

In human body, the kidney receives 20% of cardiac output and consumes 10% of the body’s oxygen to perform its functions. The kidney is composed of multiple cell populations which are involved in several vital functions that maintain body’s homeostasis such as acid-base and electrolytes balance, blood pressure regulation, nutrients reabsorption and hormone secretion [1]. The human adult kidney is composed with about 3 million functional units, or “nephrons”, and their primary functions and structures are subdivided into a glomerular filtration unit and several tubular segments. The glomerular filtration is a process of filtering blood circulation, retaining circulating cells and useful macromolecules. The tubular segments are involved in active transport processes to reabsorb water, electrolytes, and nutrients to maintain fluid and osmolarity homeostasis. The coordination of both processes maintains body homeostasis and results in metabolic waste excretion with only trace amount of proteins in the urine output [2]. Hence, kidney dysfunction often results in kidney disease with systemic complications.
Kidney diseases can be classified into two categories- acute kidney injury (AKI) or chronic kidney disease (CKD). Renal disease and complications are the current global health concerns due to the vital roles of kidney in body homeostasis [3–5]. AKI is characterized by rapid renal function decline along with accompanying electrolytes abnormalities, fluid overload, severe acidosis, hematologic abnormalities (e.g. anemia, uremic platelet dysfunction), and possibly multi-organs failure and poor clinical outcomes [6]. CKD is linked to gradual loss of renal function over time. CKD could derive from defects in glomerular filtration units or chronic tubular injuries which could lead to multiple complications including cardiovascular disease, high blood pressure, bone loss, and malnutrition. Both AKI and CKD are closely integrated and could serve as a risk factor for one another which often linked with increase cardiovascular risk and uprising mortality and morbidity rates [7, 8]. Additionally, kidney functions are susceptible to certain inherited genetic diseases and aging processes [9, 10]. All different forms of renal injuries could ultimately progress into severe end-stage renal disease (ESRD) with renal replacement as the only therapeutic option. Therefore, therapeutic strategies are in dire need to prevent renal diseases progression.
Mitochondria is the cell’s energy-producing organelle that maintains cellular redox and energy homeostasis, and therefore a major source of intracellular oxidative stress [11]. In addition to its role in adenosine triphosphate (ATP) generation through oxidative phosphorylation (OXPHOS), the mitochondria also play an essential role in metabolic signaling such as pyrimidine, heme biosynthesis, TCA cycle and fatty acid β-oxidation pathways, in calcium ion (Ca2+) homeostasis, thermogenesis, proliferation and regulating intrinsic apoptotic pathway. Mitochondria are heterogeneous and dynamic organelles. The mitochondria populations can be different in size, mass, metabolic activity, and membrane potential within a cell. Additionally, mitochondria constantly change shape, dynamics and turnover to maintain cellular homeostasis. Cellular stress can induce compromised mitochondria membrane integrity or dysfunctions, leading to cell death via different mechanisms. This could involve the release of apoptotic molecules such as cytochrome C (cyt C), and apoptosomes from mitochondrial inter-membrane spaces. Alternatively, activation of mitochondrial permeability transition (MPT) can trigger mitochondrial inner membrane potential (ΔΨm) dissipations and subsequent loss of energy production [12].
Mitochodrial damages and dysfunction are recognized as a leading factor to many chronic and acute renal diseases [11]. Therefore, it is remarkably important to understand mitochondrial biology and pathophysiology for effective therapeutics discoveries in renal diseases. In this chapter, we discuss evidence supporting mitochondrial dysfunction in the pathogenesis of kidney disease, and summarize the recent development of mitochondria-targeted therapies which hold high promise alleviating renal injury.
Mitochondria in Kidney Health
Mitochondria are especially important in metabolically active organs such as brain, heart, kidney and muscle. Kidney consumes roughly 7% of the body’s daily ATP energy expenditure [13, 14]. Due to various energy demands, different nephron segments have different mitochondria densities and distributions. It is generally accepted that renal tubule cells are rich in mitochondria, with the S1 segment containing the highest mitochondria density. These renal tubule cells require this large amount of mitochondria due to their high-energy demand for reabsorption and secretion against chemical gradients, which heavily rely on normal mitochondrial oxidative phosphorylation to supply ATP as an energy source [15]. Nevertheless, the high-energy requirement of the podocytes was only recently highlighted for possible mechanisms in structural stability, organization of cytoskeletal and extracellular matrix proteins, motility, remodeling of foot process, uptake of filtered proteins, and some other more obscure mechanisms [16]. Bioenergetic profiles studies of mitochondrial function confirmed that podocytes are very susceptible to dysfunction in energy supply during stress condtions [17]. Several factors such as mitochondrial biogenesis and turnover, bioenergetics, dynamics, and autophagy (mitophagy) regulate the conditions of mitochondria.
Mitochondrial Biogenesis
Mitochondrial turnover is an exquisitely coordinated process to maintain mitochondrial homeostasis. In this regard, dysfunctional mitochondria are selectively eliminated and replaced through biogenesis, a coordinated process between nuclear and mitochondrial genomes, to increase the mass and number of functional mitochondria in compensation for the lost or damaged mitochondria [18]. The mitochondrial DNA (mtDNA) encodes 37 genes which include 13 structural subunits of the mitochondrial respiratory chain. The molecular mechanism of mitochondrial biogenesis during tissue injury and repair are actively studied topics [19, 20] (Fig. 27.1a, b). The mitochondrial biogenesis program involves mito-nuclear communication and several key regulators have been identified such as: peroxisome proliferator-activated receptor (PPAR), PPARγ coactivator 1α (PGC-1α), sirtuin-1 and 3 (SIRT 1/3) family deacetylase, AMP-activated protein kinase (AMPK), and nuclear respiratory factors 1 and 2 (NRF1 and NRF2) [21]. AMPK and SIRT1 positively regulate PGC-1α, the master regulator of mitochondria biogenesis, through post-translational modifications with phosphorylation or deacetylation respectively. In addition, AMPK and SIRT1 are important modulators of energy metabolism. In kidney, the PGC-1α transcriptional coactivator is predominantly expressed in proximal tubules, and therefore is pivotal to tubular homeostasis. PGC-1α regulates the expression of NRF1 and NRF2 and increases the biosynthesis of nicotinamide adenine dinucleotide (NAD+), a central metabolic coenzyme/cosubstrate involved in cellular energy metabolism, to link oxidative metabolism to renal protection [22–24]. Both NRF1 and NRF2 are nuclear DNA (nDNA)-encoded transcription factors that activate genes coding for the OXPHOS system and genes involved in mtDNA transcription and replication [25, 26].
Fig. 27.1.
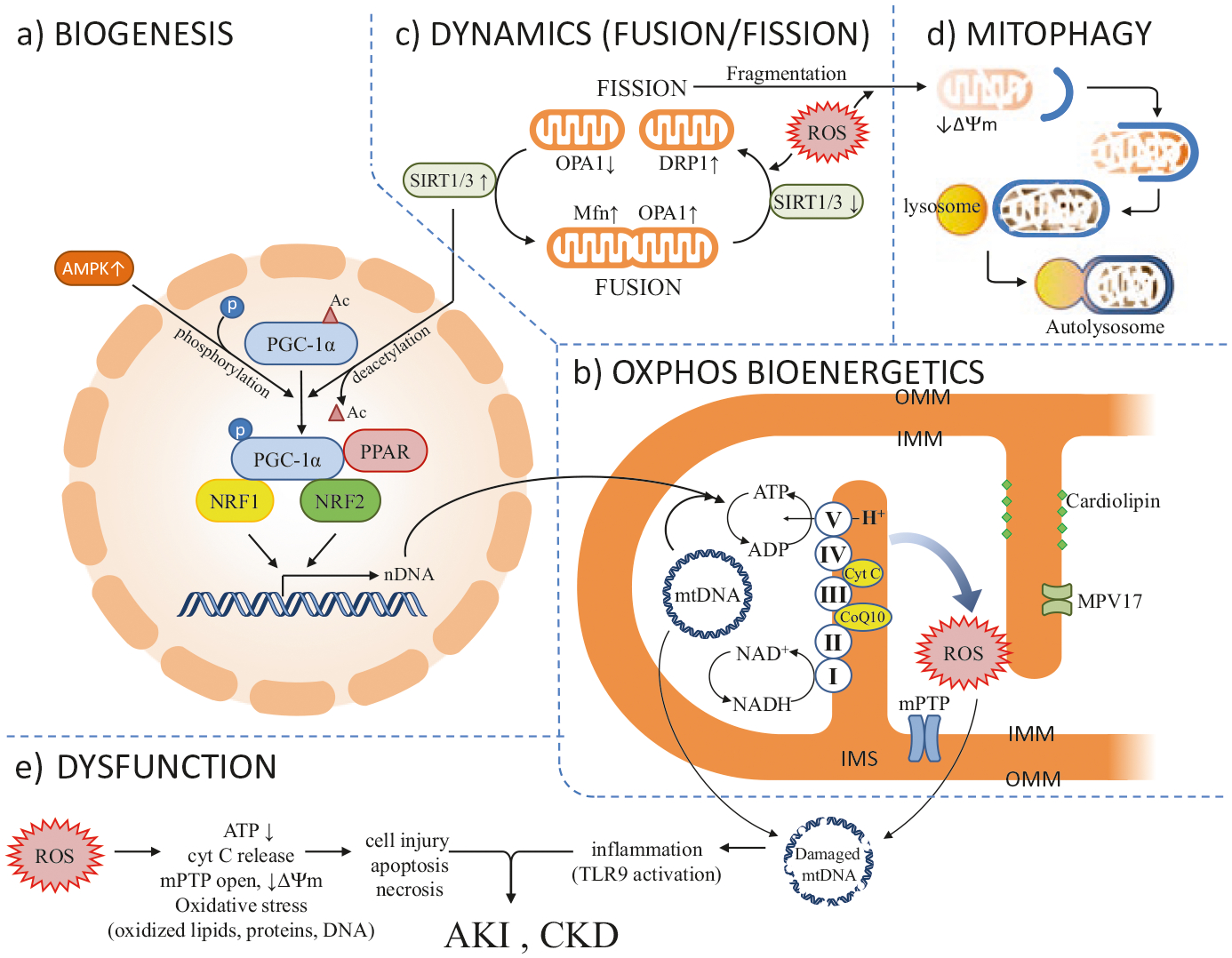
The health and disease of kidney are regulated by the pathophysiological conditions of mitochondria. Several processes regulate the health and disease states of mitochondria which include (panel a) mitochondrial biogenesis – a process requires coordinated expression of both nDNA and mtDNA and several transcriptional factors and co-activators; (panel b) OXPHOS bioenergetics – an electrochemical gradient and ATP synthesis process which depends on respiratory electron transfer chain (complexes I–V), cofactors (MPV17 and others), and cardiolipin to maintain normal oxidative phosphorylation; (panel c) mitochondrial dynamics – a process requires functional balance between fusion and fission; and (panel d) mitochondrial turnover by autophagy (or mitophagy) – an important process to clear the dysfunctional mitochondria. (Panel e) Disturbance in any steps of the processes in panels a–d will cause ROS overproduction, decrease in ATP generation, loss of IMM potential (ΔΨm), mPTP opening and cyt C release. The oxidative stress could lead to cell apoptosis or cell injury and the damaged mtDNA, nDNA could stimulate innate immune response through activation of Toll-like receptor (TLR) and inflammation which, all together, lead to AKI or CKD. See Text and Abbreviations for details
Mitochondrial Bioenergetics
The mitochondrial OXPHOS respiratory chain is composed of four protein complexes which are known as complexes I–IV. These complexes transfer electrons and protons (H+) across inner mitochondrial membrane (IMM) to generate an electrochemical gradient for ATP synthesis in a fifth protein complex, known as complex V (ATP synthase) (Fig. 27.1b). Some important factors were identified in this process. For example, NAD concentrations determine the rate-limiting process [27, 28] and Coenzyme Q10 (coQ10) shuttles electrons in the respiratory chain [29]. Additionally, depending on its interaction with cardiolipin, the IMM resident protein cytochrome C (cyt C) regulates an intricate balance between mitochondrial respiration and apoptosis. Cardiolipin was identified as a phospholipid that was exclusively expressed on the IMM, where it forms microdomains and plays a central structural role in cristae formation, a property of cardiolipin on membrane curvature, organization of the electron transport chain (ETC) complexes into supra-complex for OXPHOS activity, and as a platform for initiation of apoptosis [30, 31]. The interaction of cyt C and cardiolipin determines whether or not cyt C functions as an electron carrier or a peroxidase.
During normal electron transfer, some (<4%) of the consumed oxygen is converted into superoxide radicals such as reactive oxygen free radical species (ROS) and reactive nitrogen free radical species (RNS) via electron leakage, which constitutes different forms of oxidative stress [32]. However, under ischemic conditions, Ca2+ overflows into mitochondria during rapid loss of ATP and aggravates ROS production. These reactive radicals cause modification of biomolecules of DNAs (nDNAs and mtDNAs) and proteins, lead to lipid peroxidation, and may impair their bioactivities which eventually lead to the opening of the mitochondrial permeability transition pore (mPTP) and loss of mitochondrial membrane potential (ΔΨm) [12].
The importance of the IMM-resident MPV17 protein in mitochondrial physiology has come to light recently. MPV17 is encoded by nDNA and functions as a ΔΨm-modulating channel that contributes to mitochondrial homeostasis under different conditions. MPV17 forms weak cation-selective channesl and has shown several subconductance states in vitro [33]. Interestingly, MPV17 channel protein allows for the passage of small molecules such as deoxynucleotides triphosphates (dNTPs). It is possible that MPV17 maintains mitochondrial dNTP homeostasis, and therefore the perturbation of dNTP pools is a recognized cause of mitochondrial genomic instability [34]. Mutations of MPV17 are associated with mtDNA depletion syndrome, an inherited autosomal recessive disease in humans. Additionally, MPV-like proteins are identified as epithelial and neuronal restricted protein and implicated in ROS metabolism and apoptosis regulation through its binding and functional interaction with mitochondrial serine protease, HTRA2 [35]. Altogether, excess oxidative stress accumulation, mtDNA instability, and imbalance of bioenergetics will lead to mitochondrial dysfunction which could be important in the pathogenesis of kidney diseases [36].
Mitochondrial Dynamics
Mitochondria are highly dynamic organelles – their number, size and locations change constantly in response to energy demands. They constantly move, fuse and divide – switching between elongated interconnected networks or fragmented discrete morphologies via coordinated fusion and fission. These dynamics are essential to their size, morphology, energy biogenesis, function, and maintenance of cellular homeostasis and viability [37–39]. Perturbation of mitochondrial dynamics results in mitochondrial dysfunction and is linked to aging, end organ injury, and human diseases [40, 41]. Mitochondrial dynamics are regulated by a complex relationship between fission proteins (the large GTPase, dynamin related protein 1 (DRP1), and mitochondrial fission 1 (Fis1)) and fusion proteins (mitofusins 1 and 2 (Mfn1, Mfn2) and optical atrophy (OPA1)) [5, 42] (Fig. 27.1c). The NAD+-dependent SIRT family deacetylases, especially the mitochondrial matrix-resided SIRT3 protein, play an important role in regulating mitochondrial dynamics and function. In this role, the SIRT3 deacetylates and activates the mitochondrial fusion protein OPA1 to improve mitochondrial function under stress conditions [43] or acute kidney injury [44].
Mitochondrial Turnover: Autophagy and Mitophagy
Autophagy is a non-selective process where cytoplasmic contents, e.g. damaged organelles and aged protein aggregates, are sequestered into autophagosomes for lysosome delivery and bulk degradation within the cell. Autophagy occurs during nutrient stress for ATP preservation, amino acid recycling, and anabolic protein synthesis. In certain stress conditions, selective autophagy occurs to efficiently remove toxic cellular materials such as damaged organelles. Mitophagy is one of the selective autophagy processes to remove accumulated dysfunctional mitochondria. Mitophagy clearance encompasses several steps – the damaged mitochondria are first marked by a loss of mitochondrial inner membrane potential (ΔΨm) via intricate protein interactions involving kinases, E3 ubiquitin ligase, and proteins regulating mitochondrial dynamics and transportation [45] (Fig. 27.1d). The damaged mitochondria are eventually encapsulated into autophagosomes and degraded in autolysosomes. Autophagy (and mitophagy) is ROS-dependent, which is heavily regulated by cellular redox activity. Growing evidences support the notion that disturbance of autophagy/mitophagy is associated with the pathogenesis of renal diseases such as AKI [5], diabetic nephropathy [45] and glomerulosclerosis [46].
Mitochondria Damage in Kidney Diseases
Renal diseases encompassing both acute and chronic conditions of kidneys injury with declined renal functions are current global health concerns with tremendous medical burdens [3–5]. A common link between all forms of acute and chronic kidney injuries is the generation of toxic ROS and RNS when the disease manifests. This oxidative stress injury could be derived from ischemia/reperfusion, energy shortage from impaired mitochondrial biogenesis or ATP energetics, or defective clearance of damaged mitochondria (mitophagy). All of the listed conditions could link to recruitment of immune cells, inflammatory cytokines accumulation, apoptosis, and tissue injury. The renal phenotypes of mitochondrial dysfunction may manifest as proximal tubular dysfunction, tubularinterstitial disease, cystic kidney disease, podocytopathy, and nephrotic syndromes. All different forms of renal injuries might increase cardiovascular risk with uprising mortality and morbidity rates.
Acute Kidney Injury (AKI)
AKI has been a global concern with worldwide prevalence of 13 million people and death toll roughly 1.7 million annually [47]. AKI is a clinical condition that is commonly related to an acute episode of systemic injuries that occur in cases such as: kidney transplantation and septic and trauma patients. In the USA, AKI is the source of 1–2% of all hospital admissions and the mortality rates in hospitalized ICU patients with AKI could reach 50–70% [48]. Several etiologies such as ischemic, toxic, septic, and hypertensive injuries cause AKI with symptoms of abrupt reduction in kidney functions. AKI symptoms are manifested as acute decline in glomerular filtration rate, concomitant decreased urinary output, tubular necrosis, tubular interstitial inflammation and vascular permeability changes [5, 49].
Interestingly, in a polymicrobial septic AKI model, Tsuji and colleagues recently identified circulating mtDNA that stimulate systemic inflammation via Toll-like receptor 9 (TLR9) signaling as a novel pathogenic mechanism for AKI (Fig. 27.1e). The source of circulating mtDNA has been speculated to be derived from immune cells upon septic clearance or from spleen immune response [50, 51]. This study suggests that timely protection and removal of damaged mitochondria would reduce inflammation and could be an effective therapeutic strategy against AKI.
Ischemic-Reperfusion (I/R) Induced AKI
Renal ischemia-reperfusion injury (IRI) is a common cause of AKI and post-transplantation kidney allograft dysfunction resulting from an acute decline in general or localized renal oxygen and nutrients supplies to the affected tissue, and impairment of timely removal of metabolite wastes in kidney cells. Extensive oxidative stress generated during this process injures the tubular epithelial cells, causing histologically characteristic inflammation and necrotic cell death (necrosis) which was referred as acute tubule necrosis previously [52]. Emerging evidence supports the notion that multiple signaling pathways are involved in the pathobiology of AKI [52, 53]. Of these, damaged mitochondria with disrupted matrix cristae and mitochondrial permeability-mediated necrosis are crucial to ischemic AKI. Proper functional autophagy (mitophagy) is reno-protective during renal tissue regeneration if the injury is under a certain threshold, whereas defective mitophagy impedes tissue regeneration [5].
Nephrotoxic AKI
Exposure to nephrotoxins, such as some chemotherapy drugs, medications, intravascular contrast media, trace heavy metals, drug abuse, or certain chemicals/drugs combinations could induce AKI, especially in the elderly, young children, and high-risk patients [54, 55]. Drug nephrotoxicity was found to be responsible for 19% of all AKI cases on critically ill patients. The kidney is susceptible to intrinsic renal toxin injury which could result in various clinical syndromes ranging from minimal changes in tubular function to fulminant kidney failure. Injury may also occur to altered hemodynamic AKI. Some examples include: NSAIDs and renin-angiotensin system (RAS) antagonism, tubular epithelium direct cell injury from chemotherapy drugs like cisplatin, tubular obstruction of urinary flow due to precipitates of toxins or their metabolites, and angiopathogenesis from vascular injury and interstitial inflammation. Several research efforts have been dedicated to identify biomarkers of nephrotoxic AKI [56]. Cisplatin-induced renal mitochondrial structural, functional and homeostatic changes include swollen mitochondria, ultra-structurally disrupted IMM cristae, reduced mitochondrial mass, mtDNA depletion, and diminished cyt C oxidase activity in animal model [57]. Permeability of mitochondrial OMM upregulates the expression of mitochondrial dynamin related protein 1 (DRP1), an OMM resident fission protein, which induces mitochondrial fragmentation, cyt C release and apoptosis [58]. Reduction of mitochondria biogenesis regulators such as PGC-1α and SIRT3 were also documented [44]. Moreover, kidney specific overexpression of SIRT1 was protective against cisplatin-induced AKI [59]. The nephrotoxic effect and its pathophysiology of cisplatin-induced AKI had been a seminary topic of a recent review [60].
Septic AKI
Septic shock is one of the most frequent injury which could account for nearly half of the cause of AKI in critical ill patients [61]. Mitochondrial damage is an important contributing factor in the pathogenesis of septic AKI [62]. Clinical biopsies obtained from intensive care of human sepsis subjects also support this notion [63]. Apparent mitochondrial ultrastructure changes and biochemical studies revealed mitochondrial pathogenesis includes loss of mitochondrial mass, swelling, fragmentation due to aberrant mitochondrial dynamics, PGC-1α reduction, extensive mitochondrial cristae remodeling, and cyt C release during apoptosis. As aforementioned, mitochondrial damage and the release of mtDNA is linked to systemic inflammation and tissue injury. The intricate interplays between mitochondrial energy precursor NAD/NADH, mitochondrial health and kidney function were shown in several studies of septic AKI animal model [23, 24]. In summary, mitochondrial NAD concentration regulates the rate-limiting process of mitochondrial energy production and, hence, PGC-1α causes an increase of NAD biogenesis, mitochondrial numbers and subsequent renoprotection.
Chronic Kidney Disease (CKD)
CKD is characterized by persistent renal dysfunction for more than 3 months. The causes of CKD have been commonly associated with a number of comorbid conditions such as type 2 diabetes and hypertension [64]. The prevalence of CKD is estimated to be roughly 8–16% globally. According to the United States Renal Data System report, more than 13% of adults develop CKD and 30% of the population at the age of 65 and above experience some form of kidney failure in the USA [65]. CKD is staged based on estimated glomerular filtration rate (eGFR) and albuminuria with conditions ranging from asymptomatic stage to end-stage renal disease (ESRD) [66]. Different from AKI, CKD is recognized as a separate, irreversible and progressive pathologic condition which often inevitably leads to ESRD [7, 8, 49]. Individuals with diabetes or hypertension have high risk to develop CKD. Recent findings support the close relation between AKI and CKD and confirm, clinically, that one could serve as the risk factor to the other in maladaptive recovery conditions [7, 8].
Diabetic Nephropathy (DN)
Diabetic Nephropathy (DN), a progressive microvascular complication that arises from diabetes mellitus (DM) which affects individuals with both type 1 or type 2 DM, is the leading cause of CKD. DN is a chronic disease characterized by decreased glomerular filtration, proteinuria, glomerular hypertrophy, and renal fibrosis with gradual decline in renal function which contributes to 40% kidney failure and eventually requires renal replacement therapy [67–69].
The pathogenesis of DN is complex due to all four renal compartments-glomeruli, tubules, interstitium and blood vessel being involved. Moreover, etiologies of DN originated from hyperglycemia mediated microvasculate abnormalities which affect several signaling pathways. These include: increased glucose metabolite flux, advanced glycation end-products formation, endoplasmic reticulum stress, ROS overproduction, pro-inflammation and apoptotoc cell death in podocytes [68, 69]. It is well documented that aberrant mitochondrial homeostasis plays pivotal roles in DN progression [11, 68–73]. Hyperglycemia induced increased oxidative stress derived from renal ROS over-production in conditions like impaired mitoglutathione transport [74], increased mitochondrial activation of an ubiquitous NAD(P)H oxidase (Nox4) in respiratory chain [75], decreased mtDNA stability to against ROS production [76] and podocytes apoptosis [77] all contribute to cell damage and lethal effects linked to diabetic complications. Interestingly, hyperglycemia triggers mitochondrial fission and is mediated by Rho-associated coiled coil-containing protein kinase 1 (ROCK1)-dependent phosphorylation of dynamin related protein 1 (DRP1) and its recruitment to mitochondria [78]. These results suggest that mitochondrial dynamics play an important role in the pathogenesis of DN. Additionally, defects in mitochondrial biogenesis with downregulated expression or activity of PGC-1α is recognized to contribute to DN [71, 72]. It appears that the protective role of PGC-1α is associated with the inhibition of DRP1-mediated mitochondrial dynamics remodeling and ROS production [79]. Growing evidence also suggests that autophagy or mitophagy is altered in DN [45]. It is plausible that impairment in autophagic flux would lead to the DN complication associated with extracellular matrix deposition and fibrosis.
Glomerulonephritis (GN)
Chronic glomerulonephritis (GN), characterized by dysfunction in the glomerular filtration barrier, is a major feature of CKD. GN accounts for roughly 10% of CKD. GN is the inflammation of the glomeruli with clinical symptoms which may include hematuria, proteinuria, edema, and hypertension. Congenital GN can occur as genetic mutations on mitochondrial proteins (see section on “Genetic Mitochondrial Disease Affecting Kidney Functions”). Immune diseases may also cause chronic GN. Abnormal-shaped mitochondria are often found before the disease progress into secondary (or acquired) focal segmental glomerulosclerosis (FSGS), a severe form of GN [46]. Therefore, these evidence support the notion that impaired autophagic mitochondrial turnover is responsible for FSGS phenotype in mice.
Recently, Kruppel-like factors 6 (KLF6), a subfamily of DNA-binding zinc finger protein, was identified as an inducible early injury response gene that encodes a crucial transcriptional regulator of mitochondrial function in podocytes under stress response [80–83]. Podocyte-specific loss of Klf6 reduced mitochondrial function and increased the susceptibility to FSGS in mice. Likewise, podocyte-specific KLF6 expression was significantly reduced in FSGS patients when compared to healthy individuals. Mechanistic studies indicated KLF6 prevents mitochondrial injury via enhancing cyt C assembly [82].
The function roles of MPV17 were explored further as discussed earlier. Growing body of evidence indicated that the glomerulosclerosis gene mpv17 and its encoded MPV17 protein as important factors in regulating peroxisomal metabolism of ROS [84]. MPV17 was identified as an IMM- localized protein in podocyte. Loss of Mpv17 was found to be associated with glomerulosclerosis and knockout mice developed renal failure later on in their lifespan (9–12 months). The glomerular lesions were caused by toxic oxidative stress and lipid peroxidation adducts. Interestingly, expression of MPV17 is diminished in several glomerular injury models and also in human FSGS subjects [85]. Altogether, the evidence further confirmed that MPV17 is important for mitochondrial homeostasis in podocytes.
Genetic Mitochondrial Disease Affecting Kidney Functions
Genetic disorders that affect mitochondrial function can be mutations related to either the nuclear or mitochondrial genomes which encode mitochondrial proteins. They represent genetically and clinically heterogeneous disorders which affect mostly metabolic-active organs. Genetic mitochondrial mutations could be derived from primary (inherited) or secondary to environmental predisposition to drugs or oxidative stress injury. Kidney phenotypes are often manifested as tubular dysfunction, interstitial nephritis, glomerular dysfunction, and renal tumors [9, 86].
MELAS and tRNA-Leu (m.A3243G) Transition
MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke like symptoms) is a mitochondrial syndrome manifested in patients that are 40-years old or younger with symptoms like seizure and stroke-like episodes, muscle myopathy, lactic acidosis, and maternally inherited diabetes with deafness. A base transition (A3243G) in mitochondrial gene tRNA-Leu accounts for 80% of MELAS cases with occasional renal phenotypes of FSGS, or less commonly with tubular interstitial nephritis, which results in progressive renal insufficiency [87, 88]. Electron microscopic images from renal biopsies of MELAS patient revealed abnormal mitochondria in podocytes and tubular cells, indicating dysfunctional mitochondria is central to disease pathogenesis.
Mitochondrial DNA Deletion
Different from the mtDNA point mutations, few mitochondrial disease syndromes which are often associated with large mtDNA deletion at different genome locations and affect tubular pathologies, are Fanconi [89], Kearns-Sayre, and Pearson syndromes [90]. The clinical features of Pearson syndrome are manifested with pancreatic fibrosis with insulin-dependent diabetes, whereas the kidney phenotype of Kearns-Sayre syndrome includes renal tubular acidosis (proximal or distal) which occasionally progress to ESRD. Although occurrences may be rare, these syndromes involve renal tubulopathy (proximal or distal) manifested as electrolyte abnormalities and fibrosis. Renal biopsies of patients with Kearns-Sayre syndrome revealed impaired cyt C oxidase activity, defect in energy metabolism, and mitochondrial ultrastructures alterations [90].
Mutations in Nuclear-Encoded Genes for Mitochondrial Proteins
The mitochondria proteome is composed with about 1,000 proteins which are mostly encoded by nDNA. nDNA mutations with prominent tubular dysfunction include mutations that affect energy metabolism, often affecting complex III and IV, and mutations that affect fatty acid metabolism. Podocyte dysfunction is often correlated with multi-mutations in coenzyme Q10 biosynthesis pathways and the pathogenic mechanism was confirmed in pdss2 gene encoding prenyl diphosphate synthase subunit 2, the first enzyme of coQ10 pathway [29, 91].
Aging in Kidney Disease
Older age is associated with an elevated risk of kidney disease and increased population of older renal transplant recipients and poor graft survival. The renal aging phenotype includes profound functional alterations leading to increased renal vascular resistance and reduced repair capacity. Aging kidney is often associated with histological changes such as tubular atrophy, interstitial fibrosis and glomerulosclerosis. Clinically, renal aging is associated with an increased susceptibility to AKI [92], and CKD may share accelerated vascular disease phenotype similar to the condition of premature aging [93]. The kidney suffers from increasing oxidative stress and disturbance in autophagy with aging [94, 95]. The molecular mechanism of renal cell senescence has been explored to certain extent. Downregulation of PGC-1α may link telomere-dysfunction to compromised renal mitochondrial function through p53 activation and alteration of transcription [96]. The renin–angiotensin–aldosterone system (RAAS) is a key regulator of blood pressure, fluid homeostasis and cardiovascular physiology. Angiotensin II and the age-dependent switch of mitochondrial expression of the two types of cell surface angiotensin receptors (AT1R vs AT2R) are important factor in determining respiratory activity during aging [97]. Further studies are needed to explore the role of the telomere–mitochondrial axis in renal physiology, aging and disease.
Mitochondria-Targeted Therapeutics
As protection of mitochondria could serve as a potential effective therapeutic strategy, it has been research endeavors focused on this development toward kidney diseases [49, 98–101]. These agents include antagonizing mitochondria oxidants, promoting mitochondrial biogenesis and ATP synthesis, regulating ROS metabolism, cardiolipin protection, inhibitors of mPTP, or inhibitors of mitochondrial fragmentation as detailed below (Table 27.1).
Table 27.1.
Mitochondria-targeted therapeutics
| Therapeutics | Chemicals | Action mechanisms | Experimental model |
|---|---|---|---|
| Antioxidants | |||
| MitoQa | Mitoquinone | Anti-oxidant concentrate at matrix in a ΔΨm-dependent manner; ROS scavenger | IRI-AKI, cisplatin-AKI, sepsis-AKI |
| Biogenesis activator | |||
| Resveratrol (SRT501) | Small peptides | AMPK / SIRT-1 / PGC-1α axis activator | Hemorrhagic shock, sepsis-AKI |
| AICAR | SIRT3 activator, AMPK agonist | Cisplatin-AKI, IRI-AKI | |
| Formoterol | β2-adrenoceptor agonist, increase PGC-1α synthesis | IRI-AKI | |
| ROS metabolism and bioenergetics | |||
| Mitochonic acid (MA-5) | Synthetic compound | OXPHOS-independent increase of ATP synthesis, reduce ROS | Sepsis-AKI |
| Cardiolipin protection | |||
| Bendavia (SS-31)a | Szeto-Schiller tetrapeptide | Protect cardiolipin from peroxidation; increase ATP and reduce ROS | AKI and CKD |
| Prevent cyt C transformation into peroxidase | |||
| mPTP inhibitor | |||
| cyclosporin A (CsA)a | Small molecule | Interact with cyclophilin D (a mPTP structural protein) | Adriamycin-induced podocyte injury |
| TDZD-8 | mPTP inhibitors, GSK3β inhibitor | Podocyte injury or drug induced AKI | |
| Fission inhibitor | |||
| Mdivil | Small molecule | Inhibitor of mitochondrial division; induce mitochondria fusion | IRI-AKI |
| Block DRP1 assembly and translocation | |||
| K(ATP) channel opener | |||
| Simdax (Levosimendan)a | Small molecule | Calcium sensitizer, K(ATP) channel opener | IRI-AKI |
| cyt C assembly | |||
| KLF6 | DNA-binding zinc finger transcriptional regulator | enhance cyt C assembly | Podocytes FSGS (animal study) |
Clinical trials. https://clinicaltrials.gov
Mitochondria-Targeted Antioxidants
Mitochondria-targeted antioxidants (e.g. MitoQ, MitoTEMPO, MitoE, Mito-CP, SkQ1 and SkQR1) have been a part of the efforts to reduce mitochondrial oxidative stress. These agents are based on the mechanism of delivery of known redox agents to mitochondrial matrix through conjugation with TPP+ (triphenylalkylphosphonium cation) moiety [100]. These ROS scavenger compounds could cross the membrane lipid bilayer and concentrate at the matrix in a membrane potential (ΔΨm) – dependent manner. MitoQ was shown to be safe in clinical trial with Parkinson’s disease, fatty acid disease, and is currently under clinical trial (NCT02364648) for CKD. In summary, several animal models have confirmed that mitochondrial-targeted antioxidants represent an effective strategy to prevent or attenuate different forms of kidney diseases.
Activator of Mitochondria Biogenesis
Activation of Mitochondria biogenesis is required for the increased metabolic and energy demands during the recovery phase of acute organ injury. The AMPK/SIRT/PGC-1α axis plays crucial roles in mitochondrial biogenesis. PGC-1α is a pivotal factor that coordinately regulates NAD biogenesiss, reduces oxidative stress, and facilitates recovery from AKI [24]. SIRT1 is a NAD-dependent deacetylase that positively regulates PGC-1α expression and activity. Agents in this category include resveratrol, AICAR, and formoterol. Treatment with resveratrol, a natural plant phytoalexin, in a rat hemorrhagic shock model increases mitochondria mass, restores mitochondrial respiratory capacity, and reduces oxidative stress [102]. Moreover, resveratrol modulates immune response by suppressing inflammation driven by macrophage in a lipopolysaccharide -induced septic AKI model [103]. Pretreatment with AICAR, an AMPK activator, attenuated injury and tubular necrosis, decreased nitrosative stress, and ameliorated renal function in a rat I/R induced AKI [104]. Formoterol, a potent agonist of β2-adrenoreceptor, induces mitochondrial biogenesis by increasing mtDNA copy numbers, oxygen consumption rate, and PGC-1α synthesis. In doing so it restores renal function, rescues renal tubules from injury, and diminishes necrosis in an I/R-induced AKI animal model [105]. In summary, agents affect mitochondrial biogenesis and NAD modulation hold great promise in treating kidney disease, but their clinical translation still awaits further investigation.
Modulation of ROS Metabolism and ATP Synthesis
Mitochonic acid 5 (MA-5), a synthetic derivative of indole acetic acid plant hormone, was identified as a compound to enhance ATP production. MA-5 was found to improve the survival of fibroblasts collected from patients with mitochondrial diseases, including Leigh syndrome and MELAS [106]. Evidence supports that MA-5 enhances ATP production by promoting assembly and oligomerization of complex V at mitochondrial crista junction, and therefore preserving the mitochondria dynamics and preventing its fragmentation. These new findings provide new mechanisms in renal and cardiac protection [106, 107].
Cardiolipin Protections
As cardiolipin regulates IMM structural and functional plasticity, peroxidation or loss of cardiolipin has been associated with aging and several forms of AKI and CKD. Development of cardiolipin-target compound to optimize efficiency of the ETC and thereby restore cellular bioenergetics has been an innovative discovery. SS-31 peptide (Szeto-Schiller peptide, also named MTP-131 or Bendavia) is a cardiolipin-targeted tetrapeptide (D-Arg-dimethylTyr-Lys-Phe-NH2) that stabilizes cardiolipin, scavenges mitochondrial ROS, regulates cyt C activity, and inhibits the mitochondrial permeability transition (MPT) pore in AKI and CKD models [108–110]. SS-31 treatment demonstrated mitochondrial protective effects on swine stenotic-kidney injury and improved renal hemodynamics and function [111]. SS-31 has entered several clinical trials for treatment with AKI renal microvascular dysfunction in hypertension, and I/R injury of myocardial infarction [108].
mPTP Inhibitors
Opening of the IMM localized mitochondrial permeability transition pore (mPTP), a Ca2+-dependent nonselective pore, under certain pathological conditions such as Ca2+ overload or oxidative stress leads to the IMM permeability to small molecules (<1,500 Da in molecular weight), loss of proton motive force, uncoupling of OXPHOS, and mitochondrial swelling which often followed by cell death. Opening of mPTP appears to be a pivotal event in AKI [112]. Cyclosporin A (CsA) is a potent inhibitor of mPTP through interaction with cyclophilin D, a structural component and mediator of mPTP which critically regulates Ca2+- and ROS-dependent opening. Low dose of CsA (at submicromolar concentration) suppress mPTP opening and mitochondria swelling, and is currently under clinical trial for its effect on reperfusion injury on acute myocardial infarction (NCT01595958). CsA reduced podocyte damage through mitochondrial protection in an adriamycin-induced podocyte injury animal model [113]. A clinical trial (NCT02397213) is underway to test CsA renal protection in cardiac surgery (potential AKI condition). High dose of CsA is renotoxic and thus limits its clinical use.
Evidence suggests that glycogen synthase kinase (GSK)-3β resides at the nexus of multiple signaling pathways implicated in the regulation of mitochondrial permeability transition (MPT). The selective GSK-3β inhibitor TDZD-8, a thiadiazolidinone derivative, works as a non-ATP competitive inhibitor to prevent MPT and improve cell viability during AKI [112, 114].
Fission Inhibitors
Mitochondrial fusion is required for homogenous distribution of mtDNA, matrix proteins, and lipids across all fused mitochondria. However, mitochondria fission is important in mitochondrial proliferation following mitosis and is also involved in damaged (fragmented) mitochondrial removal by mitophagy. Mdivi-1 (mitochondrial fission inhibitor-1) was identified as a small molecule that selectively and reversibly inhibits the assembly and the GTPase activity of fission protein DRP1 [115]. Mdivi-1 induces rapid and reversible formation of fused mitochondria in wide varieties of cells and several animal disease models including I/R-injury in the heart, liver, kidney, and brain [116]. Mdivi-1 ameliorates rhabdomyolysis-induced AKI in rat by reducing ROS stress and tubular epithelial cell apoposis with simultaneous increase of ATP production [117] and protection from mPTP opening in acute cardiorenal syndrome [118]. Despite its therapeutic potential, Mdivi-1 exhibits divergent functions which could be cytoprotective or cytotoxic and the reno-protective effect of Mdivi-1 in humans is still lacking [116].
K(ATP) Channel Opener
Levosimendan is a Ca2+ sensitizing smooth muscle vasodilator clinically used for the treatment of heart failure. Levosimendan exerts pleiotropic beneficial effects including mitochondria protection in cardiomyocytes during ischemic heart disease [119]. Possible molecular mechanisms of Levosimendan might involve its function as a mitochondrial ATP-sensitive potassium channel to favorably conserve mitochondrial energy in cardiomyocytes. Pre-clinical and clinical data also indicated positive circulatory effects in the brain, lung, and renoprotection under potentially lethal stress conditions. Currently, a clinical trial (NCT01720030) with Levosimendan in an AKI study is ongoing.
Concluding Remarks and Future Perspectives
Mitochondria are the “power house” of the high-energy demanding kidney cells. The mitochondrial networks also affect the cross-talk with other cellular organelles such as the endoplasmic reticulum, nucleus, peroxisomes, and thus impact many cellular functions. Renal mitochondrial dysfunction could derive from defects on mitochondrial biogenesis, bioenergetics, dynamics, turnover and genetic mutations which may contribute to respiratory chain-derived oxidative stress, abnormal ultra-structures, increased sensitivity to apoptosis, and accumulation of damaged mitochondria with unstable mitochondrial DNA. Disturbance of renal mitochondrial homeostasis could lead to damages to microvasculatures, inflammation, fibrosis, and kidney failure. Although kidney mitochondrial dysfunctions have been exquisitely demonstrated to be tightly linked to pathobiology in many preclinical animal studies of CKD, AKI, aging and genetic defected mitochondrial disease, many direct cause-effect mechanisms remain elusive before development of clinical translations.
The mitochondria-targeting therapeutics as effective interventions to preserve mitochondria structures and functions have been demonstrated in several animal models of kidney injuries. Among these, MitoQ and SS-31 have shown promising potential in clinical trials towards certain kidney injuries and diseases. With better understandings of mitochondrial genome and its physiological functions, advances in in vivo animal studies, and human clinical trials to confirm efficacy and safety of mitochondria-targeting therapeutics, we have high hope to translate these discoveries into therapeutic strategies to ameliorate kidney injuries and diseases in the future.
Acknowledgments
PHL is supported, in part, by an OSU intramural Lockwood Fund.
Abbreviations
- AKI
Acute kidney injury
- AMPK
AMP-activated protein kinase
- ATP
Adenosine triphosphate
- Ca2+
Calcium ion
- CKD
Chronic kidney disease
- coQ
Coenzyme Q
- CsA
Cyclosporin A
- cyt C
Cytochrome C
- DN
Diabetic nephropathy
- dNTPs
Deoxynucleotides triphosphates
- DRP1
Dynamin related protein 1
- ESRD
End-stage renal disease
- ETC
Electron transport chain
- FSGS
Focal and segmental glomerulosclerosis
- GN
Glomerulonephritis
- GSK
Glycogen synthase kinase
- H+
Proton
- I/R
injury or IRI Ischemic reperfusion injury
- IMM
Inner mitochondrial membrane
- KLF-6
Krüppel-like factor 6
- MELAS
Myopathy encephalopathy lactic acidosis and stroke-like episodes
- Mfn1 and 2
Mitofusins 1 and 2
- MPT
Mitochondrial permeability transition
- mPTP
Mitochondrial permeability transition pore
- mtDNA
Mitochondrial DNA
- MA-5
Mitochonic acid 5
- NAD+
Nicotinamide adenine dinucleotide
- nDNA
Nuclear DNA
- NRF
Nuclear respiratory factors
- OMM
Outer mitochondrial membrane
- OPA1
Optical atrophy 1
- OXPHOS
Oxidative phosphorylation
- PGC-1α
PPARγ-coactivator −1α
- PPAR
Peroxisome proliferator-activated receptor
- ROS
Reactive oxygen species
- SIRT
Sirtuin
- TLR
Toll-like receptor
- ΔΨm
Mitochondrial inner membrane potential
Footnotes
Conflict of Interest No competing interest.
Contributor Information
Pu Duann, Department of Surgery, Baylor College of Medicine, Houston, TX 77030, USA.
Pei-Hui Lin, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA; Department of Surgery, The Ohio State University, Columbus, OH 43210, USA.
References
- 1.Skorecki K, Chertow GM, Marsden PA, Taal MW, Yu ASL. Brenner and Rector’s the kidney. 10th ed. Philadelphia: Saunders; 2015. 2748 p [Google Scholar]
- 2.Scott RP, Quaggin SE. Review series: the cell biology of renal filtration. J Cell Biol. 2015;209(2):199–210. Epub 2015/04/29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382(9887):170–9. Epub 2013/06/04 [DOI] [PubMed] [Google Scholar]
- 4.Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, et al. Global prevalence of chronic kidney disease – a systematic review and meta-analysis. PLoS One. 2016;11(7):e0158765. Epub 2016/07/08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duann P, Lianos EA, Ma J, Lin PH. Autophagy, innate immunity and tissue repair in acute kidney injury. Int J Mol Sci. 2016;17(5):E662, p1–19. Epub 2016/05/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weisbord SD, Palevsky PM. Design of clinical trials in acute kidney injury: lessons from the past and future directions. Semin Nephrol. 2016;36(1):42–52. Epub 2016/04/18 [DOI] [PubMed] [Google Scholar]
- 7.Ferenbach DA, Bonventre JV. Acute kidney injury and chronic kidney disease: from the laboratory to the clinic. Nephrol Ther. 2016;12(Suppl 1):S41–8. Epub 2016/03/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11(5):264–76. Epub 2015/02/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016;12(5):267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Divers J, Freedman BI. Genetics in kidney disease in 2013: susceptibility genes for renal and urological disorders. Nat Rev Nephrol. 2014;10(2):69–70. Epub 2013/12/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ratliff BB, Abdulmahdi W, Pawar R, Wolin MS. Oxidant mechanisms in renal injury and disease. Antioxid Redox Signal. 2016;25:119. Epub 2016/02/26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. Epub 2007/01/24 [DOI] [PubMed] [Google Scholar]
- 13.Hoenig MP, Zeidel ML. Homeostasis, the milieu interieur, and the wisdom of the nephron. Clin J Am Soc Nephrol. 2014;9(7):1272–81. Epub 2014/05/03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forbes JM. Mitochondria-power players in kidney function? Trends Endocrinol Metab. 2016;27(7):441–2. Epub 2016/05/25 [DOI] [PubMed] [Google Scholar]
- 15.Hall AM, Unwin RJ, Parker N, Duchen MR. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol. 2009;20(6):1293–302. Epub 2009/05/28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller-Deile J, Schiffer M. The podocyte power-plant disaster and its contribution to glomerulopathy. Front Endocrinol (Lausanne). 2014;5:209. Epub 2015/01/08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stieger N, Worthmann K, Teng B, Engeli S, Das AM, Haller H, et al. Impact of high glucose and transforming growth factor-beta on bioenergetic profiles in podocytes. Metabolism. 2012;61(8):1073–86. Epub 2012/03/01 [DOI] [PubMed] [Google Scholar]
- 18.Weinberg JM. Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol. 2011;22(3):431–6. Epub 2011/03/01 [DOI] [PubMed] [Google Scholar]
- 19.Funk JA, Schnellmann RG. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol Appl Pharmacol. 2013;273(2):345–54. Epub 2013/10/08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stallons LJ, Funk JA, Schnellmann RG. Mitochondrial homeostasis in acute organ failure. Curr Pathobiol Rep. 2013;1(3):169–77. Epub 2014/01/05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quiros PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17(4):213–26. Epub 2016/03/10 [DOI] [PubMed] [Google Scholar]
- 22.Tran M, Parikh SM. Mitochondrial biogenesis in the acutely injured kidney. Nephron Clin Pract. 2014;127(1–4):42–5. Epub 2014/10/25 [DOI] [PubMed] [Google Scholar]
- 23.Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121(10):4003–14. Epub 2011/09/02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531(7595):528–32. Epub 2016/03/17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88(Pt B):179–88. Epub 2015/05/16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoja C, Benigni A, Remuzzi G. The Nrf2 pathway in the progression of renal disease. Nephrol Dial Transplant. 2014;29(Suppl 1):i19–24. Epub 2013/06/14 [DOI] [PubMed] [Google Scholar]
- 27.Verdin E NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208–13. Epub 2016/01/20 [DOI] [PubMed] [Google Scholar]
- 28.Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22(1):31–53. Epub 2015/06/30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha SP, Whayne TF Jr. Coenzyme Q-10 in human health: supporting evidence? South Med J. 2016;109(1):17–21. Epub 2016/01/08 [DOI] [PubMed] [Google Scholar]
- 30.Ascenzi P, Coletta M, Wilson MT, Fiorucci L, Marino M, Polticelli F, et al. Cardiolipin-cytochrome c complex: switching cytochrome c from an electron-transfer shuttle to a myoglobin- and a peroxidase-like heme-protein. IUBMB Life. 2015;67(2):98–109. Epub 2015/04/11 [DOI] [PubMed] [Google Scholar]
- 31.O’Brien ES, Nucci NV, Fuglestad B, Tommos C, Wand AJ. Defining the apoptotic trigger: the interaction of cytochrome c and cardiolipin. J Biol Chem. 2015;290(52):30879–87. Epub 2015/10/22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–69. Epub 2012/06/26 [DOI] [PubMed] [Google Scholar]
- 33.Antonenkov VD, Isomursu A, Mennerich D, Vapola MH, Weiher H, Kietzmann T, et al. The human mitochondrial DNA depletion syndrome gene MPV17 encodes a non-selective channel that modulates membrane potential. J Biol Chem. 2015;290(22):13840–61. Epub 2015/04/12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dalla Rosa I, Camara Y, Durigon R, Moss CF, Vidoni S, Akman G, et al. MPV17 loss causes deoxynucleotide insufficiency and slow DNA replication in mitochondria. PLoS Genet. 2016;12(1):e1005779. Epub 2016/01/14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krick S, Shi S, Ju W, Faul C, Tsai SY, Mundel P, et al. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc Natl Acad Sci U S A. 2008;105(37):14106–11. Epub 2008/09/06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Phys Renal Phys. 2014;306(4):F367–78. Epub 2013/12/07 [DOI] [PubMed] [Google Scholar]
- 37.Putti R, Sica R, Migliaccio V, Lionetti L. Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol. 2015;6:109. Epub 2015/04/24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491–506. Epub 2013/04/09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–31. Epub 2015/12/17 [DOI] [PubMed] [Google Scholar]
- 40.McDonnell E, Peterson BS, Bomze HM, Hirschey MD. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol Metab. 2015;26(9):486–92. Epub 2015/07/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benigni A, Perico L, Macconi D. Mitochondrial dynamics is linked to longevity and protects from end-organ injury: the emerging role of sirtuin 3. Antioxid Redox Signal. 2016;25(4):185–99. Epub 2016/03/15 [DOI] [PubMed] [Google Scholar]
- 42.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83(4):568–81. Epub 2013/01/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34(5):807–19. Epub 2013/12/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015;125(2):715–26. Epub 2015/01/22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol. 2014;171(8):1917–42. Epub 2014/04/12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, et al. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol. 2015;26(5):1040–52. Epub 2014/11/20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mehta RL, Cerda J, Burdmann EA, Tonelli M, Garcia-Garcia G, Jha V, et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. 2015;385(9987):2616–43. Epub 2015/03/18 [DOI] [PubMed] [Google Scholar]
- 48.Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380(9843):756–66. Epub 2012/05/24 [DOI] [PubMed] [Google Scholar]
- 49.Eirin A, Lerman A, Lerman LO. The Emerging role of mitochondrial targeting in kidney disease. Handbook experimental pharmacology. Berlin/Heidelberg: Springer; 2016. p. 1–22. Copyright Holder Springer International Publishing Switzerland. Epub 2016/06/19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang C, Dong Z. Mitochondria in kidney injury: when the power plant fails. J Am Soc Nephrol. 2016;27(7):1869–72. Epub 2016/01/09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H. Role of mitochondrial DNA in septic AKI via toll-like receptor 9. J Am Soc Nephrol. 2016;27(7):2009–20. Epub 2015/11/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;67:293–307. Epub 2016/01/16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kers J, Leemans JC, Linkermann A. An overview of pathways of regulated necrosis in acute kidney injury. Semin Nephrol. 2016;36(3):139–52. Epub 2016/06/25 [DOI] [PubMed] [Google Scholar]
- 54.Perazella MA, Luciano RL. Review of select causes of drug-induced AKI. Expert Rev Clin Pharmacol. 2015;8(4):367–71. Epub 2015/06/26 [DOI] [PubMed] [Google Scholar]
- 55.Pendergraft WF 3rd, Herlitz LC, Thornley-Brown D, Rosner M, Niles JL. Nephrotoxic effects of common and emerging drugs of abuse. Clin J Am Soc Nephrol. 2014;9(11):1996–2005. Epub 2014/07/19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferguson MA, Vaidya VS, Bonventre JV. Biomarkers of nephrotoxic acute kidney injury. Toxicology. 2008;245(3):182–93. Epub 2008/02/26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mukhopadhyay P, Horvath B, Zsengeller Z, Zielonka J, Tanchian G, Holovac E, et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med. 2012;52(2):497–506. Epub 2011/11/29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119(5):1275–85. Epub 2009/04/08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem. 2010;285(17):13045–56. Epub 2010/02/09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014:967826. Epub 2014/08/29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813–8. Epub 2005/08/18 [DOI] [PubMed] [Google Scholar]
- 62.Parikh SM, Yang Y, He L, Tang C, Zhan M, Dong Z. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol. 2015;35(1):108–19. Epub 2015/03/22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takasu O, Gaut JP, Watanabe E, To K, Fagley RE, Sato B, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187(5):509–17. Epub 2013/01/26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382(9888):260–72. Epub 2013/06/04 [DOI] [PubMed] [Google Scholar]
- 65.SYSTEM USRD. USRDS annual data report: epidemiology of kidney disease in the United States. Bethesda: USRDS- United States Renal Data System; 2015. [Google Scholar]
- 66.KDIGO 2012, CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 2013;2012 Suppl:1–150. [DOI] [PubMed] [Google Scholar]
- 67.D’Agati VD, Chagnac A, de Vries AP, Levi M, Porrini E, Herman-Edelstein M, et al. Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol. 2016;12(8):453–71. Epub 2016/06/07 [DOI] [PubMed] [Google Scholar]
- 68.Badal SS, Danesh FR. New insights into molecular mechanisms of diabetic kidney disease. Am J Kidney Dis. 2014;63(2 Suppl 2):S63–83. Epub 2014/01/28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93(1):137–88. Epub 2013/01/11 [DOI] [PubMed] [Google Scholar]
- 70.Blake R, Trounce IA. Mitochondrial dysfunction and complications associated with diabetes. Biochim Biophys Acta. 2014;1840(4):1404–12. Epub 2013/11/20 [DOI] [PubMed] [Google Scholar]
- 71.Obesity Sharma K., oxidative stress, and fibrosis in chronic kidney disease. Kidney Int Suppl (2011). 2014;4(1):113–7. Epub 2014/11/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Obesity Sharma K. and diabetic kidney disease: role of oxidant stress and redox balance. Antioxid Redox Signal. 2016;25(4):208–16. Epub 2016/03/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hallan S, Sharma K. The role of mitochondria in diabetic kidney disease. Curr Diabetes Rep. 2016;16(7):61. Epub 2016/05/10 [DOI] [PubMed] [Google Scholar]
- 74.Lash LH. Mitochondrial glutathione in diabetic nephropathy. J Clin Med. 2015;4(7):1428–47. Epub 2015/08/05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106(34):14385–90. Epub 2009/08/27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Savu O, Sunkari VG, Botusan IR, Grunler J, Nikoshkov A, Catrina SB. Stability of mitochondrial DNA against reactive oxygen species (ROS) generated in diabetes. Diabetes Metab Res Rev. 2011;27(5):470–9. Epub 2011/04/13 [DOI] [PubMed] [Google Scholar]
- 77.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–33. Epub 2005/12/29 [PubMed] [Google Scholar]
- 78.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15(2):186–200. Epub 2012/02/14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo K, Lu J, Huang Y, Wu M, Zhang L, Yu H, et al. Protective role of PGC-1alpha in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS One. 2015;10(4):e0125176. Epub 2015/04/09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mallipattu SK, He JC. KLF 6: a mitochondrial regulator in the kidney. Oncotarget. 2015;6(18):15720–1. Epub 2015/07/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mallipattu SK, He JC. The podocyte as a direct target for treatment of glomerular disease? Am J Phys Renal Phys. 2016;311(1):F46–51. Epub 2016/04/22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mallipattu SK, Horne SJ, D’Agati V, Narla G, Liu R, Frohman MA, et al. Kruppel-like factor 6 regulates mitochondrial function in the kidney. J Clin Invest. 2015;125(3):1347–61. Epub 2015/02/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kopp JB. Loss of Kruppel-like factor 6 cripples podocyte mitochondrial function. J Clin Invest. 2015;125(3):968–71. Epub 2015/02/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zwacka RM, Reuter A, Pfaff E, Moll J, Gorgas K, Karasawa M, et al. The glomerulosclerosis gene Mpv17 encodes a peroxisomal protein producing reactive oxygen species. EMBO J. 1994;13(21):5129–34. Epub 1994/11/01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Casalena G, Krick S, Daehn I, Yu L, Ju W, Shi S, et al. Mpv17 in mitochondria protects podocytes against mitochondrial dysfunction and apoptosis in vivo and in vitro. Am J Phys Renal Phys. 2014;306(11):F1372–80. Epub 2014/03/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Toole JF. Renal manifestations of genetic mitochondrial disease. Int J Nephrol Renov Dis. 2014;7:57–67. Epub 2014/02/12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guery B, Choukroun G, Noel LH, Clavel P, Rotig A, Lebon S, et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. J Am Soc Nephrol. 2003;14(8):2099–108. Epub 2003/07/23 [DOI] [PubMed] [Google Scholar]
- 88.Tzen CY, Tsai JD, Wu TY, Chen BF, Chen ML, Lin SP, et al. Tubulointerstitial nephritis associated with a novel mitochondrial point mutation. Kidney Int. 2001;59(3):846–54. Epub 2001/03/07 [DOI] [PubMed] [Google Scholar]
- 89.Carney EF. Tubular disease: mistargeted protein disrupts mitochondrial metabolism in inherited Fanconi syndrome. Nat Rev Nephrol. 2014;10(3):125. Epub 2014/01/22 [DOI] [PubMed] [Google Scholar]
- 90.Goto Y, Itami N, Kajii N, Tochimaru H, Endo M, Horai S. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J Pediatr. 1990;116(6):904–10. Epub 1990/06/01 [DOI] [PubMed] [Google Scholar]
- 91.Peng M, Falk MJ, Haase VH, King R, Polyak E, Selak M, et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008;4(4):e1000061. Epub 2008/04/26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, et al. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol. 2006;17(4):1135–42. Epub 2006/02/24 [DOI] [PubMed] [Google Scholar]
- 93.Kooman JP, Kotanko P, Schols AM, Shiels PG, Stenvinkel P. Chronic kidney disease and premature ageing. Nat Rev Nephrol. 2014;10(12):732–42. Epub 2014/10/08 [DOI] [PubMed] [Google Scholar]
- 94.Romano AD, Serviddio G, de Matthaeis A, Bellanti F, Vendemiale G. Oxidative stress and aging. J Nephrol. 2010;23(Suppl 15):S29–36. Epub 2010/11/27 [PubMed] [Google Scholar]
- 95.Fougeray S, Pallet N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol. 2015;11(1):34–45. Epub 2014/11/12 [DOI] [PubMed] [Google Scholar]
- 96.Schmitt R, Susnik N, Melk A. Molecular aspects of renal senescence. Curr Opin Organ Transplant. 2015;20(4):412–6. Epub 2015/07/01 [DOI] [PubMed] [Google Scholar]
- 97.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. 2011;108(36):14849–54. Epub 2011/08/20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tabara LC, Poveda J, Martin-Cleary C, Selgas R, Ortiz A, Sanchez-Nino MD. Mitochondria-targeted therapies for acute kidney injury. Expert Rev Mol Med. 2014;16:e13. Epub 2014/08/12 [DOI] [PubMed] [Google Scholar]
- 99.Granata S, Dalla Gassa A, Tomei P, Lupo A, Zaza G. Mitochondria: a new therapeutic target in chronic kidney disease. Nutr Metab (Lond). 2015;12:49. Epub 2015/11/28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kezic A, Spasojevic I, Lezaic V, Bajcetic M. Mitochondria-targeted antioxidants: future perspectives in kidney ischemia reperfusion injury. Oxidative Med Cell Longev. 2016;2016:2950503. Epub 2016/06/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ishimoto Y, Inagi R. Mitochondria: a therapeutic target in acute kidney injury. Nephrol Dial Transplant. 2016;31(7):1062–9. Epub 2015/09/04 [DOI] [PubMed] [Google Scholar]
- 102.Wang H, Guan Y, Karamercan MA, Ye L, Bhatti T, Becker LB, et al. Resveratrol rescues kidney mitochondrial function following hemorrhagic shock. Shock. 2015;44(2):173–80. Epub 2015/04/22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen L, Yang S, Zumbrun EE, Guan H, Nagarkatti PS, Nagarkatti M. Resveratrol attenuates lipopolysaccharide-induced acute kidney injury by suppressing inflammation driven by macrophages. Mol Nutr Food Res. 2015;59(5):853–64. Epub 2015/02/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lempiainen J, Finckenberg P, Levijoki J, Mervaala E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br J Pharmacol. 2012;166(6):1905–15. Epub 2012/02/14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol. 2014;25(6):1157–62. Epub 2014/02/11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Suzuki T, Yamaguchi H, Kikusato M, Matsuhashi T, Matsuo A, Sato T, et al. Mitochonic Acid 5 (MA-5), a derivative of the plant hormone Indole-3-acetic acid, improves survival of fibroblasts from patients with mitochondrial diseases. Tohoku J Exp Med. 2015;236(3):225–32. Epub 2015/06/30 [DOI] [PubMed] [Google Scholar]
- 107.Suzuki T, Yamaguchi H, Kikusato M, Hashizume O, Nagatoishi S, Matsuo A, et al. Mitochonic acid 5 binds mitochondria and ameliorates renal tubular and cardiac myocyte damage. J Am Soc Nephrol. 2016;27(7):1925–32. Epub 2015/11/27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171(8):2029–50. Epub 2013/10/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014;96(6):672–83. Epub 2014/09/05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24(8):1250–61. Epub 2013/07/03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eirin A, Ebrahimi B, Zhang X, Zhu XY, Woollard JR, He Q, et al. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res. 2014;103(4):461–72. Epub 2014/06/21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang Z, Ge Y, Bao H, Dworkin L, Peng A, Gong R. Redox-sensitive glycogen synthase kinase 3beta-directed control of mitochondrial permeability transition: rheostatic regulation of acute kidney injury. Free Radic Biol Med. 2013;65:849–58. Epub 2013/08/27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guan N, Ren YL, Liu XY, Zhang Y, Pei P, Zhu SN, et al. Protective role of cyclosporine A and minocycline on mitochondrial disequilibrium-related podocyte injury and proteinuria occurrence induced by adriamycin. Nephrol Dial Transplant. 2015;30(6):957–69. Epub 2015/02/04 [DOI] [PubMed] [Google Scholar]
- 114.Wang Z, Bao H, Ge Y, Zhuang S, Peng A, Gong R. Pharmacological targeting of GSK3beta confers protection against podocytopathy and proteinuria by desensitizing mitochondrial permeability transition. Br J Pharmacol. 2015;172(3):895–909. Epub 2014/09/30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14(2):193–204. Epub 2008/02/13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rosdah AA, Holien JK, Delbridge LM, Dusting GJ, Lim SY. Mitochondrial fission – a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect. 2016;4(3):e00235. Epub 2016/07/20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tang WX, Wu WH, Qiu HY, Bo H, Huang SM. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J Nephrol. 2013;26(6):1073–82. Epub 2013/04/05 [DOI] [PubMed] [Google Scholar]
- 118.Sumida M, Doi K, Ogasawara E, Yamashita T, Hamasaki Y, Kariya T, et al. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol. 2015;26(10):2378–87. Epub 2015/02/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Papp Z, Edes I, Fruhwald S, De Hert SG, Salmenpera M, Leppikangas H, et al. Levosimendan: molecular mechanisms and clinical implications: consensus of experts on the mechanisms of action of levosimendan. Int J Cardiol. 2012;159(2):82–7. Epub 2011/07/26 [DOI] [PubMed] [Google Scholar]