

Abstract
The combination of pulsed dipolar electron paramagnetic resonance spectroscopy (PDS) with site‐directed spin labelling is a powerful tool in structural biology. Rational design of trityl‐based spin labels has enabled studying biomolecular structures at room temperature and within cells. However, most current trityl spin labels suffer either from aggregation with proteins due to their hydrophobicity, or from bioconjugation groups not suitable for in‐cell measurements. Therefore, we introduce here the highly hydrophilic trityl spin label Ox‐SLIM. Engineered as a short‐linked maleimide, it combines the most recent developments in one single molecule, as it does not aggregate with proteins, exhibits high resistance under in‐cell conditions, provides a short linker, and allows for selective and efficient spin labelling via cysteines. Beyond establishing synthetic access to Ox‐SLIM, its suitability as a spin label is illustrated and ultimately, highly sensitive PDS measurements are presented down to protein concentrations as low as 45 nm resolving interspin distances of up to 5.5 nm.
Keywords: biophysical chemistry, electron paramagnetic resonance, pulsed dipolar spectroscopy, spin labelling, trityl radicals
A hydroxylated trityl spin label for proteins, called OxSLIM was synthesized. Given its high hydrophilicity, outstanding bioresistance, and short linkage, it combines the most recent advances in just a single molecule. Moreover, its improved relaxation behaviour allows for highly sensitive electron paramagnetic resonance (EPR)‐based distance measurements down to concentrations as low as 45 nm.
Introduction
Since the discovery of the triphenylmethyl radical by Gomberg in 1900, [1] stable carbon‐centred radicals received rising attention. This especially applies to trityl radicals of the tetrathioaryl‐type (Scheme 1), derived from the so‐called Finland trityl 1..[ 2 , 3 ] Within the past two decades, 1. and its derivatives found widespread application in in vivo imaging, [4] oximetry,[ 5 , 6 ] pH‐sensing, [7] viscosity measurements, [8] and as polarizing agents in dynamic nuclear polarization (DNP).[ 9 , 10 ] Moreover, trityl radicals emerged as spin labels[ 11 , 12 , 13 , 14 ] for pulsed dipolar electron paramagnetic resonance spectroscopy (PDS)[ 15 , 16 , 17 ] to elucidate structures of biomolecules.[ 18 , 19 ] Exploiting their long TM relaxation times, [20] trityl radicals like 2 [13] paved the way to PDS measurements at physiological temperatures.[ 13 , 14 , 21 ] Recent trends in the development of trityl spin labels focused on their suitability for in‐cell measurements, where the reductive intracellular environment presents additional challenges with respect to the stability of the radical centre. Initial studies with 3. [18] proved the suitability of trityl radicals for this purpose in general, though the bioconjugation proceeded with low efficiency and inseparable aggregates. Trityl spin labels with functional groups for bioconjugation based on maleimides (4., 5., 6.)[ 22 , 23 , 24 ] coped with this challenge by providing highly selective and efficient linkage to cysteines. While 2.–5. are simple esters/amides of 1., the benzylic CH2‐linker used to construct the so‐called SLIM‐trityl 6. was shown to improve the stability towards intracellular reduction by shifting its redox potentials. Concomitantly, a short linker is introduced giving rise to narrow distance distributions. [24] Another inherent drawback caused by the lipophilic core of these first generation trityl spin labels is aggregation with themselves[ 25 , 26 ] and hydrophobic interactions with proteins.[ 18 , 19 , 22 , 23 ] This complicates the spin labelling of proteins and can adversely influence the PDS‐derived distance distributions. With bovine serum albumin for instance, aggregation of 1. occurred already at protein concentrations above 60 μm. [27] Therefore, efforts have been undertaken to increase the hydrophilicity of trityl radicals. For imaging and sensing purposes, conjugation of 1. to dendritic PEG‐esters,[ 28 , 29 ] dextrans, [30] or oligopeptides [31] addressed this issue. However, the resulting radicals become very large and are therefore not suitable as spin labels. Other approaches aimed for hydrophilic trityl cores by hydroxylation of the thioketal's methyl‐substituents. [32] Accordingly, the Ox063 radical 7. was reported early in patent literature but efficient synthetic access was disclosed only recently. [33] Based on this core, the hydrophilic spin label 8. has been introduced, [34] and utilized for distance measurements on outer membranes of E. coli just recently. [35] Despite its high water‐solubility, the methanethiosulfonate bioconjugation site is not suitable for in‐cell applications [36] and such long linking groups lead to unnecessarily broad distance distributions.[ 22 , 35 ] Therefore, we report here the modular synthesis of the hydroxylated short linked maleimide trityl (Ox‐SLIM) 9. that combines the reduction resistance and short linkage of 6. with the hydrophilicity of 7.. The hydroxyl‐groups on two of the bisthioketalaryl moieties provide the hydrophilicity, whereas the third, not hydroxylated bisthioketalaryl‐unit carries the benzylic maleimide and ensures high accessibility for labelling. Finally, it is shown that Ox‐SLIM 9. enables highly sensitive PDS measurements down to protein concentrations as low as 45 nm at a distance of 5.5 nm.
Scheme 1.
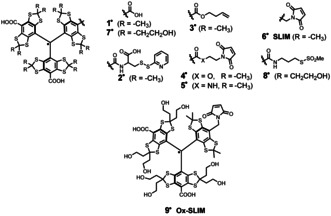
Lewis structures of trityl spin labels and their parent compounds.
Results and Discussion
The synthesis of Ox‐SLIM 9. needs aryl building block 10, which can be obtained via two alternative routes from bisthioketal 11[ 3 , 37 ] (Scheme 2). The first route starts with aromatic trimethylsilylation of 11 by reaction with lithium tetramethylpiperide (LiTMP) and in situ quenching with trimethylsilyl chloride. Noteworthy, the basicity of LiTMP (pK a = 37) [38] is not sufficient for quantitative deprotonation of 11, but it does not react with Me3SiCl either due to its low nucleophilicity. [38] This transformation avoids the use of stronger yet more nucleophilic bases such as lithium alkyls, which were shown to cleave the carbon−sulfur bond in 11.[ 32 , 40 ] An ipso‐iododesilylation with ICl then afforded 12 in a yield of 86 %. Lithium‐halogen exchange with nBuLi at −95 °C runs cleanly without any evidence for thioketal‐cleavage, and quenching with DMF gave aldehyde 13 in a yield of 73 % after acidic workup. Subsequent carbonyl reduction with NaBH4 and transformation of the benzylic alcohol to the TBS‐ether with TBS‐Cl under classical Corey conditions [41] provided 10 in a yield of 82 % over the last two steps, and of 51 % with respect to 11. However, the poor solubility of aldehyde 13 in most organic solvents rendered a scale‐up of this route cumbersome. Therefore, alternative access to 10 was sought for, and achieved via aldehyde 14, the synthesis of which has been described recently. [40] Reduction of 14 with NaBH4 gave alcohol 15 and subsequent silylation yielded TBS‐ether 16, both in yields of 93 % and 95 %, respectively. Next, 16 was iodinated adapting a recent procedure by Poncelet et al. [33] Though the iodination proceeded with a conversion of only 65 % on a 2 gram scale, simple recrystallization from CH3CN allowed for isolation of pure 10 in a yield of 56 %, or 39 % with respect to 11.
Scheme 2.
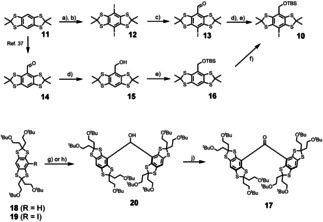
Synthesis of building blocks 10 and 17. a) 6.5 equiv. LiTMP, 0.1 equiv. Et3NHCl, 12 equiv. Me3SiCl, THF, −95 °C to r.t., 16 h. b) 3.0 equiv. ICl, CH2Cl2, r.t., 3 h, 86 % over two steps. c) 1.1 equiv. nBuLi, THF, −95 °C, 45 min, then 15.0 equiv. DMF, to r.t., 16 h. 73 % yield. d) 2.0 equiv. NaBH4, CH2Cl2/MeOH 2:1, r.t., 30 min, 93 % from 14. e) 1.2 equiv. tBuMe2SiCl, 2.5 equiv. Imidazole, DMF, r.t., 16 h, 82 % over two steps from 13, 91 % from 15. f) 1. LiTMP, THF, −78 °C, 2 h. 2. I2, to r.t., 16 h, 56 %. g) 0.95 equiv. nBuLi, THF, −95 °C, then 0.4 equiv. HCO2Me, r.t., 16 h, 36 %, for 12. h) 1.95 equiv. tBuLi, 0.48 equiv., THF, −95 °C, 45 min, then 0.48 equiv. HCO2Me, r.t., 16 h, 88 %, for 13. i) 1.25 equiv. Dess–Martin‐periodinane, CH2Cl2, r.t., 60 min, 93 %.
In the second branch of the converging synthesis towards 9., ketone 17 was synthesized from thioketals 18 and 19, which are available in three and four steps, respectively, following a recent protocol. [33] Deprotonation of 18 with nBuLi and reaction of the resulting anion with methyl formate provided diarylmethanol 20, if performing the lithiation at −95 °C. However, the yield of 36 % was fairly low. This was also the case when using MeLi. [40] By contrast, generating the lithiumaryl from 19 via lithium–halogen exchange with tBuLi at −95 °C increased the yield to 89 %. In the final step of this branch, Dess–Martin‐oxidation of 20 led to ketone 17 in a yield of 93 %.
Within the further synthesis, the most critical step is the formation of the triarylmethanol scaffold in 21 (Scheme 3). Considering the DFT‐optimized structure of ketone 17 (Figures S71 and 72), the Bürgi–Dunitz trajectory appears blocked by the bulky tert‐butoxy substituents and this steric congestion requires tremendous structural reorganization during the transformation. Ultimately, side‐reactions such as thioketal cleavage[ 32 , 40 ] or decomposition of lithium organyls through reaction with solvent molecules makes this transformation additionally cumbersome. To cope with these issues, the required nucleophile was generated from 10 by lithium‐halogen exchange with exactly 2.0 equiv. tBuLi at −95 °C. While other lithium alkyls leave the corresponding alkyl halide behind, no electrophiles remain in the solution using tBuLi, so that the aryl lithium can only react with the ketone. In this way, 21 was obtained in an isolated yield of 83 %, while the use of sBuLi [33] resulted in our hands in a yield of only 31 %. Moreover, metal–halogen exchange performed superior compared to direct lithiation of 16 with conditions proposed by Hintz et al., [40] where the yield dropped to 11 %. Noteworthy, the choice of solvent seemed important: No conversion was observed within n‐hexane, presumably related to lacking stabilization of the ionic intermediate of the nucleophilic addition, while diethyl ether proved suitable for this transformation. Advantageous of the benzylic TBS‐ether in 10 and 21 is that it paves the way for the late‐stage introduction of the concealed maleimide as outlined below. In addition, any functionality compliant with the metal–halogen exchange conditions (e.g. protected alkynes) [40] can be incorporated at this stage, highlighting the versatility of the approach chosen here for accessing asymmetric trityl radicals with high hydrophilicity.
Scheme 3.
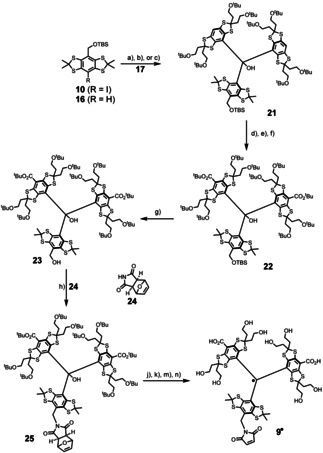
Final steps for the synthesis of 9.. a) tBuLi, −95 °C, Et2O, 45 min, then 17, to r.t., 16 h, 83 %; b) sBuLi, −95 °C, Et2O, 45 min, then 17, to r.t., 16 h, 31 %; c) MeLi, THF, r.t., 100 min, exchange for Et2O, then 18, 16 h, 11 %. a, b) for 10, c) for 16. d) sBuLi, −20 °C, TMEDA, 120 min. e) CO2, to r.t., 16 h. f) O‐tert‐butyl‐N,N’‐diisopropylisourea, PhMe, 60 °C, 4 h, 47 % over three steps. g) nBu4NF, THF, 45 °C, 3 h, 82 %. h) ADDP, nBu3P, 24, THF, r.t., 5 h, 71 %. i, 1) CF3SO3H, CH3CN, r.t., 4 h. 2) SnCl2, THF, r.t., 20 min. j) HCO2H, 45 °C, 16 h. l) NaHCO3, MeOH, r.t., 16 h. m) DMF, 100 °C, 16 h, 60 % over four steps.
Next, the carboxylation of trityl alcohol 21 was carried out by deprotonation with sBuLi in TMEDA/n‐hexane using gaseous CO2 as electrophile. Other frequently applied carboxylation reagents such as Boc2O[ 16 , 24 , 40 ] more likely contain traces of water, severely diminishing the yield of the dicarboxylated product. The obtained dicarboxylic acid was then converted into the corresponding tert‐butyl diester 22 using O‐tert‐butyldiisopropylisourea, [42] since classical Steglich conditions [43] (DCC, DMAP) or the combination Boc2O/DMAP [44] yielded only traces of product. Subsequently, the TBS‐ether in 22 was cleaved with commercially available nBu4NF (TBAF) in THF yielding 23. Here, 10 equiv of TBAF and a slightly elevated temperature of 45 °C were required for sufficient reactivity. The C−N bond required for 9. was introduced via a Mitsunobu reaction in analogy to our previous work, where the maleimide was concealed as a thermally labile tetrahydroisoindolinone. [24] However, since elevated temperatures are required in the later route, the exo‐Diels–Alder adduct of furane and maleimide 24 was used, which does not undergo retro‐Diels–Alder fragmentation up to 50 °C (Supporting Information section 3.3). Carrying out the transformation with the classical reagents Ph3P and diethylazodicarboxylate (DEAD), 25 was obtained in a yield of only 32 % among many unknown by‐products (Supporting Information section 3.1). Using instead tri‐n‐butylphosphine and 1,1’‐azodicarbonyldipiperidine (ADDP) increased the yield of the Mitsunobu‐type C−N bond formation and afforded 25 in a yield of 71 %. Both, the acceleration of the C−N bond formation owing to the higher pK a of the intermediate betaine and steric factors avoiding interactions with the central OH‐group are assumed to contribute to this improved yield. In the further course towards 9., the corresponding trityl radical was generated with CF3SO3H and reduction of the generated tritylium cation with SnCl2 in situ.
The resulting product was immediately treated with neat formic acid at 45 °C for 16 h in order to convert the remaining tert‐butyl ethers to formate esters and ensure the cleavage of the tert‐butyl esters. The formate esters were then subjected to very mild hydrolysis with NaHCO3 in methanol at room temperature. It should be noted that other hydrolysing conditions involving LiOH or Ba(OH)2 endangered the integrity of the concealed maleimide (Supporting Information section 3.4). Finally, the maleimide group was deprotected by means of a retro‐Diels–Alder reaction at 100 °C in degassed DMF giving the fully characterized (cw‐EPR, HRMS, HPLC; cf. Supporting Information) and water‐soluble spin label 9..
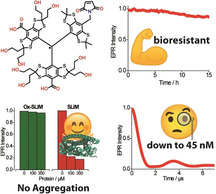
The room temperature continuous wave (cw) X‐band EPR spectrum of 9. is shown in Figure 1 a. In analogy to the spectrum of 6., [24] it consists of nine resolved lines which arise from hyperfine coupling to the benzylic nitrogen (A N = 1.48 MHz) and the two magnetically inequivalent benzylic hydrogen atoms (A H1 = 3.02 MHz; A H2 = 6.09 MHz). Interestingly, recording the spectrum in frozen solution does not only reveal the expected apparent doublet of 9. (in analogy to 6.), but also exhibits a superimposed Pake pattern (Figure 1 b) in a ratio of 69:31. Analysis of the Pake pattern provides a dipolar coupling constant of 19.4 G corresponding to an interspin distance of 9.9 Å. [22] The Pake pattern is attributed to a noncovalent dimer (9.)2, which was experimentally observed in ESI(−)‐MS (Figure S51). Additional evidence for (9.)2 was obtained through a computational study involving a conformer search by the CREST [45] algorithm at the GFN‐FF [46] level of theory. The conformation of lowest energy found by CREST was further optimized by B97‐3c [47] and is shown in Figure 2. Hybrid‐DFT single‐point calculations (PBE0, [48] def2‐TZVPP [49] ) combined with GFN2‐xTB [50] thermostatistical contributions and COSMO‐RS(H2O) [51] solvation free energies revealed that the homodimer is stabilized by ΔG = −14.8 kcal mol−1 due to the formation of hydrogen bonds involving one maleimido substituent (SI section 8.1). For the Ox063‐radical 7., a similar dimerization was shown to be facilitated by Me4N+ as a template, [25] a role fulfilled here by the maleimide moiety. Interestingly, high concentrations of glycerol suppressed dimerization to (9.)2, presumably due to competitive hydrogen bonding, underpinning the non‐covalent nature of the dimer (Supporting Information section 6.4). As mentioned above, the hydrophobicity of the first‐generation trityl spin labels 2.–6. leads to their aggregation with biomolecules. The concomitant immobilization triggers extensive line‐broadening and consequently peak‐to‐peak amplitude reduction in the cw EPR spectra. This signal depletion has been implemented as a semi‐quantitative measure of non‐specific aggregation with proteins.[ 27 , 29 , 32 ] In order to test this, a cysteine‐free construct (C219A) of yersinia outer protein O (YopO) was expressed, purified, [24] and added in increasing amounts to solutions of spin labels 4., 6., and 9. (Figure 3 a). For both 4. and 6., significant signal depletion occurs, indicating aggregation with the protein. By contrast, virtually no signal depletion was encountered for 9., highlighting the hydrophilic nature of 9. which impedes the aggregation.
Figure 1.
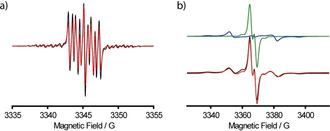
CW X‐band EPR spectra of 50 μm 9. in aqueous PBS buffer recorded at a) room temperature and at b) 100 K after addition of 20 % glycerol; simulations are overlaid in red. For (b), the experimental spectrum was simulated as a sum of a monomer (green) and a dimer of 9. (blue).
Figure 2.
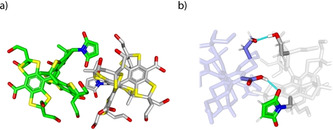
DFT structure of (9.)2. a) Complete view, and b) close‐up with hydrogen bonds highlighted in light blue.
Figure 3.
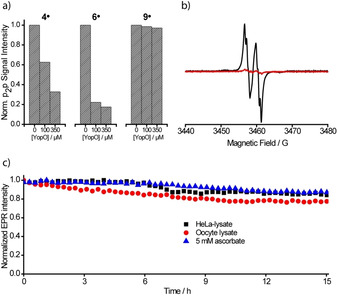
Properties of Ox‐SLIM 9.. a) CW X‐band EPR peak‐to‐peak amplitude of trityl spin labels 4., 6., and 9. (50 μm in PBS‐buffer) at increasing concentration of cysteine‐free YopO C219A. b) CW X‐band EPR spectra of YopO Y588Tox/N624Tox (black) and YopO C219A (red) after labelling and workup. The spectra were recorded at 298 K in PBS‐buffer. c) EPR double integral intensity of Y588Tox/N624Tox in HeLa lysate, X. laevis oocyte lysate, and 5 mm sodium ascorbate monitored over time.
Next, the performance of 9. as a spin label was evaluated. Performing the labelling experiment on the cysteine‐free YopO construct with 4 equivalents of 9., a minor extent of unspecific labelling (7 %, Figure 3 b and Supporting Information section 5.2) was observed. Applying the same labelling conditions to the double‐cysteine mutant (Y588C/N624C), YopO could be doubly labelled with 9. with a labelling efficiency of 85 % (Figure 3 b and Supporting Information section 5.3). Since the dimer (9.)2 is only formed in appreciable amounts upon freezing it does not interfere with the labelling reaction, and once bound to a cysteine the maleimide group cannot be involved into the dimer formation anymore. In the following, the side chain generated by binding 9. to a cysteine residue is called Tox.
In order to pave the way for future in‐cell applications, the reduction stability of Y588Tox/N624Tox was assessed within aqueous solutions of 5 mm ascorbate and lysates of HeLa cells and Xenopus laevis oocytes. As shown in Figure 3 c, YopO Y588Tox/N624Tox exhibits a high stability in these media, attributed to the imidomethylene‐motif of 9., which destabilizes the anionic species resulting from reduction. [24]
Finally, a PDS measurement was performed on YopO Y588Tox/N624Tox. Exploiting the high sensitivity of the double quantum coherence (DQC) experiment, [24] the time trace shown in Figure 4 a was obtained. It exhibits an SNR [52] of 133 h−1/2, exceeding the value obtained with 6. (46 h−1/2) due to a longer TM‐relaxation time (Supporting Information chapter 7). The corresponding distance distribution shows a bimodal shape with most probable distances at 4.60 and 5.48 nm (Figure 4 b), which coincide very well with the results obtained for 6. on this mutant, and supports the idea that the reason for the bimodality are two different structure of the α‐helix, at which the labels are bound. [24] Slight changes in the intensity distribution of the two peaks is attributed to different time trace lengths and different conformer clouds of the different labels. The high SNR prompted us to perform DQC measurements on a 45 nm sample of YopO Y588Tox/N624Tox, which still yielded an SNR of 1.24 h−1/2 at a dipolar evolution time of 4.5 μs (Supporting Information chapter 7). This implies a sensitivity improvement exceeding a factor of 2 compared to our recent publication. [24]
Figure 4.
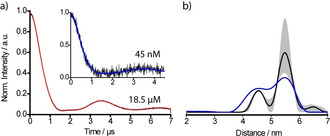
PDS measurements with 9.. a) Background‐corrected DQC time traces obtained for YopO Y588Tox/N624Tox (black), overlaid with their fits by DeerAnalysis (red/blue), shown at both 18.5 μm and 45 nm protein concentrations. b) Distance distribution derived from the time trace of the 18.5 μm sample with the background validation indicated in grey. The distance distribution obtained from the 45 nm samples is overlaid in blue.
Conclusions
In this study, the highly hydrophilic trityl spin label Ox‐SLIM 9. was introduced through a streamlined synthesis without a statistical step. The chosen approach is highly versatile and the intermediates presented in this study can be utilized as precursors to various hydrophilic trityl radicals. Additionally, an improved Mitsunobu/retro‐Diels–Alder sequence is used for the introduction of the maleimide. Overall, label 9. combines most recent developments on trityl spin labels in a single molecule, i.e., a short linker, bioresistancy, and no aggregation with proteins. Demonstrated for the protein YopO, 9. allows efficient labelling of cysteines in a selective fashion. Finally, distance measurements with protein concentrations down to 45 nm were viable, thus setting a new benchmark.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funding by the DFG via SPP1601 is gratefully acknowledged. Open access funding enabled and organized by Projekt DEAL.
N. Fleck, C. Heubach, T. Hett, S. Spicher, S. Grimme, O. Schiemann, Chem. Eur. J. 2021, 27, 5292.
References
- 1. Gomberg M., J. Am. Chem. Soc. 1900, 22, 757–771. [Google Scholar]
- 2. Andersson S., Rydbeck A., Mahno R. S., US Patent 5728370, 1999.
- 3. Reddy T. J., Iwama T., Halpern H. J., Rawal V. H., J. Org. Chem. 2002, 67, 4635–4639. [DOI] [PubMed] [Google Scholar]
- 4. Williams B. B., Halpern H. J., Biomed. EPR, Part A: Free Radicals, Metals, Medicine and Physiology (Eds.: Eaton S. R., Eaton G. R., Berliner L. J.), Springer US, Boston, 2005, pp. 283–319. [Google Scholar]
- 5. Epel B., Haney C. R., Hleihel D., Wardrip C., Med. Phys. 2010, 37, 2553–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bobko A. A., Dhimitruka I., Eubank T. D., Marsh C. B., Zweier J. L., Khramtsov V. V., Free Radic. Biol. Med. 2009, 47, 654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boś-Liedke A., Walawender M., Woźniak A., Flak D., Gapiński J., Jurga S., Kucińska M., Plewiński A., Murias M., Elewa M., Lampp L., Imming P., Tadyszak K., Cell Biochem. Biophys. 2018, 76, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poncelet M., Driesschaert B., Angew. Chem. Int. Ed. 2020, 59, 16451–16454; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 16593–16596. [Google Scholar]
- 9. Mathies G., Caporini M. A., Michaelis V. K., Liu Y., Hu K. N., Mance D., Zweier J. L., Rosay M., Baldus M., Griffin R. G., Angew. Chem. Int. Ed. 2015, 54, 11770–11774; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11936–11940. [Google Scholar]
- 10. Ardenkjær-Larsen J. H., Fridlund B., Gram A., Hansson G., Hansson L., Lerche M. H., Servin R., Thaning M., Golman K., Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reginsson G. W., Kunjir N. C., Sigurdsson S. T., Schiemann O., Chem. Eur. J. 2012, 18, 13580–13584. [DOI] [PubMed] [Google Scholar]
- 12.”Trityl Radicals as Spin Labels”: Krumkacheva O., Elena B., in Electron Paramagnetic Resonance, The Royal Society of Chemistry, Cambridge, 2017, pp. 35–60. [Google Scholar]
- 13. Yang Z., Liu Y., Borbat P., Zweier J. L., Freed J. H., Hubbell W. L., J. Am. Chem. Soc. 2012, 134, 9950–9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shevelev G. Y., Krumkacheva O. A., Lomzov A. A., Kuzhelev A. A., Rogozhnikova O. Y., Trukhin D. V., Troitskaya T. I., Tormyshev V. M., Fedin M. V., Pyshnyi D. V., Bagryanskaya E. G., J. Am. Chem. Soc. 2014, 136, 9874–9877. [DOI] [PubMed] [Google Scholar]
- 15. Reginsson G. W., Schiemann O., Biochem. Soc. Trans. 2011, 39, 128–139. [DOI] [PubMed] [Google Scholar]
- 16. Jeschke G., Annu. Rev. Phys. Chem. 2012, 63, 419–446. [DOI] [PubMed] [Google Scholar]
- 17. Borbat P. P., Freed J. H., in EMagRes, Wiley, Chichester, 2017, pp. 465–494. [Google Scholar]
- 18. Jassoy J. J., Berndhäuser A., Duthie F., Kühn S. P., Hagelueken G., Schiemann O., Angew. Chem. Int. Ed. 2017, 56, 177–181; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 183–187. [Google Scholar]
- 19. Joseph B., Tormyshev V. M., Rogozhnikova O. Y., Akhmetzyanov D., Bagryanskaya E. G., Prisner T. F., Angew. Chem. Int. Ed. 2016, 55, 11538–11542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11710–11714. [Google Scholar]
- 20. Kuzhelev A. A., Trukhin D. V., Krumkacheva O. A., Strizhakov R. K., Rogozhnikova O. Y., Troitskaya T. I., Fedin M. V., Tormyshev V. M., Bagryanskaya E. G., J. Phys. Chem. B 2015, 119, 13630–13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Z., Jiménez-Osés G., López C. J., Bridges M. D., Houk K. N., Hubbell W. L., J. Am. Chem. Soc. 2014, 136, 15356–15365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jassoy J. J., Heubach C. A., Hett T., Bernhard F., Haege F. R., Hagelueken G., Schiemann O., Molecules 2019, 24, 2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giannoulis A., Yang Y., Gong Y.-J., Tan X., Feintuch A., Carmieli R., Bahrenberg T., Liu Y., Su X.-C., Goldfarb D., Phys. Chem. Chem. Phys. 2019, 21, 10217–10227. [DOI] [PubMed] [Google Scholar]
- 24. Fleck N., Heubach C. A., Hett T., Haege F. R., Bawol P. P., Baltruschat H., Schiemann O., Angew. Chem. Int. Ed. 2020, 59, 9767–9772; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 9854–9859. [Google Scholar]
- 25. Marin-Montesinos I., Paniagua J. C., Vilaseca M., Urtizberea A., Luis F., Feliz M., Lin F., Van Doorslaer S., Pons M., Phys. Chem. Chem. Phys. 2015, 17, 5785–5794. [DOI] [PubMed] [Google Scholar]
- 26. Marin-Montesinos I., Paniagua J. C., Peman A., Vilaseca M., Luis F., Van Doorslaer S., Pons M., Phys. Chem. Chem. Phys. 2016, 18, 3151–3158. [DOI] [PubMed] [Google Scholar]
- 27. Song Y., Liu Y., Liu W., Villamena F. A., Zweier J. L., RSC Adv. 2014, 4, 47649–47656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu W., Nie J., Tan X., Liu H., Yu N., Han G., Zhu Y., Villamena F. A., Song Y., Zweier J. L., Liu Y., J. Org. Chem. 2017, 82, 588–596. [DOI] [PubMed] [Google Scholar]
- 29. Song Y., Liu Y., Hemann C., Villamena F. A., Zweier J. L., J. Org. Chem. 2013, 78, 1371–1376. [DOI] [PubMed] [Google Scholar]
- 30. Poncelet M., Driesschaert B., Tseytlin O., Tseytlin M., Eubank T. D., Khramtsov V. V., Bioorg. Med. Chem. Lett. 2019, 29, 1756–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Driesschaert B., Leveque P., Gallez B., Marchand-Brynaert J., Tetrahedron Lett. 2013, 54, 5924–5926. [Google Scholar]
- 32. Qu Y., Li Y., Tan X., Zhai W., Han G., Hou J., Liu G., Song Y., Liu Y., Chem. Eur. J. 2019, 25, 7888–7895. [DOI] [PubMed] [Google Scholar]
- 33. Poncelet M., Huffman J. L., Khramtsov V. V., Dhimitruka I., Driesschaert B., RSC Adv. 2019, 9, 35073–35076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tormyshev V. M., Chubarov A. S., Krumkacheva O. A., Trukhin D. V., Rogozhnikova O. Y., Spitsyna A. S., Kuzhelev A. A., Koval V. V., Fedin M. V., Godovikova T. S., Bowman M. K., Bagryanskaya E. G., Chem. Eur. J. 2020, 26, 2705–2712. [DOI] [PubMed] [Google Scholar]
- 35. Ketter S., Gopinath A., Rogozhnikova O., Trukhin D., Tormyshev V. M., Bagryanskaya E. G., Joseph B., Chem. Eur. J. 2021, 27, 2299–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Igarashi R., Sakai T., Hara H., Tenno T., Tanaka T., Tochio H., Shirakawa M., J. Am. Chem. Soc. 2010, 132, 8228–8229. [DOI] [PubMed] [Google Scholar]
- 37. Kopp K., Schiemann O., Fleck N., Molecules 2020, 25, 3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Campbell M., Snieckus V., Baxter E. W., in e-EROS, Wiley, 2008, 10.1002/047084289X.rl143.pub2. [DOI] [Google Scholar]
- 39. Krizan T. D., Martin J. C., J. Am. Chem. Soc. 1983, 105, 6155–6157. [Google Scholar]
- 40. Hintz H., Vanas A., Klose D., Jeschke G., Godt A., J. Org. Chem. 2019, 84, 3304–3320. [DOI] [PubMed] [Google Scholar]
- 41. Corey E. J., Venkateswarlu A., J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar]
- 42. Burk R. M., Berger G. D., Bugianesi R. L., Girotra N. N., Parsons W. H., Ponpipom M. M., Tetrahedron Lett. 1993, 34, 975–978. [Google Scholar]
- 43. Neises B., Steglich W., Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524; [Google Scholar]; Angew. Chem. 1978, 90, 556–557. [Google Scholar]
- 44. Takeda K., Akiyama A., Nakamura H., Takizawa S. I., Mizuno Y., Takayanagi H., Harigaya Y., Synthesis 1994, 1994, 1063–1066. [Google Scholar]
- 45. Pracht P., Bohle F., Grimme S., Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [DOI] [PubMed] [Google Scholar]
- 46. Spicher S., Grimme S., Angew. Chem. Int. Ed. 2020, 59, 15665–15673; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 15795–15803. [Google Scholar]
- 47. Brandenburg J. G., Bannwarth C., Hansen A., Grimme S., J. Chem. Phys. 2018, 148, 064104. [DOI] [PubMed] [Google Scholar]
- 48. Perdew J. P., Ernzerhof M., Burke K., J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar]
- 49. Schäfer A., Huber C., Ahlrichs R., J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]
- 50. Bannwarth C., Ehlert S., Grimme S., J. Chem. Theory Comput. 2019, 15, 1652–1671. [DOI] [PubMed] [Google Scholar]
- 51. Reinisch J., Diedenhofen M., Wilcken R., Udvarhelyi A., Glöß A., J. Chem. Inf. Model. 2019, 59, 4806–4813. [DOI] [PubMed] [Google Scholar]
- 52. Abdullin D., Brehm P., Fleck N., Spicher S., Grimme S., Schiemann O., Chem. Eur. J. 2019, 25, 14388–14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary