

Abstract
The relationship between the extrinsic environment and the internal transcriptional network is circular. Naive T cells first engage with antigen‐presenting cells to set transcriptional differentiation networks in motion. In turn, this regulates specific chemokine receptors that direct migration into distinct lymph node niches. Movement into these regions brings newly activated T cells into contact with accessory cells and cytokines that reinforce the differentiation programming to specify T cell function. We and others have observed similarities in the transcriptional networks that specify both CD4+ T follicular helper (TFH) cells and CD8+ central memory stem‐like (TSCM) cells. Here, we compare and contrast the current knowledge for these shared differentiation programs, compared to their effector counterparts, CD4+ T‐helper 1 (TH1) and CD8+ short‐lived effector (TSLEC) cells. Understanding the interplay between cellular interactions and transcriptional programming is essential to harness T cell differentiation that is fit for purpose; to stimulate potent T cell effector function for the elimination of chronic infection and cancer; or to amplify the formation of humoral immunity and longevity of cellular memory to prevent infectious diseases.
Keywords: cellular interactions, lymphoid niche, migration, stem‐like memory, T cell differentiation, T follicular helper, transcriptional regulation
1. INTRODUCTION
The activation and differentiation of T cells play an essential role in orchestrating adaptive immune responses. These interactions determine how responses are tailored toward the pathogen and the success of T cell responses; whether pathogens or cancer cells are eliminated by effector cells, and if immunological memory is generated for long‐lasting protection. The paths to each of these outcomes are determined by the cells that T cells interact with during their activation.
A key feature of immune cells is their motility. T cell migration is primarily driven by chemokines and their chemokine receptors, which co‐ordinate cell migration into specific sites and promote T cell interactions. 1 , 2 Naive T cells that are single positive for either CD4 or CD8 enter circulation following their maturation in the thymus. 3 Upon their arrival in the secondary lymphoid organs, such as the spleen and lymph nodes, T cells seek out interactions with dendritic cells to search for the presentation of their cognate antigen. 4 , 5 , 6 Work from our group, and others has demonstrated that where these interactions occur, and the dendritic cell subsets involved, plays a critical role in establishing the gene networks that determine effector and memory differentiation for both CD4+ and CD8+ T cells. 1 , 15
Following activation, CD4+ T cells can differentiate into a diverse set of effector helper cells. Each helper lineage is specified by distinct transcriptional regulators and cytokines that orchestrate a coordinated attack for the eradication of a specific type of pathogen. 16 , 17 T follicular helper (TFH) cells are a distinct subset that promotes B cell maturation into high‐affinity plasma and memory cells. 18 , 19 , 20 Unlike the other CD4+ T cell effectors, TFH differentiation occurs irrespective of the type of pathogen to support germinal center formation, and thus, these cells are an essential link between the cellular and humoral responses.
In contrast to CD4+ T cell differentiation, the diversity in CD8+ T cells has only been more recently appreciated. In addition to the varied effector responses, CD8+ T cell differentiation leads to a heterogeneous pool of memory subsets. CD8+ T cell memory can be broadly grouped into either central memory or effector memory populations, which were originally defined by their distinct homing potential. 21 , 22 Central memory cells continually recirculate in blood and lymph and are found in secondary lymphoid organs, while effector memory cells can recirculate through, or reside within peripheral tissues. Each of these pools contains specialized memory populations. Recently, a subpopulation of central memory cells, termed stem‐like memory cells (TSCM), has been described in humans and mice. 23 TSCM are programmed to promote self‐renewal and repress terminal differentiation. 24 , 25 , 26 By seeding the effector cell pool, TSCM mediate long‐term immunity to chronic infection, provide superior recall responses to secondary infection, and control tumor growth following immunotherapy. 23
From the initial description of CD8+ TSCM cells, comparisons have been made concerning their relatedness to CD4+ TFH cells. 24 , 27 These similarities are not only reflected in the cell‐surface markers that define these subsets, but also the transcriptional regulators that determine their specific differentiation. Understanding these similarities may highlight the cellular interactions, cytokines, and lymphoid niches that promote these differentiation pathways. Investigating this potential could reveal new avenues to harness CD4+ TFH and CD8+ TSCM cells and steer vaccine design to focus on the T cell responses that would provide the greatest humoral response combined with long‐lived, robust cellular memory. In this review, we compare and contrast our current knowledge for the differentiation of CD4+ TFH and CD8+ TSCM cells and their effector counterparts TH1 and TSLEC cells. We discuss how the details of one system may inform the other, and the emerging technologies that may help address remaining questions. Combined with recent discoveries, future work which aims to understand the environmental and cellular interactions that support one or both of these T cell differentiation fates will drive the formulation of target vaccines that establish reliable, protective, and long‐lived immunological memory. In addition, as both TFH and TSCM responses have been shown to promote the clearance of tumors following checkpoint blockade, this knowledge has the potential to inform and optimize the revival of T cell responses during chronic infection or for cancer immunotherapy. 25 , 28 , 29
2. DIVERSITY OF CD4+ AND CD8+ T CELL DIFFERENTIATION
2.1. Pathogen‐guided tailoring of CD4+ T cell differentiation
Following infection, CD4+ T cells differentiate into distinct T‐helper subsets that mediate clearance of and protection against diverse pathogens. 17 , 30 This dynamic T cell differentiation permits an exceptional flexibility to tailor responses for the clearance of distinct pathogens and protection from reinfection. TH1 cells secrete interferon‐γ (IFN‐γ) and tumor necrosis factor‐α (TNF‐α) and promote cellular immune responses mainly to intracellular pathogens, such as viruses and tumors. TH2 cells are formed during helminth and allergy challenges and secrete interleukin (IL)‐4, IL‐5, and IL‐13. TH17 cells are identified through their production of IL‐17A and IL‐17F and are increased in the context of extracellular bacteria or fungi challenges. 16 , 17 It is important to note that not every immune response contains a single lineage, but rather there is a rich and diverse continuum of CD4+ T cell effectors. In addition, considerable plasticity exists between TH subsets to fine‐tune responses. However, as the differentiation of these lineage pathways is determined by the cytokine milieu present during infection, CD4+ T cell responses are skewed toward a TH population that is tuned to respond to a distinct class of pathogen. Thus, flexibility within TH functions insures immune responses are context‐appropriate to facilitate host protection against a wide range of pathogens.
2.2. Diversity within the TFH population
Another essential role for CD4+ cells is to support and instruct the germinal center response, leading to the production of high‐affinity antibody‐producing cells and memory B cells. This task is performed by a distinct population known as TFH cells. The differentiation of TFH is distinct from the other TH lineages, as this population is required to differentiate alongside each of the effector TH subsets irrespective of the class of pathogen challenge. Still, there exists critical cytokine and transcriptional tipping points that can lead to a preference for differentiation toward either TFH and CD4+ T cell effectors. 9 Functional heterogeneity within TFH populations is a newly established concept. During B cell interactions within the germinal center, TFH cells can secrete multiple cytokines, IL‐21, IL‐4, IL‐2, IL‐9, IL‐10, IL‐13, and IFN‐γ. 19 , 31 , 32 , 33 , 34 The potential to secrete a specific cytokine combination represents functionally distinct TFH subpopulations not only between distinct infectious and pathogenic settings but also over the course of an infection. 32 , 35 , 36 In particular, TFH subpopulations defined by their distinct cytokine expression have been shown to influence the production of pathogenic antibodies in allergy and asthma in both humans and mice. 32 , 36
Heterogeneity within TFH populations was first appreciated in human studies, through the investigation of circulating peripheral blood TFH subsets. 37 , 38 While these cells exhibit lower expression of TFH identifying proteins CXCR5 and PD‐1, they still resemble those found within the tonsil and lymph nodes. 39 , 40 , 41 Interestingly, circulating TFH cells are defined by their expression of the chemokine receptors CCR6 and CXCR3. Studies assessing circulating TFH in patients with inborn errors of immunity have shown that these populations are independently transcriptionally regulated. 42 In these patients, STAT1 deficiency leads to a loss in the CCR6+ population and a reciprocal accumulation of the CXCR3+ circulating TFH cells. Several studies have investigated the relationship between TFH subpopulations and the development of vaccine response or immune protection following infection. While some indicate that CXCR3+ circulating TFH cells can be used as a biomarker of immune protection, 43 , 44 others demonstrate that protection of humoral responses is negatively correlated with CXCR3+ subpopulations. 38 , 42 , 45 , 46 Still in other settings, the relationship between TFH heterogeneity and humoral response is less clear. This is the case in convalescent COVID‐19 patients, where CCR6+ TFH cells were negatively correlated with SARS‐CoV‐2 neutralizing antibodies, while the remaining CCR6‐CXCR3−/+ population correlated with antibody levels. 47 It is currently unknown if the heterogeneity of human TFH changes over time; however, it is more likely that this is imprinted by their developmental path and determined by the infection or vaccination environment. In animal models, we have demonstrated that diversity exists in the formation of TFH cells between different viral infections. 8 This diversity is transcriptionally regulated. Specifically, we have identified a T‐bet‐dependent and T‐bet‐independent process for TFH differentiation using the experimental systems of acute lymphocytic choriomeningitis virus (LCMV) and influenza. 8
2.3. Diversity within CD8+ effector T cell differentiation
Until recently, the diversity within CD8+ T cell differentiation was restricted to a bifurcation between effector and memory. However, it is now appreciated that substantial heterogeneity exits in each of these broad paths. 22 , 48
The most common definition of CD8+ T cell effectors is found following viral infections and resemble CD4+ TH1 cells, both in terms of their transcriptional requirements and effector molecule production. These cells are identified via their surface expression of killer cell lectin‐like receptor subfamily G, member 1 (KLRG1). CD8+ T cell effector cells are critical for the clearance of viral‐infected cells due to their high production of effector molecules (such as IFN‐γ and granzyme B). 22 , 49 This population of effector cells undergoes considerable contraction following pathogen clearance and are therefore referred to as short‐lived effector T cells (TSLEC).
As TSLEC represent the CD8+ T cell equivalent of TH1 CD4+ T cells, so too do the less common TC2 and TC17 cells relate to TH2 and TH17 cells, respectively. However, both TC2 differentiation and TC17 differentiation are more strongly associated with aberrant and pathogenic immune responses than targeting responses toward diverse pathogenic infections. 50 Indeed, differentiation toward IL‐5‐producing‐TC2 cells is associated with the development of severe asthma following infection. 50 In contrast, the population of IL‐17‐producing TC17 cells were originally suspected to be an abnormality of genetic modifications in mice. TC17 cells are enriched with the compound deletion of the T‐box transcription factors T‐bet and EOMES or T‐bet and Blimp, or the deletion of TCF‐1. 51 , 52 , 53 However, in recent years, knowledge of this subset has increased to define its role in protection against fungal infection and microbial colonization of the skin. 51 , 52 , 53 In addition, TC17 cells have also been found associated with gastrointestinal cancers and responses during influenza viral infection, making them novel targets for immunotherapy. 54
In recent years, a population of CXCR5+ follicular cytotoxic T cells was identified during chronic infection. 37 , 55 Many of the key cell‐surface markers and characteristic properties of these cells resemble that of a newly described TSCM memory population. Follicular cytotoxic T cells were observed within B cell follicles, while TSCM accumulate in the T cell zone. The literature has coalesced behind the TSCM nomenclature and this population will be discussed in greater detail in the following sections. 24
2.4. Diversity within CD8+ memory T cell differentiation
CD8+ T cell memory cells are critical mediators of long‐lived immunity and provide dynamic protection against intracellular pathogens and cancer. While these memory cells differentiate during the initial exposure to a pathogen, they do not undergo the same contraction seen in TSLEC cells and are instead maintained long term, in an antigen‐independent manner. Distinct from TSLEC cells, memory can be distinguished during infection by the lack of KLRG1 expression and expression of the IL‐7 receptor subunit‐α, CD127, and CD27. 22
The central memory population is identified via the expression of the CCR7+ CD44+, CD127+, and CD62L+. The expression of both CCR7+ and CD62L+ enforces their homing to secondary lymphoid organs and bone marrow. Within the central memory population, a particularly potent memory cell has been identified. Termed stem‐like memory cells, TSCM cells are marked by their expression of PD‐1, Slamf6, SCA‐1, CXCR5, ICOS, and the transcription factor TCF‐1, although the expression of these may vary between acute and chronic settings. 56 TSCM cells provide a greater proliferative boost, compared to other central memory cells, during recall responses and following PD‐1 checkpoint blockade in chronic infection. 24 , 37 Further, TSCM exhibit enhanced renewal capabilities and are maintained long‐term. 25 , 27 , 57 While the mechanisms that underlie the enhanced multipotency of TSCM are undefined, this population is of exceptional interest for its therapeutic potential. 23 , 58
The effector memory population contains both CD127hi and CD127lo populations with low CD62L expression. These markers, along with the absence of CCR7, allow these cells to enter and circulate through peripheral tissues. 22 Previously, this confounded the identification of resident memory T cells, which are permanently retained in the periphery. 59 Furthermore, a recent report has transcriptionally dissected the CD8+ T effector memory population to identify distinct, terminally differentiated effector memory cells, termed terminal‐TEM in both humans and mice. 60 Despite their persistence for over 70 days postinfection, these cells display some hallmarks of TSLEC cells, including KLRG1 expression. Importantly, this study showed TEM exhibit potent cytotoxicity, but have limited recall potential. This population was depleted after 3‐5 months postinfection. As the TEM population has been previously conflated with the effector memory, this has redefined the classical characteristic attributed to this memory population.
In addition to effector memory cells that are found recirculating in both lymphoid and non‐lymphoid tissue, a tissue resident memory (TRM) population also exists that is restrained within peripheral tissue, local to the site of initial infection. 59 In these sites, TRM cells dominate the local response upon re‐challenge infection. 61 , 62 In addition, the scanning behavior of TRM cells in the skin can prevent the outgrowth of melanoma and is associated with a better prognosis in breast cancer patients. 63 , 64 The diversity in cell‐surface markers and transcriptional signatures observed in CD8+ T cell TRM populations, located in distinct anatomical sites, is imprinted by the pathogen and the tissue environment. 65
Unlike the pathogen‐driven differentiation of CD4+ T cells, for CD8+ T cells it is less clear what the determinants of memory heterogeneity are and what determines the predominance of one population over the other in central and effector memory populations. However, diversity in this regard does insure durable protection at multiple sites, allowing flexibility of responses upon recalled pathogen encounters.
3. SHARED TRANSCRIPTIONAL REGULATION BETWEEN CD4+ AND CD8+ T CELL SIGNATURES
In addition to the correlation between cell‐surface markers, there exist common attributes consistent within the transcriptional networks that are established between various subsets of CD4+ and CD8+ T cells. These networks act in concert with the extrinsic environment; first to move cells into specific niches, then to re‐enforce differentiation based on the cytokines present in these regions (Figure 1). Here, we explore some of the central nodes of the transcriptional programs that are shared between CD4+ TH1 and CD8+ TSLEC effector cells and alternatively, those that specify differentiation for CD4+ TFH and CD8+ central memory cells and TSCM cells. Within each of these CD4+ and CD8+ T cell branches, these regulators act to establish cell ontogeny and define functional outcomes. Understanding these shared transcriptional pathways may reveal shared environmental cues, such as cytokines and cellular interactions, which promote T cell differentiation. Here, we focus on central transcriptional nodes with implications for cell location and position.
FIGURE 1.
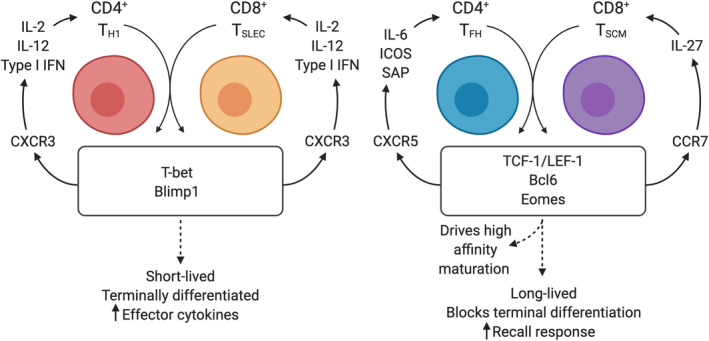
Circular relationship between transcriptional and environmental regulators of T cell differentiation. Following initial contact with dendritic cells, T cells begin to upregulate the transcription factors that regulate diverging fates. This leads to the upregulation or maintenance of chemokine regulators such as CXCR3, CXCR5, and CCR7. According to this expression, newly activated cells move into new regions and are exposed to a cytokine milieu that further feeds forward differentiation to re‐enforce the transcriptional program and function of each cell subset
3.1. Transcriptional dichotomy of Bcl6 and Blimp1
Bcl6 (BTB‐POZ; bric‐a‐bric, tramtrack, broad complex–poxvirus zinc finger) and Blimp1 (B lymphocyte‐induced maturation protein 1; encoded by Prdm1) are a pair of reciprocal and antagonistic transcription factors which play a key role in determining memory and effector fate decisions in T cells. 66 Specifically, the transcriptional repressor Blimp1 drives terminal effector differentiation in both CD8+ and CD4+ T cells. 66 , 67 Conversely, Bcl6 is vital for the generation and maintenance of CD8+ T cell memory and also promotes TFH development in CD4+ T cells. 27 , 68 , 69 , 70 , 71
Bcl6 is the master regulator of TFH cell fate. It is vital for humoral immunity, regulating B cell help and germinal center formation. 69 , 70 Early Bcl6 expression is induced by CD28 activation and cytokines such as IL‐6, which activates STAT3 signaling to enhance Bcl6 expression and drive TFH development. Bcl6 acts primarily to downregulate Blimp1 and to prevent effector TH fates. 9 , 69 , 72 Bcl6 also represses Gata3, the TH2 master regulator, RORγt, required for TH17 differentiation, and T‐bet, the TH1 master transcription factor, to further limit alternate TH fates. 19 , 69 , 71 , 73 As discussed in detail below, Bcl6 controls cell migration by silencing promoter regions of genes such as Ccr7, Ccr6, S1pr1, and Psgl1 to promote retention within the lymph node and migration toward the T:B border. 74 Bcl6 further delays T cell egress by repressing KLF2, a transcription factor which initiates expression of S1PR1 and CD62L, revealing a similar mechanism observed in tissue resident CD8+ memory T cells to prevent lymphoid tissue homing. 75 , 76 Furthermore, overexpression of Bcl6 led to increased PD‐1, CXCR5, CXCR4, and SAP which are critical for TFH migration to the B cell follicles and interaction with B cells. 77 , 78 , 79 These interactions in turn reinforce Bcl6 expression as they progress into the B cell follicle and participate in germinal center reactions.
The role of Bcl6 in TFH cells has informed studies investigating its role in the development and maintenance of CD8+ T cell memory. Here, as for CD4+ T cells, Bcl6 is inversely expressed to Blimp1, with higher levels of Bcl6 found in memory cells, particularly in TSCM cells compared to effectors. As memory precursors mature into central memory cells, Bcl6 expression progressively accumulates while Blimp1 expression decreases. 80 Interestingly, in CD8+ T cells, this expression is dependent on IL‐21, while in TFH differentiation, the requirement for IL‐21 remains controversial. 9 In memory precursors, Bcl6 binds directly to the Tcf7 locus, driving TCF‐1 expression, which is critical for memory differentiation. 27 Further, the forced expression of TCF‐1 in Bcl6‐deficient mice restores generation of memory precursors, demonstrating that Bcl6 functions upstream of TCF‐1 as a critical regulator of memory formation during acute viral infection. 81
In contrast to the expression of Bcl6, Blimp1 is highly expressed in CD8+ and CD4+ effector T cells. In CD8+ T cells, this expression drives terminal differentiation, migration to sites of inflammation and cytolytic TSLEC function. 82 , 83 Blimp1 expression is induced by IL‐2 and IL‐12 and promotes the production of effector molecules IFN‐γ and granzyme B. 84 , 85 , 86 , 87 Consistent with the counterbalancing roles of Bcl6 and Blimp1, central memory CD8+ T cells express low Blimp1 levels. 67 Blimp1 appears to repress transcriptional networks driving central memory formation, as loss of Blimp1 leads to the expansion of CD62L+ CD127+ memory cells during infection. 67 In line with the essential role in effector function, Blimp1‐deficient mice fail to clear influenza infection due to the reduced migration of TSLEC cells to the lung. 83 However, this function may be in concert with expression of T‐bet, which directly regulates CXCR3. 52 , 88
Unlike CD8+ effector T cells, early development of effector CD4+ T cells does not depend on Blimp1 expression. 89 , 90 Rather, Blimp1 is expressed later in CD4+ T cell differentiation and as such is associated with highly committed, cytokine‐producing CD4+ TH cell subsets. 87 , 90 This profile is similar to its expression profile in differentiated CD8+ effector cells and in plasma cells, both which have an increased secretion capacity. 66 CD4+ T cell effectors demonstrate reduced proliferation, again supporting the notion that Blimphi CD4+ T cells may have similar transcriptional wiring to CD8+ effectors. Additionally, overexpression of Blimp1 in CD4+ T cells leads to hyperproliferation and consequent autoimmunity. 90 Combined, the dichotomy of Bcl6 and Blimp1 expression reveals a key branching point between CD4+ and CD8+ effectors and CD8+ memory and CD4+ TFH fates.
3.2. T‐box factor regulation of T cell fates
T‐box transcription factors, T‐bet, and eomesodermin (Eomes) are essential in the differentiation and function of effector and memory T cells. Similar to Blimp1 and Bcl6, the relative expression of T‐bet and Eomes acts to tip the balance between terminally differentiated effector and memory fates. In CD8+ T cells, increasing T‐bet expression promotes effector lineages early in T cell development, while Eomes is required to promote and sustain memory fate. 49 , 91 , 92 Similarly, in the CD4+ lineage, T‐bet is considered a master regulator of driving formation of TH1 cells, while restricting non‐TH1 effector fates. 93
In both precursor and committed cell subsets, T‐bet plays a role repressing alternative T fates. In addition to direct repression, this is achieved by forming complexes with other lineage‐defining transcription factors, such as RORgt, GATA3, and Bcl6, to block TH17, TH2, and TFH differentiation, respectively. 9 , 94 , 95 Formation of these complexes sequesters these other lineage‐defining factors away from their DNA binding sequences and thus blocks their action. 91 , 96 Additionally, T‐bet cooperates with RUNX3 to antagonize GATA3 activity and also silences the Il4 locus to repress IL‐4 expression. 97 , 98 In CD8+ T cells, T‐bet expression is graded according to the level of inflammation. 49 High levels of T‐bet expression instruct a transcriptional program that is distinct from cells with moderate T‐bet expression. In this setting, T‐bet collaborates with the transcription factor ZEB2 to turn on the cytotoxic, terminally differentiated TSLEC differentiation program. 99 Combined, these studies show that in both CD4+ and CD8+ T cells, T‐bet is a master at driving effector differentiation through the co‐operation and coercion of other transcriptional regulators.
A central role of T‐bet is the direct activation of genes that are essential for effector T cell function. As such, T‐bet is specifically binding directly to the Ifng locus to drive expression of the canonical TH1 cytokine IFN‐γ. 93 , 100 , 101 This source of IFN‐γ is an essential primer for the immune system and can subsequently induce T‐bet expression in naive CD4 T cells, which do not express T‐bet, in a STAT1‐dependent manner. 102 Thus, TH1‐produced IFN‐γ production functions as a positive feed‐forward loop to drive further TH1 differentiation and induce IFN‐inducible chemokines to form peripheral and lymph node inflammatory niches. 7 , 103 Further to this, T‐bet directly binds and regulates the expression of the chemokine receptor, CXCR3. This expression further re‐enforces the inflammatory loop generated by T‐bet, as it moves cells into regions of inflammation via CXCR3. In these regions, T‐bet via IFN‐γ production induces local expression of the CXCR3 ligands, CXCL9, and CXCL10, leading to further recruitment to the site. 7 , 88
Expression of T‐bet is induced by multiple inflammatory cytokines. The most characterized of these is IL‐12 through the activation of STAT4. 30 , 49 , 94 , 95 , 101 For CD4+ T cells, multiple cytokines can tip the balance toward T‐bet expression and TH1 differentiation including IL‐2, type I IFN, and IL‐12. 8 , 11 , 104 Alternatively, others such as IL‐6 reduce T‐bet expression and increase Bcl6 to promote TFH development. 9
In CD8+ T cells, T‐bet and Eomes are expressed by distinct populations, representing reciprocal differentiation paths. 105 T‐bet is highest early in CD8+ effectors but is downregulated in memory cells, while Eomes expression is low in early effectors but upregulated later as memory cells emerge. 106 As such, the phenotype, function, and persistence of memory and effector CD8+ T cells are sensitive to the relative expression of Eomes and T‐bet. 49 , 105 , 106 , 107 T‐bet and Eomes share significant homology and perform partially redundant roles in CD8+ T cells, including regulation of IFN‐γ and granzyme B. 51 , 108 Initial T‐bet expression in naive CD8+ T cells is induced by T cell receptor (TCR) and IFN signaling. As mentioned, this is further amplified in effector T cells by IL‐12‐mediated signals in addition to mTOR activity. 49 , 109 , 110 In contrast, Eomes is induced in early effectors in a Runx‐dependent manner, enhanced by IL‐2 signaling, but is limited by IL‐12 and mTOR. 109 , 110 , 111 Eomes expression increases further in memory cells in response to WNT signaling and TCF‐1. 26 , 85 , 106 , 112 In memory CD8+ T cells, Eomes plays a critical role in cell survival and homeostatic regulation by increasing expression of IL2RB (CD122) and IL‐15 signaling to promote homeostatic proliferation. 49 , 85 , 105 Importantly, during secondary challenge, Eomes‐deficient memory precursors elicit an impaired recall response, generate less central memory, and demonstrate reduced secondary expansion. 112 In chronic infection, both the T‐bet+ and the distinct Eomes+ populations are required to keep viral load in check. The Eomes+ cells provide a reservoir which replenishes the terminally differentiated T‐bet expression pool. 113 , 114 , 115 Thus, in this setting, the Eomes‐expressing CD8+ population in chronic infection is marked by CXCR5 expression and falls into the TSCM subset of central memory. 24
3.3. TCF‐1 and LEF‐1, the master regulators of stemness
T cell factor‐1 (TCF‐1, encoded by Tcf7) is a high mobility group box (HMG) family member, and transcription factor essential for T cell development and differentiation. 116 , 117 TCF‐1, along with HMG family member lymphoid enhancer‐binding factor‐1 (LEF‐1), is a key player in establishing the T cell lineage in thymocytes, specifying distinct CD4+ T cell lineages, and regulating CD8+ T cell fate decisions. Unlike the previously mentioned transcriptional nodes, TCF‐1 expression is established in thymocytes and its expression is maintained in CD8+ central memory cells and CD4+ TFH cells following activation. 54
TCF‐1 and LEF are also essential during early TFH development. Overexpression of TCF‐1 amplifies TFH cell differentiation and increases expression of TFH associated genes, such as CXCR5 and PD‐1. 27 , 118 , 119 Mechanistically, TCF‐1 promotes early TFH differentiation in CD4+ T cells by binding directly to the Bcl6 promoter to enhance BCL6 expression, as well as acting downstream of Bcl6 during TFH differentiation. Further, TCF‐1 directly restricts TH1 differentiation by binding to the Prdm1 and IL‐2Ra loci in TFH cells to repress Blimp1 and IL2Ra expression. The blocking of IL‐2 signaling promotes TFH over TH1 cell differentiation. 69 , 119 , 120 In addition, TCF‐1 is enriched at the IL‐6 receptor gene locus, which suggests a role for TCF‐1 in increasing IL‐6 responsiveness, a vital cytokine for TFH differentiation. 121 Indeed, loss of TCF‐1 leads to a decrease in IL‐6Ra and IL‐6. 118 While the mechanism is unclear, TCF‐1 deficiency also reduces the expression of Achaete‐scute homologue‐2 (ASCL2). ASCL2 is required to downregulate CCR7, allowing migration to the T:B border. 122 Additionally, TCF‐1 directly targets costimulatory marker inducible T cell co‐stimulator (ICOS), another signaling molecule important for TFH cell commitment and growth. 118 Thus, TCF‐1 acts in multiple ways to both initiate and reinforce TFH differentiation.
Distinct from other TH subsets, TFH cells form a critical component of the memory pool, that repress exhaustion in chronic infection and maintain stem‐like potential. 27 , 123 , 124 Upon re‐exposure, this population re‐initiated B cell helper function. 125 The generation of these memory cells was impaired by the loss of TCF‐1, suggesting that TCF‐1 plays an essential role for CD4+ T cell memory stemness, particularly in TFH cells. 126
In the last decade, many studies have revealed that TCF‐1 plays a key role in the formation and persistence of memory CD8+ T cells. 26 , 127 The Tcf7 gene is expressed highly in naive and memory CD8+ T cells, but is decreased in TSLEC cells. 54 The rapid downregulation of TCF‐1 during effector differentiation is mediated, in part, by IL‐12. 128 Early studies proposed that memory CD8+ T cell differentiation required Wnt signaling and β‐catenin binding to TCF‐1 to establish cell fate; however, the role of Wnt ligands in vivo remains disputed. 26 , 127 , 129 In the absence of TCF‐1 during infection, memory formation is restricted while effector‐associated genes, including Blimp1, T‐bet, ID2, are reciprocally upregulated. 26 , 127 , 128 In keeping with this, several studies have found that TCF‐1 and the effector molecule granzyme B are reciprocally expressed. 56 Loss of TCF‐1 further impairs the expansion of memory CD8+ T cells upon secondary challenge, indicating that TCF‐1 is essential for both memory formation and secondary expansion. 26 , 127 Mechanistically, TCF‐1 directly binds to the regulatory region of Eomes and increases its expression to drive development of CD8+ central memory precursors. Based on the similar expression of TCF‐1 in CD8+ TSCM and TFH cell differentiation, the mechanisms TCF‐1 utilizes to regulate each of these compartments are likely similar. This has been demonstrated by TCF‐1‐mediated repression of Tbx21 and Prdm1 transcription and induction of Bcl6 in CD8+ memory precursors. TCF‐1 also establishes functional similarities between these cells, in particular the longevity of memory persistence is conserved in these TCF‐1‐expressing populations. 27 , 123 , 130 As such, TCF‐1 has been identified as playing a critical role in repressing exhaustion and maintaining T cell stemness during chronic and acute infection. 23 , 24 , 27 , 57 , 131 High expression of TCF‐1 in CD8+ TSCM cells is essential in chronic LCMV infection, as loss of TCF‐1 leads to a failure to generate stem‐like progenitor cells. This in turn results in impaired viral control and a rapid loss of CD8+ T cells. 23 , 24 , 131 Combined these studies suggest a shared role of TCF‐1 between TFH and central memory TSCM cells, promoting long‐term survival and proliferation which are essential in chronic infection and recall responses.
4. THE ROOM WHERE IT HAPPENS
The majority of T cell priming occurs within secondary lymphoid organs, which are organized into specific niches (Figure 2). These regions promote T cell priming of naive and newly activated cells in contact with other cells that steer T cell differentiation in a manner tailored toward the infecting pathogen. While most of these processes occur in either draining lymph nodes or the spleen, peripheral sites such as the skin, liver, and lung can mediate T cell differentiation: either by priming naive cells, or tuning and enhancing T cell function and memory locally.
FIGURE 2.
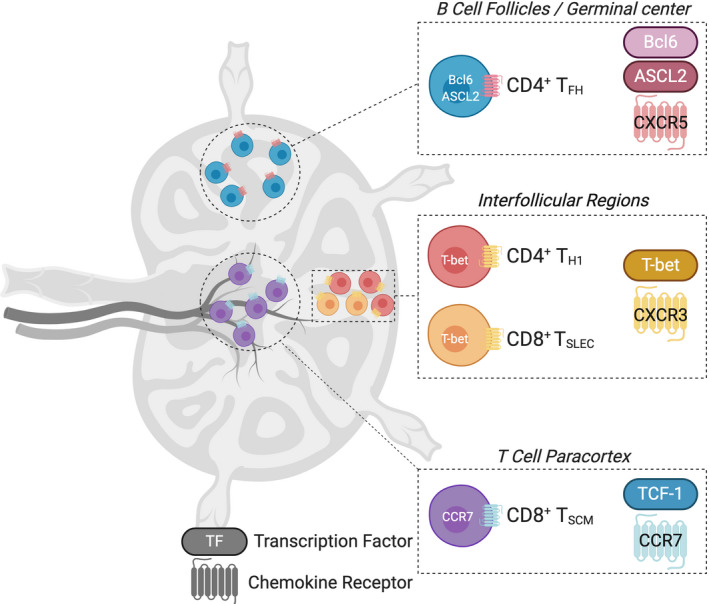
The lymph node compartments where T cell differentiation occurs. The specific transcriptional program of TH1 or TSLEC effectors, TFH, and TSCM cells moves cells into spatially distinct regions of draining lymph nodes
Due to their accessibility for dynamic imaging, much of the work detailing the organization of T cell priming and differentiation has been performed in lymph nodes. Both CD4+ and CD8+ T cells have been shown to make multiple contacts with dendritic cells on their way through the differentiation process. 5 , 6 , 132 Upon early activation following the engagement of antigen presented in the context of MHC, T cells rapidly expand and upregulate key defining transcription factors that determine where, within secondary lymphoid organs, they will move to in order to encounter further polarizing differentiation signals. 1 , 74 Conventional dendritic cells (cDCs) are professional antigen‐presenting cells and have been classified into two major branches. 133 Type 1 cDCs (cDC1) are considered efficient in presenting cell‐associated antigens through cross‐presentation, while type 2 (cDC2) dendritic cells can perform some presentation but are traditionally thought to be focused on activation of CD4+ T cells. 133 These distinct subsets are strategically located within the different lymphoid regions. 13 , 134 Thus, within these distinct niches or “rooms” T cells engage with dendritic cells and other accessory cells of influence to co‐ordinate the efficient eradication of pathogens or cancer and to establish a pool of memory cells for long‐lived protection.
4.1. Effector differentiation in the outer limits of lymph nodes
The effector differentiation of both CD4+ and CD8+ cells occurs in parallel, with strong correlations in the transcription factors, cellular partners, and migration cues that co‐ordinate this differentiation path. 1 CXCR3 is a chemokine receptor that is highly expressed on both CD4+ TH1 and CD8+ TSLEC cells. Previously, it was thought that this chemokine moves fully differentiated cells out of secondary lymphoid organs and into peripheral sites of inflammation. 103 Following the observation that CXCR3 is upregulated well before CD4+ T cells leave the lymph node, we identified a counter intuitive role for this receptor in TH1 differentiation. 7 In this setting, newly activated cells moved from the T cell paracortex located in the center of lymph nodes, into the interfollicular regions (IFRs), between B cell follicles at the lymph node periphery (Figure 2). The IFR provides a unique lymph node niche that has a rich stromal cell network. 135 Here, antigen can be deposited, either during particulate viral infection or by using specific vaccination strategies that target antigen to this region. 1 , 136 , 137 Using a dual CXCR3 ligand reporter mouse, we demonstrated that CXCR3+ CD4+ T cell movement is directed by the expression of CXCL9 and CXCL10 in this region. 7 While this study did not reveal the precise dendritic cell or stromal cell population that move T cells into this area, we have now demonstrated that type 2 conventional dendritic cells (cDC2) express high levels CXCL10 in the IFR. 138 Similar to CD4+ TH1 differentiation, CD8+ effectors are also found in IFRs and this migration correlates with robust formation of effector function. 139 CD8+ T cells in IFRs form clusters around cDC2 cells consistent with their expression of CXCL10. 134 A potential splenic correlate of this niche is the marginal zone or red pulp, where CD8+ TSLEC cells have also been shown to position in a CXCR3‐dependent manner. 140 , 141
Multiple studies have demonstrated that expression of CXCR3 is essential for CD8+ effector differentiation. 141 , 142 , 143 As outlined above, CXCR3 expression is principally regulated by the transcription factor T‐bet. 103 Consistent with this relationship, T‐bet deficiency resembles the loss of effector differentiation observed with loss of CXCR3. 49 , 144 We have shown the first observable expression of T‐bet in CD8+ T cells is found within pockets of IFRs; however, it is likely that moderate T‐bet induction is sufficient for CXCR3 expression and migration into this region. 138 While CD8+ T cells and cDC2s cluster in IFRs, it is unclear if this interaction is mediated through cDC2‐derived CXCL10, or if stromal cell expression moves T cells into this region. We have shown that CD4+ T cells preferentially interact with CXCL10+ expressing DCs, but the specific location of these interactions and the DC subset was not defined in this study. 7 In addition, there are other accessory cells located in this region that may drive the differentiation toward TH1 and TSLEC formation. While relatively scarce in secondary lymphoid organs, monocyte‐derived dendritic cells (moDCs) can induce both TH1 and TH17 responses. 133 , 145 , 146 Further, moDC promotion of TH1 differentiation is associated with their location in IFRs following vaccination. 137 Recently, Bosteel et al provided an important advance to the dendritic cell literature, revealing that these cells are not capable of antigen processing; however, they can express high levels of IL‐12 to polarize effector T cells. 147 , 148 , 149 In addition to moDCs, an inflammatory subpopulation of cDC2 cells, named infDCs, has recently been characterized. 145 Given the inflammatory milieu in IFRs following viral infection, this type I IFN‐dependent population may be a critical regulator of T cell effector fate. 145
The identity of the key cytokine or cytokines produced by dendritic cells or accessory cells in this region that promotes effector differentiation remains to be fully elucidated. IL‐12 is a good candidate for this as it has been demonstrated to steer T cell differentiation toward both CD4+ TFH and CD8+ TSLEC cell formation and away from other fates, in a T‐bet dependent manner. 128 , 146 , 150 Despite not being located within draining lymph node IFRs, cDC1 cells are commonly associated with TH1 responses due to their high production of IL‐12. 16 Indeed, in ex vivo cultures, cDC1s promote greater formation of TH1 cells. 151 This suggests that within IFRs in vivo, it is other accessory cells, either infDCs or moDCs that are present that can produce the high levels of IL‐12 needed to skew CD4+ and CD8+ T cell effector differentiation. In addition to IL‐12, other cytokines such as IL‐2, type I, and II IFNs have been demonstrated to promote TH1 and/or TSLEC differentiation. 9 The neighbor cell producing these additional cytokines is not clear, but may include other T cells with opposing fates as well as specific dendritic cell populations. 11 , 104
4.2. TFH differentiation: a stepwise progression to the follicle
The differentiation of TFH cells is dynamically regulated through time and in spatially distinct regions of secondary lymphoid organs. 74 This process takes place in three distinct steps and each involves a unique set of interacting partners that imprint and reinforce differentiation into functional, germinal center resident TFH cells. 16 , 152 , 153
Interacting dendritic cells are generally considered essential for directing the initial phases of TFH differentiation. 154 Several studies have shown that depletion of cDC2s reduced the amount of TFH differentiation, suggesting they are the prominent dendritic cell regulator of this lineage. Early contact between CD4+ T cells and dendritic cells leads to the upregulation of Bcl6; however, during early stages of differentiation, Bcl6 can be co‐expressed with T‐bet, the TH1 defining factor. 95 , 101 Thus, this initial activating event is not sufficient to define early fate bifurcation. These contacts do, however, result in the directed movement from the paracortex to the T:B cell border. 153 In addition to Bcl6, this migration is instructed by the ASCL2 helix‐loop‐helix transcription factor that acts in multiple ways to drive TFH ontogeny. 122 Expression of ASCL2 leads to the upregulation of CXCR5 and downregulation of CCR7. It is worth noting that the initial location of pre‐TFH cells at the T:B border occurs independently of Bcl6 and CXCR5, suggesting the downregulation of CCR7 and loss of retention in the paracortex is critical for migration. 155 , 156 Instead early TFH migration to the T:B border requires the G protein‐coupled receptor, EBI2 (GPR183). 157 In these regards, the initial stage of pre‐TFH migration resembles that of TH1 cells, although it is clear that TFH cells do not reach the outer limits of the IFRs, as shown for TH1 cells. 7 , 101 , 158
The T:B border is a critical site where decisions between TH1 and TFH differentiation are made. 11 , 74 , 152 , 153 Distinct infections can elicit a unique set of inflammatory signals and recruitment of interacting partners that can shift the balance of differentiation either toward or away from TFH differentiation. 11 Viral infections that induce a high expression of type I IFNs lead to increased dendritic cell production of IL‐6 and skew differentiation toward TH1 cells. Further at the T:B border, the upregulation of EBI2 promotes CD4+ T cell interactions with IL‐2 quenching cDC2s, which favors TFH differentiation. 157 Again, this work suggests that cDC2s are more permissive for TFH differentiation than for TH1, and that for the latter additional accessory cells are required to enter this site and direct effector polarization. Critically, in this location, pre‐TFH cells form long‐lived contacts with B cells through ICOS:ICOS‐ligand interactions, which leads to further upregulation of the canonical TFH genes, Bcl6, Cxcr5, and Il21. 77 , 78 , 153 , 156 Interestingly, these ICOS:ICOSL interactions are formed with bystander and not antigen‐specific B cells at the T:B border. 156 Although the importance of engagements with both cDC2s and B cells is well established, it is unclear if the three cells are required to interact at the same time at the T:B border. Further, one study has demonstrated that, during Plasmodium infection, dendritic cell interactions were dispensable, and B cells were solely responsible for the generation of TFH responses. 159 In contrast, augmenting antigen presentation can make TFH cell generation independent of B cells, highlighting the context‐dependent nature of this process. 160
Increasing CXCR5 expression directs TFH cells into the CXCL13‐expressing B cell follicles, to take part in germinal center reactions. 78 , 155 Interestingly, once CXCR5 permits entry into the germinal center, it is no longer required for the maintenance of cells in this location. 161 This is the final step in the differentiation process and allows TFH cells to accomplish their role to select high‐affinity B cells. 74 , 77 Unlike B cell interactions at the T:B border, within the germinal center T:B contacts are short‐lived, cognate interactions that maintain these microstructures within the B cell follicle via SAP:Ly108, CD40L:CD40, and ICOS:ICOSL mediated interactions. 23 , 78 , 79 , 153 , 162 , 163 Despite the multistep process for a TFH cell to enter a germinal center, this location can be further influenced by decreased expression of Bcl6, PD‐1, or increased CXCR3, which will move established CXCR3 cells out of the germinal center structure. 161 , 164
4.3. Differentiation of TSCM cells in the center of lymph nodes
Despite the shared transcriptional regulation, there are significant distinctions in location of CD4+ TFH cells and CD8+ TSCM cells. The first description of TSCM (formerly known as follicular cytotoxic) cells was found due to their expression of CXCR5. Some researchers found these cells positioned according to CXCR5 expression in the B cell follicles, in both models of chronic infection and in the spleens of HIV patients. 37 , 55 In contrast to this, others showed that they instead remained in the T cell paracortex of secondary lymphoid organs. 24 To clarify this discrepancy, we have shown that the TSCM precursors are retained in the T cell zone following viral infection, and their differentiation is enhanced in the absence of CXCR3. 138 While the transcriptional regulation of T cell effector molecules is well established, less attention has been placed on the transcriptional regulators of T cell location. 17 , 30 , 53 Delving into this further, we showed that TCF‐1, the key transcriptional regulator of TSCM cells, binds to the Ccr7 locus. 138 Supporting this, multiple studies have demonstrated that CCR7 is downregulated in the absence of TCF‐1 or in T cell populations that lack TCF‐1. 27 , 53 , 165 The retained expression of CCR7 on TSCM is in contrast to TFH cells, where this receptor is downregulated rapidly following the upregulation of Bcl6. 155 For TFH cells, the expression of CCR7 acts as a counterbalance retention signal, such that when CCR7 expression is maintained, cells are not capable of entering the B cell follicle, even when CXCR5 is overexpressed. 155 Therefore, it is likely that as TSCM cells co‐express both CCR7 and CXCR5, the retention within the paracortex is maintained to block migration out of this region. As discussed above, the key transcriptional difference between these populations could be the expression of ASCL2 in TFH cells, which orchestrates the upregulation of CXCR5 and downregulation of CCR7. 122 The role of ASCL2 in TSCM has so far not been investigated, but, unlike CCR7 it has not been reported to be expressed in transcriptional characterization studies. 24 , 25 , 56 , 165 , 166
Despite TSCM being positioned in the lymph node paracortex or splenic T cell zone, 24 it remains unclear if these precursors require contact with B cells at the T:B border in a similar manner to that shown for TFH cells. Initial characterization of these cells demonstrated they co‐expressed TFH cell‐surface markers ICOS and Slamf6, both of which facilitate TFH cell engagement with B cells. 55 A recent report showed transcriptional expression of EBI2, a molecule that is similarly expressed by pre‐TFH cells and directs them to the T:B border. 157 , 166 Again, it is unclear if in this region TSCM cells contact B cells or the IL‐2 quenching dendritic cells producing the EBI2 ligand. 157 Further, when TSCM cells were transferred into B cell‐deficient μMT recipients they were less capable of inhibiting viral replication than those transferred into wild‐type mice. 37 These data suggest that while TSCM cells may not need B cells to form, they potentially provide essential signals to function.
The specific dendritic cell subset that promotes differentiation toward TSCM remains unknown. Conventional DC1 cells reside in the paracortex of lymph nodes, positioning themselves as interacting partners during TSCM differentiation. 134 Dynamic imaging studies have shown that CD8+ T cells interact with cDC1 cells. 10 , 12 In these interactions, cDC1 cells act as conduits to facilitate CD8+ and CD4+ T cell interactions. 10 , 12 This three‐cell interaction facilitates the formation of memory, although TSCM differentiation was not assessed in either of these studies. While these cells highly express CXCL9, loss of CXCL9 does not appear to alter TSCM differentiation in vivo. 138 Previous work has shown that these interactions are alternatively instructed by CCR5. 167 Unlike cDC2s, cDC1s are thought to be a more homogeneous population 133 ; however, it could be that subsets of dendritic cells with a defined imprinting potential reside in the paracortex. In addition, TSCM may be maintained in the T cell zone by directly contacting the gp38+ CCL21‐expressing fibroblastic reticular cells (FRCs) located here. 140 IL‐27 is one cytokine that has been described to specifically promote TSCM expansion and coincidentally also promotes the expansion of TFH cells. 168 Although there is more work to be done to identify other factors that specifically direct differentiation toward TSCM, an alternative hypothesis is that positioning in the T cell paracortex instead protects these precursors away from other regions in lymph nodes where they may come into contact with inflammatory accessory cells or cytokines that promote effector and terminal differentiation. Supporting this hypothesis, it is the absence of inflammatory singles such as IL‐12 and type I IFN that steer differentiation away from TSCM. 27 , 128 We have recently shown that altering the location of CD8+ T cells by blocking CXCR3 responsiveness restricts cells to the paracortex. This change in T cell position, from the IFRs to the center of the lymph node, coincides with a decrease in effector differentiation and increased TSCM precursor formation. 138
Combined, it is clear that the migration of CD4+ and CD8+ T cells is driven by the dynamic regulation of chemokine receptors. This is controlled at the transcriptional level where early T‐bet expression leads to the upregulation of CXCR3 in TH1 and TSLEC cells; TCF‐1 insures the retention of TSCM in the T cell paracortex via CCR7 expression; and TFH cells localize in the B cell follicle due to expression of Bcl6 which instructs CXCR5 upregulation. Curiously, we have shown that early precursors of both TFH cells and TSCM cells can also upregulate CXCR3. 8 , 138 In these instances, it appears that the CCR7 and CXCL13 migration cues counterbalance CXCR3 ligands to override the movement of non‐effector cells into the locations (paracortex and B cell follicles) where these cells further differentiate.
5. CELLS OF INFLUENCE
As discussed, the location where CD4+ and CD8+ T cell differentiation occurs and been elucidated. Within these regions, several accessory cells are associated with skewing toward a particular cell type; however, the precise interactions remain poorly defined. Interactions that lead to TFH differentiation are the most clearly defined to require staged interactions with cDC2 cells and B cells. 16 , 74 , 152 Effector T cell differentiation can also be associated with cDC2 engagement and in some conditions with the presence of inflammatory moDCs. In contrast, TSCM cells likely require contact with cDC1 cells within the T cell paracortex. Additionally, simultaneous contact with CD4+ T cells and cDC1 cells is required for promotion of CD8+ T memory formation. The serial engagement of T cells with distinct immune populations, which has been dynamically studied by multi‐photon microscopy, suggests distinct roles of dendritic cell subsets during T cell differentiation. Instead, it is likely that these events work together in an additive manner to guide the transcriptional programming of T cells and determine the balance between effector and memory subsets. In this regard, movement of T cells into specific regions facilitates additive interactions that promote a single fate.
5.1. Stromal cells as non‐immune cells of influence
In addition to DC, moDC and B cell interactions, non‐immune cells are emerging as important modulators of T cell fate. Lymphoid stromal cells represent important cell organizers that enable T cell and B cell recruitment to secondary lymphoid tissues. 169 FRCs are found in close contact with T cells and act as guiding paths for dynamic scanning of naive T cells in search of their cognate antigen. 169 , 170 Previously, we have described the upregulation of CXCR3 ligands in the IFRs following vaccination. 7 FRCs express CXCL9, CXCL10, and CXCL11 within the paracortex and IFRs of resting mice and following infection. 171 , 172 These studies demonstrate that FRCs not only guide the location of cells in steady state, but can dynamically redistribute cells during an immune response. This role for stromal cells in facilitating interactions between immune cells is well established. As the stromal cell compartment becomes increasingly disorganized with age, dysfunction in both the dendritic cell and stromal cell compartment may both contribute to the dampened efficacy of vaccination with age. 173 , 174
Emerging studies suggest that in addition to this structural role, stromal cells, in particular FRCs, may directly influence T cell differentiation. When added to human T cell activation cultures, FRCs led to decreased proliferation and memory cell formation of both CD4+ and CD8+ T cells. 175 Interestingly, this inhibition was relieved in combination with IL‐6 and TGF‐β and PD‐L2 blockade, factors known to regulate either TFH or TSCM cell differentiation. Another study demonstrated that FRC co‐culture limits the production of CD8+ T cell IFN‐γ, but increased T cell production of IL‐2. 176 Importantly, this was independent of the block in T cell proliferation that is mediated through FRC iNOS production. Consistent with the IL‐2 production, FRCs altered the epigenome of T cells leading to heightened differentiation to memory and persistence in vivo. 176 A contrasting study demonstrated FRCs provided pro‐survival and proliferation signals to T cells. 177 Compared to FRC from human tonsil, follicular lymphoma stromal cells expressed the adhesion molecule ICAM and several chemokine ligands including CCL2, CCL5, CCL11, and CXCL10. Further, recent work has demonstrated that lymph node stromal cells are a checkpoint for peripheral tolerance and present self‐peptide to convert CD4+ T cells into T regulatory cells and restrict germinal center formation. 178
Much remains to be understood about how the stromal compartment directly regulates T cell differentiation. As outlined, some studies have shown contrary effects, and this may be due to different sites of isolation of FRCs and potentially distinct isolation protocols. Overlapping adhesion molecules, chemokine profiles, and skewing cytokines between stromal and dendritic cells additionally complicate the role of each cell type in vivo. To separate these influences, most of the studies to date have revealed interactions between T cells and stromal cells in in vitro co‐culture systems. Although genetic models of FRCs exist, these make it difficult to determine a direct role in T cell differentiation, as the cellular niches of secondary lymphoid organs are disrupted in these mice and the survival of naive T cells is severely impacted. 179 , 180 More work needs to be done to validate that the interactions defined with these genetic models are indeed critical during a normal immune response. To help overcome this issue, a novel human tissue in situ assay has been developed to dissect the regulators of T cell differentiation in live tissue explants. 175
6. HARNESSING TFH AND STEM‐LIKE MEMORY
Vaccines are one of public health's greatest achievements, preventing many millions of illnesses and deaths every year. 181 Traditional vaccination strategies have focused on the overall magnitude of the immune response produced. However, we can now appreciate how to manipulate immune responses to promote the most valuable T cell protection for future infectious challenge. Indeed, a preference toward CD4+ TFH cells to boost humoral responses and long‐lived CD8+ TSCM cell differentiation is preferred rather than exuberant effector response. While all current vaccine strategies provide protection via humoral immunity, many of the recalcitrant pathogens for which no vaccine is currently available [including human immunodeficiency virus (HIV), Mycobacterium tuberculosis, and Plasmodium species] are resistant to humoral immunity. 182 Strategies that specifically direct the generation of memory CD8+ T cells could aid in eliminating these intracellular pathogens. 181 , 182 , 183 Furthermore, CD8+ T cells have the potential to be utilized in the development of therapeutic vaccines to establish potent effector function against chronic infection and cancer. 181 In contrast, rapid proliferative responses with robust cytokine and effector molecule production of CD4+ TH1 and CD8+ TSLEC populations may be preferred where strategies are focused on overcoming an immediate challenge such as these settings. Therefore, the type of T cells required in each of these vaccination contexts is distinct. Thus, there is potential to exploit the shared transcriptional and cellular interactions that determine CD4+ and CD8+ effector T cells, or those that determine TFH and TSCM. Some new vaccination methods may influence the location of T cells within secondary lymphoid organs, which could, in turn, generate T cells that are fit for purpose depending on the response required.
The structure and anatomy of lymph nodes, along with the distinct positioning of T cells within them, allow for the delivery of drugs or antigens directly into specific sites. 184 The ability of these platforms to target distinct lymph node niches relies on specific design factors. Several vaccine vehicles can be used to target delivery to lymph nodes; in doing so, this improves the efficacy of T cell responses. 185 A key approach for this is via the use of nanoparticles, such as silica and protein nanoparticles, for targeted lymph node delivery of subunit vaccines. 184 , 185 , 186 , 187 , 188 These platforms increase the size of antigen which assists in the uptake and deposition within lymph nodes. We and others have demonstrated that these systems favorably affect lymphatic uptake and preferentially lodge in the lymph node IFRs. 136 , 189 , 190 While this strategy enhanced germinal center formation, this was done in the absence of increased TFH differentiation in both animal and non‐human primates. 191 Consistent with the increased recruitment of T cells to the IFR being associated with effector differentiation, this system enhanced the efficacy of anti‐cancer vaccines, compared to soluble vaccination. 186 A recent study has demonstrated that the route used to deliver nanoparticle vaccines dramatically alters T cell differentiation. 166 Specifically, intravenous delivery heightened differentiation toward TSCM, while subcutaneous delivery resulted in more terminally differentiated effector cells. Therefore, it appears that the route of nanoparticle entry into secondary lymphoid organs may alter where antigens are deposited. This demonstrates that it is not the magnitude of response postvaccination that matters, but the specific path of T cell differentiation promoted. In this system, despite the lower amplification of antigen‐specific CD8+ T cells following intravenous administration, the differentiation toward TSCM differentiation was essential to clear tumors cells more rapidly following checkpoint blockade. 166 Compared to subcutaneous delivery where antigen landed in IFRs with extended up take of MoDCs, intravenous delivery in the spleen was very transiently presented by cDC1s and MoDCs and the expression of IL‐12 and IFN‐α again was transient compared to settings where TSLEC cells were preferentially made. 166
The increased efficacy and safety of these nanoparticle vaccines lie in the ability to recapitulate the deposition of viral particles in the IFR. In addition, the structure‐based design of self‐assembling protein nanoparticles allows multiple epitopes to be complexed in a highly immunogenic manner, allowing multivalent presentation of antigen. This system has been used to elicit potent neutralizing immunity using a dual‐antigen self‐assembling SARS‐CoV‐2 nanoparticle in mice and non‐human primates. 192 Excitingly, these vaccines also provide a structure‐based design for the generation of broadly reactive universal vaccine platforms that target complete classes of viral strains, such as coronavirus or influenza. 188 , 192
An alternative strategy is the investigation of how different adjuvants deposit antigen into spatially distinct regions. This has recently been investigated for the promotion of TH1 cell differentiation in vivo. 137 This study compared soluble Toll‐like receptor and antigen delivered in the mineral oil emulsion, incomplete Freund's adjuvant (IFA). The use of the oil emulsion formulation altered the antigen deposition, moving it to the IFRs, rather than the medulla of draining lymph nodes. In addition, this approach promoted the recruitment of inflammatory monocytes which expressed CXCL10 to drive antigen‐specific CD4+ T cells into this region. 137 Although not tested, presumably this would have also been associated with increased CD8+ T cell effector differentiation, but come at a loss of TFH and TSCM. Other newer adjuvant formulations, such as GLA‐SE, have been shown to heighten TFH and memory T cell formation; however, as yet how these alter the deposition of antigen or inflammatory signals has not been demonstrated. 193
A final approach that aims to exploit specific T cell:DC interactions is via the targeting of specific dendritic cell populations. 194 This strategy exploits our understanding of T:DC interactions to target antigen to restrict presentation to a particular dendritic subtype. The optimal way to do this is to generate a genetic fusion of the antigen to the Fc portion of the antibody heavy chain to target a dendritic cell‐specific surface molecule. The two molecules that have shown the most potential for this strategy are using Fc fusions proteins for targeting CLEC‐9A, which is specifically expressed on cDC1s or targeting to DEC‐205 which is more broadly expressed dendritic cells and langerin cells. 194 Targeting DEC‐205 with adjuvants results in potent effector responses for both CD4+ and CD8+ T cells with strong humoral responses. The addition of adjuvant in this system is critical as without it, there are minimal effector responses and tolerance mechanisms related to regulatory T cells are induced. 195 Similarly, without adjuvant, targeting CLEC‐9A also results in the induction of CD8+ T cell tolerance. As discussed above, the choice of adjuvant can steer and tailor immune responses and this is true also, in combination with CLEC‐9A targeting. 195 However, in the absence of adjuvant, CLEC‐9A drives potent CD4+ TFH differentiation, germinal formation, and humoral responses, that exceed that seen with DEC‐205 targeting. 197 , 198 Interestingly, targeting cDC1s via CLEC‐9A positions these cells at the T:B border of lymph nodes and allows more contact with antigen‐specific B cells and TFH progenitors in this region. 199 Combined, these two targeting approaches hold promise to promote specific T cell engagement with both dendritic cells and/or antigen‐specific B cells. While the focus of studies to date has investigated CD4+ and CD8+ effector T cell differentiation and function, it remains to be seen how these individual targeting strategies promote the generation of long‐lived memory. Again, it is likely that adjuvant selection with each of these approaches will determine outcomes, highlighting the flexibility of these strategies.
7. CONTACT‐TRACING T CELL INTERACTIONS
Given the potential of harnessing both TFH and TSCM cells, the next questions in the field will be to more completely understand and identify the cellular contacts that promote formation of these subsets. In recent years, a host of new technologies and analysis pipelines have been developed to overcome some of the challenges for understanding the cellular interactions that govern transcriptional signatures of T cell fates. The most powerful of these incorporate advancements in imaging modalities combined with innovative transcriptional analysis.
One technology that is particularly suitable to determine cell interactions in a particular region is NICHE‐seq. 200 This combines a photoactivatable fluorescent marker to color‐in regions of interest using two‐photon laser scanning microscopy. Following dissociation of tissues, cells are sorted to identify niche components which are then analyzed by single cell RNA‐seq (scRNA‐seq). This method has been used to exceptional effect to characterize the T:B border CD4+ T cell priming niche during viral infection. 11 This revealed increased proportions of monocytes and dendritic cells following LCMV infection, when TFH differentiation is inhibited, compared to recombinant VSV where TFH differentiation is promoted. As discussed above, gene signatures within the niche also demonstrated increased type I IFN, which was subsequently shown to control TFH differentiation in this setting. 11 A further application of this technology would be to combine NICHE‐seq with a comprehensive ligand:receptor bioinformatic analysis to broadly identify the cellular interactome within a specific region. 201 The current tool kit available to bioinformatically resolve cellular interactions has recently been reviewed. 202 These analyses could also be complemented assessing the physically interacting cells (PIC‐seq) approach. 203 This method calibrates tissue dissociation protocols to insure interacting cell conjugates are preserved prior to scRNA‐seq analysis. Following conjugate RNA‐seq, the interacting partners are bioinformatically separated. Of interest, some of the receptor:ligand and downstream transcriptional events can be associated with specific interacting cells. 203 Combining these approaches within a specific lymphoid niche offers new opportunities to understand how inflammatory cues and directly interacting cells facilitate T cell differentiation.
More advanced methods have been developed to analyze specific T cell interactions. A proximity‐dependent labeling system that crosses cell:cell interfaces known as LIPSTIC (Labelling Immune Cell Partnerships by SorTagging Intercellular Contacts) has been developed to investigate CD40:CD40L interactions. 204 In this system, ligands and receptors are genetically fused to the Staphylococcus aureus transpeptidase sortase A (SrtA), or a tag residue. When the ligand and receptors interact, Srt A catalyzes the transfer of substrate onto the tagged receptor, recording the history of interaction that can be detected by flow cytometry. This study revealed that initial vaccine‐induced interactions occur between 24 and 72 hours following T cell transfer and these interactions were primarily with cDC2 as opposed to cDC1s. 204 This approach is highly adaptable to other receptor:ligand interactions and cell types of interest. An extension of this approach, termed FucolD, provides another option for interaction‐dependent labeling of T cells using cell‐surface enzymatic fucosyl‐biotinylation. 205 In this system, antigen‐specific T cells were labeled by their interacting dendritic cell partners within the tumor environment. Importantly, this approach is a proximity‐based labeling system, meaning that, unlike LIPSTIC, the receptor mediating the interaction is not required to be known prior to experimentation.
8. CONCLUSION
Given the transcriptional similarities between CD4+ and CD8+ effectors, as well as the similarities between TFH and TSCM, we have discussed in this review what is currently known for these subsets and contrasted the spatial location and environmental cues that promote specific differentiation toward each of these paths. While the secondary lymphoid niches that imprint specific T cell differentiation have been elucidated, there remains much left to understand regarding the specific interactions within these sites that direct cell fate. This information is key to understanding the ways to harness T cell differentiation for therapeutic and vaccine interventions and will allow the generation of T cells that are fit for purpose; either to stimulate potent T cell effector function for the elimination of chronic infection and cancer, or to amplify the formation of humoral immunity and longevity of cellular memory to prevent current and emerging infectious diseases.
CONFLICT OF INTEREST
The author has no conflict of interest to declare.
ACKNOWLEDGEMENTS
We thank Stephen L Nutt for critical reading of the manuscript. This work was supported by National Health and Medical Research Council (NHMRC) Ideas grant (GNT1182649) and Project grant (GNT1137989) grants to JRG. BCD is supported by a WEHI Academic Excellence scholarship. JRG is supported by an Australian Research Council Future Fellowship 130100708 and by a Walter and Eliza Hall Centenary Fellowship sponsored by CSL. Figures were generated using Biorender.
Duckworth BC, Groom JR. Conversations that count: Cellular interactions that drive T cell fate. Immunol Rev. 2021;300:203–219. 10.1111/imr.12945
[Correction added on 07 March, after first online publication: the main and sub titles for this article were swapped and have been updated in this version.]
This article is part of a series of reviews covering Genome Regulation in Innate and Adaptive Immune Cells appearing in Volume 300 of Immunological Reviews.
DATA AVAILABILITY STATEMENT
Data sharing not applicable – no new data generated, or the article describes entirely theoretical research
REFERENCES
- 1. Groom JR. Regulators of T‐cell fate: integration of cell migration, differentiation and function. Immunol Rev. 2019;289(1):101‐114. [DOI] [PubMed] [Google Scholar]
- 2. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659‐702. [DOI] [PubMed] [Google Scholar]
- 3. Miller J. The function of the thymus and its impact on modern medicine. Science. 2020;369(6503):1–8. [DOI] [PubMed] [Google Scholar]
- 4. Geijtenbeek TBH, Torensma R, van Vliet SJ, et al. Identification of DC‐SIGN, a novel dendritic cell‐specific ICAM‐3 receptor that supports primary immune responses. Cell. 2000;100(5):575‐585. [DOI] [PubMed] [Google Scholar]
- 5. Mempel TR, Henrickson SE, Von Andrian UH. T‐cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427(6970):154‐159. [DOI] [PubMed] [Google Scholar]
- 6. Miller MJ, Safrina O, Parker I, Cahalan MD. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J Exp Med. 2004;200(7):847‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Groom J, Richmond J, Murooka T, et al. CXCR3 chemokine receptor‐ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. 2012;37(6):1091‐1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sheikh AA, Cooper L, Feng M, et al. Context‐dependent role for T‐bet in T follicular helper differentiation and germinal center function following viral infection. Cell Rep. 2019;28(7):1758‐1772 e1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheikh AA, Groom JR. Transcription tipping points for T follicular helper cell and T‐helper 1 cell fate commitment. Cell Mol Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity. 2015;43(3):554‐565. [DOI] [PubMed] [Google Scholar]
- 11. De Giovanni M, Cutillo V, Giladi A, et al. Spatiotemporal regulation of type I interferon expression determines the antiviral polarization of CD4(+) T cells. Nat Immunol. 2020;21(3):321‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eickhoff S, Brewitz A, Gerner M, et al. Robust anti‐viral immunity requires multiple distinct T cell‐dendritic cell interactions. Cell. 2015;162(6):1322‐1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerner MY, Casey KA, Kastenmuller W, Germain RN. Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J Exp Med. 2017;214(10):3105‐3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kastenmuller W, Brandes M, Wang Z, Herz J, Egen JG, Germain RN. Peripheral prepositioning and local CXCL9 chemokine‐mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity. 2013;38(3):502‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kastenmuller W, Torabi‐Parizi P, Subramanian N, Lammermann T, Germain RN. A spatially‐organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell. 2012;150(6):1235‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hilligan KL, Ronchese F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell Mol Immunol. 2020;17(6):587‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327(5969):1098‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50(5):1132‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramiscal RR, Vinuesa CG. T‐cell subsets in the germinal center. Immunol Rev. 2013;252(1):146‐155. [DOI] [PubMed] [Google Scholar]
- 21. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708‐712. [DOI] [PubMed] [Google Scholar]
- 22. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12(11):749‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23(1):18‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature. 2016;537(7620):417‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siddiqui I, Schaeuble K, Chennupati V, et al. Intratumoral Tcf1(+)PD‐1(+)CD8(+) T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50(1):195‐211 e110. [DOI] [PubMed] [Google Scholar]
- 26. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33(2):229‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu T, Ji Y, Moseman EA, et al. The TCF1‐Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. 2016;1(6):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Helmink BA, Reddy SM, Gao J, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lüthje K, Kallies A, Shimohakamada Y, et al. The development and fate of follicular helper T cells defined by an IL‐21 reporter mouse. Nat Immunol. 2012;13(5):491‐498. [DOI] [PubMed] [Google Scholar]
- 32. Gowthaman U, Chen JS, Zhang B, et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science. 2019;365(6456):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reinhardt RL, Liang HE, Locksley RM. Cytokine‐secreting follicular T cells shape the antibody repertoire. Nat Immunol. 2009;10(4):385‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wichner K, Stauss D, Kampfrath B, et al. Dysregulated development of IL‐17‐ and IL‐21‐expressing follicular helper T cells and increased germinal center formation in the absence of RORgammat. FASEB J. 2016;30(2):761‐774. [DOI] [PubMed] [Google Scholar]
- 35. Weinstein JS, Herman EI, Lainez B, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol. 2016;17(10):1197‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang B, Liu E, Gertie JA, et al. Divergent T follicular helper cell requirement for IgA and IgE production to peanut during allergic sensitization. Sci Immunol. 2020;5(47):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He R, Hou S, Liu C, et al. Follicular CXCR5‐ expressing CD8(+) T cells curtail chronic viral infection. Nature. 2016;537(7620):412‐428. [DOI] [PubMed] [Google Scholar]
- 38. Morita R, Schmitt N, Bentebibel S‐E, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34(1):108‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brenna E, Davydov AN, Ladell K, et al. CD4(+) T follicular helper cells in human tonsils and blood are clonally convergent but divergent from non‐Tfh CD4(+) cells. Cell Rep. 2020;30(1):137‐152 e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heit A, Schmitz F, Gerdts S, et al. Vaccination establishes clonal relatives of germinal center T cells in the blood of humans. J Exp Med. 2017;214(7):2139‐2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vella LA, Buggert M, Manne S, et al. T follicular helper cells in human efferent lymph retain lymphoid characteristics. J Clin Invest. 2019;129(8):3185‐3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma CS, Wong N, Rao G, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol. 2015;136(4):993‐1006 e1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bentebibel S‐E, Lopez S, Obermoser G, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. 2013;5(176):176ra132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koutsakos M, Wheatley AK, Loh L, et al. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza‐specific B cell immunity. Sci Transl Med. 2018;10(428):1–15. [DOI] [PubMed] [Google Scholar]
- 45. Locci M, Havenar‐Daughton C, Landais E, et al. Human circulating PD‐1+CXCR3‐CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39(4):758‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Obeng‐Adjei N, Portugal S, Tran T, et al. Circulating Th1‐Cell‐type Tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep. 2015;13(2):425‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Juno JA, Tan H‐X, Lee WS, et al. Immunogenic profile of SARS‐CoV‐2 spike in individuals recovered from COVID‐19. medRxiv. 2020. Preprint. [Google Scholar]
- 48. Kaech SM, Wherry EJ. Heterogeneity and cell‐fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27(3):393‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Joshi NS, Cui W, Chandele A, et al. Inflammation directs memory precursor and short‐lived effector CD8(+) T cell fates via the graded expression of T‐bet transcription factor. Immunity. 2007;27(2):281‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hinks TSC, Hoyle RD, Gelfand EW. CD8(+) Tc2 cells: underappreciated contributors to severe asthma. Eur Respir Rev. 2019;28(154):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Intlekofer AM, Banerjee A, Takemoto N, et al. Anomalous type 17 response to viral infection by CD8+ T cells lacking T‐bet and eomesodermin. Science. 2008;321(5887):408‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xin A, Masson F, Liao Y, et al. A molecular threshold for effector CD8(+) T cell differentiation controlled by transcription factors Blimp‐1 and T‐bet. Nat Immunol. 2016;17(4):422‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mielke LA, Liao Y, Clemens EB, et al. TCF‐1 limits the formation of Tc17 cells via repression of the MAF‐RORgammat axis. J Exp Med. 2019;216(7):1682‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raghu D, Xue HH, Mielke LA. Control of lymphocyte fate, infection, and tumor immunity by TCF‐1. Trends Immunol. 2019;40(12):1149‐1162. [DOI] [PubMed] [Google Scholar]
- 55. Leong YA, Chen Y, Ong HS, et al. CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol. 2016;17(10):1187‐1196. [DOI] [PubMed] [Google Scholar]
- 56. Yao C, Sun H‐W, Lacey NE, et al. Single‐cell RNA‐seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol. 2019;20(7):890‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gautam S, Fioravanti J, Zhu W, et al. The transcription factor c‐Myb regulates CD8(+) T cell stemness and antitumor immunity. Nat Immunol. 2019;20(3):337‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Galletti G, De Simone G, Mazza EMC, et al. Two subsets of stem‐like CD8(+) memory T cell progenitors with distinct fate commitments in humans. Nat Immunol. 2020;21:1552‐1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mueller SN, Mackay LK. Tissue‐resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2016;16(2):79‐89. [DOI] [PubMed] [Google Scholar]
- 60. Milner JJ, Nguyen H, Omilusik K, et al. Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc Natl Acad Sci USA. 2020;117(41):25667‐25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Beura LK, Mitchell JS, Thompson EA, et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue‐autonomous recall responses that amplify secondary memory. Nat Immunol. 2018;19(2):173‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Park SL, Zaid A, Hor JL, et al. Local proliferation maintains a stable pool of tissue‐resident memory T cells after antiviral recall responses. Nat Immunol. 2018;19(2):183‐191. [DOI] [PubMed] [Google Scholar]
- 63. Savas P, Virassamy B, Ye C, et al. Single‐cell profiling of breast cancer T cells reveals a tissue‐resident memory subset associated with improved prognosis. Nat Med. 2018;24(7):986‐993. [DOI] [PubMed] [Google Scholar]
- 64. Park SL, Buzzai A, Rautela J, et al. Tissue‐resident memory CD8(+) T cells promote melanoma‐immune equilibrium in skin. Nature. 2018;565:366‐371. [DOI] [PubMed] [Google Scholar]
- 65. Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103(+)CD8+ tissue‐resident memory T cells of skin. Nat Immunol. 2013;14(12):1294‐1301. [DOI] [PubMed] [Google Scholar]
- 66. Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl‐6 and Blimp‐1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11(2):114‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rutishauser RL, Martins GA, Kalachikov S, et al. Transcriptional repressor Blimp‐1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31(2):296‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ichii H, Sakamoto A, Arima M, Hatano M, Kuroda Y, Tokuhisa T. Bcl6 is essential for the generation of long‐term memory CD4+ T cells. Int Immunol. 2007;19(4):427‐433. [DOI] [PubMed] [Google Scholar]
- 69. Johnston RJ, Poholek AC, DiToro D, et al. Bcl6 and Blimp‐1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325(5943):1006‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nurieva RI, Chung Y, Hwang D, et al. Generation of T follicular helper cells is mediated by interleukin‐21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29(1):138‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yu DI, Rao S, Tsai LM, et al. The transcriptional repressor Bcl‐6 directs T follicular helper cell lineage commitment. Immunity. 2009;31(3):457‐468. [DOI] [PubMed] [Google Scholar]
- 72. Tunyaplin C, Shaffer AL, Angelin‐Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl‐6 inhibits plasmacytic differentiation. J Immunol. 2004;173(2):1158‐1165. [DOI] [PubMed] [Google Scholar]
- 73. Choi J, Diao H, Faliti CE, et al. Bcl‐6 is the nexus transcription factor of T follicular helper cells via repressor‐of‐repressor circuits. Nat Immunol. 2020;21(7):777‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Qi H. T follicular helper cells in space‐time. Nat Rev Immunol. 2016;16(10):612‐625. [DOI] [PubMed] [Google Scholar]
- 75. Lee J‐Y, Skon CN, Lee YJ, et al. The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity. 2015;42(2):252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14(12):1285‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Choi Y, Kageyama R, Eto D, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34(6):932‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liu D, Xu H, Shih C, et al. T‐B‐cell entanglement and ICOSL‐driven feed‐forward regulation of germinal centre reaction. Nature. 2015;517(7533):214‐218. [DOI] [PubMed] [Google Scholar]
- 79. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP‐controlled T‐B cell interactions underlie germinal centre formation. Nature. 2008;455(7214):764‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin‐21‐interleukin‐10‐STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35(5):792‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu Z, Guo Y, Tang S, et al. Cutting edge: transcription factor BCL6 is required for the generation, but not maintenance, of memory CD8(+) T cells in acute viral infection. J Immunol. 2019;203(2):323‐327. [DOI] [PubMed] [Google Scholar]
- 82. Shin CJ, Wong S, Davis MJ, Ragan MA. Protein‐protein interaction as a predictor of subcellular location. BMC Syst Biol. 2009;3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kallies A, Xin A, Belz GT, Nutt SL. Blimp‐1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. 2009;31(2):283‐295. [DOI] [PubMed] [Google Scholar]
- 84. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin‐2Ralpha expression on virus‐specific CD8+ T cells favors terminal‐effector differentiation in vivo. Immunity. 2010;32(1):91‐103. [DOI] [PubMed] [Google Scholar]
- 85. Pipkin ME, Sacks JA, Cruz‐Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin‐2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32(1):79‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kwon H, Thierry‐Mieg D, Thierry‐Mieg J, et al. Analysis of interleukin‐21‐induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. 2009;31(6):941‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gong D, Malek TR. Cytokine‐dependent Blimp‐1 expression in activated T cells inhibits IL‐2 production. J Immunol. 2007;178(1):242‐252. [DOI] [PubMed] [Google Scholar]
- 88. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317(5):620‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kallies A, Hawkins ED, Belz GT, et al. Transcriptional repressor Blimp‐1 is essential for T cell homeostasis and self‐tolerance. Nat Immunol. 2006;7(5):466‐474. [DOI] [PubMed] [Google Scholar]
- 90. Martins GA, Cimmino L, Shapiro‐Shelef M, et al. Transcriptional repressor Blimp‐1 regulates T cell homeostasis and function. Nat Immunol. 2006;7(5):457‐465. [DOI] [PubMed] [Google Scholar]
- 91. Lazarevic V, Glimcher LH. T‐bet in disease. Nat Immunol. 2011;12(7):597‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol. 2016;16(2):102‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T‐bet, directs Th1 lineage commitment. Cell. 2000;100(6):655‐669. [DOI] [PubMed] [Google Scholar]
- 94. Oestreich KJ, Weinmann AS. T‐bet employs diverse regulatory mechanisms to repress transcription. Trends Immunol. 2012;33(2):78‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Oestreich KJ, Huang AC, Weinmann AS. The lineage‐defining factors T‐bet and Bcl‐6 collaborate to regulate Th1 gene expression patterns. J Exp Med. 2011;208(5):1001‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase‐mediated interaction of T‐bet with GATA‐3. Science. 2005;307(5708):430‐433. [DOI] [PubMed] [Google Scholar]
- 97. Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T‐bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8(2):145‐153. [DOI] [PubMed] [Google Scholar]
- 98. Usui T, Preiss JC, Kanno Y, et al. T‐bet regulates Th1 responses through essential effects on GATA‐3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203(3):755‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dominguez CX, Amezquita RA, Guan T, et al. The transcription factors ZEB2 and T‐bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med. 2015;212(12):2041‐2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T‐bet in TH1 lineage commitment and IFN‐gamma production in CD4 and CD8 T cells. Science. 2002;295(5553):338‐342. [DOI] [PubMed] [Google Scholar]
- 101. Nakayamada S, Kanno Y, Takahashi H, et al. Early Th1 cell differentiation is marked by a Tfh cell‐like transition. Immunity. 2011;35(6):919‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Afkarian M, Sedy JR, Yang J, et al. T‐bet is a STAT1‐induced regulator of IL‐12R expression in naive CD4+ T cells. Nat Immunol. 2002;3(6):549‐557. [DOI] [PubMed] [Google Scholar]
- 103. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89(2):207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. DiToro D, Winstead CJ, Pham D, et al. Differential IL‐2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science. 2018;361(6407):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Intlekofer AM, Takemoto N, Wherry EJ, et al. Effector and memory CD8+ T cell fate coupled by T‐bet and eomesodermin. Nat Immunol. 2005;6(12):1236‐1244. [DOI] [PubMed] [Google Scholar]
- 106. Joshi NS, Cui W, Dominguez CX, Chen JH, Hand TW, Kaech SM. Increased numbers of preexisting memory CD8 T cells and decreased T‐bet expression can restrain terminal differentiation of secondary effector and memory CD8 T cells. J Immunol. 2011;187(8):4068‐4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Intlekofer AM, Takemoto N, Kao C, et al. Requirement for T‐bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med. 2007;204(9):2015‐2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pearce EL, Mullen AC, Martins GA, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302(5647):1041‐1043. [DOI] [PubMed] [Google Scholar]
- 109. Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting edge: IL‐12 inversely regulates T‐bet and eomesodermin expression during pathogen‐induced CD8+ T cell differentiation. J Immunol. 2006;177(11):7515‐7519. [DOI] [PubMed] [Google Scholar]
- 110. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T‐bet and Eomesodermin. Immunity. 2010;32(1):67‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T‐bet‐mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36(3):374‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Banerjee A, Gordon SM, Intlekofer AM, et al. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol. 2010;185(9):4988‐4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Paley MA, Kroy DC, Odorizzi PM, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Crawford A, Angelosanto J, Kao C, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity. 2014;40(2):289‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kao C, Oestreich KJ, Paley MA, et al. Transcription factor T‐bet represses expression of the inhibitory receptor PD‐1 and sustains virus‐specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011;12(7):663‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Staal FJ, Sen JM. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur J Immunol. 2008;38(7):1788‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Xue HH, Zhao DM. Regulation of mature T cell responses by the Wnt signaling pathway. Ann N Y Acad Sci. 2012;1247:16‐33. [DOI] [PubMed] [Google Scholar]
- 118. Xu L, Cao YI, Xie Z, et al. The transcription factor TCF‐1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol. 2015;16(9):991‐999. [DOI] [PubMed] [Google Scholar]
- 119. Choi YS, Gullicksrud JA, Xing S, et al. LEF‐1 and TCF‐1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. 2015;16(9):980‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ballesteros‐Tato A, León B, Graf B, et al. Interleukin‐2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36(5):847‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL‐6‐mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol. 2013;190(7):3049‐3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu X, Chen X, Zhong BO, et al. Transcription factor achaete‐scute homologue 2 initiates follicular T‐helper‐cell development. Nature. 2014;507(7493):513‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Choi YS, Yang JA, Yusuf I, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. 2013;190(8):4014‐4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Künzli M, Schreiner D, Pereboom TC, et al. Long‐lived T follicular helper cells retain plasticity and help sustain humoral immunity. Sci Immunol. 2020;5(45):1–16. [DOI] [PubMed] [Google Scholar]
- 125. Hale J, Youngblood B, Latner D, et al. Distinct memory CD4+ T cells with commitment to T follicular helper‐ and T helper 1‐cell lineages are generated after acute viral infection. Immunity. 2013;38(4):805‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gullicksrud JA, Li F, Xing S, et al. Differential requirements for Tcf1 long isoforms in CD8(+) and CD4(+) T cell responses to acute viral infection. J Immunol. 2017;199(3):911‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf‐1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci USA. 2010;107(21):9777‐9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Danilo M, Chennupati V, Silva JG, Siegert S, Held W. Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep. 2018;22(8):2107‐2117. [DOI] [PubMed] [Google Scholar]
- 129. Cobas M, Wilson A, Ernst B, et al. Beta‐catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199(2):221‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. 2011;208(5):987‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Utzschneider DT, Charmoy M, Chennupati V, et al. T cell factor 1‐expressing memory‐like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity. 2016;45(2):415‐427. [DOI] [PubMed] [Google Scholar]
- 132. Itano AA, McSorley SJ, Reinhardt RLee, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell‐mediated immunity. Immunity. 2003;19(1):47‐57. [DOI] [PubMed] [Google Scholar]
- 133. Nutt SL, Chopin M. Transcriptional networks driving dendritic cell differentiation and function. Immunity. 2020;52(6):942‐956. [DOI] [PubMed] [Google Scholar]
- 134. Gerner MY, Torabi‐Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph‐borne particulate antigens. Immunity. 2015;42(1):172‐185. [DOI] [PubMed] [Google Scholar]
- 135. Katakai T, Hara T, Lee JH, Gonda H, Sugai M, Shimizu A. A novel reticular stromal structure in lymph node cortex: an immuno‐platform for interactions among dendritic cells, T cells and B cells. Int Immunol. 2004;16(8):1133‐1142. [DOI] [PubMed] [Google Scholar]
- 136. Kelly HG, Tan H‐X, Juno JA, et al. Self‐assembling influenza nanoparticle vaccines drive extended germinal center activity and memory B cell maturation. JCI Insight. 2020;5(10):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lian J, Ozga AJ, Sokol CL, Luster AD. Targeting lymph node niches enhances type 1 immune responses to immunization. Cell Rep. 2020;31(8):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Duckworth B, Lafouresse F, Wimmer V, et al. (2021). Effector and stem‐like memory cell fates are imprinted in distinct lymph node niches directed by CXCR3 ligands. Nature Immunology. 10.1038/s41590-021-00878-5 [DOI] [PubMed] [Google Scholar]
- 139. Hickman HD, Takeda K, Skon CN, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. 2008;9(2):155‐165. [DOI] [PubMed] [Google Scholar]
- 140. Jung YW, Rutishauser RL, Joshi NS, Haberman AM, Kaech SM. Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J Immunol. 2010;185(9):5315‐5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kurachi M, Kurachi J, Suenaga F, et al. Chemokine receptor CXCR3 facilitates CD8(+) T cell differentiation into short‐lived effector cells leading to memory degeneration. J Exp Med. 2011;208(8):1605‐1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hu JK, Kagari T, Clingan JM, Matloubian M. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T‐cell generation. Proc Natl Acad Sci USA. 2011;108(21):E118‐E127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kohlmeier JE, Reiley WW, Perona‐Wright G, et al. Inflammatory chemokine receptors regulate CD8(+) T cell contraction and memory generation following infection. J Exp Med. 2011;208(8):1621‐1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Marshall H, Chandele A, Jung Y, et al. Differential expression of Ly6C and T‐bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity. 2011;35(4):633‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bosteels C, Neyt K, Vanheerswynghels M, et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity. 2020;52:1039‐1056.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hilligan KL, Tang S‐C, Hyde EJ, et al. Dermal IRF4+ dendritic cells and monocytes license CD4+ T helper cells to distinct cytokine profiles. Nat Commun. 2020;11(1):5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Chow KV, Lew AM, Sutherland RM, Zhan Y. Monocyte‐derived dendritic cells promote Th polarization, whereas conventional dendritic cells promote Th proliferation. J Immunol. 2016;196(2):624‐636. [DOI] [PubMed] [Google Scholar]
- 148. Nakano H, Lin KL, Yanagita M, et al. Blood‐derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol. 2009;10(4):394‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lonnberg T, Svensson V, James KR, et al. Single‐cell RNA‐seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci Immunol. 2017;2(9):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Elsner RA, Shlomchik MJ. IL‐12 blocks Tfh cell differentiation during Salmonella infection, thereby contributing to germinal center suppression. Cell Rep. 2019;29(9):2796‐2809 e2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Maldonado‐Lopez R, De Smedt T, Michel P, et al. CD8alpha+ and CD8alpha‐ subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med. 1999;189(3):587‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Groom JR. Moving to the suburbs: T‐cell positioning within lymph nodes during activation and memory. Immunol Cell Biol. 2015;93(4):330‐336. [DOI] [PubMed] [Google Scholar]
- 153. Kerfoot S, Yaari G, Patel J, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. 2011;34(6):947‐960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Krishnaswamy JK, Alsen S, Yrlid U, Eisenbarth SC, Williams A. Determination of T follicular helper cell fate by dendritic cells. Front Immunol. 2018;9:2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene‐1 high germinal center‐associated subpopulation. J Immunol. 2007;179(8):5099‐5108. [DOI] [PubMed] [Google Scholar]
- 156. Xu H, Li X, Liu D, et al. Follicular T‐helper cell recruitment governed by bystander B cells and ICOS‐driven motility. Nature. 2013;496(7446):523‐527. [DOI] [PubMed] [Google Scholar]
- 157. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL‐2‐quenching dendritic cells. Nature. 2016;533(7601):110‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Woodruff MC, Heesters BA, Herndon CN, et al. Trans‐nodal migration of resident dendritic cells into medullary interfollicular regions initiates immunity to influenza vaccine. J Exp Med. 2014;211(8):1611‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Arroyo EN, Pepper M. B cells are sufficient to prime the dominant CD4+ Tfh response to Plasmodium infection. J Exp Med. 2020;217(2):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Deenick EK, Chan A, Ma CS, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. 2010;33(2):241‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Alterauge D, Bagnoli JW, Dahlström F, et al. Continued Bcl6 expression prevents the transdifferentiation of established Tfh cells into Th1 cells during acute viral infection. Cell Rep. 2020;33(1):108232. [DOI] [PubMed] [Google Scholar]
- 162. Shulman Z, Gitlin AD, Weinstein JS, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science. 2014;345(6200):1058‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Kräutler NJ, Suan D, Butt D, et al. Differentiation of germinal center B cells into plasma cells is initiated by high‐affinity antigen and completed by Tfh cells. J Exp Med. 2017;214(5):1259‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD‐1 controls follicular T helper cell positioning and function. Immunity. 2018;49(2):264‐274 e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Jadhav RR, Im SJ, Hu B, et al. Epigenetic signature of PD‐1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD‐1 blockade. Proc Natl Acad Sci USA. 2019;116(28):14113‐14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Baharom F, Ramirez‐Valdez RA, Tobin KKS, et al. Intravenous nanoparticle vaccination generates stem‐like TCF1(+) neoantigen‐specific CD8(+) T cells. Nat Immunol. 2021;22:41‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Castellino F, Huang AY, Altan‐Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell‐dendritic cell interaction. Nature. 2006;440(7086):890‐895. [DOI] [PubMed] [Google Scholar]
- 168. Huang Z, Zak J, Pratumchai I, et al. IL‐27 promotes the expansion of self‐renewing CD8(+) T cells in persistent viral infection. J Exp Med. 2019;216(8):1791‐1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Alexandre YO, Mueller SN. Stromal cell networks coordinate immune response generation and maintenance. Immunol Rev. 2018;283(1):77‐85. [DOI] [PubMed] [Google Scholar]
- 170. Denton AE, Linterman MA. Stromal networking: cellular connections in the germinal centre. Curr Opin Immunol. 2017;45:103‐111. [DOI] [PubMed] [Google Scholar]
- 171. Malhotra D, Fletcher AL, Astarita J, et al. Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol. 2012;13(5):499‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Rodda LB, Lu E, Bennett ML, et al. Single‐cell RNA sequencing of lymph node stromal cells reveals niche‐associated heterogeneity. Immunity. 2018;48(5):1014‐1028 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Masters AR, Hall A, Bartley JM, et al. Assessment of lymph node stromal cells as an underlying factor in age‐related immune impairment. J Gerontol A Biol Sci Med Sci. 2019;74(11):1734‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Stebegg M, Bignon A, Hill DL, et al. Rejuvenating conventional dendritic cells and T follicular helper cell formation after vaccination. eLife. 2020;9:e52473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Knoblich K, Cruz Migoni S, Siew SM, et al. The human lymph node microenvironment unilaterally regulates T‐cell activation and differentiation. PLoS Biol. 2018;16(9):e2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Brown FD, Sen DR, LaFleur MW, et al. Fibroblastic reticular cells enhance T cell metabolism and survival via epigenetic remodeling. Nat Immunol. 2019;20(12):1668‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Misiak J, Jean R, Rodriguez S, et al. Human lymphoid stromal cells contribute to polarization of follicular T cells into IL‐4 secreting cells. Front Immunol. 2020;11:559866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Nadafi R, Gago de Graça C, Keuning ED, et al. Lymph node stromal cells generate antigen‐specific regulatory T cells and control autoreactive T and B cell responses. Cell Rep. 2020;30(12):4110‐4123 e4114. [DOI] [PubMed] [Google Scholar]
- 179. Cremasco V, Woodruff MC, Onder L, et al. B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nat Immunol. 2014;15(10):973‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Link A, Vogt TK, Favre S, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8(11):1255‐1265. [DOI] [PubMed] [Google Scholar]
- 181. Ahmed R, Burton DR. Viral vaccines: past successes and future challenges. Curr Opin Virol. 2013;3(3):307‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Seder RA, Hill AV. Vaccines against intracellular infections requiring cellular immunity. Nature. 2000;406(6797):793‐798. [DOI] [PubMed] [Google Scholar]
- 183. Nolz JC, Harty JT. Strategies and implications for prime‐boost vaccination to generate memory CD8 T cells. Adv Exp Med Biol. 2011;780:69‐83. [DOI] [PubMed] [Google Scholar]
- 184. Schudel A, Francis DM, Thomas SN. Material design for lymph node drug delivery. Nat Rev Mater. 2019;4(6):415‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Jiang H, Wang Q, Sun X. Lymph node targeting strategies to improve vaccination efficacy. J Control Release. 2017;267:47‐56. [DOI] [PubMed] [Google Scholar]
- 186. Liu H, Moynihan KD, Zheng Y, et al. Structure‐based programming of lymph‐node targeting in molecular vaccines. Nature. 2014;507(7493):519‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. An M, Li M, Xi J, Liu H. Silica nanoparticle as a lymph node targeting platform for vaccine delivery. ACS Appl Mater Interfaces. 2017;9(28):23466‐23475. [DOI] [PubMed] [Google Scholar]
- 188. Marcandalli J, Fiala B, Ols S, et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell. 2019;176(6):1420‐1431 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Hanson MC, Crespo MP, Abraham W, et al. Nanoparticulate STING agonists are potent lymph node‐targeted vaccine adjuvants. J Clin Invest. 2015;125(6):2532‐2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Kim S‐Y, Noh Y‐W, Kang TH, et al. Synthetic vaccine nanoparticles target to lymph node triggering enhanced innate and adaptive antitumor immunity. Biomaterials. 2017;130:56‐66. [DOI] [PubMed] [Google Scholar]
- 191. Kelly HG, Kent SJ, Wheatley AK. Immunological basis for enhanced immunity of nanoparticle vaccines. Expert Rev Vaccines. 2019;18(3):269‐280. [DOI] [PubMed] [Google Scholar]
- 192. Walls AC, Fiala B, Schäfer A, et al. Elicitation of potent neutralizing antibody responses by designed protein nanoparticle vaccines for SARS‐CoV‐2. Cell. 2020;183:1367‐1382.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Hill DL, Pierson W, Bolland DJ, et al. The adjuvant GLA‐SE promotes human Tfh cell expansion and emergence of public TCRbeta clonotypes. J Exp Med. 2019;216(8):1857‐1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Macri C, Dumont C, Johnston AP, Mintern JD. Targeting dendritic cells: a promising strategy to improve vaccine effectiveness. Clin Transl Immunology. 2016;5(3):e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Mahnke K, Qian Y, Knop J, Enk AH. Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood. 2003;101(12):4862‐4869. [DOI] [PubMed] [Google Scholar]
- 196. Joffre OP, Sancho D, Zelenay S, Keller AM, Reis e Sousa C. Efficient and versatile manipulation of the peripheral CD4+ T‐cell compartment by antigen targeting to DNGR‐1/CLEC9A. Eur J Immunol. 2010;40(5):1255‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Kato YU, Zaid A, Davey GM, et al. Targeting antigen to Clec9A primes follicular Th cell memory responses capable of robust recall. J Immunol. 2015;195(3):1006‐1014. [DOI] [PubMed] [Google Scholar]
- 198. Lahoud MH, Ahmet F, Kitsoulis S, et al. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J Immunol. 2011;187(2):842‐850. [DOI] [PubMed] [Google Scholar]
- 199. Kato YU, Steiner TM, Park H‐Y, et al. Display of native antigen on cDC1 that have spatial access to both T and B cells underlies efficient humoral vaccination. J Immunol. 2020;205(7):1842‐1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Medaglia C, Giladi A, Stoler‐Barak L, et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA‐seq. Science. 2017;358(6370):1622‐1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Cohen M, Giladi A, Gorki A‐D, et al. Lung single‐cell signaling interaction map reveals basophil role in macrophage imprinting. Cell. 2018;175(4):1031‐1044 e1018. [DOI] [PubMed] [Google Scholar]
- 202. Armingol E, Officer A, Harismendy A, Lewis NE. Deciphering cell–cell interactions and communication from gene expression. Nat Rev Genet. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Giladi A, Cohen M, Medaglia C, et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat Biotechnol. 2020;38(5):629‐637. [DOI] [PubMed] [Google Scholar]
- 204. Pasqual G, Chudnovskiy A, Tas JMJ, et al. Monitoring T cell‐dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature. 2018;553(7689):496‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Liu Z, Li JP, Chen M, et al. Detecting tumor antigen‐specific T cells via interaction‐dependent fucosyl‐biotinylation. Cell. 2020;183:1117‐1133.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable – no new data generated, or the article describes entirely theoretical research