

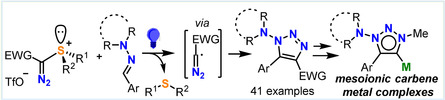
Abstract
The one‐pot synthesis of a series of sulfonium salts containing transferable diazomethyl groups is described, and the structure of these compounds is elucidated by X‐ray crystallography. Under photochemical conditions, reaction of these salts with N,N‐dialkyl hydrazones affords 1‐(dialkylamino)‐1,2,3‐triazoles via diazomethyl radical addition to the azomethine carbon followed by intramolecular ring closure. The straightforward transformation of the structures thus obtained into mesoionic carbene–metal complexes is also reported and the donor properties of these new ligands characterized.
Keywords: 1,2,3-triazoles; diazo compounds; mesoionic carbenes; photoredox catalysis; sulfonium salts
A radically new approach to the synthesis of 1‐(dialkylamino)‐1,2,3‐triazoles via addition of diazomethyl radicals to hydrazones is described. This reactivity confirms the utility of the α‐diazosulfonium salts herein introduced as synthetic equivalents of the diazomethyl cation.
Sulfonium ions are defined as positively charged organosulfur species of general formula [R3S]+, in which a central sulfur(IV) atom is attached to three organic rests while keeping a non‐shared electron pair. [1] The reactivity of these salts is dominated by the positive charge that they bear, mainly located at sulfur. [2] Thus, on treatment with bases, α‐CH deprotonation occurs on alkyl sulfonium salts leading to the formation of sulfur ylides; a class of compounds of demonstrated utility for the synthesis of three‐membered rings such as cyclopropanes, epoxides and aziridines. [3] Also as consequence of their positive charge, the R2S fragment in aryl sulfonium salts behaves as an excellent leaving group making possible the direct attack of nucleophiles to form Nu‐R coupling products, [4] or the oxidative addition of low oxidation‐state metals into the Ar‐S bond. This reactivity makes sulfonium salts suitable electrophilic partners for typical Pd‐ or Ni‐catalyzed coupling reactions. [5] One electron reduction is also a relatively facile process in sulfonium salts; it leads to a fast mesolytic scission of one of the C−S bonds, generating thioethers and organic radicals; [6] being the latter able to further engage in synthetically useful transformations such as cross coupling, oxygenation, amination or fluorination reactions among others (Figure 1 a). [7]
Figure 1.
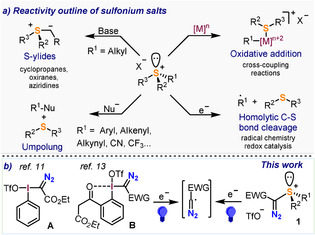
Reactivity outline of sulfonium salts.
The reactivity pattern just described shows clear overlaps between the chemistry of sulfonium salts and that of hypervalent I(III) reagents. [8] The selection of one or the other for a determined transformation often depends on factors such as the ease of synthesis of the necessary reagent, their relative stability, and the balancing between the levels of reactivity and selectivity that they provide for the specific process under study.[ 9 , 10 ] Considering these precedents and being aware that α‐diazo iodonium salts such as A, [11] and B have been used as diazo transfer reagents under thermal [12] and photochemical conditions, [13] we questioned ourselves whether sulfonium equivalents of these reagents could be efficiently prepared, and if so, what kind of reactivity would they exhibit. [14] Herein, the one pot synthesis of a series of such reagents from sulfoxides and diazo compounds is described, and their ability to transfer the diazo moiety to hydrazones under photochemical conditions is preliminary evaluated (Figure 1 b).
Our initial efforts were focused on the synthesis of the parent sulfonium salt 1 a, derived from the dibenzothiophene platform and containing an ethyldiazoacetate substituent. Sulfonium salts of related structure have been previously reported; however, they were prepared without exception by reaction of dialkyl sulfides with iodonium salt A.[ 12c , 12d ] Hence, alternative routes which avoid the use of that IIII‐reagent were explored. Considering the natural nucleophilicity of diazo compounds at the carbon adjacent to the dinitrogen moiety, we hypothesized that ethyldiazoacetate might directly react with sulfoxide 2 after its activation with triflic acid anhydride. Gratifyingly, the desired reaction took place in acceptable yield (61 %), and compound 1 a was isolated as a pale yellow crystalline solid (Scheme 1 a). Moreover, the reaction could be scaled up to 30 mmol without drop in the isolated yield (see the Supporting Information). This simple protocol also allowed the preparation of 1 b starting from trifluoromethyl diazomethane; or 1 c, 1 d and 1 e, which are based on the thianthrene, phenoxathiine and tetrahydrothiophene platforms, respectively (Scheme 1 b).
Scheme 1.
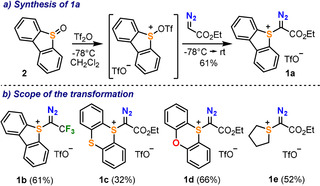
Synthesis of α‐diazo sulfonium triflates 1 a–e.
Monocrystals suitable for X‐ray diffraction analysis were grown by slow diffusion of Et2O into saturated CH2Cl2 solutions of 1 a–d. The thus obtained molecular diagrams confirmed the expected connectivity (Figure 2 and the Supporting Information). In 1 a the sulfur atom remains at the plane defined by the dibenzothiophene skeleton and adopts a trigonal‐pyramidal coordination environment, being the sum of the bond angles around S1 303.8(1)°. The S1‐C1 bond distance of 1.7357(12) Å falls within the range for S‐C(sp2) single bonds, while the C1‐N1 and N1‐N2 lengths (1.3367(15) Å and 1.1111(2) Å) are identical, within the experimental error, to those found in non‐charged diazo compounds. [15] Very similar parameters are found in the structures of 1 b–d; the only difference of note is the increase in the sum of bond angles around S1 in 1 c (306.4(1) Å) and 1 d (311.5(1) Å), which is a consequence of the geometry imposed by the heterocyclic skeleton. It is also worth noting that all compounds 1 a–d are found to form dimeric aggregates in the solid state where two triflate anions bridge simultaneously the sulfur atoms of two sulfonium moieties. In all cases the S⋅⋅⋅O distances are definitely shorter than the sum of the van der Waals radii of both elements (3.32 Å), revealing a remarkable electrophilicity at S1.
Figure 2.
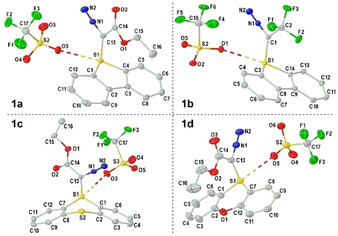
Molecular structures of compounds 1 a–d in the solid state. Anisotropic displacements shown at 50 % probability level. Hydrogen atoms and solvent molecules removed for clarity. Selected bond lengths [Å]: 1 a: S1–C1 1.7357(12), C1–N1 1.3367(15), S1–O3 2.893(1); 1 b: S1–C1 1.7309(11), C1–N1 1.3252(13), S1–O1 2.970(1); 1 c: S1–C1 1.7386(11), C1–N1 1.3138(14), S1–O3 2.938(1); 1 d: S1–C1 1.7386(2), C1–N1 1.339(3), S1–O4 3.073(2). [16]
Differential scanning calorimetry (DSC) analysis for 1 a shows decomposition starting at 130 °C that leads to an energy release of 384 J g−1 (Figure 3 a). Similar values were obtained for 1 c (396 J g−1) and 1 d (348 J g−1), all significantly lower than those reported for IIII analogues (549 J g−1). [13] On the basis of these results, the use of the sulfonium salts herein described should be safer; we carefully recommend however not to expose multigram quantities 1 a–e to temperatures above 80 °C.
Figure 3.
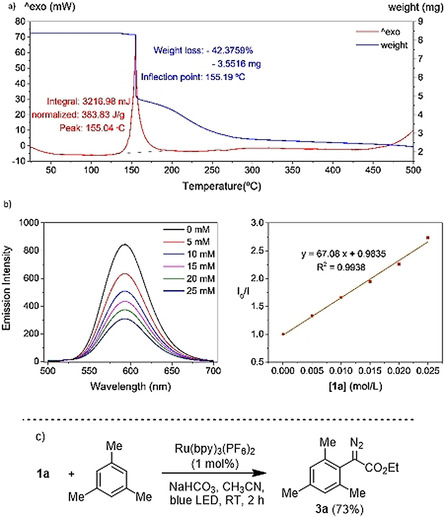
a) DSC analysis for 1 a in air; b) Quenching of the fluorescence of [Ru(bipy)3][PF6]2 following the addition of 1 a; c) trapping the postulated diazomethyl radical intermediate with 1,3,5‐trimethylbenzene to afford product 3 a and formation of diethyl acetylenedicarboxylate via radical dimerization and dinitrogen elimination.
At this stage we evaluated the possibility to generate diazomethyl radicals through single electron reduction of 1 a–e; specifically, via photoredox catalysis. An experiment often used to determine the feasibility of the desired activation consists in the evaluation of fluorescence intensity quenching of the photoexcited state of a photocatalyst in the presence of the reaction substrate (Stern Volmer relation). In fact, this analysis revealed an effective oxidative quenching of the photoexcited [Ru(bipy)3][PF6]2 catalyst (E 1/2 (II)*/(III)=−0.81 V vs. SCE) in the presence of 1 a (E red=−0.50 V vs. SCE). The formation of the envisaged diazomethyl radical was subsequently evidenced by testing 1 a in the benchmark C−H diazomethylation of arenes recently reported by Suero. [13] Gratifying, irradiation of 1 a in the presence of [Ru(bpy)3][PF6]2 (1 mol %) and 1,3,5‐trimethylbenzene (2 equiv) afforded the diazomethylated arene product 3 a in 73 % isolated yield (Figure 3). Under identical conditions salts 1 c and 1 d also promoted the formation of 3 a, albeit with lower isolated yields; when employing reagent 1 e not even traces of 3 a were detected.
Having confirmed that possibility, we set about determining the potential of the photocatalytic system just developed on the C(sp2)‐H diazomethylation of aldehyde‐derived hydrazones 4. It was reasoned at that stage that in case of a successful transfer of the diazo group, the initially obtained α‐diazohydrazones 5′ should tautomerize to the corresponding 1,2,3‐triazoles 5 providing not only a synthetically useful entry to N‐dialkylamino triazoles of potential pharmacological interest, [17] but also easy to handle precursors for α‐imino metal carbenoids in case a ring to chain equilibrium between 5′ and 5 could be established. [18]
We were pleased to observe that under slightly modified reaction conditions to those employed for the obtention of 3 a, the triazole products 5 a–v were formed in good to excellent yields from the corresponding N,N‐dibenzyl hydrazones, being those slightly better for hydrazone substrates derived from electron poor aldehydes. The cyclic nature of the products isolated was unambiguously assigned from the single‐crystal structures of 5 i and 5 s (Scheme 2 and the Supporting Information).
Scheme 2.
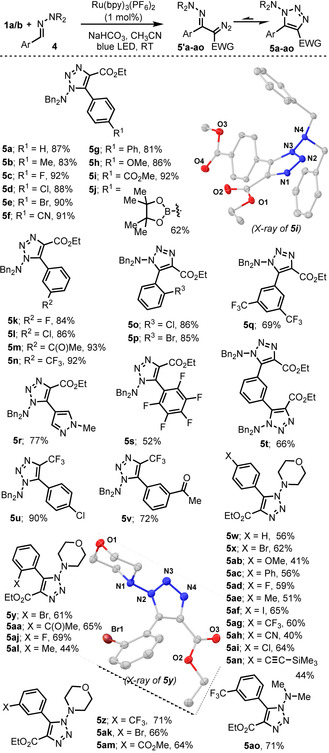
Synthesis of 1,2,3‐triazoles 5 a–ao and molecular structure of compounds 5 i and 5 y in the solid state. Anisotropic displacements shown at 50 % probability level. [16]
Interestingly, in these structures the dialkylamino nitrogen is pyramidal. As illustrative example, the sum of angles around N(4) is 329.4° in 5 i; therefore, at least in the solid state the exo‐amino group is not conjugated with the heterocyclic ring. Morpholine derived hydrazones also get involved in this transformation; yields however are slightly lower, 5 w–5 an. Scheme 2 also depicts compound 5 ao derived from 1,1‐dimethylamino hydrazine, which has been included in our product scope table for completeness.
Note that the most commonly used route to prepare 1,2,3‐triazoles precursors is a [2+3] dipolar cycloaddition of alkynes and alkyl/aryl azides. [19] This synthetic approach however, is not suitable for the preparation of the corresponding 1‐(dialkylamino) derivatives due to the unavailability of di(alkylamino)azides, which are known to be explosive compounds.[ 20 , 21 ]
A plausible reaction pathway for the formation of 5 from 1 a/b and 4 is depicted in Scheme 3. Initially, irradiation of [Ru(bpy)3]2+ generates photo exited ([Ru(bpy)3]2+)*, which reduces reagent 1 a/b via single‐electron transfer and generates the transient diazomethyl radical (Int‐I) via homolytic S−C bond cleavage. Addition of the latter intermediate to the azomethine carbon of 4 delivers an aminyl radical intermediate (Int‐II), which is subsequently oxidized by [Ru(bpy)3]3+ (E 1/2 (iii)/(ii)=1.29 V vs. SCE). [22] Deprotonation would re‐establish the C‐N double bond of the hydrazone moiety leading to 5′, which ultimately cyclizes to the observed 1,2,3‐triazole 5. Alternatively, deprotonation of Int‐II may occur first leading to the corresponding radical anion, which would undergo oxidation to 5′. [23] A light on/off experiment confirmed the necessity of continuous irradiation for the reaction to proceed; moreover, measurement of the quantum yield of the reaction to form 5 c (Φ=0.18) suggests that a radical chain process cannot be a predominant path. [24]
Scheme 3.
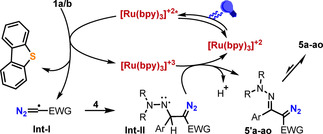
Plausible reaction mechanism for the formation of 5 a–ao.
Finally, the plausible utility of the triazole products obtained as precursors for mesoionic 1‐(dialkylamino)‐1,2,3‐triazol‐4‐ylidene ligands was evaluated. [25] Hence, 5 a,d,w,x were submitted to reaction with one equivalent of methyl triflate, affording triazolium salts 6 a,d,w,x. Methylation selectively occurs at position 3‐ of the heterocycle (Scheme 4). Subsequent saponification of these species in the presence of LiOH took place with concomitant loss of CO2 delivering azolium salts 7 a,d,w,x. The connectivity of 7 w and 7 x was additionally confirmed by X‐ray analysis. This opened the stage to the reaction with [RhCl(COD)]2 in the presence of KHMDS to afford the desired carbene‐metal complexes 8 a,d,w,x as air stable yellow solids. Compounds 8 w and 8 x were also crystallized and their structures in the solid state are depicted in Scheme 4 and the Supporting Information.
Scheme 4.
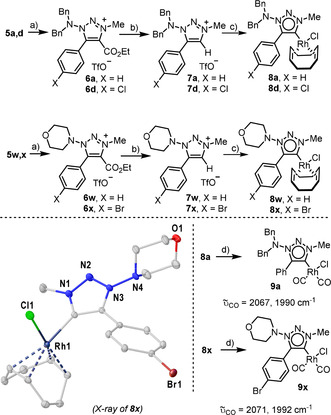
Synthesis of Rh complexes 8 a,d,w,x via decarboxylative metalation, evaluation of the donor properties of the new ligands and molecular structure of 8 x. Anisotropic displacements shown at 50 % probability level and H atoms removed for clarity. [16] Reaction conditions: a) MeOTf (1.2 equiv), DCM, 8 h, 0 °C → r.t.; b) (LiOH 3.0 equiv), THF/H2O/MeOH (1:1:1), 1 h, 60 °C, 7 a, 89 %; 7 d, 89 %;7 w, 71 %; 7 x, 61 %, (two steps yield); c) KHMDS (1.2 equiv), [RhCl(COD)]2 (0.5 equiv), THF, 0 °C → r.t., 8 a, 60 %, 8 d, 60 %; 8 w, 70 %; 8 x, 81 %; d) CO, DCM, 20 min., r.t., 9 a, 75 %, 9 x, 72 %.
Rhodium dicarbonyl complexes 9 a and 9 x were synthesized by reaction of precursors 8 a and 8 x with CO, respectively, and their CO stretching vibrations (υav=2029 cm−1, 9 a and υav=2032 cm−1, 9 x) compared with that of 1‐alkyl analogues (υav=2027 cm−1). [26] This analysis suggests that the introduction of the dialkylamino group at N(1) slightly reduces the donor ability exhibited by the original 1,2,3‐triazol‐4‐ylidenes probably due to the inductive effect of a dialkylamino moiety, which is not conjugated with the ring. [27] Both carbene ligands remain anyway stronger donors than conventional NHCs (υav=2039–2041 cm−1). [28]
In conclusion, diazomethyl radicals can be efficiently generated under photochemical conditions from α‐diazosulfonium triflates, whose synthesis is described along this paper. Addition of these species to hydrazones followed by intramolecular cyclization affords 1‐(dialkylamino)‐1,2,3‐triazoles in moderate to excellent yields. Finally, these heterocyclic compounds were further elaborated into mesoionic carbene ligands having an unprecedented dialkylamino rest at position 1. The development of additional synthetic applications of sulfonium salts is an active area of investigation in our laboratory and further progress on the topic will be reported in due course.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support from the European Research Council (ERC CoG 771295) and the DFG (INST 186/1237‐1 and INST 186/1324‐1) is gratefully acknowledged. Open access funding enabled and organized by Projekt DEAL.
X. Li, C. Golz, M. Alcarazo, Angew. Chem. Int. Ed. 2021, 60, 6943.
References
- 1.
- 1a. Kaiser D., Klose I., Oost R., Neuhaus J., Maulide N., Chem. Rev. 2019, 119, 8701–8780; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.“Arylsulfonium Salts and Derivatives”: Fernández I., Khiar N. in Science of Synthesis, Vol. 31a (Ed.: Ramsden C. A.), Thieme, Stuttgart, 2007, pp. 1001–1014. [Google Scholar]
- 2. Kozhushkov S. I., Alcarazo M., Eur. J. Inorg. Chem. 2020, 2486–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.“Sulfur Ylides in Organic Synthesis and Transition Metal Catalysis in Modern Ylide Chemistry”: Oost R., Neuhaus J. D., Merad J., Maulide N., Structure and Bonding, Vol. 177 (Ed.: Gessner V.), Springer, Cham, 2017. [Google Scholar]
- 4.
- 4a. Ming X.-X., Tian Z.-Y., Zhang C.-P., Chem. Asian J. 2019, 14, 3370–3379; [DOI] [PubMed] [Google Scholar]
- 4b. Zhao J.-N., Kayumov M., Wang D.-Y., Zhang A., Org. Lett. 2019, 21, 7303–7306; [DOI] [PubMed] [Google Scholar]
- 4c. Sander K., Galante E., Gendron T., Yiannaki E., Patel N., Kalber T. L., Badar A., Robson M., Johnson S. P., Bauer F., Mairinger S., Stanek J., Wanek T., Kuntner C., Kottke T., Weizel L., Dickens D., Erlandsson K., Hutton B. F., Lythgoe M. F., Stark H., Langer O., Koepp M., Årstad E., J. Med. Chem. 2015, 58, 6058–6080; [DOI] [PubMed] [Google Scholar]
- 4d. Mu L., Fischer C. R., Holland J. P., Becaud J., Schubiger P. A., Schibli R., Ametamey S. M., Graham K., Stellfeld T., Dinkelborg L. M., Lehmann L., Eur. J. Org. Chem. 2012, 889–892. [Google Scholar]
- 5.
- 5a. Kafuta K., Korzun A., Böhm M., Golz C., Alcarazo M., Angew. Chem. Int. Ed. 2020, 59, 1950–1955; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1966–1971; [Google Scholar]
- 5b. Engl P. S., Häring A. P., Berger F., Berger G., Pérez-Bitrián A., Ritter T., J. Am. Chem. Soc. 2019, 141, 13346–13351; [DOI] [PubMed] [Google Scholar]
- 5c. Aukland M. H., Talbot F. J. T., Fernández-Salas J. A., Ball M., Pulis A. P., Procter D. J., Angew. Chem. Int. Ed. 2018, 57, 9785–9789; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9933–9937; [Google Scholar]
- 5d. Uno D., Minami H., Otsuka S., Nogi K., Yorimitsu H., Chem. Asian J. 2018, 13, 2397–2400; [DOI] [PubMed] [Google Scholar]
- 5e. Tian Z.-Y., Wang S.-M., Jia S.-J., Song H.-X., Zhang C.-P., Org. Lett. 2017, 19, 5454–5457; [DOI] [PubMed] [Google Scholar]
- 5f. Wang S. M., Wang X. Y., Qin H. L., Zhang C. P., Chem. Eur. J. 2016, 22, 6542–6546; [DOI] [PubMed] [Google Scholar]
- 5g. Wang X.-Y., Song H.-X., Wang S.-M., Yang J., Qin H.-L., Jiang X., Zhang C.-P., Tetrahedron 2016, 72, 7606–7612; [Google Scholar]
- 5h. Cowper P., Jin Y., Turton M. D., Kociok-Köhn G., Lewis S. E., Angew. Chem. Int. Ed. 2016, 55, 2564–2568; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2610–2614; [Google Scholar]
- 5i. Wang S.-M., Song H.-X., Wang X.-Y., Liu N., Qin H.-L., Zhang C.-P., Chem. Commun. 2016, 52, 11893–11896; [DOI] [PubMed] [Google Scholar]
- 5j. Vasu D., Yorimitsu H., Osuka A., Synthesis 2015, 47, 3286–3291; [Google Scholar]
- 5k. Vasu D., Yorimitsu H., Osuka A., Angew. Chem. Int. Ed. 2015, 54, 7162–7166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7268–7272; [Google Scholar]
- 5l. Zhang S., Marshall D., Liebeskind L. S., J. Org. Chem. 1999, 64, 2796–2804; [DOI] [PubMed] [Google Scholar]
- 5m. Srogl J., Allred G. D., Liebeskind L. S., J. Am. Chem. Soc. 1997, 119, 12376–12377. [Google Scholar]
- 6.For seminal work see:
- 6a. Van Bergen T. J., Hedstrand D. M., Kruizinga W. H., Kellogg R. M., J. Org. Chem. 1979, 44, 4953–4962; [Google Scholar]
- 6b. Hedstrand D. M., Kruizinga W. H., Kellogg R. M., Tetrahedron Lett. 1978, 19, 1255–1258; For recent reviews see: [Google Scholar]
- 6c. Péter A., Perry G. J. P., Procter D. J., Adv. Synth. Catal. 2020, 362, 2135–2142; [Google Scholar]
- 6d. Otsuka S., Nogi K., Yorimitsu H., Top. Curr. Chem. 2018, 376, 199–237, and ref. [1a]. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Li J., Chen J., Sang R., Ham W.-S., Plutschack M. B., Berger F., Chabbra S., Schnegg A., Genicot C., Ritter T., Nat. Chem. 2020, 12, 56–62; [DOI] [PubMed] [Google Scholar]
- 7b. Aukland M. H., Šiaučiulis M., West A., Perry G. J. P., Procter D. J., Nat. Catal. 2020, 3, 163–169; [Google Scholar]
- 7c. Sang R., Korkis S. E., Su W., Ye F., Engl P. S., Berger F., Ritter T., Angew. Chem. Int. Ed. 2019, 58, 16161–16166; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16307–16312; [Google Scholar]
- 7d. Ye F., Berger F., Jia H., Ford J., Worthman A., Börgel J., Genicot C., Ritter T., Angew. Chem. Int. Ed. 2019, 58, 14615–14619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14757–14761. [Google Scholar]
- 8. Zhdankin V. V., Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, Wiley, Chichester, 2014. [Google Scholar]
- 9.For a direct comparison on electrophilic cyanation see:
- 9a. Li X., Golz C., Alcarazo M., Angew. Chem. Int. Ed. 2019, 58, 9496–9500; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9596–9600; [Google Scholar]
- 9b. Declas N., Le Vaillant F., Waser J., Org. Lett. 2019, 21, 524–528; [DOI] [PubMed] [Google Scholar]
- 9c. Le Vaillant F., Wodrich M. D., Waser J., Chem. Sci. 2017, 8, 1790–1800; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Frei R., Courant T., Wodrich M. D., Waser J., Chem. Eur. J. 2015, 21, 2662–2668. [DOI] [PubMed] [Google Scholar]
- 10.For a direct comparison on electrophilic alkynylation see:
- 10a. Waldecker B., Kafuta K., Alcarazo M., Org. Synth. 2019, 96, 258–276; [Google Scholar]
- 10b. Waldecker B., Kraft F., Golz C., Alcarazo M., Angew. Chem. Int. Ed. 2018, 57, 12538–12542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12718–12722; [Google Scholar]
- 10c. Frei R., Wodrich M. D., Hari D. P., Borin P.-A., Chauvier C., Waser J., J. Am. Chem. Soc. 2014, 136, 16563–16573; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Chen C. C., Waser J., Chem. Commun. 2014, 50, 12923–12926; [DOI] [PubMed] [Google Scholar]
- 10e. Fernández-González D., Brand J. P., Waser J., Chem. Eur. J. 2010, 16, 9457–9461; [DOI] [PubMed] [Google Scholar]
- 10f. Fernández-González D., Brand J. P., Mondière R., Waser J., Adv. Synth. Catal. 2013, 355, 1631–1639; [Google Scholar]
- 10g. Frei R., Waser J., J. Am. Chem. Soc. 2013, 135, 9620–9623. [DOI] [PubMed] [Google Scholar]
- 11. Weiss R., Seubert J., Hampel F., Angew. Chem. Int. Ed. Engl. 1994, 33, 1952–1953; [Google Scholar]; Angew. Chem. 1994, 106, 2038–2039. [Google Scholar]
- 12.
- 12a. Podrugina T. A., Pavlova A. S., Vinogradov D. S., Shuvalov M. V., Potapov I. D., Levina I. I., Mironov A. V., Gleiter R., Russ. Chem. Bull. 2019, 68, 284–292; [Google Scholar]
- 12b. Taylor M. T., Nelson J. E., Suero M. G., Gaunt M. J., Nature 2018, 562, 563–568; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Schnaars C., Hennum M., Bonge-Hansen T., J. Org. Chem. 2013, 78, 7488–7497; [DOI] [PubMed] [Google Scholar]
- 12d. Weiss R., Handke M., Reichel S., Hampel F., Z. Naturforsch. 1998, 53b, 599–619. [Google Scholar]
- 13. Wang Z., Herraiz A. G., del Hoyo A. M., Suero M. G., Nature 2018, 554, 86–91. [DOI] [PubMed] [Google Scholar]
- 14.α-Diazo sulfonium salts have been reported to get reduced and benzylated after SET. See reference [12b].
- 15. Allen F. H., Kennard O., Watson D. G., Brammer L., Orpen A. G., Taylor R., J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar]
- 16. Deposition Numbers 2009414, 2009415, 2009416, 2009417, 2009418, 2041657, 2041658, 2041659, 2041660, 2041661, 2041662, 2041663, and 2041664 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 17.
- 17a. Santos T. F., de Jesus J. B., Neufeld P. M., Jordão A. K., Campos V. R., Cuhna A. C., Castro H. C., de Souza M. C. B. V., Ferreira V. F., Rodrigues C. R., Abreu P. A., Med. Chem. Res. 2017, 26, 680–689; [Google Scholar]
- 17b. Sezer Ö., Dabak K., Akar A., Anaç O., Helv. Chim. Acta 1996, 79, 449–453; [Google Scholar]
- 17c. Hauptmann S., Wilde H., Moser K., Tetrahedron Lett. 1967, 8, 3295–3297. [Google Scholar]
- 18.For selected references see:
- 18a. Miura T., Tanaka T., Biyajima T., Yada A., Murakami M., Angew. Chem. Int. Ed. 2013, 52, 3883–3886; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3975–3978; [Google Scholar]
- 18b. Miura T., Funakoshi Y., Morimoto M., Biyajima T., Murakami M., J. Am. Chem. Soc. 2012, 134, 17440–17443; [DOI] [PubMed] [Google Scholar]
- 18c. Chuprakov S., Malik J. A., Zibinsky M., Fokin V. V., J. Am. Chem. Soc. 2011, 133, 10352–10355; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18d. Chattopadhyay B., Gevorgyan V., Org. Lett. 2011, 13, 3746–3749; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18e. Lourdusamy E., Yao L., Park C.-M., Angew. Chem. Int. Ed. 2010, 49, 7963–7967; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8135–8139; [Google Scholar]
- 18f. Grimster N., Zhang L., Fokin V. V., J. Am. Chem. Soc. 2010, 132, 2510–2511; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18g. Miura T., Yamauchi M., Murakami M., Chem. Commun. 2009, 1470–1471; [DOI] [PubMed] [Google Scholar]
- 18h. Chuprakov S., Kwok S. W., Zhang L., Lercher L., Fokin V. V., J. Am. Chem. Soc. 2009, 131, 18034–18035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18i. Horneff T., Chuprakov S., Chernyak N., Gevorgyan V., Fokin V. V., J. Am. Chem. Soc. 2008, 130, 14972–14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meldal M., Tornøe C. W., Chem. Rev. 2008, 108, 2952–3015. [DOI] [PubMed] [Google Scholar]
- 20. Bock H., Kompa K. L., Angew. Chem. Int. Ed. Engl. 1962, 1, 264; [Google Scholar]; Angew. Chem. 1962, 74, 327. [Google Scholar]
- 21.1-(Dialkylamino)-1,2,3-triazoles have been previously prepared by a different route, reaction of vinyldiazonium salts with hydrazines: Saalfrank R. W., Weiß B., Chem. Ber. 1985, 118, 2626–2634. [Google Scholar]
- 22.The oxidation of hydrazone-derived aminyl radical intermediates has been proposed for similar transformations. See, for example:
- 22a. Huang B., Bu X.-S., Xu J., Dai J.-J., Feng Y.-S., Xu H.-J., Asian J. Org. Chem. 2018, 7, 137–140; [Google Scholar]
- 22b. Ji H., Ni H.-q., Zhi P., Xi Z.-w., ang W., Shi J.-j., Shen Y.-m., Org. Biomol. Chem. 2017, 15, 6014–6023; [DOI] [PubMed] [Google Scholar]
- 22c. Xie J., Zhang T., Chen F., Mehrkens N., Rominger F., Rudolph M., Hashmi A. S. K., Angew. Chem. Int. Ed. 2016, 55, 2934–2938; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2987–2991. [Google Scholar]
- 23.For a mechanistic proposal following this path, see: Janhsen B., Studer A., J. Org. Chem. 2017, 82, 11703–11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.See references [7a,b] and:
- 24a. Hatchard C. G., Parker C. A., Proc. R. Soc. 1956, A235, 518–536; [Google Scholar]
- 24b. Kuhn H. J., Braslavsky S. E., Schmidt R., Pure Appl. Chem. 2004, 76, 2105–2146; [Google Scholar]
- 24c. Cismesia M. A., Yoon T. P., Chem. Sci. 2015, 6, 5426–5434; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24d.“Chemical Actinometry”: Montalti M., Credi A., Prodi L., Gandolfi M. T., Handbook of Photochemistry , 3rd ed., Taylor & Francis Group, Boca Raton, 2016, pp. 601–616. [Google Scholar]
- 25. Vivancos Á., Segarra C., Albrecht M., Chem. Rev. 2018, 118, 9493–9586. [DOI] [PubMed] [Google Scholar]
- 26. Mendoza-Espinosa D., González-Olvera R., Osornio C., Negrón-Silva G. E., Santillán R., New J. Chem. 2015, 39, 1587–1591. [Google Scholar]
- 27.For the synthesis and electronic properties of related N-dialkylamino and N-alkoxy-1,2,4-triazol-3-ylidene ligands see: Alcarazo M., Fernández R., Álvarez E., Lassaletta J. M., J. Organomet. Chem. 2005, 690, 5979–5988. [Google Scholar]
- 28. Fürstner A., Alcarazo M., Krause H., Lehmann C. W., J. Am. Chem. Soc. 2007, 129, 12676–12677. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary