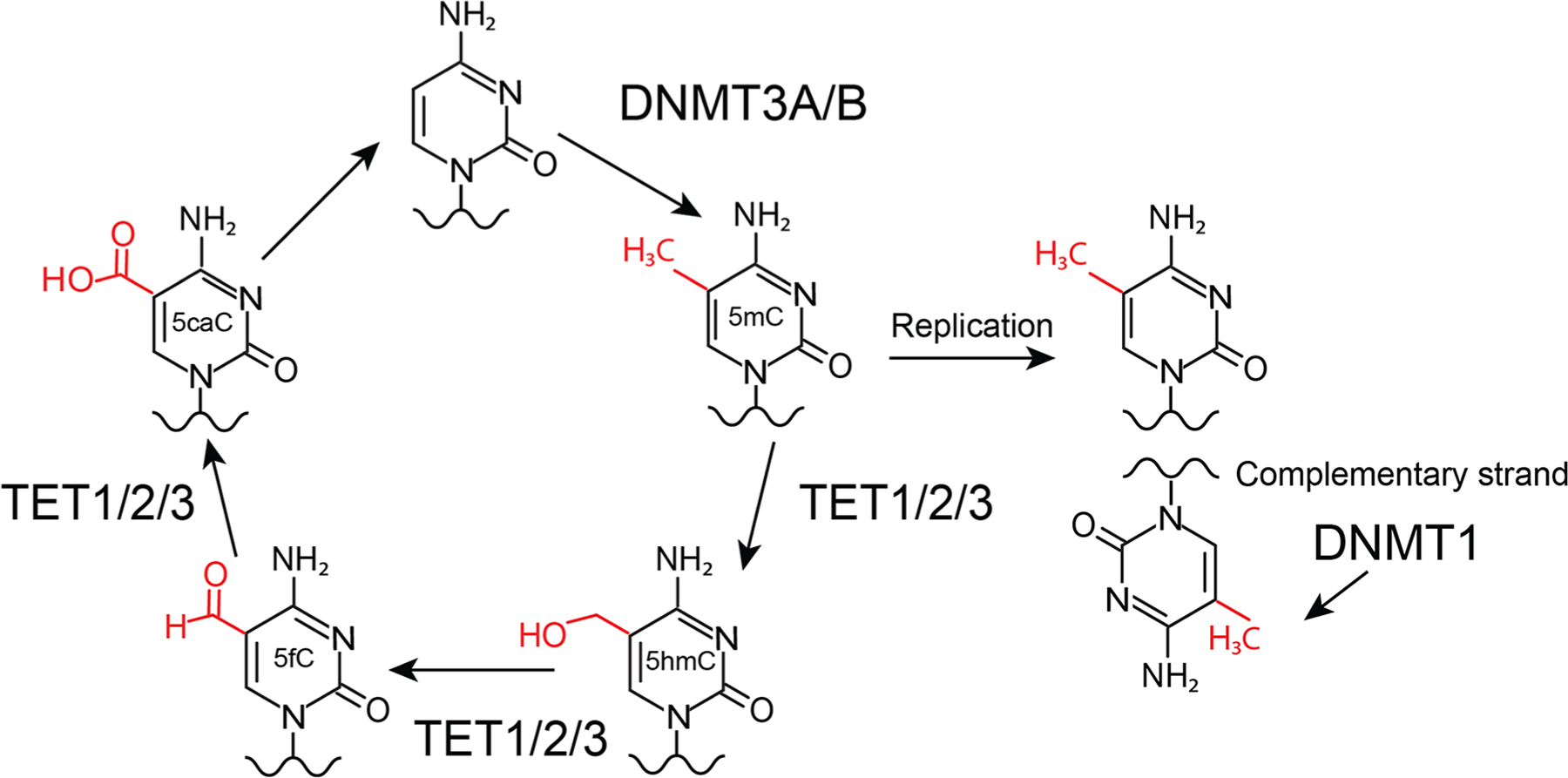
DNA methylation and demethylation. DNMT3A/B deposit a methyl group on unmodified cytosine bases to generate 5mC. During replication, DNMT1 methylates daughter strands to maintain 5mC. 5mC can be sequentially oxidized by TET enzymes to 5hmC, 5fC and 5caC. 5fC and 5caC can then be converted to an unmodified cytosine.
Despite similar amounts of 5mC across different cell types, 5hmC levels have been shown to vary; neuronal cells contain the most, upward of nearly 40% of 5hmC in postmitotic Purkinje neuronal cells.33,46–48 Due to this accumulation, 5hmC is frequently viewed as a relatively stable epigenetic mark, and 5hmC, and possibly other oxi-methylcytosine intermediates, may serve as distinct epigenetic marks rather than simply intermediates in the DNA demethylation process. Several groups have identified proteins that selectively bind 5hmC-containing DNA,49,50 and 5hmC may prevent binding of some 5mC readers40,51,52; however, the functional consequences of 5hmC as a distinct epigenetic mark remain unclear. In T-cells, 5hmC is enriched at cell-type specific enhancer regions and in the gene bodies of actively transcribed genes,53 suggesting that regulation of this epigenetic mark may have functional consequences for gene expression. Although DNA methylation at promoter regions is widely accepted as a repressive mark for gene transcription, how the presence of 5mC or its oxidized intermediates at other cis-regulatory regions, such as enhancers, affects gene expression is not yet fully understood.
TET2 and DNMT3A loss-of-function mutations occur at high frequency in hematologic malignancies. Originally identified in human myeloid neoplasms,54–58 later studies in T-cell lymphomas found that 50%–70% of certain T-cell lymphomas59–62 contained TET2 and/or DNMT3A mutations, suggesting that these enzymes play a critical role in immune cell development and homeostasis.
III. T-CELL DEVELOPMENT
Thymopoiesis is a tightly regulated process involving multiple developmental stages that ultimately results in mature T lymphocytes. Common lymphoid progenitor (CLP)-derived early thymic progenitors (ETPs) seed the thymus from the bone marrow. Developing thymocytes then progress through multiple stages defined by their expression of CD4 and CD8 coreceptors: (1) double-negative (DN; CD4−CD8−), (2) double-positive (DP; CD4+CD8+), and (3) CD4 single-positive (CD4SP; CD4+CD8−) or CD8 single-positive (CD8SP; CD4−CD8+). DN thymocytes are subsequently subdivided into four groups according to surface expression of CD44 and CD25: DN1 (CD44+CD25−), DN2 (CD44+CD25+), DN3 (CD44−CD25+), and DN4 (CD44−CD25−). As developing thymocytes transit through these stages, they pass through several developmental checkpoints. T lineage commitment, associated with concomitant loss of alternative lineage potential, occurs stepwise through the DN1 and DN2 stages (see review by Yui and Rothenberg63). In the DN3 stage, thymocytes undergo β-selection, which requires αβ T-cell precursors to signal through a functional pre–T-cell receptor (TCR) consisting of a productively rearranged TCRβ and invariant pre-Tα chain. Following β selection, the developing thymocytes initiate CD4 and CD8 transcription and upregulate TCRα expression to transition into the DP stage. At this point, thymocytes must navigate positive and negative selection, during which time DP cells are rescued from programmed cell death if they have the ability to interact with self-MHC presenting self-peptides (positive selection) and to avoid TCR-induced cell death if they react too avidly with self-peptide (negative selection) (reviewed in Stritesky et al.64). The small proportion of DP thymocytes that successfully survive these checkpoints downregulate CD4 or CD8 coreceptor expression to become CD8SP or CD4SP cells, respectively. Clearly, a multitude of factors, including transcription factors, chemokines, and cytokines, regulate these developmental processes, and more recent studies have shed light on how DNA methylation contributes to thymocyte development.
Genome-wide DNA methylation studies in murine hematopoietic lineage commitment revealed differential methylation at multiple stages of thymocyte commitment,65 suggesting that DNA methylation is dynamically regulated during T-cell development. At the CLP to DN1 stage, there were more differentially methylated regions with gain-of-methylation versus loss-of-methylation, potentially suggesting that de novo methyltransferase activity may play a key role at these stages. Overall, this study noted a skewing toward greater genomic methylation in lymphoid compared to myeloid progenitors. Consistent with this observation, the addition of 5-azacytidine, a DNA methyltransferase inhibitor, to the OP9-DL1 stromal co-culture system (an in vitro system that promotes both lymphoid and myeloid commitment from progenitors) led to an increase in myeloid commitment from multipotent progenitors (MPPs), CLPs, DN1, and DN2 cells at the expense of lymphoid commitment. These data indicate that DNA methylation plays a functional role in determining T lineage commitment and are consistent with the findings that hypomorphic DNMT1 alleles can sustain myeloid but not lymphoid commitment.66 Examination of genome-wide 5hmC in later stages of thymocyte development (DP, CD4SP, and CD8SP) revealed enrichment in thymus-specific active enhancers (defined by co-occurring H3K27ac and H3K4me1 marks) compared to embryonic stem cell and murine embryonic fibroblast-specific enhancers.53 Additionally, this study noted dynamic changes in 5hmC distribution during differentiation stages and a strong correlation between 5hmC intragenic enrichment and gene expression. Together, these data suggest that the TET enzymes may play a role in thymopoiesis. During lineage commitment, the CD8 and CD4 coreceptors facilitate TCR signaling during selection as coreceptors for MHC class I and class II, respectively. Positive selection of cells with TCRs specific for MHC class I develop into CD8SP cells, and cells with MHC class II-specific TCRs develop into CD4SP. The expressions of the CD4 and CD8 coreceptors have been shown to be epigenetically regulated (see review by Issuree et al.67), and DNA methylation mediated via the DNMT family was identified as a mechanism for heritable silencing of the CD4 locus in mature CD8+ T-cells68 and TET1/TET3-mediated methylation as controlling enhancers that regulate CD4 expression in mature CD4+ T-cells.69
During human αβ T-cell development, global DNA methylation reprogramming occurs during several key developmental checkpoints: T-cell lineage commitment, β-selection, TCRαβ expression, and positive selection with both gain and loss of DNA methylation.70 Notably, more differentially methylated regions underwent demethylation with fewer undergoing de novo methylation at all differentiation steps except for initial T-cell lineage commitment, in line with the gain-of-methylation seen in the CLP and DN1 stages in murine cells.65 These methylation changes remained mostly stable at later differentiation stages; however, notable exceptions to this finding included the CD8a, PTCRA, and Rag1 loci. Changes in DNA methylation tended to occur at genomic loci associated with T-cell differentiation or T-cell receptor function based on gene ontology analysis, supporting a regulatory role for DNA methylation during thymocyte development. When the same researchers examined the relationship between differentially methylated regions and differentially expressed genes, ~85% of the demethylation events correlated with an increase in gene expression during concurrent differentiation step. This finding may reflect the fact that promoter methylation correlates well with gene repression.
To begin to understand how these global changes in DNA methylation occur during T-cell development, several studies have focused on the enzymes responsible for regulating the methylated state of DNA (Table 1; Fig. 3). Early studies examined the role of DNMT family members in T-cell development. Inducible loss of DNMT1 just prior to the DN to DP transition resulted in a profound loss of DP and SP thymocytes due to increased apoptosis.71 Once past the DP transition, loss of DNMT1 (mediated by CD4Cre) resulted in essentially normal T-cell development with a slight reduction in CD44hi populations of peripheral CD4+ and CD8+ T-cells.71 These data support a critical role for DNMT1 in early T-cell development.
TABLE 1:
Summary of T-cell phenotypes in knockout mice lacking DNMT or TET enzymes
| Gene | Cre/mutation type | T-cell phenotype | Reference |
|---|---|---|---|
| DNMT1 | Hypomorph |
|
Broske et al., 200966 |
| LckCre |
|
Lee et al., 200171 | |
| CD4Cre |
|
||
| DNMT3A | Germline KO |
|
Okano et al., 19994 Gamper et al., 200972 |
| CD4Cre |
|
Gamper et al., 200972 | |
| TET1/TET2 | TET1 KO; TET2 CD4Cre |
|
Yang et al., 2015106 |
| TET2/TET3 | CD4Cre |
|
Tsagaratou et al., 201776 |
| FoxP3Cre |
|
Yue et al., 2019105 | |
| TET2 | CD2Cre |
|
Ichiyama et al., 201578 |
| CD4Cre |
|
Carty et al., 201836 |
FIG. 3:
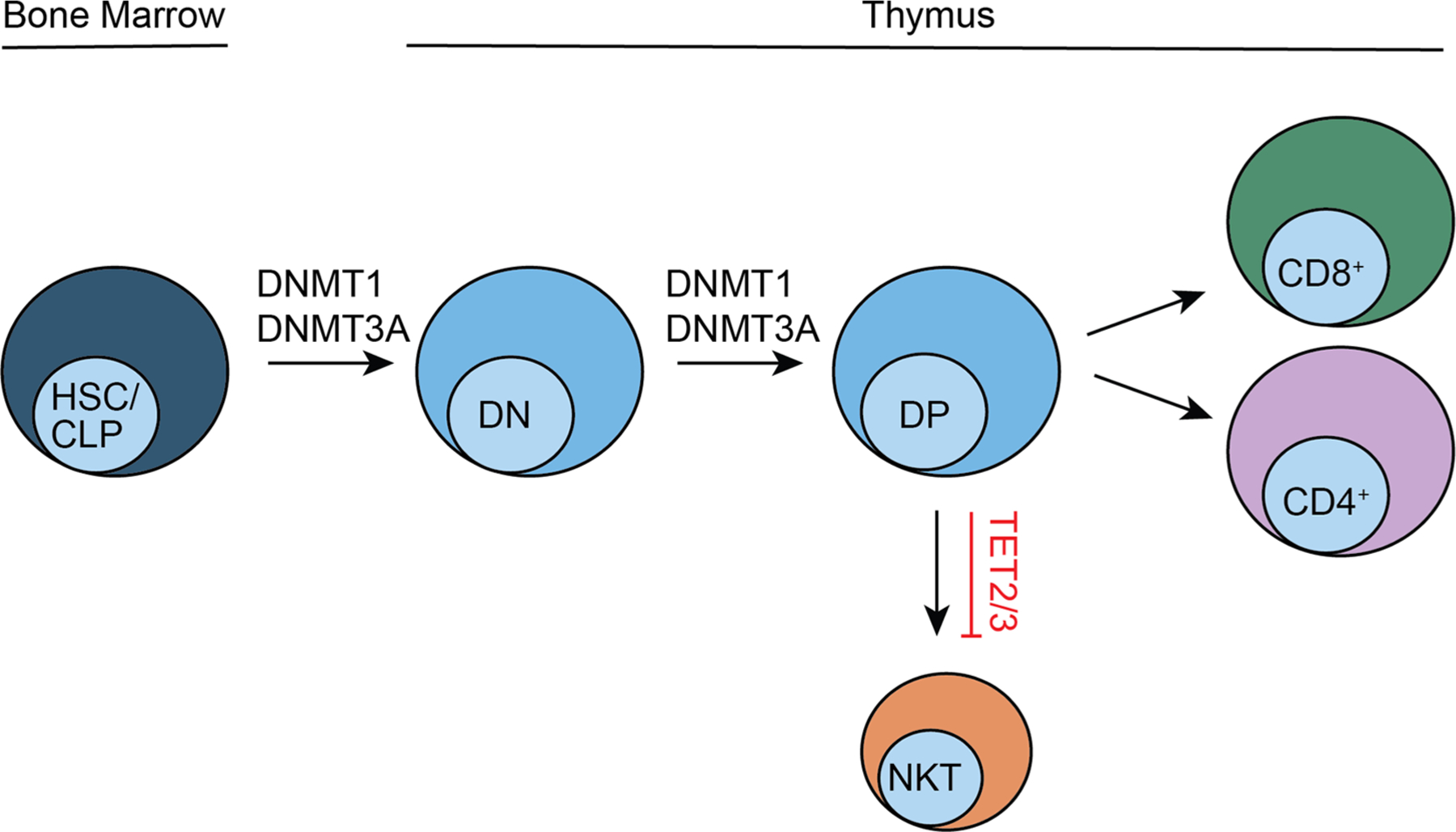
DNA methylation enzymes in T-cell development. A schema of T-cell development with the requirement of DNA methylation enzymes noted. Arrows indicate the requirement of the enzyme to proceed through individual developmental transitions. The TET2/3 line indicates suppression of the lineage.
A role for de novo DNA methylation by DNMT3A in thymopoiesis has also been identified. DNMT3A knockout mice develop normally but become stunted and die by approximately 4 weeks of age.4 Evaluation of thymocyte development in these neonates revealed a marked decrease in thymic cellularity with a partial DN to DP block in the DNMT3A-deficient mice and splenic hypocellularity.72 However, the stunted phenotype in these mice makes it difficult to draw clear conclusions regarding the role of DNMT3A in T-cell development, thus making targeted knockout mice essential for the study of DNMT3A in thymopoiesis. Inducible loss of DNMT3A in hematopoietic stem cell (HSC) progenitors can lead to an accumulation of DN2 thymocytes but only after secondary transplantation into recipient hosts and only in ~40% of these recipients.73 The partial penetrance of this effect and fact that it was not seen in primary transplant recipients suggest that other selection events may need to occur for DNMT3A loss to significantly affect thymocyte development. When DN2 expansion did occur, it was associated with downregulation of Nur77 and a decrease in apoptosis. In the same study, DNMT3A loss was shown to cooperate with Notch gain-of-function mutants to promote T-cell acute lymphoblastic leukemia (T-ALL). Importantly, using different Cre drivers, the authors determined that DNMT3A loss had to occur at the ETP stage to cooperate with Notch gain-of-function driving leukemic transformation. The resulting experimental DNMT3A-deficient T-ALLs had a gene expression signature that resembled human ETP-ALL, which has been noted to have a high rate of DNMT3A mutations.74 Together, these data suggest that DNMT3A loss in early thymic progenitors may prime developing thymocytes for cellular transformation, but it does not play a critical role in early thymopoiesis under most physiologic conditions. The role of DNMT3B has not been assessed in developing T-cells, though it is expressed.75
The TET family of enzymes also play a role in T-cell development (Table 1). Combined loss of TET2 and TET3 (either Tet2−/−TET3fl/flCD4Cre+ or TET2fl/flTET3fl/flCD4Cre+) led to a striking lymphoproliferation of invariant natural killer T (iNKT) cells, ultimately resulting in death by 8 weeks of age.76 iNKT cells are a rare subset of innate-like lymphocytes that develop in the thymus, express a semi-invariant αβ TCR that recognizes glycolipid antigen and have a mature effector phenotype. Similar to CD4+ T-cells, they can be subdivided into functional subsets (NKT1, NKT2, and NKT17) that express unique transcription factors and cytokine profiles (IFNγ, IL-4, and IL-17, respectively).77 In TET2/TET3-deficient mice, the iNKT cell expansion occurred in the thymus, presumptively during T-cell development. The expanded TET2/TET3-deficient iNKT cells were of the NKT17 phenotype, with RORγt expression and IL-17 production, in contrast to TET2 loss in conventional CD4+ T-cells, which results in a decrease in TH17 differentiation compared to wild-type.78 Although 5hmC levels were substantially reduced in the double-knockout iNKT cells, they were not abolished, suggesting that TET1 was active in these cells. A key experiment in which the TET2/TET3-deficient iNKT cells were transferred into immunocompetent wild-type or CD1d-deficient hosts (which lack the MHC that presents glycolipid antigen for iNKT cells) demonstrated the following: (1) The lethal lymphoproliferative disorder was transferable to wild-type hosts, thus supporting that these were malignant cells and wild-type Tregs could not suppress their proliferation. (2) The CD1d-deficient recipients remained healthy demonstrating that antigen was required for expansion. The critical finding that TET2/TET3 restricted an antigen-driven process suggests that the TET proteins may be regulated and function downstream of antigen activation. No studies have been published to date on the role of TET1 in thymocyte development.
IV. CD4+ T-CELL DIFFERENTIATION
CD4+ T-cells orchestrate immune responses through provision of critical help to CD8+ T-cells, B cells, or innate immune cells, typically through cytokine production and/or cell-to-cell interactions. Upon antigen recognition, naïve CD4+ T-cells are activated, proliferate, and differentiate into T helper (TH) subsets with distinct functional capabilities in response to environmental stimuli. These effector TH subsets are classically defined based on expression of ‘master’ transcription factors and their unique cytokine production. TH1 cells express T-bet and produce IFNγ. TH2 cells express GATA-3 and produce IL-4. ROR-γt is critical for TH17 differentiation and IL-17A/IL-17F production. T follicular helper (TFH) cells are defined by Bcl-6 expression and IL-21 production. Regulatory T-cells (Tregs) express Foxp3 and produce IL-10. Although the field has traditionally categorized these TH subsets based on transcriptional regulators and prototypical cytokine production, it has been increasingly recognized that they also undergo extensive epigenomic re-programming during differentiation, including changes in DNA methylation.
A. TH1/TH2 Differentiation
One of the first observations suggesting that DNA methylation plays a critical role in CD4+ T-cell differentiation came from early studies of TH1 versus TH2 differentiation. Several groups noted that the ifng genomic locus is hypomethylated in naïve T-cells and the il4 locus is hypermethylated. Under in vitro TH1-polarizing conditions, this pattern is maintained; however, under TH2 conditions, the ifng locus becomes hypermethylated and the il4 locus is hypomethylated in both human and murine CD4+ T-cells.79–87 Moreover, treatment of low IL-4–expressing TH2 clones with 5-azacytidine, a hypomethylating agent, resulted in increased IL-4 expression, suggesting that the degree of DNA methylation can regulate the magnitude of cytokine production in differentiated CD4+ cells.88 More recently, genome-wide DNA methylation/hydroxymethylation studies demonstrated that murine and human CD4+ T-cells gain 5hmC primarily at intragenic sites during in vitro T-helper differentiation.53,89 In murine cells, gain of 5hmC occurred at lineage-specific gene bodies and correlated with increased expression: TH2 cells had increased 5hmC and decreased 5mC at Gata3 and il4 loci whereas TH1 cells displayed increased 5hmC at ifng and Tbx21 loci.53,78 Together, these data suggest that dynamic DNA methylation remodeling occurs at key lineage-specific loci, supporting a role for DNA methylation in the regulation of TH1 versus TH2 differentiation.
Whether DNA methylation at lineage-specific loci serves as a mechanism to establish TH1 and TH2 cell fates or is a consequence of differentiation was addressed with mechanistic studies examining the loss of DNA methylation regulators. The DNMT family has been implicated in various aspect of TH1 versus TH2 differentiation. DNMT1 loss (mediated by CD4Cre) led to decreased peripheral T-cell proliferation and enhanced expression of IFNγ, IL-2, IL-3, and IL-4 in activated CD4+ (and CD8+) T-cells,71 suggesting that DNMT1 serves to repress cytokine production. Under TH2 polarizing conditions, DNMT1 undergoes dissociation (or decreased recruitment) from the il4 locus, permitting demethylation of the locus and increased expression of IL-4.90 Together, these data indicate that DNMT1 loss at effector cytokine loci is a critical step to allowing IL-4 expression during TH2 differentiation.
In contrast, de novo methylation by DNMT3A does not affect initial TH1 versus TH2 differentiation but does alter cell fate plasticity. Powell and colleagues demonstrated that DNMT3A-deficient naïve CD4+ T-cells develop a population of cells capable of coexpressing IL-4 and IFNγ when they are activated through the TCR in neutral conditions in vitro. These changes were associated with demethylation of both loci. However, under polarizing conditions, loss of DNMT3A did not significantly alter TH1 or TH2 differentiation and no double-producing cytokine population was noted.72 DNMT3A is recruited to the ifng promoter in murine TH2, TH17 and iTreg cells but not in TH1 cells,91 and the ifng locus is unmethylated in DNMT3A-deficient TH2 and TH17 cells.72, 91 However, lack of DNMT3A in these cells was not sufficient to allow IFNγ expression under non-TH1 conditions, suggesting that methylation alone was not enough to suppress IFNγ expression under these conditions. When DNMT3A-deficient TH2 or TH17 cells were re-cultured under TH1 biasing conditions,72,91 there was a consistent increase in IFNγ expression compared to the wild-type, suggesting that de novo methylation by DNMT3A restricts TH plasticity.
TET family members have also been implicated in directing TH1 versus TH2 differentiation. Deletion of TET2, driven by CD2Cre, resulted in increased methylation of an IFNγ enhancer with decreased IFNγ expression following in vitro TH1 polarization, with no effect on Tbx21 transcription (encoding T-bet); however, TET2 loss did not lead to alteration in TH2 differentiation.78 Using chromatin immunoprecipitation, TET2 association and 5hmC was diminished on the ifng locus in T-bet deficient cells,78 suggesting that T-bet may recruit TET2 to mediate hydroxymethylation at the ifng locus and promote IFNγ expression in TH1 cells. Although TET1 regulation of TH1 versus TH2 cell fate has not been tested, overexpression of full-length TET1 (though not TET1 catalytic domain) in human CD4+ T-cells under TH1 polarizing conditions led to a downregulation of IFNγ,89 suggesting a methylation-independent role of TET1 in regulating TH1 cytokine production. The role of TET enzymes in limiting T-helper plasticity has not been evaluated.
B. Treg Differentiation
Regulatory T-cells (Tregs) are a subset of CD4+ T-cells that limit inflammatory reactions. Expression of the Treg “master regulator” FoxP3 is essential for the development and function of Tregs and is controlled through three distinct intronic conserved noncoding sequences (CNS).92–94 CNS2 is rich in CpG elements and particularly controlled by DNA methylation. Originally, it was identified as a highly conserved region that is methylated in conventional CD4+ T-cells but demethylated in Tregs.95,96 Although CNS2 is not required for the development of Tregs, it is required for FoxP3 stability and the maintenance of Treg cell fate.92–94 CNS2 contains cis-regulatory elements that orchestrate the recruitment of transcription factors that promote or inhibit FoxP3 expression.96–98 In addition to the FoxP3 locus, additional Treg specific demethylated regions (TSDRs) have been identified to control expression of Treg-associated genomic loci in thymically derived Tregs, including Tnfsrf18, Ctla4, and Ikzf4 loci, in a FoxP3-independent manner, which occur during development and are not found in induced Tregs (i.e., those that arise in the peripheral lymphoid organs from conventional CD4+ T-cells).99 Thus, DNA methylation patterns control FoxP3 expression and promote the cell fate of thymically derived Tregs.
Given the critical role of DNA methylation in controlling FoxP3 expression and Treg cell fate, the naturally arising question is which epigenetic modifiers play a role in promoting and maintaining the methylation state of key cis-regulatory regions. Pharmacologic inhibition of DNMT activity with 5-azacytidine or genetic loss of DNMT1 promoted the expression of FoxP3 in thymic and peripheral FoxP3-negative CD4+ T-cells upon TCR stimulation.96,100,101 Together, these data support the notion that DNA methylation, likely through DNMT1, plays a key role in maintaining suppression of FoxP3 in non-Tregs. In vivo, male mice with a Treg-specific deletion of DNMT1 developed lethal autoimmunity by 3–4 weeks of age.102 These mice had similar absolute numbers of thymic Tregs but decreased number of peripheral Tregs. The DNMT1-deficient Tregs had a marked loss of suppressive capacity in vitro and in vivo. Although methylation of the FoxP3 CNS2 was unchanged by DNMT1 loss in Tregs, global changes in DNA methylation were associated with loss of expression of several genes critical for Treg function and gain of inflammatory gene expression. Together, these studies support a role for DNMT1 in maintaining appropriate repression of FoxP3 expression in conventional CD4+ T-cells and controlling a Treg gene expression program. Notably, DNMT3A was not required in Tregs in vivo,102 suggesting that maintenance of DNA methylation rather than de novo methylation plays a more critical role in Treg development and function.
Active DNA demethylation through the TET family also serves to regulate Treg cell fate and stability. Initial experiments examining the role of TET in ‘induced’ Treg generation utilized ascorbic acid to promote TET enzymatic function. These studies demonstrated that ascorbic acid promoted FoxP3 expression and stability via demethylation of the Foxp3 CNS2 enhancer in a TET2/TET3 dependent manner.103,104 Mice with a Treg-specific deletion of TET2 and TET3 develop a lethal inflammatory disease, likely due to decreased long-term (but not short-term) suppressive function of thymic Tregs.105 Mechanistically, Tregs from these mice had blunted demethylation at CNS2 of the FoxP3 locus and other TSDRs, suggesting that TET2/TET3 are responsible for demethylation of FoxP3 CNS2 and TSDRs of other Treg lineage genes. Additionally, TET2/TET3 loss led to an increase in ‘ex-Tregs’ (Tregs that had lost FoxP3 expression) consistent with the role of CNS2 FoxP3 and other TSDR methylation in maintaining FoxP3 stability and Treg function. It is likely that all TET family members cooperatively play a role in CNS2 demethylation and FoxP3 stability because TET1/2 double-deficient Tregs also have increased methylation and concomitant loss of Treg stability and function, resulting in increased inflammation and autoimmunity.106 Together, these data point to unequivocal role for TET family members in regulating FoxP3 stability and Treg cell-fate maintenance.
In recent years, it has been increasingly appreciated that Tregs can undergo differentiation into specialized tissue-resident subsets that have distinct tissue-specific gene-expression profiles and functions (see review by Panduro et al.107). Recent work has explored the role of DNA methylation in tissue Treg programming. Genome-wide methylation analysis found that skin and adipose tissue Tregs have distinct DNA methylation profiles compared to Treg and conventional CD4+ T-cells isolated from lymph nodes.108 Interestingly, more differentially methylated regions were noted between tissue Tregs and lymph node Tregs than between lymph node Tregs and conventional CD4+ T-cells. Many of the DMRs were shared between skin and fat Tregs, suggesting either a tissue Treg specific methylation program or effector Treg program. The role of the DNMT and TET families in regulating tissue-specific Treg methylation programming has not yet been explored.
C. TH17 Differentiation
CD4+ T helper cells that secrete IL-17 are termed TH17 cells and are considered proinflammatory. During in vitro TH17 differentiation, CD4+ T-cells undergo dynamic changes in methylation states at lineage-associated loci.78,109 Examining the il17 locus, Wells and colleagues found lineage-specific DNA demethylation in TH17 cells at the il17a and il17f loci. Specifically, demethylation was noted in a conserved enhancer region and at the promoters, and methylation-sensitive binding of STAT3 to the promoter enhanced transcriptional activity.110 Stability of the TH17 program seems to require DNMT3A, as this DNA methyltransferase was critical to suppressing IFNγ production in TH17 cells repolarized under TH1 conditions.91
During in vitro TH17 differentiation, TET2 loss blunted IL-17A expression and was associated with decreased 5hmC deposition and RORγt binding to il17 cis-regulatory regions. There was also a concomitant increase in IL-10 expression in these cells.78 Similar to TH1 cells, deletion of the TH17 lineage-defining transcription factor RORc (encoding RORγt) led to decreased TET2 binding and 5hmC deposition at the il17 locus, suggesting that TET2 recruitment may be mediated by lineage-specific transcriptional regulators.78 Another group found that during in vitro TH17 differentiation, TH17 cells contain increased amounts of the metabolite 2-hydroxyglutarate (2-HG), which inhibits TET1/TET2-mediated demethylation of FoxP3 CNS2, leading to decreased FoxP3 expression and increased IL-17A production under TH17 conditions.111 Taken together, data suggest that TET2 activity can impact TH17 cell differentiation by multiple mechanisms.
D. TFH Differentiation
TFH cells reside in the lymphoid follicle and interact with B cells to facilitate activation and germinal-center B-cell differentiation. At the transcriptional level, TFH development is well characterized, but how epigenetic mechanisms facilitate commitment and maintenance of TFH cells is not as well understood. Genome-wide methylation and hydroxymethylation profiling in naïve and TFH CD4+ T-cells, in combination with Bcl6 chromatin immunoprecipitation, exhibited a reduction in 5hmC at Bcl6 binding site in TFH, but not naïve cells.112 These differences were associated with decreased TET1 recruitment as assessed by chromatin immunoprecipitation, suggesting that Bcl6 may inhibit TET1 localization and activity at target genes; however, there was no assessment of the functional role of TET1 loss or other TET family members in TFH differentiation.
A role for DNA methylation in TFH cell fate is strongly suggested by the high frequency of TET2 and DNMT3a loss-of-function mutations in human TFH-derived lymphomas.59–62,113 Work in murine models has supported a role for TET2 loss being permissive but not sufficient for lymphomagenesis since expression of the dominant negative RhoA G17V mutation frequently found in these lymphomas in the setting of TET2 deficiency drove lymphomagenesis in these models.114–116 Further work needs to be done to elucidate the contributions of TET and DNMT family members to TFH differentiation.
E. CD4+ T-Cell Memory
Given that DNA methylation can frequently be a heritable epigenetic mark, it is intriguing to examine how it may play a role in CD4+ T-cell memory. Although we do not have a complete understanding of how DNA methylation regulates CD4+ T-cell memory, comprehensive epigenomic and transcriptional profiling of human CD4+ T-cell populations has demonstrated progressive loss of DNA methylation in heterochromatic regions across naïve, central memory, effector memory and terminally differentiated effector memory cells.117 In this study, gene-specific differentially methylated regions were localized to enhancer or promoter regions and correlated with differences in the expression of memory-associated genes.117 Murine studies indicate that differentiated CD4+ memory cells (specifically TH1- and TFH- committed cells) can be distinguished based on differential methylation profiles.118 Another study using TCR transgenic CD4+ T-cells assessed genome-wide methylation of in vitro generated “memory” CD4+ T-cells and found that differentially methylated regions were associated with enhancer activity of associated genes, rather than promoter activity.119 Together, these data strongly support a role for DNA methylation globally directing CD4+ memory differentiation; however, the role of individual TET or DNMT family members in directing or maintain CD4+ T-cell memory remains unclear.
V. CD8+ T-CELL DIFFERENTIATION
In response to microbes, naïve CD8+ T-cells proliferate and differentiate into a heterogeneous pool of antigen-specific cells having divergent cell fates. Following pathogen clearance, most antigen-specific effector CD8+ T-cells are terminally differentiated and undergo programmed cell death. A small subset persists to become long-lived memory cells, which are capable of rapidly responding to rechallenge. Elegant work by multiple groups has identified different cell surface proteins that can be used to distinguish cells with differing memory potential. CD8+ T-cells that are CD127hi and KLRG1lo preferentially differentiate into long-lived memory cells; whereas CD127lo KLRG1hi cells are primarily short-lived, terminally differentiated effector cells.120–122 Memory CD8+ T-cells have been further subdivided into CD62L+ ‘central memory’ (TCM) and CD62L− ‘effector memory’ (TEM), with TCM cells having the ability to self-renew and confer long-term immune protection.123
As murine and human CD8+ T-cells differentiate, they undergo genome-wide epigenetic reprogramming, including changes in the DNA methylation landscape. Using methylated DNA immunoprecipitation, Scharer and colleagues demonstrated that antigen-specific CD8+ T-cells undergo dynamic DNA remodeling during the naïve to effector CD8+ T-cell transition following LCMV-Armstrong infection.124 These researchers demonstrated that thymic enhancers (as marked by H3K4me1 and H3K27ac in ENCODE) underwent both methylation and demethylation, and they noted more differentially methylated regions occurred at active thymic enhancers. Additionally, DNA methylation at proximal promoter regions was inversely correlate with gene expression during the naïve to effector transition. The differentially methylated regions at both putative enhancer regions and gene promoters for differentially expressed genes were enriched for functional transcription factor motifs. Together, these data suggest that remodeling of DNA methylation plays a functional role in CD8+ T-cell differentiation.
Building on these findings, Youngblood and colleagues utilized whole-genome bisulfite sequencing to examine the methylation programs of viral-specific naïve, short-lived effector CD8+ T-cells and memory precursor CD8+ T-cells at days 4.5 and 8 following LCMV-Armstrong infection. These studies revealed that memory precursor CD8+ T-cells acquire a methylation program similar to terminally differentiated effector cells within the first several days of infection, supporting the hypothesis that memory precursor cells transition through an effector phase as they undergo memory differentiation.125 Additionally, they found that DNMT3A-deficient CD8+ T-cells underwent more rapid transition to memory cells compared to the wild-type. These data are consistent with an earlier report demonstrating DNMT3A loss promotes early CD8+ T-cell memory development following viral infection. In this report, TCF-1 was proposed as a potential DNMT3a target responsible for directing wild-type CD8+ T-cells toward an effector phenotype, and although knockdown of TCF7 (which encodes TCF-1) did restore some effector T-cell differentiation in DNMT3A-deficient CD8+ T-cells, the restoration was only partial, indicating that DNMT3A may target a wider array of genes that work together to direct CD8+ effector T-cell differentiation.126
Whole-genome bisulfite sequencing of human naïve and memory CD8+ T-cell subsets revealed that naïve, stem-cell memory, central memory, and effector memory subsets are epigenetically distinct, with a progressive decline in DNA methylation related to terminally differentiated state of cells (i.e., TEM with the most demethylated regions).127 To examine the stability of human memory CD8+ T-cells in vivo, these researchers examined donor-derived memory CD8+ T-cells before and after CD45RA+ depleted haploidentical stem-cell transplants. Cells were isolated from the pre-transplant donor sample and then in vivo expanded donor-derived cells two months post transplant, isolated from the recipient. Methylation status at effector loci (e.g., IFNγ, perforin) was stable in the expanded memory cells supporting the notion that transcriptionally permissive epigenetic programs are maintained at effector loci during in vivo homeostasis. Interestingly, most of the expanded memory cells had a TEM phenotype with increased methylation at differentially methylated regions in loci encoding CD62L and CCR7 despite the pre-transfer samples having a mix of TEM and TCM cells. In conjunction with their in vitro findings, these data suggest that homeostatic proliferation may lead to an interconversion of TCM to TEM cells and that regulators of DNA methylation play a role in the active maintenance of CD8+ T-cell memory subsets.
CD8+ T-cell differentiation is also regulated by active DNA demethylation. TET2 conditional knockout mice (deletion driven by CD4Cre), show no apparent differences in thymocyte or peripheral T-cell subsets, suggesting that TET2 does not play a critical role in T-cell homeostasis. However, TET2 transcripts are rapidly upregulated by TCR signaling and TET2 loss dramatically altered antigen-driven CD8+ T-cell differentiation. We recently demonstrated that the loss of TET2 alters the DNA methylation landscape in activated viral-specific CD8+ T-cells and regulates CD8+ T-cell differentiation in a cell-intrinsic manner. Intriguingly, despite opposing enzymatic function, TET2, like DNMT3A, also represses memory CD8+ T-cell development. Specifically, TET2 deficiency led to enhanced TCM CD8+ T-cell differentiation and promoted secondary recall responses.36 Enhanced reduced-representation bisulfite sequencing of viral-specific CD8+ T-cells revealed differential methylation largely of intronic regions across the genome. Several known transcriptional regulators of CD8+ T-cell effector versus memory differentiation were associated with differentially methylated regions. Although interesting potential targets have been identified, the disruption of chromatin modifying genes, such as TET2 (and DNMT3A), is likely to impact a wide number of genes that contribute to CD8+ T-cell differentiation. In contrast to the finding that TET2-deficient CD4+ T-cells had decreased TH1 differentiation and decreased IFNγ production,78 TET2-deficient CD8+ T-cells had increased IFNγ production,36 suggesting possible cell-type specific or contextual effects. The phenomenon that DNMT3A and TET2 loss promotes similar (although not identical) cellular phenotypes despite opposite roles in regulating DNA methylation has also been observed in HSCs.128,129 The explanation for this phenomenon has not yet been elucidated, although recent work has explored the epigenetic underpinning of this observation. In HSCs, Zhang and colleagues examined wild-type, DNMT3A-deficient, TET2-deficient, and double-knockout cells to evaluate the interaction between these epigenetic regulators. The study identified a complex interaction with functional, genomic, and methylation/hydroxymethylation profiling suggesting a combination of independent and interdependent roles.130 Another study examined methylation patterns in different TET-deficient cell types and found that hypermethylation typically occurred in the active euchromatic compartment, but paradoxically, they noted hypomethylation in the heterochromatic regions.131 In comparing single TET2 or DNMT3A-deficient HSCs to double-deficient HSCs, both TET2 loss and DNMT3A loss resulted in hypomethylation in the heterochromatin genome-wide. Bioinformatic reanalysis of published chromatin-immunoprecipitation data of tagged DNMT3A1 (a splice variant of DNMT3A) in wild-type and TET1-deficient murine embryonic stem cells132 revealed that DNMT3A and TET1 occupy mutually exclusive positions in the euchromatin and that DNMT3A1 relocalizes from the heterochromatic compartment to the euchromatin compartment in TET1-deficient cells,131 suggesting that DNMT3A and TET proteins may compete for localization to active euchromatic regions.
Importantly, murine studies of T-cell TET2-deficiency share characteristics with human T-cells that have lost expression of functional TET2 protein. Recently, chance disruption of TET2 in a CAR–T-cell infusion product was associated with near clonal expansion of the TET2-disrupted T-cell clone. The appearance of this expanded population correlated with tumor clearance and long-term survival.133 Similar to murine studies, these cells displayed a TCM phenotype in vivo, and experimental knock-down of TET2 in control human T-cells in vitro also promoted central memory differentiation.133
These data are also in line with studies utilizing the metabolite 2-HG, which inhibits α-ketoglutarate dependent enzymes, including TET family members.134 This metabolite is made by activated CD8+ T-cells following T-cell activation135 and treatment of CD8+ T-cells with the S-enantiomer of 2-HG resulted in upregulation of memory-associated molecules CD62L, CD127, and eomesodermin,135 similar to the in vivo phenotype in TET2-deficient CD8+ T-cells. Additionally, S-2-HG treatment resulted in superior persistence and antitumor efficacy of CD8+ T-cells in a model of adoptive cellular therapy. Together, these data raise the possibility that CD8+ T-cells rely on a metabolic mechanism to suppress TET2 activity to promote memory CD8+ T-cell fates.
Given the important role of DNA methylation and DNA methylation-modifying enzymes in T-cell development and differentiation in response to acute antigen exposure and viral challenge, it is not surprising that DNA methylation also impacts CD8+ T-cell responses to chronic infections. In the face of continuous antigen exposure, for instance in the setting of chronic infection or malignancy, CD8+ T-cells undergo a process termed ‘T-cell exhaustion’ (see review by McLane et al.136). This state is characterized by expression of multiple inhibitory receptors coupled with progressive loss of effector function, including cytokine production, cytolytic capacity, and ability to proliferate. Exhausted T-cells also have a chromatin landscape that is distinct from other T-cell subsets, with several exhaustion-specific regions having been identified.137,138 Whole-genome bisulfite sequencing (WGBS) of T-cells during chronic viral infection also revealed that DNA methylation is re-established at specific loci (e.g., ifng and myc) in exhausted T-cells and that much of this remethylation is dependent on de novo methylation, inasmuch as these changes are substantially reduced in DNMT3A-deficient T-cells. Consistent with loss of methylated regions, some features of exhaustion are less severe in the absence of DNMT3A, including cytokine loss and proliferative capacity.139
It is well known that immune checkpoint blockade, such as treatment with anti–PD-1 or anti–PL-L1 antibodies, can reverse T-cell exhaustion and lead to clinical responses in a subset of patients with certain cancers; however, for most patients, immune checkpoint blockade does not establish long-term control of their cancer. One contributing factor to this failure is likely the inability of PD-1 blockade to substantially alter the epigenetic landscape of exhausted T-cells as assessed by ATAC-seq137 and by WGBS.139 These findings have fueled interest in determining whether modulating epigenetic factors might be beneficial in combination with immune checkpoint blockade. In the case of DNA methylation and DNMT3A, combination of DNMT3A deficiency and PD-1 blockade therapy, or DNMT3A deficiency and treatment with the DNA methyltransferase inhibitor decitabine prior to PD-1 blockade, reinvigorated exhausted CD8+ T-cell to a greater extent than PD-1 blockade alone in chronic viral infection and murine tumor models, respectively.139 Ongoing clinical trials will determine whether this approach can improve clinical efficacy in human cancer patients. Early results from one early-phase clinical trial examining the combination of low-dose decitabine with an novel PD-1 blocking antibody versus PD-1 inhibition alone in patients with relapsed/refractory Hodgkin lymphoma found a similar response rate but improved complete response rate (71% vs. 32%) in patients treated with dual DNA methylation inhibition and PD-1 inhibition.140 Long-term results will be needed to determine duration of response and whether the mechanism functions through enhanced reinvigoration of exhausted antitumor T-cells. Together, these data support a role for DNA methylation in enforcing T-cell exhaustion and may present an attractive therapeutic target to help improve the clinical efficacy of immune checkpoint blockade.
VI. CONCLUDING REMARKS
T-cells are critical mediators of immunity and immunologic memory. Their cell fates are regulated in part through epigenetic mechanisms, including DNA methylation. Recent genome-wide methylation analyses have revealed dynamic alterations in the methylome at various stages of development and differentiation. Additionally, individual DNA modifiers have been implicated in directing different aspects of cell-fate decision, function, and stability. Because T-cells are dysregulated in various disease states (e.g., autoimmune disorders, chronic infections, and cancer), a more complete understanding of how epigenomic programming contributes to these pathologic states is essential. Targeting different DNA modifying enzymes may have potential for modulating immune responses in various clinical settings, including enhancing T-cell regeneration after myeloablative stem-cell transplant and improving immunotherapeutic responses for the treatment of cancer.
ACKNOWLEDGMENTS
We thank J. Paige Gronevelt for careful reading of this manuscript. This work was supported in part through grants from the National Institutes of Health R21AI144732 (MSJ), K08 AI1011008 (S.A.C), Emerson Collective (MSJ) and the American Society of Hematology (SAC). LOC is partially supported by a training grant from the NIAID T32 AI 007413.
REFERENCES
- 1.Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 2010. May 11;107(19):8689–94. Epub 2010/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010. May 14;328(5980):916–19. Epub 2010/04/17. [DOI] [PubMed] [Google Scholar]
- 3.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998. July;19(3):219–20. Epub 1998/07/14. [DOI] [PubMed] [Google Scholar]
- 4.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999. October 29;99(3):247–57. Epub 1999/11/11. [DOI] [PubMed] [Google Scholar]
- 5.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001. September 15;29(18):3784–95. Epub 2001/09/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeltsch A Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002. April 2;3(4):274–93. Epub 2002/04/05. [DOI] [PubMed] [Google Scholar]
- 7.Gowher H, Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol. 2001. June 22;309(5):1201–8. Epub 2001/06/12. [DOI] [PubMed] [Google Scholar]
- 8.Bashtrykov P, Jankevicius G, Smarandache A, Jurkowska RZ, Ragozin S, Jeltsch A. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem Biol. 2012. May 25;19(5):572–78. Epub 2012/05/29. [DOI] [PubMed] [Google Scholar]
- 9.Fatemi M, Hermann A, Pradhan S, Jeltsch A. The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J Mol Biol. 2001. June 22;309(5):1189–99. Epub 2001/06/12. [DOI] [PubMed] [Google Scholar]
- 10.Song J, Teplova M, Ishibe-Murakami S, Patel DJ. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012. February 10;335(6069):709–12. Epub 2012/02/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeshita K, Suetake I, Yamashita E, Suga M, Narita H, Nakagawa A, Tajima S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc Natl Acad Sci U S A. 2011. May 31;108(22):9055–59. Epub 2011/04/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song J, Rechkoblit O, Bestor TH, Patel DJ. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011. February 25;331(6020):1036–40. Epub 2010/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishiyama S, Nishiyama A, Saeki Y, Moritsugu K, Morimoto D, Yamaguchi L, Arai N, Matsumura R, Kawakami T, Mishima Y, Hojo H, Shimamura S, Ishikawa F, Tajima S, Tanaka K, Ariyoshi M, Shirakawa M, Ikeguchi M, Kidera A, Suetake I, Arita K, Nakanishi M. Structure of the Dnmt1 reader module complexed with a unique two-mono-ubiquitin mark on histone H3 reveals the basis for DNA methylation maintenance. Mol Cell. 2017. October 19;68(2):350–60 e7. Epub 2017/10/21. [DOI] [PubMed] [Google Scholar]
- 14.Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007. September 21;317(5845):1760–64. Epub 2007/08/04. [DOI] [PubMed] [Google Scholar]
- 15.Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, Koseki H. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007. December 6;450(7171):908–12. Epub 2007/11/13. [DOI] [PubMed] [Google Scholar]
- 16.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997. September 26;277(5334):1996–2000. Epub 1997/09/26. [DOI] [PubMed] [Google Scholar]
- 17.Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992. November 27;71(5):865–73. Epub 1992/11/27. [DOI] [PubMed] [Google Scholar]
- 18.Pradhan M, Esteve PO, Chin HG, Samaranayke M, Kim GD, Pradhan S. CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry. 2008. September 23;47(38):10000–9. Epub 2008/08/30. [DOI] [PubMed] [Google Scholar]
- 19.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994. January 28;76(2):357–69. Epub 1994/01/28. [DOI] [PubMed] [Google Scholar]
- 20.Qiu C, Sawada K, Zhang X, Cheng X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat Struct Biol. 2002. March;9(3):217–24. Epub 2002/02/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purdy MM, Holz-Schietinger C, Reich NO. Identification of a second DNA binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion. Arch Biochem Biophys. 2010. June 1;498(1):13–22. Epub 2010/03/17. [DOI] [PubMed] [Google Scholar]
- 22.Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010. August 20;285(34):26114–20. Epub 2010/06/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge YZ, Pu MT, Gowher H, Wu HP, Ding JP, Jeltsch A, Xu GL. Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J Biol Chem. 2004. June 11;279(24):25447–54. Epub 2004/03/05. [DOI] [PubMed] [Google Scholar]
- 24.Chen T, Tsujimoto N, Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol. 2004. October;24(20):9048–58. Epub 2004/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirohzu H, Kubota T, Kumazawa A, Sado T, Chijiwa T, Inagaki K, Suetake I, Tajima S, Wakui K, Miki Y, Hayashi M, Fukushima Y, Sasaki H. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am J Med Genet. 2002. September 15;112(1):31–37. Epub 2002/09/20. [DOI] [PubMed] [Google Scholar]
- 26.Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009. November;10(11):1235–41. Epub 2009/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001. May 15;20(10):2536–44. Epub 2001/05/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C, Gutierrez A, Danovi D, Bernard D, Boon T, Pelicci PG, Amati B, Kouzarides T, de Launoit Y, Di Croce L, Fuks F. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005. January 26;24(2):336–46. Epub 2004/12/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tajima S, Suetake I, Takeshita K, Nakagawa A, Kimura H. Domain structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA Methyltransferases. Adv Exp Med Biol. 2016;945: 63–86. Epub 2016/11/09. [DOI] [PubMed] [Google Scholar]
- 30.Guo X, Wang L, Li J, Ding Z, Xiao J, Yin X, He S, Shi P, Dong L, Li G, Tian C, Wang J, Cong Y, Xu Y. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature. 2015. January 29;517(7536):640–44. Epub 2014/11/11. [DOI] [PubMed] [Google Scholar]
- 31.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia. 2003. March;17(3):637–41. Epub 2003/03/21. [DOI] [PubMed] [Google Scholar]
- 32.Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002. July 15;62(14):4075–80. Epub 2002/07/19. [PubMed] [Google Scholar]
- 33.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009. May 15;324(5929):930–35. Epub 2009/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009. June 1;8(11):1698–710. Epub 2009/05/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H, Hogan PG, Aravind L, Rao A. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013. May 02;497(7447):122–26. Epub 2013/04/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E, Wells AD, Wherry EJ, Koretzky GA, Jordan MS. The loss of TET2 promotes CD8(+) T-cell memory differentiation. J Immunol. 2018. January 1;200(1):82–91. Epub 2017/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011. September 2;333(6047):1303–7. Epub 2011/08/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011. September 2;333(6047):1300–3. Epub 2011/07/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010. August 26;466(7310):1129–33. Epub 2010/07/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012. June;40(11):4841–49. Epub 2012/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otani J, Kimura H, Sharif J, Endo TA, Mishima Y, Kawakami T, Koseki H, Shirakawa M, Suetake I, Tajima S. Cell cycle-dependent turnover of 5-hydroxymethyl cytosine in mouse embryonic stem cells. PLoS One. 2013;8(12):e82961. Epub 2013/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seiler CL, Fernandez J, Koerperich Z, Andersen MP, Kotandeniya D, Nguyen ME, Sham YY, Tretyakova NY. Maintenance DNA methyltransferase activity in the presence of oxidized forms of 5-methylcytosine: structural basis for ten eleven translocation-mediated DNA demethylation. Biochemistry. 2018. October 23;57(42):6061–69. Epub 2018/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011. October 14;286(41):35334–38. Epub 2011/08/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weber AR, Krawczyk C, Robertson AB, Kusnierczyk A, Vagbo CB, Schuermann D, Klungland A, Schar P. Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun. 2016. March 2;7:10806. Epub 2016/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, Luo C, Jiang H, He C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012. February 12;8(4):328–30. Epub 2012/02/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009. May 15;324(5929):929–30. Epub 2009/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011. January;29(1):68–72. Epub 2010/12/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010. December 23;5(12):e15367. Epub 2011/01/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Munzel M, Wagner M, Muller M, Khan F, Eberl HC, Mensinga A, Brinkman AB, Lephikov K, Muller U, Walter J, Boelens R, van Ingen H, Leonhardt H, Carell T, Vermeulen M. Dynamic readers for 5-(hydroxy) methylcytosine and its oxidized derivatives. Cell. 2013. February 28;152(5):1146–59. Epub 2013/02/26. [DOI] [PubMed] [Google Scholar]
- 50.Iurlaro M, Ficz G, Oxley D, Raiber EA, Bachman M, Booth MJ, Andrews S, Balasubramanian S, Reik W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14(10):R119. Epub 2013/10/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mellen M, Ayata P, Heintz N. 5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. Proc Natl Acad Sci U S A. 2017. September 12;114(37):E7812–E7821. Epub 2017/08/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010. June;38(11):e125. Epub 2010/04/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsagaratou A, Aijo T, Lio CW, Yue X, Huang Y, Jacobsen SE, Lahdesmaki H, Rao A. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A. 2014. August 12;111(32):E3306–E3315. Epub 2014/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, Huberman K, Thomas S, Dolgalev I, Heguy A, Paietta E, Le Beau MM, Beran M, Tallman MS, Ebert BL, Kantarjian HM, Stone RM, Gilliland DG, Crispino JD, Levine RL. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009. July 2;114(1):144–47. Epub 2009/05/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, Hanson CA, Pardanani A, Gilliland DG, Levine RL. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009. July;23(7):1343–45. Epub 2009/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, Maciejewski JP, Rao A. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010. December 09;468(7325):839–43. Epub 2010/11/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O’Laughlin M, McMichael JF, Delehaunty KD, Mc-Grath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ, Swift GW, Reed JP, Alldredge PA, Wylie T, Walker J, Kalicki J, Watson MA, Heath S, Shannon WD, Varghese N, Nagarajan R, Westervelt P, Tomasson MH, Link DC, Graubert TA, DiPersio JF, Mardis ER, Wilson RK. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010. December 16;363(25):2424–33. Epub 2010/11/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O’Laughlin M, Kandoth C, Baty J, Westervelt P, DiPersio JF, Mardis ER, Wilson RK, Ley TJ, Graubert TA. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011. July;25(7):1153–58. Epub 2011/03/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, Tournillac O, Rousset T, Fabiani B, Cairns RA, Mak T, Bastard C, Bernard OA, de Leval L, Gaulard P. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012. August 16;120(7):1466–69. Epub 2012/07/05. [DOI] [PubMed] [Google Scholar]
- 60.Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012. January 5;366(1):95–96. Epub 2012/01/06. [DOI] [PubMed] [Google Scholar]
- 61.Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, Jiang X, Lossos IS, Nicolas C, Balbin M, Bastard C, Bhagat G, Piris MA, Campo E, Bernard OA, Rabadan R, Ferrando AA. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T-cell lymphomas. Nat Genet. 2014. February;46(2):166–70. Epub 2014/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, Kim S, van Bodegom D, Bolla S, Schatz JH, Teruya-Feldstein J, Hochberg E, Louissaint A, Dorfman D, Stevenson K, Rodig SJ, Piccaluga PP, Jacobsen E, Pileri SA, Harris NL, Ferrero S, Inghirami G, Horwitz SM, Weinstock DM. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014. February 27;123(9):1293–96. Epub 2013/12/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T-cell identity. Nat Rev Immunol. 2014. August;14(8):529–45. Epub 2014/07/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T-cells in the thymus. Annu Rev Immunol. 2012;30:95–114. Epub 2011/12/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, Rossi DJ, Inlay MA, Serwold T, Karsunky H, Ho L, Daley GQ, Weissman IL, Feinberg AP. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010. September 16;467(7313):338–42. Epub 2010/08/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, Nerlov C, Leutz A, Andrade-Navarro MA, Jacobsen SE, Rosenbauer F. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009. November;41(11):1207–15. Epub 2009/10/06. [DOI] [PubMed] [Google Scholar]
- 67.Issuree PD, Ng CP, Littman DR. Heritable gene regulation in the CD4:CD8 T-cell lineage choice. Front Immunol. 2017;8:291. Epub 2017/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sellars M, Huh JR, Day K, Issuree PD, Galan C, Gobeil S, Absher D, Green MR, Littman DR. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T-cell lineages. Nat Immunol. 2015. July;16(7):746–54. Epub 2015/06/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Issuree PD, Day K, Au C, Raviram R, Zappile P, Skok JA, Xue HH, Myers RM, Littman DR. Stage-specific epigenetic regulation of CD4 expression by coordinated enhancer elements during T-cell development. Nat Commun. 2018. September 5;9(1):3594. Epub 2018/09/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodriguez RM, Suarez-Alvarez B, Mosen-Ansorena D, Garcia-Peydro M, Fuentes P, Garcia-Leon MJ, Gonzalez-Lahera A, Macias-Camara N, Toribio ML, Aransay AM, Lopez-Larrea C. Regulation of the transcriptional program by DNA methylation during human alphabeta T-cell development. Nucleic Acids Res. 2015. January;43(2):760–74. Epub 2014/12/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T-cell development, function, and survival. Immunity. 2001. November;15(5):763–74. Epub 2001/12/01. [DOI] [PubMed] [Google Scholar]
- 72.Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyltransferase 3a as a T-cell receptor-induced regulator of Th1 and Th2 differentiation. J Immunol. 2009. August 15;183(4):2267–76. Epub 2009/07/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kramer AC, Kothari A, Wilson WC, Celik H, Nikitas J, Mallaney C, Ostrander EL, Eultgen E, Martens A, Valentine MC, Young AL, Druley TE, Figueroa ME, Zhang B, Challen GA. Dnmt3a regulates T-cell development and suppresses T-ALL transformation. Leukemia. 2017. November;31(11):2479–90. Epub 2017/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D, Konstandin NP, Ksienzyk B, Vosberg S, Graf A, Krebs S, Blum H, Raff T, Bruggemann M, Hofmann WK, Hecht J, Bohlander SK, Greif PA, Baldus CD. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013. June 6;121(23):4749–52. Epub 2013/04/23. [DOI] [PubMed] [Google Scholar]
- 75.Heng TS, Painter MW, Immunological Genome Project C. The immunological genome project: networks of gene expression in immune cells. Nat Immunol. 2008. October;9(10):1091–94. Epub 2008/09/19. [DOI] [PubMed] [Google Scholar]
- 76.Tsagaratou A, Gonzalez-Avalos E, Rautio S, Scott-Browne JP, Togher S, Pastor WA, Rothenberg EV, Chavez L, Lahdesmaki H, Rao A. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat Immunol. 2017. January;18(1):45–53. Epub 2016/11/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol. 2013. November;14(11):1146–54. Epub 2013/10/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, Tian Q, Aifantis I, Wei L, Dong C. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T-cells. Immunity. 2015. April 21;42(4):613–26. Epub 2015/04/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jones B, Chen J. Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J. 2006. June 7;25(11):2443–52. Epub 2006/05/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Young HA, Ghosh P, Ye J, Lederer J, Lichtman A, Gerard JR, Penix L, Wilson CB, Melvin AJ, McGurn ME. Differentiation of the T helper phenotypes by analysis of the methylation state of the IFN-gamma gene. J Immunol. 1994. October 15;153(8):3603–10. Epub 1994/10/15. [PubMed] [Google Scholar]
- 81.Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T-cell differentiation. Immunity. 1998. December;9(6):765–75. Epub 1999/01/09. [DOI] [PubMed] [Google Scholar]
- 82.Bird JJ, Brown DR, Mullen AC, Moskowitz NH, Mahowald MA, Sider JR, Gajewski TF, Wang CR, Reiner SL. Helper T-cell differentiation is controlled by the cell cycle. Immunity. 1998. August;9(2):229–37. Epub 1998/09/05. [DOI] [PubMed] [Google Scholar]
- 83.Lee DU, Agarwal S, Rao A. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity. 2002. May;16(5):649–60. Epub 2002/06/07. [DOI] [PubMed] [Google Scholar]
- 84.Winders BR, Schwartz RH, Bruniquel D. A distinct region of the murine IFN-gamma promoter is hypomethylated from early T-cell development through mature naive and Th1 cell differentiation, but is hypermethylated in Th2 cells. J Immunol. 2004. December 15;173(12):7377–84. Epub 2004/12/09. [DOI] [PubMed] [Google Scholar]
- 85.Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, Stamatoyannopoulos JA, Wilson CB. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol. 2007. July;8(7):732–42. Epub 2007/06/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim ST, Fields PE, Flavell RA. Demethylation of a specific hypersensitive site in the Th2 locus control region. Proc Natl Acad Sci U S A. 2007. October 23;104(43):17052–57. Epub 2007/10/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Santangelo S, Cousins DJ, Winkelmann NE, Staynov DZ. DNA methylation changes at human Th2 cytokine genes coincide with DNase I hypersensitive site formation during CD4(+) T-cell differentiation. J Immunol. 2002. August 15;169(4):1893–903. Epub 2002/08/08. [DOI] [PubMed] [Google Scholar]
- 88.Guo L, Hu-Li J, Zhu J, Watson CJ, Difilippantonio MJ, Pannetier C, Paul WE. In TH2 cells the Il4 gene has a series of accessibility states associated with distinctive probabilities of IL-4 production. Proc Natl Acad Sci U S A. 2002. August 6;99(16):10623–28. Epub 2002/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nestor CE, Lentini A, Hagg Nilsson C, Gawel DR, Gustafsson M, Mattson L, Wang H, Rundquist O, Meehan RR, Klocke B, Seifert M, Hauck SM, Laumen H, Zhang H, Benson M. 5-hydroxymethylcytosine remodeling precedes lineage specification during differentiation of human CD4(+) T-cells. Cell Rep. 2016. July 12;16(2):559–70. Epub 2016/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Makar KW, Perez-Melgosa M, Shnyreva M, Weaver WM, Fitzpatrick DR, Wilson CB. Active recruitment of DNA methyltransferases regulates interleukin 4 in thymocytes and T-cells. Nat Immunol. 2003. December;4(12):1183–90. Epub 2003/11/05. [DOI] [PubMed] [Google Scholar]
- 91.Thomas RM, Gamper CJ, Ladle BH, Powell JD, Wells AD. De novo DNA methylation is required to restrict T helper lineage plasticity. J Biol Chem. 2012. June 29;287(27):22900–9. Epub 2012/05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li X, Liang Y, LeBlanc M, Benner C, Zheng Y. Function of a Foxp3 cis-element in protecting regulatory T-cell identity. Cell. 2014. August 14;158(4):734–48. Epub 2014/08/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010. February 11;463(7282):808–12. Epub 2010/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T-cell identity by a cis element in the Foxp3 locus. Cell. 2014. August 14;158(4):749–63. Epub 2014/08/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J. Epigenetic control of the foxp3 locus in regulatory T-cells. PLoS Biol. 2007. February;5(2):e38. Epub 2007/02/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim HP, Leonard WJ. CREB/ATF-dependent T-cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007. July 9;204(7):1543–51. Epub 2007/06/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008. February;9(2):194–202. Epub 2007/12/25. [DOI] [PubMed] [Google Scholar]
- 98.Ogawa C, Tone Y, Tsuda M, Peter C, Waldmann H, Tone M. TGF-beta-mediated Foxp3 gene expression is cooperatively regulated by Stat5, Creb, and AP-1 through CNS2. J Immunol. 2014. January 1;192(1):475–83. Epub 2013/12/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, Osaki M, Tanaka Y, Yamashita R, Nakano N, Huehn J, Fehling HJ, Sparwasser T, Nakai K, Sakaguchi S. T-cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012. November 16;37(5):785–99. Epub 2012/11/06. [DOI] [PubMed] [Google Scholar]
- 100.Josefowicz SZ, Wilson CB, Rudensky AY. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J Immunol. 2009. June 1;182(11):6648–52. Epub 2009/05/21. [DOI] [PubMed] [Google Scholar]
- 101.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008. June;38(6):1654–63. Epub 2008/05/22. [DOI] [PubMed] [Google Scholar]
- 102.Wang L, Liu Y, Beier UH, Han R, Bhatti TR, Akimova T, Hancock WW. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood. 2013. May 2;121(18):3631–39. Epub 2013/02/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sasidharan Nair V, Song MH, Oh KI. Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. J Immunol. 2016. March 1;196(5):2119–31. Epub 2016/01/31. [DOI] [PubMed] [Google Scholar]
- 104.Yue X, Trifari S, Aijo T, Tsagaratou A, Pastor WA, Zepeda-Martinez JA, Lio CW, Li X, Huang Y, Vijayanand P, Lahdesmaki H, Rao A. Control of Foxp3 stability through modulation of TET activity. J Exp Med. 2016. March 7;213(3):377–97. Epub 2016/02/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yue X, Lio CJ, Samaniego-Castruita D, Li X, Rao A. Loss of TET2 and TET3 in regulatory T-cells unleashes effector function. Nat Commun. 2019. May 1;10(1):2011. Epub 2019/05/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, Chen C, Liu S, Liu D, Chen Y, Zandi E, Chen W, Zhou Y, Shi S. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T-cell differentiation and maintain immune homeostasis. Immunity. 2015. August 18;43(2):251–63. Epub 2015/08/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol. 2016. May 20;34:609–33. Epub 2016/05/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Trager U, Hofer AC, Kagebein D, Wang Q, Frauhammer F, Mallm JP, Bauer K, Herrmann C, Lang PA, Brors B, Plass C, Feuerer M. Genome-wide DNA-methylation landscape defines specialization of regulatory T-cells in tissues. Nat Immunol. 2017. October;18(10):1160–72. Epub 2017/08/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang BH, Floess S, Hagemann S, Deyneko IV, Groebe L, Pezoldt J, Sparwasser T, Lochner M, Huehn J. Development of a unique epigenetic signature during in vivo Th17 differentiation. Nucleic Acids Res. 2015. February 18;43(3):1537–48. Epub 2015/01/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thomas RM, Sai H, Wells AD. Conserved intergenic elements and DNA methylation cooperate to regulate transcription at the il17 locus. J Biol Chem. 2012. July 20;287(30):25049–59. Epub 2012/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, Li K, Ma T, Wang H, Ni L, Zhu S, Cao N, Zhu D, Zhang Y, Akassoglou K, Dong C, Driggers EM, Ding S. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature. 2017. August 10;548(7666):228–33. Epub 2017/08/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu X, Lu H, Chen T, Nallaparaju KC, Yan X, Tanaka S, Ichiyama K, Zhang X, Zhang L, Wen X, Tian Q, Bian XW, Jin W, Wei L, Dong C. Genome-wide analysis identifies Bcl6-controlled regulatory networks during T-follicular helper-cell differentiation. Cell Rep. 2016. February 23;14(7):1735–47. Epub 2016/02/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y, Sakata S, Kamada Y, Nakamoto-Matsubara R, Tran NB, Izutsu K, Sato Y, Ohta Y, Furuta J, Shimizu S, Komeno T, Sato Y, Ito T, Noguchi M, Noguchi E, Sanada M, Chiba K, Tanaka H, Suzukawa K, Nanmoku T, Hasegawa Y, Nureki O, Miyano S, Nakamura N, Takeuchi K, Ogawa S, Chiba S. Somatic RHOA mutation in angioimmunoblastic T-cell lymphoma. Nat Genet. 2014. February;46(2):171–75. Epub 2014/01/15. [DOI] [PubMed] [Google Scholar]
- 114.Zang S, Li J, Yang H, Zeng H, Han W, Zhang J, Lee M, Moczygemba M, Isgandarova S, Yang Y, Zhou Y, Rao A, You MJ, Sun D, Huang Y. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T-cell homeostasis. J Clin Invest. 2017. August 1;127(8):2998–3012. Epub 2017/07/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cortes JR, Ambesi-Impiombato A, Couronne L, Quinn SA, Kim CS, da Silva Almeida AC, West Z, Belver L, Martin MS, Scourzic L, Bhagat G, Bernard OA, Ferrando AA, Palomero T. RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell. 2018. February 12;33(2):259–73 e7. Epub 2018/02/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ng SY, Brown L, Stevenson K, deSouza T, Aster JC, Louissaint A Jr, Weinstock DM. RhoA G17V is sufficient to induce autoimmunity and promotes T-cell lymphomagenesis in mice. Blood. 2018. August 30;132(9):935–47. Epub 2018/05/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Durek P, Nordstrom K, Gasparoni G, Salhab A, Kressler C, de Almeida M, Bassler K, Ulas T, Schmidt F, Xiong J, Glazar P, Klironomos F, Sinha A, Kinkley S, Yang X, Arrigoni L, Amirabad AD, Ardakani FB, Feuerbach L, Gorka O, Ebert P, Muller F, Li N, Frischbutter S, Schlickeiser S, Cendon C, Frohler S, Felder B, Gasparoni N, Imbusch CD, Hutter B, Zipprich G, Tauchmann Y, Reinke S, Wassilew G, Hoffmann U, Richter AS, Sieverling L, Consortium D, Chang HD, Syrbe U, Kalus U, Eils J, Brors B, Manke T, Ruland J, Lengauer T, Rajewsky N, Chen W, Dong J, Sawitzki B, Chung HR, Rosenstiel P, Schulz MH, Schultze JL, Radbruch A, Walter J, Hamann A, Polansky JK. Epigenomic profiling of human CD4(+) T-cells supports a linear differentiation model and highlights molecular regulators of memory development. Immunity. 2016. November 15;45(5):1148–61. Epub 2016/11/17. [DOI] [PubMed] [Google Scholar]
- 118.Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, Wu T, Iyer SS, Ahmed R. Distinct memory CD4+ T-cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity. 2013. April 18;38(4):805–17. Epub 2013/04/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hashimoto S, Ogoshi K, Sasaki A, Abe J, Qu W, Nakatani Y, Ahsan B, Oshima K, Shand FH, Ametani A, Suzuki Y, Kaneko S, Wada T, Hattori M, Sugano S, Morishita S, Matsushima K. Coordinated changes in DNA methylation in antigen-specific memory CD4 T-cells. J Immunol. 2013. April 15;190(8):4076–91. Epub 2013/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T-cells that give rise to long-lived memory cells. Nat Immunol. 2003. December;4(12):1191–98. Epub 2003/11/20. [DOI] [PubMed] [Google Scholar]
- 121.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T-cell fates via the graded expression of T-bet transcription factor. Immunity. 2007. August;27(2):281–95. Epub 2007/08/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T-cell subsets with distinct memory fates. J Exp Med. 2008. March 17;205(3):625–40. Epub 2008/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999. October 14;401(6754):708–12. Epub 1999/10/28. [DOI] [PubMed] [Google Scholar]
- 124.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T-cell effector function. J Immunol. 2013. September 15;191(6):3419–29. Epub 2013/08/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, Araki K, West EE, Ghoneim HE, Fan Y, Dogra P, Davis CW, Konieczny BT, Antia R, Cheng X, Ahmed R. Effector CD8 T-cells dedifferentiate into long-lived memory cells. Nature. 2017. December 21;552(7685):404–9. Epub 2017/12/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ladle BH, Li KP, Phillips MJ, Pucsek AB, Haile A, Powell JD, Jaffee EM, Hildeman DA, Gamper CJ. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc Natl Acad Sci U S A. 2016. September 20;113(38):10631–36. Epub 2016/09/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, Triplett BM, Sekaly RP, Youngblood B. Human memory CD8 T-cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. 2017. June 5;214(6):1593–606. Epub 2017/05/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011. August 30;108(35):14566–71. Epub 2011/08/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, Liang S, Lu Y, Darlington GJ, Meissner A, Issa JP, Godley LA, Li W, Goodell MA. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011. December 4;44(1):23–31. Epub 2011/12/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, Guzman A, Lei Y, Huang YH, Rao A, Li W, Goodell MA. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016. September;48(9):1014–23. Epub 2016/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lopez-Moyado IF, Tsagaratou A, Yuita H, Seo H, Delatte B, Heinz S, Benner C, Rao A. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc Natl Acad Sci U S A. 2019. August 20;116(34):16933–42. Epub 2019/08/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gu T, Lin X, Cullen SM, Luo M, Jeong M, Estecio M, Shen J, Hardikar S, Sun D, Su J, Rux D, Guzman A, Lee M, Qi LS, Chen JJ, Kyba M, Huang Y, Chen T, Li W, Goodell MA. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018. July 12;19(1):88. Epub 2018/07/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, Cogdill AP, Morrissette JJD, DeNizio JE, Reddy S, Hwang Y, Gohil M, Kulikovskaya I, Nazimuddin F, Gupta M, Chen F, Everett JK, Alexander KA, LinShiao E, Gee MH, Liu X, Young RM, Ambrose D, Wang Y, Xu J, Jordan MS, Marcucci KT, Levine BL, Garcia KC, Zhao Y, Kalos M, Porter DL, Kohli RM, Lacey SF, Berger SL, Bushman FD, June CH, Melenhorst JJ. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T-cells. Nature. 2018. June;558(7709):307–12. Epub 2018/06/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011. January 18;19(1):17–30. Epub 2011/01/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, You J, Chia GS, Sim J, Doedens A, Abelanet A, Evans CE, Griffiths JR, Poellinger L, Goldrath AW, Johnson RS. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature. 2016. December 8;540(7632):236–41. Epub 2016/11/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T-cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019. April 26;37:457–95. Epub 2019/01/25. [DOI] [PubMed] [Google Scholar]
- 137.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, Wherry EJ. Epigenetic stability of exhausted T-cells limits durability of reinvigoration by PD-1 blockade. Science. 2016. December 2;354(6316):1160–65. Epub 2016/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, Tonnerre P, Chung RT, Tully DC, Allen TM, Frahm N, Lauer GM, Wherry EJ, Yosef N, Haining WN. The epigenetic landscape of T-cell exhaustion. Science. 2016. December 2;354(6316):1165–69. Epub 2016/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG, Youngblood B. De novo epigenetic programs inhibit PD-1 blockade-mediated T-cell rejuvenation. Cell. 2017. June 29;170(1):142–57 e19. Epub 2017/06/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nie J, Wang C, Liu Y, Yang Q, Mei Q, Dong L, Li X, Liu J, Ku W, Zhang Y, Chen M, An X, Shi L, Brock MV, Bai J, Han W. Addition of low-dose decitabine to anti-PD-1 antibody camrelizumab in relapsed/refractory classical hodgkin lymphoma. J Clin Oncol. 2019. June 10;37(17):1479–89. Epub 2019/05/01. [DOI] [PubMed] [Google Scholar]