

Summary
Most RNA processing occurs co-transcriptionally. We interrogated nascent pol II transcripts by chemical and enzymatic probing, and determined how the “nascent RNA structureome” relates to splicing, A-I editing and transcription speed. RNA folding within introns and steep structural transitions at splice sites are associated with efficient co-transcriptional splicing. A slow pol II mutant elicits extensive remodeling into more folded conformations with increased A-I editing. Introns that become more structured at their 3’ splice sites get co-transcriptionally excised more efficiently. Slow pol II altered folding of intronic Alu elements where cryptic splicing and intron retention are stimulated, an outcome mimicked by UV which decelerates transcription. Slow transcription also remodeled RNA folding around alternative exons in distinct ways that predict whether skipping or inclusion is favored, even though it occurs post-transcriptionally. Hence co-transcriptional RNA folding modulates post-transcriptional alternative splicing. In summary the plasticity of nascent transcripts has widespread effects on RNA processing.
Keywords: pol II transcription elongation, nascent RNA structureome, co-transcriptional splicing, RNA editing, alternative splicing, splice site structure
eTOC Blurb
How does the folding of growing pre-mRNAs affect their co-transcriptional processing? By probing of nascent RNA pol II transcripts Saldi et al identify structural features associated with efficient co-transcriptional splicing and A-I editing. Slow transcription extensively remodels nascent RNA structures in ways that predict its effects on alternative splicing.
Graphical Abstract
Introduction
Most introns are spliced at least partly co-transcriptionally, meaning that the splicing substrate is often a nascent pol II transcript (Ameur et al., 2011; Beyer and Osheim, 1988; Carrillo Oesterreich et al., 2010; Khodor et al., 2011; Neugebauer, 2019; Tilgner et al., 2012). Splicing can be completed either co-transcriptionally or post-transcriptionally but little is known about what governs the decision between these alternative paths (Takashima et al., 2011; Vargas et al., 2011). Co- versus post-transcriptional splicing of a particular intron is could influence protein binding and inter-dependent splicing of neighboring introns (Drexler et al., 2020; Fededa et al., 2005; Herzel et al., 2018; Kim et al., 2017).
In growing transcripts, RNA structures fold rapidly and can control the proximity of 5’ and 3’ splice sites and the accessibility of splice sites and cis-elements to the spliceosome and regulatory RNA binding proteins (RBPs) (Goguel and Rosbash, 1993; Taliaferro et al., 2016; Warf and Berglund, 2010). Many studies implicate RNA secondary structures as effectors of splicing extending back to the self-splicing Group II intron ancestors of nuclear introns (Pyle, 2016). Structures predicted to bring 5’ and 3’ splice sites into proximity can promote efficient splicing (Charpentier and Rosbash, 1996; Goguel and Rosbash, 1993; Howe and Ares, 1997; Meyer et al., 2011; Rogic et al., 2008) and bypass the requirement for U2AF (Lin et al., 2016). Secondary structures affect splicing by presenting sequence elements in loops (Taliaferro et al., 2016) or masking them in stems (Buratti and Baralle, 2004; Eperon et al., 1988; Saha et al., 2020) and can thereby dictate alternative splicing outcomes (Solnick, 1985) (McManus and Graveley, 2011)(Buratti and Baralle, 2004; Schwartz et al., 2009; Tomezsko et al., 2020) (Saha et al., 2020). Furthermore, alternative exons are associated with conserved sequences predicted to form local and long range RNA structures (Pervouchine et al., 2012; Raker et al., 2009; Shepard and Hertel, 2008).
Whether the effects of nascent RNA structure are limited to a subset of alternatively spliced exons, or extend generally among constitutive and alternative splicing events is a major unresolved question. The role of nascent RNA structure in pre-mRNA maturation is poorly understood in part because the structure of most primary pol II transcripts is unknown. Furthermore, it has not been possible to manipulate pre-mRNA structure widely and ask how it affects mRNA processing. Seminal RNA structure mapping studies of nuclear RNA showed that introns are generally more highly structured than exons (Gosai et al., 2015; Sun et al., 2019)(Zafrir and Tuller, 2015). Mature mRNAs, on the other hand, are relatively unstructured presumably due to unwinding by the ribosome (Rouskin et al., 2015). Whether highly structured introns and the transitions to less structured exon sequences are of functional significance for splicing is not known.
The speed of transcription influences sequential folding of the nascent transcript by controlling the delay between synthesis of proximal and distal sequence elements that compete to make mutually exclusive base pairing interactions (Pan and Sosnick, 2006; Saldi et al., 2018; Zhang and Landick, 2016). Pol II elongation rates vary in vivo by over ten-fold (Jonkers et al., 2014), but it is not known whether this variation affects pre-mRNA folding and maturation by splicing and A-I editing. In yeast slow transcription increases the efficiency of specific constitutive splicing events (Aslanzadeh et al., 2018; Braberg et al., 2013; Howe et al., 2003) and in mammalian cells transcription speed has widespread effects on alternative exon inclusion and intron retention (de la Mata et al., 2003; Fong et al., 2014; Ip et al., 2011). Why slow transcription enhances inclusion of some exons and skipping of others is not well understood. According to the “window of opportunity” model (de la Mata et al., 2003) the delay between synthesis of splice sites in the nascent transcript governs alternative splicing (AS) outcomes but only a small fraction of elongation rate-dependent splicing changes are accounted for by this model (Dujardin et al., 2014; Fong et al., 2014). Another possibility is that co-transcriptional RNA folding pathways change with elongation rate, and thereby modulate co-transcriptional versus post-transcriptional splicing and the competition between AS reactions. If nascent RNA folding were affected by elongation rate, it would be expected to change A-I editing by ADARs as this modification occurs co-transcriptionally (Rodriguez et al., 2012) and is specific to dsRNA elements (Eggington et al., 2011). The extent of A-I editing within the nascent transcriptome is unknown, but deep sequencing of steady state RNA that contains pre-mRNAs identified millions of editing sites within intronic Alu elements (Bazak et al., 2014).
We report the first global analysis of the structure of nascent pol II transcripts which we determined by chemical and enzymatic probing together with mapping of A-I edits. We found that the structure of nascent pre-mRNAs is strongly affected by transcription speed and that alternative structures formed during transcription are closely related to the extent of co-transcriptional splicing and the outcome of alternative splicing decisions.
Results
tNET-Structure-seq maps nascent RNA structure
The structure of the nascent transcriptome has yet to be described in detail. To address this gap, we developed tNET-StructureSeq (Fig. 1A) that combines RNA sequencing of nascent transcripts immunoprecipitated by anti-pol II (tNET-seq, total Nascent Elongating Transcript sequencing)(Fong et al., 2017) with enzymatic and chemical probing as well as identification of A-I edits. Enzymatic probing of nascent RNA (tNET-RNAse-seq) was performed by a combination of ssRNA-seq, dsRNA-seq (Li et al., 2012) and Protein Interaction Profile sequencing (PIPseq) (Gosai et al., 2015) (Fig. 1A) to identify regions of single and double stranded RNA that are distinct from protein footprints. For enzymatic probing, HEK293 cells expressing α-amanitin resistant (Amr) pol II large subunit Rpb1 were lightly cross-linked with formaldehyde to stabilize RNA/protein associations, then RNA pol II was immunoprecipitated, and associated nascent transcripts were treated ex-vivo with a single-strand specific RNAse (RNase I), a double-strand specific RNase (RNase V1), or a combination of both RNases, and resistant fragments were sequenced (743M mapped reads, RNase I, V1 and I+V1 combined). As predicted for nascent RNA, these libraries were enriched for introns, sequences downstream of poly(A) sites and divergent transcripts upstream of genes compared to mRNA (Fig. 1B). Sequences protected from both RNAse I and VI correspond to sites of co-transcriptional protein binding or unusual RNAse resistant sequences. RNAse digestion after proteinase K digestion showed that the background of RNAse resistant nascent transcripts is low (Fig. S1A). That genuine protein footprints are being identified on nascent RNA by PIPseq is suggested by the fact that they are enriched at exon-exon boundaries as expected for exon junction complexes (EJC’s) which are deposited co-transcriptionally (Fig. 1C). Regions corresponding to RNAse resistant putative protein footprints were removed from our analysis which is confined to sequences that are accessible to RNAses. We calculated a Structure Score at each base as described (Gosai et al., 2015; Li et al., 2012). Structure Score is the difference in normalized ds RNA seq (RNAseI resistant) coverage minus ss RNAseq coverage (RNAseVI resistant) after arsinh transformation to stabilize the variance between regions with high and low sequence coverage (Huber et al., 2002). Positive Structure Scores therefore represent regions that are predominantly structured as indicated by resistance to RNAse I. Negative Structure Scores do not reflect the absence of structure; rather the more negative the score, the greater the fraction of single-stranded conformations at that position in the ensemble of structures. Because Structure Score is not determined for regions bound by proteins, the results are limited to sequences that are not engaged in stable protein-RNA complexes. As in previous work (Gosai et al., 2015) we did not detect unambiguous spliceosome footprints which are large and heterogeneous (Chen et al., 2018) and may not withstand the nucleases used here. Much of the stable structure in the human transcriptome comprises pairs of sense/antisense Alu elements (Bazak et al., 2014) and, as expected Structure Score was strongly elevated at expressed Alu’s (Fig. S1B).
Figure 1. tNET-Structureseq mapping of nascent RNA structure.

A. Enzymatic (tNet-RNAse-seq ) and DMS (tNet-MaPseq ) probing of nascent pol II transcripts.
B. Distribution of tNet-RNAse-seq and tNet-MaPseq compared to mRNAseq reads in HEK293 WT Amr pol II cells.
C. Metaplot of putative exon junction complex footprints (RNAse I+VI resistant, blue arrow) upstream of splice sites (SS).
D. Predicted structures with DMS reactivities of the non-canonical XBP1 intron (arrows) (Yoshida et al., 2001) (chr22:29,192,089–29,192,172) and XIST exon 4 (chrX :73,050,979–7,3051,093) within nascent RNA.
E. Nascent RNA structure across an intron (chr16:89,867,703–89,871,732). Normalized DMS reactivity, RNAse I, RNAse VI and RNAse I+VI resistant reads, fraction of A-I editing per base and nascent RNA seq reads (tNET-seq) are shown.
We validated structured RNA regions mapped by tNET-RNAse-seq by probing by in vivo DMS modification in HEK293 Amr Rpb1 cells followed by anti-pol II immunoprecipitation to purify nascent transcripts (Fig. 1A). DMS identifies single-stranded RNA in vivo by methylating unpaired A, C, and to a lesser extent G and U, residues (Mustoe et al., 2019)(Fig. S1C). These RNA modifications are detected as substitutions, insertions and deletions introduced by reverse transcription in the presence of Mn2+ using Mutational Profile sequencing (tNET-MaP-seq, 87M mapped reads)(Smola et al., 2015; Zubradt et al., 2017). Mutations caused by DMS modification are scored relative to a DMSO-treated sample that controls for RT errors and A-I edits that are more common in nascent RNA than mRNA. To facilitate detection of DMS-dependent mutations in a transcriptome as large as human pol II nascent RNA, we employed the variant calling approach (Li, 2011) (see Methods) used previously on a small scale (Saldi et al., 2018). Base pairing and protein binding protect from DMS methylation, so unmodified regions are inferred as either double-stranded or bound by RBPs. We removed the protein footprint regions determined by enzymatic probing from the DMS reactivity calculations in order to unambiguously distinguish dsRNA regions. DMS reactivity assignments were compared with several known RNA structures (Fig. 1D, S1D, E) which confirmed that most modifications were in single-stranded regions, as expected. The complementary results of independent enzymatic and chemical probing are illustrated in Fig. 1E by intron 7 of the FANCA gene. In this example the upstream region of low DMS reactivity (blue arrow), spans a protein footprint resistant to both RNAses, whereas the downstream region of DMS protection (red arrow) corresponds to structured RNA that is resistant to RNAse I, and sensitive to RNAse VI.
Structured sequences identified by enzymatic (tNet-RNAse-seq) and DMS (tNet-MAPseq) probing were further validated by cross-checking. As expected, peaks of Structure Score, which reflect double-strandedness, were anti-correlated with DMS reactivity, which reflects single-strandedness (Fig. 2A). In addition, we confirmed that DMS reactivity was decreased in regions of putative protein footprints identified by enzymatic probing (Fig. 2B).
Figure 2. Validation of nascent RNA structure by DMS probing, RNAse probing and A-I editing.

A, B. Reduced DMS reactivity at regions of high Structure Score (A) and Protein Footprints (B) (15 base bins). The reactivities differ because most regions of high Structure Score are intronic whereas most Protein Footprints are exonic.
C. A-I edits in nascent pol II transcripts from HEK293 WT Amr Rpb1 cells. Note the large fraction of edits in intergenic regions and introns.
D. Nascent RNA and mRNA reads and fraction of A-I editing per base. Note extensive editing in introns, 3’ flanking regions in divergent antisense transcripts upstream of ACAD9 (blue arrows, see Fig. S3B).
E. A-I editing is enriched in nascent RNA around peaks (>1.9) of Structure Score. Metaplot with 15 base bins.
F. Coincidence of Structure Score with A-I editing at inverted Alu elements (see Fig. S3C, D).
Because A-I editing is specific to dsRNA, it serves as an internal read out of RNA sequences that are structured in vivo and complements the identification of ds regions by ex vivo digestion with RNAses. A-I edits in nascent RNA (36.8M-209.5M reads, 3 replicates) were mapped using Redi-tools (Picardi and Pesole, 2013). As expected, most editing occurred in introns, but surprisingly, almost as many edited sequences were found in intergenic sequences transcribed in the termination zones downstream of genes and in antisense transcripts upstream of divergent promoters (Fig. 2C, D, S2B) A large fraction (72%) of edits within introns and intergenic sequences occurred within LINE and SINE repeats. A-I edits are enriched in regions of high Structure Score including intronic Alu elements, thereby validating the identification of dsRNA regions by RNAse probing (Fig. 1E. 2E, F S2C, D). In sum, the combination of three independent measures of RNA structure incorporated in tNET-Structureseq permits high confidence identification of nascent RNA structures.
Structural motifs associated with efficient co-transcriptional splicing
To ask how nascent RNA structure and co-transcriptional splicing are related, we first determined the co-transcriptional splicing efficiency (SE) at several thousand introns using replicate published tNETseq datasets (Fong et al., 2017) (GSE97827) that were extended by deeper sequencing of the libraries. These nascent RNA-seq datasets are enriched for intron sequences and transcripts upstream of promoters and uncleaved transcripts that span poly(A) sites (Fig. 2D, S2E). To calculate SE we developed a method called F-cov (see Methods, Fig. 3A) that quantifies unspliced transcripts using read density throughout introns rather than just at exon intron boundaries. SE values for some individual introns were validated by RT-PCR (Fig. S3A). The median SE for all introns was ~0.75, in agreement with previous work (Tilgner et al., 2012) (Fig. 3B, Table S1). SE values agreed with known properties of co-transcriptional splicing including reduced splicing of first and last introns, and very long introns (Khodor et al., 2011; Tilgner et al., 2012)(Fig. S3B, D). SE also correlated with splice site strength (Fig. S3C). Furthermore, the frequency of protein footprints at the expected position of co-transcriptionally deposited EJC’s correlated well with SE (Fig. S3E). Co-transcriptionally well-spliced and poorly-spliced introns frequently occurred within the same transcript (Fig. 3C, S3G) and the median range of SE values for introns in the same gene (>3 introns) is 0.57 (Fig. S3F). Consistent with previous work, we also found evidence of splicing coordination within clusters of adjacent introns (Fig. 3C, S3G) (Drexler et al., 2020; Fededa et al., 2005; Herzel et al., 2018; Kim et al., 2017).
Figure 3: RNA structural signature of efficient co-transcriptional splicing.

A. Calculation of co-transcriptional splicing efficiency, SE by the Fcov method (see Methods). The blue line represents idealized nascent RNA coverage.
B. Probability density plot of SE from tNET-seq of HEK293 WT Amr pol II cells based on three biological replicates for introns with ≥1 read/bp and/or ≥3 reads/bp across both flanking exons. Introns with standard deviation > 0.2 were removed. See Table S1.
C. mRNA and tNET-seq showing co-transcriptionally well-spliced (black arrow SE=.91) and poorly spliced (gray arrow SE=.009) introns and clustering of introns (grey box) with coordinated splicing (SE < 0.4) (see Fig. S4G).
D. Well-spliced introns (SE > 0.8, n=5739) are more structured than poorly spliced introns (SE < 0.2, n=1001). Mean Structure Score per nucleotide for introns >100bases with coverage >9 reads for at least 10% of the length of the intron. P value determined by unpaired t-test
E. Structure Score across well-spliced (SE>0.8) and poorly spliced (SE<0.2) introns >200 bases long (50 bins). Putative protein footprint regions were removed. Bottom panels −loge adjusted P values per bin (unpaired Wilcoxon-rank test). Blue line: −loge 0.05.
F, G. Efficient co-transcriptional splicing associated with peaks of intronic structure (blue arrows) and steep structural transitions at 5’ and 3’SS’s. Metaplots (5 base bins) of Structure Score, F, and DMS reactivity, G, at well-spliced (SE> 0.8) and poorly spliced (SE < 0.2,) introns. FDR values as in E. See Fig. S5.
H. Correlation between median calculated 3’ SS strength (Yeo and Burge, 2004) and experimentally determined structural transitions across 3’ splice sites. (Mean Structure Score of bases −1 to −100 minus bases +1 to +40 relative to 3’SS, n=18455).
There is a marked structural contrast between introns that are well spliced (SE ≥ 0.8) and those that are poorly spliced co-transcriptionally (SE ≤ 0.2). Co-transcriptionally well-spliced introns are more structured than poorly spliced introns on average throughout their length as evidenced by their Structure Scores (Fig. 3D, E). The G/C content of well-spliced and poorly spliced introns is similar, 42% and 45%, respectively so the enhanced structure of well-spliced introns is not attributable simply to nucleotide composition. At introns with high SE, there are steep transitions across 5’ and 3’ splice sites (after removal of protein footprints), that we designate the 5’ and 3’SS-steps where intron sequences were more highly structured than adjacent exons. In contrast, at introns with low SE, the structural transitions across splice sites were smaller. This result is shown for metaplots of merged Structure Score results from enzymatic probing in Figure 3F and for individual replicate data sets in Figure S4A. Examples of 3’ splice sites from introns with high and low SE are shown in Figure S4B. The structural distinction at splice sites between co-transcriptionally well-spliced and poorly spliced introns is confirmed by DMS probing in Figure 3G. Note that the structures reported here are for unspliced pre-mRNAs, and not excised introns which do not co-purify with immunoprecipitated pol II (Sheridan et al., 2019). The correlation between the size of 3’ SS structural step and co-transcriptional SE was much more marked for longer introns suggesting this structural feature is of greater functional significance when 5’ and 3’ splice sites are distant or when exon definition rather than intron definition operates (Fig. S4C). Remarkably, the size of the structural transition at the 3’SS correlated well with splice site strength calculated by the MaxEnt method (Yeo and Burge, 2004) (Fig. 3H) suggesting that the functionality of the 3’ SS is determined in part by RNA structure and not only by primary sequence. In contrast, we did not detect a clear correlation between the structural transition at 5’ splice sites and their calculated strength. This discrepancy may reflect the fact that 5’SS strength is based on only 9 bases spanning the exon intron junction whereas 3’SS strength is based on 20 bases upstream and 3 bases downstream of the junction (Yeo and Burge, 2004).
Together the chemical and enzymatic probing approaches demonstrate that RNA structure throughout the length of introns, and abrupt structural transitions at 5’ and 3’ splice sites, correlate widely with co-transcriptional SE. Similar structural transitions at splice sites were reported previously in plant and mouse nuclear RNA, but their relation to splicing activity was not known (Gosai et al., 2015; Sun et al., 2019). The association of these structural signatures with co-transcriptional splicing, together with their conservation between plants and mammals suggests that they are functionally significant.
Slow transcription alters nascent RNA folding and A-I editing
The results in Figure 3 suggest that in addition to splice site sequences, the conformation of the nascent pre-mRNA at intron-exon junctions may influence the decision between rapid co-transcriptional splicing and delayed post-transcriptional splicing. It is not possible currently to test this idea by directed manipulation of RNA folding on a large scale and asking how RNA processing is affected. Instead, based on previous work (Pan et al., 1999; Pan and Sosnick, 2006), we asked whether slowing the speed of transcription alters nascent pre-mRNA folding, and if so, how such structural changes relate to splicing and editing. For these experiments we employed HEK293 cells expressing inducible an α-amanitin resistant mutant Rpb1 (R749H) that slows transcription by about 3-fold on average (Fong et al., 2014) to ~0.5kb/min, which is still within the normal physiological range of transcription rates (Jonkers et al., 2014). We assayed RNAse sensitivity and DMS reactivity of nascent transcripts co-immunoprecipitated with the mutant pol II. These experiments revealed a widespread re-structuring with increased folding of nascent transcripts in both exons and introns when transcription slows down, with introns remaining more structured than exons (Fig. 4A, B, S5A). Slow transcription also reduced the density of putative protein footprints in introns determined by PIPseq (8.5 footprints/100kb in WT vs. 5.2 footprints/100kb in slow), consistent with the anti-correlation between protein binding and RNA structure (Taliaferro et al., 2016). In summary these findings suggest that slow transcription shifts the balance between alternative RNA folding pathways in a way that generally favors more highly folded states.
Figure 4. Slow transcription extensively modifies nascent RNA structure and A-I editing.

A. Slow transcription increases nascent RNA structure. The fractions of bases (minimum 15 reads/base) with Structure Score >1.9 are plotted in cells expressing WT and slow (R749H) pol II (see Fig. S6A).
B. Metaplots as in 3E showing increased structure throughout introns transcribed by slow pol II.
C. A-I edits in nascent RNA from HEK293 expressing slow pol II. Note increased editing relative to WT pol II (Fig. 1C).
D. A-I editing, Structure Score and nascent RNA coverage downstream of SRSF3. Note elevated editing (blue arrows) and Structure Score (green arrows) in the slow pol II mutant.
E. Slow transcription increases intronic A-I editing independent of expression level. Introns contain >3 edited sites in WT or slow pol II mutant in three replicates. Red and blue dots have FDR<0.05 and fold change >1.3. See Table S3.
F. A-I editing in nascent RNA (see Methods) in introns, UTRs, and intergenic regions upstream (US) and downstream (DS) of genes. Note de novo editing (red bars) with slow pol II.
We next investigated the relation between transcription rate dependent changes in RNA folding and co-transcriptional RNA processing. To examine how editing is affected, we performed nascent RNA seq on the slow pol II mutant (Rpb1 R749H tNET-seq 67.9M-133.8M reads, 3 replicates) and compared the frequencies of A-I editing with those in nascent RNA made by WT pol II. This analysis revealed that slow transcription increased the overall number of positions that become A-I edited in nascent transcripts by approximately two fold (compare Fig. 2C and 4C) with most edited sites lying in introns and intergenic regions (Fig. 4D). Slow pol II strongly enhanced A-I editing within introns relative to WT pol II regardless of expression level (Fig. 4E). Among sequences expressed in nascent RNA in both the WT and slow pol II mutant cells, many regions within introns and intergenic sequences became edited de novo in the slow mutant (Fig. 4F). Enhanced A-I editing was not the result of increased ADAR1 expression in slow pol II mutant cells (Fig. S5B) and therefore is presumably due to formation of new dsRNA structures. In summary, RNA structure and A-I editing are highly plastic in response to changes in transcription speed.
Enhanced co-transcriptional splicing and RNA structural changes associated with slow transcription
We compared co-transcriptional splicing in nascent RNA synthesized by WT and slow mutant pol II using published tNET-seq results (GSE97827) (Fong et al., 2017) that were extended by deeper sequencing (Fig. 5A). The extent of mRNA contamination did not differ substantially between the WT and slow mutant nascent RNA samples as determined by the fraction of unprocessed reads that span poly(A) sites (Fig. S2E). Splicing of most introns was not significantly affected by slow pol II showing that longer transcription times do not necessarily promote co-transcriptional splicing. On the other hand, slow transcription caused significant changes in co-transcriptional splicing (SE) of several thousand introns and in over 90% of cases it increased splicing efficiency (Fig. 5B, Table S2) consistent with previous observations in yeast (Aslanzadeh et al., 2018; Braberg et al., 2013). The fact that slow pol II increases rather than decreases co-transcriptional splicing and A-I editing suggests that its effects are not the result of a non-specific reduction in gene expression or cell viability, consistent with our previous finding that expression of most splicing factors is not significantly affected (Fong et al., 2014). The median range of SE values within genes (>3 introns) under slow transcription conditions is slightly less than for WT (0.49 vs 0.57) (Fig. S3F). The median length of introns whose splicing was enhanced by slow transcription (1383 bases) did not differ significantly from unaffected introns (1051 bases, p-value=0.17 t-test). We compared the structural features of introns that are affected and unaffected by slow transcription. Notably, those introns with enhanced co-transcriptional splicing had significantly steeper 5’ and 3’ SS steps when transcription was slow (Fig. 5C, pink and grey lines). In contrast, at introns where splicing was unaffected, there was less effect of slow transcription on the structure of splice sites. This result is shown for merged Structure Score data sets in Figure 5C (upper panel, pink and gray lines) and for individual replicates in Figure S5C, D. The effect of slow transcription on splice site structures at introns with enhanced SE was confirmed by DMS probing of transcripts immunoprecipitated with the R749H mutant pol II (183.4M mapped reads, Fig. 5C lower panel). Structural changes at 3’ SS’s with increased splicing in the slow mutant are also evident at individual sequences (Fig. 5D, S6). Predicted local RNA folding of 3’SS regions that become more structured in the slow mutant revealed that residues at branchpoints and splice sites can be presented in loops or bulges which might enhance their reactivity (Fig. 5D, S6). It is also possible that double stranded structures might enhance splicing by preventing binding of inhibitory RBPs. Reduced elongation rates in both exons and introns could contribute to the structural changes associated with altered splicing.
Figure. 5. Slow transcription specifically alters RNA structure at splice sites with increased co-transcriptional splicing.

A. Slow transcription increases co-transcriptional splicing. Probability density plot of SE (Fig. 3A, n=3) in HEK293 expressing WT and slow pol II.
B. Slow transcription preferentially increases co-transcriptional splicing of a subset of introns. Pink and blue points indicate altered SE (FDR<0.05, >1.25-fold change) in slow vs WT pol II.
C. Metaplots (5 base bins) of Structure Score and DMS reactivity across splice sites of introns whose SE is increased (grey and pink) or unaffected (black and red) by slow transcription. Note elevated structure and steeper 5’ and 3’ SS steps where splicing is stimulated by slow transcription (blue arrows). See Fig. S6C, D.
D. Elevated RNA structure (blue arrow) at the 3’ SS of an intron (chr19:41,209,740–41,211,039) where SE is increased by slow pol II. tNET-seq, Structure Score and predicted RNA folding around the 3’SS and branch point (b.pt.). See Fig. S7.
E. Elevated 3’SS structure in the slow pol II mutant predicts increased co-transcriptional splicing. Change in Structure Score of bases −10 to −70 relative to 3’SS between slow and WT pol II was ranked for 3994 introns and SE of the top and bottom 10% is plotted. Note increased SE of introns with the greatest increase in 3’SS structure.
If rate-sensitive 3’SS structures affect splicing, then introns whose 3’SS steps are enhanced are predicted to be better co-transcriptionally spliced when transcription is slow. Conversely, introns whose 3’SS steps are less affected by slow transcription are predicted to have their co-transcriptional splicing relatively unaffected. To test this prediction, we ranked 3’ splice site regions (−70 to −10 bases relative to splice junction) based on the increase in structure caused by slow transcription. We compared SE of introns with the greatest increase (top 10%) versus those with the smallest increase (bottom 10%) in the slow mutant. This analysis revealed that introns with the greatest increase in 3’SS structure had significantly increased SE with slow transcription relative to those where 3’SS structure was relatively unaffected (Fig. 5E). This observation therefore indicates that steeper structural transitions at 3’ splice sites contribute to faster, and more co-transcriptional splicing. On the other hand, we did not detect a significant correlation between the magnitude of changes in 3’SS structure and SE among all introns indicating that many structural effects of slow pol II do not change splicing enough for us to detect. In summary, the effects of slow transcription on RNA structure and co-transcriptional splicing argue that nascent RNA structure can modulate numerous splicing reactions and that the strength of a splice site is determined not only by its sequence, but also by how it is folded.
Transcription rate sensitive cryptic splicing and the response to UV radiation
Nascent RNA sequencing revealed abundant co-transcriptional cryptic splicing including almost 12,000 splice junctions and over 8,000 cryptic exons (Fig. 6A). Slow pol II affected ~2500 (FDR<0.05, >2-fold change) such splicing events with a strong bias (81%) in favor of cryptic exon inclusion (Fig. 6A, Fig. S7A, Table S3), particularly at intronic Alu elements (Fig. 6B) (Gal-Mark et al., 2008). Cryptic non-coding exons have been proposed to act as “decoys” that enhance intron retention (IR) by competing with canonical splice sites (Parra et al., 2018). Consistent with the “decoy exon” model, introns harboring cryptic Alu exons activated by slow pol II were significantly more retained in mRNA relative to cells expressing WT pol II (Fig. 6C).
Figure 6. Nascent RNA structure, Alu exonization and intron retention in response to slow pol II and UVR.

A. Slow transcription increases cryptic splicing in nascent RNA. PSI is the ratio of junctions supporting the cryptic exon divided by those supporting cryptic and canonical splices. Red and blue points indicate FDR<0.05, fold change >1.5. Arrows mark cryptic splices unique to WT or slow pol II mutant.
B. Alu’s are enriched among cryptic splices increased by slow transcription (up arrow) relative to those decreased (down arrow) and all cryptic splices.
C. Slow pol II enhances retention of introns where cryptic splicing is stimulated (see A). Retention is the intron coverage normalized to coverage over the adjacent 30bp of flanking exons. P-values in B, C calculated by Chi2.
D. Structure Score and protein footprint metaplots (5 base bins) at cryptic splice sites within Alu’s exonized in nascent RNA by slow pol II. Note increased structure associated with cryptic splice site use (blue arrows). Map shows antisense Alu with polyU sequences (yellow boxes) and hnRNPC binding site (hnC) (Zarnack et al., 2013). Note protein footprints at hnRNPC binding sites are lost with slow pol II (red arrow).
E. Cryptic splicing is stimulated in mRNA following UV which slows transcription. mRNAseq 8 hr post UVR (Williamson et al., 2017). PSI as in A. Red and blue points indicate FDR<0.05, fold change >1.2. Arrows mark splices unique to control or UV treated samples.
F. Overlap of cryptic Alu exons included in response to slow pol II (HEK293 cells) and UVR (MRC5V5cells) (Williamson et al., 2017).
G. Alu exonization (blue arrows) and intron retention (black arrows) (chr10:1,123,843–1,126,008) are stimulated by slow pol II (HEK293) and UVR (MRC5VA) (Williamson et al., 2017) which slows transcription. RT-PCR validation of Alu exonization in nascent RNA (HEK293) at lower left.
Unlike most coding exons, Alu’s are highly structured. We investigated whether slow transcription altered RNA structure near Alu’s that become exonized. RNA structure upstream of the cryptic 3’SS, and downstream of the cryptic 5’SS in these Alu’s was increased by slow transcription as shown by Structure Score and DMS reactivity (Fig. 6D, S7B). These changes makes the cryptic splice sites in Alu’s more closely resemble well-spliced canonical sites (see Fig. 3G). We speculate that this remodeling activates Alu cryptic splice sites by making them better substrates for the spliceosome and/or inhibiting hnRNPC binding, which antagonizes recognition of their 3’SS’s (Zarnack et al., 2013). Consistent with the latter possibility, protein footprints were reduced in the slow pol II mutant at the 3’SS’s of exonized Alu’s (Fig 6D), but not near putative 3’SS’s of expressed Alu’s generally (Fig. S7C). In summary the results in Figures 5C–E and 6A–D suggest that alternative nascent RNA structures resulting from slow transcription can enhance co-transcriptional splicing at both canonical and cryptic splice sites.
To ask whether elongation rate dependent changes in nascent RNA structure could be physiologically relevant, we examined mRNA-seq data from cells exposed to UV radiation (UVR) which provokes a strong transcriptional deceleration comparable to that caused by the R749H slow mutant (Munoz et al., 2009; Williamson et al., 2017). Cryptic splicing was altered at ~1500 introns following UVR, and similar to slow mutant pol II, cryptic exon inclusion increased in over 80% of cases (Fig. 6E, Table S5). UV regulated cryptic splicing was highly enriched for inclusion of Alu elements (Fig. S8A) and remarkably, a significant fraction of Alu exons included in response to UV was also included in response to slow mutant pol II (Fig. 6F). We note that inclusion of Alu cryptic exons in HEK293 cells expressing slow pol II was only evident in nascent RNA, whereas in UV treated MRC5V5 cells their inclusion was readily apparent in mRNA, perhaps due to stability differences. As in the slow pol II mutant (Fig. 6C), cryptic exon inclusion in response to UVR was also associated with intron retention as predicted by the “decoy exon” model (Parra et al., 2018) (Fig. S8B). Examples of cryptic Alu exon inclusion and intron retention common to slow pol II and UVR are shown in Figs. 6G and S8C. Based on the structural changes in nascent RNA at Alu elements caused by slow pol II (Fig. 6D, S7B), we speculate that slow transcription in UV treated cells elicits a similar effect with consequent activation of Alu decoy exons and intron retention.
RNA structure of competing 3’ splice sites predicts exon skipping
We asked how RNA structure is related to whether exon inclusion or skipping is favored by slow transcription (Dujardin et al., 2014; Fong et al., 2014). Splicing of over 2000 cassette exons were significantly (FDR <0.05, >1.3-fold change) affected by slow transcription in mature mRNA, with slightly more skipping than inclusion consistent with previous work (Fong et al., 2014) (Fig. 7A, Table S5). Curiously, only a small fraction (247/2017) of the rate-sensitive splicing changes in mRNA displayed significant inclusion of alternative exons in nascent RNA (Fig. S8D, E Table S7). This observation reflects the low level of co-transcriptional splicing around alternative exons generally (Fig. 7B) and agrees with previous reports (Ameur et al., 2011; Tilgner et al., 2012) On the other hand, the low level of co-transcriptional AS that does occur among these exons was mostly concordant with the outcome in mRNA (Fig. S8D). These results beg the question, how can transcription speed affect alternative splicing of exons that are processed predominantly post-transcriptionally?
Figure 7. Post-transcriptional alternative cassette exon splicing controlled by co-transcriptional RNA folding.

A. Slow transcription stimulates cassette exon skipping and inclusion in mRNA (n=2) (Fong et al., 2014). PSI as in Fig. 6A. Red and blue points indicate rate-sensitive exons (FDR<0.05, fold change >1.3). Alternative splices unique to WT or slow pol II are not shown but are included in the n values. See Table S6.
B. Low SE of rate-sensitive alternative exons from A (blue arrow) relative to constitutive exons shows they are largely spliced post-transcriptionally.
C. Metaplots (5 base bins) of Structure Score 5’ and 3’ SS’s 1 and 4 around rate-sensitive alternative exons. Note the complementary changes in structure caused by slow transcription at splice site 4 for exons with increased and decreased inclusion in the slow pol II mutant (blue arrows). See Fig. S9.
D. Elevated RNA structure (blue arrow) at the 3’ SS downstream of a cassette exon (green arrow) with enhanced skipping when transcribed by slow pol II (Chr20:62,504,807–62,514,338). See Fig. S9.
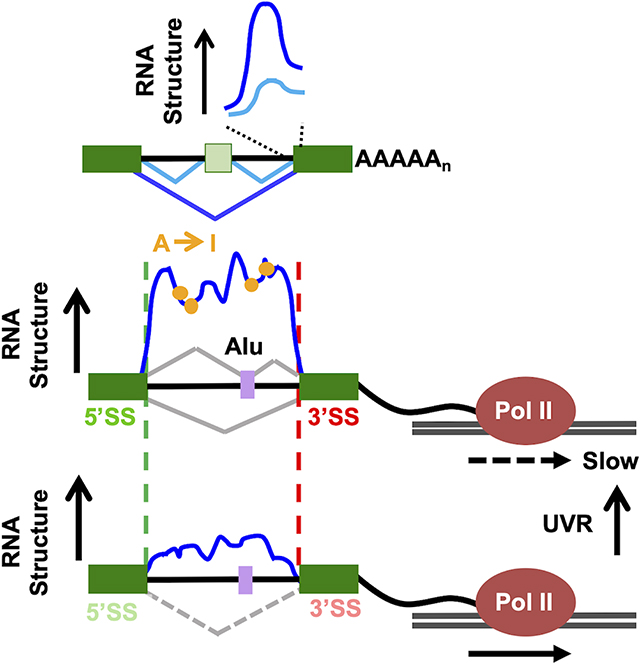
E. Model of the relation between nascent RNA structure, A-I editing, splice site strength, co-transcriptional splicing, post-transcriptional alternative splicing and transcription speed.
If nascent RNA structure helps determine the strength of splice sites, then a change in structure at a splice site might alter the competition between alternative splice sites and thereby affect the inclusion/exclusion decision. To test this prediction, we asked whether RNA structures determined by RNAse probing were affected by slow pol II at splice sites around rate-sensitive cassette exons. (The depth of our DMS MaP-seq data sets was not sufficient to provide coverage of enough alternative splice sites for this analysis). We observed no significant effects of slow transcription at the 5’splice sites that flank cassette exons themselves (Fig. S9A site 3). However, consistent with the 3’SS competition model, the structural signature at the 3’SS flanking cassette exons was strengthened, albeit not to a statistically significant extent, specifically at exons where slow transcription favored inclusion (Fig. S9A site 2). In contrast, slow transcription was associated with significant changes in the structures of the distal splice sites upstream and downstream of cassette exons (Fig. 7C sites 1 and 4). Importantly, distinct structural changes occurred depending on whether the exon is included or skipped as a result of slow transcription. At exons where slow transcription favored skipping, the step transition at proximal 5’splice site 1 was specifically enhanced, suggesting that a structurally strong 5’ splice site might favor pairing to the distal 3’ splice site 4. The most marked structural distinction between exon skipping and inclusion occurred at the distal 3’ splice site 4. Where slow pol II increased inclusion, intron structure at site 4 was modestly reduced consistent with weakening of the site and less effective competition with splice site 2 (Fig. 7C). Conversely where slow transcription increased skipping, the 3’ splice site 4 assumed a steeper profile with more intronic structure consistent with strengthening of the site and more effective competition with 3’ splice site 2 (Fig. 7C). These alternative structures are evident in metaplots (Fig. 7C) and at individual 3’ splices (Fig. 7D, S9B–D). In summary, the structural changes at 3’ splice site 4 in the slow mutant are consistent with a model where the decision between exon inclusion and skipping is determined by a competition between 3’ splice sites (Fig. 7C, sites 2 and 4) (Shao et al., 2014; Sohail and Xie, 2015) that is influenced by formation of alternative RNA structures at those sites. These results suggest that RNA structures established co-transcriptionally influence alternative splicing reactions that are only completed post-transcriptionally. Hence co-transcriptional RNA folding may regulate post-transcriptional splicing.
Discussion
We report the first global structural analysis of nascent pol II transcripts that are the substrates of most splicing reactions. The structure of nascent transcripts co-purifying with RNA pol II was interrogated by three methods that gave concordant results: in vivo DMS probing, ex vivo RNAse probing and mapping of A-I edits that are specific to folded dsRNA elements. This approach revealed common structural features of mRNA precursors that are strongly associated with the efficiency of co-transcriptional splicing. Efficient co-transcriptional splicing is associated with more structure within introns, and steep structural transitions at splice sites, particularly 3’ splice sites (Fig. 3F, G). Conversely less intron structure and flatter transitions at splice sites are associated with splicing that is less co-transcriptional. RNA structure can facilitate splicing by bringing splice sites into proximity as in Group II introns that are the ancestors of spliceosomal introns (Pyle, 2016). Nascent RNA folding could also affect splicing kinetics by modifying how splice sites, branch points, silencers and enhancers are presented to snRNPs and RBPs. Notably, we observed a correlation between predicted 3’SS strength based on primary sequence (Yeo and Burge, 2004) and RNA Structure Score based on enzymatic probing of nascent RNA (Fig. 3H). This relationship suggests that the functionality of 3’ SS sequences is determined in part by their ability to form structures that promote co-transcriptional splicing.
A major conclusion of this investigation is that the pol II nascent RNA structure-ome is highly plastic and extensively re-configured in response to altered transcription speed in agreement with observations in other systems (Pan et al., 1999; Pan and Sosnick, 2006; Wong et al., 2007). Moreover many RNA structural changes associated with slow transcription are relevant to splicing. Slow transcription amplified the structural steps at 5’ and 3’ splice sites specifically at introns where co-transcriptional splicing was enhanced (Fig. 5C–E S5C,D). The re-structuring that results from slow transcription is associated with abundant de novo A-I editing (Fig. 4C–F) consistent with the more compact folding predicted for slowly elongating transcripts (Pan and Sosnick, 2006). Transcription speed therefore has enormous potential to affect transcript function by shifting the balance between alternative RNA folding pathways. While our results suggest that altered RNA structure is a major effector, direct or indirect, of the splicing and editing changes that result from altered elongation rate, we cannot exclude other mechanisms that might also contribute to these effects.
Comparison of nascent transcripts made by WT and slow pol II also revealed alternative RNA structures related to cryptic splicing of Alu elements (Fig. 6) and alternative splicing of cassette exons, which is completed predominantly post-transcriptionally (Fig. 7). The example of cassette exon skipping versus inclusion is particularly informative. Formation or dissolution of structures at the 3’ splice site downstream of the alternative exon is associated with opposite outcomes, skipping or inclusion, that are predicted by competition between alternative 3’ splice sites (Fig. 7C, S9A). Hence formation of a strong structural signature at the downstream 3’ splice site, which is predicted to make it a strong competitor, is associated with exon skipping. Conversely unfolding of a strong structural signature is predicted to make that 3’splice site compete poorly, resulting in exon inclusion. We propose that co-transcriptional RNA folding influences post-transcriptional splicing of alternative exons through formation of alternative structures that control the strength of competing 3’ splice sites flanking cassette exons. This model can resolve the paradox that much alternative splicing is affected by transcription speed even though it usually happens after transcription is completed. The structural plasticity of the nascent transcriptome could regulate splicing, editing and other RNA modifications in response to cellular and viral elongation factors and physiological stimuli like UVR that alter transcription speed. In this context it is interesting to note that m6A deposition, which occurs co-transcriptionally, is strongly induced by UV (Xiang et al., 2017). Consistent with this idea, we found that UV, which severely slows transcription, results in inclusion of a subset Alu exons that overlaps with those where the cryptic 3’ splice site is remodeled by a slow pol II mutant (Fig. 6F). We speculate that other stimuli that influence transcription speed generally, or within specific genes, could also regulate splicing by affecting folding of nascent transcripts. In summary, these results suggest that in addition to differential RBP binding, the plastic structure of pre-mRNAs also plays a major role in splicing regulation.
Limitations
It is important to investigate the potential functional consequences for the mRNA of co-transcriptional versus post-transcriptional splicing. The approach we used distinguishes more and less structured sequences and how they can change under different conditions but it does not identify specific base-pairing interactions which could be studied in future using cross-linking approaches (Lu et al., 2016). In addition the short-read sequencing methods can not address how RNA structure influences long range coordination between splicing events in the same transcript.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Bentley (david.bentley@cuanschutz.edu)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The Sequencing Data generated during this study are available at GSE149018.
The DMS reactivity pipeline code is available at https://github.com/rnabioco/rnastruct
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Cell Lines
Flp-In-293 TREX cells (Female, Invitrogen) expressing inducible α-amanitin resistant WT and slow mutant Rpb1 (R749H) have been described (Fong et al., 2014). All experiments were performed after induction with 2.0 μg/ml doxycycline for 12–24 hr. and treatment with α-amanitin (2.5 μg/ml) for a further 42–45hr at which time all cell lines were viable and endogenous pol II is inactive.
METHOD DETAILS
tNet-MaPseq and tNet-RNAse-seq
For tNet-MaPseq WT and slow mutant expressing pol II cell lines were treated in vivo with 2% DMS or equal volume of DMSO for five minutes at 37° and stopped by addition of β-mercaptoethanol (30% final). Cells were washed twice with cold PBS and nascent RNA was isolated as described (Fong et al., 2017). Briefly, nuclei were isolated and treated with DNase I (50U/ml) for 1–1.5 hours at 4°, lysed in RIPA buffer and pol II complexes were precipitated using rabbit anti- pol II pan CTD antibody (Schroeder et al., 2000). Co-immunoprecipitated RNA was purified using Trizol, Dnased, reverse transcribed with Superscript II in the presence of 6mM MnCl2 which causes mutations at modified bases (Smola et al., 2015). The cDNA was used as input for the Kapa stranded RNAseq kit (catalogue no. KK8400) beginning at the second-strand synthesis step.
For tNet-RNAse-seq (Silverman and Gregory, 2015), WT and slow mutant pol II expressing cells were rinsed with PBS and crosslinked with 0.1% formaldehyde for 3 mins followed by quenching for 5 mins with 125mM glycine and washed twice with cold PBS. Nascent RNA was immunoprecipitated as described above with the following modifications:, 1) the RIPA buffer was supplemented with a final concentration of 0.3% SDS and 0.3% Empigen to facilitate nuclear lysis and 2) the immunoprecipitated Pol II complexes were washed four times in IP buffer (Fong et al., 2017) twice with IP wash buffer containing 500mM KCl and twice with IP wash buffer containing 500mM NaCl. While still attached to magnetic beads, the immunoprecipitated material was divided into three tubes, resuspended in 100ul RNA structure buffer (10mM Tris-HCL, pH 7.5, 100mM KCL, 10mM MgCl2) and treated with a 0.5U of RNAse I (Promega), or 0.1U of RnaseV1 (Ambion) or both Rnases at room temperature for 15mins with periodic agitation. Proteinase K was added to 1mg/ml to each reaction and incubated for 15mins at 37° and then 65 ° for 30mins to reverse crosslinks. Trizol was used to isolate nascent RNA. tNet-RNAse-seq samples were Dnased, phosphorylated using T4 polynucleotide kinase (Invitrogen) and used as input for the Lexogen small RNAseq kit (catalogue no. 058).
Semi Quantitative RT-PCR
Immunoprecipitated nascent RNA (100ng) was reverse transcribed using Superscript IV and random hexamer primers and amplified (30 Cycles) with primers (Table S8) spanning cryptic Alu exons or across well and poorly spliced introns. Products were analyzed on Agilent Tape station.
DMS reactivity calculation
Following sequencing of tNet-MaPseq libraries, duplicates were removed and adaptors trimmed using bbTools dedup and deduk functions (BBMap – Bushnell B. – sourceforge.net/projects/bbmap). Trimmed reads were mapped uniquely against the hg19 genome using hisat2 (Pertea et al., 2016). Unmapped reads were remapped using bowtie2 on the local setting. Substitutions, insertions and deletions in the DMS libraries were identified by post-processing the output of samtools mpileup (v1.9) (Li, 2011) after subtracting the background in the DMSO control with custom scripts (https://github.com/rnabioco/rnastruct) to generate mutation frequencies per nucleotide. Nucleotides were required to have a read depth of 15, and only primary alignments with MAPQ > 0 were processed. Deletions spanning greater than 4 nucleotides were not counted. Indels were left-aligned and assigned to the first nucleotide of the indel. The RNA-seq libraries were stranded and therefore the alignments were partitioned into those deriving from sense or antisense orientations prior to processing to maintain strand information. DMS reactivities were then calculated by subtracting the mutation frequencies from the untreated controls from the DMS treated samples to generate background corrected reactivity. Positions with reactivity values of less than zero were set to 0.001 and positions with reactivity greater than 0.1 were set to 0.1.
Structure Score calculation
tNet-RNAse-seq reads were trimmed and duplicates were removed. Reads were mapped in one of two ways. 1) For reads representing Structure Score calculations across splice-sites and introns, PCR duplicates were removed using bbtools (version 38.86) clumpify and adaptors trimmed using cutadapt (version 1.16)(Martin, 2011). Filtered reads were mapped uniquely against hg19 using hisat2 and unmapped reads were remapped with bowtie2 on local setting requiring a quality score of 2 or greater. Reads mapping to rRNA or mitochondria RNA were removed. 2) Reads representing Structure Scores across Alu elements were mapped with bowtie2 on local allowing multi-mapped reads to map to a single position in the genome. For both unique and multi-mapped files, strand-specific coverage values per nucleotide in RNase treated files was determined using bedtools (V2.26.0) genomecov function and only positions covered by >9 reads in either RNAse I treated or RNaseV1 treated were considered. Structure scores were calculated on per nucleotide basis as previously described (Li et al., 2012). Structure score (Si) equals normalized ds RNA seq (RNAseI resistant) coverage (dsi) (Li et al., 2012) minus ss RNAseq coverage (RNAseVI resistant) (ssi) after arsinh transformation for variance stabilization (Huber et al., 2002).
Replicates were merged to maximize read depth except where individual replicates are shown. Structure peaks were defined as regions with a Structure Score of ≥ 1.9 and an average coverage of greater than >9 reads. This stringent cutoff represents the top 3% or 2.8 standard deviations above the mean. Positions within 10bp of another structure peak were merged.
Footprint analysis and Exon junction complexes (EJCs)
tNet-RNAse-seq reads resistant to both RNAse I and RNase V1 were considered sites of potential RNA binding protein footprints. Footprints were defined as having an average coverage of greater than 15 reads and enriched 1.5-fold or more over normalized total nascent RNA coverage (three replicates combined). Normalization was done using total uniquely mapped reads from each dataset after subtraction of reads mapping to rRNA and mitochondrial sequences. The relatively low 1.5 fold enrichment over normalized input (total nascent RNA sequence coverage) was chosen as the threshold for protein footprint detection in order to be inclusive of all potential footprints that were then excluded from RNA structure analysis. Footprints within 5bp of one another were merged into a single region EJCs were defined as RNAse I and V1 resistant reads spanning a splice junction and covering greater than 15bp of the upstream exon with a depth greater than 15.
DMS reactivities overlaid on known structures
DMS reactivities were plotted onto secondary structures using the VARNA RNA secondary structure visualization tool (v3–93) (Darty et al., 2009).
Metaplots
Metaplots show mean counts or score per bin and include all regions in common between the datasets for which a minimum signal was obtained. For metaplots showing regions around splice-sites, plotted regions were required to be covered by at least 5 reads in at least 25% of the represented regions. Plotted introns were required to be covered >5% of the total length and spanning 50bp or more. Only introns greater than 200bp are shown in metaplots. P values were calculated using the R stats package v.3.6.2 using the unpaired wilcox.test() function for the counts or scores per bin and adjusted for multiple testing by the Holm method with the p.adjust() function and the −loge transformed data per bin was plotted below the metaplot. P value calculations could be inaccurate if positions within a bin that are closer to one other are more strongly correlated than sites that are more distant however this effect is expected to be small as the 5 base non-overlapping bins we used (Figs. 3F–G, 5C, 6D, 7C) are small relative to the folded structural elements being detected.
Feature Coverage method (F-cov) to measure co-transcriptional splicing efficiency
Features of co-transcriptional splicing efficiency calculation were defined as introns annotated in Refseq and supported by > 10 junction reads in mRNAseq from both WT and slow pol II GSE63375 (Fong et al., 2014). Strand-specific, mean coverage was calculated across features consisting of the last 30bp of an upstream exon, an intron and the first 30bp of the downstream exon. Mapped bam files were converted to bedfiles using bedtools bamtobed -split and coverage was calculated with bedtools coverage -mean in a strand-specific manner. Reads were only counted as covering exons if they read overlapped completely with the 30bp exon interval. For intron coverage, reads perfectly overlapping exons (annotated and cryptic – see below) and non-coding RNAs were removed and at least 10% of the read length was required to overlap the intron interval to be counted. Introns were defined by being present in both Refseq and having 10 or more junction reads (determined using subread/1.6.2 featureCounts -J option) spanning that intron in mRNAseq from both wild type and slow pol II mutant. In the case of junctions with alternative 3’ or 5’ splice sites, the intron with the highest number of junction supporting reads was selected to represent that region. Only features with introns greater than 60bp were considered. Exons were defined as being present in Refseq and having start and/or end coordinates overlapping the start and/or end of at least 10 junction reads in mRNAseq from both Rpb1 WT and Rpb1 R749H mutant cells. Only features with exons 30bp or greater were counted. To calculate splicing efficiency (SE) the following equation was used: SE = 1−[mean intron coverage/mean coverage (last 30bp of upstream exon, first 30bp of downstream exon)]. A small fraction of introns (4999/81768) had negative SE values and these were set to zero. Regions with less than 0.5 mean read depth across the intron and/or less than 3 reads mean coverage across exons were removed from the analysis. Significant differences in SE between wild type and slow was calculated using a paired t-test across three biological replicates and corrected for false discovery rate using the Benjamini-Hochberg method.
Alternative splicing analysis
Changes in alternative splicing between wild and slow mutant in mRNA and nascent RNA were calculated as a percent splicing index (PSI). Only alternative, cassette exons annotated in HEXEvent database (Busch and Hertel, 2013) were considered. For mRNA, a minimum of 10 junction reads supporting the skipping or inclusion of cassette exons in both replicates of either wild type or slow pol II and having an inclusion ratio of 0.05 or greater in either wild type or slow pol II datasets was required to consider that exon as alternatively spliced. Due to the decreased representation of splicing junctions in nascent RNA, cassette exons identified as spliced in mRNAseq were considered in nascent RNA if the feature containing the cassette exon had mean read depth across the introns >0.5 and > 3 reads mean coverage across the exon. Alternative splicing events with a PSI >0.05 in either wild type or slow pol II and having more than 3 junctions supporting exon inclusion (at least one per replicate) in either wild type or slow pol II were considered alternatively spliced co-transcriptionally. Significant changes in cassette exon usage were calculated using the R Bioconductor package edgeR (v. 3.28.1).
Cryptic Splicing analysis
Junctions were quantified in three replicates of nascent RNA from wild type and slow pol II mutant using subread/1.6.2 featureCounts -J --splitOnly --primary. Junctions spanning introns annotated in Refseq or represented at a frequency of greater than 1% in mRNA were removed and cryptic junctions were required to have consensus (GT:AG) nucleotides adjacent to splice sites. Cryptic junctions were defined from this subset as those whose 5’ or 3’ termini overlapped perfectly with the beginning or end of an intron (as defined in the F-cov method above) and the other side of the cryptic junction was located >50 bases from a constitutive 5’ or 3’ splice-site. Additionally, cryptic junctions smaller than 100bp and/or falling within a parent intron smaller than 200bp were removed. Junctions splicing in and out of a single putative cryptic exon were combined to calculate PSI values. Cryptic splicing events with a total junction count of less than 5 and/or a PSI value of less than 0.05 in both wild type and slow pol II mutant nascent RNA were filtered out. Only cryptic exons within regions considered expressed in the F-cov method described above were considered. Cryptic splicing analysis in poly(A) select RNA from control and UVR treated cells was calculated as in nascent RNA with several modifications. Due to the decreased representation of cryptic events in mRNA, cassette exon inclusion events with a PSI of greater than 0.01 were considered and events with a total junction count of less than 10 across both replicates with control and UVR treated were removed. Significant changes were calculated used R Bioconductor package edgeR as for cassette exon inclusion.
Intron retention
Intron retention in mRNA was defined at the mean coverage of the intronic region normalized to the mean coverage of 30bp of upstream and downstream flanking exon sequence. Introns were defined as regions with ten or more supporting junction reads in mRNA and present in Refseq. For mean intron coverage, regions spanning annotated alternative exons and cryptic exons identified in this study were removed prior to analysis such that only true intronic coverage was considered. Statistically significant differences in mRNA from wild type and slow pol II or control and UVR treated were calculated from two biological replicates using R Bioconductor package edgeR.
A-I Editing analysis
A-I editing was identified on three biological replicates of wild type and slow pol II nascent RNA. Reads were trimmed and mapped to hg19 using bowtie2 −local. Reads containing more than three non A/G or T/C mismatches compared to the genome were removed using an in house program. Sites of A-I editing were identified using REDItools (Picardi et al., 2015) and positions were required to have >4 reads coverage in each replicate (3 biological replicates of wild type and slow mutant), an editing frequency of >5% and having no mismatches besides ones consistent with an A-I conversion. Positions with A/G or T/C mismatches in total DNA sequencing (taken from ChIPseq libraries made from wild type or slow pol II expressing HEK293 cells) were removed. To calculate editing frequency across introns, we identified introns containing more than three edited positions (as defined above) in either wild type or slow pol II mutant. Edited positions within each intron were required to be covered by more than a total of 50 reads with an editing frequency of greater than 0.09 in either sample. Statistically significant differences were calculated using edgeR. To identify edited regions (Fig. 4F), individual edited positions within 150bp of each other were merged and edited regions were required to have 3 or more unique edited positions that exist in at least 2 of the 3 biological replicates of either WT or slow mutant pol II tNet-seq. Edited regions were required to be covered by an average read depth of > 9 after merging replicates for both the WT and slow pol II mutant data sets.
Quantification of unprocessed reads at poly(A) sites
The number of uncleaved tNET-seq reads were determined that completely overlap a 10 base region spanning cleavage sites annotated in Refseq. This value was normalized by dividing into the number of reads in the region from −1000 to −500bp upstream of the poly(A) site.
Quantification and Statistical Analysis
Statistical details including the number of replicates (n) are provided in figure legends. For metaplots of Structure Score and DMS reactivity, P values were calculated using the R stats package v.3.6.2 using the unpaired wilcox.test() function for the counts or scores per bin and adjusted for multiple testing by the Holm method with the p.adjust() function. P values for differences in mRNA levels and PSI for alternative exon inclusion were calculated using R Bioconductor package edgeR (v. 3.28.1). P values for differences in SE between wild type and slow were calculated using a paired t-test and corrected for false discovery rate using the Benjamini-Hochberg method.
Supplementary Material
Supplemental Table S1. Related to Fig. 3
Co-transcriptional splicing efficiency (SE) in HEK293 expressing WT Rpb1Amr for all expressed introns in nascent RNA.
Supplemental Table S3. Related to Fig. 4
A-I editing frequency in introns in HEK293 expressing Rpb1 WTAmr and the Rpb1 R749H slow mutant. n=3.
Supplemental Table S4. Related to Fig. 6
PSI for cryptic splicing events affected by slow mutant pol II.
Supplemental Table S5. Related to Fig. 6
PSI for cryptic splicing events affected by UV irradiation. mRNAseq data from (Williamson et al., 2017).
Supplemental Table S6. Related to Fig. 7
PSI for cassette exons affected in mRNA from wild type and slow pol II mutant.
Supplemental Table S7. Related to Fig. 7
PSI for rate sensitive cassette exons in mRNA compared with nascent RNA.
Supplemental Table S8. Related to Figures 3, 6, S3, S7.
Oligonucleotide primers used in this study.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-pan pol II CTD | Schroeder et al., 2000 | N/A |
| Critical Commercial Assays | ||
| Small RNA-seq kit | Lexogen | 058 |
| Stranded RNA-seq kit | KAPA | KK8400 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α-amanitin | SantaCruz | Sc2024405 |
| Protein A Dynabeads | ThermoFisher | 10002D |
| Dimethyl sulphate | Sigma | D186309 |
| DNAse I | NEB | M0303 |
| ProteoGuard | Takara | 635673 |
| Formaldehyde 16% methanol free | ThermoFisher | 28908 |
| RNAseVI | Ambion | N/A |
| RNAse One | Promega | M4261 |
| Model cell lines | ||
| HEK293 Flp-in T-Rex pcDNA5 Rpb1 αAmr WT | Fong et al 2014 | N/A |
| HEK293 Flp-in T-Rex pcDNA5 Rpb1 αAmr R749H | Fong et al 2014 | N/A |
| Oligonucleotides | ||
| See Table S8 for primer sequences | ||
| Software and Algorithms | ||
| R version 4.0.2 | https://www.r-project.org/ | |
| Python version 3.7 | https://www.python.org | |
| Bowtie2 version 2.3.2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Hisat2 version 2.1.0 | Pertea et al., 2016 | https://daehwankimlab.github.io/hisat2/ |
| BBmap version 38.86 | https://sourceforge.net/projects/bbmap/ | |
| REDI tools | Picardi et al., 2015 | https://github.com/BioinfoUNIBA/REDItools |
| Cutadapt version 1.16 | Martin M, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| Picard Tools version 2.18.7 | https://broadinstitute.github.io/picard/ | |
| Samtools version 1.9 | Li et al., 2009 | http://www.htslib.org/ |
| DMS reactivity calling | https://github.com/rnabioco/rnastruct | |
| VARNA (v3–93) | Darty et al., 2009 | http://varna.lri.fr/index.php?lang=en&page=downloads&css=varna |
Highlights.
The nascent RNA structureome shows structures that predict co-transcriptional splicing
Nascent RNA structure is extensively remodeled in response to slow transcription
Co-transcriptional RNA folding affects post-transcriptional alternative splicing
Nascent RNA structures are highly plastic with numerous effects on splicing and editing
Acknowledgments:
We thank R. Zhao, O. Rissland, T. Blumenthal, N. Mukherjee, M. Taliaferro, M. Johnston, K. Conklin and H. Shenasa for helpful suggestions, and A. Hofler for technical help. We thank K. Diener, T. Shade and B. Gao and the UC Denver Sequencing facility for sequencing. D.B thanks A. Cohan and I. Pretty for their hospitality.
Funding: T.S. was supported by an American Cancer Society fellowship PF-15-188-01-RMC. K. R is supported by the UC Denver RNA Bioscience Initiative. This work was supported by NIH grant R35GM118051 to D.B.
Footnotes
Competing interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, and Feuk L (2011). Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat Struct Mol Biol 18, 1435–1440. [DOI] [PubMed] [Google Scholar]
- Aslanzadeh V, Huang Y, Sanguinetti G, and Beggs JD (2018). Transcription rate strongly affects splicing fidelity and cotranscriptionality in budding yeast. Genome Research 28, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. (2014). A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer AL, and Osheim YN (1988). Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev 2, 754–765. [DOI] [PubMed] [Google Scholar]
- Braberg H, Jin H, Moehle EA, Chan YA, Wang S, Shales M, Benschop JJ, Morris JH, Qiu C, Hu F, et al. (2013). From Structure to Systems: High-Resolution, Quantitative Genetic Analysis of RNA Polymerase II. Cell 154, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, and Baralle FE (2004). Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol 24, 10505–10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, and Hertel KJ (2013). HEXEvent: a database of Human EXon splicing Events. Nucleic Acids Res 41, D118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo Oesterreich F, Preibisch S, and Neugebauer KM (2010). Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol Cell 40, 571–581. [DOI] [PubMed] [Google Scholar]
- Charpentier B, and Rosbash M (1996). Intramolecular structure in yeast introns aids the early steps of in vitro spliceosome assembly. RNA 2, 509–522. [PMC free article] [PubMed] [Google Scholar]
- Chen W, Moore J, Ozadam H, Shulha HP, Rhind N, Weng Z, and Moore MJ (2018). Transcriptome-wide Interrogation of the Functional Intronome by Spliceosome Profiling. Cell 173, 1031–1037.e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K, Denise A, and Ponty Y (2009). VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25, 1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, and Kornblihtt AR (2003). A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12, 525–532. [DOI] [PubMed] [Google Scholar]
- Drexler HL, Choquet K, and Churchman LS (2020). Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nanopores. Molecular Cell 77, 985–998.e988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin G, Lafaille C, de la Mata M, Marasco LE, Munoz MJ, Le Jossic-Corcos C, Corcos L, and Kornblihtt AR (2014). How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell 54, 683–690. [DOI] [PubMed] [Google Scholar]
- Eggington JM, Greene T, and Bass BL (2011). Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eperon LP, Graham IR, Griffiths AD, and Eperon IC (1988). Effects of RNA secondary structure on alternative splicing of pre-mRNA: is folding limited to a region behind the transcribing RNA polymerase? Cell 54, 393–401. [DOI] [PubMed] [Google Scholar]
- Fededa JP, Petrillo E, Gelfand MS, Neverov AD, Kadener S, Nogues G, Pelisch F, Baralle FE, Muro AF, and Kornblihtt AR (2005). A polar mechanism coordinates different regions of alternative splicing within a single gene. Mol Cell 19, 393–404. [DOI] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, and Bentley DL (2014). Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev 28, 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Saldi T, Sheridan RM, Cortazar MA, and Bentley DL (2017). RNA Pol II Dynamics Modulate Co-transcriptional Chromatin Modification, CTD Phosphorylation, and Transcriptional Direction. Mol Cell 66, 546–557 e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Mark N, Schwartz S, and Ast G (2008). Alternative splicing of Alu exons--two arms are better than one. Nucleic Acids Research 36, 2012–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goguel V, and Rosbash M (1993). Splice site choice and splicing efficiency are positively influenced by pre-mRNA intramolecular base pairing in yeast. Cell 72, 893–901. [DOI] [PubMed] [Google Scholar]
- Gosai SJ, Foley SW, Wang D, Silverman IM, Selamoglu N, Nelson AD, Beilstein MA, Daldal F, Deal RB, and Gregory BD (2015). Global analysis of the RNA-protein interaction and RNA secondary structure landscapes of the Arabidopsis nucleus. Mol Cell 57, 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel L, Straube K, and Neugebauer KM (2018). Long-read sequencing of nascent RNA reveals coupling among RNA processing events. Genome Research 28, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KJ, and Ares M (1997). Intron self-complementarity enforces exon inclusion in a yeast pre-mRNA. Proceedings of the National Academy of Sciences of the United States of America 94, 12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KJ, Kane CM, and Ares M Jr. (2003). Perturbation of transcription elongation influences the fidelity of internal exon inclusion in Saccharomyces cerevisiae. RNA 9, 993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, von Heydebreck A, Sultmann H, Poustka A, and Vingron M (2002). Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1, S96–104. [DOI] [PubMed] [Google Scholar]
- Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, and Blencowe BJ (2011). Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Research 21, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, and Lis JT (2014). Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3, e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodor YL, Rodriguez J, Abruzzi KC, Tang C-HA, Marr MT, and Rosbash M (2011). Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev 25, 2502–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Taggart AJ, Heintzelman C, Cygan KJ, Hull CG, Wang J, Shrestha B, and Fairbrother WG (2017). Widespread intra-dependencies in the removal of introns from human transcripts. Nucleic Acids Research 45, 9503–9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Zheng Q, Vandivier LE, Willmann MR, Chen Y, and Gregory BD (2012). Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant Cell 24, 4346–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-L, Taggart AJ, Lim KH, Cygan KJ, Ferraris L, Creton R, Huang Y-T, and Fairbrother WG (2016). RNA structure replaces the need for U2AF2 in splicing. Genome Research 26, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, et al. (2016). RNA Duplex Map in Living Cells Reveals Higher- Order Transcriptome Structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 3. [Google Scholar]
- McManus CJ, and Graveley BR (2011). RNA structure and the mechanisms of alternative splicing. Current Opinion in Genetics & Development 21, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Plass M, Perez-Valle J, Eyras E, and Vilardell J (2011). Deciphering 3’ss selection in the yeast genome reveals an RNA thermosensor that mediates alternative splicing. Mol Cell 43, 1033–1039. [DOI] [PubMed] [Google Scholar]
- Munoz MJ, Perez Santangelo MS, Paronetto MP, de la Mata M, Pelisch F, Boireau S, Glover-Cutter K, Ben-Dov C, Blaustein M, Lozano JJ, et al. (2009). DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 137, 708–720. [DOI] [PubMed] [Google Scholar]
- Mustoe AM, Lama NN, Irving PS, Olson SW, and Weeks KM (2019). RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proc Natl Acad Sci U S A 116, 24574–24582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer KM (2019). Nascent RNA and the Coordination of Splicing with Transcription. Cold Spring Harbor Perspectives in Biology 11, a032227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterreich FC, Herzel L, Straube K, Hujer K, Howard J, and Neugebauer KM (2016). Splicing of Nascent RNA Coincides with Intron Exit from RNA Polymerase II. Cell 165, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Artsimovitch I, Fang XW, Landick R, and Sosnick TR (1999). Folding of a large ribozyme during transcription and the effect of the elongation factor NusA. Proc Natl Acad Sci U S A 96, 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, and Sosnick T (2006). RNA folding during transcription. Annu Rev Biophys Biomol Struct 35, 161–175. [DOI] [PubMed] [Google Scholar]
- Parra M, Booth BW, Weiszmann R, Yee B, Yeo GW, Brown JB, Celniker SE, and Conboy JG (2018). An important class of intron retention events in human erythroblasts is regulated by cryptic exons proposed to function as splicing decoys. RNA 24, 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, and Salzberg SL (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervouchine DD, Khrameeva EE, Pichugina MY, Nikolaienko OV, Gelfand MS, Rubtsov PM, and Mironov AA (2012). Evidence for widespread association of mammalian splicing and conserved long-range RNA structures. RNA 18, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, D’Erchia AM, Montalvo A, and Pesole G (2015). Using REDItools to Detect RNA Editing Events in NGS Datasets. Current protocols in bioinformatics / editoral board, Baxevanis Andreas D. … [et al. ] 49, 12.12.11–12.12.15. [DOI] [PubMed] [Google Scholar]
- Picardi E, and Pesole G (2013). REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814. [DOI] [PubMed] [Google Scholar]
- Pyle AM (2016). Group II Intron Self-Splicing. Annu Rev Biophys 45, 183–205. [DOI] [PubMed] [Google Scholar]
- Raker VA, Mironov AA, Gelfand MS, and Pervouchine DD (2009). Modulation of alternative splicing by long-range RNA structures in Drosophila. Nucleic Acids Research 37, 4533–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Menet JS, and Rosbash M (2012). Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell 47, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogic S, Montpetit B, Hoos HH, Mackworth AK, Ouellette BF, and Hieter P (2008). Correlation between the secondary structure of pre-mRNA introns and the efficiency of splicing in Saccharomyces cerevisiae. BMC Genomics 9, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, and Weissman JS (2015). Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo Nature 505, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K, England W, Fernandez MM, Biswas T, Spitale RC, and Ghosh G (2020). Structural disruption of exonic stem-loops immediately upstream of the intron regulates mammalian splicing. Nucleic Acids Res, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldi T, Fong N, and Bentley DL (2018). Transcription elongation rate affects nascent histone pre-mRNA folding and 3’ end processing. Genes & Development 32, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder SC, Schwer B, Shuman S, and Bentley D (2000). Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev 14, 2435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Gal-Mark N, Kfir N, Oren R, Kim E, and Ast G (2009). Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput Biol 5, e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao C, Yang B, Wu T, Huang J, Tang P, Zhou Y, Zhou J, Qiu J, Jiang L, Li H, et al. (2014). Mechanisms for U2AF to define 3′ splice sites and regulate alternative splicing in the human genome. Nature Structural & Molecular Biology 21, 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, and Hertel KJ (2008). Conserved RNA secondary structures promote alternative splicing. RNA 14, 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan RM, Fong N, D’Alessandro A, and Bentley DL (2019). Widespread Backtracking by RNA Pol II Is a Major Effector of Gene Activation, 5′ Pause Release, Termination, and Transcription Elongation Rate. Molecular Cell 73, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman IM, and Gregory BD (2015). Transcriptome-wide ribonuclease-mediated protein footprinting to identify RNA protein interaction sites. Methods 72, 76–85. [DOI] [PubMed] [Google Scholar]
- Smola MJ, Rice GM, Busan S, Siegfried NA, and Weeks KM (2015). Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc 10, 1643–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail M, and Xie J (2015). Diverse regulation of 3′ splice site usage. Cellular and Molecular Life Sciences 72, 4771–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnick D (1985). Alternative splicing caused by RNA secondary structure. Cell 43, 667–676. [DOI] [PubMed] [Google Scholar]
- Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, Huang W, Kool ET, Chang HY, and Zhang QC (2019). RNA structure maps across mammalian cellular compartments. Nature Structural & Molecular Biology 26, 322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y, Ohtsuka T, Gonzalez A, Miyachi H, and Kageyama R (2011). Intronic delay is essential for oscillatory expression in the segmentation clock. Proc Natl Acad Sci U S A 108, 3300–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro JM, Lambert NJ, Sudmant PH, Dominguez D, Merkin JJ, Alexis MS, Bazile C, and Burge CB (2016). RNA Sequence Context Effects Measured In Vitro Predict In Vivo Protein Binding and Regulation. Mol Cell 64, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR, and Guigo R (2012). Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res 22, 1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomezsko PJ, Corbin VDA, Gupta P, Swaminathan H, Glasgow M, Persad S, Edwards MD, McIntosh L, Papenfuss AT, Emery A, et al. (2020). Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 582, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas DY, Shah K, Batish M, Levandoski M, Sinha S, Marras SA, Schedl P, and Tyagi S (2011). Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell 147, 1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, and Berglund JA (2010). Role of RNA structure in regulating pre-mRNA splicing. Trends in Biochemical Sciences 35, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson L, Saponaro M, Boeing S, East P, Mitter R, Kantidakis T, Kelly GP, Lobley A, Walker J, Spencer-Dene B, et al. (2017). UV Irradiation Induces a Non-coding RNA that Functionally Opposes the Protein Encoded by the Same Gene. Cell 168, 843–855.e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TN, Sosnick TR, and Pan T (2007). Folding of noncoding RNAs during transcription facilitated by pausing-induced nonnative structures. Proc Natl Acad Sci U S A 104, 17995–18000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Laurent B, Hsu C-H, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al. (2017). RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, and Mori K (2001). XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 107, 881–891. [DOI] [PubMed] [Google Scholar]
- Zafrir Z, and Tuller T (2015). Nucleotide sequence composition adjacent to intronic splice sites improves splicing efficiency via its effect on pre-mRNA local folding in fungi. RNA 21, 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnack K, König J, Tajnik M, Martincorena I, Eustermann S, Stevant I, Reyes A, Anders S, Luscombe Nicholas M., and Ule J (2013). Direct Competition between hnRNP C and U2AF65 Protects the Transcriptome from the Exonization of Alu Elements. Cell 152, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Landick R (2016). A Two-Way Street: Regulatory Interplay between RNA Polymerase and Nascent RNA Structure. Trends Biochem Sci 41, 293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, and Rouskin S (2017). DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature methods 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. Related to Fig. 3
Co-transcriptional splicing efficiency (SE) in HEK293 expressing WT Rpb1Amr for all expressed introns in nascent RNA.
Supplemental Table S3. Related to Fig. 4
A-I editing frequency in introns in HEK293 expressing Rpb1 WTAmr and the Rpb1 R749H slow mutant. n=3.
Supplemental Table S4. Related to Fig. 6
PSI for cryptic splicing events affected by slow mutant pol II.
Supplemental Table S5. Related to Fig. 6
PSI for cryptic splicing events affected by UV irradiation. mRNAseq data from (Williamson et al., 2017).
Supplemental Table S6. Related to Fig. 7
PSI for cassette exons affected in mRNA from wild type and slow pol II mutant.
Supplemental Table S7. Related to Fig. 7
PSI for rate sensitive cassette exons in mRNA compared with nascent RNA.
Supplemental Table S8. Related to Figures 3, 6, S3, S7.
Oligonucleotide primers used in this study.
Data Availability Statement
The Sequencing Data generated during this study are available at GSE149018.
The DMS reactivity pipeline code is available at https://github.com/rnabioco/rnastruct