

Abstract
Imeglimin is an investigational first‐in‐class novel oral agent for the treatment of type 2 diabetes (T2D). Several pivotal phase III trials have been completed with evidence of statistically significant glucose lowering and a generally favourable safety and tolerability profile, including the lack of severe hypoglycaemia. Imeglimin's mechanism of action involves dual effects: (a) amplification of glucose‐stimulated insulin secretion (GSIS) and preservation of β‐cell mass; and (b) enhanced insulin action, including the potential for inhibition of hepatic glucose output and improvement in insulin signalling in both liver and skeletal muscle. At a cellular and molecular level, Imeglimin's underlying mechanism may involve correction of mitochondrial dysfunction, a common underlying element of T2D pathogenesis. It has been observed to rebalance respiratory chain activity (partial inhibition of Complex I and correction of deficient Complex III activity), resulting in reduced reactive oxygen species formation (decreasing oxidative stress) and prevention of mitochondrial permeability transition pore opening (implicated in preventing cell death). In islets derived from diseased rodents with T2D, Imeglimin also enhances glucose‐stimulated ATP generation and induces the synthesis of nicotinamide adenine dinucleotide (NAD+) via the ‘salvage pathway’. In addition to playing a key role as a mitochondrial co‐factor, NAD+ metabolites may contribute to the increase in GSIS (via enhanced Ca++ mobilization). Imeglimin has also been shown to preserve β‐cell mass in rodents with T2D. Overall, Imeglimin appears to target a key root cause of T2D: defective cellular energy metabolism. This potential mode of action is unique and has been shown to differ from that of other major therapeutic classes, including biguanides, sulphonylureas and glucagon‐like peptide‐1 receptor agonists.
Keywords: Imeglimin, mechanism, mitochondria, therapeutic, type 2 diabetes
1. INTRODUCTION
Imeglimin is the first in a new tetrahydrotriazine‐containing class of oral antidiabetic agents referred to as ‘glimins’. 1 Its discovery was enabled by an in vivo phenotypic screen (based on antihyperglycaemic activity in rodents), followed by additional chemical modification of a lead molecule (Figure 1 depicts the chemical structure). Imeglimin is under investigation, with three pivotal phase III clinical trials having been recently completed in Japan. Clinical experience with Imeglimin in Japanese and Caucasian patients with type 2 diabetes (T2D) to date has shown significant and durable antihyperglycaemic activity, generally favourable safety and tolerability, and a lack of severe hypoglycaemia in multiple trials, including combinations with metformin, dipeptidyl peptidase‐4 inhibitors, insulin and other classes. 1 , 2 , 3 , 4
FIGURE 1.
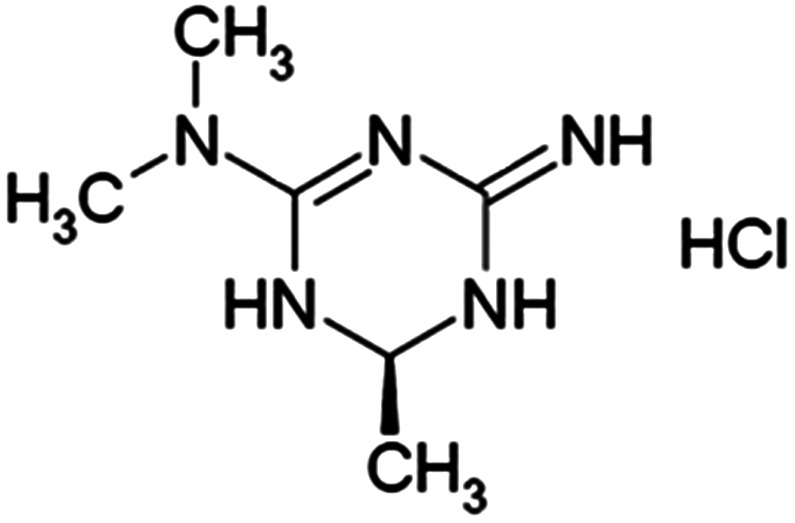
Chemical structure of Imeglimin hydrochloride salt
As described in this review, the mode of action of Imeglimin is unique and distinct compared with other major classes of therapeutic agents. It involves dual effects, both to enhance insulin action and to reverse pancreatic β‐cell dysfunction. 5 The mode of action of Imeglimin is generally well aligned with our current understanding of the pathophysiology of T2D. A genetic predisposition plus key environmental factors, principally overnutrition and reduced energy expenditure, are drivers of disease. 6 Consequently, defects in insulin action plus pancreatic islet β‐cell dysfunction develop and conspire to yield glucose intolerance and, subsequently, the onset of overt diabetes. 7 , 8 , 9
Insulin resistance occurs in several tissues including skeletal muscle 10 and in the liver, where the effect of insulin to suppress glucose production is attenuated. 11 Deficient β‐cell function is evident from a marked reduction in the acute insulin secretory response to glucose—both in vivo and in islets isolated from patients—and partial loss of the response to other stimuli including glucagon‐like peptide‐1 (GLP‐1). 8 , 12 , 13 , 14 A clear loss of β‐cell mass has also been documented in humans with T2D. 15 , 16
At the molecular level, mitochondrial dysfunction is a prominent feature of T2D pathology that contributes to both β‐cell defects 17 , 18 , 19 and insulin resistance. 20 , 21 , 22 , 23 The importance of mitochondrial function is underscored by the existence of rare inherited forms of T2D that result from mutations in mitochondrial DNA. 24 , 25 Mitochondrial dysfunction is manifested in several ways. Deficient oxidative metabolism and reduced ATP generation have been described in several tissues, along with more specific defects leading to reduced or incomplete fatty acid oxidation that are implicated as causes of cellular lipid accumulation. 23 , 26 Reduced mitochondrial content and structural damage are also reported. 23 Defects in function of the mitochondrial respiratory chain also cause excessive reactive oxygen species (ROS) formation, 23 which has a clearly causative role in T2D pathophysiology. 27 , 28 Nicotinamide adenine dinucleotide (NAD+) is a critical co‐factor that is required for mitochondrial function and several other core cellular functions, including cell survival. 29 , 30 , 31 Importantly, metabolic disorders, including obesity and T2D, are associated with altered NAD+ metabolism and generally reduced levels. 32 Moreover, exogenous nicotinamide, an NAD+ precursor, has been shown to augment islet β‐cell function. 33 , 34 , 35
As discussed below, Imeglimin's dual mechanism of action may result from more specific effects that target the above noted molecular aspects of diabetes pathophysiology. This includes the reversal of β‐cell dysfunction via amplification of glucose‐stimulated insulin secretion (GSIS) and augmentation of insulin action in liver and skeletal muscle. Underlying these effects are improvements in mitochondrial function that have been shown in multiple cell types. In addition, Imeglimin induces an increase in the cellular NAD+ pool (in islets) that has also been linked to Ca++ mobilization and enhanced GSIS.
There are several existing classes of therapeutic agents available today for the treatment of T2D. 36 , 37 However, these medicines are often only partially effective and may also confer safety risks or are poorly tolerated in some patients. 37 , 38 Therefore, there is an ongoing need for new therapies that target core aspects of pathophysiology and could potentially address persistent unmet medical needs. Imeglimin's unique mechanism of action is consistent with its known clinical profile and may support its potential inclusion in the future treatment paradigm.
2. REVERSAL OF PANCREATIC ISLET β‐CELL DYSFUNCTION
2.1. Clinical evidence
Direct evidence of increased GSIS in patients with T2D was obtained from a translational medicine study where the response to hyperglycaemia during a clamp procedure was substantially enhanced after 7 days of treatment with Imeglimin versus placebo. 39 The homeostatic model assessment (HOMA)‐β score, an index of β‐cell function, 40 was also increased versus placebo in a phase III monotherapy trial (Poxel, unpublished data). In addition, Imeglimin significantly reduced the proinsulin/insulin (or proinsulin/C‐peptide) ratio in phase II studies, 3 further suggesting an improvement in β‐cell function.
2.2. Improved glucose‐stimulated insulin secretion in animal models of T2D
A robust effect of Imeglimin to attenuate β‐cell dysfunction and to amplify GSIS in rodent models of T2D has been described in several studies. Imeglimin improved hyperglycaemia in both streptozotocin (STZ) diabetic rats and Goto‐Kakizaki (GK) rats, models that are characterized by a primary defect in β‐cell mass and function. 41 Increases in insulinogenic index during oral glucose tolerance tests were also noted in these models 41 and in additional studies performed in Zucker diabetic fatty (ZDF) rats. 42 As illustrated in the example depicted in Figure 2, Imeglimin treatment markedly potentiated in vivo GSIS in both lean and high‐fat–fed (HFF) rats 43 ; a similar effect was noted in high‐fat high‐sucrose diet (HFHSD) mice. 44 Increased in vivo GSIS was also evident in hyperglycaemic clamp studies performed in both non‐diabetic and STZ‐diabetic rats. 41
FIGURE 2.
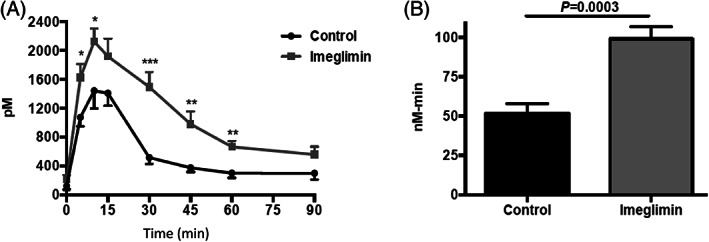
In vivo effect of Imeglimin (150 mg/kg) to enhance glucose‐stimulated insulin secretion (GSIS) in a rodent model of type 2 diabetes. A, Plasma insulin levels and B, insulin area under the curve (AUC) obtained during an oral glucose tolerance test in HFF rats are shown. Although there is a trend towards higher plasma insulin levels at the end of this experiment (90 minutes time point), glucose levels returned to baseline at this time point and no hypoglycaemia was observed. Additional details are described in Perry et al. 43 Copyright © 2016 The American Physiological Society
An acute and direct effect of Imeglimin to affect pancreatic islet β‐cell function has been shown in several contexts. Firstly, first phase glucose‐dependent insulin secretion was increased up to 6‐fold in an isolated perfused pancreas model derived from STZ‐diabetic rats. 41 Secondly, a direct effect to amplify insulin secretion in the presence of high glucose was observed using islets isolated from normal rats. 43 Thirdly, in more recent studies, we determined that in vitro incubation with Imeglimin could acutely, albeit partially (relative to non‐diabetic control animals), restore glucose‐responsive insulin secretion in isolated islets derived from diseased GK‐ and STZ‐diabetic rats, an effect that was also similar to GLP‐1 studied as an active comparator. 45 Note that the variant of the STZ‐diabetic rat used here has a small number of residual islet β‐cells, which allowed for an effect on GSIS to be seen. Importantly, no effect of Imeglimin on insulin secretion in the presence of low glucose was detected in these studies or in the clamp studies described above; by contrast, a sulphonylurea (tolbutamide) was shown to stimulate insulin release from GK rat islets incubated in low glucose in parallel experiments.
Therefore, multiple preclinical experiments involving both in vivo and in vitro approaches have revealed a consistent and strong effect of Imeglimin to improve pancreatic β‐cell function by amplifying insulin release in an exclusively glucose‐dependent fashion.
2.3. Improvement in islet β‐cell mass
In addition to direct effects to amplify GSIS, previous in vitro studies performed with isolated human or rat islets or cultured rat insulinoma (INS1) cells showed a protective effect of Imeglimin to reduce cell death in response to cytokines or high glucose incubation. 41 , 46 These effects suggested that Imeglimin might have longer term benefits to prevent the loss of functional β‐cell mass in T2D. To address this hypothesis, chronic effects of Imeglimin have also been characterized in male ZDF rats, an extreme model of obesity‐driven T2D associated with inadequate β‐mass and increased β‐cell death. 47 In this context, Imeglimin treatment for 5 weeks attenuated the decline in β‐cell loss normally seen in this model; this effect appeared to be caused by a modest increase in β‐cell proliferation and a decline in cell death from apoptosis. 42 These newer preclinical findings suggest the possibility of disease‐modifying benefits on islets in patients with T2D. Whether there might be some potential for greater durability than is usually the case with other oral therapies will require longer term clinical studies in the future.
3. ENHANCED INSULIN ACTION
3.1. Clinical evidence
In the TIMES 1 phase III monotherapy trial, Imeglimin treatment for 24 weeks produced a significant effect on the Quantitative Insulin Sensitivity Check Index (QUICKI), a measure of insulin sensitivity that correlates with results from glucose clamp studies. 48 Specifically, mean QUICKI values were increased by 0.0093 in Imeglimin versus placebo‐treated patients (P = .005) after 24 weeks (Poxel, unpublished data). A similar effect on the related Stumvoll Index—an alternative calculated estimate of insulin sensitivity 49 —was noted in an additional unpublished phase II trial. Future studies are being planned to more directly measure the effect of Imeglimin on insulin sensitivity in humans.
3.2. Functional effects in animal and cellular models
In addition to prominent effects to reverse β‐cell dysfunction, several lines of evidence indicate that Imeglimin can augment insulin action.
In a HFHSD mouse model of T2D, treatment with Imeglimin enhanced the glucose‐lowering effect of exogenous insulin. 44 An insulin‐sensitizing effect of the molecule in both liver and skeletal muscle was also reported; this was determined by measuring the degree of PKB (Akt) phosphorylation via Western blot—a key component of the insulin signalling pathway—in response to exogenous insulin. 44 In the same model, chronic treatment with Imeglimin also reduced hepatic steatosis (decreasing triglycerides, cholesterol and diacyglycerol), an effect that would predict improved hepatic insulin sensitivity. 44 After Imeglimin treatment for 45 days, increased in vivo 14C‐2‐deoxy glucose uptake into skeletal muscles of STZ‐diabetic rats was also observed 41 ; this effect is consistent with improved insulin sensitivity but was also probably influenced by improvements in insulin secretion. Using cultured (H‐2Kb) skeletal muscle cells, Imeglimin incubation was also shown to mediate an insulin‐like effect on glucose uptake in vitro. 41
Effects of Imeglimin to exert insulin‐like effects on the liver include dose‐dependent inhibition of glucose production using primary cultured rat (Wistar) hepatocytes; a similar effect that was also shown with in vitro incubation of liver slices derived from insulin‐resistant ZDF rats. 41 The above noted effect of Imeglimin to inhibit gluconeogenesis in isolated rat hepatocytes has also been replicated in additional experiments 50 and appears to be similar to that of metformin; however, as discussed below and summarized in Table 1, the underlying mechanisms for Imeglimin and metformin are distinct.
TABLE 1.
Imeglimin's mode of action is distinct versus metformin
| Imeglimin | Metformin |
|---|---|
| In vivo (clinical) | |
| ↑Glucose‐stimulated insulin secretion (hyperglycaemic clamp) 39 | No reported effect on insulin secretion 39 , 71 |
| Evidence of insulin sensitivity ‐ QUICKI a , Stumvoll a | No clear increase in insulin sensitivity 71 , 72 |
| In vivo (preclinical) | |
| ↑Glucose‐stimulated insulin secretion (GTT) 44 ; hyperglycaemic clamp 41 , 43 | No effect on insulin secretion 71 |
| ↑Glucose disposal; ↑insulin sensitivity (Figure 3); ↑insulin signalling 41 , 44 | ± Insulin sensitization 71 , 72 |
| Cell and organ | |
| ↑Glucose‐stimulated insulin secretion (islets/perfused pancreas) 41 , 43 , 45 | No effect on glucose‐stimulated insulin secretion 71 , 73 |
| Islet β‐cell protection; preserved β‐cell mass 41 , 42 | In vitro β‐cell protection 74 , 75 ; no known in vivo effects on β‐cell mass 71 |
| ↑Muscle glucose uptake 41 | ± ↑Muscle glucose uptake 71 |
| ↓Gluconeogenesis (hepatocytes) 41 | ↓Gluconeogenesis (hepatocytes) 71 |
| Intracellular | |
| Competitive/partial mitochondrial Complex I inhibition; no decrease in mitochondrial respiration; decreased ROS b formation 44 , 51 , 55 | Uncompetitive mitochondrial Complex I inhibition; decreased respiration 55 , 71 ; decreased ROS formation 76 , 77 |
| No effect on mitochondrial glycerophosphate a | ↓Mitochondrial glycerophosphate dehydrogenase 58 |
| Increased NAD+ synthesis; potentially via NAMPT c ; increased glucose‐responsive intracellular Ca++ 45 | No increase in Ca++; 73 no effect on NAD+ reported 3 |
Poxel, unpublished data.
Reactive oxygen species.
Nicotinamide phosphoribosyltransferase.
Abbreviations: GTT, glucose tolerance test; NAD+, nicotinamide adenine dinucleotide; NAMPT, nicotinamide phosphoribosyltransferase; ROS, reactive oxygen species.
In additional experiments, an euglycaemic hyperinsulinaemic clamp was performed to assess the effects of Imeglimin treatment on insulin sensitivity (Figure 3). After 2 weeks’ treatment of STZ‐diabetic rats, the total glucose infusion rate required to maintain euglycaemia was significantly increased (+215%; P < .01), indicating a substantial improvement in whole body insulin sensitivity. Basal endogenous glucose production was not significantly affected; however, in the presence of hyperinsulinaemia, Imeglimin‐treated rats showed a significant decrease in hepatic glucose production compared with controls (−40%; P < .05). By contrast, Perry et al. did not observe significant effects of Imeglimin on whole body or hepatic insulin sensitivity in HFF rats after 2 weeks of treatment with Imeglimin. 43 The reason(s) for this discrepancy are unknown but may involve several factors. In addition to strain (Wistar vs. Sprague‐Dawley) and model differences, there are possible differences in drug exposure because of different modes of administration (oral gavage vs. admixture with food). It should also be noted that HFF and STZ rats were clamped at different levels of glucose. In addition, longer treatment periods may be needed to produce clear effects on insulin sensitivity because improvements in HFHSD mice with insulin resistance were observed after 6 weeks of treatment. 44
FIGURE 3.
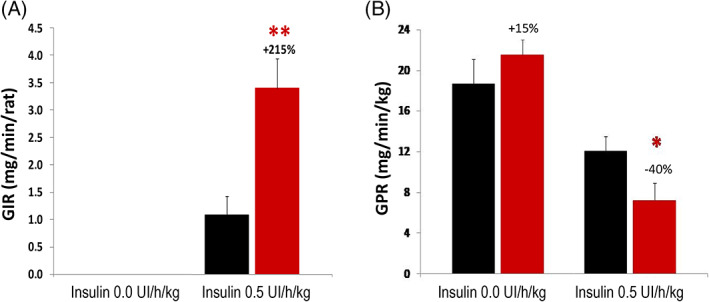
Imeglimin improves insulin sensitivity in vivo. Male Wistar rats (age 40‐42 weeks, weighing 200‐225 g) were treated with streptozotocin (STZ) (50 mg/kg in citrate buffer via intraperitoneal injection) to produce mild diabetes (mean basal fasting glucose 5‐6 mM); after 1 week, animals were randomized into two groups based on their fasting glucose levels. Beginning 10 days post‐STZ, animals were treated with Imeglimin (150 mg/kg BID; red bars ) or vehicle (0.5% methylcellulose; black bars
) or vehicle (0.5% methylcellulose; black bars  ) for 15 days. An euglycaemic hyperinsulinaemic clamp was then conducted, beginning 45 minutes after the last administration of Imeglimin or vehicle in overnight fasted rats (15 hours). Plasma insulin was increased to a constant level via primed, continuous infusion of exogenous insulin (0.5 UI/kg/h); plasma glucose was maintained at a constant euglycaemic level by varying the infusion of exogenous glucose. Two key variables were assessed during the clamp: A, The steady state glucose infusion rate (GIR) was measured as an index of whole body insulin sensitivity. B, [3‐3H]‐glucose was infused to assess endogenous glucose production (glucose production rate [GPR]). Both basal and hyperinsulinaemic conditions were studied. *P < .05; **P < .01 vs. vehicle control (Student t‐test); n = 10 rats per group
) for 15 days. An euglycaemic hyperinsulinaemic clamp was then conducted, beginning 45 minutes after the last administration of Imeglimin or vehicle in overnight fasted rats (15 hours). Plasma insulin was increased to a constant level via primed, continuous infusion of exogenous insulin (0.5 UI/kg/h); plasma glucose was maintained at a constant euglycaemic level by varying the infusion of exogenous glucose. Two key variables were assessed during the clamp: A, The steady state glucose infusion rate (GIR) was measured as an index of whole body insulin sensitivity. B, [3‐3H]‐glucose was infused to assess endogenous glucose production (glucose production rate [GPR]). Both basal and hyperinsulinaemic conditions were studied. *P < .05; **P < .01 vs. vehicle control (Student t‐test); n = 10 rats per group
On balance, the available data indicate that Imeglimin enhances insulin action in vivo. Direct effects to suppress gluconeogenesis in hepatocytes and promote glucose uptake in skeletal muscle cells have also been described.
4. MOLECULAR MECHANISMS
4.1. Improved mitochondrial function
Given the effects of Imeglimin on multiple organs and cell types, it is not surprising that effects related to mitochondrial dysfunction—which involves multiple tissues in T2D 17 , 20 , 50 —might underlie the pleiotropic beneficial phenotypic changes imparted by Imeglimin treatment. As depicted in Figure 4, there are several documented effects of Imeglimin leading to modulation of mitochondrial function and to potentially favourable downstream sequelae.
FIGURE 4.
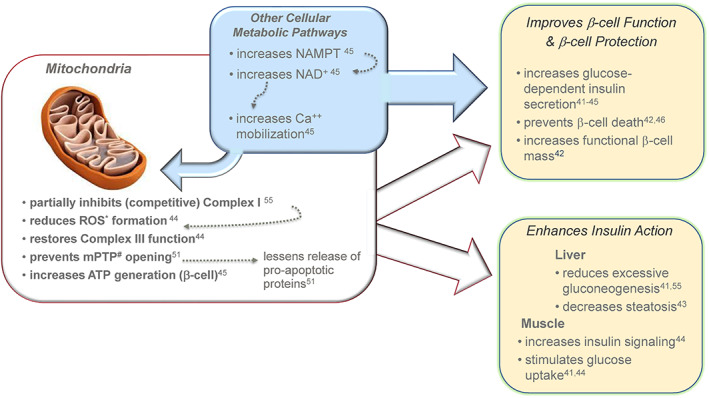
Summary of Imeglimin's dual mode of action. Imeglimin has direct effects on islet β‐cells to enhance glucose‐stimulated insulin secretion (GSIS) (and to potentially prevent loss of β‐cell mass); enhanced insulin action occurs in both liver and skeletal muscle. Underlying cellular effects include modulation of mitochondrial function and an additional effect in islet β‐cells to enhance generation of NAD+ that contributes to Ca++ mobilization in the insulin secretion amplification pathway. *reactive oxygen species; #permeability transition pore;  Nicotinamide Phosphoribosyl‐transferase;
Nicotinamide Phosphoribosyl‐transferase;  Nicotinamide Adenine Dinucleotide (NAD+)
Nicotinamide Adenine Dinucleotide (NAD+)
In diseased pancreatic islets, Imeglimin enhances ATP generation and increases the ATP/ADP ratio implicating a net improvement in mitochondrial function. 45 In addition, the effect of Imeglimin to amplify GSIS is maintained when obligate mitochondrial fuels such as leucine or succinate are employed. 43 As further described below, Imeglimin also promotes the synthesis of NAD+ in diabetic GK rat islets. 45 In this context there was no change in cellular NADH content but the NAD/NADH ratio was significantly increased by 30%‐31%. 45 Despite the lack of a net increase in total islet NADH, an expansion of the NAD+ pool suggests the potential for an increased supply of reducing equivalents needed to fuel oxidative phosphorylation via the mitochondrial electron transport chain. 29
Improvements in respiration were measured using mitochondria isolated from liver tissue of diet‐induced diabetic mice after Imeglimin treatment. 44 More specifically, Imeglimin restored deficient Complex III activity while partially inhibiting Complex I. This rebalancing effect was associated with a substantial decrease in excess disease‐associated ROS formation, as measured by H2O2 production with succinate as a substrate. Taken together, a decrease in ROS generated from reverse electron flux via Complex I could potentially be implicated by these results. Potentially beneficial effects of Imeglimin on components of mitochondrial structure (e.g. cardiolipin content) were also documented in this study. 44 Using human endothelial cells (HMEC‐1), the effect of Imeglimin to suppress ROS formation via decreasing reverse electron transport through Complex I was replicated; and no net reduction in cellular oxygen consumption was noted. 51
Elevated ROS production is known to enhance opening of the mitochondrial permeability transition pore (PTP). This occurs in a variety of pathological states and can result in the release of pro‐apoptotic proteins into the cytosol, resulting in cell death. 52 , 53 In HMEC‐1 cells, Imeglimin effectively prevented PTP opening and this was associated with reduced cell death. 51 This finding raises an intriguing possibility that the aforementioned effects of Imeglimin to deter cell death in cultured islet β‐cells 41 might be attributable to the effect on PTP opening; however, as these are very different cell types, further studies would be needed to confirm this hypothesis.
Finally, the effect of Imeglimin to inhibit gluconeogenesis in hepatocytes—and potentially its effect to also suppress hepatic glucose output in vivo—can be tentatively linked to partial inhibition of respiratory chain Complex I. Metformin is also known to inhibit gluconeogenesis via mild inhibition of Complex I. 54 In isolated rat hepatocytes, both Imeglimin and metformin inhibit gluconeogenesis; however, their effects on Complex I appear to be divergent. By measuring the affinity of NADH for the respiratory chain in permeabilized hepatocytes, Vial et al. recently observed 55 that Imeglimin decreased the affinity of NADH for the respiratory chain but did not affect its Vmax (competitive inhibition), whereas metformin decreased both the Vmax and the affinity (uncompetitive inhibition). Given that Imeglimin produces mild, competitive Complex I inhibition without affecting overall cellular oxygen consumption, the authors speculated that it induces a kinetic constraint on the respiratory chain that does not affect its maximal activity but which could be sufficient to affect gluconeogenesis in hepatocytes. By contrast, metformin is an uncompetitive inhibitor of the respiratory chain and significantly reduces the cell's oxygen consumption rate. 55 These data are also consistent with other results obtained using HMEC‐1 cells where metformin, but not Imeglimin, produced a modest decrease in cellular oxygen consumption. 51
The above noted findings raise some important questions that remain to be answered in the future. What molecular mechanism(s) might account for the observed effects on Complex I (or Complex III)? Are there tissue‐selective differences in the effects of Imeglimin on mitochondrial function? If ATP levels are suppressed in hepatocytes, why were they observed to increase in islets? In hepatocytes, high concentrations (100‐1000 μM) were studied; in islets, effects of Imeglimin were observed at 25‐100 μM. In addition, no detailed characterization of the effects of Imeglimin on mitochondria was performed in islets (or cell types other than hepatocytes and HMEC‐1 cells). In addition, the increase in cellular NAD+ has to date only been observed in islets. The effect of Imeglimin to increase Complex III activity also suggests that net effects on ATP synthesis may relate to the degree of baseline dysfunction in different components of the respiratory chain (i.e. whether there is an underlying imbalance in Complex I vs. Complex III that could be restored towards normal). The extent to which Imeglimin's effects on ROS also translate to islets or to in vivo situations also remains to be established. Most importantly, a direct link between Imeglimin's effects on mitochondrial function and in vivo improvements in GSIS or insulin action have yet to be elucidated.
Increased plasma lactate and even lactic acidosis can occur rarely with metformin overdose or in the setting of renal failure. 56 , 57 Because inhibition of Complex I may contribute to this risk, 57 it is important to consider whether Imeglimin might lead to increases in lactate. Interestingly, metformin has also been shown to inhibit mitochondrial glycerophosphate dehydrogenase (mGPD), an effect that could also lead to both inhibition of gluconeogenesis and lactate accumulation. 58 We recently determined that Imeglimin had no effect to inhibit rat mGPD in comparison with metformin, which performed as expected. Furthermore, additional in vivo studies were conducted in two animal models to show that metformin, but not Imeglimin, could provoke substantial elevations of plasma lactate (Poxel, unpublished data).
Although there are some similarities in their respective chemical structures, most of the in vivo and in vitro effects of Imeglimin are distinctly different from metformin, based both on head‐to‐head experiments and a review of the available literature (Table 1). Both metformin and Imeglimin have related effects to inhibit Complex I; however, the specific mechanisms are divergent. Therefore, available evidence strongly suggests that Imeglimin does not provoke increases in lactate and has a lower potential risk of lactic acidosis compared with that associated with metformin.
4.2. Potential role of other pathways in improvement of β‐cell function
As mentioned above, Imeglimin increases the cellular pool of NAD+ in isolated islets from GK rats. Additional experiments suggested that Imeglimin increases NAD+ synthesis by inducing the expression and levels of nicotinamide phosphoribosyltransferase (NAMPT), a key enzyme in the ‘salvage’ synthesis pathway. 45 In addition to its role to enhance mitochondrial function, 59 NAD+ is metabolized by CD38 60 to generate a second messenger, cyclic ADP‐ribose (cADPR), which is implicated in enhancing Ca++ mobilization from intracellular stores via its interaction with the ryanodine receptor (RyR). 29 , 30 , 61 Increases in intracellular Ca++ are required for GSIS 62 and potentiated by Imeglimin as well as other stimuli, including incretin hormones. 45 To further interrogate the possible link between Imeglimin's effects to increase NAD+ and promote Ca++ mobilization, additional experiments were conducted. 45 Partial knockdown (siRNA) of CD38 appeared to block Imeglimin's effects on GSIS; excess ryanodine was also used to block RyR, resulting in a reduced ability of Imeglimin to augment GSIS. Taken together, these results suggested that Imeglimin's effects on GSIS are potentially mediated via this pathway. 45 Although increased NAD+ synthesis may contribute to enhancement of GSIS, it is improbable that this pathway accounts for other effects of Imeglimin (e.g. insulin sensitization in other cells or tissues). Moreover, possible effects of Imeglimin on NAD+ (or NAD+ metabolism and actions) in other tissues have yet to be interrogated.
Importantly, Imeglimin's effects on GSIS were also shown to be diazoxide‐resistant, whereas sulphonylurea action was fully inhibited by diazoxide as expected. 45 Thus, the predominant action of Imeglimin in islets appears to be independent from the classical ‘triggering’ pathway involving K+‐ATP channel closure. 63 There are clear similarities between Imeglimin's effect to amplify GSIS and the effects of GLP‐1, which are also diazoxide‐resistant 64 ; however, Imeglimin had no effect to increase cAMP, which is an obligate mediator of incretin action. 45
Overall, the molecular basis for Imeglimin's strong effect to improve β‐cell function appears to be consistent with its ability to modulate mitochondrial function and to also increase Ca++ mobilization, which can be tentatively linked to the synthesis and metabolism of NAD+. Although increased mitochondrial respiration might be expected to trigger insulin release via K+‐ATP channel closure, it is well known that additional anaplerotic mitochondrial metabolic pathways can result in K+‐ATP–independent GSIS amplification. 62 , 65
In addition to the potential impact of this acute mechanism on β‐cell function, it is interesting to also speculate that the aforementioned effects of Imeglimin on β‐cell protection might also be driven, in part, by increases in cellular NAD+ levels. Indeed, NAD+ depletion is known to potentiate apoptotic cell death, 66 whereas exogenous NAD+ is reportedly cytoprotective. 31 , 67
5. ADDITIONAL EFFECTS AND POTENTIAL BENEFITS
In light of evidence showing that Imeglimin modulates mitochondrial function, reduces ROS and protects cells from death in response to insults that include cytokines or hyperglycaemia, the potential for organ protection may extend to other tissues that are damaged in the context of diabetes. Using a Zucker fa/fa rat model of metabolic syndrome, Lachaux et al. recently described an effect of Imeglimin treatment to ameliorate cardiac dysfunction. 68 Specific improvements included decreased left ventricle (LV) end‐diastolic pressure and increased LV tissue perfusion; moreover, these potential benefits were associated with a parallel reduction in LV ROS production. In the same experiment, Imeglimin was also shown to increase coronary artery endothelium‐dependent relaxation. Importantly, 90 days of Imeglimin treatment in this model partially normalized elevated albuminuria, from a mean of 385 in untreated rats to 251 mg/24 hours (P < .05); the mean value in lean control rats was 108 mg/24 hours. This apparent renal benefit was also associated with a significant decrease in kidney interstitial fibrosis; however, tubular injury and interstitial inflammation were not significantly ameliorated. 68 Thus, Imeglimin may have additional potential to address important complications of diabetes, including cardiac dysfunction and nephropathy; additional clinical studies are needed to assess the extent to which these findings may translate to benefits for patients. Regarding cardiovascular safety, no adverse effects have been observed to date and recent clinical data also indicate a lack of QT prolongation (or other ECG abnormalities) with Imeglimin treatment. 69
6. CONCLUSIONS
Imeglimin is a first‐in‐class novel oral agent designed to target key components of T2D pathophysiology. Its mechanism of action (Figure 4) involves dual effects to improve pancreatic β‐cell function and to enhance insulin action in key tissues, including liver and skeletal muscle. At the cellular molecular level, the molecule modulates mitochondrial function, as documented in several cell types, leading to improvements in cellular energy metabolism and protection from cell death associated with excess ROS formation or other insults. An additional effect via increased NAD+ synthesis and enhanced Ca++ mobilization is operative in islet β‐cells.
Importantly, Imeglimin's mechanism is distinct from existing therapies used in the treatment of T2D (Table 2). Like GLP‐1 receptor agonists, Imeglimin amplifies insulin secretion in an exclusively glucose‐dependent fashion; however, cellular pathways employed by incretins versus Imeglimin are divergent. Sulphonylureas and glinides function to release insulin via K+‐ATP closure in a glucose‐independent way that clearly differs from Imeglimin. In comparison with metformin, Imeglimin's effects are also quite distinct, as described above and in Table 1. Other major classes such as sodium‐glucose co‐transporter‐2 inhibitors, Peroxisome proliferator‐activated receptor gamma (PPARγ) agonists (thiazolidinediones) and α‐glucosidase inhibitors, mediate glucose lowering via pathways that appear not to overlap with Imeglimin's actions. Given these mechanistic distinctions, Imeglimin may have complementary effects when added to existing therapeutics, as has been previously reported with sitagliptin 2 and with metformin. 3 In addition, the pharmacokinetic properties of Imeglimin appear to generally support combination therapy use. The molecule is well absorbed with a Tmax of 4 hours and an elimination t1/2 of 5‐6 hours; it is largely cleared via the kidney but there are no clinically meaningful interactions with either metformin or sitagliptin. 70
TABLE 2.
Comparison of Imeglimin with other major therapeutic classes
| Therapeutic class | Clinical HbA1c‐lowering effect b | Other key attributes or liabilities | Mechanism(s) of action |
|---|---|---|---|
| Biguanides (metformin) | −0.7% to 1.2% 78 | Possible risk of lactic acidosis; GI adverse effects 78 | See Table 1 |
| Sulphonylureas, Glinides | ≈−1.0% with potential loss of effect over time 79 | Hypoglycaemia; weight gain; increased risk of CV mortality 78 | K+‐ATP subunit (SUR) binding → channel closure → glucose‐independent insulin secretion 63 |
| Sodium‐glucose co‐transporter‐2 inhibitors | −0.6% to 0.9% 80 | Renal and CV benefits; mild weight loss; reduced glycaemic efficacy with renal insufficiency; volume depletion; genito‐urinary infections | Inhibits renal glucose reabsorption; secondary ↑ insulin sensitivity 81 |
| α‐Glucosidase inhibitors | −0.44% to 1.0% (−0.78% at 100 mg TID) 82 | GI adverse effects (up to 74%); modest weight loss 82 | Inhibits intestinal carbohydrate digestion 83 |
| GLP‐1 receptor agonists (incretin mimetics) a | −0.8% to 1.4% 84 , 85 | Weight loss; CV risk reduction 84 , 85 | cAMP signalling → increase GSIS; decrease gastric emptying; other 86 |
| DPP4 inhibitors | −0.6% to 0.8% 87 | Potential loss of efficacy after 9‐12 mo | Stabilize and increase incretin levels leading to ↑ GSIS 88 |
| Thiazolidinediones | −1.0% to 1.6% 89 | Weight gain; oedema; increased bone fracture risk | PPARγ agonists → insulin sensitization 90 |
| Imeglimin | −0.94% to 1.0% 4 c | ‐ | Modulate mitochondrial function; increase ATP and NAD+ synthesis (islets) → increase GSIS; augment insulin action |
HbA1c data reported for dulaglutide and semaglutide (weekly injection).
Data are reported as difference from placebo in 12‐26 week clinical trials (as monotherapy or add‐on to metformin in most cases).
Poxel, unpublished data.
Abbreviations: CV, cardiovascular; DPP4, dipeptidyl peptidase‐4; GI, gastrointestinal; GLP‐1, glucagon‐like peptide‐1; GSIS, glucose‐stimulated insulin secretion; NAD+, nicotinamide adenine dinucleotide; PPARγ, Peroxisome proliferator‐activated receptor gamma.
Given its tolerability profile and consistent glucose‐lowering efficacy in a range of clinical trials, 4 Imeglimin may be suitable for use in certain difficult‐to‐treat patients, such as the elderly or patients with renal insufficiency. Although Imeglimin could be useful in a broad range of patients with T2D, there may also be situations where it should not be used. Patients who are well managed with other agents or who are in need of additional effects (e.g. weight loss) that other classes can provide may not be optimal candidates. Occasional patients may also experience gastrointestinal symptoms (such as diarrhoea) although the frequency of such effects is substantially lower than observed with metformin 1 ; thus, patients experiencing such symptoms may prefer alternative regimens. In addition, Imeglimin has not been adequately characterized in selected patient populations, including paediatric T2D or hepatic insufficiency.
The unique mechanism of action of this new investigational agent is consistent with existing clinical data and suggests several key features: there are no apparent risks of severe hypoglycaemia and the risk of lactic acidosis appears to be very low; no mechanism‐based adverse effects are evident; longer term effects relating to durability and disease modification in islets are possible; finally, direct organ protection in heart and kidney have also been suggested as possible effects.
CONFLICT OF INTEREST
S.H.B., P.F., S.B. and D.E.M. are employees of Poxel SA and own stock in the company. M.K. and E.F. have nothing to declare. G.V. owns Poxel SA stock. A.L.B. has nothing to declare regarding the present work.
AUTHOR CONTRIBUTIONS
S.H.B., P.F., A.L.B., G.V., E.F. and S.B. designed and implemented and/or supervised experiments described herein. D.E.M. and all the other authors engaged in data analysis and interpretation. All the named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14277.
ACKNOWLEDGEMENTS
We thank all of our scientific advisors and collaborators for their outstanding efforts and support.
This work was funded by Poxel SA.
Hallakou‐Bozec S, Vial G, Kergoat M, et al. Mechanism of action of Imeglimin: A novel therapeutic agent for type 2 diabetes. Diabetes Obes Metab. 2021;23:664–673. 10.1111/dom.14277
Funding information This work was funded by Poxel SA.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request
REFERENCES
- 1. Pirags V, Lebovitz H, Fouqueray P. Imeglimin, a novel glimin oral antidiabetic, exhibits a good efficacy and safety profile in type 2 diabetic patients. Diabetes Obes Metab. 2012;14:852‐858. [DOI] [PubMed] [Google Scholar]
- 2. Fouqueray P, Pirags V, Diamant M, et al. The efficacy and safety of imeglimin as add‐on therapy in patients with type 2 diabetes inadequately controlled with sitagliptin monotherapy. Diabetes Care. 2014;37:1924‐1930. [DOI] [PubMed] [Google Scholar]
- 3. Fouqueray P, Pirags V, Inzucchi SE, et al. The efficacy and safety of imeglimin as add‐on therapy in patients with type 2 diabetes inadequately controlled with metformin monotherapy. Diabetes Care. 2013;36:565‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crabtree TSJ, DeFronzo RA, Ryder REJ, Bailey CJ. Imeglimin, a novel, first in‐class, blood glucose‐lowering agent: a systematic review and meta‐analysis of clinical evidence. Brit J Diabetes. 2020;20:28‐31. [Google Scholar]
- 5. Yaribeygi H, Maleki M, Sathyapalan T, Jamialahmadi T, Sahebkar A. Molecular mechanisms by which Imeglimin improves glucose homeostasis. J Diabetes Res. 2020;2020:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herder C, Roden M. Genetics of type 2 diabetes: pathophysiologic and clinical relevance. Eur J Clin Invest. 2011;41:679‐692. [DOI] [PubMed] [Google Scholar]
- 7. Moller DE. Insulin Resistance. London, UK: John Wiley and Sons, Ltd.; 1993. [Google Scholar]
- 8. Kasuga M. Insulin resistance and pancreatic β cell failure. J Clin Invest. 2006;116:1756‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383:1068‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeFronzo RA, Simonson D, Ferrannini E. Hepatic and peripheral insulin resistance: a common feature of type 2 (non‐insulin‐dependent) and type 1 (insulin‐dependent) diabetes mellitus. Diabetologia. 1982;23:313‐319. [DOI] [PubMed] [Google Scholar]
- 11. DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non‐insulin‐dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism. 1989;38:387‐395. [DOI] [PubMed] [Google Scholar]
- 12. Kahn SE, Zraika S, Utzschneider KM, Hull RL. The β cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia. 2009;52:1003‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng S, Vatamaniuk M, Huang X, et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. 2004;53:624‐632. [DOI] [PubMed] [Google Scholar]
- 14. Del Guerra S, Lupi R, Marselli L, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727‐735. [DOI] [PubMed] [Google Scholar]
- 15. Butler AE, Janson J, Bonner‐Weir S, Ritzel R, Rizza RA, Butler PC. Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102‐110. [DOI] [PubMed] [Google Scholar]
- 16. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic β‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(suppl 4):32‐42. [DOI] [PubMed] [Google Scholar]
- 17. Anello M, Lupi R, Spampinato D, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. 2005;48:282‐289. [DOI] [PubMed] [Google Scholar]
- 18. Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and beta‐cell failure in type 2 diabetes mellitus. Exp Diabetes Res. 2012;2012:703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haythorne E, Rohm M, van de Bunt M, et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β‐cells. Nat Commun. 2019;10:2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384‐387. [DOI] [PubMed] [Google Scholar]
- 21. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gonzalez‐Franquesa A, Patti ME. Insulin resistance and mitochondrial dysfunction. Adv Exp Med Biol. 2017;982:465‐520. [DOI] [PubMed] [Google Scholar]
- 23. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98:2133‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maassen JA, 'T Hart LM, Van Essen E, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53(suppl 1):S103‐S109. [DOI] [PubMed] [Google Scholar]
- 25. Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ‐based analysis. Am J Physiol Endocrinol Metab. 2019;316:E268‐E285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159:1253‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944‐948. [DOI] [PubMed] [Google Scholar]
- 28. Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katsyuba E, Romani M, Hofer D, Auwerx J. NAD+ homeostasis in health and disease. Nat Metab. 2020;2:9‐31. [DOI] [PubMed] [Google Scholar]
- 31. Yang H, Yang T, Baur JA, et al. Nutrient‐sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okabe K, Yaku K, Tobe K, Nakagawa T. Implications of altered NAD metabolism in metabolic disorders. J Biomed Sci. 2019;26:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otonkoski T, Beattie GM, Mally MI, Ricordi C, Hayek A. Nicotinamide is a potent inducer of endocrine differentiation in cultured human fetal pancreatic cells. J Clin Invest. 1993;92:1459‐1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zawalich WS, Dye ES, Matschinsky FM. Nicotinamide modulation of rat pancreatic islet cell responsiveness in vitro. Horm Metab Res. 1979;11:469‐471. [DOI] [PubMed] [Google Scholar]
- 35. Deery DJ, Taylor KW. Effects of azaserine and nicotinamide on insulin release and nicotinamide‐adenine dinucleotide metabolism in isolated rat islets of Langerhans. Biochem J. 1973;134:557‐563.16742817 [Google Scholar]
- 36. Buse JB, Wexler DJ, Tsapas A, et al. 2019 update to: management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of diabetes (EASD). Diabetologia. 2020;63:221‐228. [DOI] [PubMed] [Google Scholar]
- 37. Clapham JC. Sixty years of drug discovery for type 2 diabetes: where are we now? Methods Mol Biol. 2020;2076:1‐30. [DOI] [PubMed] [Google Scholar]
- 38. Araki E, Haneda M, Kasuga M, et al. New glycemic targets for patients with diabetes from the Japan diabetes society. J Diabetes Investig. 2017;8:123‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pacini G, Mari A, Fouqueray P, Bolze S, Roden M. Imeglimin increases glucose‐dependent insulin secretion and improves beta‐cell function in patients with type 2 diabetes. Diabetes Obes Metab. 2015;17:541‐545. [DOI] [PubMed] [Google Scholar]
- 40. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412‐419. [DOI] [PubMed] [Google Scholar]
- 41. Fouqueray P, Leverve X, Fontaine E, et al. Imeglimin ‐ a new oral anti‐diabetic that targets the three key defects of type 2 diabetes. J Diabetes Metab. 2011;2(4). [Google Scholar]
- 42. Hallakou‐Bozec S, Kergoat M, Moller DE, Bolze S. Imeglimin preserves islet β‐cell mass in type 2 diabetic ZDF rats. Endocrinol Diabetes Metab. 2020:e00193. 10.1002/edm2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Perry RJ, Cardone RL, Petersen MC, et al. Imeglimin lowers glucose primarily by amplifying glucose‐stimulated insulin secretion in high‐fat‐fed rodents. Am J Physiol Endocrinol Metab. 2016;311:E461‐E470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vial G, Chauvin MA, Bendridi N, et al. Imeglimin normalizes glucose tolerance and insulin sensitivity and improves mitochondrial function in liver of a high‐fat, high‐sucrose diet mice model. Diabetes. 2015;64:2254‐2264. [DOI] [PubMed] [Google Scholar]
- 45. Hallakou‐Bozec S, Kergoat M, Fouqueray P, Bolze S, Moller DE. Imeglimin amplifies glucose‐stimulated insulin release from diabetic islets via a distinct mechanism of action. PLOS ONE 2020. 10.1101/2020.10.20.346841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J, Shirakawa J, Togashi Y, et al. Imeglimin Modulated ER Stress to Prevent ß‐Cell Apoptosis Induced by High Glucose or Thapsigargin. San Francisco, CA: American Diabetes Association Annual Scientific Sessions; 2019. [Google Scholar]
- 47. Pick A, Clark J, Kubstrup C, et al. Role of apoptosis in failure of beta‐cell mass compensation for insulin resistance and beta‐cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998;47:358‐364. [DOI] [PubMed] [Google Scholar]
- 48. Katz A, Nambi SS, Mather K, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402‐2410. [DOI] [PubMed] [Google Scholar]
- 49. Stumvoll M, Van Haeften T, Fritsche A, Gerich J. Oral glucose tolerance test indexes for insulin sensitivity and secretion based on various availabilities of sampling times. Diabetes Care. 2001;24:796‐797. [DOI] [PubMed] [Google Scholar]
- 50. Vial G, Dubouchaud H, Leverve XM. Liver mitochondria and insulin resistance. Acta Biochim Pol. 2010;57:389‐392. [PubMed] [Google Scholar]
- 51. Detaille D, Vial G, Borel AL, et al. Imeglimin prevents human endothelial cell death by inhibiting mitochondrial permeability transition without inhibiting mitochondrial respiration. Cell Death Discov. 2016;2:15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626‐629. [DOI] [PubMed] [Google Scholar]
- 53. Javadov S, Karmazyn M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol Biochem. 2007;20:1‐22. [DOI] [PubMed] [Google Scholar]
- 54. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti‐diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(pt 3)):607‐614. [PMC free article] [PubMed] [Google Scholar]
- 55. Vial G, Lamarche F, Cottet C, et al. The mechanism by which imeglimin inhibits gluconeogenesis in rat liver cells. Endocrinol Diabetes Metab. 2020; in press. 10.1002/edm2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dell'Aglio DM, Perino LJ, Kazzi Z, Abramson J, Schwartz MD, Morgan BW. Acute metformin overdose: examining serum pH, lactate level, and metformin concentrations in survivors versus nonsurvivors: a systematic review of the literature. Ann Emerg Med. 2009;54:818‐823. [DOI] [PubMed] [Google Scholar]
- 57. DeFronzo R, Fleming GA, Chen K, Bicsak TA. Metformin‐associated lactic acidosis: current perspectives on causes and risk. Metabolism. 2016;65:20‐29. [DOI] [PubMed] [Google Scholar]
- 58. Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Katsyuba E, Mottis A, Zietak M, et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature. 2018;563:354‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kato I, Takasawa S, Akabane A, et al. Regulatory role of CD38 (ADP‐ribosyl cyclase/cyclic ADP‐ribose hydrolase) in insulin secretion by glucose in pancreatic beta cells. Enhanced insulin secretion in CD38‐expressing transgenic mice. J Biol Chem. 1995;270:30045‐30050. [DOI] [PubMed] [Google Scholar]
- 61. Takasawa S, Nata K, Yonekura H, Okamoto H. Cyclic ADP‐ribose in insulin secretion from pancreatic beta cells. Science. 1993;259:370‐373. [DOI] [PubMed] [Google Scholar]
- 62. Newgard CB. Mechanisms controlling pancreatic islet alpha and beta cell function. Nat Rev Mol Cell Biol. 2020; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51(suppl 3):S368‐S376. [DOI] [PubMed] [Google Scholar]
- 64. Yajima H, Komatsu M, Schermerhorn T, et al. cAMP enhances insulin secretion by an action on the ATP‐sensitive K+ channel‐independent pathway of glucose signaling in rat pancreatic islets. Diabetes. 1999;48:1006‐1012. [DOI] [PubMed] [Google Scholar]
- 65. Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB. Metabolic cycling in control of glucose‐stimulated insulin secretion. Am J Physiol Endocrinol Metab. 2008;295:E1287‐E1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Takeuchi M, Yamamoto T. Apoptosis induced by NAD depletion is inhibited by KN‐93 in a CaMKII‐independent manner. Exp Cell Res. 2015;335:62‐67. [DOI] [PubMed] [Google Scholar]
- 67. Pittelli M, Felici R, Pitozzi V, et al. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol. 2011;80:1136‐1146. [DOI] [PubMed] [Google Scholar]
- 68. Lachaux M, Soulié M, Hamzaoui M, et al. Short‐and long‐term administration of imeglimin counters cardiorenal dysfunction in a rat model of metabolic syndrome. Endocrinol Diabetes Metab. 2020;3:e00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dubourg J, Perrimond‐Dauchy S, Felices M, Bolze S, Voiriot P, Fouqueray P. Absence of QTc prolongation in a thorough QT study with imeglimin, a first in class oral agent for type 2 diabetes mellitus. Eur J Clin Pharmacol. 2020;76:1393‐1400. [DOI] [PubMed] [Google Scholar]
- 70. Chevalier C, Fouqueray P, Bolze S. In vitro investigation, pharmacokinetics and disposition of imeglimin, a novel oral antidiabetic drug, in preclinical species and humans. Drug Metab Dispos. 2020;48:1330‐1346. [DOI] [PubMed] [Google Scholar]
- 71. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Giannarelli R, Aragoona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003;29:6528‐6535. [DOI] [PubMed] [Google Scholar]
- 73. McKiney JM, Irwin N, Flatt PR, Bailey CJ, McClenaghan N. Acute and long‐term effects of metformin on the function and insulin secretory responsiveness of clonal beta‐cells. Biol Chem. 2010;391:1451‐1459. [DOI] [PubMed] [Google Scholar]
- 74. Marchetti P, Del Guerra S, Marselli L, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89:5535‐5541. [DOI] [PubMed] [Google Scholar]
- 75. Lablanche S, Cottet‐Rousselle C, Lamarche F, et al. Protection of pancreatic INS‐1 beta‐cells from glucose‐ and fructose‐induced cell death by inhibiting mitochondrial permeability transition with cyclosporin a or metformin. Cell Death Dis. 2011;2:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vial G, Detaille D, Guigas B. Role of mitochondria in the mechanism(s) of action of metformin. Front Endocrinol. 2019;10:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guigas B, Detaille D, Chauvin C, et al. Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem J. 2004;382:877‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. US Food and Drug Administration . Glucophage Product Label.
- 79. US Food and Drug Administration . Glimeperide Product Label Volume 2020, 2016.
- 80. US Food and Drug Administration . Jardiance (Empagliflozin) Product Label Volume 2020, 2014.
- 81. Idris I, Donnelly R. Sodium‐glucose co‐transporter‐2 inhibitors: an emerging new class of oral antidiabetic drug. Diabetes Obes Metab. 2009;11:79‐88. [DOI] [PubMed] [Google Scholar]
- 82. US Food and Drug Administration . Acarbose Product Label; 2020, 2011. [Google Scholar]
- 83. Derosa G, Maffioli P. Efficacy and safety profile evaluation of acarbose alone and in association with other antidiabetic drugs: a systematic review. Clin Ther. 2012;34:1221‐1236. [DOI] [PubMed] [Google Scholar]
- 84. US Food and Drug Administration . Dulaglutide Product Label; 2020, 2017.
- 85. US Food and Drug Administration . Ozempic (Semaglutide) Product Label; 2020, 2017.
- 86. Nauck MA, Meier JJ. Incretin hormones: their role in health and disease. Diabetes Obes Metab. 2018;20(suppl 1):5‐21. [DOI] [PubMed] [Google Scholar]
- 87. US Food and Drug Administration . Januvia (Sitagliptin) Product Label; 2020, 2010.
- 88. Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase‐4 inhibitors. Endocr Rev. 2014;35:992‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. US Food and Drug Administration . Actos (Pioglitazone Hydrochloride) Product Label; 2020, 1999.
- 90. Berger J, Moller DE. Mechanism of action of PPARs. Ann Rev Med. 2002;53:409‐435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request