

Summary
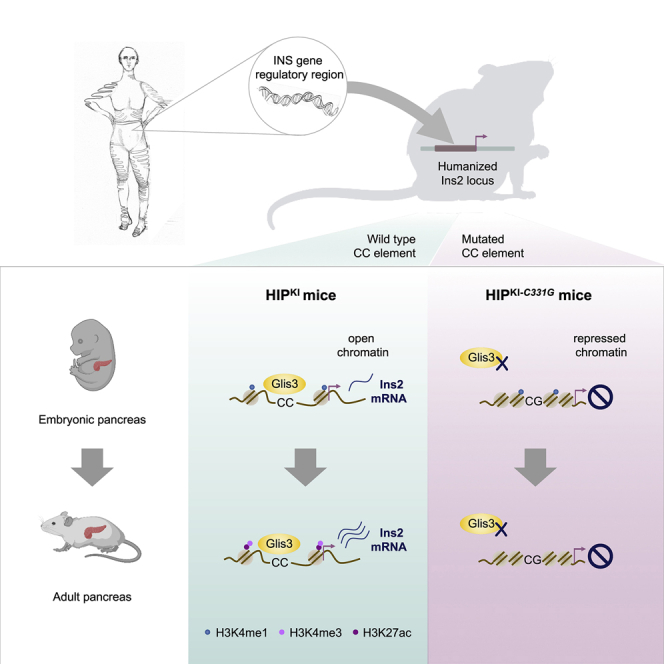
Despite the central role of chromosomal context in gene transcription, human noncoding DNA variants are generally studied outside of their genomic location. This limits our understanding of disease-causing regulatory variants. INS promoter mutations cause recessive neonatal diabetes. We show that all INS promoter point mutations in 60 patients disrupt a CC dinucleotide, whereas none affect other elements important for episomal promoter function. To model CC mutations, we humanized an ∼3.1-kb region of the mouse Ins2 gene. This recapitulated developmental chromatin states and cell-specific transcription. A CC mutant allele, however, abrogated active chromatin formation during pancreas development. A search for transcription factors acting through this element revealed that another neonatal diabetes gene product, GLIS3, has a pioneer-like ability to derepress INS chromatin, which is hampered by the CC mutation. Our in vivo analysis, therefore, connects two human genetic defects in an essential mechanism for developmental activation of the INS gene.
Keywords: INS promoter, regulatory element, mouse model, neonatal diabetes, GLIS3, HIP
Graphical abstract
Highlights
-
•
Mutations of a CC dinucleotide in the human INS promoter cause neonatal diabetes
-
•
We humanized ∼3.1 kb of mouse Ins2 and created a CC mutant version
-
•
Humanized Ins2, but not the CC mutant, recapitulates developmental chromatin activation
-
•
GLIS3, also mutated in diabetes, activates INS chromatin and requires an intact CC
Mutations in the CC element of the INS promoter or the transcription factor GLIS3 cause neonatal diabetes. Akerman et al. humanize a 3.1-kb region upstream of the mouse Ins2 gene and show that GLIS3 and the CC element form a pioneering mechanism that activates INS chromatin during pancreas development.
Introduction
Most sequence variants underlying Mendelian diseases affect coding sequences, although several patients are known to harbor causal cis-regulatory mutations (Benko et al., 2009; Garin et al., 2010; Hansen et al., 2002; Lettice et al., 2003; Weedon et al., 2014). This number is expected to rise as millions of human genomes are sequenced and the field learns to discriminate pathogenic noncoding mutations from a vast number of inconsequential variants (Chong et al., 2015; Huang et al., 2017; Kowalski et al., 2019; Ward and Kellis, 2012). In polygenic diseases, common and rare cis-regulatory variants play a central role in the genetic susceptibility (Cowper-Sal lari et al., 2012; Maurano et al., 2012; Pasquali et al., 2014).
Despite the relevance of cis-regulatory variants, they have been largely studied outside their chromosomal context. Current experimental models usually test noncoding variants with episomal DNA constructs, ectopically located transgenes, or in vitro protein-DNA interaction assays. The extent to which these models reflect the true impact of cis-regulatory variants is unknown. Expression quantitative trait loci can provide insights into which genes are affected by regulatory variants in their native genome context yet often fail to distinguish causal from linked variants. It is now also possible to directly edit mutations in stem cells and differentiate them in vitro, but this does not always allow modeling the mutational impact in relevant developmental or physiological in vivo contexts. There is a need, therefore, to develop complementary tools that facilitate understanding of the in vivo impact of noncoding variants.
One approach to address this need is to engineer human genomic sequences in mice. Several examples of human knockins in the mouse genome have been created to model human protein-coding mutations (Zhu et al., 2019). One study successfully edited a 5-bp noncoding sequence in mice to model a common human regulatory variant (Pashos et al., 2017). However, the extent to which mice can be used to study human cis-regulatory mutations in more-extended orthologous genomic contexts is poorly explored.
Modeling noncoding mutations in model organisms poses major challenges, because the consequence of mutations that disrupt transcription factor-DNA interactions can be influenced by essential combinatorial interactions with nearby transcription factor binding sites or by the existence of redundant binding sites (Biggin, 2011; Heinz et al., 2013; Junion et al., 2012). Unlike coding sequences, which are often highly conserved, noncoding DNA sequences can maintain functions despite substantial evolutionary turnover, and conserved noncoding sequences can acquire divergent functions (Khoueiry et al., 2017; Villar et al., 2015). It is thus difficult to predict pathological consequences of human cis-regulatory mutations from mouse models unless the broader sequence context has been humanized.
We have now examined a cis-regulatory mutation in the INS gene that causes diabetes mellitus. A subset of patients with neonatal diabetes harbors recessive loss-of-function coding or promoter mutations in INS, encoding for insulin (Garin et al., 2010). Single-nucleotide mutations of the INS promoter published so far are located in a CC dinucleotide 331 bp upstream of the INS start codon (Bonnefond et al., 2011; Deeb et al., 2016; Demirbilek et al., 2015; Garin et al., 2010). Functional studies in tumoral β cells using episomal luciferase assays showed that c.-331C > G, the most common of these mutations, causes partial disruption of INS promoter activity (Garin et al., 2010). However, decades of work have shown that artificial mutations in various other elements of the INS 5′ flanking region that disrupt binding by transcription factors, such as MAFA, PDX1, or NEUROD1, also lead to reduced transcriptional activity in episomal assays (Docherty et al., 2005; German et al., 1995; Le Lay and Stein, 2006; Melloul et al., 2002; Odagiri et al., 1996). This raises the question of whether the CC element is specifically vulnerable due to an essential role in the in vivo regulation of the INS gene that cannot be examined in episomal assays.
In this study, we establish the selectivity of CC element mutations in an extended cohort of patients. We humanized a large noncoding region of the mouse Ins2 gene and used a mutant version of this model to show that the c.-331C > G mutation disrupts active chromatin formation during pancreas development. We then linked the CC element to GLIS3, a zinc finger transcription factor that also carries neonatal diabetes mutations, as well as type 1 and type 2 diabetes risk variants (Barrett et al., 2009; Dupuis et al., 2010; Senée et al., 2006). GLIS3, one of several transcription factors known to regulate the insulin gene (Kang et al., 2009; Yang et al., 2009), showed a singular capacity to create INS gene active chromatin in non-pancreatic cells, and this was inhibited in the CC mutant. This in vivo analysis of two regulatory defects has therefore revealed a pioneering mechanism of the human INS gene. These insights are relevant to the mechanisms of diabetes and for regenerative strategies that aim to activate the INS gene in non-pancreatic cells.
Results
Human genetics establishes a unique role of the INS promoter CC element
To further define the importance of the INS promoter CC element, we re-examined the selectivity of mutations in a large cohort of previously published (n = 19) and unpublished (n = 41) patients with diabetes caused by recessive INS promoter mutations. This showed that, among 60 patients from 44 families, all single-base-pair mutations located 5′ of the transcriptional start site resided in the CC element. This included 42 patients with c.-331C > G mutations (40 homozygous; two compounds heterozygous), two with c.-332C > G mutations (one homozygous; another compound heterozygous), and 13 with c.-331C > A mutations (12 homozygous; one compound heterozygous with a c.-331C deletion; Figure 1; Table S1). This newly described c.-331C single-base-pair deletion is interesting, because it strongly favors a loss- rather than gain-of-function mechanism for CC mutations. The only other pathogenic INS upstream mutation was a larger homozygous deletion that disrupts MAFA and NEUROD1 binding sites, which was observed in four patients from two families. Most patients had characteristic clinical features of insulin-deficient neonatal diabetes, with low birth weight and diabetes onset soon after birth, although 2 patients were diagnosed outside the neonatal period and 12 (including both patients with -332 mutations) showed transient neonatal diabetes (Table S1). No associated clinical abnormalities were consistently noted in this patient series. These extended human genetic findings, therefore, establish that the CC dinucleotide sequence of the INS promoter is unusually sensitive to mutations, suggesting a singular function of this cis-regulatory element.
Figure 1.

Location of INS promoter mutations
The top panel is a schematic of the human INS 5′ flanking sequence, showing approximate locations of established cis-regulatory sequences based on published mutational analyses of episomal sequences. The bottom panel shows a zoomed-in sequence that contains previously characterized MAFA, NEUROD1, and PDX1-bound cis-elements (C1, E1, and A1, respectively), as well as the CC element, with a graph that depicts the location of all INS promoter recessive mutations from our analysis. Each dot represents a patient, and the color represents the genotype. All single-base-pair mutations are located in the CC element, including a distinct 24-bp deletion that disrupts MAFA and NEUROD1 binding sites.
Generation of a human cis-regulatory mutation mouse model
To study the function of the CC element in vivo, we modeled the c.-331C > G mutation in a humanized sequence context. We generated mice in which homologous recombination was used to replace the endogenous 3.17-kb mouse genomic region containing the Ins2 gene and its upstream regions with a human INS upstream DNA fragment. This genomic region showed high-sequence conservation to the mouse counterpart in an ∼350-bp region upstream of INS TSS and another ∼500-bp region downstream of the TH gene but contained intervening sequences lacking identifiable orthology with the mouse genome, including a primate-specific variable number tandem repeat region previously reported to influence INS transcription (Kennedy et al., 1995; Figure S1A). The knocked in allele thus contained (1) this 3.10-kb upstream region of human INS, including a transcribed INS 5′ untranslated sequence; (2) mouse Ins2 exon1, intron, and exon 2; and (3) an internal ribosome entry site (IRES) followed by green fluorescent protein (GFP) sequence (Figure 2). The resulting transcript was thus translated into two proteins: mouse preproinsulin-2 and GFP. In parallel, we generated a mouse model harboring the same humanized sequence and the INS c.-331C > G single point mutation (Figure 2). We named mice carrying the two human INS upstream knockin alleles HIPKI and HIPKI-C331G.
Figure 2.

Generation of HIPKI and HIPKI-C331G mouse alleles
A rectangle with dotted lines in the top two panels depicts the 3.1-kb human sequence located between the human TH and INS genes (including INS 5′ untranslated transcribed sequences), which was cloned into a targeting vector. This targeting vector contained the 3.1-kb human INS upstream region followed by Ins2-IRES-GFP, which includes mouse Ins2 exons and intron and an IRES, GFP, and Ins2 3′ UTR and was flanked by mouse Ins2 homology arms. Targeted replacement of the indicated mouse Ins2 sequence with this human upstream INS sequence followed by Ins2-IRES-GFP was carried out by homologous recombination. The same process was used to create two allelic versions carrying the normal human INS sequence or the c.-331C > G mutation. A neomycin cassette flanked by LoxP sites was excised in vivo and is omitted for simplicity. The sequence of the neomycin-excised targeted allele is provided in File S1.
Homozygous HIPKI and HIPKI-C331G mice were born at expected Mendelian ratios and appeared healthy without overt hyperglycemia (Figure S1B), consistent with the fact that HIP knockin mice retained an intact copy of Ins1, a retroposed mouse gene that also encodes insulin (Duvillié et al., 1997; Leroux et al., 2001).
HIPKI recapitulates, and HIPKI-C331G abrogates, cell-specific INS transcription
To determine whether the human INS 5′ flanking region inserted into its orthologous mouse chromosomal context was able to drive β-cell-specific expression in HIPKI mice, we used dual immunofluorescence analysis of GFP and islet hormones in tissues from newborn mice (postnatal day 1 [P1]–P3). This showed that GFP expression was restricted to insulin-positive pancreatic islet core β cells of HIPKI mice, whereas it was not detected in mantle glucagon- or somatostatin-positive islet cells or surrounding exocrine cells (Figures 3A and 3B). Moreover, GFP fluorescence was readily detected in live islets isolated from 3- to 5-month-old HIPKI mice (Figure 3C). Ins2-IRES-GFP transcript was also restricted to islets across a panel of mouse tissues (Figure S1C).
Figure 3.

HIPKI recapitulates and HIPKI-C331G abrogates β cell-specific Ins2 expression
(A) Immunofluorescence imaging of insulin, glucagon, and GFP, showing selective GFP expression in HIPKI insulin-positive core islet cells, but not in the surrounding glucagon islet mantle, or in extra-islet exocrine cells. In contrast, no GFP expression is detected in HIPKI-C331G islet cells.
(B) Immunofluorescence imaging of glucagon, somatostatin, and GFP, showing lack of GFP expression in somatostatin or glucagon-positive cells.
(C) Bright-field and fluorescence imaging of islets isolated from HIPKI and HIPKI-C331G mice.
(D) Quantitative PCR analysis of Ins2 and Ins1 mRNAs in pancreatic islets from 3- to 5-month-old C57BL/6 (n = 4), HIPKI (n = 6), and HIPKI-C331G (n = 6) mice. Values were normalized to Actb mRNA. ∗∗∗p < 0.0001 (Student’s t test).
In sharp contrast to HIPKI islets, HIPKI-C331G islets showed no detectable GFP fluorescence or immunoreactivity (Figures 3A–3C). Thus, Ins2-IRES-GFP expression in HIPKI mice recapitulates expected β-cell-specific patterns of insulin expression, whereas the HIPKI-C331G mutation disrupts this expression pattern.
To further assess the function of human INS flanking regions, we measured Ins2 mRNA in islets isolated from HIPKI and HIPKI-C331G mice. Quantitative RT-PCR analysis revealed Ins2 mRNA in islets isolated from both control C57BL/6 and HIPKI mice, but not in HIPKI-C331G mouse (Figure 3D), thus confirming that the c.-331C > G single point mutation abrogates transcriptional activity of the humanized INS/Ins2 locus in mice. We note, however, that Ins2 transcripts in HIPKI islets, which form part of the larger Ins2-IRES-GFP transcript, were reduced in comparison to control C57BL/6 islets (Figure 3C). This resulted from a combined effect of ∼10-fold reduced transcription and ∼14-fold reduced stability of the Ins2-IRES-GFP transcript, as compared to the intact Ins2 mRNA from C57BL/6 islets (Figures S1D and S1E).
Further analysis revealed normal levels of Ins1, Pdx1, Glis3, NeuroD1, MafA, Glut2, and Gck mRNAs in HIPKI and HIPKI-C331G islets, suggesting that humanization of regulatory sequences in the mouse Ins2 locus did not impact these key pancreatic β cell identity markers (Figures 3C and S1F).
Mice with homozygous null Ins2 mutations display mild transient hyperglycemia (Duvillié et al., 1997; Leroux et al., 2001). Consistently, HIPKI-C331G mice, which were Ins2-deficient, showed mildly increased glycemia (119[32] versus 145[24] mg/dL; mean [IQR]; Student’s t test p = 0.0031; Figure S1A).
These findings indicate that an ∼3-kb human INS upstream region can replace its mouse orthologous region and still direct cell-specific transcription in mouse β cells. Furthermore, they show that the human INS c.-331C > G point mutation abrogates the function of this humanized region, consistent with the severe phenotype observed in humans with neonatal diabetes.
HIPKI, but not HIPKI-C331G, islets mirror human islet INS chromatin
Nucleosomes that flank promoters of transcriptionally active genes are typically enriched in trimethylated histone H3 lysine 4 (H3K4me3) and acetylated histone H3 lysine 27 (H3K27ac), and this was expectedly also observed in the human INS promoter in human islets (Morán et al., 2012; Pasquali et al., 2014; Figure S2). We thus examined these histone modifications at the INS promoter in HIPKI and HIPKI-C331G adult mouse islets by chromatin immunoprecipitation (ChIP) assays. The INS proximal promoter showed enriched H3K4me3 and H3K27ac in adult HIPKI mouse islets (Figures 4A–4C). By contrast, HIPKI-C331G islets showed 5-fold and 7-fold lower signals (Student’s t test; p < 0.001; Figures 4B and 4C). HIPKI-C331G islets also showed markedly reduced INS promoter chromatin accessibility as measured by formaldehyde-assisted isolation of regulatory elements (FAIRE) (Figure 4D) and reduced binding of PDX1, an islet transcription factor that binds to multiple sites in the INS promoter (Figure 4E). These findings, therefore, demonstrate that the c.-331C > G point mutation does not only prevent transcriptional activity conferred by the human INS 5′ flanking region but also the formation of accessible active chromatin and thereby occupancy by a key INS gene transcription factor.
Figure 4.

HIPKI recapitulates and HIPKI-C331G abrogates formation of active chromatin at the INS locus
(A) Schematic showing approximate primer locations for ChIP assays. INS CC primers encompass the CC element.
(B–D) ChIP quantification of H3K4me3, H3K27ac, and FAIRE accessibility at the humanized INS locus and control regions in HIPKI or HIPKI-C331G adult mouse islets. The Actb promoter was used as a positive control, and a gene desert lacking active histone modifications in islets was used as a negative control. ChIP and FAIRE DNA were expressed as a percentage of the total input DNA. n = 3 independent experiments.
(E) ChIP for the transcription factor PDX1 in HIPKI or HIPKI-C331G adult mouse islets at a known PDX1-bound enhancer near Pdx1 as a positive control. Data are expressed as a percentage of input DNA. n = 3 independent experiments. Error bars indicate SEM; asterisks indicate Student’s t test p values (∗p < 0.05; ∗∗p < 0.001; ∗∗∗p < 0.0001).
Activation of humanized INS locus during development
We next used HIPKI and HIPKI-C331G models to assess how the c.-331C > G mutation influences chromatin activation at the INS gene during pancreas development. As a reference, we analyzed ChIP sequencing (ChIP-seq) maps of H3K4me1 and H3K4me3 histone modifications in human fetal pancreas at Carnegie stage 23 (Cebola et al., 2015), which precedes INS gene activation, and compared them with analogous maps from adult human islets (Morán et al., 2012; Pasquali et al., 2014). In the human fetal pancreas, we observed that the histone modification H3K4me1 demarcated a broad region that encompasses the TH and INS genes as well as downstream regions, without detectable H3K4me3, a histone modification associated with active promoters (Figure 5A). This combination, H3K4me1 enrichment and absence of H3K4me3, has been described in genomic regions that are poised for gene activation (Creyghton et al., 2010) and is consistent with the virtual absence of INS transcription in the early fetal pancreas. In adult human islets, by contrast, H3K4me1 in the INS locus was largely replaced with H3K4me3, in keeping with active INS transcription (Figure 5A).
Figure 5.

Developmental activation of INS promoter chromatin during development
(A) H3K4me1 and H3K4me3 profiles at the INS locus showing H3K4me1, but not H3K4me3, enrichment in human fetal pancreas (Carnegie stage 23; Cebola et al., 2015) across the INS region and H3K4me3, but not H3K4me1, enrichment in adult human islets (Morán et al., 2012; Pasquali et al., 2014).
(B) Top: approximate location of oligonucleotides for ChIP assays. Bottom: ChIP H3K4me1 enrichments in pancreas from HIPKI or HIPKI-C331G E12.5 embryos are shown. n = 2 independent experiments from pools of 8–12 embryos. Data are expressed as a percentage of input DNA.
(C) Quantitative PCR analysis for Ins2 and Ins1 mRNA from HIPKI or HIPKI-C331G in E12.5 and E15.5 embryonic pancreas or E15.5 liver (n = 3). Error bars indicate SEM; asterisks indicate Student’s t test p values; ∗∗∗p < 0.0001.
We next examined whether the chromatin environment that precedes transcriptional activation of the INS locus is recapitulated in HIPKI embryos. As in human fetal pancreas, we observed deposition of H3K4me1 in the humanized INS flanking regions in HIPKI embryonic day 12.5 (E12.5) mouse fetal pancreas (Figure 5B). Ins2 mRNA was expectedly not detected above background levels at this stage (Figure 5C). Interestingly, both HIPKI and HIPKI-C331G displayed similar H3K4me1 profiles at E12.5 (Figure 5B), indicating that the humanized INS sequences recapitulate poised chromatin preceding gene activity, and this is not impeded by the c.-331C > G mutation.
By contrast, the c.-331C > G mutation prevented the transcriptional activation of the humanized locus at early stages of β cell differentiation. Thus, Ins2 mRNA was detectable in HIPKI E15.5 fetal pancreas, but it was undetectable in HIPKI-C331G embryos (Figure 5C), whereas Ins1 mRNA was readily detected in both HIPKI and HIPKI-C331G E15.5 pancreas (Figure 5C). Thus, mice carrying humanized regulatory regions in the Ins2 locus can recapitulate salient developmental features of the chromatin landscape of the human INS locus and show that, although the c.-331C > G mutation does not affect INS chromatin poising, it disrupts activation of promoter chromatin in differentiated cells.
GLIS3-dependent activation of the INS gene is prevented by c.-331C > G
We next sought to identify factors whose DNA binding activity is influenced by the c.-331C > G mutation. We performed stable isotope labeling by amino acids in cell culture (SILAC) experiments and identified four zinc finger transcription factors (MAZ, ZFP37, KLF13, and KLF16) that showed decreased in vitro binding to the c.-331C > G mutation and additionally selected 5 zinc finger transcription factors expressed in human islets that were predicted to show differential binding based on in silico and/or literature analysis (Figure S3A). Of these 9 candidate transcription factors, GLIS3, which was previously shown to bind to this element in vitro (Kang et al., 2009), induced significant luciferase activity in a human insulin promoter episomal construct, and the c.-331C > G mutation suppressed this effect (Student’s t test p = 0.02; Figure 6A). Systematic DNA binding site selection studies predict that the C > G mutation impairs GLIS3 binding (Beak et al., 2008), and this was confirmed with electromobility shift assays (Figure S3B). These findings, therefore, indicated that the c.-331C > G mutation disrupts GLIS3 in vitro binding to the CC element as well as activation of an episomal INS promoter.
Figure 6.

GLIS3 activates the endogenous INS gene and requires an intact CC element
(A) Transfection of candidate transcription factors or PDX1 as a positive control, along with INS promoter (wild-type or c.-331C > G) luciferase reporter plasmids in ENDOCβ-H1 cells. Statistical comparisons correspond to GLIS3 versus empty expression vectors, both with a wild-type reporter plasmid, or for hGLIS3 vector, the wild-type versus c.-331C > G reporter plasmid.
(B) ChIP-seq shows GLIS3 binding to the INS promoter (arrow) in human islets.
(C) GLIS3 activates INS in heterologous cell types. HEK293T, MCF7, or SW480 cells were transfected with plasmids encoding indicated transcription factors or left untreated (unt). INS mRNA was calculated as INS to GAPDH mRNA ratios × 1,000. Significance was calculated relative to untreated samples in 3 independent experiments.
(D) FAIRE assessment of accessible chromatin in transfected HEK293T cells. FAIRE DNA was quantified by PCR and expressed as percentage of input DNA and fold enrichment over untreated cells. Statistical significance was calculated relative to untreated samples (n = 3 independent experiments). Expectedly, signal from NANOG and GAPDH did not change.
(E) GLIS3-dependent activation of INS in fibroblasts obtained from HIPKI (n = 7) and HIPKI-C331G (n = 10) embryos. Fibroblasts were transduced with mouse GLIS3, MAFA, PDX1, and NEUROD1 lentivirus, and RNA was analyzed after 2 days. Two independent experiments were performed with cells from 7 HIPKI and 10 HIPKI-C331G embryos from two litters. Error bars are SEM; asterisks are Student’s t test; ∗p < 0.05; ∗∗p < 0.001; ∗∗∗p < 0.0001.
Recently, GLIS3 was shown to bind in vivo to the Ins2 promoter, and pancreatic inactivation of Glis3 in adult mice specifically depletes Ins2 mRNA in islet cells (Scoville et al., 2019; Yang et al., 2013). To test whether GLIS3 is a direct in vivo regulator of the human INS gene, we performed ChIP-seq of GLIS3 in human islets. This showed 746 high-confidence binding sites, which displayed marked enrichment of canonical GLIS3 binding sequences, including a de novo motif matching the INS promoter CC element and a specific binding site in the INS promoter (Figures 6B and S4A; Table S2). Additional GLIS3-bound regions were observed in genes known to be important for islet cells, including PDX1, MAFA, CREB1, and DLL1 (Figure S4B). Furthermore, transduction of two independent short hairpin RNAs (shRNAs) that reduced GLIS3 mRNA in human EndoCβ-H1 β cells led to ∼2-fold lower INS mRNA levels (Student’s t test p < 0.05; Figure S4C). This provided in vivo evidence that GLIS3 is a direct regulator of the human INS gene.
To understand GLIS3-mediated regulation of INS locus chromatin, we assessed the ability of GLIS3 to activate INS in three immortalized non-pancreatic human cell lines in which the INS gene is repressed. Combinations of islet cell transcription factors, such as PDX1, MAFA, and NEUROG3 or NEUROD1, have been used to activate β cell programs in pancreatic acinar or liver cells (Yechoor et al., 2009; Zhou et al., 2008), in line with the knowledge that PDX1, MAFA, and NEUROD1 directly bind and regulate the insulin gene promoter in various species (Le Lay and Stein, 2006; Zhao et al., 2005). We found that expression of various combinations of such transcription factors failed to elicit major changes in INS mRNA in cell lines from distant lineages, namely HEK293T, MCF7, or SW480 cells (Figures 6C and S5A). By contrast, co-transfection of these factors with GLIS3 led to ectopic activation of the endogenous INS gene (Figures 6C and S5A). Other candidate transcription factors that were predicted to bind to the CC element failed to activate INS in the presence of PDX1 and NEUROD1 (Figure S5A). Microarray analysis of HEK293T cells transfected with GLIS3, PDX1, NEUROD1, and MAFA showed that INS was the most highly induced gene relative to cells that were only transfected with PDX1, MAFA, and NEUROD1 (Figure S5B). These results indicate that GLIS3 has a specific ability to activate the INS gene in cell lines from distant lineages, which requires other INS promoter-binding transcription factors that do not elicit this effect on their own.
To further understand this unique effect of GLIS3 on the INS gene, we examined chromatin accessibility using FAIRE. Misexpression of GLIS3, PDX1, and NEUROD1 created accessible chromatin at the endogenous INS gene in HEK293T cells, whereas this was not elicited by PDX1 and NEUROD1 alone (Figure 6D). This effect was direct, as ChIP experiments showed that transfected GLIS3 was bound to the INS promoter (Figure S5C). Thus, GLIS3 has a singular ability to bind and activate the INS promoter in repressed chromatin cellular environments.
Because GLIS3 activation of episomal INS promoter constructs was impaired by the c.-331C > G mutation, we next tested whether the GLIS3-dependent chromatin pioneering function was also mediated by the CC element. We thus prepared fibroblasts from HIPKI and HIPKI-C331G embryos. Given that other islet transcription factors are required for the GLIS3 effect, we transduced HIPKI and HIPKI-C331G mouse embryonic fibroblasts with PDX1, NEUROD1, MAFA, and GLIS3. This combination of transcription factors activated transcription from the humanized locus in HIP fibroblasts, whereas the effect was inhibited by the c.-331C > G mutation (Figure 6E).
These findings, therefore, indicate that GLIS3 has a distinct pioneering function in the activation of human INS in an endogenous chromosomal context and indicate that c.-331C > G mutation impairs this function.
Discussion
We have examined the in vivo consequences of two gene-regulatory defects that cause diabetes mellitus. We studied an extended cohort of patients with neonatal diabetes and provide firm human genetic evidence that the CC element of the human INS gene is selectively vulnerable to loss-of-function mutations. We humanized a large genomic regulatory region in mice and demonstrated that a neonatal diabetes point mutation in this element disrupts an essential pioneering step in the activation of the human INS gene. We further demonstrate that GLIS3, which is also mutated in neonatal diabetes and harbors polygenic diabetes risk variants (Barrett et al., 2009; Dupuis et al., 2010; Senée et al., 2006), has a unique role in the activation of INS gene chromatin mediated by the CC element that is mutated in neonatal diabetes. These results revealed cis and trans regulators of an essential mechanism for developmental activation of the endogenous INS gene.
Transcriptional regulatory DNA variants play a central role in human disease (Miguel-Escalada et al., 2015), yet so far, most efforts have investigated their function in experimental systems that do not consider their in vivo impact (Cannon and Mohlke, 2018; Corradin and Scacheri, 2014; Kircher et al., 2019). This is a major limitation, because it is currently clear that gene regulation entails a complex interplay between elements that are difficult to reproduce outside of an in vivo context, including chromatin structure, epigenetic chemical modifications, or noncoding RNAs. Such factors are highly dynamic throughout development and physiological settings. Importantly, in vitro models cannot easily examine transcription factor-DNA interactions that overcome repressed chromatin states. Many transcription factors can only bind recognition sequences in accessible DNA, whereas a subset of transcription factors have the ability to bind to nucleosomal-bound DNA and to reprogram silent chromatin (Soufi et al., 2015; Zaret and Carroll, 2011). Such pioneer functions play a major role in differentiation and cellular programming and should thus be studied with in vivo approaches that recapitulate salient genomic and chromatin contexts.
Previous work with a trans-species aneuploid model showed that the human chromosome 21 can recapitulate human cell-specific regulatory landscapes in mice (Wilson et al., 2008). Our study has now shown that a human ∼3.1-kb genomic region could be integrated into an orthologous region of another mammal and recapitulate stage- and cell-specific functions. Importantly, we have modeled a human noncoding mutation in an extended human regulatory sequence integrated in an orthologous mouse locus, thereby extended earlier models that introduced human mutations in an orthologous mouse sequence (Zhu et al., 2019). This in vivo strategy was essential to model CC element mutations, because systematic in vitro binding studies and episomal reporter assays have disclosed >16 binding activities and functional elements in the human insulin promoter that do not appear to harbor mutations that are pathogenic in humans (Bonnefond et al., 2011; Docherty et al., 2005; Garin et al., 2010; German et al., 1995; Melloul et al., 2002; Odagiri et al., 1996). Interestingly, a mutation of a 34-bp region that contains the CC element in randomly integrated human INS promoter transgenics did not significantly alter promoter activity (Itier et al., 1996). Our humanized model showed that the c.-331C > G mutation did not prevent INS chromatin priming in pre-differentiated cells but disrupted H3K4 trimethylation, chromatin accessibility, and transcription factor binding to the INS promoter in differentiated β cells. This indicates that the CC element acts as an essential seeding site for chromatin opening and transcriptional activation of the INS promoter in β cell development. It also shows that the essential function of this element can only become fully apparent in a natural chromatinized environment, such as that of human patients or mutant HIP mice.
Our studies also show that the Krüppel-like zinc finger protein GLIS3 activates the human INS gene in vivo. This extends studies showing that GLIS3 binds the mouse Ins2 promoter in vivo and the human INS CC element in vitro (Kang et al., 2009; ZeRuth et al., 2013). Furthermore, pancreatic Glis3-deficient and Glis3+/− mice show severe abnormalities in insulin expression (Scoville et al., 2019; Yang et al., 2013). Importantly, our studies now show that GLIS3 has a selective ability among known INS gene regulators to activate INS in cellular environments in which this locus is repressed. It is interesting to note that GLIS1, a GLIS3 paralog, has a major impact on reprogramming of pluripotent cells from somatic cells in the presence of other pluripotency transcription factors (Maekawa et al., 2011), and GLIS3 has a similar reprogramming function in some somatic cell lineages (Lee et al., 2017). It is therefore likely that GLIS3 can derepress diverse target genes through combinatorial interactions with cell-specific transcription factors. We have further shown that the c.-331C > G mutation prevented GLIS3-dependent activation of both episomal and integrated human INS. These findings, therefore, provide a common mechanism for genetic defects in GLIS3 and the INS CC element.
Our studies, therefore, uncover an unanticipated protagonism of GLIS3 and the CC element of the INS promoter to initiate an active chromatin state at the human INS gene. Our findings are relevant to understanding genetic mechanisms underlying diabetes, as well as for efforts to use cis-acting sequences and transcription factors to activate β cell genes for replacement therapies (Bakhti et al., 2019; Ding et al., 2013; Zhou and Melton, 2018).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GLIS3 | Yang et al. (2011); L. Chan lab, Baylor College of Medicine, Texas, USA | PMID 21786021 |
| H3 mono methyl K4 | Abcam | ab8895 |
| Histone H3 (acetyl K27) | Abcam | ab4729 |
| H3 tri-methyl K4 | Millipore | 04-745 |
| FLAG M2 (against GLIS3-FLAG) | Sigma FLAG-M2 | F3165 |
| PDX1 | BCBC | Ab2027 |
| Insulin | Dako | A0564 |
| gunie pig anti-Glucagon | MilliporeSigma | 4031-01F |
| rabbit anti glucagon | Dako | A0565 |
| Somatostatin | Dako | A0566 |
| GFP | R&D Systems | AF4240 |
| Immunoflourescence secondary antibodies | Jackson immunoResearch | Cy3, Cy5 and Alexa flour 488 |
| Biological samples | ||
| Human Islets | European Consortium on Islet Transplantation (ECIT) AND Islets for Basic Research Program supported by the Juvenile Diabetes Research Foundation | https://ecit.dri-sanraffaele.org and JDRF-program 2-RSC-2019-724-I-X |
| Chemicals, peptides, and recombinant proteins | ||
| Antibody diluent | DAKO | S3022 |
| β-2-mercaptoethanol | Sigma-Aldrich | M3148-25ML |
| BSA | Sigma-Aldrich | A3059 |
| BSA (for Endo-C culture only) | Roche | 10775835001 |
| Calf Serum | Sigma-Aldrich | C8056-100ML |
| Collagenase | Roche | 11215809103 |
| Diethylaminoethyl (DEAE) | Sigma-Aldrich | 67578-5G |
| DMEM | Lonza | be12-604f |
| DMEM 5.5mM Glucose | Thermofisher Scientific | 31885023 |
| dsDNA BR assay kit | Thermofisher Scientific | Q32853 |
| dsDNA HS assay kit | Thermofisher Scientific | Q32854 |
| ECM gel | Sigma-Aldrich | E1270-10ML |
| EDTA | Thermofisher Scientific | AM9260G |
| Ethanol (immunoflourescence) | Panreac Applichem, Spain | 141086.1211 |
| Ethanol | Merck Millipore | 108543 |
| Fetal Calf Serum (FBS) | Cambrex | 14-801/ 91s1810-500 |
| Fibronectin | Sigma-Aldrich | F1141 |
| Formaldehyde | Calbiochem | 344198 |
| Glucose | Sigma-Aldrich | G-8270-1KG |
| Glycine | Sigma-Aldrich | 50046-250G |
| HBSS | Invitrogen | 14060-040 |
| HEPES | Sigma-Aldrich | H7523-50G |
| Histopaque 1077 | Sigma-Aldrich | 10771-500ml |
| Histopaque 1119 | Sigma-Aldrich | 11191-100ml |
| L-Glutamine | Cambrex | BE17-605E |
| LiCl | Sigma-Aldrich | L9650-100G |
| Lipofectamine 2000 | Invitrogen | 11668-019 |
| NEBNext Ultra DNA Library Prep Kit | New England Biolabs | E7370L |
| Newborn calf serum (Endo-C during passage) | GIBCO | 16010-159 |
| Nicotinamide | Sigma-aldrich | 481907 |
| Normal donkey serum | Jackson Immunoresearch | 017-000-121 |
| NP-40 substitute IGEPAL CA-630 | Sigma-Aldrich | I8896 |
| Paraformaldehyde | Agar Scientific | R1026 |
| PBS | Sigma-Aldrich | D-8537 |
| Penicillin | Lonza | DE17-602E/ 17-745E |
| Phenol chloroform | Sigma-Aldrich | 77617-100ML |
| Power SYBR green mastermix | Applied Biosystems | 4368702 |
| Protease inhibitor cocktail | Roche | 4693132001 |
| Protein A Sepharose beads | Ge Healthcare | 17-0780-01 |
| Protein G Sepharose beads | Ge Healthcare | 17-0618-01 |
| Proteinase K | Fermentas | EO0492 |
| Qiaquick columns | QIAGEN | 28106 |
| RNase A | Sigma-Aldrich | R4875-100MG |
| RPMI 1640 medium | Lonza | BE12-702F |
| RPMI 1640 medium- without glucose | Lonza | BE12-752F |
| SDS | Sigma-Aldrich | 71736-500ML |
| SDS (ChIP) | Invitrogen | 15553-027 |
| Sodium selenite | Sigma-Aldrich | S9133 |
| Sodium-chloride | Sigma-Aldrich | S3014 |
| Sodium-deoxycholate | Sigma-Aldrich | 30970-100G |
| Stratagene Site directed mutagenesis kit | Stratagene (currently Thermofisher) | A13282 |
| Streptomycin | Lonza | DE17-602E/ 17-745E |
| Superscript III Reverse Transcriptase | Invitrogen | 18080093 |
| SYBR green mastermix | Applied Biosystems | 4368708 |
| TaqMan Probes | Applied Biosystems | costom |
| Transferrin | Sigma-Aldrich | T8158-100MG |
| TriPure reagent | Invitrogen/ Roche | 11667165001 |
| Tris-HCL | Life Technologies | 15568-025 |
| Trypsin-EDTA 0.05% | GIBCO | 25300062 |
| Xylene | PanReac/AppliChem | 211769.1714 |
| Critical commercial assays | ||
| Dual-LuciferaseReporter Assay System | Promega | E1960 |
| Deposited data | ||
| GLIS3 ChIP-seq in human islets | This paper | GSE151405 |
| H3K4me1, Human fetal pancrease | Cebola et al., 2015 | PMID: 25915126 |
| H3K4me3, Human fetal pancrease | Cebola et al., 2015 | PMID:25915126 |
| H3K4me1, Human donor-derived islet | Pasquali et al., 2014 | PMID:24413736 |
| H3K4me3, Human donor-derived islet | Pasquali et al., 2014 | PMID:24413736 |
| H3K27ac, Human donor-derived islet | Pasquali et al., 2014 | PMID:24413736 |
| PDX1, Human donor-derived islet | Pasquali et al., 2014 | PMID:24413736 |
| NKX6-1, Human donor-derived islet | Pasquali et al., 2014 | PMID:24413736 |
| FOXA2, Human donor-derived islet | Pasquali et al., 2014 | PMID:24413736 |
| MAFB, Human donor-derived islet | Pasquali et al., 2014 | PMID:24413736 |
| RNA-seq, Human donor-derived islet | Akerman et al., 2017 | PMID:28041957 |
| Experimental models: cell lines | ||
| EndoCβ-H1 | Philippe Ravassard Laboratory | N/A |
| Mouse embryonic fibroblasts (MEFs) | Generated in this study | N/A |
| HEK293T | ATCC | ATCC CRL-11268 |
| MCF7 | ATCC | HTB-22 |
| SW480 | ATCC | CCL-228 |
| Experimental models: organisms/strains | ||
| C57BL/6 | Charles River | C57BL/6 (inbred) |
| HIPKI, HIPKI-C331G | Generated in this study | N/A |
| Oligonucleotides | ||
| Primers used in this study - Table S3 | IDT | N/A |
| Taqman Probe Ins1 | Applied Biosystems | Mm01259683_g1 |
| Taqman Probe Ins2 | Applied Biosystems | Mm00731595_gh |
| Taqman Probe INS | Applied Biosystems | Hs02741908_m1 |
| Taqman Probe Actb | Applied Biosystems | Mm00607939_s1 |
| Taqman Probe ACTB | Applied Biosystems | Hs01060665_g1 |
| Software and algorithms | ||
| FASTQC | Babraham institute | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS/wiki/Install-macs2 |
| Picard Suite | Broad Institute | https://broadinstitute.github.io/picard/ |
| Rstudio | RStudio Team (2020). RStudio: Integrated Development for R. | https://www.rstudio.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Ildem Akerman (i.akerman@bham.ac.uk).
Materials availability
Materials generated in this study are available upon request from the lead contact.
Data and code availability
Accession number for GLIS3 ChIP-seq data is GEO: GSE151405 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151405).
All code generated during this study is available upon request.
Experimental model and subject details
Patients with monogenic diabetes
The study was conducted in accordance with the Declaration of Helsinki principles with informed parental consent given on behalf of children. Patients reported in this study comprise all probands and family members with INS promoter homozygous or compound heterozygous mutations sequenced in the Exeter Genomics laboratory, and 3 additional patients from the original joint description of recessive INS mutations (DM1293.1, DM1293.2 and DM1265 in Table S1). The coding, flanking intronic regions and up to 450 bp upstream of the INS transcriptional start site (NM_000207.2) were analyzed by Sanger sequencing as previously described (Garin et al., 2010). Clinical information was provided by the referring clinicians via a neonatal diabetes request form (available at https://www.diabetesgenes.org) and from clinical notes.
Generation of HIPKI and HIPKI-C331G mice
Mouse experiments were conducted following procedures approved by the Ethical Committee of Animal Experimentation of the University of Barcelona. Targeted replacement of Ins2 with human INS 5′ flanking sequences driving Ins2 and GFP was performed as schematized in Figure 1 (genOway). The exact sequences of these regions are provided in File S1. In brief, targeting vectors were generated with a 3.10 kb unmodified or mutated (c.C331G) 5′ flanking INS region, Ins2 exon 1, intron, and exon 2, an IRES-GFP reporter cassette inserted in the 3′ untranslated region of Ins2 exon 2, followed by a neomycin selection cassette flanked by loxP sites. This construct was flanked by 5.3 kb and 1.8 kb C57BL/6J mouse homology arms. Targeting of this vector was carried out by homologous recombination in C57BL/6J embryonic stem cells, using a diphtheria toxon A cassette for negative selection. Recombinants were verified by PCR screening and Southern blotting. Four suitable clones resulted, which were used for blastocyst injections and generation of the HIP knock-in mouse strains.-This was followed by CRE-mediated excision of the neomycin cassette in vivo, and again verified by PCR screening and Southern blotting. The sequence of the replaced region was verified by Sanger sequencing. Oligonucleotides used for genotyping are shown in Table S3.
Human islets
Human pancreatic islets were obtained through the European Consortium on Islet Transplantation (ECIT), Islets for Basic Research Program supported by the Juvenile Diabetes Research Foundation (program 2-RSC-2019-724-I-X). Pancreatic islets were isolated from multiorgan donors without a history of glucose intolerance (Nano et al., 2015), shipped in culture medium and re-cultured at 37°C in a humidified chamber with 5% CO2 in glucose-free RPMI 1640 supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 U/ml streptomycin and 11mM glucose for three days before analysis.
EndoCβ-H1 cell line and culture
EndoCβ-H1 cells were obtained from P. Ravassard and cultured on plastic tissue culture vessels coated with 2 μg/ml Fibronectin and 1% extracellular matrix (ECM) (Sigma-Aldrich) and with the following media: DMEM containing 5.5 mM glucose (GIBCO), 2% bovine serum albumin (BSA, Roche), 2 mM glutamine, 10 mM nicotinamide, 100 international units (U)/ml penicillin, 100 μg/ml streptomycin (P/S), 50 μM β-2-mercaptoethanol, 5.5 μg/ml transferrin and 6.6 ng/ml sodium selenite at 37°C in a humidified chamber with 5% CO2.
MEF, HEK293T, MCF7 and SW480 cultures and cell lines
Mouse embryonic fibroblasts (MEFs) were obtained from E15.5 HIPKI and HIPKI-C331G mouse embryos by clipping the tail tips and culturing in media (DMEM (with glucose) supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 U/ml streptomycin at 37°C in a humidified chamber with 5% CO2). HIPKI and HIPKI-C331G MEFs, HEK293T, MCF7 and SW480 cells were maintained in DMEM (Lonza) and supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 U/ml streptomycin at 37°C in a humidified chamber with 5% CO2.
Method details
Immunofluorescence
Pancreases were processed for immunofluorescence as previously described (Maestro et al., 2003).Tissues were fixed in 4% paraformaldehyde-PBS overnight at 4°C with gentle rotation, then washed once in 20 mL cold PBS and embedded in parrafin. Paraffin blocks were cut into 4 μm sections, deparaffinized with xylene by washing twice with a 15 minutes incubation. Xylene was removed and the blocks were rehydrated through serial washes with 5 mL ethanol-water (100% Ethanol incubated for 5 min; 95% Ethanol incubated for 5 min; 75% Ethanol incubated for 5 min; 100% Water incubated for 5 min) followed by an incubation with PBS for 5 min. Sections were blocked for 30 min at room temperature in antibody diluent (DAKO Corporation) with 3% normal serum-PBS solution, using serum from the same species as the secondary antibody, and incubated overnight at 4°C with primary antibodies diluted in 100 μL of blocking buffer per section. Antibody dilutions used were: anti-insulin 1:200, anti somatostatin 1:200, rabbit anti glucagon 1:200, guinea pig anti glucagon 1:1000, anti-GFP 1:200. We then incubated one hour at room temperature with secondary antibodies also diluted in blocking buffer (100 μl/section) at the manufacturer’s recommended concentration (1:400 for fluorochromes Cy3 and Cy5 and 1:800 for Alexa 488 fluorochrome). Images were acquired using Leica TSE confocal microscope for immunofluorescence. Antibodies used are shown in Table S3.
Mouse islets and embryonic pancreas isolation and processing
Mouse islets were isolated using previously described protocols (Párrizas et al., 2001; van Arensbergen et al., 2010). C57BL/6, HIPKI or HIPKI-C331G mice were anesthetized with urethane (15% solution, 1 ml/kg) and sacrificed. Pancreatic islets were isolated using the following procedure: a cannula tube was inserted in the main pancreatic duct and the pancreas was inflated with Hanks’ balanced salt solution (HBSS) buffer freshly added with 3U Collagenase P per ml. The pancreas was then removed, placed in a 15 mL falcon tube and digested for 10 min at 37°C in a water bath with constant, gentle agitation. The cell suspension was washed twice in 15 mL cold HBSS–0.5% bovine serum albumin (HBSS-BSA), resuspended in 5 mL cold HBSS–BSA and gently passed through a needle (0.8 mm diameter, 25 mm gauge). Islets were then washed by adding 10 mL of HBSS-BSA and sedimented for 3 minutes followed by removal of 10 mL from the top half of the solution (islets sediment to the bottom 5 ml). Wash procedure was repeated 3 times. Islets were then sedimented at 1800 rpm (580 g) for 1 min at 4°C and supernatant was removed. Islets were resuspended in 7 mL of HBSS-BSA and gently placed on top of 7 mL of histopaque mixture. Histopaque mixture is made freshly by mixing a 7:3 ratio of Histopaque 1077 and Histopaque 1119 (Sigma Aldrich). Histopaque mixture is heavier than HBSS-BSA and thus two distinct layers are formed. The islets were collected at the interphase of the two layers through centrifugation at 2300 rpm (950 g) for 20 min at room temperature (Acceleration = 5, Deceleration = 0). The islet-enriched fraction was aspirated from the interface, washed three times in cold HBSS–0.5% BSA, and further purified by handpicking under a stereomicroscope. Isolated islets were incubated in 11 mM glucose RPMI 1640 medium, supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 U/ml streptomycin at 37°C in a humidified chamber with 5% CO2 for 48 hours after isolation. For ChIP experiments, mouse islets were immediately crosslinked with formaldehyde after isolation and snap frozen at −80°C until use.
For isolation of embryonic tissue, embryos from timed-pregnancies were placed in cold PBS and dissected under the microscope. Entire embryonic pancreas or liver tissue was removed and placed in 1 mL Tripure (Invitrogen) reagent for RNA harvest.
FAIRE, ChIP and ChIP-Seq
ChIP, ChIP-seq (Chromatin Immunoprecipitation and sequencing) and FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) were performed essentially as previously published protocols (Gaulton et al., 2010; Nammo et al., 2011; Pasquali et al., 2014). ChIP experiments using antibodies against H3K4me1 and H3K4me3 were performed on ∼250 mouse islet equivalents (IEQ) per sample, while ChIP experiments using antibodies against PDX1 were performed on ∼400 mouse IEQ per sample. FAIRE experiments were performed on ∼250 mouse IEQ. GLIS3 ChiP-seq experiments were performed with ∼3000 human IEQ, using a previously described antibody (Yang et al., 2011; Table S3).
For ChIP, all cells were fixed in 1% formaldehyde-PBS for 10 minutes at room temperature with gentle agitation. Fixation was stopped with addition of glycine to a final concentration of 125 mM for 5 minutes with gentle agitation. Fixed cells were washed twice in cold PBS. Fixed islets were then suspended in sonication buffer and sonicated using Bioruptor (Diagenode) to a length of 200–1000 bp (∼12 minutes, 45sec ON, 45 s OFF). Samples were diluted with dilution buffer (1:4 ratio) to obtain 1% Triton X-100, 0.1% SDS and 130 mM NaCl and were precleared with high speed spin (12,000 rcf., 5 minutes) followed by incubation with protein A+G-Sepharose beads (1:1) (20 μl/sample) for 2 hours at 4°C. Input DNA (1% of volume of sample) was collected and frozen. Samples were then subjected to immunoprecipitation with the indicated antibodies (1 μL of H3K4me1/3 and PDX1 and 3 μL of GLIS3 antibodies, Antibody sources are provided in Table S3) overnight at 4°C with gentle rotation. Immune-complexes were collected by incubation with protein A or G-Sepharose (15 μl/sample) for 2 h at 4°C with gentle rotation. Beads were washed using standard ChIP wash buffers (1x 1 mL low salt wash buffer, 1x 1 mL high salt wash buffer, 1x 1 mL LiCl buffer, 3x 1 mL TE buffer) and immunocomplexes eluted with 300 μL elution buffer (incubation at room temperature for 15 minutes with gentle agitation). Input and eluted ChIP DNAs were subjected to reverse cross-linking (65°C for 12 hours), RNase A (20 μg) and Proteinase K digestion (90 μg), and purification with Qiaquick columns (QIAGEN).
For FAIRE assays, sonicated chromatin was subjected to phenol chloroform extraction (2x) without immunoprecipitation and wash steps62,63.
ChIP Buffers
Sonication Buffer: (2% Triton X-100, 0.5 % SDS, 100 mM NaCl, 1 mM EDTA,10 mM Tris-HCl pH 8.0 and 1x protease inhibitor cocktail (freshly added)
Dilution Buffer: 0.75 % Triton X-100, 140 mM NaCl, 1 mM EDTA, 0.1% Na-deoxycholate, 50 mM HEPES pH8.0, 1x protease inhibitor cocktail (freshly added)
Low Salt Wash Buffer: 20 mM Tris-HCl, pH 8.0, 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na Deoxycholate, 0.1% SDS, 1x protease inhibitor cocktail (freshly added)
High Salt Wash Buffer: 20 mM Tris-HCl, pH 8.0, 500mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na Deoxycholate, 0.1% SDS, 1x protease inhibitor cocktail (freshly added)
LiCl Buffer: 20mM Tris-HCl (pH 8.0), 250mM LiCl, 1mM EDTA, 0.5% Na Deoxycholate, 0.5% NP-40 substitute (IGEPAL), 1x protease inhibitor cocktail (freshly added)
TE Buffer: 10mM Tris-HCl (pH 8.0), 1mM EDTA, 1x protease inhibitor cocktail (freshly added)
Elution Buffer: 10mM Tris-HCl (pH 8.0), 1mM EDTA, 1% SDS, 1x protease inhibitor cocktail (freshly
ChIP and FAIRE Library Preparation and sequencing
Sequencing and processing were performed as described (Pasquali et al., 2014). Input DNA samples were quantified with QUBIT dsDNA BR assay kit (Q32853), and ChIP DNA samples were quantified with QUBIT dsDNA HS assay kit (Q32854). Quantitation was performed using Qubit 2.0 fluorometer. ChIP-Seq DNA libraries were prepared from 5-10 ng of Input DNA or ChIP DNA using NEBNext Ultra DNA Library Prep Kit for Illumina sequencing (New England BioLabs, # E7370L) by the Imperial College Genomics Facility. Libraries were sequenced using Illumina HiSeq2500.
ChIP and FAIRE data analysis
Reads were subjected to quality control analysis using FastQC (v0.11.5) followed by trimming by the Picard suite (default options). Trimmed reads were aligned to the human genome (hg19) using bowtie (Langmead and Salzberg, 2012) (v2.2.6, default options). Peaks were called using MACS2 (Zhang et al., 2008) (v2.2.1, q-value cutoff 0.05, band width 300) as enrichment over ChIP Input DNA. GLIS3-bound regions (peaks) are provided in Table S2.
References for new and previously reported human islet and fetal pancreas ChIP-seq datasets used in this study (Cebola et al., 2015; Morán et al., 2012; Pasquali et al., 2014) are provided in Table S4.
Quantitative PCR analysis
RNA was isolated using TriPure reagent as described (Akerman et al., 2017) following manufacturer’s instructions. RNA quality was ascertained with a 2100 Agilent Bioanalyzer (RIN > 8). Ins2, INS, ActB and Ins2 intronic transcripts were measured using TaqMan (Applied Biosystems) probes, while other regions were quantified using oligonucleotide primers and SYBR green mastermix (Illumina). cDNA synthesis was carried out using Superscript III (Invitrogen) and real-time PCR was performed with the ABI 7300 Real Time PCR system using the Power SYBR Green reagent (Applied Biosystems). Serial dilutions of genomic DNA (5 data points) were used to establish a standard curve. SDS software (Applied Biosystems) was used to generate quantitative values based on the standard curve with arbitrary units. All mRNA levels were normalized to ActB or Hprt as indicated in Figure legends. Oligonucleotides and TaqMan probe sequences can be found in Table S3.
In silico motif analysis
To identify candidate transcription factors that bind the CC element, we performed in silico motif searches using the MEME suite (Bailey et al., 2009) using the normal and mutated (c.331 C > G) sequences. Analysis of de novo and previously characterized position weight matrixes (PWM) was performed with HOMER (v3.12) (Heinz et al., 2010) using peak summit coordinates and flags -size −50,50 -len 6,8,10,12.
GLIS3 knockdown in EndoCβ-H1 cells
Lentiviral-mediated knockdown of GLIS3 in the human pancreatic β-cell line EndoCβ-H1 was performed with two independent shRNAs placed in artificial miRNAs as described in detail (Akerman et al., 2017). In brief, five non-targeting and two GLIS3-targeting amiRNAs were designed using BLOCK-IT software (Invitrogen) and cloned into pTRIP-CMV gateway vectors as described (Akerman et al., 2017). These were then used to produce lentiviruses (Scharfmann et al., 2014), which were transduced into the EndoC-βH1 cells. For knockdown experiments, cells were dissociated with trypsin (0.05%), washed twice with room temperature PBS. 105 cells per well (24 wells) were incubated with lentivirus (60 ng of p24 capsid protein) and 10μg/ ml Diethylaminoethyl (DEAE) in 400 μL of medium for 1 hour at 37°C. Cells were harvested at 80 hours post-transduction, and RNA levels were assessed using real time PCR. The efficiency of transduction was judged to be > 95% based on GFP expression. Oligonucleotides used for shRNAs are shown in Table S3.
Luciferase assays
INS promoter activity was measured by transfection of a reporter plasmid: pSOUAPRL-251hINS-Luc (Roland Stein, Vanderbilt University), which contains 251 bp of human INS proximal promoter DNA located in front of the Firefly luciferase cDNA. The c.C331G mutation was introduced using site directed mutagenesis (Garin et al., 2010) using Stratagene site directed mutagenesis kit, following manufacturer’s instructions. The presence of the mutation was verified by Sanger sequencing. The plasmids (0.5 μg/24 well) were transfected into EndoCβ-H1 with Lipofectamine 2000 at 4:1 luciferase:overexpression vector ratio as previously described (Allen et al., 2011), following manufacturer’s instructions. We co-transfected a Renilla expressing construct (pGL4.75, 0.02 ng) as a normalizer to correct for differences in transfection efficiency. Expression vectors used in co-transfections are given in Table S3. Luciferase activity was measured at 48 hours post- transfection using a luminometer (Promega Veritas Microplate luminometer) with the reagents of the Promega Dual-Luciferase Reporter Assay System.
Activation of endogenous INS in heterologous cell types
MEFs were transduced with lentiviral vectors, one encoding the transcription factors PDX1, NEUROD1 and MAFA in a polycistronic transcript, and the other mouse GLIS3-ΔN155 (Beak et al., 2008; Table S3). Cells were harvested 48 hours post transduction and subjected to quantitative PCR analysis. HEK293T, MCF7 and SW480 cells were transfected using Lipofectamine 2000, following manufacturer’s instructions. We note that the omission of MAFA or substitution of NEUROD1 with NEUROG3 in such experiments resulted in similar INS induction levels. For microarray hybridization experiments described in Figure S5, harvested RNA was hybridized to Gene ST 1.0 Affymetrix arrays and the data was analyzed on Affymetrix TAC (v1.0.24) software as described (Pasquali et al., 2014).
SILAC
We used Stable Isotope Labeling by Amino acids in Cell culture (SILAC) for mass spectrometry identification of proteins that specifically bound to unmodified or mutated (c.C331G) double stranded oligonucleotides containing a 5′ linker and PstI restriction site (Table S3), exactly as described in detail (Mittler et al., 2009), using MIN6 immortalized mouse beta-cells (Miyazaki et al., 1990). MIN6 cells were maintained in DMEM (4.5g/L Glucose), 15% fetal calf serum, 100 U/ml penicillin, 100 U/ml streptomycin at 37°C in a humidified chamber with 5% CO2) and 50 μM beta-mercaptoethanol. SILAC identified 4 proteins whose binding was affected by the mutation: Krueppel-like factor 13 (UniProtKB/Swiss-Prot Q9Y2Y9), Krueppel-like factor 16 (UniProtKB/Swiss-Prot Q9BXK1), MAZ (UniProtKB/TrEMBL Q8IUI2) and ZFP37 (UniProtKB/Swiss-Prot Q9Y6Q3).
Quantification and statistical analysis
Boxplots were drawn using Rstudio (v3.5). Line within the boxplot represents median, whereas the bounds of the box define the first and third quartiles. Bottom and top of whiskers represent minimum and maximum numbers respectively for each boxplot. Dot plots and bar plots were drawn using Excel. Where indicated, Student’s t test was used to calculate statistically significant differences between samples.
Acknowledgments
This research was supported by the Birmingham Fellowship Programme, RD Lawrence Fellowship (Diabetes UK, 20/0006136), and Academy of Medical Sciences Springboard (SBF006\1140) to I.A. Other main funding sources (to J.F.) are Ministerio de Ciencia e Innovación (BFU2014-54284-R and RTI2018-095666-B-I00), Medical Research Council (MR/L02036X/1), a Wellcome Trust Senior Investigator Award (WT101033), European Research Council Advanced Grant (789055), and FP6-LIFESCIHEALTH 518153. E.D.F. is a Diabetes UK RD Lawrence Fellow (19/005971). A.T.H. and S.E. are the recipients of a Wellcome Trust Senior Investigator award (WT098395/Z/12/Z), and A.T.H. is employed as a core member of staff within the NIHR-funded Exeter Clinical Research Facility and is an NIHR senior investigator. S.E.F. has a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (105636/Z/14/Z). Human islets for research were supported by the Juvenile Diabetes Research Foundation (2-RSC-2019-724-I-X). Work in CRG was supported by the CERCA Programme, Generalitat de Catalunya, and Centro de Excelencia Severo Ochoa (SEV-2015-0510). CRG acknowledges the support of the Spanish Ministry of Science and Innovation to the EMBL partnership. We thank the University of Barcelona School of Medicine animal facility, Center of Genomic Regulation and Imperial College London Genomics Units, and Larry Chan (Baylor College), Roland Stein (Vanderbilt University), Anton Jetten (NIEHS, NIH, North Carolina), Doris Stoffers (University of Pennsylvania), Jochen Seufert (University of Freiburg), Marko Horb (Marine Biological Laboratory), Alpana Ray (University of Missouri), and Tatsuya Tsurimi (Aichi Cancer Center, Ngoya, Japan) for generous gifts of valuable reagents and Kader Thiam (genOway) for overseeing the design of mouse models. We thank Diego Balboa and Mirabai Cuenca for critical comments on the manuscript. Graphical abstract drawings were by Yasemin Ezel with clipart from Biorender.
Author contributions
I.A. and J.F. conceived and coordinated the study. I.A. performed cell-based and computational studies and supervised mouse analysis. I.A. and M.A.M. performed image analysis of mouse mutants and isolated islets. L.P. purified human islets. V.G. maintained mouse colonies, G.M. performed SILAC experiments, P.R. designed and created lentiviral vectors, and E.D.F., S.F., S.E., and A.T.H. identified and studied patient mutations. I.A. and J.F. wrote the manuscript with input from the remaining authors.
Declaration of interests
P.R. is a shareholder and consultant for Endocells/Unicercell Biosolutions. The other authors declare no competing interests.
Published: April 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108981.
Contributor Information
Ildem Akerman, Email: i.akerman@bham.ac.uk.
Jorge Ferrer, Email: jorge.ferrer@crg.eu.
Supplemental information
References
- Akerman I., Tu Z., Beucher A., Rolando D.M.Y., Sauty-Colace C., Benazra M., Nakic N., Yang J., Wang H., Pasquali L. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017;25:400–411. doi: 10.1016/j.cmet.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen H.L., Flanagan S.E., Shaw-Smith C., De Franco E., Akerman I., Caswell R., Ferrer J., Hattersley A.T., Ellard S., International Pancreatic Agenesis Consortium GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 2011;44:20–22. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhti M., Böttcher A., Lickert H. Modelling the endocrine pancreas in health and disease. Nat. Rev. Endocrinol. 2019;15:155–171. doi: 10.1038/s41574-018-0132-z. [DOI] [PubMed] [Google Scholar]
- Barrett J.C., Clayton D.G., Concannon P., Akolkar B., Cooper J.D., Erlich H.A., Julier C., Morahan G., Nerup J., Nierras C., Type 1 Diabetes Genetics Consortium Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beak J.Y., Kang H.S., Kim Y.S., Jetten A.M. Functional analysis of the zinc finger and activation domains of Glis3 and mutant Glis3(NDH1) Nucleic Acids Res. 2008;36:1690–1702. doi: 10.1093/nar/gkn009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benko S., Fantes J.A., Amiel J., Kleinjan D.J., Thomas S., Ramsay J., Jamshidi N., Essafi A., Heaney S., Gordon C.T. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009;41:359–364. doi: 10.1038/ng.329. [DOI] [PubMed] [Google Scholar]
- Biggin M.D. Animal transcription networks as highly connected, quantitative continua. Dev. Cell. 2011;21:611–626. doi: 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Bonnefond A., Lomberk G., Buttar N., Busiah K., Vaillant E., Lobbens S., Yengo L., Dechaume A., Mignot B., Simon A. Disruption of a novel Kruppel-like transcription factor p300-regulated pathway for insulin biosynthesis revealed by studies of the c.-331 INS mutation found in neonatal diabetes mellitus. J. Biol. Chem. 2011;286:28414–28424. doi: 10.1074/jbc.M110.215822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon M.E., Mohlke K.L. Deciphering the emerging complexities of molecular mechanisms at GWAS loci. Am. J. Hum. Genet. 2018;103:637–653. doi: 10.1016/j.ajhg.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebola I., Rodríguez-Seguí S.A., Cho C.H., Bessa J., Rovira M., Luengo M., Chhatriwala M., Berry A., Ponsa-Cobas J., Maestro M.A. TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat. Cell Biol. 2015;17:615–626. doi: 10.1038/ncb3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J.X., Buckingham K.J., Jhangiani S.N., Boehm C., Sobreira N., Smith J.D., Harrell T.M., McMillin M.J., Wiszniewski W., Gambin T., Centers for Mendelian Genomics The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am. J. Hum. Genet. 2015;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin O., Scacheri P.C. Enhancer variants: evaluating functions in common disease. Genome Med. 2014;6:85. doi: 10.1186/s13073-014-0085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowper-Sal lari R., Zhang X., Wright J.B., Bailey S.D., Cole M.D., Eeckhoute J., Moore J.H., Lupien M. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 2012;44:1191–1198. doi: 10.1038/ng.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb A., Habeb A., Kaplan W., Attia S., Hadi S., Osman A., Al-Jubeh J., Flanagan S., DeFranco E., Ellard S. Genetic characteristics, clinical spectrum, and incidence of neonatal diabetes in the Emirate of AbuDhabi, United Arab Emirates. Am. J. Med. Genet. A. 2016;170:602–609. doi: 10.1002/ajmg.a.37419. [DOI] [PubMed] [Google Scholar]
- Demirbilek H., Arya V.B., Ozbek M.N., Houghton J.A., Baran R.T., Akar M., Tekes S., Tuzun H., Mackay D.J., Flanagan S.E. Clinical characteristics and molecular genetic analysis of 22 patients with neonatal diabetes from the South-Eastern region of Turkey: predominance of non-KATP channel mutations. Eur. J. Endocrinol. 2015;172:697–705. doi: 10.1530/EJE-14-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Gysemans C., Mathieu C. β-cell differentiation and regeneration in type 1 diabetes. Diabetes Obes. Metab. 2013;15(Suppl 3):98–104. doi: 10.1111/dom.12164. [DOI] [PubMed] [Google Scholar]
- Docherty H.M., Hay C.W., Ferguson L.A., Barrow J., Durward E., Docherty K. Relative contribution of PDX-1, MafA and E47/beta2 to the regulation of the human insulin promoter. Biochem. J. 2005;389:813–820. doi: 10.1042/BJ20041891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis J., Langenberg C., Prokopenko I., Saxena R., Soranzo N., Jackson A.U., Wheeler E., Glazer N.L., Bouatia-Naji N., Gloyn A.L., DIAGRAM Consortium. GIANT Consortium. Global BPgen Consortium. Anders Hamsten on behalf of Procardis Consortium. MAGIC investigators New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvillié B., Cordonnier N., Deltour L., Dandoy-Dron F., Itier J.M., Monthioux E., Jami J., Joshi R.L., Bucchini D. Phenotypic alterations in insulin-deficient mutant mice. Proc. Natl. Acad. Sci. USA. 1997;94:5137–5140. doi: 10.1073/pnas.94.10.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin I., Edghill E.L., Akerman I., Rubio-Cabezas O., Rica I., Locke J.M., Maestro M.A., Alshaikh A., Bundak R., del Castillo G., Neonatal Diabetes International Group Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl. Acad. Sci. USA. 2010;107:3105–3110. doi: 10.1073/pnas.0910533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton K.J., Nammo T., Pasquali L., Simon J.M., Giresi P.G., Fogarty M.P., Panhuis T.M., Mieczkowski P., Secchi A., Bosco D. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010;42:255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German M., Ashcroft S., Docherty K., Edlund H., Edlund T., Goodison S., Imura H., Kennedy G., Madsen O., Melloul D. The insulin gene promoter. A simplified nomenclature. Diabetes. 1995;44:1002–1004. doi: 10.2337/diab.44.8.1002. [DOI] [PubMed] [Google Scholar]
- Hansen S.K., Párrizas M., Jensen M.L., Pruhova S., Ek J., Boj S.F., Johansen A., Maestro M.A., Rivera F., Eiberg H. Genetic evidence that HNF-1alpha-dependent transcriptional control of HNF-4alpha is essential for human pancreatic beta cell function. J. Clin. Invest. 2002;110:827–833. doi: 10.1172/JCI15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Romanoski C.E., Benner C., Allison K.A., Kaikkonen M.U., Orozco L.D., Glass C.K. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.F., Gulko B., Siepel A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 2017;49:618–624. doi: 10.1038/ng.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier J.M., Douhet P., Desbois P., Joshi R.L., Dandoy-Dron F., Jami J., Bucchini D. Human insulin gene expression in transgenic mice: mutational analysis of the regulatory region. Differentiation. 1996;60:309–316. doi: 10.1046/j.1432-0436.1996.6050309.x. [DOI] [PubMed] [Google Scholar]
- Junion G., Spivakov M., Girardot C., Braun M., Gustafson E.H., Birney E., Furlong E.E. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012;148:473–486. doi: 10.1016/j.cell.2012.01.030. [DOI] [PubMed] [Google Scholar]
- Kang H.S., Kim Y.-S., ZeRuth G., Beak J.Y., Gerrish K., Kilic G., Sosa-Pineda B., Jensen J., Pierreux C.E., Lemaigre F.P. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol. Cell. Biol. 2009;29:6366–6379. doi: 10.1128/MCB.01259-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy G.C., German M.S., Rutter W.J. The minisatellite in the diabetes susceptibility locus IDDM2 regulates insulin transcription. Nat. Genet. 1995;9:293–298. doi: 10.1038/ng0395-293. [DOI] [PubMed] [Google Scholar]
- Khoueiry P., Girardot C., Ciglar L., Peng P.C., Gustafson E.H., Sinha S., Furlong E.E. Uncoupling evolutionary changes in DNA sequence, transcription factor occupancy and enhancer activity. eLife. 2017;6:e28440. doi: 10.7554/eLife.28440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M., Xiong C., Martin B., Schubach M., Inoue F., Bell R.J.A., Costello J.F., Shendure J., Ahituv N. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 2019;10:3583. doi: 10.1038/s41467-019-11526-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski M.H., Qian H., Hou Z., Rosen J.D., Tapia A.L., Shan Y., Jain D., Argos M., Arnett D.K., Avery C., NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. TOPMed Hematology & Hemostasis Working Group Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15:e1008500. doi: 10.1371/journal.pgen.1008500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lay J., Stein R. Involvement of PDX-1 in activation of human insulin gene transcription. J. Endocrinol. 2006;188:287–294. doi: 10.1677/joe.1.06510. [DOI] [PubMed] [Google Scholar]
- Lee S.Y., Noh H.B., Kim H.T., Lee K.I., Hwang D.Y. Glis family proteins are differentially implicated in the cellular reprogramming of human somatic cells. Oncotarget. 2017;8:77041–77049. doi: 10.18632/oncotarget.20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux L., Desbois P., Lamotte L., Duvillié B., Cordonnier N., Jackerott M., Jami J., Bucchini D., Joshi R.L. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes. 2001;50(Suppl 1):S150–S153. doi: 10.2337/diabetes.50.2007.s150. [DOI] [PubMed] [Google Scholar]
- Lettice L.A., Heaney S.J., Purdie L.A., Li L., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- Maekawa M., Yamaguchi K., Nakamura T., Shibukawa R., Kodanaka I., Ichisaka T., Kawamura Y., Mochizuki H., Goshima N., Yamanaka S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature. 2011;474:225–229. doi: 10.1038/nature10106. [DOI] [PubMed] [Google Scholar]
- Maestro M.A., Boj S.F., Luco R.F., Pierreux C.E., Cabedo J., Servitja J.M., German M.S., Rousseau G.G., Lemaigre F.P., Ferrer J. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum. Mol. Genet. 2003;12:3307–3314. doi: 10.1093/hmg/ddg355. [DOI] [PubMed] [Google Scholar]
- Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melloul D., Marshak S., Cerasi E. Regulation of insulin gene transcription. Diabetologia. 2002;45:309–326. doi: 10.1007/s00125-001-0728-y. [DOI] [PubMed] [Google Scholar]
- Miguel-Escalada I., Pasquali L., Ferrer J. Transcriptional enhancers: functional insights and role in human disease. Curr. Opin. Genet. Dev. 2015;33:71–76. doi: 10.1016/j.gde.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittler G., Butter F., Mann M. A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements. Genome Res. 2009;19:284–293. doi: 10.1101/gr.081711.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- Morán I., Akerman I., van de Bunt M., Xie R., Benazra M., Nammo T., Arnes L., Nakić N., García-Hurtado J., Rodríguez-Seguí S. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16:435–448. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nammo T., Rodríguez-Seguí S.A., Ferrer J. Mapping open chromatin with formaldehyde-assisted isolation of regulatory elements. Methods Mol. Biol. 2011;791:287–296. doi: 10.1007/978-1-61779-316-5_21. [DOI] [PubMed] [Google Scholar]
- Nano R., Bosco D., Kerr-Conte J.A., Karlsson M., Charvier S., Melzi R., Ezzouaoui R., Mercalli A., Hwa A., Pattou F. Human islet distribution programme for basic research: activity over the last 5 years. Diabetologia. 2015;58:1138–1140. doi: 10.1007/s00125-015-3536-5. [DOI] [PubMed] [Google Scholar]
- Odagiri H., Wang J., German M.S. Function of the human insulin promoter in primary cultured islet cells. J. Biol. Chem. 1996;271:1909–1915. doi: 10.1074/jbc.271.4.1909. [DOI] [PubMed] [Google Scholar]
- Párrizas M., Maestro M.A., Boj S.F., Paniagua A., Casamitjana R., Gomis R., Rivera F., Ferrer J. Hepatic nuclear factor 1-alpha directs nucleosomal hyperacetylation to its tissue-specific transcriptional targets. Mol. Cell. Biol. 2001;21:3234–3243. doi: 10.1128/MCB.21.9.3234-3243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashos E.E., Park Y., Wang X., Raghavan A., Yang W., Abbey D., Peters D.T., Arbelaez J., Hernandez M., Kuperwasser N. Large, diverse population cohorts of hiPSCs and derived hepatocyte-like cells reveal functional genetic variation at blood lipid-associated loci. Cell Stem Cell. 2017;20:558–570.e10. doi: 10.1016/j.stem.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali L., Gaulton K.J., Rodríguez-Seguí S.A., Mularoni L., Miguel-Escalada I., Akerman İ., Tena J.J., Morán I., Gómez-Marín C., van de Bunt M. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 2014;46:136–143. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfmann R., Pechberty S., Hazhouz Y., von Bülow M., Bricout-Neveu E., Grenier-Godard M., Guez F., Rachdi L., Lohmann M., Czernichow P., Ravassard P. Development of a conditionally immortalized human pancreatic β cell line. J. Clin. Invest. 2014;124:2087–2098. doi: 10.1172/JCI72674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville D., Lichti-Kaiser K., Grimm S., Jetten A. GLIS3 binds pancreatic beta cell regulatory regions alongside other islet transcription factors. J. Endocrinol. 2019 doi: 10.1530/JOE-19-0182. Published online July 1, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senée V., Chelala C., Duchatelet S., Feng D., Blanc H., Cossec J.C., Charon C., Nicolino M., Boileau P., Cavener D.R. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat. Genet. 2006;38:682–687. doi: 10.1038/ng1802. [DOI] [PubMed] [Google Scholar]
- Soufi A., Garcia M.F., Jaroszewicz A., Osman N., Pellegrini M., Zaret K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 2015;161:555–568. doi: 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J., García-Hurtado J., Moran I., Maestro M.A., Xu X., Van de Casteele M., Skoudy A.L., Palassini M., Heimberg H., Ferrer J. Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 2010;20:722–732. doi: 10.1101/gr.101709.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar D., Berthelot C., Aldridge S., Rayner T.F., Lukk M., Pignatelli M., Park T.J., Deaville R., Erichsen J.T., Jasinska A.J. Enhancer evolution across 20 mammalian species. Cell. 2015;160:554–566. doi: 10.1016/j.cell.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward L.D., Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012;30:1095–1106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon M.N., Cebola I., Patch A.M., Flanagan S.E., De Franco E., Caswell R., Rodríguez-Seguí S.A., Shaw-Smith C., Cho C.H., Allen H.L., International Pancreatic Agenesis Consortium Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat. Genet. 2014;46:61–64. doi: 10.1038/ng.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.D., Barbosa-Morais N.L., Schmidt D., Conboy C.M., Vanes L., Tybulewicz V.L., Fisher E.M., Tavaré S., Odom D.T. Species-specific transcription in mice carrying human chromosome 21. Science. 2008;322:434–438. doi: 10.1126/science.1160930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Chang B.H., Samson S.L., Li M.V., Chan L. The Krüppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. 2009;37:2529–2538. doi: 10.1093/nar/gkp122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Chang B.H., Yechoor V., Chen W., Li L., Tsai M.J., Chan L. The Krüppel-like zinc finger protein GLIS3 transactivates neurogenin 3 for proper fetal pancreatic islet differentiation in mice. Diabetologia. 2011;54:2595–2605. doi: 10.1007/s00125-011-2255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Chang B.H., Chan L. Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol. Med. 2013;5:92–104. doi: 10.1002/emmm.201201398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yechoor V., Liu V., Espiritu C., Paul A., Oka K., Kojima H., Chan L. Neurogenin3 is sufficient for transdetermination of hepatic progenitor cells into neo-islets in vivo but not transdifferentiation of hepatocytes. Dev. Cell. 2009;16:358–373. doi: 10.1016/j.devcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret K.S., Carroll J.S. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZeRuth G.T., Takeda Y., Jetten A.M. The Krüppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol. Endocrinol. 2013;27:1692–1705. doi: 10.1210/me.2013-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Guo M., Matsuoka T.A., Hagman D.K., Parazzoli S.D., Poitout V., Stein R. The islet beta cell-enriched MafA activator is a key regulator of insulin gene transcription. J. Biol. Chem. 2005;280:11887–11894. doi: 10.1074/jbc.M409475200. [DOI] [PubMed] [Google Scholar]
- Zhou Q., Melton D.A. Pancreas regeneration. Nature. 2018;557:351–358. doi: 10.1038/s41586-018-0088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Brown J., Kanarek A., Rajagopal J., Melton D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F., Nair R.R., Fisher E.M.C., Cunningham T.J. Humanising the mouse genome piece by piece. Nat. Commun. 2019;10:1845. doi: 10.1038/s41467-019-09716-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession number for GLIS3 ChIP-seq data is GEO: GSE151405 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151405).
All code generated during this study is available upon request.