

Summary
Bi-species, fusion-mediated, somatic cell reprogramming allows precise, organism-specific tracking of unknown lineage drivers. Fusion of Tcf7l1−/− murine embryonic stem cells with EBV-transformed human B-cell lymphocytes, leads to generation of bi-species heterokaryons. Human mRNA transcript profiling at multiple timepoints allows to track reprogramming of B-cell nuclei to a multipotent state. Interrogation of a human B-cell regulatory network with gene expression signatures identifies 8 candidate Master Regulator proteins. Out of these 8 candidates, ectopic expression of BAZ2B, from the bromodomain family – efficiently reprograms hematopoietic committed progenitors into a multipotent state, and significantly enhances their long-term clonogenicity, sternness and engraftment in immune compromised mice. In conclusion, unbiased systems biology approaches let us to identify the early driving events of human B-cell reprogramming.
Introduction
Early events driving somatic cell reprogramming to pluripotency can be effectively elucidated using cell-to-cell fusion approaches (Lluis et al., 2008; Sanges et al., 2011; Soza-Ried and Fisher, 2012; Tada et al., 1997; Ying et al., 2002). Consistently, cell fusion was shown to play a physiological role during in vivo regeneration of several tissues (Altarche-Xifro et al., 2016; Alvarez-Dolado et al., 2003; Pedone et al., 2017; Sanges et al., 2013; Wang et al., 2003). In particular, bi-species heterokaryons, derived from fusion between cells of two different species, have been used to study nuclear reprogramming by monitoring species-specific gene expression changes in time-dependent fashion (Bhutani et al., 2010; Brady et al., 2013; Foshay et al., 2012; Pereira et al., 2008).
A drawback of such studies is that identification of functionally relevant genes relies mainly on differential expression analysis, thus preventing clear differentiation of causally relevant driver-genes, responsible for mechanistic activity controlling reprograming events (Dong et al., 2015; Lefebvre et al., 2012). The VIPER (Virtual Inference of protein activity by Enriched Regulon analysis) algorithm (Alvarez et al., 2016), an extension of the Master Regulator Inference Algorithm (MARINa) (Lefebvre et al., 2010), addresses this issue by accurately inferring the differential activity of transcriptional regulators from the differential expression of their transcriptional targets. Both algorithms were highly effective in identifying Master Regulator (MR) proteins, whose coordinated activity is necessary and/or sufficient to induce lineage differentiation/maturation (Kushwaha et al., 2016; Kushwaha et al., 2015; Lefebvre et al., 2010), cellular reprogramming (Carro et al., 2010; Talos et al., 2017).
To elucidate early drivers of B-cell reprogramming, we fused Tcf7l1−/− murine embryonic stem cells (ESCs) with human Epstein Barr Virus-immortalized human B-lymphocytes (EBV-B) and isolated the resulting bi-species heterokaryons. Tcf7l1—a key effector of the Wnt pathway (Hackett and Surani, 2014; Merrill, 2012)—plays a crucial role in pluripotency maintenance. Moreover, Tcf7l1 significantly enhances the efficiency of reprogramming of the somatic nucleus in cell-fusion hybrids (Lluis et al., 2011), which are therefore an ideal cellular context to study reprogramming initiation.
VIPER analysis identified two distinct MR protein sets that causally regulate the transcriptional signature of the reprogramming event, including an “early” and a “late” regulatory program. These were sequentially activated during somatic B-cell reprogramming. We identified and experimentally tested 8 “late” MRs—then refined to 5—as drivers of committed progenitor reprogramming to a multipotent state. Ectopic expression of a single MR, BAZ2B, significantly enhanced sternness and clonogenicity of human cord blood-derived CD34+ cells as well as reprogramming of human hematopoietic committed progenitors. BAZ2B remodeled chromatin in distal elements of committed progenitors and the resulting multipotent hematopoietic stem cells could repopulate the bone marrow of immunocompromised mice after long term engraftment. These results confirm BAZ2B’s ability to mechanistically control the reprogramming signature of human hematopoietic cells and suggest that the proposed approach is effective in prioritizing key functional drivers of cell state reprogramming events.
Results
Two distinct master regulator repertoires are sequentially activated in heterokaryons during human EBV-B reprogramming
To identify unknown MRs that drive initiation of reprogramming, we fused murine Tcf7l1−/− ESCs with EBV-B cells, thus yielding bi-species heterokaryons. The hybrid cells were FACS-sorted and sequenced at different time-points after fusion (Figure 1A, S1A, S1B, S1C and Table S1). Sequencing reads were mapped to mouse and human genomes (Figure S1C, Table S1, see STAR Methods for details) to efficiently track the human somatic cell reprogramming, with high reproducibility among replicates (Figure S2A, S2B).
Figure 1. Fusion of mouse ESCs with human EBV-B lymphocytes, induces genome-wide transcriptional changes in the human nuclei.
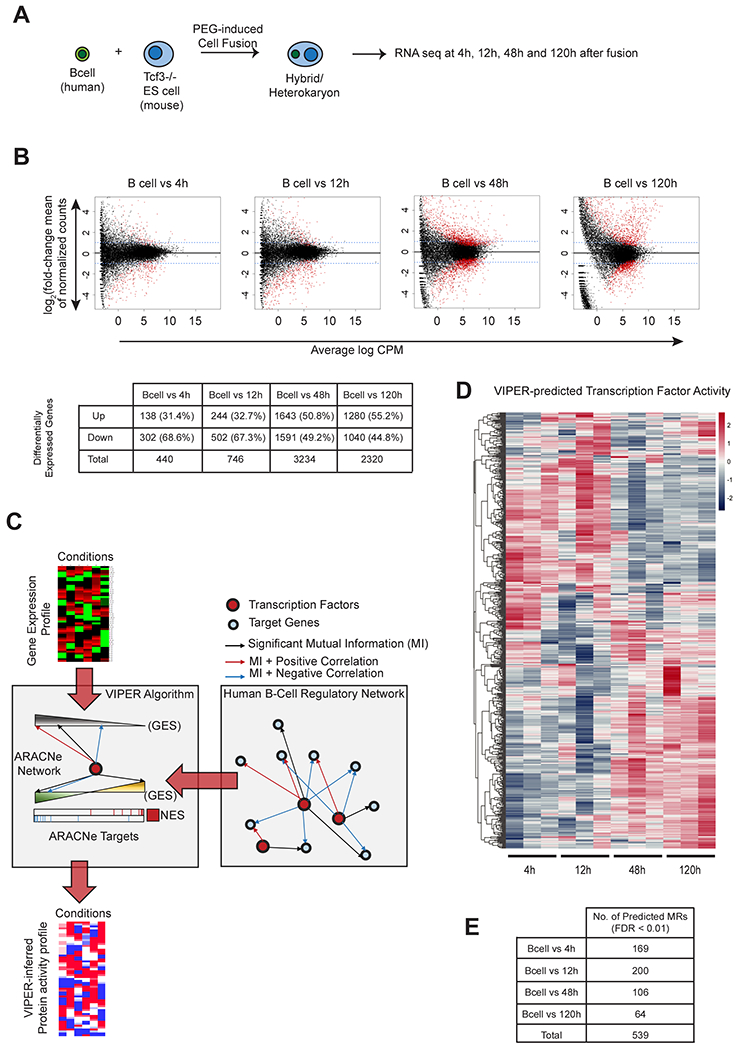
(A) Schematic for generation of heterokaryons and paired-end sequencing. (B) Differential expression of human genes after fusion of murine Tcf7l1−/− ESCs with human EBV-B lymphocytes. x-axis : Log2 fold-change of normalized counts and y-axis : mean of normalized counts. Blue-dashed line indicates fold-change of 1. Genes with significant (FDR < 0.05) change in expression are shown in red. Average log CPM (Count Per Million) values were calculated from 3 biological replicates for each time-point. Table shows upregulated and downregulated genes. See also Figure S1. (C) Schematic for the generation of protein activity profile using the VIPER algorithm and ARACNe network (See STAR Methods for details). (D) Heatmap of VIPER-predicted Normalized Enrichment Score values (NES values) of 539 MRs that had significant predicted activities (FDR < 0.01) in the heterokaryon samples. NES values were calculated using the VIPER algorithm by comparing each heterokaryon sample against the unfused PEG-treated B-cells. Positive NES values indicating active MRs are shown in red, and negative NES values indicating silenced MRs are shown in blue. (E) Number of MRs predicted in each timepoint (FDR < 0.01).
Differential expression analyses showed significant global changes in gene expression in the human genome, both early after cell fusion and at later time points (Figure 1B). Five days after fusion with Tcf7l1−/− ESCs, mRNA expression of human pluripotency markers such as NANOG, POU5F1 and KLF4 was upregulated in the heterokaryons (Figure S2C), consistent with a previous report (Pereira et al., 2008), suggesting reprogramming of human EBV-B cells toward a pluripotent state.
In order to identify MRs whose activity plays a mechanistic role in the reprogramming of human EBV-B cell nuclei, a B-cell regulatory network (BCRN) was interrogated with human gene expression signatures using the VIPER algorithm (Alvarez et al., 2016), for each sampled time point (Figure 1C and see STAR Methods for details). The BCRN was assembled by integrating two previously published datasets generated by ARACNe analysis of a large collection of normal and tumor related gene expression profiles (Basso et al., 2005; Lefebvre et al., 2010). The analysis identified 539 candidate MRs that, based on differential expression of their transcriptional targets, were significantly differentially activated in at least one sample (FDR < 0.01) (Figure 1D, 1E), suggesting a causal role in mechanistically establishing the transcriptional state of the reprogrammed B-cell nuclei at the corresponding time points.
Singular value decomposition (SVD) (Alter et al., 2000) was used to identify MRs providing the most orthogonal contribution at the 4 time points (4h, 12h, 48h and 120h). Specifically 12 principal components were identified (eigengenes), representing orthogonal linear combinations of weighted MR activity (Figure 2A). Critically, the first two principal components accounted for most of the total data variance (~85%) (Figure 2A) and showed unique and opposite, time-dependent MR activity patterns during heterokaryon reprogramming (Figure 2B).
Figure 2. Two distinct clusters of transcription factors are sequentially activated in a time-dependent manner in the human EBV-B nuclei in the heterokaryons.
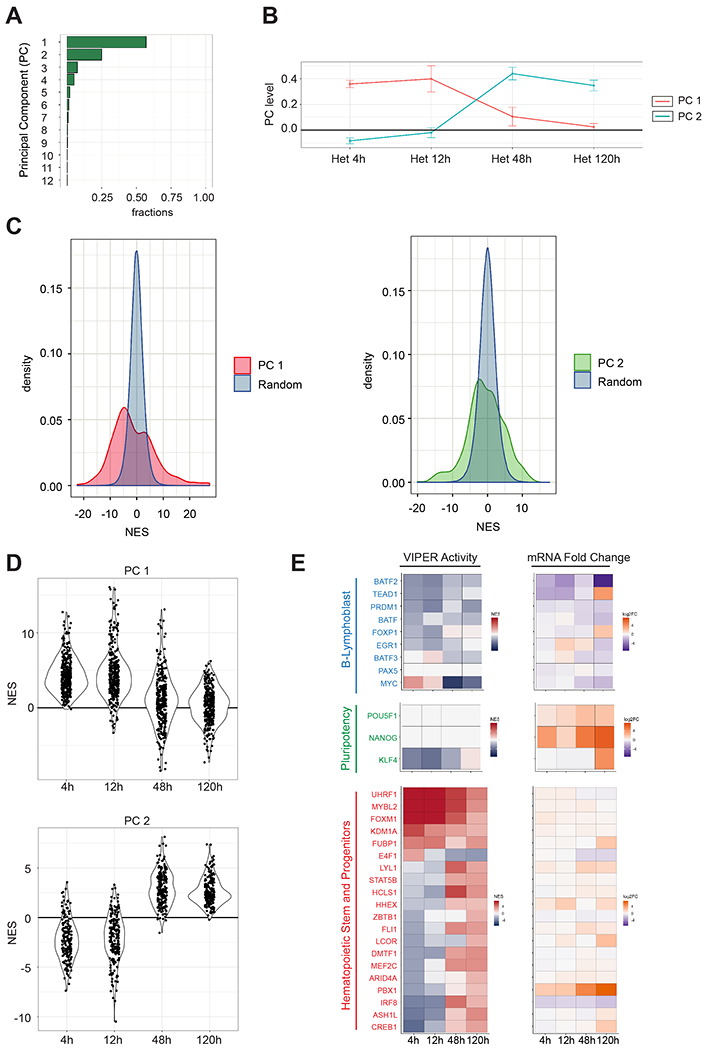
(A) Single Value Decomposition (SVD) analysis. PC are plotted on the y-axis and the proportion of variance of VIPER activity in the heterokaryon dataset is plotted on the x-axis. (B) PC levels across the sample time points for the top-2 PCs of the heterokaryon dataset. (C) Genes contributing significantly to the PCs 1 and 2 by comparing their PC coefficients with PC coefficients calculated from randomly shuffled VIPER activity levels. The random distributions are shown in blue and the coefficients PC 1 and PC 2 in red and green, respectively (p < 0.05). (D) Violin plots for VIPER-predicted activity levels represented by average NES for the 105 transcription factors (upper panel) significantly (p < 0.05) associated with PC 1 and for 64 transcription factors (lower panel) significantly associated with PC 2. VIPER-predicted activities were calculated comparing the heterokaryon samples of each time point against the unfused EBV-B cells. (E) VIPER-predicted activity and differential expression of a representative set of human genes during reprogramming. Positive NES values indicating active MRs are shown in red, and negative NES values indicating silenced MRs are shown in blue. mRNA Fold Change (Log2FC) was calculated using EdgeR by comparing the heterokaryon samples with unfused B-cells. Upregulated genes are shown in orange, and downregulated genes are shown in dark purple.
Based on the first two principal components we could broadly classify relevant MRs into 2 distinct clusters – one associated with MRs activated from Oh to 12h (early) and one associated with MRs activated after 12h (late). Key MRs were identified as those with coefficients corresponding to a statistically significant p-value (p ≤ 0.05) based on a null model assembled by sample shuffling (Figure 2C). Thus, the analysis identified two distinct, highly coordinated programs controlling reprogramming, including 105 MRs significantly associated with the first principal component (early reprogramming events) (Figure 2D, top panel, Table S2) and 64 MRs significantly associated with the second principal component (late reprogramming events) (Figure 2D bottom panel, Table S3).
VIPER analysis showed rapid inactivation of established B-cell related lineage proteins (,BATF2, TEAD1, PRDM1, BATF, FOXP1, EGR1, BATF3, PAX5), included in the two programs (Figure 2E). These are known to maintain B-cell commitment and differentiation (Dinkel et al., 1998; Laurenti et al., 2013; Nutt et al., 1999), B-cell activation (Briana et al., 2010; Turner et al., 1994; Wataru et al., 2011), and B-cell survival (Martine van et al., 2014).
EBV-mediated human B-cell immortalization is driven by the oncogene MYC (Kaiser et al., 1999; Wood et al., 2016). At 48 hours and 5 days after fusion, VIPER-inferred MYC activity was significantly decreased (FDR < 0.01) (Figure 2E). Critically, while the mRNA expression levels of pluripotency genes, such as POU5F1, NANOG and KLF4, were significantly upregulated (adjusted p-value < 0.05) at later time-points they showed no differential activity until 5 days after fusion (Figure 2E). Moreover, genome-wide comparison of heterokaryon gene expression profiles with those of human induced Pluripotent Stem Cells (iPSC) and human ESCs (Choi et al., 2015) did not show significant similarity (Figure S2D), suggesting that even five days after fusion, the reprogramming state of heterokaryons differs from those of human ESCs and iPSCs.
Finally, MRs such as UHRF1, MYBL2, F0XM1 and KDM1A, known to regulate hematopoietic stem cell (HSC) and progenitor cell function (Baker et al., 2014; Hou et al., 2015; Wang et al., 2018; Zhao et al., 2017), were significantly activated at the early time points (Figure 2E). Likewise, LYL1, DMTF1 and ASH1L known to play a key role in HSC survival (Souroullas et al., 2009), quiescence (Kobayashi and Srour, 2011) and maintenance (Jones et al., 2015), respectively, were exclusively activated at 48h and 120h late time points (Figure 2E). Taken together, these data suggest that, upon fusion with mouse Tcf7l1−/− ESCs, human EBV-B cell nuclei may be reprogrammed toward an HSC-like state.
Human EBV-B lymphocytes are reprogrammed to a hematopoietic stem and progenitor-like state
To determine the transcriptional identity of the human nucleus within heterokaryons, we compared their human transcriptome with those of a publicly available human hematopoietic lineage dataset (Figure S3A, S3B, S3C) (Laurenti et al., 2013). Two major clusters were identified in the latter, one including stem-like cells (HSCs, MPPs and MLPs), and one representing lineage-committed cells (CMPs, GMPs and MEPs), (see Figure S3B for non-abbreviated names), albeit with cross-replicate variability (Figure S3C), possibly due to cellular heterogeneity between individual cord blood donors.
VIPER analysis of genes differentially expresses in each Laurenti et al. sample, compared to human B-cells, identified 445 candidate MRs with statistically significant differential activity in at least one sample (FDR < 0.01) (Figure 3A). Interestingly, MR activity in the hematopoietic lineage showed a bimodal pattern similar to the early and late transcriptional programs in the heterokaryons (Figure 1D, 2B), with “stem-cell” related MRs active in HSC, MPP, and MLP cells and then inactivated in committed progenitors (CMP, GMP, and MEP), where they were replaced by a second wave of activated MRs (Figure 3A). Consistent with previous data (Laurenti et al., 2013), MLP expression profiles were similar to those of HSCs and MPPs.
Figure 3. The human EBV-B nuclei are reprogrammed to a multipotent hematopoietic stem progenitor-like state 5 days after fusion.
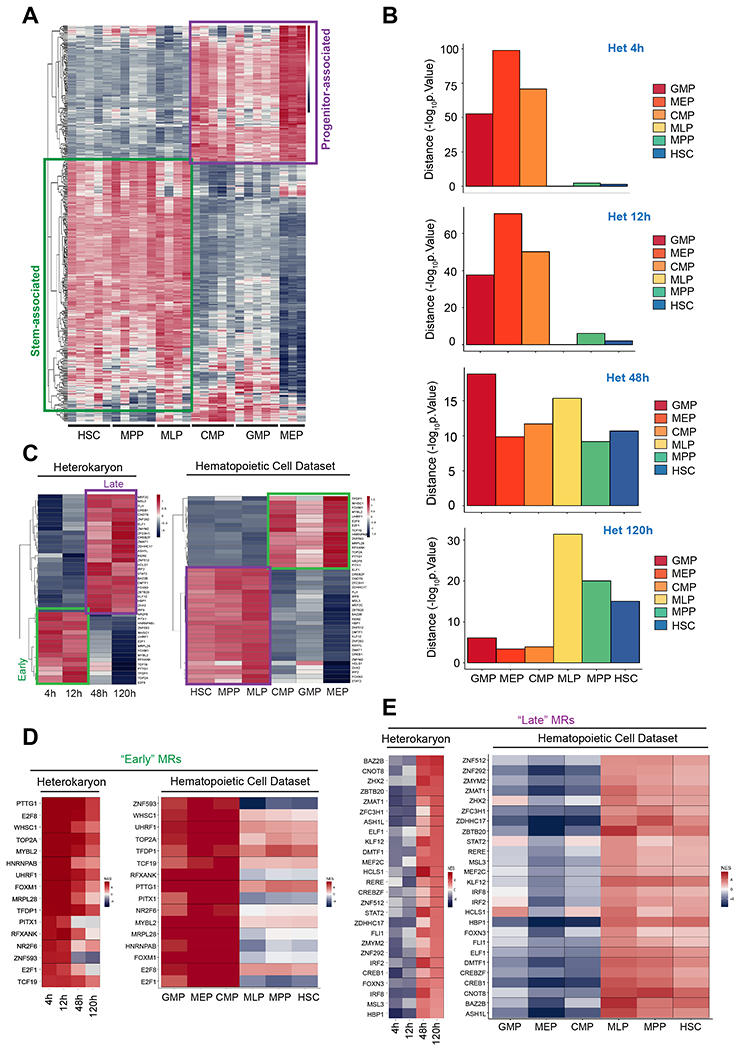
(A) Heatmap of predicted activity for significant hematopoietic Master Regulators (MRs). Heatmap shows NES values of 445 MRs that were significant (FDR < 0.01) in each sample from the Laurenti et al., dataset compared to unfused B-cell samples. (B) Correlation of transcription factor with positive activity (FDR < 0.05) between heterokaryons and the human hematopoietic cells using Fisher’s Exact Test (FET). −Log10 of the p-values of the overlap are shown. (C) Heatmap of the early and late MR programs in the Heterokaryons and in the hematopoietic cell dataset. (D, E) Heatmap showing the activity of the “Early” MRs (D) and “Late” MRs (E) in the heterokaryon and the hematopoietic cell dataset. (A, C-E) Positive NES values indicating active MRs are shown in red, and negative NES values indicating silenced MRs are shown in blue.
VIPER was effective in recapitulating established, physiologically relevant MRs in the hematopoietic system. MYBL2 and E4F1 were significantly activated in the myeloid progenitor population (Figure S3D), consistent with knockout studies in mice where depletion of Mybl2 or E4f1 led to myeloid progenitor cell apoptosis (Baker et al., 2014; Grote et al., 2015). HHEX had the highest activity in MLP (Figure S3D), consistent with the role of HHEX for lymphoid lineage specification(Goodings et al., 2015; Jackson et al., 2015). LCOR was significantly activated in HSC, MPP and MLP populations, in agreement with its role in inducing the reprogramming of hemogenic endothelium cells into hematopoietic stem and progenitor cells (Sugimura et al., 2017). ASH1L was significantly activated in HSC and MPP (Figure S3D), consistent with studies showing that depletion of Ash 11 severely affects HSC self-renewal potential (Jones et al., 2015).
We then assessed the overlap of statistically significant activated MRs (FDR < 0.05) in both datasets (Figure 3B). The analysis showed that MR activity profiles of early heterokaryons (4h and 12h after fusion) significantly overlapped with those of lineage-committed progenitors, which comprise myeloid progenitors (GMP, MEP, CMP) (Figure 3B) (p < 1E-5, by FET). Conversely, MR activity profiles of late heterokaryons (120h after fusion) significantly overlapped with those of stem and multipotent-progenitors (HSC, MPP and MLPs) (Figure 3B) (p < 1 E-5, by FET), while heterokaryons at 48h after fusion, showed a significant overlap with all populations (p < 1 E-5, by Fisher’s Exact Test (FET)).
Of these, 16 MRs had comparable differential activity in early heterokaryons and in lineage committed progenitors (Figure 3C, 3D) and 26 TFs in late heterokaryons and stem/multipotent progenitors (Figure 3C, 3E) (FDR < 0.01). These two sets were also associated with the first and second principal components, respectively (Figure 2A and Table S2 and S3), suggesting a causal role in implementing the transcriptional programs associated with EBV-B cell reprogramming.
Taken together, these data suggest that, following fusion, human EBV-B cell nuclei are first (4h/12h) reprogrammed to a state resembling that of a proliferative, lineage-committed progenitor (Figure S3E), which is mechanistically regulated by the concerted activity of a first wave of activated TFs (Early-MRs). Following this initial transition, human nuclei are then further reprogrammed toward a hematopoietic/multipotent state (48h/120h) by a second wave of activated TFs (Late-MRs).
A combination of 8 MRs can enhance the clonogenicity and stemness of human CD34+ hematopoietic progenitor cells.
Since Late-MRs were predicted to reprogram EBV-B cells toward an HSC-like state in the heterokaryon system, we chose to investigate their ability to induce stemness of human CD34+ hematopoietic progenitor cells towards an HSC-like state. We first ranked the 26 MRs in the Late-MR cluster (Figure 3C and 3E) based on their VIPER-inferred activity in each of the stem fractions (HSC, MPP and MLP) and in the heterokaryon 120h time point (Table S4). We then selected the 7 MRs with highest VIPER-inferred activity in both late heterokaryons (48h/120h) and HSC/MPP/MLP cells (DMTF1, BAZ2B, ZBTB20, ZMAT1, CNOT8, KLF12, HBP1), as well as FLI1 (Figure 3C, 3E and Table S4) also enriched as a significantly active MR and previously shown to control HSC formation during mouse development (Gottgens et al., 2002; Schutte et al., 2012). We cloned these MRs into a doxycycline inducible lentiviral vector, carrying a constitutive GFP reporter, and then infected CD34+ human hematopoietic progenitor cells with all 8 MRs and with all possible combinations of 7 out of 8 (Figure S4A and 4A). Finally, we tested the effect on stemness and clonogenicity of the transduced CD34+ cells.
Figure 4. A combination of the predicted MRs enhances the stemness and long-term clonogenicity of CD34+ human hematopoietic cells.
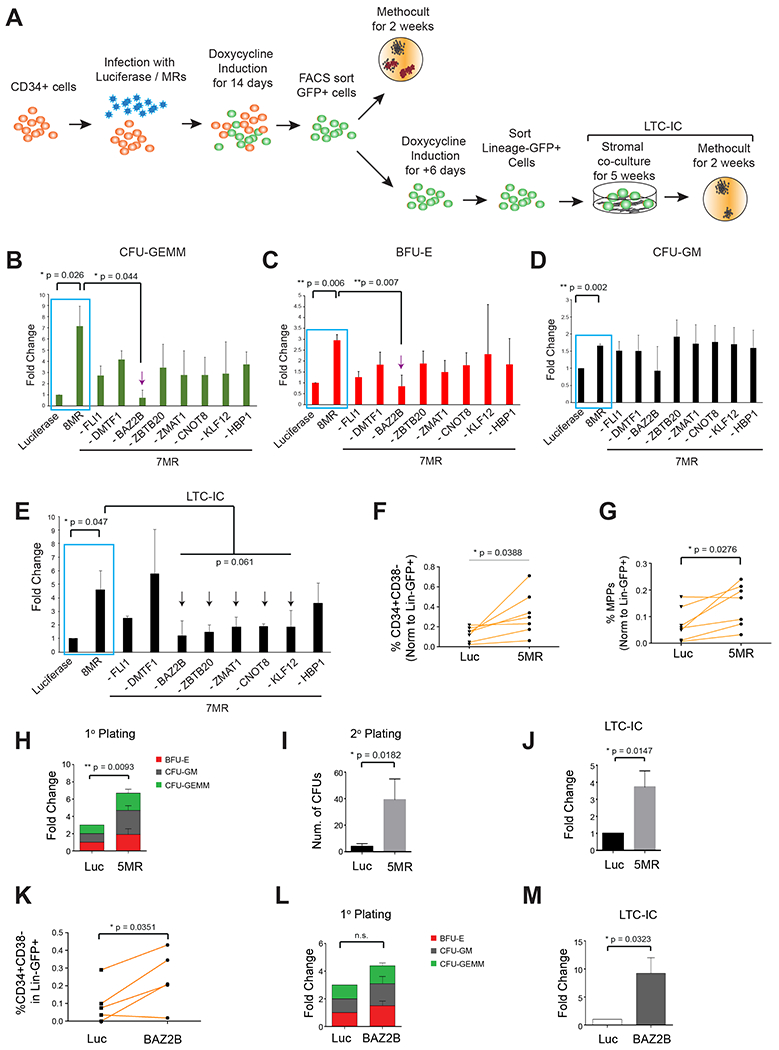
(A) Schematic showing the experimental workflow of the first screen with human CD34+ hematopoietic stem and progenitor cells. (B-D) Human CD34+ cells infected with 8 MRs, or 8 combinations of 7 MRs were plated on methocult assays to count colonies of lineages (B) CFU-GEMM, (C) BFU-E and (D) CFU-GM based on morphology. N = 3 donors. Data represented as mean ± SD. (E) Sorted Lineage-GFP+ cells plated on LTC-IC assay followed by counting of colonies. N = 3 donors. Data represented as mean ± SD. Luc vs 8-MR: two-tailed paired t-test **P<0.01, *P<0.05. 8-MR vs 7-MR combinations: two-tailed unpaired t-test with unequal variance. (F-J) Overexpression of a combination of 5 MRs in human CD34+ cells. Lineage-GFP+ cells were FACS analyzed or characterized by in vitro colony assays. (F) Quantification of Lin-CD34+CD38− Stem Progenitors represented for N = 7 donors. (G) Quantification of MPPs represented for N = 7 donors. (H) Quantification of colonies from primary CFC assay N = 6 donors. Data represented as mean ± SD. (I) Quantification of colonies from secondary CFC assay N= 5 donors. Data represented as mean ± SD. (J) Quantification of colonies from LTC-IC CFC assay N = 5 donors. Data represented as mean ± SD. (K-M) Human CD34+ cells transduced with Luciferase or BAZ2B for in vitro analysis. (K) Quantification of the CD34+CD38− multipotent stem progenitor within Lineage-GFP+ cells from N = 5 donors (L) Quantification of colonies from primary CFC assay N = 5 donors. Data represented as mean ± SD. (M) Quantification of colonies from LTC-IC CFC assay N= 5 donors. Data represented as mean ± SD. (B-M) Two-tailed paired t-test, unless specified otherwise **P<0.01, *P<0.05.
We induced expression of the 8-MR and of each 7-MR cocktail for 14 days and FACS-sorted GFP+ cells to test their colony-forming ability (Figure 4A). The 8-MR cocktail showed substantial increase of CFU-GEMM (Colony-Forming Unit-Granulocyte, Erythrocyte, Monocyte, Megakaryocyte) colonies, representing the primitive stem progenitors (Figure 4B, approximately 7-fold increase with respect to the Luciferase control). The 8-MR cocktail also increased the number of BFU-E (Burst-Forming Unit-Erythroid) and CFU-GM (CFU-Granulocyte, Monocyte) colonies (approximately 3-fold and 1.7-fold increase respectively, compared to Luciferase control) (Figure 4C, 4D). Interestingly, all 7-MR cocktails yielded a lower number of CFU-GEMM colonies (Figure 4B), thus suggesting that all candidate MRs emerging from the VIPER analysis may work in concert to induce stemness. Yet, CFU-GEMM colony formation was severely reduced for the 7-MR cocktail lacking BAZ2B (Figure 4B), suggesting that this gene may have a dominant role. Consistently, BAZ2B removal also significantly reduced the number of BFU-E colony forming units (Figure 4C) and mildly reduced CFU-GM colonies (Figure 4D).
The long-term clonogenic (LTC-IC) capacity of the progenitors cells was enhanced 5-fold by the 8-MR cocktail compared to luciferase controls (Figure 4A, 4E). However, longterm clonogenicity was compromised in 5 of the 7-MR cocktails, specifically those lacking BAZ2B, ZBTB20, ZMAT1, CNOT8 and KLF12 (Figure 4E). Interestingly these 5 MRs (BAZ2B, ZBTB20, ZMAT1, CNOT8 and KLF12) also represent the 5 most statistically significant MRs, based on VIPER-predicted activity in both reprogrammed heterokaryons at 120h and hematopoietic stem and multipotent fractions (Table S4).
To robustly validate these 5 MRs, we performed inducible, co-ectopic expression of the 5-MR cocktail in CD34+ human hematopoietic progenitor cells (Figure S4B) and found significant enrichment of the Lin-CD34+CD38− stem/progenitor cells and of the Lineage-GFP+CD34+CD38-CD45RA-90-hematopoietic multipotent cells (p = 0.038 and p = 0.027 respectively) (Figure 4F, 4G, S4C). Interestingly, GFP+ cells that were transduced with the 5-MR cocktail showed significant increase of BFU-E, CFU-GM and CFU-GEMM colonies in primary methocult assays (p = 0.009) (Figure 4H, S4D). Furthermore, overexpression of the 5-MR cocktail led to a significantly higher number of CFU compared to Luciferase control in secondary methocult assays (p = 0.018) (S4B, Figure 4I, S4E). Finally, the expression of the 5-MR cocktail also significantly increased the long-term clonogenicity of the cells (p = 0.0147) (Figure 4J, S4B and S4F). Collectively, these data suggest that the overexpression of the 5 genes (BAZ2B, ZBTB20, ZMAT1, CNOT8 and KLF12) in human CD34+ progenitors is effective in inducing stemness and clonogenicity.
BAZ2B enhances the long-term clonogenicity and stemness in human CD34+ hematopoietic progenitor cells
BAZ2B is the MR with the highest VIPER-predicted activity in both heterokaryon samples at 120h and the HSC fraction of human hematopoietic cells (Table S4, Figure 3E). Consistently, the 7-MR cocktail lacking BAZ2B severely reduced the ability of CD34+ cells to form colonies, compared to the 8-MR cocktail. Moreover, leading-edge enrichment analysis of genes differentially expressed in the heterokaryon dataset in ARACNe-inferred BAZ2B targets identified key factors such as EPC2, a Polycomb complex protein (Searle and Pillus, 2018); PRMT5, a histone methyl transferase essential for mouse preimplantation development (Stopa et al., 2015; Tee et al., 2010); VNN2/GPI-80, essential for human HSC maintenance and engraftment (Prashad et al., 2015); TPP1, associated with a critical function in telomeric protection (Nandakumar et al., 2012; Wang et al., 2007; Xin et al., 2007); GEMIN5, an RNA-binding protein that regulates global mRNA translation (Francisco-Velilla et al., 2016); LYAR, a transcription factor that targets chromatin factors (Luna-Pelaez and Garcia-Dominguez, 2018); and CUL3, an E3 ubiquitin ligase that can regulate the expression of transcriptional MRs from the bromodomain protein family (Dai et al., 2017; Janouskova et al., 2017) (Figure S4G). This suggests that BAZ2B may regulate expression of genes controlling chromatin modification, gene transcription, mRNA translational control, telomere protection and hematopoietic cell engraftment and expansion. These predictions along with the observation that the CD34+ cells displayed enhanced stemness and clonogenicity after BAZ2B expression, motivated us to further investigate this MR as a critical, single-reprogramming factor.
We expressed BAZ2B for 2 weeks in human CD34+ cells, followed by clonogenicity and stemness assays. Interestingly, we observed consistent increase in (Lineage-GFP+CD34+CD38−) hematopoietic stem and multipotent progenitors compared to Luciferase controls (Figure 4K, S4H). In the primary colony-forming assay, we found only a mild increase in the number of colony-forming units (Figure 4L, S4I). Interestingly however, the LTC-IC assays showed that the ectopic BAZ2B expression in CD34+ cells induced dramatic increase of colony-forming units, compared to Luciferase controls (p = 0.032) (Figure 4M). Furthermore, in some cases ectopic BAZ2B expression lead to both BFU-E and CFU-GEMM colony formation (Figure S4J). CFU-GM colonies from BAZ2B-expressing cells were also much larger, compared to Luciferase-treated cells (Figure S4J).
These data suggest that BAZ2B overexpression in CD34+ cells is sufficient to significantly enhance stemness and increase their long-term clonogenic potential.
BAZ2B enhances the renewal of multipotent Lin-CD34+CD38− hematopoietic progenitors
CD34+ cells represent a heterogeneous mixture of stem cells and lineage committed progenitors. Sorted Lineage-CD34+CD38− cells can differentiate into CD33+ myeloid and CD19+ B lymphoid lineages (Figure S5A–F) (Doulatov et al., 2010; Laurenti et al., 2013; Mazurier et al., 2003). In contrast, the lineage-committed progenitor fraction isolated by Lin-CD34+CD38+ surface markers was effectively depleted of HSC, MPP and MLP cells and could not engraft in the bone marrow or peripheral blood (Figure S5A–F), as shown by previous studies (Doulatov et al., 2010; Laurenti et al., 2013; Mazurier et al., 2003).
To assess whether BAZ2B could enhance hematopoietic stem and progenitor cell renewal and increase their in vivo engraftment potential, we induced expression of exogenous Luciferase or BAZ2B in Lin-CD34+CD38− stem fraction for 14 days (Figure 5A). BAZ2B overexpression significantly expanded the multipotent stem fraction of CD34+CD45RA-CD90+ within the Lineage-GFP+ population (Figure 5B and 5C). To assess long-term engraftment efficiency, we sorted Lineage-GFP+ cells at 14d following induction and transplanted them intra-femorally in irradiated NOD SCID Gamma (NSG) mice (Figure 5A). At 12 weeks after transplantation, BAZ2B-transduced cells showed significant enhancement of engraftment in the bone marrow (p = 0.018) (Figure 5D, 5E and 5F), albeit with donor-to-donor variability in engraftment efficiency (Figure 5E). BAZ2B-transduced cells also showed significant enhancement in spleen (p = 0.017) and peripheral blood engraftment (p = 0.046) (Figure 5G, S5G, S5H) compared to Luciferase-transduced controls. BAZ2B-transduced cells showed lymphoid potential with significant increase in the proportion of CD19+ B-lymphocytes within human CD45+ engrafted cells, in the bone marrow, spleen and peripheral blood (Figure 5H), which did not compromise the myeloid fraction. The proportion of the myeloid (CD33+) lineage within the engrafted human CD45+ population was similar in Luciferase and BAZ2B transduced cells (Figure 5H). We also confirmed that preferential differentiation toward lymphoid lineage was consistent with the CD19+ lymphoid-biased lineage potential of the uncultured, freshly isolated, Lineage-CD34+CD38− stem fraction transplanted in the NSG mice (Figure S5D, S5F).
Figure 5. BAZ2B enhances renewal of Lin-CD34+CD38− stem progenitor fraction.
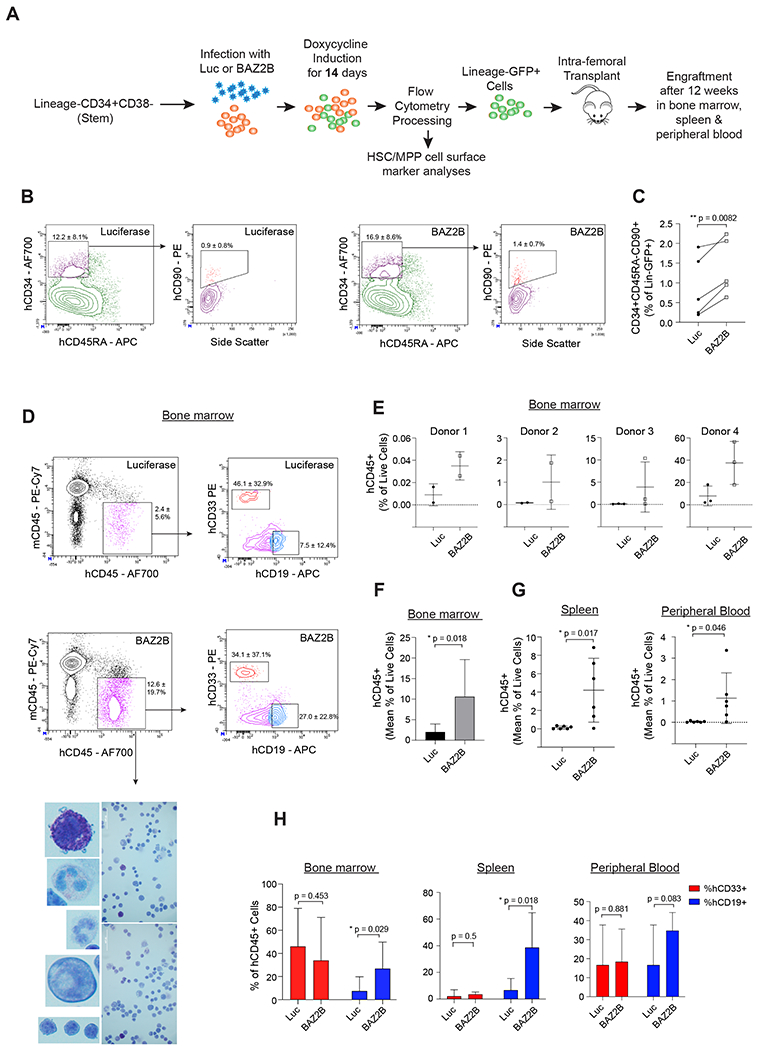
(A) Lin-CD34+CD38− cells were transduced with Luciferase or BAZ2B and intra-femoral transplanted into irradiated NSG mice (See STAR Methods, for details). (B-C) FACS analyses of the Lineage-GFP+ population after gene induction (B) Representative FACS plot showing the enrichment of CD34+CD45RA-CD90+ population in BAZ2B vs Luciferase-transduced cells. Percentage fractions for each gate normalized to Lineage-GFP+ fraction represented as mean ± SD from N = 5 donors. (C) Quantification of the CD34+CD45RA-CD90+ normalized to Lineage-GFP+ cells N = 5 donors. Two-tailed paired t-test **P<0.01, *P<0.05. (D-F) Bone marrow FACS analyses of the transplanted NSG mice after 12 weeks. (D) Representative FACS plot showing the enrichment of the engrafted human CD45+ cells in the BAZ2B vs Luciferase-transduced cells. Percentage fractions for hCD45 gate normalized to live cells represented as mean ± SD. CD33+ myeloid and CD19+ lymphoid gates show the lineage potential for the human CD45+ cells. Percentage fractions for hCD33/hCD19 gate normalized to hCD45+ cells represented as mean ± SD. N = 4 donors, 2-3 mice per donor. Bottom panel from top to bottom: basophils, eosinophils, neutrophils, monocytes and lymphocytes derived from human CD45+ cells stained with the Wright-giemsa method. Scale bar: 50 urn. (E) Quantification of engrafted human CD45+ cells within the total live cells of the mouse bone marrow. N= 4 donors; 2-3 mice transplanted per donor. (F) Mean bone marrow engraftment of human CD45+ cells. N = 4 donors. Two-tailed paired t-test **P<0.01, *P<0.05. (G) Quantification of engrafted human CD45+ cells in the spleen and peripheral blood of the transplanted NSG mice. N= 2 donors; 2-3 mice per donor. Two tailed unpaired t-test **P<0.01, *P<0.05. (H) Quantification of the CD33+ myeloid and CD19+ lymphoid fraction normalized to the total human CD45+ cells engrafted in the bone marrow, spleen and peripheral blood. Bone marrow N= 4 donors; 2-3 mice per donor. Spleen and peripheral blood N= 2 donors; 2-3 mice per donor. Two tailed unpaired t-test **P<0.01, *P<0.05.
Taken together, these data show that transient BAZ2B overexpression, during ex vivo expansion of Lineage-CD34+CD38− cells, enhances renewal of long-term engraftable multipotent hematopoietic progenitors that can differentiate into both myeloid and B-lymphoid lineages.
BAZ2B reprograms lineage-committed progenitors into multipotent hematopoietic cells.
To assess whether ectopic BAZ2B expression may be sufficient to reprogram lineage-committed progenitors toward multipotency, we FACS-sorted Lin-CD34+CD38+ committed progenitors (Figure 6A), which are unable to engraft in the bone marrow (Figure S5A–E) (Doulatov et al., 2010; Laurenti et al., 2013; Mazurier et al., 2003). Interestingly, ectopic BAZ2B expression in committed progenitors induced significant enrichment of the Lin-CD34+CD38− stem and multipotent progenitors, across four different cord blood derived donors (Figure 6B, Figure S6A), albeit with donor-to-donor variability. Moreover, ectopic BAZ2B expression induced consistent and significant increase in the total number of colony-forming units—and thus of BFU-E, CFU-GM and CFU-GEMM colonies—compared to Luciferase (p = 0.0171) (Figure 6C, S6B). Furthermore, we observed significant colony number increase in long-term clonogenicity LTC-IC assays (p=0.0109) (Figure 6D, S6C), suggesting that ectopic BAZ2B expression alone is sufficient to induce lineage-committed progenitor reprogramming into a multipotent stem cell state, with increased clonogenic capacity.
Figure 6. BAZ2B induces reprogramming of lineage committed hematopoietic progenitors to multipotent state by chromatin remodeling.
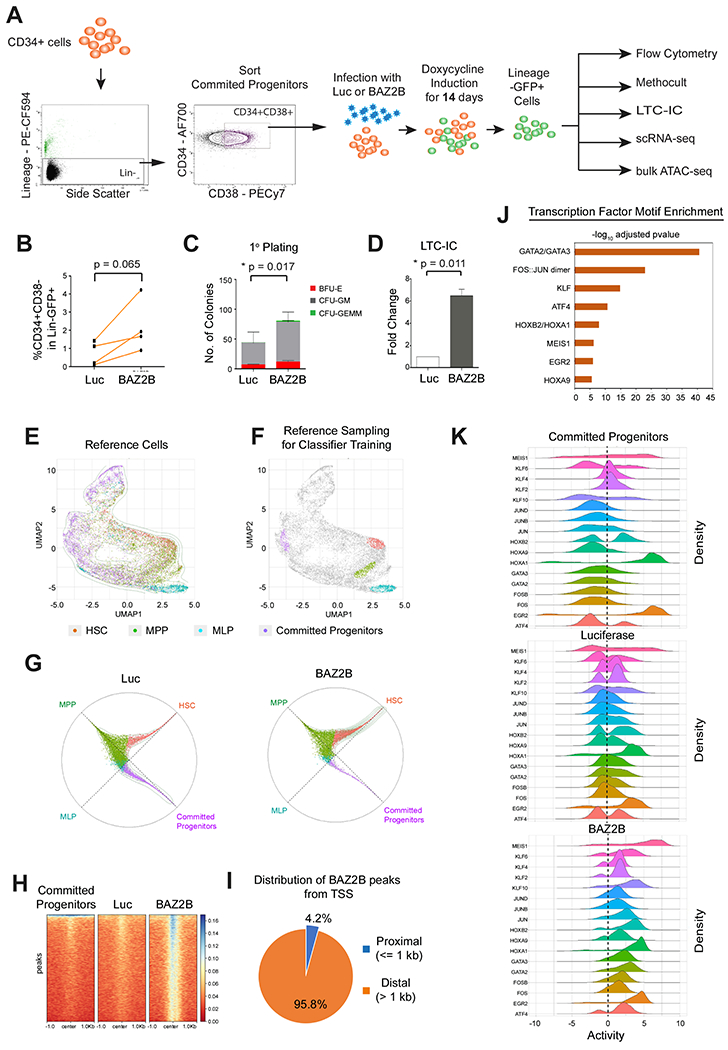
(A) Lineage-CD34+CD38+ committed progenitors were transduced with Luciferase or BAZ2B for in vitro analysis. (B) Quantification of the CD34+CD38− multipotent stem progenitor within Lineage-GFP+ cells from N = 4 donors. (C) Quantification of colonies from primary CFC assay N = 3 donors. Data represented as mean ± SD. (D) Quantification of colonies from LTC-IC CFC assay N= 3 donors. Data represented as mean ± SD. (B-D) Two-tailed paired t-test **P<0.01, *P<0.05. (E) UMAP visualization of the four reference populations FISCs, MPPs, MLPs and Lineage Committed Progenitors in VIPER space. (F) Top 1% of the cells of the four reference populations used for the random forest model, based on their differential density in the UMAP space. (G) Circular visualization of the model classification for Luciferase and BAZ2B samples. Samples closer to the circumference have a definite classification, and the plotting angle for each sample is determined by the weighted average of the model’s classification votes. (H) Heatmap of ATAC-Seq peaks showing unique SAZ2S-induced chromatin-accessible regions. (I) Distribution of BAZ2B-induced unique ATAC-Seq peaks from the transcription start sites. (J) Transcription factors with enriched motifs in the BAZ2B-induced nucleosome-free regions and a predicted VIPER activity > 1. (K) Ridgeline density plots showing VIPER-activity from single-cell RNA-Seq samples for 17 transcription factors (VIPER activity > 1) with enriched motifs in BAZ2B-induced nucleosome-free regions.
To further assess reprogramming potential of the BAZ2B-induced progenitor population, at the molecular level, we profiled single cells mRNA before and after BAZ2B expression. To establish a positive control for the stemness signature, we sorted HSCs, MPPs, MLPs and lineage-committed progenitor populations, and performed single-cell mRNA sequencing and analysis of these populations (Figure S6D). To specifically investigate BAZ2B-mediated HSC-like state induction we transduced Luciferase or BAZ2B in Lineage-CD34+CD38+ committed progenitors and sorted Lineage-GFP+ cells for singlecell expression profiling (Figure S6D).
We first used the single-cell gene expression profiles of each sorted population (HSC, MPP, MLP, Lineage committed progenitors, BAZ2B expressing Lineage-CD34+CD38+ progenitors and luciferase expressing controls) to generate an ARACNe-inferred, singlecell hematopoietic lineage regulatory networks, independent of prior knowledge. We then used metaVIPER, a single cell extension of the VIPER algorithm (Ding et al., 2018), to measure protein activity at the single-cell level, followed by UMAP dimensionality reduction, resulting into a 2D spatial map of the distinct sub-populations (Figure 6E).
To refine the reference populations to be used in the model, we performed a probability density analysis, in each of the four reference populations, to determine the UMAP regions with the highest differential probability density (peak) and selected the top 1% of cells under each peak. This provided an optimal single cell reference for each population (Figure 6F). The analysis confirmed activation of well-established, sub-population-specific lineage markers within each reference sample—e.g., GATA2 and HMGA2 in HSCs and MPPs and BCL11A in MLPs, (Figure S6E). A random forest classifier was trained using the top 46 most differentially active proteins (Table S5) in the selected reference populations (See STAR Methods for details). We then analyzed lineage-committed progenitors overexpressing Luciferase or BAZ2B using metaVIPER and used the random forest classifier to classify each single cell as either an HSC, MPP, MLP, or Committed Progenitor. The resulting classification is shown in the circle plots (Figure 6G), where the distance from the origin is inversely proportional to classification uncertainty (based on entropy analysis). Thus, cells with a definitive classification appear near the circumference, while those with more ambiguous classifications appear closer to the circle’s center. The angle at which each cell appears is determined by the average of their classification score across each of the four classes, weighted by a power of two. As expected, classification of committed progenitors overexpressing Luciferase shows a heterogeneous population with a significant proportion of lineage-committed progenitors, a few progenitors with multipotent properties (MPP- or MLP-like cell) and a negligible number of HSC-like cells. In sharp contrast, compared to Luciferase-transduced cells, ectopic BAZ2B expression induced highly statistically significant increase in the HSC-like compartment (p < 2.2E-16), as shown by a dramatic shift of the HSC-specific probability density towards the circumference of the circle plot, and concomitant depletion of committed progenitors (p < 2.2E-16) (Figure 6G). Sternness induction was associated with significant increase of both BAZ2B expression and activity within the same population (Figure S6F, S6G). Increase in HSC-like cells was also complemented by significant decrease in multipotent-primed or MPP-like cells (p = 3.2E-09) and lymphoid-primed MLP-like cells (p < 2.2E-16).
Taken together, these data suggest that, although lineage-committed progenitors from the Lineage-CD34+CD38+ fraction represent a highly heterogenous population of differentiated and multipotent primed cells, BAZ2B overexpression induces reprogramming of lineage-committed progenitors, lymphoid and multipotent-primed progenitors towards a HSC-like state.
BAZ2B induces genome-wide chromatin remodeling in distal elements
Previous reports suggest that BAZ2B might play a role in chromatin remodeling (Bortoluzzi et al., 2017; Oppikofer et al., 2017; Tallant et al., 2015). To elucidate BAZ2B’s role in chromatin accessibility, we performed ATAC-sequencing analysis of both Luciferase and BAZ2B-transduced committed Lineage-CD34+CD38+ progenitors (Figure 6A). BAZ2B overexpression increased accessibility to unique chromatin regions that were otherwise inaccessible in Luciferase-transduced progenitors and in untransduced and uncultured committed progenitors (Figure 6H, S7A). The majority (95.8%) of the chromatin accessible regions were localized in distal elements, more than 1kb away from transcription start sites (Figure 6I). We observed significant enrichment of 152 transcription factor binding motifs, classified under 30 transcription factor families, within BAZ2B-induced nucleosome-free regions (Table S6 and See STAR Methods for details). Interestingly, the analysis predicted significant differential enrichment of transcription factor motifs from the GATA family and from the activating protein 1 (AP-1) complex comprising heterodimers between FOS (FOS, FOSB, FOSL1, FOSL2) and JUN (JUN, JUNB, JUND) family proteins (Hess et al., 2004; Jochum et al., 2001) (Table S6). We then measured VIPER activity for the corresponding TFs from the single cell RNASeq data (Figure 6E, 6F, 6G). Of 152 TF identified by binding motif analysis, 57 showed differential VIPER-measured protein activity in the single cell dataset. Of these 48 were significantly activated or silenced in BAZ2B-transduced progenitors (Table S7). Interestingly, the top 17 MRs with the most significant protein activity (NES >1.0, p <= 1.25-218) (Table S7) were members of 8 TF families whose binding motifs were significantly enriched in the BAZ2B-induced nucleosome-free regions (Figure 6J, S7B). Of note, global comparison of differential protein activity in the single cell data showed a significant shift towards transcriptional activation for these 17 TFs in BAZ2B-transduced progenitors, compared to the Luciferase-transduced progenitors or the starting population of untransduced and uncultured committed progenitors (Figure 6K). Consistent with ATAC-Seq data, VIPER activity was significantly increased in BAZ2B-transduced progenitors for MEIS1 shown to enhance reprogramming efficiency of mouse hematopoietic progenitors into long-term engraftable HSCs (Riddell et al., 2014). GATA2 and GATA3 were also significantly activated in the BAZ2B-transduced progenitors. These factors play a key role in reprogramming of fibroblasts to hematopoietic progenitors (Gomes et al., 2018; Pereira et al., 2013) and in HSC renewal (Frelin et al., 2013). The transcriptional activity of FOS and FOSB were also significantly activated in BAZ2B- transduced progenitors. FOS and FOSB were previously validated as key factors to reprogram human fibroblasts (Gomes et al., 2018) or endothelial cells (Sandler et al., 2014) into hematopoietic progenitors, respectively. We also observed a concomitant increase in the activity of JUN, JUNB and JUND transcription factors, suggesting the potential activity of AP-1 complex of FOS/JUN heterodimers. AP-1 complex was previously shown to be essential for the specification of hematopoietic progenitors from mouse ESCs (Obier et al., 2016). The transcription factors with a lower VIPER activity (<1) did not show a major difference in transcriptional activity in BAZ2B-transduced progenitors compared to the Luciferase-transduced or untransduced committed progenitors (Figure S7C), confirming VIPER prediction accuracy. VIPER activity for a majority of MRs converged to zero in the Luciferase-transduced cells, compared to untransduced committed progenitors or the BAZ2B-transduced progenitors (Figure 6K, S7C). This could potentially be due to the prolonged culture condition of 14 days that leads to differentiation of Luciferase-transduced cells.
These data suggest that BAZ2B can mechanistically induce genome-wide chromatin remodeling, consistent with transcriptional regulation by MR proteins that, in turn, can induce reprogramming of committed progenitors towards a multipotent stem state.
Reprogramming of lineage committed progenitors by BAZ2B generates multipotent hematopoietic progenitors with long-term engraftment potential
To demonstrate long-term engraftment capacity of reprogrammed committed progenitors, we assessed in vivo reprograming of Lin-CD34+CD38+ committed progenitors following ectopic BAZ2B or Luciferase in vivo expression into NSG mice (Figure 7A and see STAR Methods for details) as described previously (Riddell et al., 2014; Sugimura et al., 2017). BAZ2B overexpression led to engraftment enhancement in the bone marrow, spleen and peripheral blood, albeit the reprogramming of the committed progenitors into multipotent hematopoietic progenitors showed variability of engraftment efficiency across donor samples and mice (Figure 7B, 7C, S8A and S8B; cells engrafted from one out of three donors). Furthermore, reprogrammed progenitors were able to differentiate into CD33+ myeloid and CD19+ lymphoid lineage cells (Figure 7D). Taken together, these data suggest that BAZ2B overexpression in lineage-committed progenitors was effective in reprograming these cells towards a multipotent hematopoietic progenitor state, promoting enhanced stemness, clonogenicity and long-term engraftment potential.
Figure 7. BAZ2B-induced multipotent hematopoietic progenitors possess long-term engraftment potential.
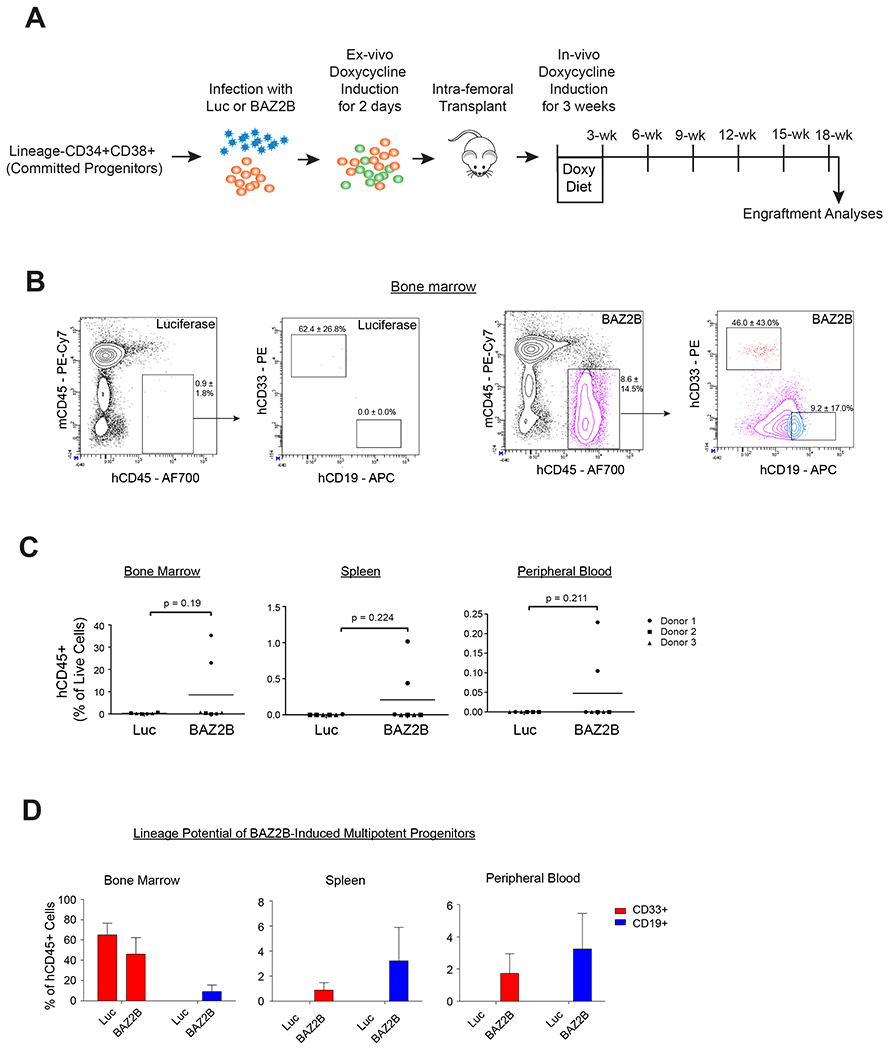
(A) Lin-CD34+CD38+ committed progenitors were sorted and transduced with Luciferase or BAZ2B followed by NSG mice transplantation (See STAR Methods, for details). Human hematopoietic engraftment was analyzed after 16 weeks. (B) FACS plot showing engraftment of human CD45+ hematopoietic cells in the mouse bone marrow upon expression of BAZ2B or Luciferase in Lin-CD34+CD38+ committed progenitors. Percentage fractions for hCD45 gate normalized to live cells represented as mean ± SD. CD33+ myeloid and CD19+ lymphoid gates show the lineage potential for the human CD45+ cells. Percentage fractions for hCD33/hCD19 gate normalized to hCD45+ cells represented as mean ± SD. N = 3 donors with 2-3 mice per donor. (C) Quantification of the engraftment of human CD45+ cells with respect to total live cells in the bone marrow, spleen and peripheral blood. Two-tailed unpaired t-test. (D) Quantification of the CD33+ myeloid and CD19+ lymphoid cells with respect to the total human CD45+ hematopoietic cells from Luciferase or BAZ2B-induced multipotent hematopoietic progenitors.
Discussion
Reprogramming of somatic cells toward a hematopoietic precursor lineage is widely studied, since the precise molecular mechanisms presiding over this process are still elusive. Given the relevance of hematopoiesis in clinical care, this also represents a critically important area of investigation for translational medicine applications. We carried out a transcription factor regulatory network analysis and identified MRs that are drivers of reprogramming. We discovered a group of early MRs that likely control, directly or indirectly the activity of the identified late MRs. Notably, our methodology led us to discover key MRs that could reprogram lymphoid cells into a multipotent hematopoietic stem state. Importantly, this method can be used to study any reprogramming event that can be followed over time either in bulk population or in single cells.
The human EBV-B lymphocytes upon fusion with mouse ESCs, are reprogrammed within the hematopoietic hierarchy to a multipotent hematopoietic stem progenitor-like state and not toward an embryonic stem cell state. This might imply that the reprogramming within the same lineage is more likely to occur. Indeed, maintenance of epigenetic markers of the cell of origin has been shown in the reprogramming toward iPSCs (Kim et al., 2010; Polo et al., 2010). Accordingly, we identified and experimentally validated one MR, BAZ2B, able to reprogram the hematopoietic lineage-committed progenitors into multipotent stem state, resulting in reprogrammed cells with an increased long-term clonogenicity, enhanced engraftment potential and ability to differentiate into multiple lineages. We observed significant variability of engraftment among the human donor samples and the transplanted mice. This was expected since both the human hematopoietic stem and committed progenitor fractions show a great level of phenotypic variability from donor to donor. Furthermore, the experimental setup for in vivo reprogramming requires maintaining a steady plasma concentration of doxycycline that has a very short elimination half-life of 3-6 hours in mice (Lucchetti et al., 2019). Moreover, the doxycycline half-life relies on the food and water consumption habits of the mice, which may vary on an individual level (Smarr et al., 2019) finally causing unavoidable variability in the in vivo reprogramming process.
Murine fibroblasts were reprogrammed to hemogenic endothelial precursor cells using a combination of 4 genes – GATA2, Gfi1b, cFos and Etv6 (Pereira et al., 2013). Another study reported the reprogramming of murine fibroblasts into multipotent hematopoietic progenitor cells using a combination of 5 genes – ERG, GATA2, LMO2, RUNX1c and SCL (Batta et al., 2014). Murine lineage-committed progenitors were reprogrammed into multipotent hematopoietic progenitors using a combination of 8 genes – Run1t1, Hlf, Lmo2, Prdm5, Pbx1, Zfp37, Mycn and Meisl (Riddell et al., 2014). In another study, human endothelial cells have been reprogrammed to multipotent hematopoietic progenitors using a combination of 4 genes - FOSB, GFI1, RUNX1 and SPI1 (Sandler et al., 2014). We confirmed the ability of one single gene, BAZ2B, to function as MR that can reprogram the committed progenitors into multipotent hematopoietic stem and progenitor cells.
The BAZ2B protein and its functional activity is not well understood. It consists of a bromodomain (BRD) and a plant homeodomain (PHD). Crystal structure studies of purified BAZ2B protein, show that the PHD domain interacts with unmodified histone H3K4 and the bromodomain can interact with the acetylated histone marks on H3K14 and H3K16 (Bortoluzzi et al., 2017; Tallant et al., 2015). Human BAZ2B protein has been identified as a component of the ISWI chromatin remodeling complex and physically interacts with the ISWI sub-components, SMARCA1 and SMARCA5 (Oppikofer et al., 2017) forming a catalytically active complex able to induce remodeling of the DNA-bound mononucleosomes (Oppikofer et al., 2017). In our heterokaryon studies, we found that the leading edge predicted targets of the BAZ2B include Polycomb factors, components of chromatin remodeling complexes and genes essential for human HSCs, among others. Therefore, we hypothesized that BAZ2B can induce reprogramming of the lineage-committed progenitors into multipotent cells through its remodeling activity by genomewide rewiring of the chromatin. Indeed, with the ATAC-Seq studies, we found that after 14 days of BAZ2B overexpression, the chromatin structure is opened to enhance accessibility to de novo genomic loci that were otherwise closed in the committed progenitors. This potentially allows for the binding of other MR genes (MEIS1, GATA2, FOS and FOSB) involved in hematopoietic reprogramming. Interestingly, a large majority of these genomic loci were in the distal elements, suggesting that BAZ2B potentially relies on long-range enhancer-promoter interactions for regulating transcription. These interactions could be investigated with Hi-C, 3C or Capture-C approaches in the future. Finally, our studies suggest the potential for BAZ2B to reprogram also different cell types, as fibroblasts and endothelial cells, a possibility that remains to be tested.
Beside its reprogramming function via a putative chromatin remodeling activity, we also propose that BAZ2B forms part of a critical transcription factor network, which enhances stemness and multipotency in hematopoietic cells. This finding might have important clinical applications. Indeed, there is a high demand of multipotent hematopoietic cells due to the lack of the availability of histocompatible donors for patients in need of transplantation. Our findings might have important applications in the major goal of generating autologous transplantable human multipotent hematopoietic cells.
Finally, our work also suggests that regulatory-network-based analysis of heterokaryon RNA profiles can provide critical biological insights, which are unlikely to emerge using more conventional gene-discovery methods based on literature mining or on differential gene expression analysis.
STAR Methods
Resource Availability
Lead Contact
Further information for resources and reagents can be obtained from the lead contact Dr. Maria Pia Cosma (pia.cosma@crg.eu).
Materials availability
All the unique/stable reagents generated from this study are available at request from the lead contact with a complete Materials Transfer Agreement.
Data and code availability
All the raw sequencing data related to the heterokaryon, hematopoietic single-cell and ATAC-seq data are available on the NCBI gene expression omnibus with the accession code GSE114240. Networks were generated using the ARACNe-AP tool from the Califano lab (https://github.com/califano-lab/ARACNe-AP)(Lachmann et al., 2016). VIPER tool is available to download from bioconductor: https://www.bioconductor.org/packages/release/bioc/html/viper.html. Implementation of all model training, validation, and testing, as well as subsequent downstream analyses and plotting can be found at https://github.com/califano-lab/COSMA.
Experimental Model and Subject Details
Cell lines
Tcf7l1−/− mouse embryonic stem cells (mESCs) are male and were a generous gift from Dr. Brad Merrill (UIC, USA). The mESCs were cultured at 37 degrees C in media supplemented with 20% serum and mLIF. The human B-cell line are EBV-immortalized human B lymphocytes that were obtained from the Corriell Institute of Medical Research (GM22647). The lymphoblast cell line was derived by Epstein-Barr Virus mediated immortalization of peripheral blood mononuclear cells (PBMCs) from a healthy Caucasian individual (Shirley et al., 2012). The genotype of the lymphoblast cell line was thoroughly assessed and showed a high concordance with the donor’s PBMCs (Shirley et al., 2012). The cell line did not show any abnormal copy number variations, or genetic mosaicism (Shirley et al., 2012). The gender information for the human EBV-B cell line is not provided by the Corriell Institute Repository. The human EBV-B cells were cultured at 37 degrees C in RPMI media supplemented with 20% foetal bovine serum.
Primary Cell Culture
Umbilical cord blood samples were purchased from the blood bank of Barcelona (Banc de Sang I Teixits) after approval from the Clinical Research Ethical Committee (CEIC, Parc de Salut Mar, Barcelona). For all of our experiments the human hematopoietic stem and progenitor cells were derived from fresh umbilical cord blood that were collected within less than 26 hours. Briefly we isolated the mononuclear cells from a fresh cord blood sample using a Ficoll gradient (Lymphoprep, Stemcell Technologies), followed by magnetic isolation of CD34+ cells using the Miltenyi human CD34 Ultrapure enrichment kit (Catalog # 130-100-453) according to the manufacturer’s instructions. For some of the experiments, we purchased frozen CD34+ human cord blood cells from Stemcell Technologies (Catalog # 70008.5). The number of cord blood samples used per study are indicated as donors in the figure legends since we ensured that cord blood samples used per study were from different donors. The genders of the cord blood samples were not provided by the blood bank of Barcelona nor from Stemcell Technologies.
Mice
Adult NOD.Cg-Prkdcscid II2rgtm1Wjl/SzJ (NSG) male or female mice at the age of 9-10 weeks were used for the transplantation experiments. The animal handling and transplant procedures were approved by the Ethical Committee of Animal Experimentation in Barcelona (CEEA). The mice were maintained in a pathogen-free facility with an automated 12-hour light/ 12-hour dark schedule and were provided with food and water ad libitum. All mice were generally maintained on a standard maintenance diet (Special Diets Services). For the in vivo reprogramming experiments, 2-3 days prior to transplantation, the NSG mice were placed on a SAFE doxycycline diet of food pellets containing 625 p.p.m of doxycycline (SAFE Diets-E8220 Version 0232) and drinking water was infused with 1 mg/ml of doxycycline. The doxycycline based diet was maintained for 3 weeks during the reprogramming and then the mice were returned to the standard maintenance diet with normal drinking water. For the transplantation experiments, we maintained an equal ratio of male and female mice whenever possible depending on the litter at the time of conducting the experiment. The number of mice used in each study are indicated in the figure legends.
Method Details
Human CD34+ Culture and Lentiviral Infection
Human CD34+ cells were cultured in serum-free enhanced media (Stemspan SFEM, StemCell Technologies) supplemented with two different formulations of recombinant human cytokines, (1) Stimulation media – contains SCF 300 ng/ml, FLT3 300 ng/ml, TPO 100 ng/ml, IL3 60 ng/ml (2) Maintenance media - SCF 100 ng/ml, FLT3 100 ng/ml, TPO 100 ng/ml, IL3 20 ng/ml, IL6 20 ng/ml and doxycycline 2 ug/ml. For experiments using the entire fraction of CD34+ cells, the cells were incubated in the stimulation media for 24 hours at 37 degrees C. The cells from each donor were then split into two separate wells and infected for a first round with lentiviral vectors containing the Luciferase Control or transcription factor cDNAs of interest and incubated overnight in the stimulation media at 37 degrees C. The cells were then washed and re-suspended in stimulation media. After approximately 4 hours the cells were re-infected for a second round with lentiviral vectors and continued incubation overnight at 37 degrees C. The cells were then washed and cultured in the maintenance media supplemented with 2 ug/ml of Doxycycline (Sigma Aldrich, Catalog # D9891) for the rest of the experiment. Every 2 days the cells were washed and re-plated in fresh media with doxycycline. For experiments associated with transplantation, we used the Stemspan SFEM II (StemCell Technologies) basal media. The maintenance media composition was changed to SCF 100 ng/ml, FLT3 100 ng/ml, TPO 50 ng/ml, UM171 35 nM (StemCell Technologies), SR1 750 nM (StemCell Technologies), LDL 10 ug/ml (StemCell Technologies) and doxycycline 2 ug/ml.
Committed progenitor isolation from CD34+ enriched cells, culture and infection
To isolate the Lineage-CD34+CD38+ lineage committed progenitors the CD34+ enriched cells were treated with anti-CD34 antibodies that targets a distinct epitope other than one used for isolation. For CD34+ cells isolated using the Miltenyi CD34 enrichment kits, we used the APC-labelled anti-human CD34 (Clone AC136). For the CD34+cells purchased from Stemcell Technologies we used the Alexa Fluor 700 labeled anti-human CD34 (Clone 581). In addition, we used a combination of anti-CD38 (Clone FIBC) antibody and a biotin-labeled cocktail of antibodies (from Miltenyi) targeting the human “Lineage” antigens CD2, CD3, CD11b, CD14, CD15, CD16, CD19, CD56, CD123, and CD235a. The cells were then sorted using BD FACS ARIA II flow cytometer. The sorted cells were cultured in the maintenance media – SFEM supplemented with SCF 100 ng/ml, FLT3 100 ng/ml, TPO 100 ng/ml, IL3 20 ng/ml, IL6 20 ng/ml and doxycycline 2 ug/ml. Approximately 2 hours after sorting, the cells from each donor were split into two separate wells and then infected with a first round of the lentiviral vectors containing Luciferase Control or transcription factor cDNAs and incubated overnight at 37 degrees C. The cells were then washed and re-suspended in the maintenance media. After approximately 4 hours the cells were re-infected with a second round of lentiviral vectors and continued incubation overnight at 37 degrees C. The cells were then washed and cultured in the maintenance media supplemented with 2 ug/ml of Doxycycline for the remainder of the experiment with fresh media changes for every 2 days.
Heterokaryon generation and RNA isolation
The human-mouse heterokaryons were generated as described previously (Pereira et al., 2008). For each heterokaryon sample, 30 million Tcf7l1−/− mESCs were labeled with Vybrant DiD (1:400) and 30 million human EBV-B lymphocytes were labeled with Vybrant DiO (1:400) for 15 mins at 37 degrees C. The labeled cells were then washed twice with PBS and resuspended in 6 ml of PBS each. The mESCs and the human EBV-B cells were then mixed in a 1:1 proportion and then centrifuged to pellet the cells. The pellet was disrupted and then resuspended in Polyethylene Glycol (PEG) in a dropwise manner with the procedure lasting a maximum of 60 seconds. They were then incubated at 37 degrees C for 90 seconds. The cells were then re-suspended slowly with serum-free DMEM in a dropwise manner, and constant shaking. The cells were then incubated for 3 min at 37 degrees C and spun down to recover a pellet. The supernatant was discarded and fresh mESC media (+LIF) was added without disrupting the pellet. The cells were then incubated at 37 degrees C for 3 mins and then plated on gelatin-coated plates. For the time points at 4 hours, 12 hours and 48 hours, the cells were harvested by collecting them in suspension in the supernatant followed by trypsinization of the remaining adherent cells on the plate surface. The cells were than washed and re-suspended in PBS (with 3%FBS and 2.5 mM EDTA) to be processed for FACS sorting. They were then sorted directly into the lysis buffer (Buffer RLT) provided in the Qiagen RNEasy mini kit (74104) using a 100 um nozzle at the flow cytometer (BD FACS ARIA II SORP). For the timepoint at day 5, we altered our sorting strategy. The cells were fused and plated on gelatin-coated plates as described above. After 4 hours, all the cells were harvested and the fused hybrids were sorted and replated on gelatin-coated plates in mESC media for 5 days. On day 5 all the cells were harvested again by trypsinization and lysed with lysis buffer (Buffer RLT) for RNA extraction using the Qiagen RNEasy mini kit (74104).
Immunofluorescence staining of sorted Heterokaryons
The fused cells were sorted as described above onto a slide. The cells were fixed with 4% PFA for 15 minutes at room temperature and then permeabilized with 0.3% triton for 20 minutes at room temperature. Blocking was performed for 30 minutes with 1% goat serum and 0.05% tween. The anti-human Lamin A/C (clone 636) was diluted 1:100 in blocking solution followed by incubation with the cells for 90 minutes at room temperature. The cells were then washed with PBS followed by incubation with the secondary antibody, goat anti-mouse Alexa Fluor 488 at 1:400 dilution for 45 mins at room temperature. The cells were washed again and then incubated with Alexa Fluor 568 Phalloidin at a dilution of 1:40 for 20 mins at room temperature. The cells were then washed and stained with the DNA-labeling dye DAPI. Confocal imaging was performed on a Leica TCS SPE inverted confocal microscope.
Sequencing of the Heterokaryon mRNA samples
RNA Samples isolated from the heterokaryons were further processed to generate sequencing libraries using a Truseq RNA library Prep Kit. The libraries were then analyzed on an Illumina HiSeq 2000 sequencer using 100 bp paired-end sequencing.
Single-cell sample processing and Sequencing
Single-cell RNA sequencing libraries were generated at the JP Sulzberger Columbia Genome Center using a 10X Genomics Chromium Controller and Single-cell 3’ Library & Gel Bead Kit v2 (10X Genomics, #120237). Single cells were sorted in a BD Influx cytometer and were pelleted by centrifugation (300rcf, 5min) followed by resuspension in DMEM at approximately 500cells/μl. Cell viability and concentration was verified using a Countess II Automated Cell Counter (ThermoFisher, #AMQAX1000). Each sample was loaded into one well of a Chromium chip (10X Genomics, #120236), following manufacturer’s instructions, and aiming for a recovery of 5,000 cells per sample. Library construction was carried out according to the manufacturer’s instructions and were sequenced on Illumina Hiseq 2000. The sequenced reads were processed through the Cell Ranger (10x Genomics) pipeline to generate the single-cell gene expression profile.
ATAC-seq sample processing and sequencing
FACS-sorted cells were pelleted and lysed in a buffer containing NP40 (0.1%), Tween-20 (0.1%) and Digitonin (0.01%). The lysis buffer was washed out to pellet the nuclei that were then resupended in a Tn5 tagmentation mix and incubated at 37 degrees C in a thermomix shaking at 1000 rpm. The DNA was then extracted using a Zymo DNA clean and concentrator kit and eluted in 22 uL ultrapure water. Appropriate sample indexing for 6-plex sequencing on an Illumina NextSeq were chosen for each library and the samples were amplified for 5 cycles to incorporate the sequencing adapters. A qPCR reaction was then performed to determine the optimal additional number of amplification cycles required for each sample to minimize PCR duplication, and each library was amplified for the appropriate number of additional cycles. No libraries exceeded a total of 11 PCR cycles. After library amplification, the samples were again purified with the Zymo kit and eluted in 22 uL ultrapure water. Next, each library was run on a Novex-TBE 4-20% PAGE 10-well non-denaturing gel and subsequently stained with SYBR Gold. The gel was imaged and fragments within the range of 100-1000 bp were excised from the gel for each library. The libraries were recovered from the PAGE gel fragments using electroelution using D-tubes. The libraries were finally purified from the recovered TBE buffer using an Isopropanol/Acetate precipitation. Following the final purification of the libraries, quality control was done by perfoming a Bioanalyzer and KAPA qPCR library quantification assay. This allowed the determination of accurate library concentrations, after which the libraries were pooled, prepped for sequencing on an Illumina Nextseq 500/550 by standard methods using a 75 cycle High-output kit.
Hematopoietic cell Dataset
The hematopoietic cell dataset used in our analysis was a previously published dataset that was generated from human HSCs and progenitor cell populations that were isolated from human cord blood (Laurenti et al., 2013). The authors obtained RNA from flow-sorted populations of human cord blood based on surface expression levels of CD34, CD38, CD45RA, Thy1 and CD49f, CD10, CD7, CD19 and CD1a. Samples were profiled using the Illumina HumanHT-12 WG-DASL v 4.0 R2 expression beadchip (Laurenti et al., 2013). The reference dataset was publicly available through the Gene Expression Omnibus (GSE42414).
Antibody staining, Flow Cytometry Analysis and Sorting
Human CD34+ cells were analyzed and sorted on a FACS ARIA II Cytometer (BD Biosciences). Prior to the FACS processing, the cells were blocked using the human Fc Block (Miltenyi) for 10 minutes on ice. Following this, the cells were washed and incubated with the specific panel of fluorescence/biotin labeled primary antibodies for 30 mins on ice. In the case of a use of biotin-labeled primary antibodies, the cells were further washed and re-incubated with PE-CF594 streptavidin for 10 mins on ice.
Antibody panel for marking the HSCs and MPPs
For the FACS analysis of FISC and MPP populations in our cell culture experiments, we used the following combination of antibodies – Alexa Fluor 700 anti-human CD34 (clone 581), PE-Cy7 anti-human CD38 (clone HB7), APC anti-human CD45RA (clone HI100), PE anti-human CD90 (clone 5E10), and antibody and a biotin-labeled cocktail of antibodies (from Miltenyi) targeting the human “Lineage” antigens CD2, CD3, CD11b, CD14, CD15, CD16, CD19, CD56, CD123, and CD235a.
Primary and Secondary Methocult Assays
For the primary colony-forming cell (CFC) assays, the 2000 FACS-sorted HSPCs were plated in Human Methocult Classic (H4434, Stemcell Technologies) on 35 mm plates and cultured for 14 days at 37 degrees C before the enumeration of colonies. For secondary CFC assays all the cells from the primary plating were collected in PBS and re-plated in Human Methocult (H4434, Stemcell Technologies) on 35 mm plates and cultured for another 14 days at 37 degrees C. The counting of colonies in both primary and secondary plating were performed using a blind method.
Long-term Culture-Initiating Cell (LTC-IC) Assay
LTC_IC assays were performed as described previously with some modifications (Liu et al., 2013). Briefly, mouse bone marrow stromal cells, M2-10B4, were irradiated at 40 Gy and plated on collagen-coated 6 well plates at a density of approximately 250,000 cells per well. After approximately 24 hours, 60,000 FACS-sorted human Lineage-GFP+ cells were plated on the irradiated feeders and cultured in Human Myelocult media (H5100, Stemcell Technologies) for 5 weeks at 37 degrees C. Every week 1 ml of the media was removed and refreshed with fresh media. At the end of 5 weeks, all the cells from each well were harvested by trypsinization and plated in Human Methocult Enriched media (H4435, Stemcell Technologies) and cultured for 2 weeks at 37 degrees C after which the colonies were enumerated by the blind method.
Blind method for counting colonies in Methocult assays
Briefly, the 35 mm plates were labeled on the side-walls of the plate on the day of plating, instead of the lids. On the day of counting, all of the control and treated plates were shuffled and the plates were given random reference numbers on the top of the lids. The colonies in each plate was counted and noted by the given reference numbers. At the end of counting all the plates, the labels on the side-walls were matched with the assigned random reference numbers on top of the lid.
Construction of the inducible Lentiviral Vector
The lentiviral vector pInducer11-miR-RUG that was purchased from Addgene (Meerbrey et al., 2011) was designed to clone and express miR based-shorthairpins under an inducible CMV promoter. The 14.7 kb vector was modified to replace the miR sequence with a RefA gateway cassette to allow Gateway cloning of human cDNAs. The vector was digested with Agel and Mlul to dropout a fragment (approximate size 2 kb) downstream of the CMV promoter that includes the miR sequence and the Turbo RFP reporter. The 5’ and 3’ ends of the remaining 12kb vector were then blunted using the Klenow polymerase. The RefA gateway cassette was then inserted into the vector by blunt-end ligation to generate the modified lentiviral vector, referred to as pInducer11-gw.
cDNA cloning
Human cDNAs were purchased from the Harvard Plasmid Database (https://plasmid.med.harvard.edu/PLASMID/Home.xhtml). The cDNAs for FLI1, KLF12 and HBP1 were in the entry vector pDONR221 that were then cloned into pInducer11-gw by Gateway LR cloning. The cDNAs for CNOT8 (originally in entry vector pOTB7) and ZBTB20 (originally in entry vector pCMV-SPORT6) were first cloned into the pDONR221 entry vector by a Gateway BP reaction. Subsequently the cDNAs were transferred from pDONR221 to pInducer11-gw by a Gateway LR reaction. The cDNA for ZMAT1 (originally in cloning vector pCR-XL-TOPO) was amplified by PCR using the forward primer (5’-GGGCCCCATCTTTATTGGAAAATGT-3’) with a 5’ attB1 gateway cloning adapter and the reverse primer (5’-ACCTCTCCTTTTCTTCATCAGGTGT-3’) with 5’ attB2 cloning adapter. The amplified PCR product was then cloned into pDONR221 by gateway LR reaction. The cDNAs for BAZ2B and DMTF1 originally in the vector pENTR223 lacked a termination codon pre-designed for C-terminal fusion cloning. Using the Quikchange II site-directed mutagenesis kit (Agilent Technologies) we first inserted a termination stop codon for BAZ2B cDNA within the pENTR223 vector using the forward primer (5’-GCAAAAAGAACAGATAACCAACTTTCTTGTAC-3’) and the reverse primer (5’-GTACAAGAAAGTTGGTTATCTGTTCTTTTTGC-3’). We used a similar site-directed mutagenesis strategy to insert a termination stop codon for DMTF1 cDNA within the pENTR223 vector, using the forward primer (5’-GGTAAACTGTCATTAGCCAACTTTCTTGTAC-3’) and the reverse primer (5’-GTACAAGAAAGTTGGCTAATGACAGTTTACC-3’). Finally, we transferred the full-length BAZ2B and DMTF1 cDNAs (with termination codons) from the pENTR223 vector to the pInducer11-gw using the gateway LR reaction.
The cDNA for Luciferase was obtained from Addgene in pDONR223 entry vector (Yang et al., 2011). The Luciferase cDNA did not have a stop codon. The cDNA was cloned in to the destination vector pInducer11-gw by Gateway LR cloning. Upon recombination, the Luciferase cDNA was in-frame with a STOP codon in the destination vector generated by the recombined vector sequence.
Lentivirus production and viral titer estimation
For production of lentiviral particles, HEK293T cells were transfected using the Calcium Phosphate Transfection Kit (Clontech). On day one 12.5 million HEK293T cells were plated on 150mm dishes and after approximately 24 hours the media was refreshed to prepare for transfection. For each plate, the plasmid cocktail was prepared by mixing the Lentiviral vector, the pCMV-dR8.9 packaging plasmid, and the VSVG plasmid expressing the envelope glycoprotein. The cells were then transfected using the Calphos Mammalian Transfection Kit (Clontech) as per the manufacturer’s instructions. The cells were then incubated at 37 degrees C overnight. On day 1 after the transfection, the cells were washed with PBS and were refreshed with fresh media. On day 2 the supernatant was collected and ultracentrifuged at in a Beckman Coulter L-100K centrifuge at 64047 g for 2 hours at 22 degrees. The cells were replenished with fresh media and incubated overnight at 37 degrees C. The virus pellet was then resuspended in PBS and stored at 4 degrees. On day 2 after transfection the supernatant was collected and once again a virus pellet was obtained by ultracentrifugation in a Beckman Coulter L-100K centrifuge at 64047 g for 2 hours at 22 degrees. The PBS suspension with the virus from day one was used to resuspend the fresh virus pellet from day 2 and stored at 4 degrees overnight. The following day the viruses were aliquoted and stored at −80 degrees C.
For estimating the viral titer, HEK293T cells were plated into 6-well plates at a density of 500,000 cells per well. The frozen viral pellets were thawed and for each lentiviral vector we prepared a dilution series from 1:10 to 1:320. The 293T cells were infected with the respective dilutions and after 48 hours the cells were processed for flow cytometry to detect GFP positive cells. The titer was calculated using the formula as described previously (Kutner et al., 2009). The formula is: Transducing Units per ml = (% of GFP positive cells x number cells at the time of transduction x Fold Dilution x 1000) x volume of diluted vector used for transduction.
Committed progenitors in vivo reprogramming and engraftment assay
Lineage-CD34+CD38+ progenitors were cultured in Stemspan SFEMII stimulation media (SCF 300 ng/ml, FLT3 300 ng/ml, TPO 100 ng/ml, IL3 60 ng/ml) and then infected with BAZ2B or Luciferase. The cells were then cultured in vitro in Stemspan SFEM II maintenance media (SCF 100 ng/ml, FLT3 100 ng/ml, TPO 50 ng/ml, UM171 35 nM (StemCell Technologies), SR1 750 nM (StemCell Technologies), LDL 10 ug/ml (StemCell Technologies) and doxycycline 2 ug/ml) for 2-days. About 47,500 to 180,000 cells were than transplanted into irradiated NSG mice. For each donor, same number of cells were transplanted for the Luciferase or BAZ2B transduced samples. The mice were maintained on a doxycycline diet for 3 weeks. Mice were then changed to a normal diet regime and engraftment efficiency was assessed at 16 weeks after resuming the normal diet.
Mouse Transplantation assays
Mice were sublethally irradiated (200-225 rads) and after 24 hours, the mice were transplanted intra-femorally with 47,500-180,000 cells per animal. The drinking water was supplemented with multi-spectral fluoroquinolone antibiotic (Roxacin 0.6 mg/ml) for one month after irradiation. For each donor, the same number of cells were transplanted for the Luciferase or BAZ2B transduced samples. Bone marrow or peripheral blood was analyzed at 12-16 weeks after transplantation.
Flow cytometry analysis of mouse bone marrow, peripheral blood and spleen
Peripheral blood was collected into EDTA-coated tubes by puncturing the tail vein or the facial vein. Mice were sacrificed to collect the bone marrow and spleen samples. Bone marrow was collected by flushing both femurs of each mice. Spleen tissues were homogenized by mincing using a surgical blade. Red blood cell (RBC) lysis was carried out using the 1x RBC lysis buffer (Thermo Scientific) for 5-10 mins on ice. Antibody panels used for the human chimerism analyses were PE-Cy7 anti-mouse CD45 (Clone 30-F11), Alexa Fluor 700 anti-human CD45 (Clone HI30), APC anti-human CD19 (Clone HIB19), PE anti-human CD33 (Clone WM53). In some of the studies we used the PE-Cy5 anti-mouse CD45 (Clone 30-F11) and/or APC-eFluor 780 anti-human CD45 (Clone HI30). The fluorophores and the surface marker gating details are provided on the X and Y axes of the FACS plots in the figures.
Mapping to Human and Mouse Genome and Multi-mapping reads
RNA-Seq reads were first mapped to the Mus musculus assembly 10 reference genome (mm10), and the human assembly 19 (hg19) reference genome using TopHat v 2.0.4 (Kim et al., 2013). Reads mapping to known genes, based on Entrez gene identifiers, were then counted using the GenomicFeatures R-system package (Bioconductor) (Lawrence et al., 2013).
Multi-mapping reads that came either from the ES Mouse nucleus or the Human EBV-B cell nucleus contributed to approximately 5% of the total reads sequenced. In order to maintain the integrity of all the sequenced reads, we attempted to include the reads into the count files into the final counts by taking the following steps. We first increased the stringency of the mapping, of the paired-end sequencing reads. More specifically, the “no–mixed” flag in TopHat assured that alignments where both reads in the pair were mapped were included. The “no–discordant” flag assured that only concordant reads were mapped, meaning the reads had the expected mate orientation and expected distance between them. Once the reads were mapped, the read names given by the lllumina Sequencer were used to separate the reads that mapped uniquely to each genome to multi-mapping reads that mapped to both genomes. First the counts were summarized using the GenomicRanges package on bioconductor (Lawrence et al., 2013).
Next, we reasoned that the multi-mapping reads would fall into one of 3 situations. In the first situation, the reads would map to both the mouse and human genomes, but would only map to a gene in one of genomes. In this case, the reads were assigned to the appropriate gene. Next, we used the CIGAR field, which is a feature of the SAM file and gives a representation of how the read mapped to the reference genome, and whether there was a match/mismatch, insertion, deletion, or if any positions were skipped. We used the CIGAR score to determine which genome a read mapped to, and if there was a difference, then the read was assigned to the genome with the higher quality read.
Finally, we considered reads that mapped perfectly to genes in both genomes. For these we chose to “fairly split” the reads between each of the genomes by considering how many unique reads had already been mapped to each gene. We reasoned, that the multi-mapped reads would follow the same overall proportion of expression that would already be modeled by the unique reads, which would be affected by differences in gene length, expression levels, or a combination of both. For example, if a read had been assigned to a mouse gene that already had 15 unique reads mapped to it, and a human gene that already had 3 unique reads mapped to it, then the mouse gene would receive 15/18th of the read and the human gene would receive 3/18th of the read. The final counts were later rounded to the nearest integer value. Differential expression analyses were performed using EdgeR (Robinson et al., 2010).
ARACNe Networks
The B-cell regulatory network (BCRN) used in this study was an integration of two previously published datasets. The first human BCRN was reverse engineered by the ARACNe algorithm from a dataset of 264 gene expression profiles that included normal (naive and germinal-center B-cells), several tumor phenotypes including, B-cell lymphomas and cell lines. Gene expression was profiled on Affymetrix U133 Plus 2.0 arrays, processed by the Cleaner algorithm, and normalized with MAS5. The resulting BCRN and contained predictions for 1,223 transcription factors regulating 13,007 target genes through 327,837 interactions. The second human BCRN was built from an additional set of 254 samples including normal cells, several tumor phenotypes and cell lines. Gene expression for this dataset was profiled on Affymetrix H-GU95Av2 arrays, and also went through processing through MAS5, Cleaner and ARACNe. This second regulatory network included 173,539 predicted interactions between 633 transcription factors and 6,403 genes. The integration was done by taking a union of the predictions of the two networks, with MR-target interactions that were predicted by both networks having their p-values integrated using Fisher’s method. The final BCRN contained predictions for 1,241 transcription factors regulating 11,770 target genes through 288,616 interactions.
VIPER
The relative activity of each transcription factor represented in the BCRN was inferred using the VIPER algorithm, available as a package through Bioconductor. Conceptually, the VIPER algorithm is similar to the Master Regulator Inference Algorithm (MARINA), which uses the MR targets inferred by the ARACNe algorithm to predict drivers of changes in cellular phenotypes. In addition to calculating the enrichment of ARACNe-predicted targets in the signature of interest, VIPER also takes into account the regulator mode of action, regulator-target gene interaction confidence and pleiotropic nature of each target gene regulation. Statistical significance, including P value and normalized enrichment score (NES), was estimated by comparison to a null model generated by permuting the samples uniformly at random 1,000 times.
Transcription Factors Classification for Network
To identify transcription factors (TFs), we selected the mouse genes annotated as ”transcription factor activity” in Gene Ontology and the list of TFs from TRANSFAC. This produced a final list of 1,794 TFs, which mapped to 3,758 probesets on the gcrma-normalized expression profile.
Transformation for HSC and Heterokaryon dataset for VIPER analysis.
Since the HSC dataset was profiled on a microarray platform and the heterokaryon samples were profiled using RNA-seq, the datasets were not directly comparable. The differences between RNA-seq and microarray data arise from the fact that microarray data is treated as a continuous measurement of the fluorescence intensity, typically modeled by a log-normal distribution. RNA-seq experiments count the number of reads that map to a particular gene or transcript, and methods that analyze RNA-seq data commonly use a Negative Binomial (NB) distribution (Soneson and Delorenzi, 2013). In order to make the two datasets comparable, both expression profiles were transformed using rank and z-transformation. More specifically, the gene expression was rank-transformed for each sample, and then each gene was z-transformed across samples. The two gene expression profiles were combined after this transformation.
Singular Value Decomposition Analysis
Singular value decomposition (SVD) was performed using the biosvd package (Daemen and Brauer, 2019). SVD is a method in linear algebra that allows for a factorization of any m x n matrix into the following form:
When applied to gene expression data, the method can be used to bring out dominant underlying behaviors in gene expression patterns (Alter et al., 2000). According to this study, SVD factorization of the gene expression data resulted in a transformation of the data from an N-genes and M-arrays space in to an M-“eigenarrays” and “M-eigengenes” space, which accounted for most of the variance, despite the great reduction in dimensionality. The proportion of variance explained by each eigengene v(ei) (or principal component) was calculated as:
Single-cell analysis
The four reference populations – HSCs, MPPs, MLPs and Lineage-Committed Progenitors – were each filtered for quality control, removing cells with high mitochondrial read percentage or two few reads as well as genes with not enough coverage to contribute to the analysis. The samples that passed these quality control filters were pooled and normalized to CPM. A distance matrix was constructed using the Pearson distance based on the 100 most variable genes in gene expression space, and this distance matrix was used to construct a k-nearest-neighbor graph with 10 neighbors. Metacells were imputed for each cell by summing the reads of the ten nearest neighbors (using the unnormalized counts) before re-normalization and sub-sampling to 1000 metacells. These metacells were then used as input to ARACNe for the inference of a regulatory network.
The original, non-imputed CPM matrix was transformed into a gene expression signature (GES) using an internal double rank transformation. This GES was then used as the input to VIPER, along with the ARACNe network described previously, inferring the protein activity for all cells in the reference populations.
Single-Cell Random Forest Classifier Model
A train-test split was performed on the reference population in a 70-30 proportion, and the feature set was optimized based on the performance on the held-out set. To identify candidate feature sets, we performed a pairwise Wilcoxon-Rank-Sum test for each protein for all six possible group-to-group comparisons. Proteins were sorted in population-specific manner by the maximum p-value of their pairwise comparisons, and one feature from each populations’ sorted list was added at each iteration. This approach was chosen in order to avoid a single population with bigger differences to the other three dominating the candidate features. Ultimately, a set of 43 proteins were found to have the optimum model performance. Similar optimization was carried out to refine the mtry (number of features to consider at each branch point in the random forest) parameter before a final, ten-thousand tree model was trained using the activity of the selected features in the entire reference population
Protein activity was then inferred for the test population. The BAZ2B population was normalized against the Luciferase control using a double-rank transformation, while the Luciferase population was normalized internally. These GES were then used as the input to VIPER along with the metacell network from the reference populations. Finally, this VIPER matrix was fed into the random forest model and classified based on the maximal vote in each cell.
Circle Plot
Random forests have the advantage of generating a class vote percentage rather than a single classification. This can be regarded as a measurement of classification confidence, a useful tool in determining how distinct members of different classes actually are. In order to visualize this, we developed a circular plotting structure where the class labels are placed at equidistant intervals along the circumference and the samples are plotted in the interior. The position of each sample within the plot is determined in polar coordinates; the radius is given by the inverse of the Shannon information entropy of the classification, while the angle (or theta) is taken as the average of the class-specific angles weighted by the squared vote percentage for each class in the given sample.
As an example, a sample where 100% of the trees in the random forest classified the sample as an HSC would be plotted on the circumference at an angle of pi / 4 (or 45 degrees). A sample where the votes were split 50 / 50 between HSCs and MPPs would appear roughly halfway between the origin and the circumference of the circle and at an angle of pi / 2 (or 90 degrees), the average of the angle for HSCs and MLPs. Finally, a sample with a totally uncertain classification – equal votes for all four classes – would appear at the origin. This method of class visualization can be extended to any number of classes or model contexts. Code is available in the Github repository associated with this paper.
ATAC-Seq data analysis
Reads filtering and alignment
Reads where adapter-trimmed, filtered for low quality ones using fastp (https://doi.org/10.1093/bioinformatics/bty560) and aligned to GRCh38 using STAR v2.5.2a (Dobin et al., 2013) in end-to-end mode and using --alignlntronMax 1 to prohibit splicing. A number of mismatches up to 5% of the paired read length where allowed and only reads mapping uniquely and not onto chromosome M and blacklisted regions where retained for the following analyses using samtools (Li et al., 2009). PCR duplicates where removed using Picard Tools (http://broadinstitute.github.io/picard/).
Peak calling
Reads from pooled replicates of each sample where converted in BEDPE format using bedTools (https://bedtools.readthedocs.io/en/latest/) and adjusted for Tn5 insertion. Peaks where called using MACS ver. 2.2.6 (Zhang et al., 2008) with “--nomodel”, “--call-summits”, “--nolambda”, “-f BEDPE” and “--keep-dup all” options and the resulting narrowPeaks where parsed with the R package chromVar (Schep et al., 2017) using a window of 500bp around the summits.
For nucleosome-free region (NFR) peaks calling we used fragments <100bp (Buenrostro et al., 2013) and parsed peaks with chromVar using a 100bp window around the summits.
Motif Enrichment
To discover motif enrichment in NFR peaks unique to BAZ2B (i.e. not overlapping with any other peaks in LUCIFERASE or PROGENITOR samples) we use the AME tool (McLeay and Bailey, 2010) from the MEME suite (Bailey et al., 2009) with default parameters and JASPAR2020 CORE motif database filtered for the Homo sapiens specie, yielding 639 motifs. As control sequences, we used those of all PROGENITOR peaks not overlapping any of the LUCIFERASE ones. The motifs found to be enriched were clustered with RSAT matrix-clustering tool (Castro-Mondragon et al., 2017) using the web interface (http://pedagogix-tagc.univ-mrs.fr/rsat/RSAT_portal.html) with correlation parameter set to 0.8 but ‘merge-matrices’ set to ‘average’.
Quantification and Statistical Analysis
The details for all the statistical tests are provided in the figure legends, results and STAR Methods. All the statistical analyses in Figures 4–7 and S5 were performed using Graphpad Prism 5 or Microsoft Excel. Some of the plots were created using doBy (Højsgaard and Halekoh, 2020), Ggplot2 (Wickham, 2016), Cowplot (Wilke, 2016), reshape (Wickham, 2018), Pheatmap (Kolde, 2019), RColorBrewer (Neuwirth, 2014).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotin mouse anti-human Lineage Cocktail | Miltenyi Biotec | Order # 130-092-211 |
| Alexa Fluor 700 mouse anti-human CD34 (Clone 581) | BD Biosciences | Cat # 561440 |
| PE-Cy7 mouse anti-human CD38 (Clone HB7) | eBioscience (Thermo Scientific) | Cat # 25-0388-41 |
| APC mouse anti-human CD45RA (Clone HI100) | BD Biosciences | Cat # - 550855 |
| PE mouse anti-human CD90 (Clone 5E10) | BD Biosciences | Cat # 561970 |
| PE-CF594 Streptavidin | BD Biosciences | Cat # 562284 |
| FcR Blocking Reagent, Human | Miltenyi | Order # 130-059-901 |
| APC mouse Anti-human CD34 (Clone AC136) | Miltenyi | Order # 130-098-139 |
| PE-Cy7 rat anti-mouse CD45 (Clone 30-F11) | BD Biosciences | Cat # 552848 |
| PE-Cy5 rat anti-mouse CD45 (Clone 30-F11) | BD Biosciences | Cat # 553082 |
| APC-eFluor 780 mouse anti-human CD45 (Clone HI30) | eBioscience(Thermo Scientific) | Cat # 47-0459-42 |
| Alexa Fluor 700 mouse anti-human CD45 (Clone HI30) | BD Biosciences | Cat # 560566 |
| PE mouse anti-human CD33 (Clone WM53) | BD Biosciences | Cat # 561816 |
| APC mouse anti-human CD19 (Clone HIB19) | BD Biosciences | Cat # 561742 |
| Mouse anti-human Lamin A/C (clone 636) | Vector Labs | Cat # VP-L550 |
| Alexa Fluor 568 Phalloidin | Molecular Probes (Thermo Scientific) | Cat # A12380 |
| Goat anti-mouse IgG Alexa Fluor 488 | Life Technologies (Thermo Scientific) | Cat # R37120 |
| Bacterial and Virus Strains | ||
| pInducer11-miR-RUG lentiviral Vector | Meerbrey et al., 2011 | Addgene (Cat # 44363) |
| pInducer11 – gw lentiviral vector | This paper | NA |
| Biological Samples | ||
| Human Cord Blood CD34+ Cells, Frozen | Stemcell Technologies | Cat # 70008.5 |
| Human Umbilical Cord Blood | Banc de Sang I Teixits | Prod. Code- BB201 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human SCF | Peprotech | Cat # 300-07 |
| Human FLT3 | Peprotech | Cat # 300-19 |
| Human TPO | Peprotech | Cat # 300-18 |
| Human IL3 | Peprotech | Cat # 200-03 |
| Human IL6 | Peprotech | Cat # 200-06 |
| StemSpan SFEM | Stemcell Technologies | Cat # 09650 |
| StemSpan SFEM II | Stemcell Technologies | Cat # 09655 |
| StemRegeninl (SR1) | Stemcell Technologies | Cat # 72342 |
| UM171 | Stemcell Technologies | Cat # 72912 |
| Human LDL | Stemcell Technologies | Cat # 02698 |
| Human Methocult Classic | Stemcell Technologies | Cat # H4434 |
| Human Methocult Enriched | Stemcell Technologies | Cat # H4435 |
| Human Myelocult Media | Stemcell Technologies | Cat # H5100 |
| Vybrant DiD Cell Labeling Solution | ThermoFisher Scientific | Cat # V22887 |
| Vybrant DiO Cell Labeling Solution | ThermoFisher Scientific | Cat # V22886 |
| Doxycycline Hyclate | Sigma-Aldrich | Cat # D9891 |
| Roxacin | Proultry | http://poultry.proultry.com/products/laboratorios-caliersa/roxacin-solucion-oral |
| CalPhos Mammalian Transfection Kit | Clontech | 631312 |
| 1X RBC Lysis buffer | Thermo Scientific | 00-4333-57 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| All RNA-seq and ATAC-seq data are publicly available. | Gene Expression Omnibus | GSE114240 |
| Experimental Models: Cell Lines | ||
| M2-10B4, mouse bone marrow stromal cells | ATCC | CRL-1972 |
| Tcf3−/− mouse embryonic stem cells | Laboratory of Bradley Merrill (Pereira L., et al 2006) | NA |
| Epstein Barr transformed human B lymphoblasts | Coriell Institute of Medical Research | GM22647 |
| HEK 293T cells | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid II2rgtm1Wjl/SzJ (NSG) mice | The Jackson Laboratory | 005557 |
| Oligonucleotides | ||
| BAZ2B Mutagenesis forward primer (GCAAAAAGAACAGATAACCAACTTTCTTGTAC) |
This paper | NA |
| BAZ2B Mutagenesis reverse primer (GTACAAGAAAGTTGGTTATCTGTTCTTTTTGC) |
This paper | NA |
| DMTF1 Mutagenesis forward primer (GGTAAACTGTCATTAGCCAACTTTCTTGTAC) |
This paper | NA |
| DMTF1 Mutagenesis reverse primer (GTACAAGAAAGTTGGCTAATGACAGTTTACC) |
This paper | NA |
| ZMAT1 PCR cloning forward primer (GGGCCCCATCTTTATTGGAAAATGT) |
This paper | NA |
| ZMAT1 PCR cloning reverse primer (ACCTCTCCTTTTCTTCATCAGGTGT) |
This paper | NA |
| Recombinant DNA | ||
| pENTR223 - BAZ2B (NCBI Acc No BC012576) | Harvard Plasmid Repository (The ORFeome collaboration) | HSCD00377199 |
| PDONR221 - FLI1 (NCBI Acc No – BC001670) | Harvard Plasmid Repository (Harvard Institute of Proteomics) | HSCD00044634 |
| PENTR223 - DMTF1(NCBI Acc No – BC070064.1) | Harvard Plasmid Repository (The ORFeome collaboration) | HSCD00365376 |
| PCMV-SPORT6 - ZBTB20 (NCBI Acc No – BC029041) | Harvard Plasmid Repository (Mammalian Gene Collection) | HSCD00335411 |
| pCR-XL-TOPO - ZMAT1 (NCBI Acc No – BC140920) | Harvard Plasmid Repository (Mammalian Gene Collection) | HSCD00342908 |
| pOTB7-CNOT8 (NCBI Acc No – BC017366) | Harvard Plasmid Repository (Mammalian Gene Collection) | HSCD00324635 |
| pDONR221 - KLF12 (NCBI Acc No – BC019680) | Harvard Plasmid Repository (Harvard Institute of Proteomics) | HSCD00043856 |
| pDONR221 - HBP1 (NCBI Acc No - BC017069) | Harvard Plasmid Repository (Harvard Institute of Proteomics) | HSCD00043659 |
| pDONR223-Luciferase | Addgene | Plasmid #25894 |
| Software and Algorithms | ||
| Tophat v2.0.4 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| STAR v2.5.2a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| bedTools | bedtools.readthedocs.i0 | https://bedtools.readthedocs.io/en/latest/ |
| Picard Tools | Broad Institute | http://broadinstitute.github.io/picard/ |
| Ggplot2 | Wickham, 2016 | http://ggplot2.org/ |
| Viper | Alvarez et al., 2016 | 10.18129/B9.bioc.viper |
| EdgeR | Robinson et al., 2010 | 10.18129/B9.bioc.edgeR |
| Cowplot | Wilke, 2016 | https://cran.r-project.org/web/packages/cowplot/index.html |
| reshape | Wickham, 2018 | https://cran.r-project.org/web/packages/reshape |
| Pheatmap | Kolde, 2019 | https://cran.r-project.org/web/packages/pheatmap |
| Biosvd | Daemen and Brauer, 2019 | https://bioconductor.org/packages/3.10/bioc/html/biosvd.html |
| doBy | Højsgaard and Halekoh, 2020 | https://cran.r-project.org/web/packages/doBy/doBy.pdf |
| GenomicFeatures | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicFeatures.html |
| GenomicRanges | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| Cell Ranger | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Graphpad Prism 5 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| RColorBrewer | Neuwirth, 2014 | https://cran.r-project.org/web/packages/RColorBrewer |
| MACS (2.2.6) | Zhang et al., 2008 | |
| chromVar (1.6.0) | Schep et al., 2017 | https://greenleaflab.github.io/chromVAR/index.html |
| AME (5.0.5) | McLeay and Bailey, 2010 | http://meme-suite.org/doc/ame.html |
| RSAT | Castro-Mondragon et al., 2017 | http://pedagogix-tagc.univ-mrs.fr/rsat/RSAT_portal.html |
| Networks were generated using the ARACNe-AP tool | Lachmann et al., 2016 | https://github.com/califano-lab/ARACNe-AP |
| Implementation of all single-cell model training, validation, and testing | This paper | https://github.com/califano-lab/COSMA |
| Other | ||
| SAFE Doxycycline diet (0.625 mg/Kg Doxycycline Hyclate) | Safe Diet | E8220 Version 0232 |
| Standard Maintenance diet RM1 (P) | Special Diets Services | 801151 |
Acknowledgements
We would like to thank the Microscopy, and Flow Cytometry Facilities of the CRG/UPF. We thank Andrea Cerutti (IMIM, Barcelona), Joao Frade (CRG), Marie Victoire Neguembor (CRG) and Shoma Nakagawa (CRG) for critical suggestions on the manuscript. This work was supported by a Human Frontier Science Program Grant 2010 (to M.P.C. and A.C.), by the European Union’s Horizon 2020 Research and Innovation Programme (CellViewer No 686637 to M.P.C); Ministerio de Ciencia e Innovación, grant BFU2017-86760-P (AEI/FEDER, UE), AGAUR grant from Secretaria d’Universitats i Recerca del Departament d’Empresa I Coneixement de la Generalitat de Catalunya (2017 SGR 689 to M.P.C.), Juan de la Cierva Fellowship (K.A.), BIST Master Fellowship (X.T.), and the R35CA197745 outstanding NCI investigator award to A.C. We acknowledge the support of the Spanish Ministry of Science and Innovation to the EMBL partnership, the Centro de Excelencia Severo Ochoa and the CERCA Programme (to M.P.C) and the two instrumentation grants S10OD012351 and S10OD021764 to A.C. supporting the analytical work.
Footnotes
Declaration of interests
A.C. is a funder and shareholder of DarwinHealth Inc. which was granted an exclusive license by Columbia University for the commercialization of algorithms used in this study, as ARACNe and VIPER. Columbia University is a shareholder in DarwinHealth. A provisional US patent application (US 63/086,265) has been filed related to this work, with M.P.C., A.C. and K.A. as inventors.
References
- Altarche-Xifro W, di Vicino U, Munoz-Martin MI, Bortolozzi A, Bove J, Vila M, and Cosma MP (2016). Functional Rescue of Dopaminergic Neuron Loss in Parkinson’s Disease Mice After Transplantation of Hematopoietic Stem and Progenitor Cells. EBioMedicine 8, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter O, Brown PO, and Botstein D (2000). Singular value decomposition for genomewide expression data processing and modeling. Proc Natl Acad Sci U S A 97, 10101–10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, and Califano A (2016). Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet 48, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K, Lois C, Morrison SJ, and Alvarez-Buylla A (2003). Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. nature 425, 968–973. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SJ, Ma’ayan A, Lieu YK, John P, Reddy MV, Chen EY, Duan Q, Snoeck HW, and Reddy EP (2014). B-myb is an essential regulator of hematopoietic stem cell and myeloid progenitor cell development. Proc Natl Acad Sci U S A 111,3122–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, and Califano A (2005). Reverse engineering of regulatory networks in human B cells. Nat Genet 37, 382–390. [DOI] [PubMed] [Google Scholar]
- Batta K, Florkowska M, Kouskoff V, and Lacaud G (2014). Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep 9, 1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, and Blau HM (2010). Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 463, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortoluzzi A, Amato A, Lucas X, Blank M, and Ciulli A (2017). Structural basis of molecular recognition of helical histone H3 tail by PHD finger domains. Biochem J 474, 1633–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady JJ, Li M, Suthram S, Jiang H, Wong WH, and Blau HM (2013). Early role for IL-6 signalling during generation of induced pluripotent stem cells revealed by heterokaryon RNA-Seq. Nature cell biology 15, 1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briana CB, Kimberly LJ-W, Chuanwu W, Seung Goo K, Juan L, Michael RL, Chang HK, and Elizabeth JT (2010). Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med 207, 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. (2010). The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Mondragon JA, Jaeger S, Thieffry D, Thomas-Chollier M, and van Helden J (2017). RSAT matrix-clustering: dynamic exploration and redundancy reduction of transcription factor binding motif collections. Nucleic Acids Res 45, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Lee S, Mallard W, Clement K, Tagliazucchi GM, Lim H, Choi IY, Ferrari F, Tsankov AM, Pop R, et al. (2015). A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol 33, 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A, and Brauer B (2019). Package for high-throughput data processing, outlier detection, noise removal and dynamic modeling (Bioconductor). [Google Scholar]
- Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, et al. (2017). Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 23, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Douglass EF Jr., Sonabend AM, Mela A, Bose S, Gonzalez C, Canoll PD, Sims PA, Alvarez MJ, and Califano A (2018). Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat Commun 9, 1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel A, Warnatz K, Ledermann B, Rolink A, Zipfel PF, Burki K, and Eibel H (1998). The transcription factor early growth response 1 (Egr-1) advances differentiation of pre-B and immature B cells. J Exp Med 188, 2215–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Yambartsev A, Ramsey SA, Thomas LD, Shulzhenko N, and Morgun A (2015). Reverse enGENEering of Regulatory Networks from Big Data: A Roadmap for Biologists. Bioinformatics and biology insights 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, and Dick JE (2010). Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol 11, 585–593. [DOI] [PubMed] [Google Scholar]
- Foshay KM, Looney TJ, Chari S, Mao FF, Lee JH, Zhang L, Fernandes CJ, Baker SW, Clift KL, Gaetz J, et al. (2012). Embryonic stem cells induce pluripotency in somatic cell fusion through biphasic reprogramming. Mol Cell 46, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco-Velilla R, Fernandez-Chamorro J, Ramajo J, and Martinez-Salas E (2016). The RNA-binding protein Gemin5 binds directly to the ribosome and regulates global translation. Nucleic Acids Res 44, 8335–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frelin C, Herrington R, Janmohamed S, Barbara M, Tran G, Paige CJ, Benveniste P, Zuniga-Pflucker JC, Souabni A, Busslinger M, et al. (2013). GATA-3 regulates the self-renewal of long-term hematopoietic stem cells. Nat Immunol 14, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AM, Kurochkin I, Chang B, Daniel M, Law K, Satija N, Lachmann A, Wang Z, Ferreira L, Ma’ayan A, et al. (2018). Cooperative Transcription Factor Induction Mediates Hemogenic Reprogramming. Cell Rep 25, 2821–2835 e2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodings C, Smith E, Mathias E, Elliott N, Cleveland SM, Tripathi RM, Layer JH, Chen X, Guo Y, Shyr Y, et al. (2015). Hhex is Required at Multiple Stages of Adult Hematopoietic Stem and Progenitor Cell Differentiation. Stem Cells 33, 2628–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottgens B, Nastos A, Kinston S, Piltz S, Delabesse EC, Stanley M, Sanchez MJ, Ciau-Uitz A, Patient R, and Green AR (2002). Establishing the transcriptional programme for blood: the SCL stem cell enhancer is regulated by a multiprotein complex containing Ets and GATA factors. EMBO J 21, 3039–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote D, Moison C, Duhamel S, Chagraoui J, Girard S, Yang J, Mayotte N, Coulombe Y, Masson JY, Brown GW, et al. (2015). E4F1 is a master regulator of CHK1-mediated functions. Cell Rep 11, 210–219. [DOI] [PubMed] [Google Scholar]
- Hackett JA, and Surani MA (2014). Regulatory principles of pluripotency: from the ground state up. Cell stem cell 15, 416–430. [DOI] [PubMed] [Google Scholar]
- Hess J, Angel P, and Schorpp-Kistner M (2004). AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 117, 5965–5973. [DOI] [PubMed] [Google Scholar]
- Højsgaard S, and Halekoh U (2020). Groupwise Statistics, LSmeans, Linear Contrasts, Utilities (CRAN R-Project; ). [Google Scholar]
- Hou Y, Li W, Sheng Y, Li L, Huang Y, Zhang Z, Zhu T, Peace D, Quigley JG, Wu W, et al. (2015). The transcription factor Foxm1 is essential for the quiescence and maintenance of hematopoietic stem cells. Nature immunology 16, 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JT, Nasa C, Shi W, Huntington ND, Bogue CW, Alexander WS, and McCormack MP (2015). A crucial role for the homeodomain transcription factor Hhex in lymphopoiesis. Blood 125, 803–814. [DOI] [PubMed] [Google Scholar]
- Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, et al. (2017). Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med 23, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochum W, Passegue E, and Wagner EF (2001). AP-1 in mouse development and tumorigenesis. Oncogene 20, 2401–2412. [DOI] [PubMed] [Google Scholar]
- Jones M, Chase J, Brinkmeier M, Xu J, Weinberg DN, Schira J, Friedman A, Malek S, Grembecka J, Cierpicki T, et al. (2015). Ash1l controls quiescence and self-renewal potential in hematopoietic stem cells. J Clin Invest 125, 2007–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, and Kempkes B (1999). The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J Virol 73, 4481–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich I, et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, and Srour EF (2011). Regulation of murine hematopoietic stem cell quiescence by Dmtf1. Blood 118, 6562–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R (2019). pheatmap: Pretty Heatmaps (CRAN R-Project; ). [Google Scholar]
- Kushwaha R, Jagadish N, Kustagi M, Mendiratta G, Seandel M, Soni R, Korkola JE, Thodima V, Califano A, Bosl GJ, et al. (2016). Mechanism and Role of SOX2 Repression in Seminoma: Relevance to Human Germline Specification. Stem cell reports 6, 772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha R, Jagadish N, Kustagi M, Tomishima MJ, Mendiratta G, Bansal M, Kim HR, Sumazin P, Alvarez MJ, Lefebvre C, et al. (2015). Interrogation of a context-specific transcription factor network identifies novel regulators of pluripotency. Stem Cells 33, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutner RH, Zhang XY, and Reiser J (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4, 495–505. [DOI] [PubMed] [Google Scholar]
- Lachmann A, Giorgi FM, Lopez G, and Califano A (2016). ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenti E, Doulatov S, Zandi S, Plumb I, Chen J, April C, Fan J-B, and Dick JE (2013). The transcriptional architecture of early human hematopoiesis identifies multilevel control of lymphoid commitment. Nature immunology 14, 756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, et al. (2010). A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol 6, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Rieckhof G, and Califano A (2012). Reverse-engineering human regulatory networks. Wiley Interdisciplinary Reviews: Systems Biology and Medicine 4, 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Miller CL, and Eaves CJ (2013). Human long-term culture initiating cell assay. Methods Mol Biol 946, 241–256. [DOI] [PubMed] [Google Scholar]
- Lluis F, Ombrato L, Pedone E, Pepe S, Merrill BJ, and Cosma MP (2011). T-cell factor 3 (Tcf3) deletion increases somatic cell reprogramming by inducing epigenome modifications. Proceedings of the National Academy of Sciences 108, 11912–11917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluis F, Pedone E, Pepe S, and Cosma MP (2008). Periodic activation of Wnt/beta-catenin signaling enhances somatic cell reprogramming mediated by cell fusion. Cell stem cell 3, 493–507. [DOI] [PubMed] [Google Scholar]
- Lucchetti J, Fracasso C, Balducci C, Passoni A, Forloni G, Salmona M, and Gobbi M (2019). Plasma and Brain Concentrations of Doxycycline after Single and Repeated Doses in Wild-Type and APP23 Mice. J Pharmacol Exp Ther 368, 32–40. [DOI] [PubMed] [Google Scholar]
- Luna-Pelaez N, and Garcia-Dominguez M (2018). Lyar-Mediated Recruitment of Brd2 to the Chromatin Attenuates Nanog Downregulation Following Induction of Differentiation. J Mol Biol. [DOI] [PubMed] [Google Scholar]
- Martine van K, Leonie JG, Michal M, Ruben van B, Jan K, Paul JC, Steven TP, and Marcel S (2014). FOXP1 directly represses transcription of proapoptotic genes and cooperates with NF-κB to promote survival of human B cells. Blood 124, 3431–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurier F, Doedens M, Gan OI, and Dick JE (2003). Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat Med 9, 959–963. [DOI] [PubMed] [Google Scholar]
- McLeay RC, and Bailey TL (2010). Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC bioinformatics 11, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill BJ (2012). Wnt pathway regulation of embryonic stem cell self-renewal. Cold Spring Harbor perspectives in biology 4, a007971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, and Cech TR (2012). The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 492, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwirth R (2014). RColorBrewer: ColorBrewer Palettes (CRAN R-Project; ). [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, and Busslinger M (1999). Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401,556–562. [DOI] [PubMed] [Google Scholar]
- Obier N, Cauchy P, Assi SA, Gilmour J, Lie ALM, Lichtinger M, Hoogenkamp M, Noailles L, Cockerill PN, Lacaud G, et al. (2016). Cooperative binding of AP-1 and TEAD4 modulates the balance between vascular smooth muscle and hemogenic cell fate. Development 143, 4324–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppikofer M, Bai T, Gan Y, Haley B, Liu P, Sandoval W, Ciferri C, and Cochran AG (2017). Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep 18, 1697–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedone E, Olteanu VA, Marucci L, Munoz-Martin MI, Youssef SA, de Bruin A, and Cosma MP (2017). Modeling Dynamics and Function of Bone Marrow Cells in Mouse Liver Regeneration. Cell Rep 18, 107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF, Chang B, Qiu J, Niu X, Papatsenko D, Hendry CE, Clark NR, Nomura-Kitabayashi A, Kovacic JC, Ma’ayan A, et al. (2013). Induction of a hemogenic program in mouse fibroblasts. Cell stem cell 13, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF, Terranova R, Ryan NK, Santos J, Morris KJ, Cui W, Merkenschlager M, and Fisher AG (2008). Heterokaryon-based reprogramming of human B lymphocytes for pluripotency requires Oct4 but not Sox2. PLoS Genet 4, e1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, et al. (2010). Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 28, 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prashad SL, Calvanese V, Yao CY, Kaiser J, Wang Y, Sasidharan R, Crooks G, Magnusson M, and Mikkola HK (2015). GPI-80 defines self-renewal ability in hematopoietic stem cells during human development. Cell stem cell 16, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell J, Gazit R, Garrison BS, Guo G, Saadatpour A, Mandal PK, Ebina W, Volchkov P, Yuan GC, Orkin SH, et al. (2014). Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell 157, 549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler VM, Lis R, Liu Y, Kedem A, James D, Elemento O, Butler JM, Scandura JM, and Rafii S (2014). Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature 511, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanges D, Lluis F, and Cosma MP (2011). Cell-fusion-mediated reprogramming: pluripotency or transdifferentiation? Implications for regenerative medicine. In Cell Fusion in Health and Disease (Springer; ), pp. 137–159. [DOI] [PubMed] [Google Scholar]
- Sanges D, Romo N, Simonte G, Di Vicino U, Tahoces AD, Fernandez E, and Cosma MP (2013). Wnt/beta-catenin signaling triggers neuron reprogramming and regeneration in the mouse retina. Cell Rep 4, 271–286. [DOI] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte J, Moignard V, and Gottgens B (2012). Establishing the stem cell state: insights from regulatory network analysis of blood stem cell development. Wiley Interdiscip Rev Syst Biol Med 4, 285–295. [DOI] [PubMed] [Google Scholar]
- Searle NE, and Pillus L (2018). Critical genomic regulation mediated by Enhancer of Polycomb. Curr Genet 64, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley MD, Baugher JD, Stevens EL, Tang Z, Gerry N, Beiswanger CM, Berlin DS, and Pevsner J (2012). Chromosomal variation in lymphoblastoid cell lines. Hum Mutat 33, 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smarr B, Rowland NE, and Zucker I (2019). Male and female mice show equal variability in food intake across 4-day spans that encompass estrous cycles. PLoS One 14, e0218935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, and Delorenzi M (2013). A comparison of methods for differential expression analysis of RNA-seq data. BMC bioinformatics 14, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souroullas GP, Salmon JM, Sablitzky F, Curtis DJ, and Goodell MA (2009). Adult hematopoietic stem and progenitor cells require either Lyl1 or Scl for survival. Cell stem cell 4, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soza-Ried J, and Fisher AG (2012). Reprogramming somatic cells towards pluripotency by cellular fusion. Current opinion in genetics & development 22, 459–465. [DOI] [PubMed] [Google Scholar]
- Stopa N, Krebs JE, and Shechter D (2015). The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci 72, 2041–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu YF, Goettel JA, Serrao E, Rowe RG, Malleshaiah M, et al. (2017). Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 545, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Tada T, Lefebvre L, Barton SC, and Surani MA (1997). Embryonic germ cells induce epigenetic reprogramming of somatic nucleus in hybrid cells. The EMBO journal 16, 6510–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallant C, Valentini E, Fedorov O, Overvoorde L, Ferguson FM, Filippakopoulos P, Svergun DI, Knapp S, and Ciulli A (2015). Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure 23, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos F, Mitrofanova A, Bergren SK, Califano A, and Shen MM (2017). A computational systems approach identifies synergistic specification genes that facilitate lineage conversion to prostate tissue. Nat Commun 8, 14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee WW, Pardo M, Theunissen TW, Yu L, Choudhary JS, Hajkova P, and Surani MA (2010). Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev 24, 2772–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CA Jr., Mack DH, and Davis MM (1994). Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell 77, 297–306. [DOI] [PubMed] [Google Scholar]
- Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, and Lei M (2007). The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 445, 506–510. [DOI] [PubMed] [Google Scholar]
- Wang J, Saijo K, Skola D, Jin C, Ma Q, Merkurjev D, Glass CK, and Rosenfeld MG (2018). Histone demethylase LSD1 regulates hematopoietic stem cells homeostasis and protects from death by endotoxic shock. Proc Natl Acad Sci U S A 115, E244–E252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Olson S, and Grompe M (2003). Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 422, 897–901. [DOI] [PubMed] [Google Scholar]
- Wataru I, Masako K, Barbara US, Tingting Z, Bjoern S, Uttiya B, Frederick WA, Jun T, Eugene MO, Theresa LM, et al. (2011). The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol 12, 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag; New York: ). [Google Scholar]
- Wickham H (2018). reshape: Flexibly Reshape Data (CRAN R-Project; ). [Google Scholar]
- Wilke CO (2016). cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’ (CRAN R-Project; ). [Google Scholar]
- Wood CD, Veenstra H, Khasnis S, Gunnell A, Webb HM, Shannon-Lowe C, Andrews S, Osborne CS, and West MJ (2016). MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, O’Connor MS, and Songyang Z (2007). TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 445, 559–562. [DOI] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q-L, Nichols J, Evans EP, and Smith AG (2002). Changing potency by spontaneous fusion. Nature 416, 545–548. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen X, Song G, Zhang J, Liu H, and Liu X (2017). Uhrf1 controls the self-renewal versus differentiation of hematopoietic stem cells by epigenetically regulating the cell-division modes. Proc Natl Acad Sci U S A 114, E142–E151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the raw sequencing data related to the heterokaryon, hematopoietic single-cell and ATAC-seq data are available on the NCBI gene expression omnibus with the accession code GSE114240. Networks were generated using the ARACNe-AP tool from the Califano lab (https://github.com/califano-lab/ARACNe-AP)(Lachmann et al., 2016). VIPER tool is available to download from bioconductor: https://www.bioconductor.org/packages/release/bioc/html/viper.html. Implementation of all model training, validation, and testing, as well as subsequent downstream analyses and plotting can be found at https://github.com/califano-lab/COSMA.