

Abstract
The development of transition metal-catalysed cross-coupling reactions has greatly influenced the manner in which the synthesis of complex organic molecules is approached. A wide variety of methods are now available for the formation of C(sp2)–C(sp2) bonds, and more recent work has focused on the use of C(sp3) electrophiles and nucleophiles. The use of secondary and tertiary alkyl nucleophiles in cross-coupling reactions remains a challenge because of the propensity of these alkyl groups to isomerize under the reaction conditions. Here, we report the development of a general Pd-catalysed process for the stereoretentive cross-coupling of secondary alkyl azastannatrane nucleophiles with aryl chlorides, bromides, iodides and triflates. Coupling partners with a wide range of electronic characteristics are well tolerated. The reaction occurs with minimal isomerization of the secondary alkyltin nucleophile, and with retention of absolute configuration. This process constitutes the first general method to use secondary alkyltin reagents in cross-coupling reactions.
The development of transition metal-catalysed carbon–carbon bond-forming reactions has greatly influenced the manner in which we approach the synthesis of complex organic molecules. Although previous studies have largely focused on the generation of C(sp2)–C(sp2) bonds, more recently, the use of C(sp3) nucleophiles and electrophiles in metal-catalysed cross-coupling reactions has been investigated1. The use of secondary and tertiary main-group organometallic nucleophiles in Pd-catalysed cross-coupling reactions is particularly challenging because of the propensity of the alkyl group to isomerize via β-hydride elimination/reinsertion sequences (Fig. 1a)2–4. Such side reactions result in the formation of inseparable isomeric products alongside reduced aryl products3. The use of electronically or sterically activated electrophiles can curb the background isomerization reactions by accelerating reductive elimination in Pd-catalysed reactions5–9. However, the overall utility of a synthetic method decreases dramatically when it is mainly limited to activated substrates.
Figure 1 |. Cross-coupling reactions using configurationally stable, optically active secondary alkyl nucleophiles.
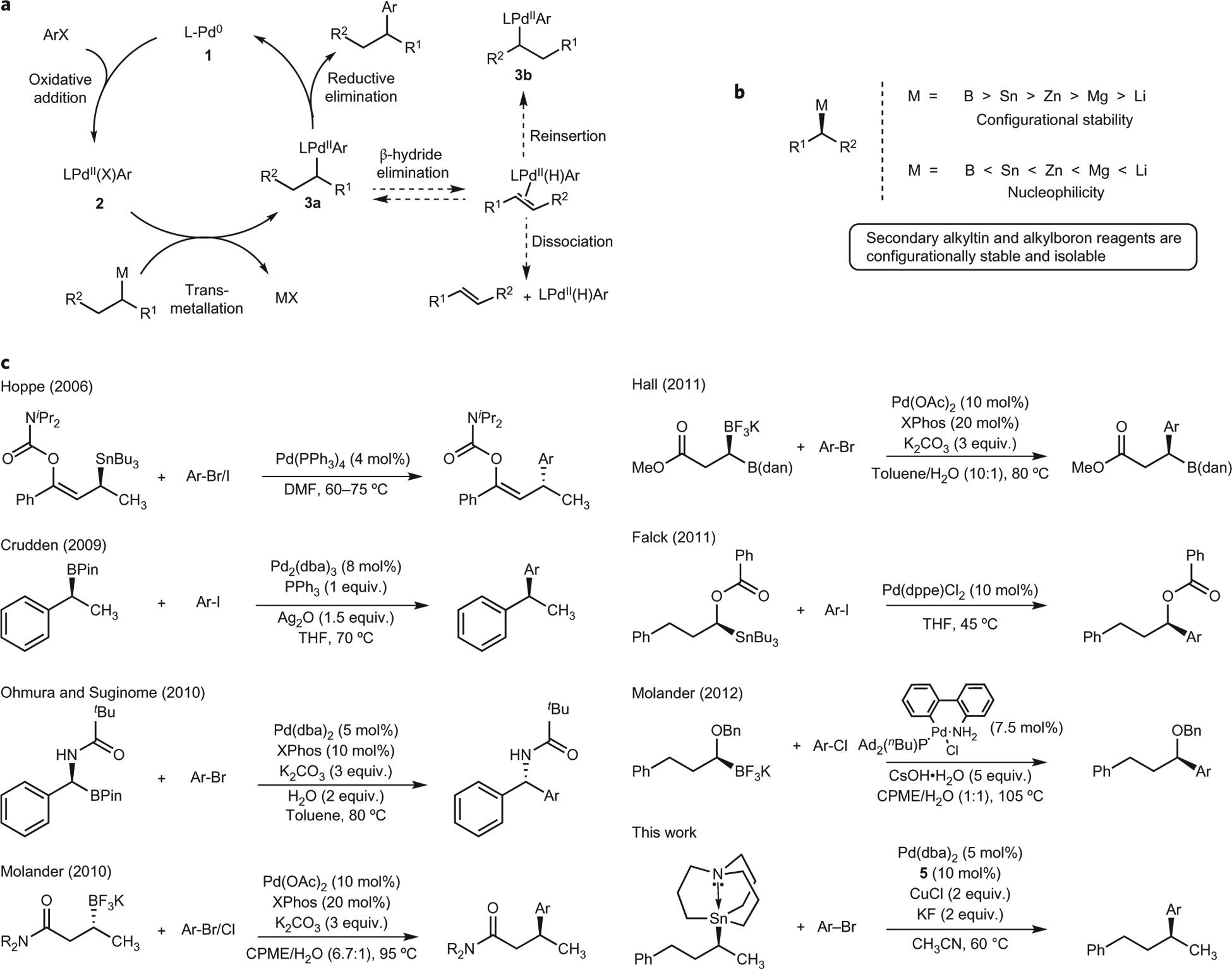
a, Proposed catalytic cycle for the Pd-catalysed cross-coupling of secondary nucleophiles and aryl electrophiles. It is necessary for reductive elimination to occur in the absence of the competing β-hydride elimination pathway to avoid the formation of isomeric by-products. b, Inverse relationship between configurational stability and nucleophilicity in secondary alkyl organometallic nucleophiles. c, Recent advances in Pd-catalysed cross-coupling reactions between isolable, optically active organometallic nucleophiles and aryl halides. The groups of Hoppe29, Crudden22, Ohmura and Suginome32, Molander31,37, Hall35 and Falck28 have demonstrated the use of isolable, optically active organometallic nucleophiles in cross-coupling reactions. However, nucleophiles in these examples require activation via the presence of a C(sp2) α-carbon, heteroatomic α-carbon and/or a strongly coordinating β-carbonyl group. DMF, dimethylformamide; THF, tetrahydrofuran; CPME, cyclopentyl methyl ether; dba, dibenzylideneacetone; XPhos, 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl.
A major focus of the research in our laboratory is the development of new methods for carbon–carbon bond formation via metal-catalysed cross-coupling reactions using alkyl nucleophiles10–13. Recently, we reported general Ni-catalysed processes for the cross-coupling of secondary alkylzinc and tertiary alkylmagnesium nucleophiles with aryl electrophiles10–12. These methods largely circumvent the β-hydride elimination/reinsertion sequences that have limited previous Pd-catalysed systems. To expand the versatility of the cross-coupling reactions that use secondary alkyl nucleophiles, we sought to extend the processes to the use of isolable, configurationally stable, optically active organometallic nucleophiles. The development of such transformations is impeded by the inverse relationship that exists between the nucleophilicity and configurational stability of carbon–metal bonds in main-group organometallic nucleophiles (Fig. 1b)14. Although increased covalency tends to coincide with enhanced configurational stability of the carbon–metal bond, it also tends to coincide with reduced nucleophilicity15–19. This trend, in addition to the inherent bulk of a secondary nucleophile, results in the prohibitively slow transmetallation of a secondary nucleophile as the covalency of the carbon–metal bond increases. Additionally, rapid reductive elimination is still required to circumvent the competing β-hydride elimination pathway.
Secondary alkyltin and alkylboron nucleophiles may be activated towards transmetallation and reductive elimination by the inclusion of an sp2-hybridized carbon atom in the α-position to the secondary alkyl centre20–23. The use of secondary alkyltin24–30 and secondary alkylboron31–37 nucleophiles containing C(sp3) α-carbons requires remote activation via a coordinating group on the nucleophile in order to accelerate transmetallation38 and/or retard the formation of isomerized products via competitive β-hydride elimination pathways. These limitations have prevented the development of a general method to make use of configurationally stable, optically active nucleophiles directly in cross-coupling reactions. Recently developed methods for the Pd-catalysed cross-coupling of activated optically active alkylboron and alkyltin nucleophiles with aryl electrophiles are shown in Fig. 1c. Because optically active organotin and organoboron reagents are stable, storable and can undergo stereospecific transmetallation/reductive elimination, the development of a general method by which to efficiently employ unactivated secondary alkyltin or secondary alkylboron nucleophiles in a cross-coupling reaction would potentially alter the paradigm by which asymmetric synthesis is approached.
Results and discussion
Development of a cross-coupling reaction using racemic secondary alkyl azastannatranes.
The use of unactivated alkylstannane reagents in cross-coupling reactions is also complicated by the need for four substituents on the tin centre. This problem is overcome in traditional Stille cross-coupling reactions by exploiting the enhanced migratory aptitude of C(sp2) and C(sp) substituents relative to C(sp3) substituents on tin1,39,40. Alkyl ligands can therefore be used as inert ‘dummy ligands’ in cases where the transfer of aryl, alkenyl or alkynyl substituents is desired. However, in cases where tetraalklylstannanes are used as a nucleophile, only one alkyl group is typically transmetallated. As a result, three equivalents of a potentially precious alkyl unit are effectively wasted. Vedejs and colleagues demonstrated an elegant solution to this problem, achieving selective alkyl transfer from an alkyl-azastannatrane reagent (4)41. Internal coordination of the nitrogen atom in the azastannatrane backbone was proposed to selectively labilize the apical alkyl substituent towards transmetallation. Although this original work was limited to the use of primary alkyl azastannatranes42,43, we felt that an analogous approach might be used to achieve selective alkyl transfer from secondary alkyl azastannatranes, which might then be expanded to the use of optically active secondary azastannatranes. Here, we report the development of a general Pd-catalysed process for the cross-coupling of unactivated secondary alkyl azastannatrane nucleophiles and aryl electrophiles. This reaction displays little dependence on the electronic characteristics of either coupling partner, and occurs with minimal concurrent isomerization of the secondary alkyltin nucleophile. Aryl chlorides, bromides, iodides and triflates are all viable electrophiles in this process. Furthermore, optically active secondary alkyl azastannatranes undergo cross-coupling reactions with complete retention of absolute configuration using this method. This process constitutes the first general method to use secondary alkyltin reagents in cross-coupling reactions, and the first cross-coupling reaction that is conducive to the broad use of isolable and optically active nucleophiles.
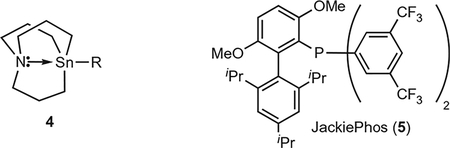
Using the Pd-catalysed coupling of bromobenzene and s-butyl azastannatrane (6) as a model system, ligand effects were investigated. Low conversions and a preponderance of rearrangement product resulted from the use of most ligands. JackiePhos (5), a bulky electron-deficient biarylphosphine, emerged as the most promising ligand for this transformation. This commercially available ligand was developed by the Buchwald group to facilitate the rate-limiting transmetallation of secondary amides in Pd-catalysed amidation reactions of aryl halides44. The efficacy of 5 as a supporting ligand in the cross-coupling of secondary alkyl azastannatranes may similarly result from the propensity of 5 to facilitate transmetallation of the secondary alkyl group while still promoting rapid reductive elimination. Systematic reaction optimizations revealed that the cross-coupling reaction proceeded most efficiently at 60 °C in acetonitrile and in the presence of CuCl and KF45.
Scope of Pd-catalysed cross-coupling reactions using racemic alkyl azastannatranes.
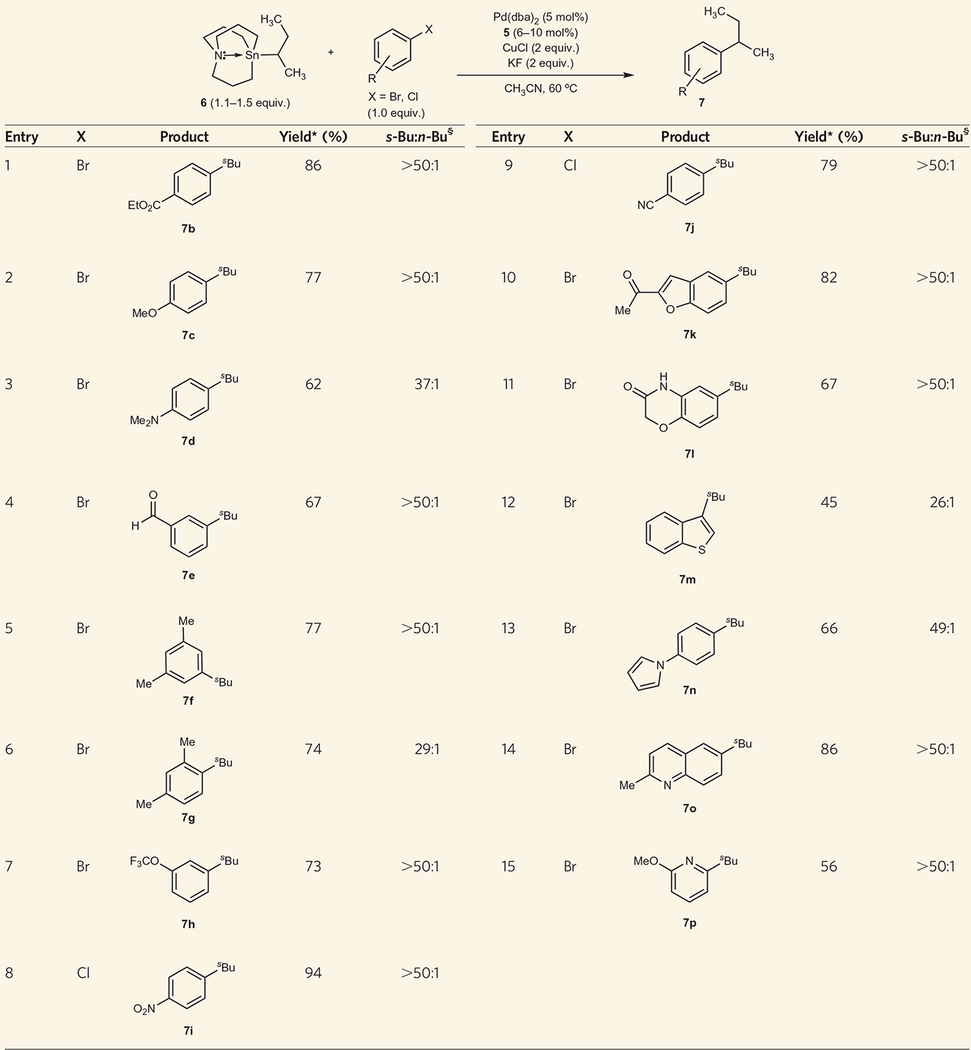
To evaluate the substrate scope accommodated in this reaction, we used 6 in cross-coupling reactions with a series of aryl and heteroaryl electrophiles (Table 1; Supplementary Information). In general, cross-coupling products were obtained with ratios of retention to isomerization greater than 50:1. A nominal dependence on the electronic properties of the aryl bromide electrophile was observed. Electron-rich, electron-neutral and electron-deficient aryl bromides all reacted to give high yields of product. Aryl bromides bearing electrophilic functional groups such as aldehydes, ketones, esters, amides and nitriles underwent successful cross-coupling reactions. The presence of an ortho-substituent was also well-tolerated. Importantly, heteroaromatic electrophiles could be broadly used without significant isomerization of the secondary alkyl nucleophile. Electron-deficient aryl chloride electrophiles also underwent efficient cross-coupling reactions under these conditions. All these reactions were performed on the benchtop, without the need for an inert-atmosphere glovebox. Inductively coupled plasma mass spectrometry analysis of a sample prepared using this cross-coupling procedure showed the presence of only 30 μg g−1 trace tin.
Table 1 |.
Pd-catalysed cross-coupling reactions using s-butyl azastannatrane (6) and aryl/heteroaryl electrophiles.
|
Reaction conditions: aryl halide (1 mmol), 6 (1.1–1.5 mmol), CuCl (2 mmol), KF (2 mmol), Pd(dba)2 (0.05 mmol), 5 (0.06–0.10 mmol), CH3CN (3 ml).
Average isolated yield of two runs.
Ratio of retention product (s-Bu) to isomerization product (n-Bu) determined by gas chromatography.
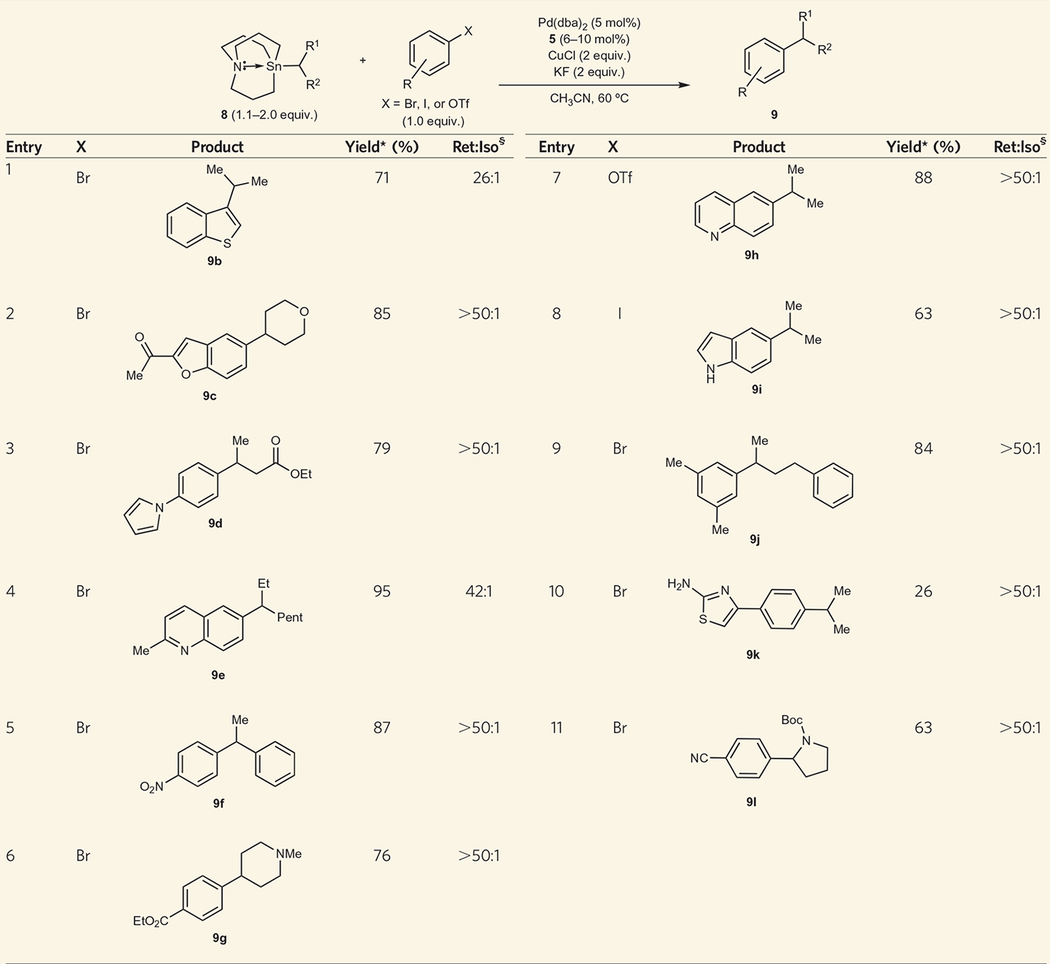
To demonstrate the general application of secondary azastannatrane nucleophiles in Pd-catalysed reactions, we used a variety of secondary azastannatranes. These substrates were readily generated from 4b using a modified version of the procedure developed by Vedejs (Fig. 2). The judicious choice of solvent allowed secondary lithium, magnesium halide and zinc halide nucleophiles to be used as precursors to the secondary azastannatrane reagents. All alkyl azastannatranes were air- and moisture-stable and were prepared in excellent yield. We successfully employed secondary alkyl groups bearing ethers, amines, esters and amides (Table 2). Secondary benzylic nucleophiles and bis-α-substituted nucleophiles underwent efficient cross-coupling reactions. During these studies, we also demonstrated that the use of aryl triflates and aryl iodides can be tolerated in these reactions. Therefore, this cross-coupling reaction appears to be highly general with respect to the nucleophile and electrophile employed.
Figure 2 |. Preparation of alkyl azastannatrane derivatives.
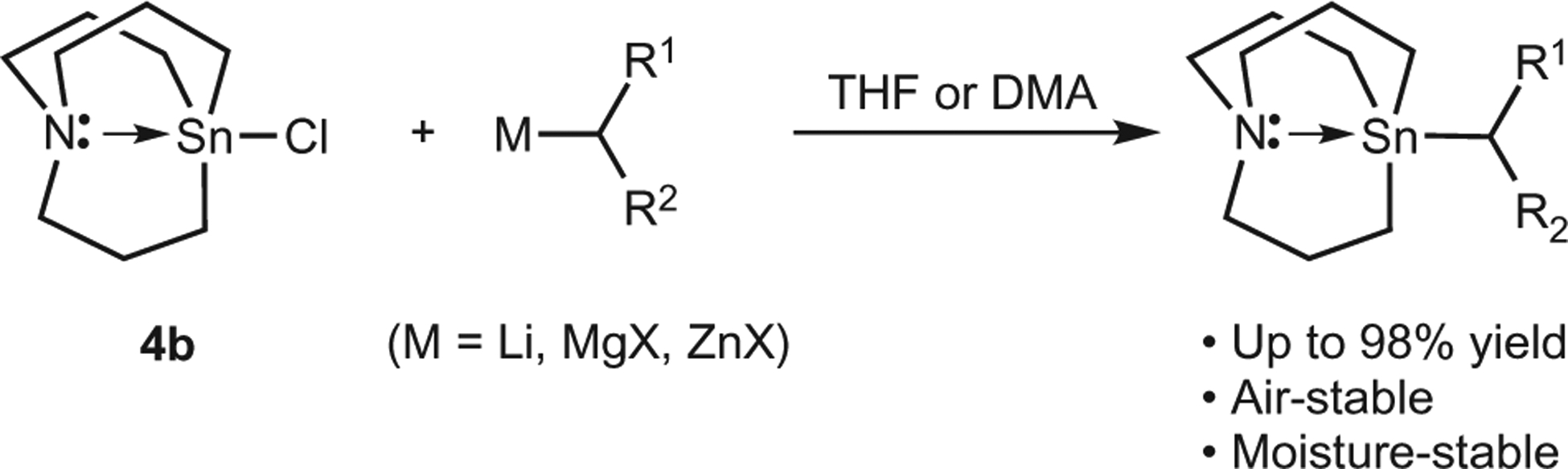
Stable and easily handled alkyl azastannatrane reagents can be prepared by reaction of a variety of organometallic nucleophiles with the azastannatrane chloride. THF, tetrahydrofuran; DMA, dimethylacetamide.
Table 2 |.
Pd-catalysed cross-coupling reactions using secondary alkyl azastannatrane (8) and aryl/heteroaryl electrophiles.
|
Reaction conditions: aryl halide (0.5 mmol), 6 (0.55–1.0 mmol), CuCl (1 mmol), KF (1 mmol), Pd(dba)2 (0.025 mmol), 5 (0.03–0.05 mmol), CH3CN (3 mL).
Average isolated yield of two runs.
Ratio of retention product to isomerization product determined by gas chromatography.
Stereospecificity of cross-coupling reactions using optically active alkylstannatranes.
In the field of drug discovery, it is particularly important that non-racemic molecules can be readily and reliably generated for evaluation as potential therapeutic agents. With this in mind, we evaluated the ability of non-racemic alkyl azastannatrane derivatives to undergo a stereospecific cross-coupling reaction without erosion of the original enantiomeric excess (e.e.). Using the asymmetric lithiation procedure described by Beak and colleagues46,47, we prepared and isolated optically active 2-stannylpyrrolidine derivative 10 with 93% e.e. (Fig. 3a). The enantiomers of racemic 10 have also been successfully separated using preparatory chiral high-performance liquid chromatography (HPLC). Under ambient conditions, 10 appears to exhibit configurational stability indefinitely. When enantiopure 10 was used in a cross-coupling reaction with 4-bromo benzonitrile, only nominal erosion of enantiomeric excess was observed in cross-coupling product 9l (Fig. 3b). To determine the absolute stereochemistry of the product, we obtained an X-ray crystal structure of a derivative of 9l containing a heavy atom (11) (Fig. 3c). Because it has been well-established that Beak’s (−)-sparteine-mediated asymmetric lithiation of pyrrolidine selectively abstracts the pro-S hydrogen of prochiral C-1, the X-ray structure confirms that transmetallation occurs with complete retention of stereochemistry.
Figure 3 |. Investigation of the stereorentention of transmetallation.
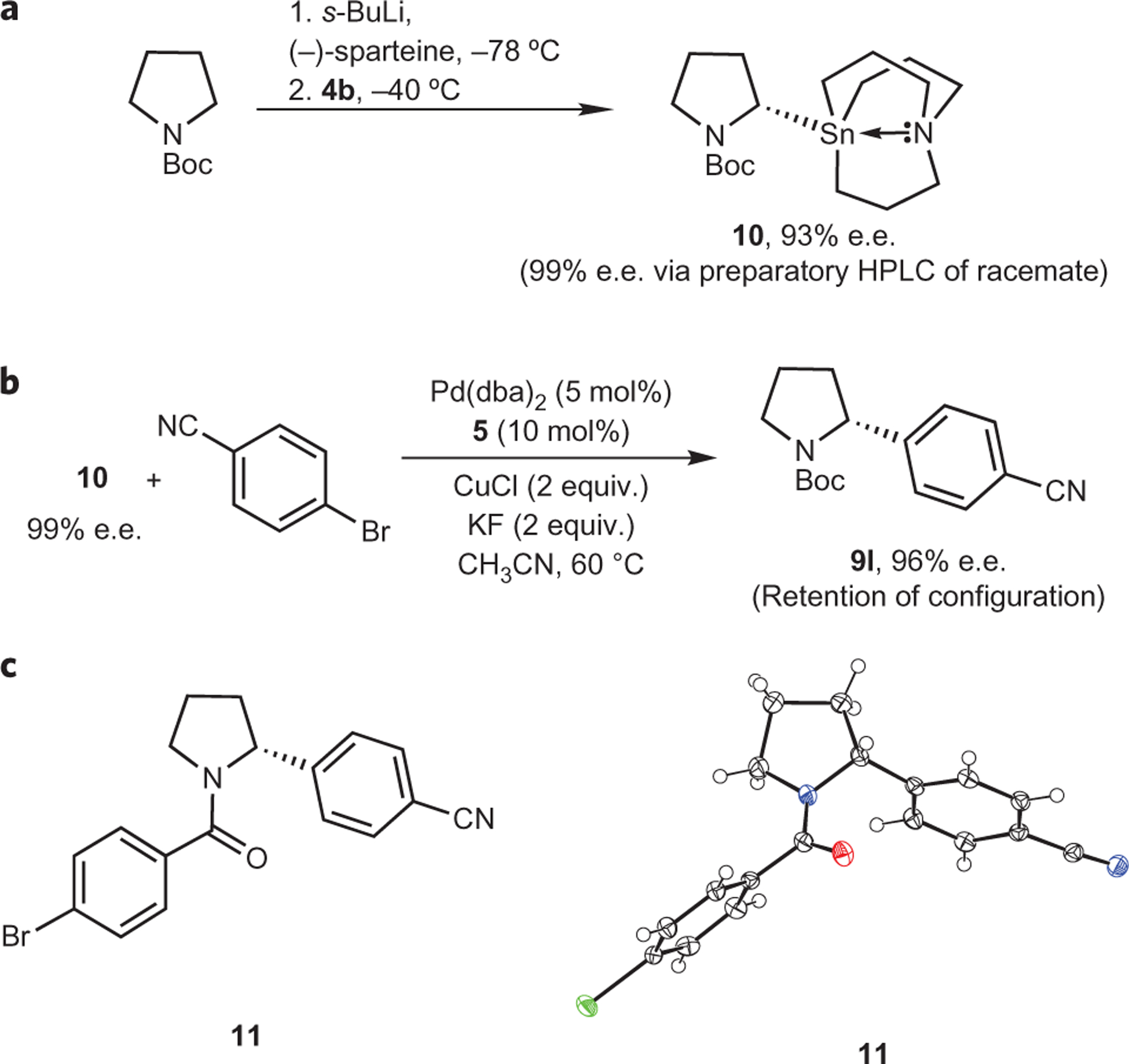
a, Preparation of enantioenriched 10. b, Transfer of stereochemistry in cross-coupling reaction using 10. c, X-ray crystal structure of 11 (thermal ellipsoids at 50% probability).
It has been shown that the presence of α-heteroatoms and carbonyl coordinating groups can activate alkyltin and alkylboron reagents towards stereospecific cross-coupling reactions23–35,37. Therefore, it is conceivable that the successful transfer of stereochemical information in 10 arises as a result of similar activation. To demonstrate that our stereospecific cross-coupling method can be used in a general manner for unactivated secondary alkyl nucleophiles, we prepared an optically active secondary alkylstannatrane not containing an α-heteroatom or a carbonyl coordinating group (12) via preparatory chiral HPLC. Because heterocyclic electrophiles are generally difficult to cross-couple, we specifically choose 2-bromopyridine and 6-bromo-2-methylquinoline as the electrophilic components in these reactions. When enantioenriched 12 was used in the cross-coupling reactions, minimal erosion of enantiomeric excess was observed (Fig. 4). This suggests that our process is indeed general and does not require the use of an activated secondary alkyl nucleophile.
Figure 4 |. Pd-catalysed cross-coupling reactions using unactivated, optically active secondary alkyl azastannatrane 12.
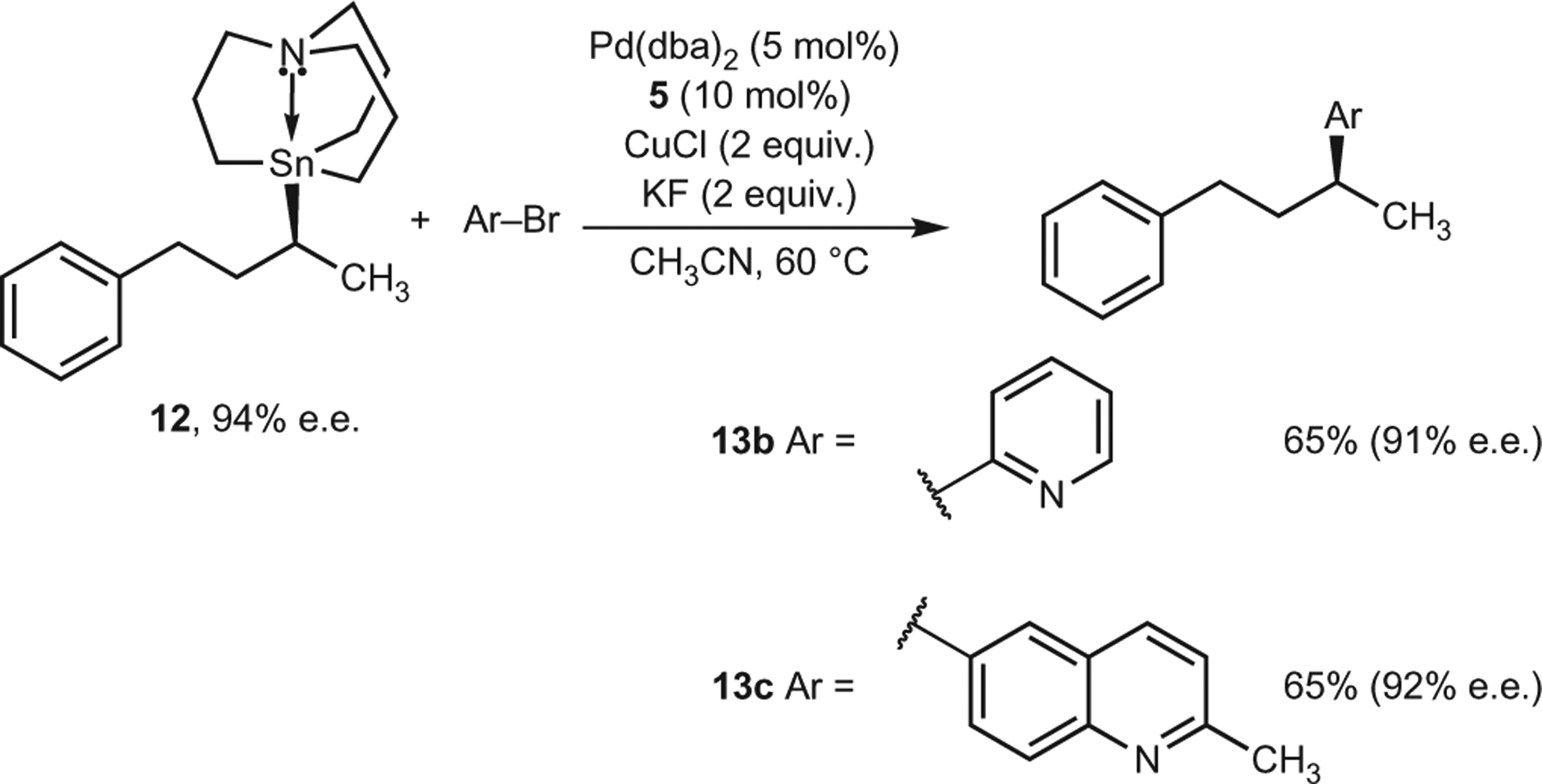
Cross-coupling can be achieved without significant loss of enantiopurity, even without the potentially stabilizing effect of an α-heteroatom.
In conclusion, we have developed the first general stereoretentive method to use secondary alkylstannane nucleophiles in cross-coupling reactions. Using an azastannatrane backbone, secondary alkyl groups underwent transmetallation to palladium with excellent fidelity, independent of the electronic properties of the alkyl nucleophile. Only nominal isomerization of the secondary alkyl nucleophile was observed using this method. Aryl chloride, bromides, iodides and triflates could be efficiently used as the electrophilic component of these cross-coupling reactions. Furthermore, retention of configuration was observed when an optically active secondary alkyl azastannatrane was used. The benchtop stability of optically active alkyl azastannatrane reagents, coupled with the ease by which stereochemical information may be transferred via Pd-catalysed cross-coupling reactions, should enable our process to be broadly applied in organic syntheses, particularly in the preparation of libraries of optically active drug candidates. Use of this method in such applications will be facilitated by the development of new methods for the generation of optically active alkyl azastannatranes. We are currently exploring the use of asymmetric catalysis for the production of enantioenriched alkyl azastannatranes.
Methods
General procedure for cross-coupling reactions using secondary alkyl azastannatranes.
Pd(dba)2 (29 mg, 0.05 mmol), JackiePhos (80 mg, 0.10 mmol), CuCl (200 mg, 2 mmol) and KF (116 mg, 2 mmol) were weighed out on the benchtop then transferred to an oven-dried Schlenk tube with stir bar. With stirring, the Schlenk tube was evacuated (50 mtorr) and backfilled with argon. This process was repeated three times. Azastannatrane (1.5 mmol) and aryl halide/triflate (1 mmol) were then added to the Schlenk tube via microsyringe, followed by degassed CH3CN (3 ml). If the aryl halide/triflate or tin reagent were a solid, it was weighed on the benchtop alongside the other solids. The Schlenk tube was sealed with a Teflon stopper and heated to 60 °C for 18 h (unoptimized reaction time). The reaction mixture was cooled to room temperature and diluted with ether. The organic layer was washed sequentially with saturated aqueous KF and brine, and then dried over Na2SO4. The dried organic layer was filtered, concentrated, and purified by column chromatography on silica gel.
X-ray crystallography data.
CCDC 928782 contains the crystallographic data for compound 11. These data can be obtained free of charge from the Cambridge Cyrstallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Supplementary Material
Acknowledgements
The authors acknowledge the National Institutes of Health (5SC2GM096932), the City College of New York (CCNY), the Alfred P. Sloan Foundation and PSC-CUNY for financial support. The authors also acknowledge the National Science Foundation for an instrumentation grant (CHE-0840498). The donors of the American Chemical Society Petroleum Research Fund (50307-DNI1) are thanked for partial support of this research. The authors thank Chunhua Hu for assistance with the X-ray analysis and acknowledge the Molecular Design Institute of NYU for purchase of the single-crystal diffractometer. The authors thank J. Norton for donation of a sample of (-)-sparteine.
Footnotes
Additional information
Supplementary information and chemical compound information are available in the online version of the paper. Reprints and permissions information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to M.R.B.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.De Meijere A & Diederich F (eds) Metal-Catalysed Cross-Coupling Reactions (Wiley-VCH, 2004). [Google Scholar]
- 2.Netherton MR & Fu GC in Topics in Organometallic Chemistry: Palladium in Organic Synthesis (ed. Tsuji J) 85–108 (Springer, 2005). [Google Scholar]
- 3.Rudolph A & Lautens M Secondary alkyl halides in transition metal-catalyzed cross-coupling reactions. Angew. Chem. Int. Ed 48, 2656–2670 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Chemler SR, Trauner D & Danishefsky SJ The β-alkyl Suzuki–Miyaura cross-coupling reaction: development, mechanistic study, and applications in natural product synthesis. Angew. Chem. Int. Ed 40, 4544–4568 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Luo X et al. Superior effect of a π-acceptor ligand (phosphine-electron-deficient olefin ligand) in the Negishi coupling involving alkylzinc reagents. Org. Lett 9, 4571–4574 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Dreher SD, Dormer PG, Sandrock DL & Molander GA Efficient cross-coupling of secondary alkyltrifluoroborates with aryl chlorides—reaction discovery using parallel microscale experimentation. J. Am. Chem. Soc 130, 9257–9259 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han C & Buchwald SL Negishi coupling of secondary alkylzinc halides with aryl bromides and chlorides. J. Am. Chem. Soc 131, 7532–7533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakao Y, Takeda M, Matsumoto T & Hiyama T Cross-coupling reactions through the intramolecular activation of alkyl(triorgano)silanes. Angew. Chem. Int. Ed 45, 4447–4450 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Calimsiz S & Organ MG Negishi cross-coupling of secondary alkylzinc halides with aryl/heteroaryl halides using Pd–PEPPSI–IPent. Chem. Commun 47, 5181–5183 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Joshi-Pangu A, Ganesh M & Biscoe MR Nickel-catalyzed Negishi cross-coupling reactions of secondary alkylzinc halides and aryl iodides. Org. Lett 13, 1218–1221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi-Pangu A, Wang C -Y. & Biscoe, M. R. Nickel-catalyzed Kumada cross-coupling reactions of tertiary alkylmagnesium halides and aryl bromides/triflates. J. Am Chem. Soc 133, 8478–8481 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi-Pangu A & Biscoe MR The use of tertiary alkyl nucleophiles in metal-catalyzed cross-coupling reactions. Synlett 23, 1103–1107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi-Pangu A et al. Palladium-catalyzed borylation of primary alkyl bromides. J. Org. Chem 77, 6629–6633 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boudier A, Bromm LO, Lotz M & Knochel P New applications of polyfunctional organometallic compounds in organic synthesis. Angew. Chem. Int. Ed 39, 4414–4435 (2000). [PubMed] [Google Scholar]
- 15.Hoffman RW, Hölzer B, Knopff O & Harms K Asymmetric synthesis of a chiral secondary Grignard reagent. Angew. Chem. Int. Ed 39, 3072–3074 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Hölzer B & Hoffman RW Kumada–Corriu coupling of Grignard reagents, probed with a chiral Grignard reagent. Chem. Commun 732–733 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Boudier A, Darcel C, Flachsman F, Micouin L, Oestreich M & Knochel P Stereoselective preparation and reactions of configurationally defined dialkylzinc compounds. Chem. Eur. J 6, 2748–2761 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Campos KR, Klapars A, Waldman JH, Dormer PG & Chen C-Y Enantioselective, palladium-catalyzed α-arylation of N-boc-pyrrolidine. J. Am. Chem. Soc 128, 3538–3539 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Thaler T et al. Highly diastereoselective Csp3–Csp2 Negishi cross-coupling with 1,2-, 1,3- and 1,4-substituted cycloalkylzinc compound. Nature Chem 2, 125–130 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Denmark SE & Werner NS On the stereochemical course of palladium-catalyzed cross-coupling of allylic silanolate salts with aromatic bromides. J. Am. Chem. Soc 132, 3612–3620 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridgway BH & Woerpel KA Transmetalation of alkylboranes to palladium in the Suzuki coupling reaction proceeds with retention of stereochemistry. J. Org. Chem 63, 458–460 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Imao D, Glasspoole BW, Laberge VS & Crudden CM Cross coupling reactions of chiral secondary organoboronic esters with retention of configuration. J. Am. Chem. Soc 131, 5024–5025 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Kells KW & Chong JM Stille coupling of stereochemically defined α-sulfonamidoorganostannanes. J. Am. Chem. Soc 126, 15666–15667 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Ye J, Bhatt RK & Falck JR Stereospecifc palladium/copper cocatalyzed cross-coupling of α-alkoxy- and α-aminostannanes with acyl chlorides. J. Am. Chem. Soc 116, 1–5 (1994). [Google Scholar]
- 25.Falck JR, Bhatt RK & Ye J Tin–copper transmetalation: cross-coupling of α-heteroatom-substituted alkyltributylstannanes with organohalides. J. Am. Chem. Soc 117, 5973–5982 (1995). [Google Scholar]
- 26.Mohapatra S, Bandyopadhyay A, Barma DK, Capdevila JH & Falck JR Chiral α, β-dialkoxy- and α-alkoxy-β-aminostannanes: preparation and copper-mediated cross-coupling. Org. Lett 5, 4759–4762 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Falck JR, Patel PK & Bandyopadhyay A Stereospecific cross-coupling of α-(thiocarbamoyl)organostannanes with alkenyl, aryl, and heteroaryl iodides. J. Am. Chem. Soc 129, 790–793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goli M, He A & Falck JR Pd-catalyzed cross-coupling of alpha-(acyloxy)-tri-n-butylstannanes with alkenyl, aryl, and heteroaryl electrophiles. Org. Lett 13, 344–346 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalkofen R & Hoppe D First example of an enantiospecific sp3–sp2 Stille coupling of a chiral allylstannane with aryl halides. Synlett 2006, 1959–1961 (2006). [Google Scholar]
- 30.Lange H, Fröhlich R & Hoppe D Cu(i)-catalyzed stereospecific coupling reactions of enantioenriched α-stannylated benzyl carbamates and their application. Tetrahedron 64, 9123–9135 (2008). [Google Scholar]
- 31.Sandrock DL, Jean-Gerard L, Chen C -Y., Dreher, S. D. & Molander, G. A. Stereospecific cross-coupling of secondary alkyl β-trifluoroboraroamides. J. Am. Chem. Soc 132, 17108–17110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohmura T, Awano T & Suginome M Stereospecific Suzuki–Miyaura coupling of chiral α-(acylamino)benzylboronic esters with inversion of configuration. J. Am. Chem. Soc 132, 13191–13193 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Daini M & Suginome M Palladium-catalyzed, stereoselective, cyclizative alkenylboration of carbon–carbon double bonds through activation of a boron–chlorine bond. J. Am. Chem. Soc 133, 4758–4761 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Awano T, Ohmura T & Suginome M Inversion or retention? Effects of acidic additives on the stereochemical course in enantiospecific Suzuki–Miyaura coupling of α-(acetylamino)benzylboronic esters. J. Am. Chem. Soc 133, 20738–20741 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Lee JCH, McDonald R & Hall DG Enantioselective preparation and chemoselective cross-coupling of 1,1-diboron compounds. Nature Chem 3, 894–899 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Partridge BM, Chausset-Boissarie L, Burns M, Pulis AP & Aggarwal VK Enantioselective synthesis and cross-coupling of tertiary propargylic boronic esters using lithiation–borylation of propargylalic carbamates. Angew. Chem. Int. Ed 51, 11795–11799 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Molander GA & Wisniewski SR Stereospecific cross-coupling of secondary organotrifluoroborates: potassium 1-(benzyloxy)alkyltrifluoroborates. J. Am. Chem. Soc 134, 16856–16868 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itami K, Kamei T & Yoshida J Unusually accelerated silylmethyl transfer from tin in Stille coupling: implication of coordination-driven transmetalation J. Am. Chem. Soc 123, 8773–8779 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Littke AF & Fu GC The first general method for Stille cross-coupling of aryl chlorides. Angew. Chem. Int. Ed 38, 2411–2413 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Labadie JW & Stille JK Mechanisms of the palladium-catalyzed couplings of acid chlorides with organotin reagents. J. Am. Chem. Soc 105, 6129–6137 (1983). [Google Scholar]
- 41.Vedejs E, Haight AR & Moss WO Internal coordination at tin promotes selective alkyl transfer in the Stille coupling reaction. J. Am. Chem. Soc 114, 6556–6558 (1992). [Google Scholar]
- 42.Jensen MS et al. Anti-methicillin-resistant Staphylococcus aureus (MRSA) carbapenem via stannatrane-mediated Stille coupling. Org. Lett 2, 1081–1084 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Sebehar HL, Yoshida K & Hegedus LS Effect of adjacent chiral tertiary and quaternary centers on the metal-catalyzed allylic substitution reaction. J. Org. Chem 67, 3788–3795 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Hicks JD, Hyde AM, Cuezva AM & Buchwald SL Pd-catalyzed N-arylation of secondary acyclic amides: catalyst development, scope, and computational study. J. Am. Chem. Soc 131, 16720–16734 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mee SPH, Lee V & Baldwin JE Stille coupling made easier—the synergistic effect of copper(i) salts and the fluoride ion. Angew. Chem. Int. Ed 43, 1132–1136 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Beak P, Kerrick ST, Wu S & Chu J Complex induced proximity effects: enantioselective syntheses based on asymmetric deprotonations of N-boc-pyrrolidines. J. Am. Chem. Soc 116, 3231–3239 (1994). [Google Scholar]
- 47.Beak P, Basu A, Gallagher DJ, Park YS & Thayumanavan S Regioselective, diastereoselective, and enantioselective lithiation–substitution sequences: reaction pathways and synthetic applications. Acc. Chem. Res 29, 552–560 (1996). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.