

Abstract
Discoidin domain receptor 1 (DDR1) inhibitors with a desired pharmacophore were designed using deep generative models (DGMs). DDR1 is a receptor tyrosine kinase activated by matrix collagens and implicated in diseases such as cancer, fibrosis and hypoxia. Herein we describe the synthesis and inhibitory activity of compounds generated from DGMs. Three compounds were found to have sub‐micromolar inhibitory activity. The most potent of which, compound 3 (N‐(4‐chloro‐3‐((pyridin‐3‐yloxy)methyl)phenyl)‐3‐(trifluoromethyl)benzamide), had an IC50 value of 92.5 nM. Furthermore, these compounds were predicted to interact with DDR1, which have a desired pharmacophore derived from a known DDR1 inhibitor. The results of synthesis and experiments indicated that our de novo design strategy is practical for hit identification and scaffold hopping.
Keywords: deep generative model, pharmacophore model, de novo design, DDR1
Helpful for scaffold hopping: DDR1 inhibitors were designed using a deep generative model that generates chemical structures with the desired pharmacophore. In structure sampling, about 10,000 generated structures from the agent network have pharmacophore scores greater than 0.8. After filtering the structures, nine compounds were synthesized and their inhibitory activities were evaluated against DDR1. The results of this study show that our de novo design strategy is practical for hit identification and scaffold hopping.
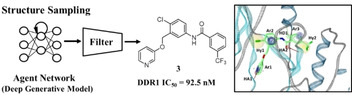
Deep generative models (DGMs) have been successfully applied to image generation, [1] language translation, [2] and others. [3] In recent years, chemical structure generation using DGMs is receiving a lot of attention in de novo drug design. [4] Successful examples of hit identification with DGMs have been reported by several groups.[ 5 , 6 ] Although DGMs are able to generate molecules with desired properties, most of the properties don't have 3D information, such as shape and pharmacophore. [4] It has been known that properties originating from 3D shape and/or pharmacophore are very useful in the drug design process. [7] Thus, we constructed DGMs for generating molecules with a desired pharmacophore. The procedure for DGM construction has been published elsewhere. [8] Briefly, the method has three steps consisting of prior network construction, agent network construction, and structure sampling (Figure 1). First, the prior network is trained using SMILES strings [9] from ChEMBL. [10] After the training, the prior network generates valid SMILES strings. Next, the agent network is trained using reinforcement learning. The training shifts the probability distribution from that of the prior network towards a distribution modulated by a pharmacophore score. In the third step, the trained agent network generates SMILES strings, which are likely to have a desired pharmacophore. The prior and agent network constructions were implemented using REINVENT [11] and the pharmacophore score was calculated using LigandScout 4.4. [12]
Figure 1.
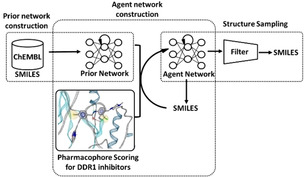
Workflow of our strategy for identification of DDR1 inhibitors.
Discoidin domain receptor 1 (DDR1) is a collagen‐activated receptor tyrosine kinase and a potential therapeutic target for a wide range of human diseases, such as cancer, [13] fibrosis [14] and hypoxia. [15]
To construct a desired pharmacophore for DDR1, the crystal structure of DDR1 kinase domain in complex with ponatinib (PDB: 3ZOS) is used (Figure 2A). [16] Here, eight pharmacophore features of the inhibitor were identified as three aromatic (Ar1, Ar2, Ar3), two hydrophobic (Hy1, Hy2), two hydrogen acceptor (HA1, HA2) and one hydrogen donor (HD1) features (Figure 2B). In addition, an ensemble of exclusion volume spheres obtained from the crystal structure was used. For pharmacophore scoring, the scoring function was set to ‘Relative Pharmacophore‐Fit’; maximum number of omitted features is set to ‘1’ and the Hy1, Hy2, Ar3 and HD1 features were set as ‘optional feature’. The Relative Pharmacophore‐Fit (rel.SFCR) was defined in Equation 1.[ 8 , 12 ]
| (1) |
Figure 2.
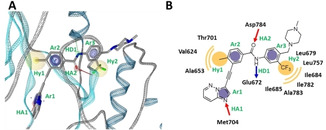
A) Pharmacophore model for DDR1 inhibitors. B) 2D depiction of the pharmacophore model for DDR1 inhibitors.
where SFCR is the feature count/RMS distance score, NMFP is the number of geometrically matched feature pairs, SRMS is the matched feature pair RMS distance score in the range [0,9], and RMSFP is the RMS of the matched feature pair distances. The parameter of conformer generation of generated structures from agent network was set as ‘iCon Fast’ option for idbgen tool [17] provided with LigandScout 4.4.
The agent network was trained based on the desired pharmacophore and prior network. Training of the agent network was done with a batch size of 64 using the Adam optimizer for 10,000 steps. All other parameters were set to default values in REINVENT. After the training, structure sampling was perfomed. 570,542 valid SMILES were generated during the structure sampling of 640,000 SMILES strings. In the same way, 588,240 valid SMILES were generated from the prior network. The pharmacophore scores of the generated structures were calculated using LigandScout 4.4. The distribution of pharmacophore scores is displayed as a histogram in Figure 3. The 137,790 structures having pharmacophore scores ≥0.5 were confirmed among the valid SMILES strings from the agent network. On the other hand, there are only 4,306 structures having pharmacophore scores ≥0.5 among the valid SMILES strings from the prior network. This result indicates that the agent network can generate structures fulfilling the desired pharmacophore of DDR1 inhibitor with high frequency. In order to perform filtering of the generated structures from agent network which have pharmacophore scores greater than 0.8 (10,694 structures), binding affinity scores were calculated using iaffinity module implemented in LigandScout 4.4. Compounds having binding affinity scores less than −37 kJ/mol were selected (4731 compounds). The selected compounds were inspected visually to determine which compounds to synthesize, taking into account their pharmacophore scores, binding scores and synthesis accessibilities. Consequently, 9 compounds (1–9) were selected which are illustrated in Figure 4. During the visual inspection, two compounds (7, 8) were modified by removing halogen atoms, that do not contribute much to binding interaction with DDR1. Compound 9 was modified by changing the position of pyridinyl nitrogen from para to meta to form hydrogen bond with the hinge region of DDR1. Accordingly, we synthesized 9 compounds (1–6 and 7 a, 8 a, 9 a). Synthesis of the nine compounds is summarized in Schemes 1–9 (see Supporting Information).
Figure 3.
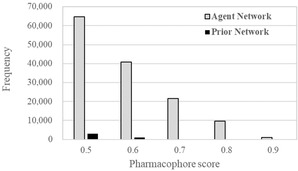
Distribution of Pharmacophore scores generated structures from the Agent and Prior network. Axis labels of pharmacophore score <0.5 were omitted because all of hit compounds have pharmacophore score ≥0.5.
Figure 4.
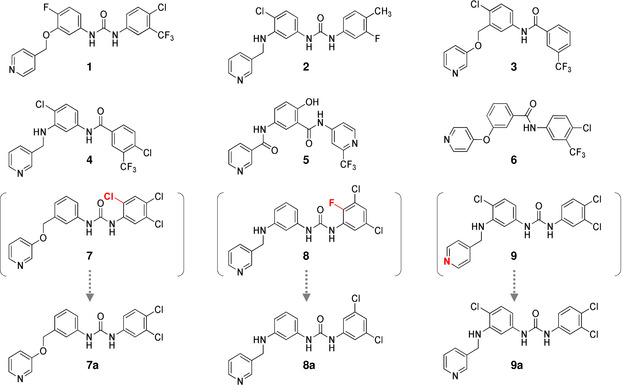
Synthesized compounds evaluated as DDR1 inhibitors.
The synthesized compounds were evaluated for their inhibitory activity against DDR1. The kinase assays were performed using Off‐chip Mobility Shift Assay which were carried out via a kinase profiling service (Carna Biosciences, Inc., Kobe, Japan) (see Supporting Information). The results are summarized in Table 1. Among the tested compounds, compound 3 exhibited interesting double‐digit nanomolar inhibitory activity against DDR1 (IC50=92.5 nM). The binding interaction of compound 3 derived from pharmacophore matching is shown in Figure 5A. Compound 3 fulfills all of the pharmacophore features of a DDR1 inhibitor, although there are slightly misaligned features.
Table 1.
DDR1 inhibitory activity of the synthesized compounds.
|
Compound |
Pharmacophore score[a] |
Binding affinity score [kJ/mol][b] |
IC50 [nM][c] |
|---|---|---|---|
|
1 |
0.96 |
−50.51 |
1005.9 |
|
2 |
0.95 |
−52.67 |
2239.4 |
|
3 |
0.83 |
−47.13 |
92.5 |
|
4 |
0.96 |
−51.97 |
186.7 |
|
5 |
0.86 |
−37.29 |
>30,000 |
|
6 |
0.84 |
−40.83 |
>30,000 |
|
7 |
0.85 |
−54.38 |
NT[d] |
|
8 |
0.85 |
−51.78 |
NT |
|
9 |
0.85 |
−49.46 |
NT |
|
7 a |
0.85 |
−53.06 |
171.3 |
|
8 a |
0.85 |
−51.15 |
1244.3 |
|
9 a |
0.85 |
−54.83 |
1111.0 |
[a] Calculated using Relative Pharmacophore‐Fit score in LigandScout 4.4. [b] Calculated using iaffnity module in LigandScout 4.4. [c] The compound concentration required for 50 % inhibition (IC50) was determined from semi‐logarithmic dose‐response plots, and the results represent the mean of duplicated samples. [d] NT=not tested.
Figure 5.
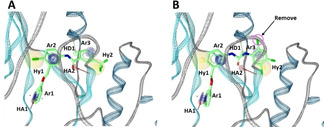
Binding interactions of A) compound 3 and B) compound 7.
Compounds 4 and 7 a were also found to have potent inhibitory activities (IC50 values: 186.7 and 171.3 nM, respectively). Compound 7 a was designed by removing a Cl atom from 1,2,4‐trichlorobenzene in compound 7 as illustrated in Figure 5B. Compounds 1, 2, 8 a, 9 a were found to moderately inhibit DDR1 activity (IC50 values: 1005.9, 2239.4, 1244.3 and 1111.0 nM, respectively). Compounds 5 and 6 did not exhibit any inhibitory activity at all. Binding affinity scores of the two compounds are high values (−37.29, and −40.83 kJ/mol), indicating low binding affinities. These results indicate that our strategy of using DGMs has worked efficiently to design DDR1 inhibitors.
To check if the generated structures (compound 1–6, 7 a–9 a) have already been registered in certain databases, structure search was performed in ChEMBL [10] and PubChem. [18] We have found that compound 3 is registered in PubChem with CID 58614959 and is annotated as Raf kinase and p38 MAP kinase inhibitor.
In conclusion, we were able to design DDR1 inhibitors with a desired pharmacophore using DGMs. Compound 3 showed potent inhibitory activity with an IC50 value of 92.5 nM against DDR1. In general, in order to predict inhibitory activities of generated compounds from DGMs, many experimental inhibitory data are needed to construct accurate prediction models. However, our strategy needs only pharmacophore information to design inhibitors against a target protein. Therefore, our strategy can be used in the early stage of drug discovery process. Ponatinib is a drug used to treat chronic myeloid leukemia and inhibits DDR1 with a Kd value of 1.3 nM. [16] In this study, our pharmacophore is derived from the crystal structure of DDR1 kinase domain in complex with ponatinib. The scaffolds of the synthesized compounds (Figure 4) were found to be different from that of ponatinib (Figure 2). Thus, our strategy can also be used for scaffold hopping.
In order to determine which compounds to synthesize, it is important to filter generated structures from the agent network efficiently. We are now trying to construct more practical filtering methods that include criteria such as drug‐likeness score, [19] ADMET properties [20] and synthesis accessibility. [21] We believe that this pharmacophore‐based DGM strategy can be applied to various drug discovery campaigns in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Hirofumi Nakano for his insightful comments and suggestions.
A. Yoshimori, Y. Asawa, E. Kawasaki, T. Tasaka, S. Matsuda, T. Sekikawa, S. Tanabe, M. Neya, H. Natsugari, C. Kanai, ChemMedChem 2021, 16, 955.
References
- 1.H. Huang, P. S. Yu, C. Wang, arXiv:1803.04469v2 2018.
- 2. Johnson M., Schuster M., Le Q. V., Krikun M., Wu Y., Chen Z., Thorat N., Viégas F., Wattenberg M., Corrado G., Hughes M., Dean J., Trans. Assoc. Comput. Linguist. 2017, 5, 339–351. [Google Scholar]
- 3. de Bem R., Ghosh A., Ajanthan T., Miksik O., Boukhayma A., Siddharth N., Torr P., Int. J. Comput. Vis. 2020, 128, 1537–1563. [Google Scholar]
- 4. Elton D. C., Boukouvalas Z., Fugea M. D., Chung P. W., Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar]
- 5. Merk D., Friedrich L., Grisoni F., Schneider G., Mol. Inf. 2018, 37, 1700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhavoronkov A., Ivanenkov Y. A., Aliper A., Veselov M. S., Aladinskiy V. A., Aladinskaya A. V., Terentiev V. A., Polykovskiy D. A., Kuznetsov M. D., Asadulaev A., Volkov Y., Zholus A., Shayakhmetov R. R., Zhebrak A., Minaeva L. I., Zagribelnyy B. A., Lee L. H., Soll R., Madge D., Xing L., Guo T., Aspuru-Guzik A., Nat. Biotechnol. 2019, 37, 1038–1040. [DOI] [PubMed] [Google Scholar]
- 7. Gao Q., Yang L., Zhu Y., Curr. Comput.-Aided Drug Des. 2010, 6, 37–49. [DOI] [PubMed] [Google Scholar]
- 8. Yoshimori A., Kawasaki E., Kanai C., Tasaka T., Chem. Pharm. Bull. 2020, 68, 227–233. [DOI] [PubMed] [Google Scholar]
- 9. Weininger D. J., J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar]
- 10. Bento A. P., Gaulton A., Hersey A., Bellis L. J., Chambers J., Davies M., Krüger F. A., Light Y., Mak L., McGlinchey S., Nowotka M., Papadatos G., Santos R., Overington J. P., Nucleic Acids Res. 2014, 42, D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a.REINVENT: Molecular de novo design using recurrent neural networks and reinforcement learning https://github.com/marcusolivecrona/reinvent;
- 11b. Olivecrona M., Blaschke T., Engkvist O., Chen H., J. Cheminf. 2017, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Wolber G., Langer T., J. Chem. Inf. Model. 2005, 45, 160–169; [DOI] [PubMed] [Google Scholar]
- 12b. Wolber G., Dornhofer A. A., Langer T., J. Comput.-Aided Mol. Des. 2007, 20, 773–788. [DOI] [PubMed] [Google Scholar]
- 13. Quan J., Yahata T., Adachi S., Yoshihara K., Tanaka K., Int. J. Mol. Sci. 2011, 12, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moll S., Desmoulière A., Moeller M. J., Pache J., Badi L., Arcadu F., Richter H., Satz A., Uhles S., Cavalli A., Drawnel F., Scapozza L., Prunotto M., BBA Mol. Cell Res. 2019, 1866, 118474. [DOI] [PubMed] [Google Scholar]
- 15. Li S., Zhang Z., Xue J., Guo X., Liang S., Liu A., Med. Sci. Monit. 2015, 21, 2433–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Canning P., Tan L., Chu K., Lee S. W., Gray N. S., Bullock A. N., J. Mol. Biol. 2014, 426, 2457–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poli G., Seidel T., Langer T., Front. Chem. 2018, 6, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B. A., Thiessen P. A., Yu B., Zaslavsky L., Zhang J., Bolton E. E., Nucleic Acids Res. 2019, 47, D1102–D1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bickerton G. R., Paolini G. V., Besnard J., Muresan S., Hopkins A. L., Nat. Chem. 2012, 4, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wenzel J., Matter H., Schmidt F., J. Chem. Inf. Model. 2019, 59, 1253–1268. [DOI] [PubMed] [Google Scholar]
- 21. Ertl P., Schuffenhauer A., J. Cheminf. 2009, 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary