

Abstract
The long-term function of transplanted therapeutic cells typically requires systemic immune suppression. Here, we show that a retrievable implant comprising of a silicone reservoir and a porous polymeric membrane protects human cells encapsulated in it after implant transplantation in the intraperitoneal space of immunocompetent mice. Membranes with pores 1 µm in diameter allowed host macrophages to migrate into the device without the loss of transplanted cells, whereas membranes with pore sizes under 0.8 µm prevented their infiltration by immune cells. A synthetic polymer coating prevented fibrosis and was necessary for the long-term function of the device. For over 130 days the device supported human cells engineered to secrete erythropoietin in immunocompetent mice as well as transgenic human cells carrying an inducible gene circuit for the on-demand secretion of erythropoietin. Pancreatic islets from rats encapsulated in the device and implanted in diabetic mice restored normoglycaemia in the mice for over 75 days. The biocompatible device provides a retrievable solution for the transplantation of engineered cells in the absence of immunosuppression.
Cell-based therapy has the potential to revolutionize the treatment of chronic diseases such as autoimmune disorders, endocrine disorders, neurological degeneration, and cancer1–3. Engineered cells can simultaneously detect pathological conditions and act as living factories to manufacture and deliver drugs that treat diseased tissue2. Cells can be programmed to deliver a variety of therapeutics in the form of small molecules, proteins4, exosomes5 and virus6. Implanted cells containing synthetic gene circuits have alleviated allergic response7, cured diabetes4 and treated Parkinson’s disease5 in rodent models. Recent advances in stem cell technology have made it possible to generate unrestricted quantities of functional pancreatic β-cells8,9 making islet replacement therapy a clinically viable option. However, a major roadblock for translation of these cell-based therapies is the lack of a safe and effective technology for transplanting these cells within patients 3,10.
Therapeutic cells can be immunogenic to the host2,3,11. Cell-encapsulation has been broadly investigated as a method to allow transplantation of cells without chronic immunosuppression10. Microencapsulation of cells in natural10 or synthetic hydrogel beads12 is one method for immune isolation. Despite the potential of microencapsulation, the development of a single macro encapsulation device may enable greater control of 3-Dimensional structure and facilitate device retrieval. This maybe particularly important when transformed cells are used with tumorigenic potential, including hESC-derived cells that can contain undifferentiated cells which may form teratomas10.
One approach of developing a retrievable encapsulation device has been by reinforcing alginate gel with polymeric fibers13,14. These millimeter-scale devices offer the advantage of high permeability to nutrients and have already shown promising results in pre-clinical models. However, the lack of rigidity may cause difficulty in maintaining device shape10, in addition to the risk of graft escaping from the alginate gel. Macrodevices made out of durable polymeric materials can overcome these limitations and offer greater flexibility with the shape and configuration of the implant but have been challenging to develop.
In a typical immune isolating macrodevice, the transplanted cells are protected by a membrane that blocks various elements of the immune system10,15,16. The membrane pore size is critical for proper macrodevice function as it should be designed to selectively block harmful immune components while offering minimum resistance to the transport of oxygen, nutrients, and the secreted therapeutic factors10. An optimal cell encapsulation device will enable a rapid response to environmental cues, such as secreting insulin in response to blood glucose change15,16. Effectors of the immune system span over four orders of magnitude in size (cytokines ~ 2 nm and cells ~ 10 μm), and there continues to be debate around which elements of the immune system need to blocked from access to the cells16,17. While it is clear that direct contact with immune cells must be prevented, there is a lack of evidence on the requirement for blocking cytokines15. Previous studies developed size-exclusion membranes with pores below 20 nm in diameter which can block cytokines and antibodies. Despite promising results in vitro, these cytokine blocking membranes did not prevent graft loss in vivo18,19.
Polymeric membranes with pores up to 400 nm diameter have been investigated in the construction of encapsulation devices because of their ability to prevent infiltration of immune cells 10,20,21. Interestingly, studies on cell migration have reported that cells are unable to squeeze through confined spaces below 7μm2 in cross-sectional area (~3μm diameter circular pores) without matrix degradation22. We reasoned that a micron-sized pores might provide cell barrier function, while allowing for high-permeability. However, the upper limit on the membrane pore-size which can maintain immune-barrier function was unclear.
Another challenge to macrodevice function in vivo is the potential of a foreign body reaction to result in fibrosis and cellular isolation19–21. Materials commonly used for fabricating macrodevices induce a foreign body reaction to the implant16. Additionally, it is generally held that immunogenic antigens shed by the encapsulated cells can intensify the fibrotic response making xenotransplants more difficult than allotransplants15,16. The foreign body reaction is a complex process which involves activation and recruitment of several types of cells such as macrophages and fibroblasts, to the implant site, eventually leading to deposition of a collagenous fibrotic capsule around the device 23. The fibrotic capsule limits the supply of oxygen and nutrients to the encapsulated cells and induces cell death due to hypoxia. Recently chemical structures have been identified that substantially limit fibrosis in rodents and non-human primates24. These improved materials have enabled long-term function of microencapsulation devices with islets in both rodents and non-human primates25,26.
Fibrosis-induced hypoxia remains a major barrier for macrodevices and various strategies have been explored to overcome this10. The Theracyte device used a composite membrane comprising of an inner immune-isolating membrane and an outer membrane with larger pores designed to induce neovascularization20. While early tests in rats were encouraging, a recent phase I clinical trial with a modified version of this device (marketed as VC-01 by ViaCyte) revealed extensive fibrosis and cell death in 17 of the 19 patients receiving an implant 27. Promising strategies for improving vascularization have been explored for hydrogel-based devices28,29, but they remain to be tested on macrodevices in fibrotic animal models. Other approaches have involved perfusing the implanted macrodevice with external oxygen to maintain normoxia within the device chambers30, and repeated filling of the implanted macrodevice with therapeutic cells through an external catheter31. While these strategies are promising, the requirements for cell maintenance and contact with the external environment limit their clinical potential. Therefore, there is a need for developing macrodevices that do not fibrose.
Here we develop a biocompatible macrodevice that can serve as a technology to deliver and maintain immunogenic therapeutic cells in vivo, over long periods. In our modular device, we sequentially improve membrane characteristics and biocompatibility to enable device performance. We establish a pore-size cut off for membranes to maintain their cell barrier function. We also develop a surface modification technique to graft anti-fibrotic molecules on our devices and demonstrate a surface coating that can mitigate the fibrotic response over long periods. Finally, we demonstrate the efficacy of the device with human cell lines and rat islets in immunocompetent mouse models.
Results
Device development and in vitro characterization
The device comprises of a cell reservoir attached to a thin porous polymeric membrane (Fig 1a). Detail of the device fabrication is described in methods section. A flexible silicone elastomer (polydimethoxysiloxane, PDMS) is used to cast the device which contains a microfabricated cell-reservoir along with microchannels for injection of cells into the device. Finally, a polycarbonate track-etched membrane (PCTE) is chemically bonded to the device32. The assembled device is loaded with cells through an access port and sealed using a UV curable adhesive. The microfabricated reservoir had a depth of 150 μm (similar to the distance between capillaries33) and allowed precise positioning of the cells close to the membrane ensuring optimal diffusion of nutrients. The reservoir contained PDMS pillars which prevented collapse of the membrane onto the PDMS surface, and aided in uniform filling of the microchamber with cells during the device loading process (see Fig S1 for a 3D model of the device). PCTE membranes are mechanically robust and can be manufactured with pores of distinct sizes and narrow size-distribution34. The device is flexible allowing safe implantation within soft tissues such as the peritoneum without the risk of injury.
Figure 1: Overview of an implantable macrodevice and its pre-clinical development.

a) Schematic showing device design and operation. The device consists of a microfabricated body sealed to a polymeric membrane with a controlled pore-size which allows exchange of macromolecules but prevents infiltration of immune cells. b) Pre-clinical realization of the macrodevice. Top left: a view of the device used in pre-clinical testing in mouse. Right: false colored SEM image of the device cross section showing the polymeric membrane, reservoir containing cells and the silicone body of the device. Bottom left: false colored SEM of the membrane cross-section. Scale bar, top left panel 5 mm, right panel 20 μm, bottom left 5 μm. c) Z-stack confocal images showing human embryonic kidney cells (HEK293T), mouse fibroblasts (3T3) and myoblasts (C2C12) within the sealed devices cultured in vitro for 7 days. Scale bar 200 μm. d) Rate of erythropoietin (EPO) production over time by devices encapsulating HEKepo cells and cultured in vitro. 0.25 million HEKepo cells were encapsulated per device and EPO concentration was measured in culture supernatants over a period of 12 hrs for each time point; N=7 devices; error bars mean ± s.e.m.
The device supports the growth of several cell lines in vitro, including human embryonic kidney cell HEK293, fibroblasts 3T3, and myoblasts C2C12 (Fig 1 b,c). Scanning Electron Microscopy (SEM) images of the device cross-section showed encapsulated cells tightly packed inside the cell reservoir (Fig 1b). Confocal imaging of the encapsulated cells though the PDMS body (see Fig S1 for experimental setup) demonstrated 3D-structures formed by the cells within the device confirming survival of the cells (Fig 1c). Since implanted human cells can invoke a xenogeneic response in rodents35, we sought to examine the utility of our device with human cells in immunocompetent mice. HEK293T cells were transformed to constitutively secrete mouse erythropoietin (HEKepo) (Fig S2). Erythropoietin (EPO) - a hormone that drives red blood cell production in the bone marrow - is used to treat anemia in chronic kidney disease and chemotherapy36. Moreover, EPO is also a useful hormone evaluating the activity of therapeutic cells. A short plasma half-life (less than 6 hours in rodents) and high bioavailability37 of EPO results in a good correlation between the EPO concentration in the serum to the amount of viable therapeutic cells secreting EPO.
HEKepo cells encapsulated in the device were cultured for 4 weeks, while the secretion rate of EPO from the device was measured periodically. Increasing EPO secretion was seen in the first 2 weeks which plateaued afterward (Fig 1d) to a steady state rate of 0.4 – 0.6 μg of EPO per day, which we expected to achieve a therapeutic effect38. We found that although the secretion rate of EPO from cells was not affected by encapsulation (Fig S2), the proliferation rate of cells rapidly dropped inside the device over the first week possibly due to metabolic restriction on cell growth due to limited available space and nutrients (Fig S3). The encapsulated cells remained confined within the device when membranes with 1 μm or smaller pore size was used, but readily escaped from pores with 3μm in diameter (Fig S4).
Determining membrane pore-size for immune protection
Membrane pore-size is a critical parameter governing immune isolation and ultimately device function. Pores with large diameters favor increased diffusion of macromolecules and other large payloads (exosomes and viruses) across the membrane, but risks compromising the immune-barrier function of the membrane. Despite several suggestions10,39, the upper limit on the membrane pore-size which can maintain immune-barrier function and prevent the destruction of encapsulated cells by the host immune system remains to be determined.
To do this, we used BALB/c mice which are immune competent and have been extensively used to study acute allo- and xenograft rejection40. Devices were fabricated using PCTE membranes with nominal pore-size of 3 μm, 1 μm, 0.8 μm, 0.6 μm, and 0.4 μm. The membrane permeability was equal amongst the groups by varying pore density to rule out differences in oxygen transport. Devices loaded with HEKepo cells were transplanted into the intraperitoneal space (i.p.) of BALB/c mice, and EPO concentrations in the serum were monitored weekly. We found that the serum EPO level more than doubled in all animals by 2 days post-transplantation and continued to increase through the first week (Fig 2a). The devices were functional in vivo allowing survival and growth of the encapsulated cells, and successfully delivered EPO. After the first week, devices with pore-size of 3μm failed as EPO levels eventually returned to baseline, while other groups remained functional with steady increase in serum EPO levels.
Figure 2: Membrane pore-size regulate immune cell infiltration and survival of xenograft in BALB/c mice.
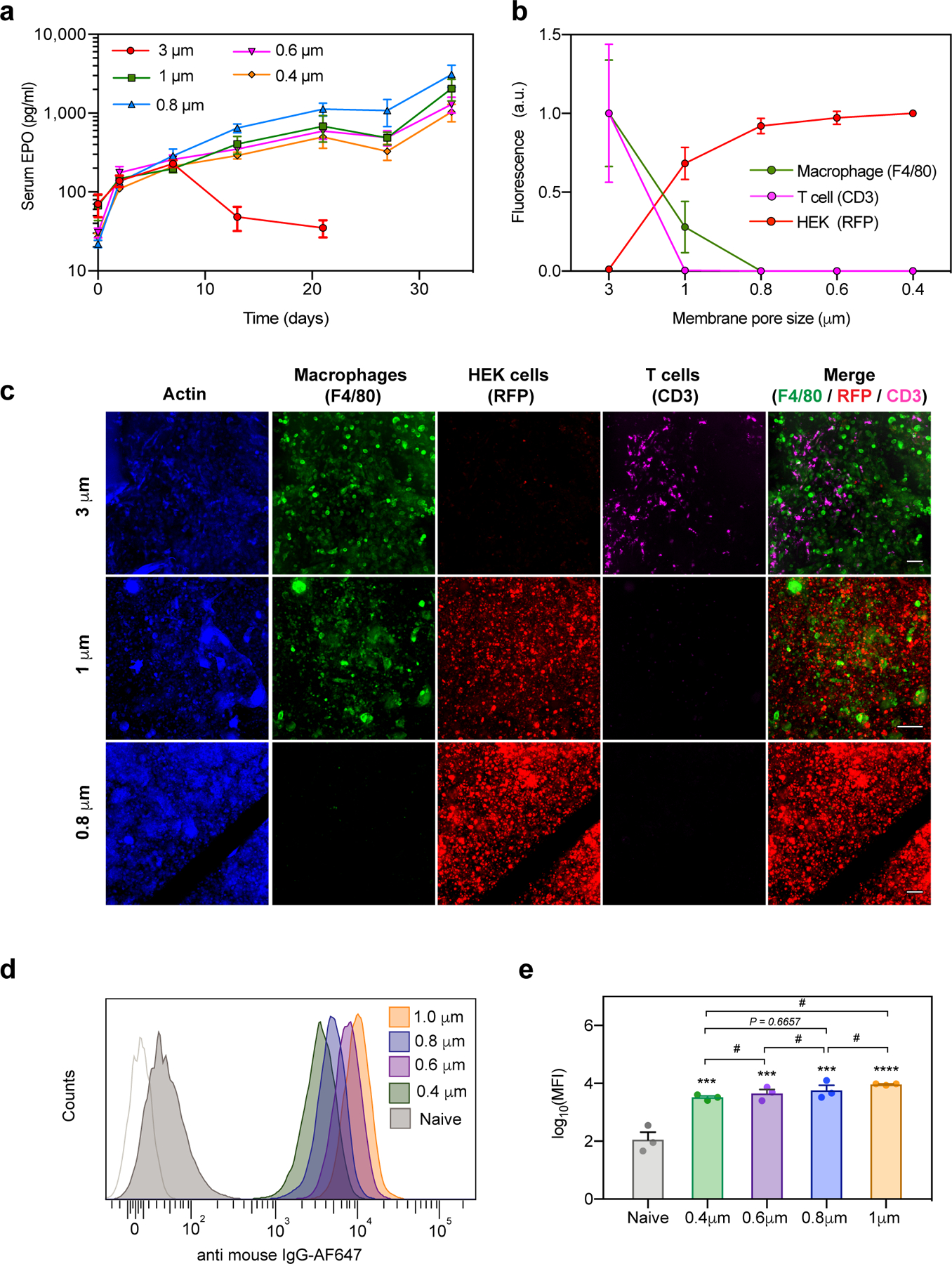
a) Time couse of serum concentration of EPO in BALB/c mice after i.p. transplantaton of HEKepo cells encapsulated within macrodevices with different membrane pore-size (n = 4 animals per group). Devices with 3 μm pores fail rapidly while pore-sizes of 1μm or less, protect the grafted cells. Error bar: mean ± s.e.m
b-c) The retrieved devices (after 35 days) were immunostained for actin (cytoskeleton, blue), macrophages (F4/80, green), HEKepo (RFP, red) and T-cells (CD3, magenta), after which confocal microscopy was used to image and identify the cells present inside the reservoir volume of the devices (n=3 animals per group). Total fluorescence (normalized) quantification (b) and representative z-stack images (c) shows complete infiltration of immune cells through the 3 μm membranes leading to widespread loss of graft, while 1 μm membranes selectively allowed infiltration of macrophages but not T-cells, preventing graft damage. All pore sizes of 800 nm and below prevented immune cell infiltration Supplementary Fig S5. Scale bar 100μm. Experiments were repeated twice with similar results.
d-e) Detection of graft specific antibody in serum. HEKepo cells incubated with animal serum (day 35) was counterstained with anti-mouse IgG-AF647 and analyzed using flow-cytometry (d) which showed the presence of cell specific antibodies in serum of all the animals receiving the devices. (e) Quantification of mean fluorescence intensity (MFI) showed significant levels of anti-graft IgG with the devices over untreated animals (naïve). n = 3 animals per group. Experiments were repeated twice. Error bars mean ± s.e.m. Statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction. ***, p<0.001; **** p<0.0001; # p>0.999. Symbols over the bar graphs represents comparison with naïve group.
We suspected that the 3μm pores allowed infiltration of immune cells, as reported previously41, causing graft rejection. To verify our hypothesis, we retrieved the implanted devices after 5 weeks and immunostained them to identify macrophages (F4/80) and T-cells (CD3). We used confocal microscopy to image the encapsulated cell reservoir within intact devices. The HEKEpo cells expressed RFP which was used to identify grafted cells. As expected, we found extensive infiltration of macrophages and T-cells, along with complete loss of the graft, inside the 3μm devices (Fig 2b,c). In contrast, devices with pore-size of 0.8μm or lower had no infiltration of immune cells and large aggregates of HEKepo cells were seen (Fig 2b,c Fig S2).
Interestingly, 1 μm pore-sized membranes prevented entry of T-cells but allowed infiltration of macrophages inside the device. The HEKepo cells were found intact despite direct contact with the infiltrated macrophages. This was a startling observation and we wanted to ascertain the size of the pores which allowed macrophage infiltration into the device. The rated pore-size of PCTE membranes are determined by the bubble point method which might underestimate the pore cross-section area – a parameter relevant for cell migration. Therefore, SEM imaging was used to analyze the membranes and determine the pore cross-sectional area. We found that all the membranes had a wide distribution in the pore cross-sectional area with considerable overlap between membranes of different groups (see supplementary discussion, and Fig S6). Based on the distribution, it seemed likely that pores with area between 1.00 – 1.25 μm2 which are unique to the 1μm membrane (not present on the 800nm membranes), are responsible for selective infiltration of macrophages. Cells have only been known to migrate through confined spaces with a minimum 7μm2 in cross-section (~ 3μm in diameter) 22 or higher42, and this report of cells migrating though pores 1.25 μm2 in area is below that limit in vivo.
Lastly, we tested for an antibody response against the graft, and found that by 5 weeks all the groups had high levels of circulating HEKepo specific IgG antibodies (Fig 2d,e). Since our membranes are not permselective for antibodies (pores are 100-fold greater in size than antibody39), our results indicate that a humoral response alone was not sufficient to cause significant graft damage. Also, a membrane pore-size cut-off of 0.8 μm (pore area < 1 μm2) was determined to be adequate for preventing immune cell infiltration and graft failure in our model. However, commercial membranes can potentially contain larger defects affecting long term experiments. Therefore, we decided to use 0.4 μm rated membranes in our further studies.
Developing a biocompatible surface coating
Biocompatibility of the implanted device is an important feature that affects in vivo. Foreign body response (FBR) to the implanted device has the potential to damage the graft in two ways – direct cytokine assault, and through deposition of a fibrotic capsule causing hypoxia10. Materials commonly used in the construction of macrodevices are poorly biocompatible and previous studies have noted significant fibrosis on implanted devices43 impacting their success. To address this issue, we set out to develop a surface coating that can enhance device biocompatibility and mitigate the FBR. Zwitterionic polymers demonstrate anti-fouling properties and coatings of zwitterionic polymers on biomaterials have reduced FBR in rodents and large animal models44–46. Zwitterionic head-groups can significantly affect biocompatibility46, so we decided to test three major classes of zwitterionic polymer coatings: poly(phosphorylcholine) (PC), poly(carboxybetaine) (CB) and poly(sulfobetaine) (SB) (Fig 3a). In another recent study, a combinatorial screen identified a small-molecule modification of alginate which demonstrated notable improvement in biocompatibility of alginate hydrogels in rodents and non-human primates24. We postulated that the small molecule - tetrahydropyran phenyl triazole (THPT) - might act as an anti-fibrotic coating and decided to test it on our devices along with the zwitterionic polymers. Devices were coated through Surface Initiated Atom Transfer Radical Polymerization (si-ATRP) which allowed controlled growth of dense polymer brushes from the surface (Fig 3a). X-ray photoelectron spectroscopy (XPS) analysis confirmed a high degree of polymer grafting on the surface of the device (Fig S7, SI table 1)47. Modification of the surface properties was confirmed with contact angle measurements which showed significant reduction in water contact angle values. The zwitterionic coatings made the devices significantly hydrophilic with contact angle values ~ 10°. SEM imaging of the device further confirmed the presence of the polymer coating on the device surface (Fig S8). The coating did not appear to affect the permeability of the membranes significantly (Fig S9).
Figure 3: A biocompatible surface coating minimized foreign body reaction and prevents device failure in C57BL/6 mice.
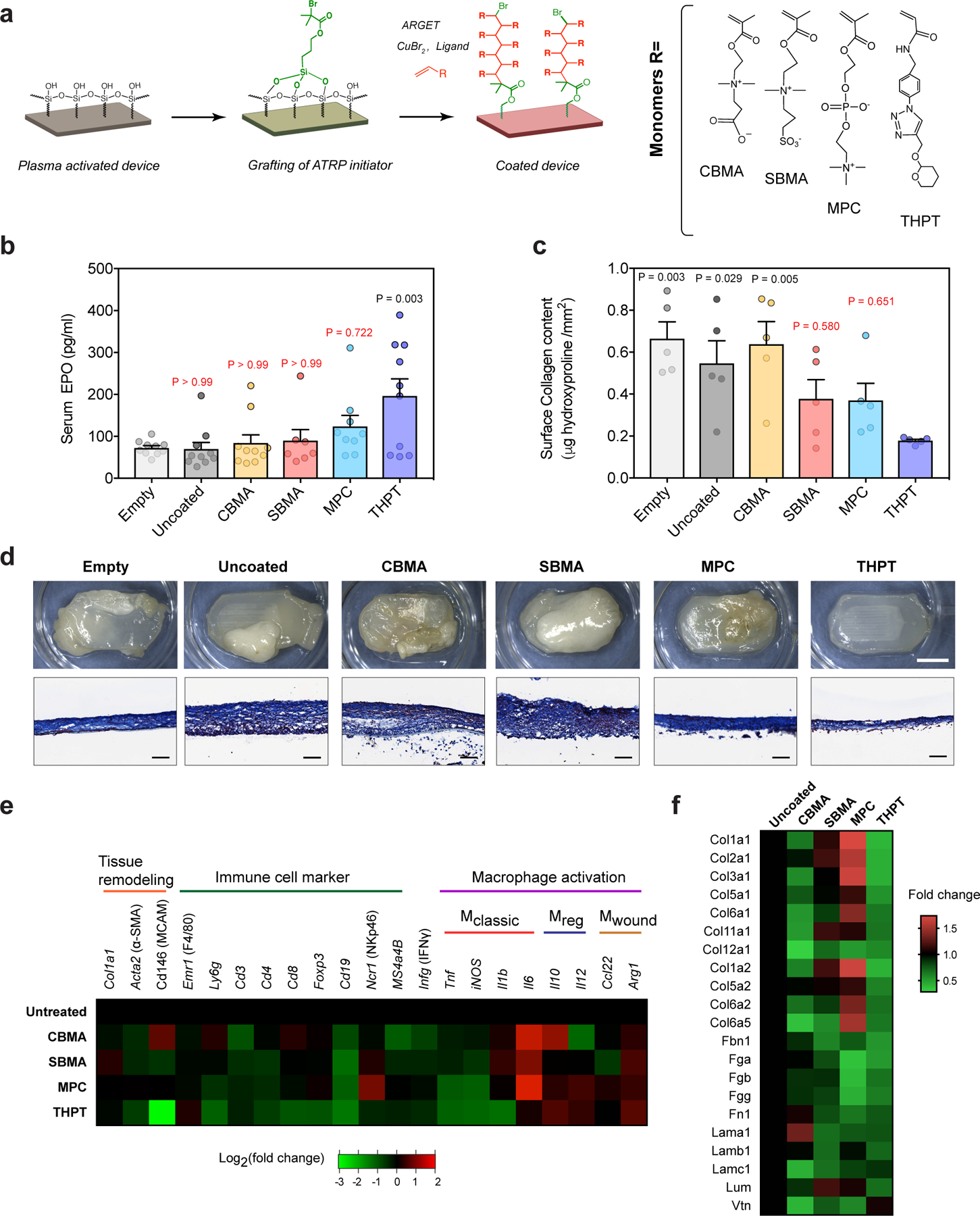
a) Schematic of the chemistry scheme used to modify device surface with biocompatible polymers. Surface initiated ARGET-ATRP was used to grow polymer brushes of four different monomers - carboxybetaine methacrylate (CBMA), sulfobetaine methacrylate (SBMA), methacryloyloxylethyl phosphorylcholine (MPC) and tetrahydropyran phenyl triazole (THPT), the structures of which are shown.
b) Serum concentration of EPO in C57BL/6 mice, 28 d after i.p. transplantation with surface-modified(coated) devices encapsulating HEKepo cells (1×106 per device). Uncoated devices with or without cells (empty) were used for comparison. The THPT-coated devices showed the highest increase in serum EPO compared to the other coatings. Error bar: mean ± s.e.m, Group size (animals) n = 7 (SBMA), 9 (MPC), and 10 (empty, uncoated and THPT) represents combined data from two independent experiments. Statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction. P values are shown for comparison with empty devices.
c) Collagen deposition on retrieved devices quantified by hydroxyproline assay of proteins extracted from the device surface, showing lowest hydroxyproline deposition on THPT-coated devices. Error bar: mean ± s.e.m (n = 5 animals). Statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction. P values are shown for comparison with THPT group.
d) Representative images of the retrieved devices. Bright-field images of intact devices (top panel) along with histological sections of the device mebranes (lower panel, masson’s trichrome) shows minimal fibrosis on the THPT coated devices. Scale bar: 5 mm (BF), 100μm (MT). Experiments were repeated twice with similar results.
e) Nanostring-based analysis for expression of phenotypic markers in cells attached to the device surface 28d post-transplant. Values are normalized to uncoated devices and presented on a base 2 logarithm scale (n = 5 animals per group).
f) Relative proteomic analysis of the protein lysates extracted from retrieved devices, showing levels of major proteins of the extracellular matrix. Values are expressed as fold change over uncoated devices.
Next, we directly tested the efficacy of each coating on improving device performance in C57BL/6 mice - a fibrotic a mouse model 48. Coated and uncoated devices loaded with HEKepo cells were implanted in the i.p. space of C57BL/6 mice, with empty devices serving as a control. We found that by 4 weeks the THPT coated group had achieved highest serum EPO levels (195.7 pg/mL), significantly more than the uncoated (69.84 pg/mL) or empty device (72.3 pg/mL) (Fig 3b). In contrast, PC-coating was most effective amongst the three zwitterionic coatings, but only modestly increased serum EPO levels. Thus, we selected THPT for the biocompatible surface coating on our devices. Since surface coatings can degrade under harsh conditions, we wanted to test the long-term stability of the THPT coating in the in vivo environment. Surface analysis of implanted materials is challenging due to the deposition of proteins and cells which can mask the actual chemical composition of the surface. To overcome this challenge, we used confocal-Raman spectroscopy to analyze surface of devices retained in vivo for 4 weeks. The characteristic peak for the triazole ring49 was used to confirm the presence of the THPT coating as this moiety is absent in physiological molecules. We found no significant change of characteristic peaks after retrieval of the device (Fig S11), confirming that the THPT coating is retained for at least 4 weeks in vivo.
The effect of the surface coatings on foreign body response
We investigated how the THPT coating might improve the survival of encapsulated cells. Visual examination of the explanted devices (after 4 weeks in vivo) revealed moderate to heavy fibrosis and extensive tissue overgrowth on uncoated and zwitterionic-coated devices, while the THPT-coated devices remained clean with only a thin collagenous capsule (Fig 3c,d). Masson’s trichrome staining of device cross-sections showed a 150 – 250 μm thick fibrotic capsule on the uncoated membranes, while it was only 30 – 50 μm on the THPT-coated membranes (Fig 3c). The zwitterionic-coated membranes had varying degree of fibrosis with PC being the best of the zwitterionic coatings. Quantification of the collagen deposition on the devices correlated with the histological findings and confirmed that THPT-coating had the best anti-fibrotic efficacy resulting in a 70% reduction in collagen buildup (Fig 3c).
To understand how the surface coatings might alter the immune response to the device, we performed gene expression analysis using nanostring assay on the RNA extracted from explanted devices and measured the relative abundance of genes associated with FBR. We found that each coating had a unique pattern of gene expression in comparison to uncoated devices (Fig 3e). Notably, the THPT-coating suppressed the expression of pro-inflammatory cytokines - TNFα and IL1β typically associated with classically activated macrophage, and CD146 – a marker for neovascularization. Proteomic analysis of proteins extracted from the device surface using the iTRAQ method detected 11 isoforms of collagen, 3 isoforms of fibrinogen, fibronectin, laminin, and other extracellular matrix proteins, all of which were substantially reduced on the THPT-coated devices (Fig 3f). Finally, we immunostained the devices for myofibroblast (αSMA), T-cells (CD3), macrophages (F4/80) and F-actin (cytoskeleton) and used confocal microscopy to analyze the cellular deposit on the device membranes. We found that the THPT-coated membranes had markedly reduced cellular overgrowth compared to untreated membrane (Fig 4a). Direct measurement of total DNA content on the device surface was significantly lower on THPT-coated device further supporting the microscopic observations (Fig 4b). Together, these results suggest that the THPT-coating possibly alters the immune response resulting in reduced secretion of pro-inflammatory cytokines, lower cellular recruitment to the device, finally leading to a mitigated fibrotic response.
Figure 4: THPT-coating reduces cellular buildup on the implanted devices.
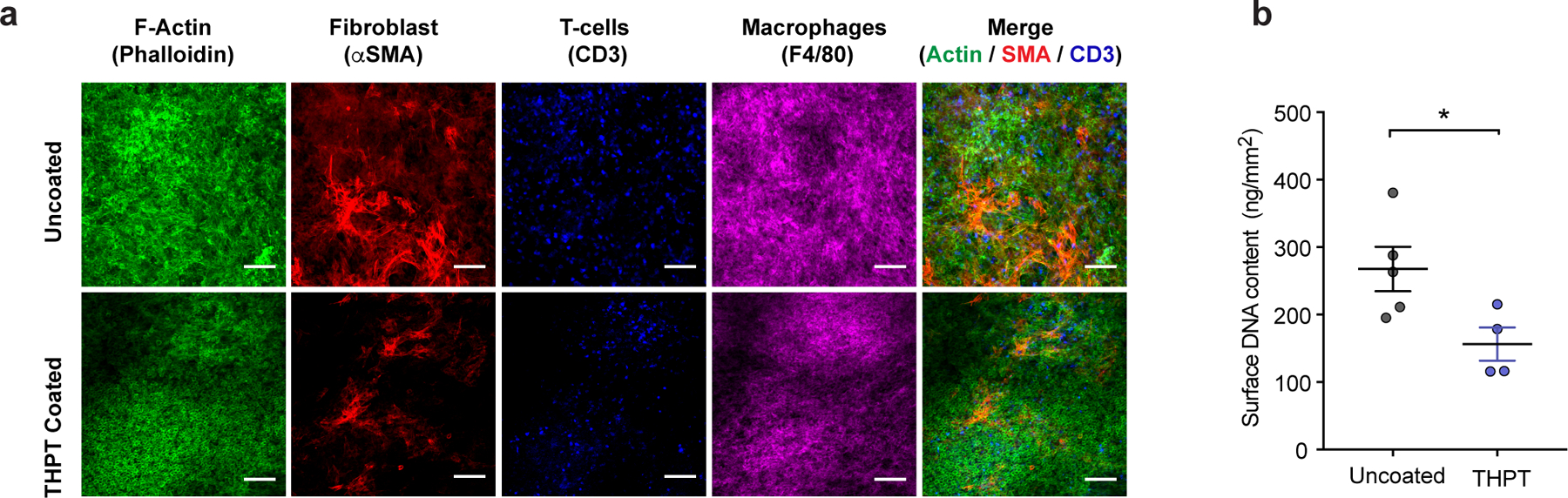
a) Representative immunofluorescent z-stack confocal images showing cytoskeleton (actin, green), fibroblast (α-SMA, red), T-cells (CD3, blue) and macrophages (F4/80, magenta), within the fibrotic layer on the membranes of uncoated and THPT-coated devices from Figure 3. Significantly lower amount of staining can be seen on THPT membranes. Scale bar: 100μm. Experiments were repeated 3 times. b) Quantification of total DNA extracted from the cells attached to the surface of THPT-coated and uncoated devices in Figure 3. Amount of extracted DNA was normalized to device surface area. Sample size n = 5 animals (uncoated) and 4 animals (THPT). Error bar: mean ± s.e.m. *, p = 0.024 comparisons by unpaired, two-tailed t-test.
Demonstration of long-term sustained release of therapeutic proteins in vivo
THPT-coated and uncoated devices were loaded with 2 ×106 HEKepo cells and transplanted in the i.p. space of C57BL/6 mice. The control group received THPT-coated devices without cells. We found that the serum EPO levels in the THPT group increased steadily over first 10 weeks reaching ~500 pg/mL (about 8-fold increase), and remained high for the duration of the study (130 days) (Fig 5a). The result shows that THPT-coated devices can successfully maintain the viability and function of xenografts for over 4 months. The biphasic trend in serum EPO levels recapitulated in vitro observations (Fig 1d) and may be attributed to the restricted growth of cells due to overcrowding inside the device and cellular aging. The THPT coating did not prevent a humoral response as high levels of circulating antibodies against the HEKepo cells were detected in the sera of all animals (Fig S12) similar to that seen previously (Fig 2d). In contrast, the uncoated device failed to increase serum EPO in the first 10 weeks, most likely due to FBR to the device, which resulted in damage to the grafted cells (Fig 5a). However, a modest rise in serum EPO was observed in the later part of the study which might indicate recovery of the surviving cells from the initial damage and their subsequent proliferation within the device.
Figure 5: Long term delivery of erythropoietin (EPO) in C57BL/6 mice in a sustained or ‘on-demand’ manner, using coated macrodevice encapsulating HEKepo cells.
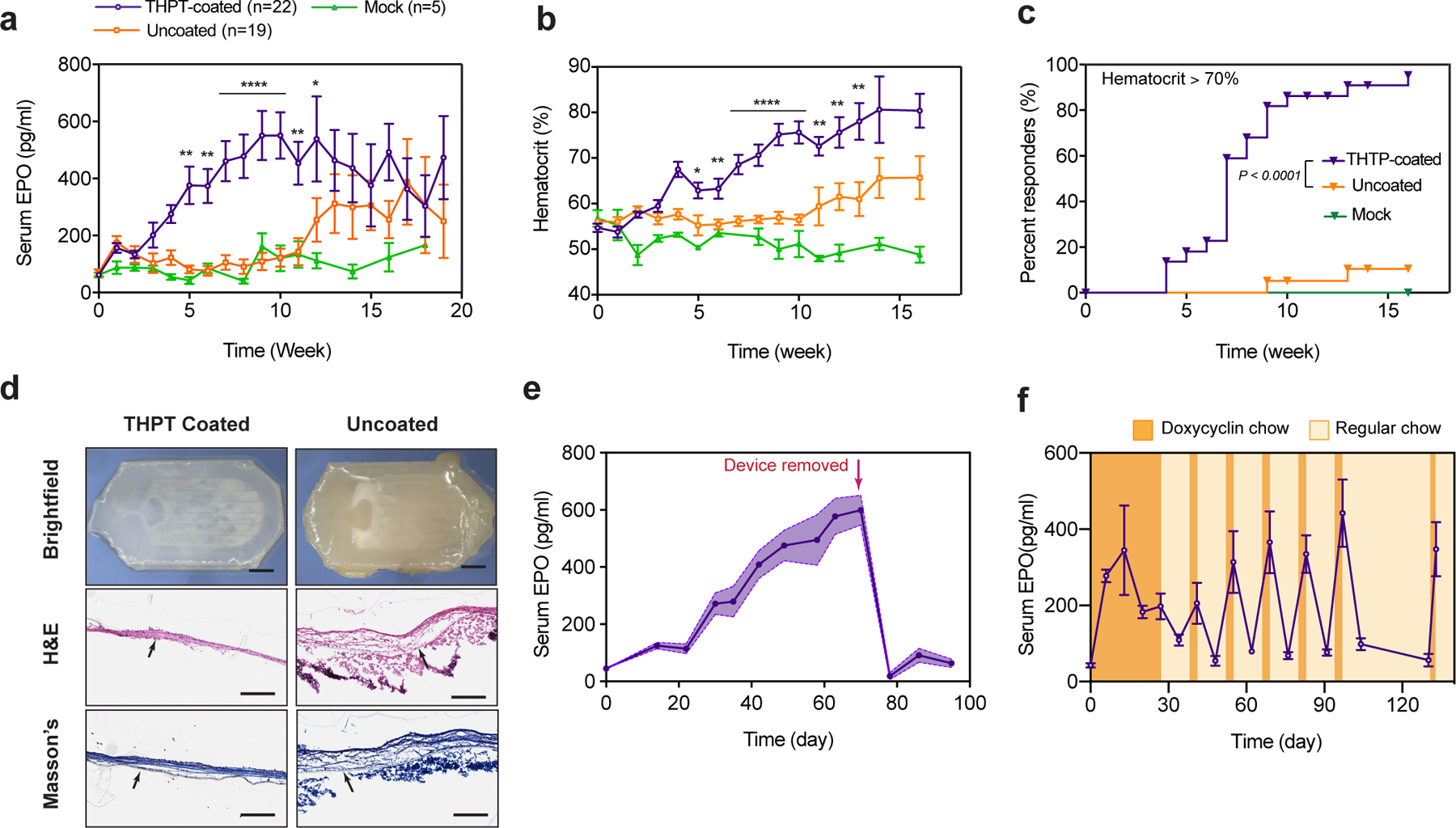
a-c) Efficacy of THPT-coated devices in long-term sustained delivery of EPO. Serum concentration of EPO (a) and hematocrit (b) over time in C57BL/6 mice after i.p. transplantation of THPT-coated or uncoated macrodevices encapsulating 2×106 HEKepo cells. Empty THPT-coated devices were used as mock transplants. THPT-coated devices showed excellent efficacy in elevating serum EPO and hematocrit, while the uncoated devices showed only limited success. Error bar: mean ± s.e.m. Statistics: EPO and hematocrit comparison between THPT and uncoated devices at each time point by unpaired, two-tailed t-test. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001. c) Kaplan–Meier plot of the fraction of animals with hematocrit ≥ 70% over the course of treatment, showing a significantly better response with THPT-coated devices. Group size (animals): THTP n=22, uncoated n=19, mock =5, represents combined data from three independent experiments. Log-rank (Mantel-Cox) test, P < 0.0001, two-sided.
d) Representative bright-field and histology images of the devices in a, retrieved after 130d, showing the THPT-coated devices remained biocompatible with only a thin fibrotic capsule. Histological sections shows fibrosis on the device membrane with H&E stain (middle panel) and masson’s trichrome (bottom panel). The membrane is indicated by a black arrow. Scale bar: Top panel - 2mm, Middle and lower panel – 200μm.
e) Time course of serum EPO concentration after C57BL/6 mice were implanted with THPT-coated devices encapsulating HEKepo cells, showing animals maintained high EPO levels until devices were explanted (day 70, indicated by an arrow), after which the EPO levels promptly returned to baseline levels. Data points represents group mean (n = 5 animals) while the shaded region is s.e.m.
f) ‘On-demand’ delivery of EPO achieved by encapsulating doxycycline-inducible HEKepo cells within THPT-coated devices and transplanting in i.p space of C57BL/6 mice. Doxycycline diet (orange shaded region) resulted in sharp rise in serum EPO concentrations, which was rapidly reversed by switching to normal diet (yellow region). Error bar: mean ± s.e.m (n = 5 animals).
Increase in blood hematocrit is another indicator of therapeutic delivery of EPO50 and can reflect long-term serum EPO levels. Measurements of hematocrit indicated a robust performance by the THPT-coated devices showing a steady rise in hematocrit values, significantly higher than the uncoated group (Fig 5b). To better evaluate the efficacy of our device in maintaining high RBC production and treating anemia, we set a therapeutic goal of maintaining the hematocrit at supraphysiologic levels (> 70%) which can only be achieved through the sustained delivery of EPO50. The THPT group showed significantly better response to treatment in comparison to the uncoated group, with >95% of animals achieving the therapeutic goal by the end of the study (Fig 5c). Examination of the retrieved devices at the end of the study (130 days) revealed the uncoated devices to be heavily fibrosed while the THPT-coated devices remained relatively clean (Fig 5d) indicating that the THPT coating can provide durable protection against fibrosis.
Demonstration of device retrieval
Macrodevices simplify the removal of the implant to allow rapid withdrawal of therapy10. To demonstrate this, we implanted devices loaded with HEKepo cells in the i.p. space of C57BL/6 mice through a laparotomy procedure. After retaining them for 10 weeks, we performed another laparotomy procedure to extract the devices. The THPT-devices could be easily located and retrieved without apparent distress to the animals. In contrast, extensive adhesion to internal organs was noted on many of the uncoated devices, preventing their safe removal.
Removal of the THPT-coated devices resulted in a sharp drop in serum EPO which returned to pre-surgery levels within a week (Fig 5e). To confirm that the explanted devices contained viable cells, we measured the EPO secretion rate of the devices in vitro after retrieval and compared to the secretion rate measured before implantation. We found that the secretion rate of the THPT devices increased over the 10 weeks period in vivo, indicating survival and proliferation of the encapsulated cells. An opposite trend was seen with the uncoated devices where the secretion rate declined over that period (Fig S13). This data provides direct evidence that the THPT-coating is essential for the survival of the encapsulated graft.
Long-term regulated delivery of therapeutic proteins using tetracycline-inducible engineered cells
For some diseases, it is desirable to have therapeutics ‘on-demand’ to more precisely follow a dosing regimen. Cells engineered to produce therapeutics that are regulated by Doxycycline offers one way to achieve this. Oral delivery of doxycycline (an antibiotic) can rapidly turn on the secretion of proteins in transplanted engineered cells harboring the inducible gene51. To demonstrate proof-of-concept, we first developed HEK cells with doxycycline-inducible EPO secretion (see methods). In vitro characterization showed excellent control over EPO secretion with minimal secretion in the absence of doxycycline and a 50-fold increase in secretion upon exposure to physiologically relevant doxycycline concentrations (Fig S14). The dox-inducible HEKepo cells were loaded inside THPT-coated devices and transplanted intraperitoneally in C57BL/6 mice, where they were retained for 130 days. To determine the ability of the device to deliver EPO ‘on-demand’, animals were switched from a regular diet to a doxycycline-containing diet (200 mg/kg) for 3 days every alternate week. EPO concentration was measured weekly. We found that while serum EPO remained at normal levels with regular diet, it increased by more than 5-fold while on a doxycycline diet (Fig 5f). The serum EPO remained responsive to doxycycline induction throughout the study (130 days) indicating that the encapsulated cells maintained their activity. Importantly, we could maintain hematocrit levels at physiological levels throughput the experiment with controlled delivery of EPO (Fig S15).
Macroenapsulation of islets
Islets isolated from Sprague-Dawley rats were loaded inside THPT-coated or uncoated devices (200–300 I.E. per device). Since naked islets do not survive well without a supporting matrix52, we loaded the islets in an alginate solution which was later crosslinked in-situ by dipping the device in a barium bath (see methods). The devices were transplanted intraperitoneally in STZ-induced diabetic C57BL/6 mice and blood glucose (BG) measurement was used to monitor islet function over time non-invasively. While both the groups achieved euglycemia (BG ≤ 200 mg/dL) immediately post-transplantation, the majority of uncoated devices failed within 3 weeks (median 19.5 days) resulting in hyperglycemia (Fig 6a, Fig S16). In contrast, the median graft survival time as determined by maintenance of normoglycemia of the THPT-coated group was 75 days post-transplantation. The BG levels and fraction-cured of the animals receiving THPT devices was significantly better compared to those receiving uncoated devices (Fig 6a, 6d).
Figure 6: Efficacy of coated macrodevice encapsulating rat islets in curing STZ-diabetic C57BL/6 mice.
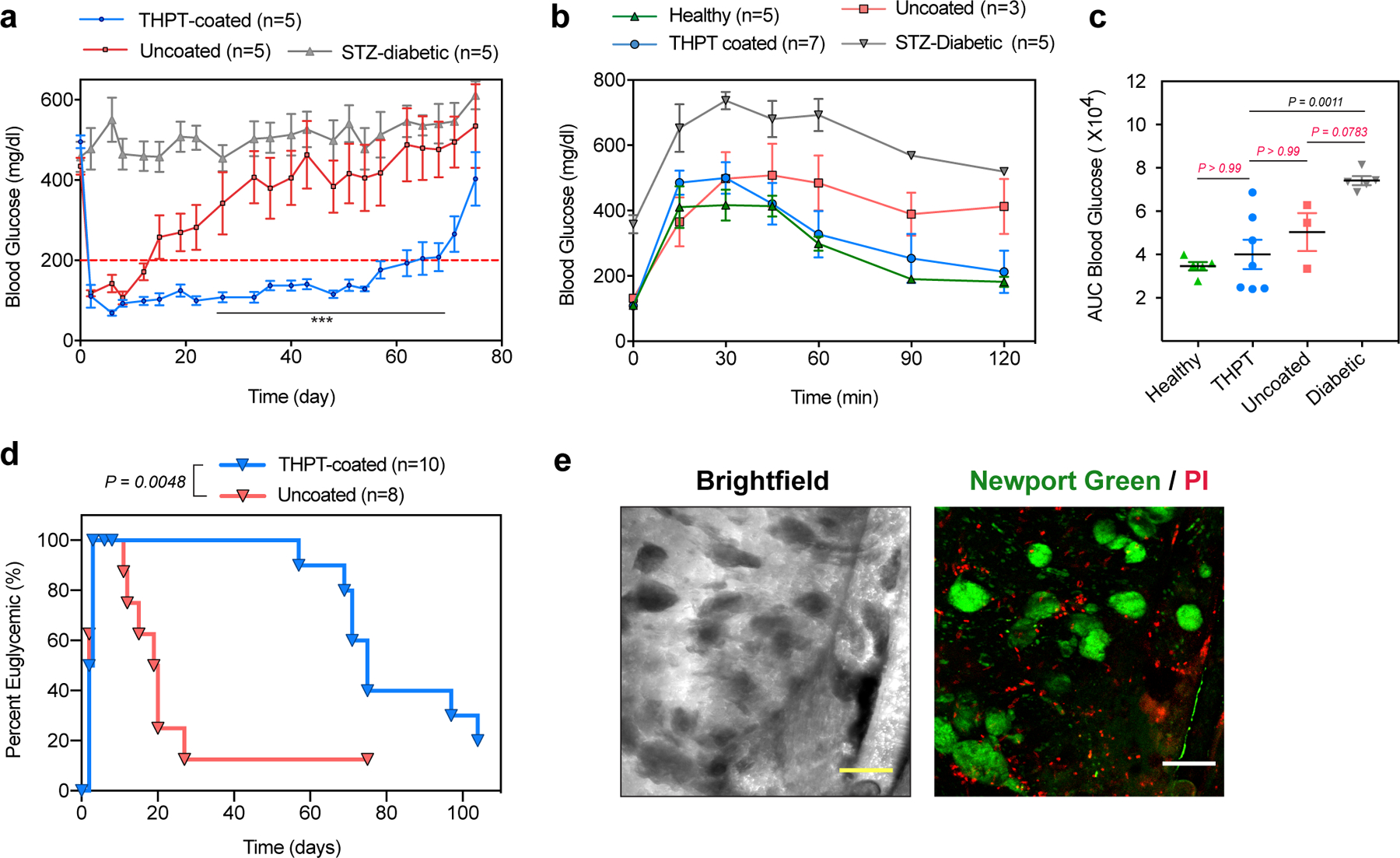
a) Blood-glucose curves showing prolonged euglycemia with THPT-coated devices, while success was short-lived with uncoated devices resulting in significantly higher blood glucose levels. Devices encapsulated 200–300 IEs of islets with n=5 animals in each group, and the experiments were repeated twice. Error bar: mean ± s.e.m, statistical comparison was performed between THPT and uncoated devices at each time point by unpaired, two-tailed t-test. ***, p <0.001.
b-c) Intravenous glucose tolerance test (IVGTT) of healthy and diabetic mice performed 15 days post-transplantation shows (b) no significant delay in BG correction with THPT-group and healthy mice, while the uncoated devices failed to correct BG during the 120 mins. Area-under-the curve (AUC) analysis (c) shows significant improvement with THPT-devices in blood glucose kinetics. Error bar: mean ± s.e.m. One-way ANOVA with Bonferroni multiple comparison correction.
d) Kaplan–Meier plot showing the fraction of euglycemic (BG<200 mg/dl) STZ-C57BL/6 mice after receiving rat islets within THPT-coated or uncoated devices. Log-rank (Mantel-Cox) test, P = 0.0048, two-sided.
e) Live/dead staining with Newport green (stains insulin granules) and PI (dead cells) confirming the viability of encapsulated islets inside THPT-coated devices retrieved from STZ-C57BL/6 mice 35 days post-transplantation. Experiments were repeated twice with similar results. Scale bar: 200μm.
To evaluate the kinetics of insulin release from the device, we performed an intravenous glucose tolerance test (IVGTT) on the animals for the first three weeks post-transplant (Fig 6b,c and Fig S17). Although both the devices functioned similarly on the first week, differences between the THPT and uncoated devices started to emerge from the second week onwards (Fig 6b, Fig S17b). The THPT devices demonstrated an excellent glycemic profile similar to that of healthy animals (Fig 6 b,c,), while the uncoated devices had a poor glycemic profile and did not achieve euglycemia over the 120 mins of the study. Rise in serum insulin levels in response to glucose challenge was also significant in THPT coated group (Fig S17). In a subset of animals, we explanted THPT-coated devices 35 days post-transplantation. All the animals saw a prompt return of diabetes immediately following the device removal confirming that the cure was device-dependent (Fig S18). Imaging of explanted devices showed viable islets clusters within the device along with a few scattered dead cells, indicating successful immune protection offered by the THPT-coated device. Although the reason for the failure of the device during the late phase of the study is unclear, device fibrosis did not appear to be the cause since the THPT-coated devices retrieved at the end of the study were found with minimal fibrotic deposition (Fig S19). It is more likely that the number of islets used was insufficient for a long-term cure53 and they could not accommodate an increase in body weight (~20–40%) of the animals over the period of the study 54.
Discussion
We have developed a modular biocompatible macrodevice that can maintain xenografts in immunocompetent animals over long periods. To demonstrate the broad potential of this microfabricated device, we first showed that it is able protect engineered human cells secreting EPO in C57BL/6 mice for at least 130 days (Fig 5a–c). Using transgenic human cells with an inducible gene circuit, the device was also adapted to provide on-demand therapeutic protein in response to orally dosed small molecules (Fig 5f). Approaches like this may enable the simple implementation of therapies where irregular dosing of protein therapeutics is required. Finally, we show that encapsulated rat islets were capable of restoring euglycemia for more than 75 days in diabetic mice (Fig 6a,d). While additional engineering maybe needed to enable longer term survival of islets, this study provides proof-of-principle that cell therapies which secrete therapeutics in response to endogenous environmental cues can also be incorporated into this device.
To optimize transport properties, we identified an upper limit on membrane pore-size for maintaining immune isolation properties. Commercial polycarbonate track-etched membranes with rated pore diameter up to 0.8μm could prevent infiltration of immune cells and offer long term protection to encapsulated human cells in vivo. Since the cells were encapsulated without any supporting matrix, the polycarbonate membrane seems sufficient for immune protection. We discovered that infiltration of immune cells could be selectively regulated by tuning membrane pore-size, with pores possibly between 1 – 1.25 μm2 in area allowing selective macrophage infiltration but blocking T-cells. To our knowledge, cell migration through a pore less than 7 μm2 in area, as well as the selective blockage of migration of different immune cells, hasn’t been reported before. Recent research has highlighted the crucial role of macrophages in graft repair and protection55. Macrophages are also important for β-cell regeneration in vivo and may actually be beneficial for islet transplantation56. Therefore, the ability to selectively recruit beneficial macrophages to the graft without rejection may provide new methods of affecting cell-based therapies.
A key component of device design was the need to reduce the fibrotic response (Fig 3b). The zwitterionic coatings – despite their potent anti-fouling properties44 – did not improve the biocompatibility of our devices. While the failure mode is unclear, we note that the presence of xeno-antigens can aggravate the immune reaction, and may have provided a confounding factor. The THPT coating described here provided durable protection against fibrosis, and was required to enable the survival of cells long term (Fig 5a). While the original chemistry was identified in a screen of modified alginate polymers, we found it surprising that the addition of a small fragment of this modification would enable resistance to fibrosis on a completely different material. Since fibrosis is a common challenge with other micro- and macro- cell encapsulation devices10,15,16, we believe the ability to use this coating on different devices may facilitate the function of other cell delivery systems. It might be possible that THPT coating allows increasing the membrane pore size while maintaining immune isolation, and needs further investigation.
We further hypothesize that this approach maybe applicable to a wide variety of medical devices such as catheters, breast implants, sensors, and drug delivery devices.
Methods
Material and Reagents
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and cell culture reagents from Life Technologies (Grand Island, NY), unless otherwise noted. Sillicone elastomer Sylgard 184 was obtained from Dow Corning Corp. (Cat. # 3097358–1004). Polycarbonate (PCTE) Membrane filters (hydrophobic, PVP-free), 25 mm in diameter, were obtained from Sterlitech corporation. Silane initiator for ATRP : (3-Trimethoxysilyl)Propyl 2- Bromo-2-methylpropionate (Cat. # SIT8397.0, Gelest Inc.). HEK292T cells were purchased from ATCC. Pre-packaged lentivirus for cell transfection was purchased from AMSBio (mouse EPO LVP496, tetracycline inducible construct LVP017-Hygro). Precoated EPO ELISA plates were purchased from Biolegend (Cat# 442780). Antibodies: AF687-conjugated anti-mouse F4/80 (Clone BM8), BV421-conjugated anti-mouse CD3 (clone 17A2) and AF647-conjugated anti-mouse IgG (Cat# 405322) was purchased from Biolegend. Cy3-conjugated anti-mouse alpha smooth muscle actin antibody (Clone# 14A) was purchased from Sigma Aldrich (St. Louis MO). Filamentous actin (F- actin)-specific Alexa Fluor 488-conjugated Phalloidin, DAPI, Propidium Iodide, Sytox blue, Newport Green, and Click-iT Edu AF647 cell proliferation kit (C10424) were purchased from Life Technologies. Cell Titer Glo Luminescent viability assay was purchased from Promega. THPT-acrylate monomer was custom synthesized from Pharmacon Inc. (Irvine, CA) and obtained with >98% purity as determined by mass spectrometry and NMR. CBMA (3-[[2-methacryloyloxy)ethyl] dimethylammonio] propionate) and MPC (2-(methacryloyloxy)ethyl 2-(trimethylammonio)ethyl phosphate) monomer was purchased from TCI America (Portland, OR). NOA81 UV Curable glue was obtained from Norland Products.
Transformation and maintenance of cell lines
HEK293T, 3T3 and C2C12 cells were purchased from ATCC and cultured in DMEM (High glucose) supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin, at 37C with 5% CO2.
HEKepo cells:
HEK293TCells were transformed with lentivirus expressing mouse erythropoietin under a modified CMV promoter also carrying a RFP- blasticidin fusion selection gene. Cells were transfected at a MOI of 50:1 in a media containing polybrene (5 ug/mL) for 24 hrs, after which cells were selected against blasticidin (20 μg/mL) for 3 weeks. Finally, the population was FACS sorted using to collect the top 10% of the RFP expressing cells in order to select for the clones with highest expression of EPO. The RFP expression and in vitro EPO secretion was found to be stable over time, even upon withdrawal of blasticidin. Therefore, we cultured the HEKepo cells in normal cell culture media.
Dox-inducible-HEKepo cells:
The modified CMV promoter used in the EPO construct carries two copies of tetracycline-operator elements, which normally does not interfere with expression, but can repress action of the promoter in the presence of tetracycline repressor protein (TetR). The HEKepo cells selected earlier, were transfected with lentivirus for constitutive expression of TetR protein according to manufacturer’s protocol, and selected in hygromycin media (800 μg/mL) for 3 weeks. The cell line was characterized in vitro for sensitivity to doxycycline and stability of protein production.
Rat islets isolation
Male Sprague-Dawley rats from Taconic (Rensselaer, NY) weighing approximately 300 grams were used for harvesting islets. All rats were anesthetized by a 1:20 xylazine (10 mg/kg) to ketamine (150 mg/kg) injection given intraperitoneally. The bile duct was cannulated and the pancreas was distended by an in vivo injection of 0.15% Liberase in RPMI 1640 media solution. Rats were sacrificed by cutting the descending aorta and the distended pancreatic organs were removed and held in 50 mL conical tubes on ice until the completion of all surgeries. All tubes were placed in a 37°C water bath for a 30 min digestion, stopped by adding 10–15 mL of cold RPMI 1640 media (with 10% HIFBS) and lightly shaking. Digested pancreases were washed twice in RPMI media, filtered through a 450 μm sieve, and then suspended in a Histopaque 1077 and centrifuged at 1,700 RCF at 4°C. Finally, the islets were collected from the gradient and further purified by gravity sedimentations. Purified islets were hand-counted by aliquot under a light microscope and then washed three times in sterile 1X phosphate-buffered saline. Islets were then cultured in RPMI 1640 media with 10% HIFBS and 1% penicillin/streptomycin, overnight for further use.
Fabrication of devices
The design was designed in AutoCAD and the photomask was printed on myler films at 50,000 dpi by a third-party vendor (Fineline Imagine, CO). Photolithography was then used to make the master-mold of the device. A 150μm think film of SU8–2050 (Microchem, USA) was spin-coated on a Silicon wafer, UV exposed in a Karl-Suss MA4 photoaligner and developed using PMAcetate. The height of structure was determined by Dektak surface profilometer and was found to be within 10% of the target height. Then, wafers were rendered inert by treating with Trichloro (1H,1H, 2H,2H-perfluorooctyl) silane in a vacuum chamber for 6 hrs. Polydimethylsiloxane (PDMS) prepolymer was mixed at a 10:1 ratio with the crosslinking agent, degassed, and poured on the Si-wafer at a fixed volume such that the thickness of the PDMS layer was between 0.8–1mm. The PDMS layer was cured at 60°C overnight, peeled from the wafer and cut to shape to produce the PDMS chips. Inlet holes were made using a 0.75 mm biopsy punch (Harris Ui-core, TedPella, USA), following which the PDMS chips were cleaned using ethanol and dried.
Polycarbonate track etched (PCTE) membrane (25mm diameter) were cut in half and silanized by heating in 5% (v/v) solution of (3-aminopropyl) trimethoxysilane in water at 90°C for 1 h. Silanized membranes were washed in DI water and then ethanol to remove excess silane. The membranes were dipped in 1% HCl solution for 30 min to generate surface silanol groups and then stored in DI water. To bond the membranes the PDMS chips were activated with air plasma (Harris Plasma chamber) for 70 second. Silanized membranes were briefly wetted with 70% ethanol and laid over the activated PDMS chips. The devices were carefully heated to 60C in a high humidity chamber for 3 hrs followed by an overnight baking in 120C for the silane crosslinking to complete and form stronger bond between the PCTE membrane and the PDMS chips.
Surface modification of devices
A special reaction chamber was custom designed, and devices were placed in a manner which ensured proper exposure of the device surfaces to the reactants, and the reaction mixture was constantly agitated for proper mixing. Devices were first grafted with an ATRP initiator by treating with air plasma for 70s and incubating in a silane bath : 95% Ethanol, 3% water (pH 4.5) and 2% (3-Trimethoxysilyl)Propyl 2- Bromo-2-methylpropionate, overnight at room temperature. The devices were washed with ethanol and dried on a hot plate before proceeding to the ATRP reaction.
ARGET-ATRP:
We optimized the concentration of monomer, ligand and Cu(II) for each reaction so as to enable maximum surface grafting. Reaction mixture was prepared by dissolving 5 μmol of copper(II) bromide (0.5 mM), 20 μmol of ligand (2 mM) and 20 μmol of free initiator (ethyl-α-bromoisobutyrate, 2 mM) in 10 mL of anhydrous methanol. The final concentration of the zwitterionic monomers (CBMA and SBMA) was 500 mM while it was 100mM for the THPT-monomer. After screening a number of commercially available ATRP-ligands, we found that TPMA (Tris(2-pyridylmethyl)amine) worked best for CBMA and SBMA coating reactions, while Me4Cyclam (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) was ideal for THPT-coating. The ATRP mixture was deoxygenated by three freeze-thaw cycles and charged into a nitrogen purged reaction chamber. The ATRP was initiated by injecting 0.3 mmoles of tin(II) 2-ethylhexanoate though a syringe, and the reaction was stirred at room temperature for 6 h. The tin(II) 2-ethylhexanoate acts as a mild reducing agent producing Cu(I)+ ions which catalyzes the polymer growth in a controlled manner. After 6 h, the devices were removed from the solution, washed 2x in methanol, followed by a wash in sterile 10X PBS (to dissolve deposited polymers), then DI water(sterile) and finally in 70% ethanol, after which they were air-dried in a sterile environment. ATRP of MPC: We found that unlike other monomers, best coating with the MPC (2-(methacryloyloxy)ethyl 2-(trimethylammonio)ethyl phosphate) monomer was obtained through a traditional ATRP approach. To modify the devices with MPC, monomer (2 g, 6.78 mmol) and 12 mL of 1:1 mixture of methanol and distilled water were added to a 25 mL schlenk flask. This reaction mixture was sealed with a rubber septum stopper and placed in an ice bath. After degassing the reaction solution with nitrogen for 20 minutes, the ice bath was removed and CuBr (9.7 mg, 0.067 mmol), and bpy (21.15 mg, 0.135 mmol) were added to reaction mixture under strong nitrogen flow. Then, the mixture was charged into a nitrogen purged reaction chamber containing the devices and allowed to react for 3 h at room temperature under continuous-weak nitrogen flow. Finally, the devices were removed, washed and prepared as descried previously.
Loading of devices
Devices were sterilized by immersion in 70% Ethanol for 1 hour and drying under sterile environment at room temperatures. Cells were loaded into pre-sterilized devices using aseptic techniques. The devices had a total fill volume of 15 μl.
HEKepo cells:
For EPO delivery, each device was loaded with 2 ×106 HEKepo or HEKepo-tetR cells (unless otherwise mentioned). The cells were harvested from culture flasks using standard cell culture procedure, and resuspended at a final concentration of 2 ×108 cells/mL in cell culture media inside a 1.5mL centrifuge tube. A Hamilton gastight glass syringe with 27G cemented needle (this reduced cell loss during injection) was used to inject 10μL of the cell suspension into each device. Following this, 5 μL of NOA81 UV glue (loaded into a plastic normjet 1mL syringe with 23 blunt needle) was injected to form a plug at the inlet. The device was then exposed to 365nm UV light (EA180 Spectroline Inc) for 10 sec with the clear PDMS side facing the UV source. This exposure was sufficient for curing the glue without causing any cell damage. Sealed devices were immediately immersed in media and cultured in 12 well plates.
Rat islets:
Our initial experiments found that rat islets required a supportive matrix for survival inside the device. Therefore, we decided to use alginate as the cell loading matrix since it is clinically approved, non-degrading, and has been extensively used in islet transplantation. Rat islets were harvested and encapsulated the same day to prevent loss. Islets were washed in HBSS (Ca/Mg free) two times to remove the media and counted. The islets were then aliquoted into individual 1.5 mL microcentrifuge tubes with each tube carrying ~2500 islets. The islets were pelleted and the supernatant carefully removed. Finally, the islets were resuspended in 100 μL of 2% solution of a Sodium alginate Pronova SLG20 (NovaMatrix, Norway) dissolved in 0.9% sterile saline (ph 7.4) to yield a final concentration of ~25 IE.q./ μL. About 15μL of the islet-alginate suspension was then loaded into each macrodevice using a Digital microdispenser (Drummond, Cat # 3-000-525) which ensured minimal islet loss during the injection process. The loaded devices were immediately immersed in a Ba2+ solution (20mM BaCl2 in HBSS) for 20 min to crosslink the alginate inside the device. Since the Barium diffuses through the membrane into the device and crosslinks the alginate, care was taken such that the membrane was wetted properly immediately after contact with the solution as any trapped bubbles would prevent the entry of barium. The devices were washed in fresh HBSS for 10 min and transferred to 12 well plates with media (RPMI 1640 and 10% FBS) and cultured overnight.
In Vitro culture of cells inside macrodevices
Sealed macrodevices loaded with cells were cultured in 12 well plate for short term culture (1–5 days or overnight before implantation) with 2 mL of media replaced every 2–3 days. For long term culture (> 1 week) the devices were cultured in 6 well plates with 5 mL of media. Culture media was replaced every 2 days. During cell culture, care was taken to orient the device such that membrane side faces towards the media (or allow supply of nutrients) while the PDMS side sits on the bottom of the plate. In some cases, 2–3 devices were cultured together in 35 mm cell culture petri-dish with 12 mL of media replaced every 3–4 days.
Implantation surgery
All animal procedures were approved by the MIT Committee on Animal Care and supervised by MIT Division of Comparative Medicine veterinary staff. Male C57BL/6 mice, STZ-induced diabetic C57BL/6 mice, and BALB/cJ mice between 6 – 8 weeks in age were obtained from Jackson Laboratory (Bar Harbor, ME) and used in experiments. Animals were anesthetized with 3% isoflurane in oxygen. To prepare for surgery, the mice had their abdomens shaved and sterilized using betadine and isopropanol and were subcutaneously administered 0.03 mg buprenorphine SR-LAB (ZooPharm) and 1 mL of 0.9% saline. A small incision was made along the midline of the abdomen and blunt dissection was used to expose the peritoneal lining. An incision was made along the linea alba, and the device was transplanted in the abdomen, away from the fat pad. During transplantation, care was taken to orient the device such that the membrane faces the intraperitoneal organs while the PDMS side faces abdominal wall. The incision was closed using 5–0 taper-tipped polydioxanone (PDS II) absorbable sutures. Skin was closed over the incision using a wound clip and tissue glue.
EPO measurement and BG monitoring
EPO producing HEK-293 cell-loaded devices were incubated overnight in cell media and washed three times with Hank’s Balanced Salt Solution (HBSS, Gibco by Life Technologies) prior to transplantation surgery in healthy C57BL/6 mice. On the day of surgery and every week thereafter, 100 μL of blood was collected from the medial saphenous vein in hematocrit tubes and (Cat. # HP8H-10, SafeCrit) blood collection tubes (Cat. # 365967, BD Microtainer). Hematocrit was processed using a micro-hematocrit centrifuge (StatSpin CritSpin, Beckman Coulter, Inc.) and digital hematocrit reader (HemoCue America). After serum separation, EPO concentration was measured using Mouse EPO ELISA kit (Cat. # 442708, Biolegend).
Healthy C57BL/6 mice were treated with STZ by the vendor (Jackson Laboratory, Bar Harbor, ME) to induce type I diabetes prior to shipment to MIT. Blood glucose levels of all mice were tested preoperatively, and only mice with post-fasted blood glucose levels above 350mg/dL were considered diabetic and underwent transplantation. Blood glucose levels were monitored daily (with no fasting) for 3 days post-surgery and twice a week the following weeks after a 2 h fasting period. A small drop of blood was collected from a tail prick and tested using commercial glucometer AlphaTRACK 2 (Zoetis Inc, US) unless otherwise mentioned. Mice with fasted blood glucose level below 200 mg/dL were considered normoglycemic. Monitoring continued until all mice had returned to hyperglycemic state at which point, they were euthanized and devices were retrieved.
In vivo Glucose tolerance test: Mice were fasted for 6 hours before the test with free access to water. Individual animals were given a bolus dose of 30% sterile glucose solution (in saline) at 1.5g/kg through a tail vein injection using a 30G insulin syringe (BD). Blood glucose was measured at 0, 15, 30, 60, 90 and 120 mins after the injection. In order to assay for insulin levels, ~50–70 uL of blood was collected from the medial saphenous vein in blood collection tubes before the glucose infusion and at an appropriate time afterwards.
Retrieval of devices, cells and tissues
For non-terminal surgeries, the same implantation surgical procedure was also used to retrieve devices. For terminal studies, mice were euthanized using carbon dioxide administration, followed by cervical dislocation, at the desired time point. Implanted devices were recovered following a necropsy, with care being taken to collect all the tissue and cells adhered to the device. In the majority of the cases, the implanted devices were found away from the site of implantation. While no clear pattern of migration of the device was noted, a notable number of devices were found either near the stomach or near the perigonadal fat pad. Upon retrieval, devices were washed with HBSS. Devices were either fixed (for use in microscopy and histology), flash frozen (for RNA/DNA/Protein analysis), or cultured in vitro to determine EPO production rate.
Imaging of retrieved materials
Light microscopy:
Retrieved devices were washed with HBSS and transferred into 35mm petri dishes for bright-field imaging using a Leica Stereoscopic microscope. For immunofluorescence imaging, fixed devices were washed twice with PBS, and cells were permeabilized with 0.1% Tween-20. After an additional wash with PBS, devices were incubated with the staining solution containing appropriate antibody cocktail (typical concentrations: F4/80 – 5ug/mL, CD3 – 3ug/mL, aSMA – 3ug/mL) in 1% BSA solution for 12 h at 4 °C. After 12 h, devices were washed with PBS, transferred to a 70% glycerol solution in a glass bottom dish, and imaged using LSM 700 point scanning confocal microscope (Carl Zeiss Microscopy, Jena Germany).
Electron microscopy:
Device was washed in PBS and fixed in 10% glutaraldehyde solution overnight. The device was then sectioned from the middle using a sharp razor and handled carefully thereafter. The samples were slowly dehydrated through a series of alcohol change (10%, 30%, 50%, 75%, 90%, 100%, 100%). The samples were then dried in a Critical point dyer (Tousimus Autosamdri-815) and loaded on aluminum pellets using carbon tape. The samples were coated with a 2 nm gold layer using a sputter coating system (Hummer 6.2) and images on a Jeol 5600LV Scanning Electron Microscope.
Histological processing for H&E and Masson’s
Retrieved devices were fixed for 20 min using 4% paraformaldehyde at room temperature and washed 2x in PBS. The devices were cut in half in the middle using a sharp razor and stored in 70% ethanol for histological processing. The blocks were processed for paraffin embedding where care was taken to orient the sample such that histological sections of the device cross-sections can be obtained to reveal the thickness of the fibrotic capsule. The paraffin blocks were sectioned and stained according to standard histological methods.
Quantification of RNA, DNA, collagen and proteins
Freshly retrieved devices were segmented into two symmetric segments along the lateral axis and immediately flash frozen and stored under liquid nitrogen till further use. One segment was processed using RNA/DNA/Protein extraction kit (Qiagen) to retrieve the total RNA, DNA and protein from the sample. Another segment was processed for quantification of hydroxyproline as detailed below. The segments were imaged using a Leica Stereoscopic microscope to measure the total surface area of each portion which was later used in normalization of the surface DNA or Hydroxyproline content.
Hydroxyproline Assay:
Devices were immersed in 1:1 mixture of water and 37% hydrochloric acid and hydrolyzed at 120 °C for 2h. The resulting solution was used to determine collagen concentration of each device using a hydroxyproline collagen assay kit (Cat # MAK008, Sigma Aldrich) according to the manufacturer’s instructions.
DNA quantification:
The total DNA extracted from the surface was measured using the Qbit dsDNA Quantification assay (Life Technology, Cat# Q32851).
Nanostring assay:
RNAs extracted from devices with different surface coatings (n = 4/group) was quantified, diluted to the appropriate concentration (100 ng/μl), and then 500 ng of each sample was processed according to NanoString manufacturer protocols for expression analysis via our customized multiplexed gene panel. RNA levels (absolute copy numbers) were obtained following nCounter (NanoString Technologies Inc., Seattle, WA) quantification, and group samples were analyzed using nSolver analysis software (NanoString Technologies Inc., Seattle, WA). Individual gene expression values were normalized to housekeeping genes (Hprt, Bact, and Cltc). Relative fold-change was calculated by normalizing with the mean expression value of the uncoated devices and log transformed.
Proteomics:
For this assay five samples were randomly selected from each device group. Equivalent amounts of protein (corresponding to similar surface areas from each sample) were reduced (10 mM dithiothreitol, 56 °C for 45 min) and alkylated (50 mM iodoacetamide, room temperature in the dark for 1 h). Proteins were subsequently digested with trypsin (sequencing grade; Promega, Madison, WI). Trypsin activity was quenched by adding formic acid to a final concentration of 5%. Peptides were desalted using C18 SpinTips (Protea, Morgantown, WV) then lyophilized and stored at −80 °C. Peptides were labeled with TMT 6plex (Thermo Scientific) as per the manufacturer’s instructions. Peptides were then loaded on a precolumn and separated by reverse-phase HPLC (Thermo Easy nLC1000) over a 140-min gradient before nanoelectrospray using a QExactive mass spectrometer (Thermo Scientific). Raw mass spectral data files (.raw) were searched using Proteome Discoverer (Thermo Scientific) and Mascot version 2.4.1 (Matrix Science). TMT quantification was obtained using Proteome Discoverer and isotopically corrected as per the manufacturer’s instructions.
FACS analysis
Anti-HEK IgG assay:
FACS was used to detect and quantify the levels of circulating IgG against transplanted HEKepo cells. Healthy and treated animals were bled to collect about 100 μl of serum. 100,000 HEK cells were incubated with 100 μl of 10-fold diluted serum (90 μl PBS and 10 μl of serum) for 2 hrs at 4°C. The cells were washed 2x with cold PBS and incubated with 100 μl of anti-mouse IgG-AF647 (5 μg/mL) for 30 min at 4C. Finally the cells were washed and analyzed on LSR Fortessa Flow cytometer. Sytox blue was included in the final cell suspension buffer and was used to exclude dead cells. HEKepo cells directly stained with anti-mouse IgG-AF647 was used as a negative control. FlowJo software was used for gating and data analysis.
Cell proliferation and viability assay:
HEKepo cells grown in well plates (<30% confluency) or inside devices were incubated with 10μM EdU-ClickIT reagent at 37C for 2 hours. The membranes of the devices were ripped using a sharp scalpel and the cells harvested using a trypsin treatment (5 min). The cells then washed with media to remove excess reagents and separated into two tubes. One tube was kept on ice and subsequently used to asses % viability using Sytox blue staining. The other tube was used to complete the EdU staining protocol (permeabilization and reaction with AF647-Azide reagent) according to manufacturer’s guideline. The samples were analyzed on a BD Fortessa Flow Cytometer and the data analyzed using the FlowJo software.
Membrane permeability measurement
To determine the permeability of the PCTE membranes, diffusion studies were conducted using a custom-built amber Valia-Chien Cell with 2 mL volumes and a 5 mm orifice (Permegear, PA). The membrane was degassed in a 50% ethanol solution to remove an air bubbles blocking the pores, washed several times in degassed deionized water, and placed in the center of the diffusion cell. One side of the cell was filled with 2 mL of deionized water, while the other was filled with 2 mL of a 500 μS/cm KCl conductivity standard. A conductivity meter (eDAQ Conductivity Isopod with Miniature Dip-In Conductivity Electrode) was placed in the water-side of the diffusion cell and recorded the conductivity of the solution every second for 10 min. To ensure diffusive-dominated transport, both sides of the diffusion cell were connected to an external bath of deionized water using 20 cm of 0.5 mm ID tubing. The conductivity values were converted to concentrations using a standard curve, and the flux was calculated as the slope of the concentration vs. time plot. The normalized permeability values K (m−1) was then calculated from the following equation:
where J (mol/s) is the flux of KCl, Am(m2) is the area of the membrane, D is the diffusivity of KCl (1.85×10−9 m2/s), and ΔC (mol/m3) is the concentration difference between the two sides of the diffusion cell.
Surface characterization
XPS: All XPS measurements were done on a Thermo Scientific K-Alpha plus X-ray photoelectron spectrometer. operated at a base pressure of less than 5 × 10−8 Torr during XPS analysis. The beam area was 400 μm and three points was analyzed for each sample. The data was analyzed and quantified on the Advantage Software (Thermo Scientific) with automatic elemental analysis feature. Confocal- Raman: Devices were washed in DI water and then sonicated in 1% SDS solution to remove proteins and cells from the surface. Finally, the devices were thoroughly rinsed in DI water and placed on the quartz coverslip. Excess liquid was absorbed by a tissue. A custom-built near-infrared confocal Raman microscopy system was previously reported (Kang, J.W. et al., Biomed. Opt. Express 2, 2011) and was used in our experiments. The detail of experimental procedure was followed as described elsewhere (Vegas et al., Nat Biotechnol, 2016). Scans were taken for a total 20 z-planes with 20 μm gaps. Afterwards, the plane were manually analyzed to identify the device interface and the spectra was plotted using Prism 7 (Graphpad).
Statistical Analysis
Details of sample size and appropriate statistical test is described for each figure. All experiments were repeated at least 2 times unless specified. Data are expressed as mean ± s.e.m. Data were analyzed for statistical significance either by unpaired, two-tailed t-test, or one-way ANOVA with Bonferroni multiple comparison correction, as implemented in GraphPad Prism 7. Survival curve was analyzed using Mantel-Cox test and P-values, two sided, have been indicated in figure legends.
Supplementary Material
Acknowledgements
This work was supported by a JDRF grant (2-SRA-2019-714-S-B) to D.G.A and R.L. S.B. was supported by the National Institutes of Health (NIH/NIBIB) K99EB025254 and a JDRF Postdoctoral Fellowship (PDF-2015-90-A-N). L.V. was supported by the NSF Graduate Research Fellowship Program, S.J. was supported by the Mazumdar-Shaw oncology fellowship, and O.V. was supported by a DOD/CDMRP postdoctoral fellowships (W81XWH-13-1-0215). D.L.G. is supported by the NIH UC4 DK104218. The authors would like to acknowledge Stephanie K Aresta-Dasilva, Casey Landry, and Amy Nguyen for assistance with the animal experiments. The authors would also like to acknowledge Sinisa Hrvatin for helpful discussions. This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, and we thank Koch Institute Swanson Biotechnology Center for technical support, specifically Histology, Proteomics and Flow Cytometry Core Facilities. We also acknowledge the use of resources at W. M. Keck Biological Imaging Facility and the Genomics core at the Whitehead Institute. This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959. CNS is part of Harvard University.
Footnotes
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
D.G.A, R.L. and O.V. are founding scientists of Sigilon Therapeutics, a biotech company based in Cambridge, MA, USA, that produce antifibrotic materials for cell-based therapies. The remaining authors declare no competing interests.
Data Availability
The main data supporting the findings in this study are available within the paper and its supplementary information. The raw and analysed datasets are too numerous to be readily shared publicly. Source data for the figures are available for research purposes from the corresponding author on reasonable request.
References
- 1.Towards advanced cell therapies. Nature Biomedical Engineering 2, 339–340 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Sedlmayer F, Aubel D, Biomedical MFN 2018. Synthetic gene circuits for the detection, elimination and prevention of disease. nature.com [DOI] [PubMed]
- 3.Aijaz A et al. Biomanufacturing for clinically advanced cell therapies. Nature Biomedical Engineering 2, 362–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie M et al. b-cell-mimetic designer cells provide closed-loop glycemic control. Science 354, 1296–1301 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kojima R et al. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat Commun 9, 5066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Power AT et al. Carrier Cell-based Delivery of an Oncolytic Virus Circumvents Antiviral Immunity. Molecular Therapy 15, 123–130 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Chassin H et al. Sensing and responding to allergic response cytokines through a genetically encoded circuit. Nat Commun 8, 1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagliuca FW et al. Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millman JR et al. Generation of stem cell-derived β-cells from patients with type 1 diabetes. Nat Commun 7, 11463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desai T & Shea LD Advances in islet encapsulation technologies. Nat Rev Drug Discov 16, 338–350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wood KJ, Issa F & Hester J Understanding Stem Cell Immunogenicity in Therapeutic Applications. Trends Immunol 37, 5–16 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Phelps EA et al. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv. Mater. Weinheim 24, 64–70-2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.An D et al. Designing a retrievable and scalable cell encapsulation device for potential treatment of type 1 diabetes. Proc. Natl. Acad. Sci. U.S.A 115, E263–E272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.An D et al. Developing robust, hydrogel-based, nanofiber-enabled encapsulation devices (NEEDs) for cell therapies. Biomaterials 37, 40–48 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Weir GC Islet encapsulation: advances and obstacles. Diabetologia 56, 1458–1461 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Hwa AJ & Weir GC Transplantation of Macroencapsulated Insulin-Producing Cells. Curr. Diab. Rep 18, 50 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Colton CK in Principles of Tissue Engineering 543–562 (Academic Press, 2014). 10.1016/B978-0-12-398358-9.00028-8 [DOI] [Google Scholar]
- 18.Nyitray CE et al. Polycaprolactone Thin-Film Micro- and Nanoporous Cell-Encapsulation Devices. ACS Nano 9, 5675–5682 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang R et al. Nanoporous Immunoprotective Device for Stem-Cell-Derived β-Cell Replacement Therapy. ACS Nano 11, 7747–7757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumagai-Braesch M et al. The TheraCyte Device Protects Against Islet Allograft Rejection in Immunized Hosts. Cell Transplant 22, 1137–1146 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Pedraza E, Coronel MM, Fraker CA, Ricordi C & Stabler CL Preventing hypoxia-induced cell death in beta cells and islets via hydrolytically activated, oxygen-generating biomaterials. Proc. Natl. Acad. Sci. U.S.A 109, 4245–4250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paul CD, Mistriotis P & Konstantopoulos K Cancer cell motility: lessons from migration in confined spaces. Nat. Rev. Cancer 17, 131–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson JM, Rodriguez A & Chang DT Foreign body reaction to biomaterials. Seminars in Immunology 20, 86–100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vegas AJ et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat. Biotechnol 34, 345–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vegas AJ et al. Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat. Med 22, 306–311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bochenek MA et al. Alginate encapsulation as long-term immune protection of allogeneic pancreatic islet cells transplanted into the omental bursa of macaques. Nature Biomedical Engineering 2, 810–821 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pullen LC Stem Cell-Derived Pancreatic Progenitor Cells Have Now Been Transplanted into Patients: Report from IPITA 2018. American Journal of Transplantation 18, 1581–1582 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Mahou R, Zhang DKY, Vlahos AE & Sefton MV Injectable and inherently vascularizing semi-interpenetrating polymer network for delivering cells to the subcutaneous space. Biomaterials 131, 27–35 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Weaver JD et al. Design of a vascularized synthetic poly(ethylene glycol) macroencapsulation device for islet transplantation. Biomaterials 172, 54–65 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ludwig B et al. Transplantation of human islets without immunosuppression. Proc. Natl. Acad. Sci. U.S.A 110, 19054–19058 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whyte W et al. Sustained release of targeted cardiac therapy with a replenishable implanted epicardial reservoir. Nature Biomedical Engineering 2, 416–428 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Lee KS & Ram RJ Plastic-PDMS bonding for high pressure hydrolytically stable active microfluidics. Lab Chip 9, 1618–1624 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Lovett M, Lee K, Edwards A & Kaplan DL Vascularization strategies for tissue engineering. Tissue Eng Part B Rev 15, 353–370 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendelsohn A & Desai T Inorganic nanoporous membranes for immunoisolated cell-based drug delivery. Adv. Exp. Med. Biol 670, 104–125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scalea J, Hanecamp I, Robson SC & Yamada K T-cell-mediated immunological barriers to xenotransplantation. Xenotransplantation 19, 23–30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leader B, Baca QJ & Golan DE Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov 7, 21–39 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Lee DE, Son W, Ha BJ, Oh MS & Yoo OJ The prolonged half-lives of new erythropoietin derivatives via peptide addition. Biochem. Biophys. Res. Commun 339, 380–385 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Régulier E, Schneider BL, Déglon N, therapy YBG 1998. Continuous delivery of human and mouse erythropoietin in mice by genetically engineered polymer encapsulated myoblasts. nature.com [DOI] [PubMed]
- 39.Schweicher J, Nyitray C, bioscience TDFI 2014. Membranes to achieve immunoprotection of transplanted islets. ncbi.nlm.nih.gov [DOI] [PMC free article] [PubMed]
- 40.Chong AS, Alegre M-L, Miller ML & Fairchild RL Lessons and limits of mouse models. Cold Spring Harb Perspect Med 3, a015495–a015495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tucker SP, Melsen LR & Compans RW Migration of polarized epithelial cells through permeable membrane substrates of defined pore size. Eur. J. Cell Biol 58, 280–290 (1992). [PubMed] [Google Scholar]
- 42.Bryers JD, Giachelli CM & Ratner BD Engineering biomaterials to integrate and heal: the biocompatibility paradigm shifts. Biotechnol. Bioeng 109, 1898–1911 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whyte W, Roche ET, Varela CE, Biomedical KMN 2018. Sustained release of targeted cardiac therapy with a replenishable implanted epicardial reservoir. nature.com [DOI] [PubMed]
- 44.Zhang L et al. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol 31, 553–556 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Weaver D, Roth LA, translational ACS 2012. Vascular catheters with a nonleaching poly-sulfobetaine surface modification reduce thrombus formation and microbial attachment. stm.sciencemag.org [DOI] [PubMed]
- 46.Xie X et al. Reduction of measurement noise in a continuous glucose monitor by coating the sensor with a zwitterionic polymer. Nature Biomedical Engineering 12, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin X, Yuan J & Shen J Zwitterionic polymer brushes via dopamine-initiated ATRP from PET sheets for improving hemocompatible and antifouling properties. Colloids Surf B Biointerfaces 145, 275–284 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Veiseh O et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature Materials 14, 643–651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tireli M et al. Solvent-free copper-catalyzed click chemistry for the synthesis of N-heterocyclic hybrids based on quinoline and 1,2,3-triazole. Beilstein Journal of Organic Chemistry 13, 2352–2363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Régulier E, Schneider BL, Déglon N, Beuzard Y & Aebischer P Continuous delivery of human and mouse erythropoietin in mice by genetically engineered polymer encapsulated myoblasts. Gene Ther 5, 1014–1022 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Sommer B et al. Long-term doxycycline-regulated secretion of erythropoietin by encapsulated myoblasts. Molecular Therapy 6, 155–161 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Weber LM & Anseth KS Hydrogel encapsulation environments functionalized with extracellular matrix interactions increase islet insulin secretion. Matrix Biology 27, 667–673 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki K et al. Function and survival of macroencapsulated syngeneic islets transplanted into streptozocin-diabetic mice. Transplantation 66, 21–28 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Neufeld T et al. The Efficacy of an Immunoisolating Membrane System for Islet Xenotransplantation in Minipigs. PLOS ONE 8, e70150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salehi S & Reed EF The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transplant 20, 446–453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris DL Minireview: Emerging Concepts in Islet Macrophage Biology in Type 2 Diabetes. Mol. Endocrinol 29, 946–962 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the findings in this study are available within the paper and its supplementary information. The raw and analysed datasets are too numerous to be readily shared publicly. Source data for the figures are available for research purposes from the corresponding author on reasonable request.