

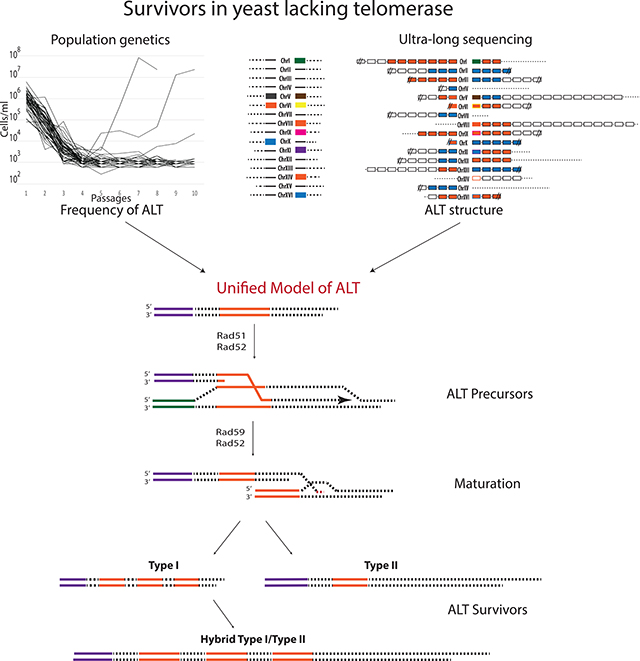
SUMMARY
Alternative lengthening of telomeres (ALT) is a recombination process that maintains telomeres in the absence of telomerase and helps cancer cells to survive. Yeast has been used as a robust model of ALT; however, the inability to determine the frequency and structure of ALT survivors hindered the understanding of ALT mechanism. Here, using populational and molecular genetics approaches we overcome these problems and demonstrate that, contrary to the current view, both RAD51-dependent and -independent mechanisms are required for a unified ALT survivor pathway. This conclusion is based on the calculation of ALT frequencies as well as on ultra-long sequencing of ALT products that revealed “hybrid” sequences containing features attributed to both recombination pathways. Sequencing of ALT intermediates demonstrate that recombination begins with Rad51-mediated strand invasion to form DNA substrates that are matured by a Rad51-independent ssDNA annealing pathway. A similar unified ALT pathway may operate in other organisms including humans.
Keywords: Alternative lengthening of telomeres (ALT), recombination, Rad51, Rad59, yeast, ultra-long sequencing, break-induced replication
IN BRIEF
Alternative lengthening of telomeres (ALT) was believed to proceed in yeast via two separate pathways. Kockler et al. combined population genetic principles and ultra-long sequencing to determine the frequency and molecular structure identity of ALT survivors, which revealed a unified pathway that includes both Rad51-dependent and -independent steps.
Graphical Abstract
INTRODUCTION
Telomeres are essential to maintain genomic integrity by protecting chromosome ends. In the absence of telomerase, telomeres progressively shorten with every round of replication until a minimum length is reached, leading to replicative senescence. Alternative lengthening of telomeres (ALT) is a pathway that, in the absence of telomerase, maintains eukaryotic telomeres by recombination (Neumann and Reddel, 2002), which is utilized by ~10–15% of cancers (reviewed in (Cesare and Reddel, 2010; Dilley and Greenberg, 2015)). Further, treatment of telomerase-positive cancers with anti-telomerase drugs has been shown to activate ALT, thus promoting cancer relapse (Hu et al., 2012). The significant role that ALT plays in human disease makes it important to understand how ALT is initiated, established, and maintained. However, current human cell models only allow the study of ALT maintenance, while ALT establishment remains obscure.
A great model to study the establishment of ALT from the beginning of telomere erosion all the way through ALT survivor formation is yeast Saccharomyces cerevisiae where ALT was originally discovered. (Lundblad and Blackburn, 1993). Yeast telomeres contain irregular (TG1–3)n short telomere repeats that are ~200–300 bp in length (reviewed in (McEachern and Haber, 2006; Wellinger and Zakian, 2012)). In addition to telomeres, many chromosome ends contain sub-telomere regions (Louis and Haber, 1990a, b), called Y’ elements that generally are separated into two types based on their sequence length (short 5.2 kb and long 6.7 kb Y’ elements) and can also participate in the protection of chromosome ends. In telomerase-deficient cells, telomeres gradually shorten until they can no longer provide protection from a DNA damage response, followed by replicative senescence. A rare minority of yeast cells escape senescence by stabilizing their telomeres through a recombination-driven pathway (Lundblad and Blackburn, 1993) that is similar to ALT in humans (reviewed in (McEachern and Haber, 2006)), which made yeast a robust model of ALT. Yet, the inability to determine the molecular structure of ALT chromosomes and to estimate ALT frequency has hindered the understanding of the ALT mechanism.
The structure of ALT chromosomes have been analyzed by Southern blot analysis producing an overall representation of all chromosomal ends at once (Chen et al., 2001; Le et al., 1999; Lundblad and Blackburn, 1993; Teng et al., 2000; Teng and Zakian, 1999), also a very small subset of ALT survivor chromosomes have been Sanger sequenced (Chang et al., 2007; Chang et al., 2011; Churikov et al., 2014). Together, this led to the general categorization of ALT surviving yeast cells into two types: Type I, that was proposed to stabilize chromosome ends by multiplying Y’ elements with telomeres remaining short (Lundblad and Blackburn, 1993; Teng and Zakian, 1999), and Type II, proposed to stabilize their ends by long telomeres (Chen et al., 2001; Le et al., 1999; Lundblad and Blackburn, 1993; Teng et al., 2000; Teng and Zakian, 1999). However, the knowledge obtained by these methods might be insufficient to fully unravel the mechanism of ALT, which requires the analysis of the features of individual ALT telomere structures. Sequencing of individual ALT telomeres has not been possible because short-read sequencing techniques do not allow sequence reads containing telomere, or Y’ regions, to be mapped to individual chromosome ends. Therefore, to accurately assess individual telomere structures requires the application of a whole genome ultra-long sequencing approach.
Current models of ALT are based on its genetic analysis. The strongest conclusions were made based on rad52Δ and pol32Δ mutants eliminating both Type I and Type II ALT outcomes (Lundblad and Blackburn, 1993; Lydeard et al., 2007). In particular, the RAD52 requirement (Lundblad and Blackburn, 1993) indicated the involvement of homologous recombination (HR), while the requirement for POL32 (Lydeard et al., 2007) suggested that this HR proceeds through break-induced replication (BIR). However, other genes have been reported to participate in only one of the ALT types. For example, strains containing deletion of RAD51, which encodes the main DNA strand invasion protein, formed only Type II survivors (Chen et al., 2001; Le et al., 1999; Teng et al., 2000). This led to the conclusion that these genes are required specifically for the formation of Type I ALT outcomes, but not for Type II. Conversely, deletion of RAD59 (Chen et al., 2001), SGS1 (Huang et al., 2001; Johnson et al., 2001), or RAD50 (Le et al., 1999; Teng et al., 2000) resulted in formation of only Type I survivors, which suggested that these genes were specifically required for Type II formation. Moreover, the double mutants rad51Δrad59Δ or rad51Δrad50Δ did not produce ALT survivors (Chen et al., 2001; Le et al., 1999), which led to the conclusion that Type I and Type II pathways not only have different genetic requirements, but also proceed independently of each other.
However, because the frequency of ALT events is not known, alternative explanations of these data are possible. For example, genes that were proposed to affect a single pathway could affect both, albeit to different extents, which could result in the predominance of one type of survivor. Another possibility is that some of these deletions could in fact stimulate a particular pathway, which would result in the predominance of a single survivor type. Distinguishing between these possibilities requires the precise estimation and comparison of ALT frequencies among mutants, which has been deemed impossible because of the stochastic nature of ALT (reviewed in (Wellinger and Zakian, 2012)). Together, the lack of quantitative analyses and the inability to study the structures of individual chromosome ends in ALT survivors has limited our understanding of the ALT mechanism.
Here, we describe a novel approach that is not hindered by, but actually exploits, ALT’s stochastic nature to determine the frequency of independent ALT survivors in multiple genetic backgrounds. Additionally, we applied ultra-long sequencing methodologies to determine the detailed structure of individual chromosomal ends in ALT survivors. Our data, from these analyses, support a novel model where ALT proceeds through a unified pathway that first relies upon RAD51-mediated strand invasion and an efficient DNA damage checkpoint to form precursors that are then matured by a RAD59-dependent pathway to stabilize telomeres. Structural analysis by ultra-long sequencing of the resulting chromosome structures revealed the presence of a “hybrid” Type I/Type II structure that supported the sequential steps of our described unified pathway. In sum, our work establishes a new paradigm of a unified ALT pathway and describes new approaches that may be employed for exploring ALT in various organisms, including humans.
RESULTS
The frequency of ALT survivors in yeast cultures can be determined.
Previous studies of ALT survivor formation involved serial passaging of tlc1Δ cells in liquid culture at high initial density (often ~105 cells per ml), which always produced at least one, and likely more than one, ALT survivors in each experiment (Chen et al., 2001; Le et al., 1999; Lydeard et al., 2007; Teng et al., 2000; Teng and Zakian, 1999) (Figure 1A). Instead, we propose that the frequency of ALT could be directly estimated using Poisson statistics when only the minority of cultures (~5%) produce a survivor. To establish such experimental conditions, we isolated tlc1Δ mutants by tetrad dissection of TLC1/tlc1Δ heterozygote diploids, and we performed multiple serial passing experiments each with a different number of cells transferred, with initial cell densities ranging from 250 to 100,000 cells/ml (STAR Methods)) (Figures 1A, 1B, S1A, and S1B). We determined that transferring ~250 cells/ml resulted in 3 out of 53 (5.6%) cultures forming ALT survivors, which ensured that each survivor resulted from a single event and allowed us to calculate the frequency of ALT using Poisson statistics (Figure 1B and 1C; Table S1). To reduce the number of experiments needed to generate statistically reliable data, we next simplified this approach such that cells were passaged in liquid twice to allow cells to enter senescence (STAR Methods) and subsequently plated to monitor formation of isogenic ALT survivor colonies (Figure 1C, S2). Using this approach, we calculated ALT survivor frequency as 2×10−5, which was similar to what we observed with liquid cultures but with much tighter confidence intervals (Figure 1C; Tables S1 and S2). Thus, our experiments have yielded the ALT survivor frequency in yeast cultures, and this approach can be used to analyze the genetic control of ALT formation quantitatively.
Figure 1. Deletions of recombination or checkpoint genes decrease ALT frequency.

(A) ALT survivors form in every tlc1Δ culture when large populations (105 cells/ml) are passaged in liquid cultures (dissection and passaging scheme above).
(B) ALT survivors form rarely when small populations (250 cells/ml) are passaged allowing use of Poisson statistics for ALT frequency calculation.
(C) Effect of deletions of recombination, checkpoint, and telomere capping genes on the frequency of ALT in tlc1Δ cells. tlc1Δ Liquid was calculated as described in (B). For all other cultures the modified passaging scheme with plating cells after second passage was used. Medians of ALT frequencies and 95% confidence intervals (in parenthesis) are shown. The arrows and folds change on the top represent reduction (or increase) of ALT frequency as compared to tlc1Δ.
Rad51, Rad59, and DNA damage checkpoints cooperate in a unified ALT pathway
The current literature supports the existence of two independent ALT pathways that yield phenotypically distinct outcomes (Chen et al., 2001; Hu et al., 2013; Le et al., 1999; Lundblad and Blackburn, 1993; McEachern and Haber, 2006; Teng et al., 2000; Teng and Zakian, 1999; Wellinger and Zakian, 2012). If this were accurate, then the sum of the frequencies of Type I plus Type II ALT outcomes should closely approximate the overall frequency of ALT survivor formation. To test this, we established cultures of RAD51- or RAD59-deleted tlc1Δ yeast strains, which should eliminate Type I or Type II ALT survivor formation, respectively (Chen et al., 2001; Le et al., 1999; Lundblad and Blackburn, 1993; Teng et al., 2000; Teng and Zakian, 1999). Deletion of either RAD51 or RAD59 markedly reduced the formation of ALT survivors (Figure 1C; Tables S1 and S2). ALT survivor formation was reduced by 18-fold in tlc1Δrad51Δ (1.2×10−6) versus tlc1ΔRAD51, and a 15-fold reduction was observed in tlc1Δ rad59Δ (1.4×10−6) versus tlc1ΔRAD59, making the sum of these two frequencies (2.6×10−6) significantly lower than the frequency observed in tlc1Δ (2.0×10−5) (Figure 1C; Table S2). These results strongly suggest that both Type I and Type II ALT survivor formation are markedly inhibited upon loss of either RAD59 or RAD51.
Based on this observation, we postulate that all ALT survivors may result from a unified pathway that requires both Rad51 and Rad59 at its various steps. Support for a common Rad51-mediated step in the formation of all ALT survivors came from the analysis of tlc1Δsrs2Δ strains, where we observed a complete elimination of ALT survivor formation (Figure 1C; Table S2). Because Srs2 deficiency was shown to induce death in yeast after Rad51-mediated strand invasion (Elango et al., 2017; Niu and Klein, 2017), we hypothesized that the absence of ALT survivors in tlc1Δsrs2Δ might also result from strand invasion. Consistently, deletion of RAD51 restored ALT survivor formation in tlc1Δsrs2Δ cells to the level of tlc1Δrad51ΔSRS2 cells (Figure 1C; Table S2), similar to (Elango et al., 2017; Liu et al., 2011). Further support came from the partial rescue of ALT survivor frequency in tlc1Δsrs2Δ cells by concurrent deletion of RAD55, which encodes a protein that stabilizes Rad51 filaments (Heyer, 2015) (Figure 1C; Table S2).
It has been shown that Srs2-deficient cells are also unable to adapt to, or recover from, DNA damage-induced cell cycle arrest (Vaze et al., 2002). Thus, to confirm that the senescence we observed in tlc1Δsrs2Δ strains was related to the ALT pathway versus a cell cycle arrest recovery defect (Harrison and Haber, 2006), we established isogenic rad24Δ or rad9Δ strains. Based on our data, genetic inactivation of the DNA damage checkpoint by either RAD24 or RAD9 deletion in tlc1Δsrs2Δ cells did not restore ALT survivor formation (Figure 1C; Table S2), supporting a critical role for Srs2 to process Rad51-mediated strand invasion during ALT formation. Strikingly, though we observed that both tlc1Δrad24Δ and tlc1Δrad9Δ strains had significantly decreased frequency of ALT survivor formation, as compared to tlc1Δ cells (Figure 1C; Table S2). This suggests a critical role for functional DNA damage checkpoints in the ALT pathway that is likely due to the need for cells to stop the cell cycle to repair eroded telomeres. Finally, we tested the effect of rif1Δrif2Δ double mutation that has been previously proposed to stimulate ALT due to its effect on telomere capping or on interactions with other proteins (Dewar and Lydall, 2012; Graf et al., 2017; Levy and Blackburn, 2004; Ribeyre and Shore, 2012; Teixeira et al., 2004) and observed a ~17-fold increase of ALT frequency (Figure 1C; Table S2). Together, we propose the existence of a unified ALT pathway where both Rad51 and Rad59, as well as DNA damage checkpoint proteins participate in the survivor formation.
The formation of early precursors of ALT requires HR and DNA damage checkpoints.
Our results suggested the existence of a common, Rad51-dependent HR step in the history of all ALT survivors. To identify the timing of this Rad51-dependent step, we started with the calculation of the number of population doublings (PD) that are required to reach senescence, defined by the point when the culture fails to double, and found that HR deficient cells (rad51Δtlc1Δ and rad52Δtlc1Δ) senesced earlier than tlc1Δ (PD27 versus PD38, respectively, (Figure 2A)), consistent with (Chen et al., 2001; Le et al., 1999; Lundblad and Blackburn, 1993; Teng et al., 2000; Teng and Zakian, 1999; Xu et al., 2015). This indicated that the Rad51-dependent HR step functions early, and we hypothesized that it occurs during telomere erosion. To gain insight, we first computationally modeled populations of cells undergoing telomere erosion. Our model begins with a distribution of telomere lengths (average 275bp, STAR Methods) that progressively shorten with every cell division until at least one telomere in a cell reaches the length that triggers senescence (Ls) that we determined to be 70bp (STAR Methods) (Figure 2B). Knowing that HR deficient cells become senescent at PD27 (Figure 2A, S3) we modeled telomere erosion in HR-defective mutants (Figures 2B–2D), which allowed us to determine the rate of telomere erosion in the absence of HR repair as ~6 bp per division (Figures 2C, 2D, and S4A) (STAR Methods). Previously, (Fallet et al., 2014; Marcand et al., 1999; Singer and Gottschling, 1994) studied telomere erosion in HR-proficient cultures, and reported a slower erosion rate of ~3–4 bp /div. We hypothesized that the reason for the difference from our results was that HR occurring in their cells counteracted telomere erosion. To account for this effect of HR on telomere length distribution, we modified our model such that telomere erosion continues until at least one telomere in a cell was shortened to a length (Lr; always Lr ≥ Ls) that stopped the cell cycle and promoted HR with a donor telomere, or Y’ pre-telomeric region (Figure 3A). In the case the donor telomere is longer than the Lr, the recipient telomere will gain a sufficient length to resume the cell cycle (Figure 3A). The modeling was performed for a set of Lr values including 75bp, a length just slightly longer than Ls, and also Lr=90 (Figure S4B). Modeling with Lr=75 bp predicted that activation of HR at telomeres delays senescence (from PD27 to ~PD41) (compare Figure 3B to Figure 2C, D). The prediction of the HR delaying senescence was also obtained for Lr=90bp (Figure S4B). Additionally, the model predicted that a telltale of HR would be a distribution of telomere lengths (which was symmetric before telomere erosion began) to become asymmetric or skewed (with a tail of long telomeres describing a positive skew or right-tailed distribution) by PD27 (Figure 3B and 3C).
Figure 2. Dynamics of telomere erosion and onset of senescence in tlc1Δ strains.

(A) Scatter plot of the number of Population Doublings (PD) that occur for the culture to become senescent (defined by no further cell divisions). # indicates a statistically significant difference of the PD as compared to tlc1Δ (Mann-Whitney, test P=<0.0001).
(B-D) Computational modeling of telomere erosion (modeling telomere dynamics in HR deficient cells following elimination of telomerase).
(B) Schematic for model of telomere erosion. (ITS= interstitial telomere sequence, Y’= Y’ region).
(C) Modeling the decrease of the 10th percentile of telomere length at various erosion rates.
(D) Modeling the entire distribution of telomere lengths with telomere erosion at 6bp/division (div). The time of senescence was defined by the 10th percentile line crossing the Ls threshold.
Figure 3. ALT Precursor formation requires Rad51 and DNA damage checkpoints.

(A-C) Computational modeling of dynamics of telomere erosion and HR repair.
(A) Schematic for model of telomere erosion with recombination at homology, erosion rate=6bp/div., Ls=70, and Lr=75.
(B) The entire distribution of telomere lengths resulting, from the model, with telomere erosion at 6bp/div., Ls=70, and Lr =75. See legend to Figure 2D for other details.
(C) Comparison of skewness of telomere length distribution in the presence or absence of recombination for the modeling shown in (B) and in Figure 2D.
(D) Illustration of chromosome end structure with XhoI cut site within the sub-telomere Y’. Representative telomere length distribution in tlc1Δ (HR proficient strain) analyzed by Southern blot analysis with XhoI digestion and hybridization with Y’ specific probe.
(E) Distribution of telomere lengths in several samples shown in (D) with population doubling (PD) and Pearson’s moment of skewness (PMs) indicated.
(F) The effect of rad52Δ (P=<0.0001), rad51Δ (P=0.0016), and rad59Δ (NS) on the skewness of telomere length distribution at PD27, # indicates statistically significant difference of PMs from tlc1Δ; Solid line indicate median skewness; dashed line indicates PMs=0.
(G) Similar to (F), but for the effect of rad59Δ (NS) and rad24Δ (P=0.043) on the skewness of telomere length distribution at PD38, # statistically significant difference of PMs from tlc1Δ. All statistical comparisons are performed by using Mann-Whitney test.
(See also Figure S4)
To test this prediction experimentally, the distribution of telomere lengths was assessed in HR-proficient tlc1Δ cultures by Southern blot that was analyzed by Pearson’s moment of skewness (PMs) (Figure 3D). As predicted, the distribution of telomeres was symmetric in the initial cultures (see example in Figure 3E, where PMs = −0.065 at PD0) but became increasingly skewed by PD27 and PD38 (an example in Figure 3E, PMs = 0.216 and PMs = 0.407, respectively; see also Figures 3F, 3G). Also, as predicted, the distribution remained symmetric in rad51Δtlc1Δ and in rad52Δtlc1Δ cultures until their senescence (Figure 3F), consistent with the notion that asymmetry reflects the formation and dynamics of HR repair intermediates leading to an extension of eroded telomeres. Notably, this asymmetry was much less pronounced in checkpoint-defective tlc1Δrad24Δ mutants, pointing out the critical role of DNA damage-induced cell cycle arrest for the formation of HR repair telomere intermediates (Figure 3G). As predicted, checkpoint deficiency also significantly reduced the Ls, while other mutations that we found to affect ALT frequency (including rad51Δ, rad52Δ, rif1Δrif2Δ) did not significantly change Ls value (Figure S3A). Based on similar genetic requirements of ALT (RAD51, RAD52, and checkpoint genes), we propose that the skewed distribution of telomere lengths that is driven by HR and DNA damage checkpoints represents early precursors of ALT that are actively repairing their shortest telomeres.
Rad59 is required for the transition from early ALT precursors to ALT survivors
Our ALT frequency analysis suggested an essential role for both Rad51 and Rad59 in the unified ALT pathway (Figure 1C), so we evaluated whether Rad59, similar to Rad51, is required for the early step of ALT of forming ALT precursors. Interestingly, RAD59 deletion did not affect the time of senescence (Figure 2A) or the distribution of telomere lengths during telomere erosion (Figure 3F and 3G), suggesting that Rad59 is not required for the formation of early ALT precursors and, therefore, is likely needed in a later step of maturing ALT precursors into ALT survivors.
To gain further insight into the role of Rad59 we considered the fact that Rad59, and Rad52, mediate recombination at microhomologies (Anand et al., 2014; Ira and Haber, 2002; Sugawara et al., 2000; Tsaponina and Haber, 2014). This property is particularly relevant because yeast telomeres consist of degenerative repeats (Shampay et al., 1984), which would create ample opportunities for an eroded telomere to find microhomology across the entirety of other telomeres to potentially maximize the length of the telomere template. To address this, we expanded our computational model of telomere erosion and repair by adding the possibility of recombination at microhomologies along telomeres (Figure 4A). Since recombination at microhomologies is known to be less efficient than HR (Anand et al., 2014; Tsaponina and Haber, 2014), we explored various frequencies of recombination at microhomologies from 5×10−6 to 5×10−2 (Figures 4B, S4C, and S4D) relative to HR. Our modeling predicted that the addition of recombination at microhomology at the intermediate frequency (5× 10−4) would allow the formation of telomeres that were significantly longer than those produced in the model with HR only (Figure 4B compared to Figure 3B), and it would occur after the onset of senescence (Figure 4B). At a higher frequency of recombination (5×10−2), the long telomeres were also predicted to form, but early in the process (Figure S4C), while at the lowest frequency tested (5× 10−6) the formation of long telomeres was not predicted at all (Figure S4C). Similar results were observed using a Lr=90 (Figure S4D). To investigate whether we could capture this lengthening of telomeres experimentally we incubated tlc1Δ cells in liquid culture, collected samples at four points across telomere erosion and ALT formation process and analyzed individual telomeres by PacBio sequencing (Figure 4C). The results of sequencing telomeres within these samples demonstrated that in the first two samples that were within telomere erosion time (PD27 and PD36) the telomeres, as expected, became shorter and did not have telomeres longer than 320bp (Figures 4Di–ii and S3E). To capture the formation of the long telomeres, the PD36 cells, which are near senescence, were incubated for an additional 48 hours (until PD44) to allow for time to become fully senescent and sequenced using PacBio (Figure 4C). Indeed, in tlc1Δ cells at PD44, a new population of 300-through 2550-bp survivor-long telomeres emerged (Figures 4Diii; see Figure S3E for additional example). This new population comprised 9% of all telomeres in the sample, while the majority (91%) of telomeres had further eroded (Figures 4Diii and S3E). In subsequent passages, the vast majority of telomeres in this population became >300bp long (Figures 4Div and S3E), which suggested that the minority population of cells with elongated telomeres observed at PD44 eventually took over the culture as ALT survivors (Figures 4Div and S3E). Overall, the emergence of long telomeres after the onset of senescence was most consistent with our model of recombination at microhomology at an intermediate frequency where long telomeres formed after the onset of senescence. Therefore, we believe that the frequency of recombination at microhomology is lower than 5×10−2 (when long telomeres formed before senescence), but higher than 5×10−6 (when long telomeres were not predicted by our modeling) relative to HR. When the same experiment was repeated in tlc1Δrad59Δ cells, we did not observe survivor-long telomeres (>300 bp) at the same passage where they were observed in tlc1Δ (compare Figure 4Eiii and Figure 4Diii), consistent with the idea that the formation of ALT survivor-long telomeres observed in tlc1Δ cells requires Rad59 (see additional examples in Figures S3F versus S3E). The appearance of longer telomeres was eventually observed in tlc1Δrad59Δ cultures following an additional 48-hour incubation, which likely resulted from the multiplication of very rare ALT survivors that were able to form in this genetic background (Figure 4Eiv) as a result of the residual annealing activity of Rad52 (Sugawara et al., 2000). Importantly, when rad51Δ culture was kept beyond senescence we were able to observe formation of survivor-long telomeres (>300 bp) (Figure S3D). Together, our genetic and genomic data support a model where early ALT precursors formed by Rad51-dependent telomere interactions mature into ALT telomeres via a Rad59-mediated pathway.
Figure 4. Rad59-mediated recombination mediates the transition from ALT precursors to survivors.

(A-B) Computational modeling of telomere erosion in the presence of recombination at homology and microhomology (see STAR Methods).
(A) Schematic.
(B) The dynamics of the entire distribution of the telomere lengths with parameters (erosion rate=6bp/div., Ls=70, and Lr=75) and with recombination at microhomology at frequency of 5×10−4.
(C) Cell passaging scheme for PacBio sequencing analysis.
(D) Analysis of telomere lengths by PacBio sequencing in tlc1Δ cells passaged, as shown in (C). Telomere length distribution is shown in 20bp bins indicated by the middle value of the bin. Red bracket indicates telomere lengths compatible with ALT survivors. (See also Figure S3)
(E) Analysis of tlc1Δrad59Δ cells like (D).
(See also Figures S3 and S4)
The unified ALT pathway produces survivors with “hybrid” structure
Our model of a unified ALT pathway predicts that structural signatures of both Rad51- and Rad59-dependent pathways should be combined within individual ALT survivors. To test this prediction, we characterized the telomeres of ALT survivors using Oxford Nanopore Technologies (ONT) to sequence each end of the chromosomes of individual ALT survivors. First, ONT was used to sequence the TLC1/tlc1Δ parental strain (AM3692), and it identified 11 Y’ sub-telomeric regions (7 short Y’ and 4 long Y’ as defined by (Louis and Haber, 1990a, b)), all present on different chromosome ends, and telomere lengths that ranged from 163 bp to 338 bp (average of 275 bp) (Figure 5A; Table S3). To correct for errors, especially for indels that were frequently present in ONT reads, we created consensus sequences for individual Y’ by taking the 6 ONT reads for each end and using a base-by-base majority rule to determine the correct bases for each sequence. The consensus sequences for individual Y’s were compared to each other using a nucleotide distance-based clustering method (STAR Methods), and this allowed us to identify eight discrete groups among 11 Y’ regions of the parental strain (Figure 5A and 5B), which demonstrated that the majority of its Y’ sequences can be distinguished from each other by sequencing.
Figure 5. Oxford Nanopore sequencing and analysis of chromosome ends in Type II ALT survivors.

(A) Structure of chromosomal ends in the parental (TLC1/tlc1Δ) strain AM3692 by Oxford Nanopore Technology (ONT) and droplet-digital PCR (ddPCR); Y’ rectangle fill color corresponds to the Y’ source classified in (B).
(B) Nucleotide distance-based clustering of individual Y’ sequences; bootstrap values greater than 95 are interpreted as a difference.
(C) Fractions of Type I and Type II among ALT survivors as estimated by Y’ copy number by ddPCR. Type II: ≤ 32Y’ and Type I: >32Y’.
(D) Structure of chromosome ends in a representative tlc1Δ ALT survivor ZK-1 by ONT. Lost Y’=dashed outline of source color with no fill; Gained Y’= black outline with fill of new Y’; Swapped Y’= original Y’ source outline with fill of new Y’; and Maintained Y’= outline and fill the same color.
(E) The structure of a representative Type II ALT survivor ZK-3 from tlc1Δrad51Δ.
(F) Like (E) but ZK-4 from tlc1Δrad59Δ.
To determine the molecular structure of chromosome ends of ALT survivors, we first obtained a representative number of ALT survivors from various genetic backgrounds in experiments similar to those described in (Figure 1B), where no more than one survivor per culture could be formed. Using droplet-digital PCR (ddPCR) analysis to quantify the copy number of Y’, we divided these survivors in two groups: survivors containing <32 Y’s (Type II), and survivors containing >32 Y’s per survivor (Type I). This threshold of 32 Y’ was selected because yeast contain 32 chromosome ends in each cell, so ≤32 Y’s can correspond to one or less Y’ per chromosome end (matching a Type II outcome (Le et al., 1999; Lundblad and Blackburn, 1993; Teng and Zakian, 1999)), while >32 Y’s would require tandems on chromosome ends (indicative of a Type I outcome) (Le et al., 1999; Lundblad and Blackburn, 1993; Teng et al., 2000; Teng and Zakian, 1999). We observed that tlc1Δ cultures form survivors of both types in equal amounts (Figure 5C), while tlc1Δrad51Δ produced only Type II survivors (Figure 5C), consistent with (Hu et al., 2013; Le et al., 1999; Teng et al., 2000). However, in contrast to previous reports of tlc1Δrad59Δ cultures forming only Type I telomere structures (Chen et al., 2001), our experimental conditions, where only one survivor per culture can occur, revealed that both types of ALT outcomes can form in the absence of Rad59 (Figure 5C). The proportions of the two survivor types in tlc1Δrad59Δ were similar to those seen in tlc1Δ cultures, but the frequency of survivors was lower overall (Figure 1C), suggesting that Rad59 plays an important role in the formation of both types of ALT survivor telomere structures.
Next, we subjected representative Type I and Type II ALT survivors to ONT sequencing. Analysis of a Type II survivor from a RAD+ background, ZK-1, revealed elongated telomeres (up to 1103 bp in length) (Figure 5D, Table S3; Supplemental Information), which is expected for Type II events (Chen et al., 2001; Le et al., 1999; Lundblad and Blackburn, 1993; Teng et al., 2000). However, in addition to the lengthened telomeres, our ONT analysis showed that the Y’ elements also underwent changes, including Y’ losses, gains, and swapping between chromosome ends (Figure 5D). Therefore, although the total number of Y’ elements was not different from the parental strain (11 Y’ elements in ZK-1 and AM3692), the chromosome regions containing Y’ elements were involved in the formation of this survivor. The structures of chromosome ends in Type II survivors obtained in tlc1Δrad51Δ (Figures 5E and S5B) and tlc1Δrad59Δ (Figure 5F) mutants were similar to those in the RAD+ strain, including the presence of elongated telomeres, as well as losses, gains, and swapping of Y’ elements.
We next performed ONT analysis on a representative Type I survivor with >32 Y’ tandems, ZK-17, from a RAD+ background. Strikingly, ZK-17 chromosome ends contained features of both ALT types: Y’ tandems and elongated telomeres (Figure 6A). In particular, the chromosome ends of ZK-17 contained multiple Y’ elements (between 2 and 16) present as tandems on individual chromosome ends, which is a feature of Type I ALT outcomes (Figure 6A; Table S3). Further analysis of the tandem Y’ elements demonstrated that Y’ elements belonging to the same chromosome end were always similar to each other, and likely originated from the same parental Y’ element (Figure 6A). Importantly, we observed that, the chromosome ends containing tandem Y’ elements were capped with long telomeres of up to 1985 bp, which is a feature of Type II ALT outcomes (Figure 6A; Table S3). Based on the co-occurrence of tandem Y’ elements and long telomeres, we propose that the two classic ALT types are not mutually exclusive and can form a “hybrid” structure. The fact that some of the chromosome ends of this survivor had no Y’ elements at all, but contained only long telomeres, further supports its classification as a “hybrid”.
Figure 6. The unified ALT pathway produces survivors with “Hybrid” structure.

(A) The ONT analysis of tlc1Δ survivor ZK-17 with >32 Y’ elements demonstrate “hybrid” chromosome end structure.
(B) Unstable (Type I) survivor ZK-2 from tlc1Δ; see the graphical legend in (A).
(C) Changes of Y’ copy number during passaging of the unstable survivor shown in (B) by ddPCR for each of 10 passages of 250 cells/ml in 4ml (similar to Figure 1B).
(D) The chromosomal end structure of the stable ALT survivor obtained after 10 passages described in (C).
We reasoned that the classical Type I survivors, which are defined as containing Y’ tandems and short telomeres, may in fact represent intermediates of ALT that can be further stabilized by telomere elongation. To test this, we analyzed telomere sequences from Type I tlc1Δ (ZK-2, ZK- 5), and tlc1Δrad59Δ (ZK-33), survivors obtained immediately after plating (similar to Figure 1C), and we found that their structure was consistent with classic Type I outcomes, including tandem Y’ elements and short telomeres (Figures 6B, S5A and S5C; Table S3). When we analyzed these survivors by ONT, we noted striking heterogeneity between the sequence reads mapping to the same chromosome ends (Supplemental Information). Such heterogeneity suggested that these chromosome ends may continue to evolve, which we tested by passaging ZK-2 further in liquid culture. This resulted in drastic changes in the copy number of Y’ elements at each passage (Figure 6C); however, after 10 passages, the resulting stabilized survivor (ZK-18) contained a “hybrid” telomere structure (Figure 6D), similar to ZK-17 described above (Figure 6A). This dynamic behavior of the chromosome ends in classical Type I survivors strongly supports that Type I ALT survivor outcomes represent intermediates of ALT establishment that require stabilization of chromosome ends by the addition of long telomeres.
DISCUSSION
In this study, we combined molecular genetics, population dynamics and ultra-long-read sequencing approaches to solve two major limitations in the field that have existed for decades: the inability to determine the frequency of ALT and the structure of ALT telomeres. In addition, our methods allowed us to follow ALT establishment in yeast from the beginning of telomere erosion all the way through the formation of ALT survivors. Our results enable us to propose a new model (Figure 7), wherein ALT survivors form through a unified pathway that proceeds via two sequential steps, beginning with the Rad51-mediated formation of precursors, followed by Rad51-independent precursor maturation into stable ALT survivors. Further, we used our combined approaches to ascribe specific roles to several key proteins within the unified ALT pathway.
Figure 7. The model of the unified ALT pathway.

(A) Following inactivation of telomerase, the telomeres erode at a rate of 6bp/div. until Lr is reached (inducing HR) or Ls is reached (inducing senescence). Rad51, Rad52-dependent HR leading to telomere elongation forms ALT precursors.
(B) Rad59/Rad52 mediates the maturation of precursors, from (A), into ALT survivors by recombination at microhomologies. Thin lines: alternative pathways in the absence of Rad51 or Rad59.
(C) Rad59, Rad52 mediate recombination at microhomologies within telomeres to form Type II survivors.
(D) Rad59, Rad52 mediate recombination between eroded telomere and ITS to form tandem Y’ that are propagated to other chromosomal ends through Rad51-dependent recombination.
(E) Rad59, Rad52 mediate maturation of products of (D) to form “hybrid” ALT outcomes containing long telomeres.
Formation of ALT precursors
We propose that telomere erosion in telomerase-deficient yeast cells proceeds at a rate of ~6 bp per division, until at least one telomere in the cell is sufficiently short (between 70 and 90 bp). Such shortening of telomeres likely compromises telomere capping, promoting the activation of DNA resection (Fallet et al., 2014; Ribeyre and Shore, 2012; Shore and Bianchi, 2009), which is likely to affect not only telomeres, but also subtelomeric regions, suggested by (Fallet et al., 2014), and supported by the frequent swapping of Y’ elements that we observed. The ssDNA regions formed by resection likely engage in Rad51-dependent BIR that can lead to telomere elongation (Figure 7A). Based on our data, the length of telomeres inducing recombination (Lr) is between 70 bp and 90 bp (the length driving cells into senescence (Ls) was about 70 bp, and 90bp represents the mode value of telomere lengths determined in our PacBio experiments for tlc1Δ senescent cells (Figure 4Diii); since the distribution includes all the telomeres in the population, including those already repaired by recombination, it is unlikely that the short telomere length in the population driving recombination will be longer than the mode). Future studies will determine the Lr with higher precision, as well as identify factors affecting the Lr. One promising mutant background to evaluate in this context would be rif1Δrif2Δ, for which our studies uncovered an increase in ALT frequency (Figure 1C; Table S2), which may result from compromised telomere capping (consistent with (Teng et al., 2000)).
Further, we propose that the cells with shortened telomeres involved in recombination events represent precursors of ALT, and that they execute two essential functions that enable their maturation into ALT survivors. The first function is to initiate the DNA damage checkpoint response to allow time for the telomeres to repair and/or become matured, which is supported by our observation of a decreased Ls in checkpoint-defective mutants (Figure S3A). The second function is to lengthen the shortest telomeres in the cell to prevent the onset of senescence, and thereby to become endowed with the ability to undergo subsequent cell divisions that present opportunities to mature the telomeres into survivor-long structures. If the shortened telomeres (Ls ~ 70 bp) fail to extend, the cells enter senescence (in a DNA damage checkpoint-dependent way).
An interesting possibility is that the ALT precursors detected in our experiments may correspond to “Type B” cell lineages (Xu et al., 2015), which, in the absence of telomerase, undergo successive cycles of cell cycle arrest and BIR-mediated recovery. Our approach to analyzing the skewness of the entire telomere length distribution may allow detection of Type B cells in large populations, making it possible to link these precursors to the formation of ALT survivors within the same cultures.
Maturation of ALT precursors into ALT survivors
According to our model, ALT precursors are matured by a Rad51-independent, Rad59/Rad52-dependent pathway into ALT survivors containing long telomeres (Figure 7B). Specifically, when Rad59/Rad52 mediate recombination at sites of microhomology within telomeres of ALT precursors, Type II ALT survivor structures are formed (Figure 7C). Alternatively, upon Rad59/Rad52-mediated recombination between eroded telomere ends and microhomologies within ITS (measured in our strains to have a median length of 165 bp (Figure S3G)), Y’ element tandems are formed, consistent with (Churikov et al., 2014), which can then be further propagated throughout the cell by Rad51-mediated recombination (Figure 7D). It is possible that the formation of long Y’ element tandems generated by Rad59/Rad52 may involve multiple reinvasions, or formation of Y’ element circles (consistent with (Larrivee and Wellinger, 2006)) that can become a substrate for rolling-circle replication. The latter mechanism is consistent with (Louis and Haber, 1990a, b), and is supported by our finding of a single Y’ element origin for each tandem, in addition to a limited number of Y’ element tandem sources in each survivor (Figure 6A). The Type I-like intermediates containing short telomeres can later be stabilized by telomere elongation mediated by the annealing function of Rad59/Rad52 to form “hybrid” chromosome ends (Figures 7E, 6A and 6D).
Unified ALT Pathway
Our quantitative genetic and structural analyses of ALT survivor formation strongly indicate that a unified ALT pathway relies upon the functions of Rad51 and Rad59 at sequential steps. Because deletion of either Rad51 or Rad59 results in a marked decrease in ALT frequency, it is likely that the unified pathway is the dominant mechanism producing ALT survivors. In addition, the formation of rare ALT survivors that was observed in the absence of Rad51 or Rad59 could still be consistent with the unified ALT model. Specifically, even in the absence of ALT precursors in rad51Δ strains, telomere substrates formed during telomere erosion could be directly annealed to the donor at microhomology by Rad59/Rad52, leading to transition to ALT. In addition, we postulate that the rare cases of survivor formation in the absence of Rad59 are mediated by the annealing function of Rad52 alone, which has been observed to occur, although with low efficiency (Sugawara et al., 2000). We cannot exclude the existence of alternative ALT pathways that function in the absence of Rad51 or Rad59; however, our data suggest that any alternative pathways would yield ALT survivors at much lower frequencies than the unified pathway.
Our quantitative genetic and structural analyses of ALT survivor formation applied in this study have defined the role of several genes in the unified ALT pathway. Similar tools can be used to determine the specific roles played by proteins that have been previously implicated in formation of either Type I or Type II survivors. For example, it will be particularly important to determine the frequency and telomere structures of ALT survivors in Rad50- or Sgsl-deficient mutants (Chen et al., 2001; Le et al., 1999; Huang et al., 2001; Johnson et al., 2001), because these proteins are both proposed to play a specific role in Type II ALT survivor formation based on the prevalence of Type I outcomes in the deletion mutants.
Limitations and future challenges
Determining the exact parameters of telomere erosion and ALT intermediates remains a challenge. Here, using a combination of genomics, computer modeling and molecular genetic analyses, we were able to characterize, or at least to estimate, some of these parameters, including the rate of telomere erosion, the frequency of recombination at microhomology governing the transition from ALT precursors to survivors, and the values of Ls and Lr. However, much more work is required to establish these parameters more precisely. In addition, their exact values might vary in different strain backgrounds and may be affected by various environmental exposures. We believe the tools we developed in this work will bolster future research in this direction.
Finally, we propose that, by advancing our knowledge of the ALT pathway in yeast, our study provides important context for the field to understand ALT in other organisms, including humans. Our results obtained in yeast provide specific, testable hypotheses to determine whether a unified ALT pathway is conserved in mammals, and whether this process also involves both Rad51-dependent and -independent steps.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Please direct requests for reagents and resources to the lead contact Anna Malkova (anna-malkova@uiowa.edu).
MATERIALS AVAILABILITY
Distribution of Yeast strains or unique reagents will require the material transfer agreement in accordance with the rules and regulations of The University of Iowa.
DATA and CODE Availability
All sequencing reads from ONT and PacBio studies reported in this study are deposited in the NCBI SRA PRJNA630168.
All raw data for Figures and Tables are available at http://dx.doi.org/10.17632/m6xzygyvit.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains
The AM3692 yeast strain (Figure 5A), TLC1/tlc1Δ, was constructed by auto-diploidization of the haploid AM939 (Malkova et al., 2004) strain (MATa ura3-Y1 ade5-y7 LYS5 MET13 HPH CYH2 crl3–2 trp-Y1 leu1-Y1). Specifically, AM939 was transformed with pJH132 (Wu and Haber, 1995) plasmid containing HO gene under control of GAL1–10 promoter. The mating type switch by galactose-induced HO break leading to auto-diploidization was confirmed by non-mater phenotype of the resulting diploid strain. Next, one copy of TLC1 was disrupted by transforming with a DNA fragment containing ~200bp homology to TLC1 gene flanking the BSD cassette (ThermoFisher). The presence of the heterozygous deletion (TLC1/tlc1::BSD) was confirmed by PCR. AM3758 is a derivative of AM3692, and was created by deleting one copy of SRS2 by replacing it with a Bleor marker (Gueldener et al., 2002). Further, heterozygous deletions of several other genes were introduced into AM3758 by replacing one of its copies with the KanMX (Wach et al., 1994) module. Specifically, the following derivatives were created: RAD9/rad9::KanMX (AM3840), RAD52/rad52::KanMX (AM3849), RAD24/rad24::KanMX (AM3857), RAD51/rad51::KanMX (AM3858). AM4950 is a derivative of AM3692 and was created by deleting one copy of RAD59 by replacing it with a KanMX module.
METHOD DETAILS
Media
Rich media (Yeast extract-peptone- dextrose (YEPD)), synthetic complete media, and sporulation media (KAc) were prepared as described (Elango et al., 2018). Antibiotics were added to YEPD after autoclaving at concentrations described in (Elango et al., 2018). All cultures were grown at 30°C.
Determining ALT frequencies by passaging yeast in liquid cultures.
To determine the frequency of ALT, we used tlc1Δ colonies obtained by tetrad analysis of AM3692 (TLC1/tlc1::BSD) selected by their resistance to BSD. Such colonies were inoculated into 1 ml of YEPD, grown overnight to cellular density ~1 ×107 cells/ml (Passage 0, Figure 1B), where the cellular density was determined by counting using a hemocytometer. Following this step, cells were passaged into 4 ml of YEPD (passage 1) and the similar passages continued every 24 hours until the formation of survivors. To determine the optimal conditions, we reduced the number of cells passaged from 105 cells/ml to the cell density of 104 cells/ml, 103 cells/ml, or 250 cells/ml (Figures 1A, 1B, S1A and S1B). The total number of cells and the number of completed population doublings (PD) were determined 24 hours after passaging (Figure S2). As expected, the growth of tlc1Δ cultures progressively declined with every doubling, and the termination of growth was usually detected between passages 3 and 4. At the end of the experiment, some cultures were able to overcome the cellular senescence and return to normal growth due to the formation of survivors. Importantly, in experiments where the standard amount of cells were passaged (105 cells/ml) (similar to (Le et al., 1999; Lydeard et al., 2007; Teng et al., 2000; Teng and Zakian, 1999)) each culture formed survivors (Figure 1A), but the reduction of cells passaged allowed us to detect cultures that did not form survivors. The goal was to achieve conditions where ~ 5% of cultures form survivors to ensure that each survivor forms through a single event, allowing the use of Poisson statistics to determine the frequency of survivor formation (formula shown below). FALT = the frequency of ALT, Co= the number of cultures that did not form survivors, Ct= total number of cultures, and PS= the total number of cells passaged in the culture.
Determining the frequency of ALT in other mutants, including tlc1Δ rad51Δ and tlc1Δ rad59Δ, was performed similarly to tlc1Δ, but with a number of cells passaged optimized for each mutant (see Table S1 for details).
Frequency determination by plating.
Since determining the frequencies by liquid transfers limits the number of obtained survivors, resulting in wide 95% confidence intervals, we developed a modified version of this method where the cultures were plated after several liquid passages performed similar to (Figures 1B and 1C). Specifically, we noticed that after growth observed in passage 2, the cells divided an additional average of 1.8 times (in passage 3) (Figures 1B and S2A), meaning that the cells at the moment of transfer from passage 2 (into passage 3) were at or near senescence. Therefore, with a tube full of senescent cells (after 24 h in passage 2) we calculated the frequency of survivor formation per senescent cell by plating a known number of these cells and identifying how many survivors formed (Figure 1C). To do this, we followed the same procedure as described for our liquid transfer experiment (Figure 1B), but only for the first three passages, where passage 2 containing senescent cells (now called tube S), were plated. The survivors that were formed on the plates were confirmed by maintenance of growth after streaking for single cells, and the number of these survivors was used to calculate the frequency of ALT using the formula below. (S= the total number of survivors (for experiments in tlc1Δ it was 428 survivors formed in 61 platings (See Table S2)). AC = the total number of cells on the plate that was calculated by using the total cells plated after passage 2 (for experiment in tlc1Δ it was 5.83 ×106 cells) corrected by the number of doublings completed on the plate that was estimated from the number of doublings (K) in liquid passage 3 (for experiment in tlc1Δ, K=1.83 (Table S2)).
For experiment in tlc1Δ FALT= 2.01 ×10−5 with a 95% CI: (1.85 ×10−5− 2.20×10−5) (see Table S2 for details).
To determine FALT in other mutants including: tlc1Δ rad51Δ, tlc1Δ rad59Δ, tlc1Δ srs2Δ, tlc1Δ rad51Δ srs2Δ, tlc1Δ rad55Δ, tlc1Δ rad55Δ srs2Δ, tlc1Δ rad9Δ, tlc1Δ rad9Δ srs2Δ, tlc1Δ rad24Δ, tlc1Δ rad24Δ srs2Δ, tlc1Δ rif1 Δ, tlc1Δ rif2 Δ, tlc1Δ rif1 Δ rif2Δ, the experiments were performed similarly to tlc1Δ, but with adjusted parameters for Tube S, K, and the number of cells transferred based on the different growth dynamics and frequencies, see Table S2 for parameters.
Distribution of telomere length determined by Southern blot analysis.
The distributions of telomere lengths were determined in cell populations prior to and after telomerase inactivation using genomic DNA extracted from several consecutive passages of tlc1Δ yeast liquid cultures grown to a specific PD. DNA was digested with XhoI, which cuts within the Y’ sequences, ~875bp from the telomere. The fragments were separated using agarose gel (similar to (Lundblad and Blackburn, 1993; Teng et al., 2000; Teng and Zakian, 1999; Xu et al., 2015)), and probed with a Y’ specific probe (Lendvay et al., 1996), which allows detection of 11/32 yeast telomeres, and determining their length (Figures 3D and 3E). Images were taken using a Typhoon Phosphoimager FLA7000, and the range of intensities corresponding to telomeres were detected similar to (Xu et al., 2015). The distribution of signal intensities specific to telomere regions were extracted (after subtracting background) by using ImageQuant TL (GE healthcare) and were analyzed by Pearson’s moment of skewness test. The comparison of skewness of telomere length distribution between different strains was performed by using non-parametric Mann-Whitney test.
Computational modeling of yeast populations undergoing telomere erosion and repair.
We established a computational model of yeast populations mimicking the full stochastic dynamics of cell division, erosion, and potential repair based on population genetics principles and forward simulations. Note that differently from more standard modeling, our in silico populations took into account within-cell dynamics (driven by stochastic differences in telomere lengths due to erosion and repair within a cell) and cell-to-cell dynamics (driven by senescent- vs- non-senescent properties, as well as by random genetic drift). Our modeling scheme fully mimics the complete experimental scheme and population doublings (PD) shown in (Figure 4C), thus allowing very large numbers of cells as well as bottlenecks. After each PD, we characterized the full distribution of telomeres in the population, as well as the fractions of senescent and repairing cells. The telomere dynamics was characterized for a minimum of 100 independent populations followed for up to 63 PD for each set of conditions (see below). Our modeling scheme assumed that at the beginning of the experiment cells started with 32 telomeres with lengths following a normal, symmetrical distribution that recapitulated our experimental data for steady telomere lengths (average 275 bp) (Figures 3E and 5A; Table S3). In that regard cells could have a different initial average and distribution of telomere lengths both within and between cells. Each round of replication, every telomere within every cell in a population was subjected to erosion based on a Poisson distribution with an average erosion rate (bp /div.). Cells were considered to be in a state of cell cycle arrest when one or more telomeres reached a critical length of senescence (Ls) that was first approximated by using Southern blot hybridization. Using a Southern blot method described above (see also Figure 3D and 3E and (Xu et al., 2015)), the 10th percentile was calculated for at least 12 independent senescent cultures and it was observed that their values varied between 55bp and 90bp and their distribution was not significantly different between tlc1Δ, rad51Δtlc1Δ, and rad52Δtlc1Δ (Figure S3A). Therefore, Southern blot hybridization allowed the Ls to be narrowed between 55–90bp, but the high variation limited the resolution and may be due to technical limitations of size determination of Southern blots (especially for the 10th percentile because it is close to background intensities). To more accurately calculate the Ls, we used PacBio sequencing using a passaging scheme outlined in (Figure 4C) to reach senescence, or near senescence, in liquid culture. Specifically, four tlc1Δ samples (two at PD36 and two at PD44) (Figure 4Dii–iii and S3E) were sequenced and the length of telomeres was determined. The shortest telomere in the sample, defined by the 10th percentile, were ~70 bp (67 and 72 for tlc1Δ PD36, 69 and 70 for tlc1Δ PD44) (Figure 4Dii–iii and S3E), which suggested that ~70bp defines a critically short telomere length (Ls) that drives cells into senescence. An additional HR-deficient (tlc1Δ rad51Δ at PD27) senescent sample was sequenced using PacBio and the 10th percentile telomere length was 72bp (Figure S3C).
Our simplest model assumed telomere erosion without repair (Figure 2B–D), similar to what happens in yeast tlc1Δ rad52Δ cells. This modeling allowed us to determine the best fit for erosion rate. Our expectation was that since in the absence of Rad52, cultures stop doubling at PD27 then the Ls=70 in our modeling experiment should be reached within PD27. Based on this modeling (Figure 2C) we observed that with an erosion rate=4 (suggested by previous studies (Marcand et al., 1999)), the Ls was not reached until PD42 even by the shortest telomeres (10th percentile) of the distribution (Figure 2C), while an erosion rate=5 reached the Ls at PD34 (Figure S4A). With an erosion rate=6 the 10th percentile of the telomere length distribution reached the Ls at PD27, which matched the expectation, while with erosion rate=9 the 10th percentile of the telomere length distribution reached Ls too early, already at PD17 (Figure 2C). Together, we conclude that an erosion rate=6 is the best fit for the erosion rate in yeast tlc1Δ.
Our next model, in addition to telomere erosion, also allowed for the repair of telomeres at positions of homology (Figure 3A–C) either between telomeres or Y’ elements that both will lead to the same outcome for telomere lengths after recombination. According to this model, cells with one or more telomeres shorter than a threshold repair length (Lr; where Lr≥Ls) stop dividing and allow telomeres shorter than Lr to randomly pair with one other (donor) telomere of the same cell, attaining the donor telomere length when the latter telomere is longer. This homology-mediated telomere recombination and repair can lead to the resumption of the cell cycle when/if all telomeres become longer than Lr. We investigated Lr values of 75 and 90 that were chosen based on the idea that Lr should be higher than the Ls value as well as being equal to or less than the value of the mode of telomere length distribution determined by PacBio sequencing at the point of senescence (Figure 4D). Lr value of 120 was also investigated but produced, as expected, telomere length distributions not compatible with sequencing (PacBio) data.
We also modeled a situation where in addition to repair at homology, telomeres were allowed to repair by recombination at microhomology between telomeres (Figure 4A–B). In this case, telomeres shorter than Lr pair randomly with one other telomere and recombination occurs at two random positions along each of the two telomeres. Since telomeres in yeast consist of degenerative repeats, microhomology for such recombination should be available. Recombination at these two shifted positions allows the chance of bigger increases or decreases in length. Since the efficiency of recombination at microhomology is known to be lower than that of HR we investigated varying relative frequencies of recombination at microhomology from 0.05 to 5×10−6 per repairing telomere.
Telomere length determination via PacBio sequencing.
To characterize telomere lengths in entire cell populations by PacBio sequencing, cell passaging was performed as described in the schematic in (Figure 4C) and genomic DNA was extracted. PacBio Sequencing was performed by the University of Washington PacBio Sequencing Services, Multiplexing the genome library prep followed by BluePippen size selection above 15kb and sequencing using Sequel II SMRT Cell 8M with 30-hour movie, yielding an average output of 4Gb of reads of each pooled sample with lengths between 15kb and 25kb. The resulting CCS sequencing reads were ascribed to chromosome ends by using BLAST (dc-megablast with -no_greedy, -perc_identity of 0.9, -gapopen 2, -gapextend 2, and -min_raw_gapped_score of 3000 parameters) to identify a 10kb unique probe sequence specific to each parental chromosomal end. The telomere lengths were defined as the lengths of C1–3 A/TG1–3 sequences on the ends of individual PacBio reads, and the telomere lengths for each cell population were plotted (Figures 4D, 4E and S3C–F).
Chromosome end structure analysis by Oxford Nanopore Technology (ONT).
To determine the telomere lengths for individual chromosomes, we used either ONT reads directly or ONT reads corrected with the Canu package (Koren et al., 2017) with the -correct option to improve the quality of the single-molecule reads. (These two approaches gave us similar results). We ascribed the reads to specific chromosome ends by using BLAST to identify reads that contained a chromosome-end-specific 10kb probe (with dc-megablast with -no_greedy, - perc_identity of 0.9, -gapopen 2, -gapextend 2, and -min_raw_gapped_score of 3000 parameters). The lengths of individual telomeres were determined by identifying terminal C1–3 A/TG1–3 sequences from the three longest ONT reads ascribed to each end (to minimize possibility of sequence termination within telomere). The number of Y’ sequences was determined based on the longest ONT sequencing read where individual Y’ sequences were identified using BLAST with Y’ specific sequence probes. Similarly, additional shorter reads were used to support the number of Y’ for individual ends.
Nucleotide distance-based clustering of Y’ sequences.
Each Oxford Nanopore read contains many mistakes within the sequence (even after use of Canu package), with a preference for indels, and therefore each read cannot be trusted for sequence comparisons. To correct these sequencing errors, a consensus sequence was created for individual Y’ by taking the 6 ONT reads for each chromosome end and using a base by base majority rule to determine which bases are correct for each sequence. This was completed by using the CLCgenomics12 function of “Map Reads to Reference” to align the 6 reads to a reference sequence of that specific end using the parameters of Match score 1, Mismatch cost of 2, insertion open cost of 2, insertion extend cost 1, deletion open cost 2, deletion extend cost 1, length fraction 0.5, and similarity fraction 0.8 (all alignments were inspected and confirmed visually). The consensus sequences were then extracted using the “Extract Consensus Sequence” function of CLCgenomics12 under the parameters of low coverage threshold of 4, noise threshold of 0.1, and minimum nucleotide count of 4. The resulting outcome was a consensus sequence in FASTA format that was used for further comparisons.
Identification of individual Y’ sequence sources was performed by creating an alignment with all parental Y’ consensus sequences along with the Y’ of unknown source using the “Create Alignment” function in CLCgenomics12 with parameters of gap open 4, gap extension of 1, and an end gap cost of free (all alignments were inspected and confirmed visually). The Alignment file was extracted as a FASTA alignment file and MEGAX (Koren et al., 2017) was used to create a phylogenetic tree, using UPGMA bootstrap method, of at least 10,000 replications, with the model of maximum composite likelihood with uniform rates among all sites, and all gap/missing data were completely deleted. Other methods to create phylogenetic trees, including maximum likelihood and neighbor joining tree, were created and produced similar results. The Y’ regions were considered to be significantly different from each other based upon bootstrap values above 95. Bootstrap values below 95 were not considered significantly different from each other.
Droplet-digital polymerase chain reaction (ddPCR) to quantify the number of Y’ copies.
Genomic DNA was extracted from yeast samples and digested with Xhol, to separate tandem Y’ sequences, then DNA concentration was determined by using Picogreen using a Turner BioSciences TBS-380 system, following manufacturers recommendations. ACT1, that is present in a single copy within haploid strains, was used as a reference for copy number. Primers were designed using PrimerQuest IDT online primer design tool to amplify DNA within the Y’ regions, as well as for primers within the ACT1 gene. DNA samples were diluted to 0.1ng/μL for the ACT1 control while multiple dilutions were used for Y’ sample (to account for the possibility of many Y’ copies in some outcomes), and then the number of PCR positive droplets were quantified by using the QX200 Droplet-digital PCR system (BioRad) according to manufactures recommendation. The total number of Y’ in each sample was quantified using the number of PCR positive droplets and taking in account the dilutions of DNA.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with GraphPad Prism v.8.1.2. Cut-off used for statistical significance P<0.05.
Supplementary Material
Table S1: Frequency of ALT measured by passaging yeast in liquid cultures. Related to Figure 1.
Table S2: Frequency of ALT measured by plating senescent cells. Related to Figure 1.
Table S3: Chromosome end structure analysis by Oxford Nanopore Technology (ONT). Related to Figures 5 and 6.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Plasmids | ||
| pJH132 | James Haber | Wu & Haber 1995 |
| Chemicals | ||
| Zymolase | US biological Co. | NC0439194 |
| Phenol/Chloroform/Isoamyl alcohol | Millipore Sigma | P3803-400ML |
| Sorbitol | Fisher | BP439-500 |
| Isopropanol | Fisher | BP2618500 |
| Potassium acetate | Sigma Aldrich | P1190-500G |
| XhoI | New England Biolabs | R0146S |
| AMPure beads | Beckman Coulter | A63880 |
| Deposited Data | ||
| Raw ONT sequencing data | This Paper | PRJNA630168 |
| Raw PacBio sequencing data | This Paper | PRJNA630168 |
| Uncropped Photographs | This Paper | http://dx.doi.org/10.17632/m6xzygyvjt.1 |
| Critical Commercial Assays | ||
| QX200 Evagreen Supermix | BioRad | 186-4034 |
| QX200 Evagreen Droplet Oil | BioRad | 186-4033 |
| NEBNext FFPE DNA repair Kit | New England Biolabs | M660S |
| NEBNext Quick T4 DNA ligase | New England Biolabs | E6056S |
| MinIon Platform | Oxford Nanopore Technologies | MIN101B |
| MinIon Flow Cell | Oxford Nanopore Technologies | R9.4.1 |
| MinIon Sample Prep Kit | Oxford Nanopore Technologies | SQK-LSK109 |
| Experimental Models: Organisms/Strains | ||
| Yeast Saccharomyces cerevisiae: AM939 | James Haber | Malkova et al. 2004 |
| Oligonucleotides | ||
| Yeast Y’ probe FWD: CGCGAATTCGCCCTACAGCACTTCTACATAGC | This Paper | OL3467 |
| Yeast Y’ probe RVS: CGAGAATTCCAGCGTTTGCGTTCCATGACG | This Paper | OL3468 |
| Software and Algorithms | ||
| CLCgenomics12 | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/ |
| MinKnow | Oxford Nanopore Technology | https://nanoporetech.com/ |
| ONT guppy | Oxford Nanopore Technology | https://nanoporetech.com/ |
| Graphpad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
HIGHLIGHTS.
ALT frequency in yeast was identified by using population genetics approach.
Ultra-long sequencing identifies “hybrid” Type I/Type II ALT survivors in yeast.
Rad51 and Rad59 define two consecutive steps in the unified ALT pathway in yeast.
Rad51 promotes formation of ALT precursors that become survivors by Rad59/Rad52.
ACKNOWLEDGMENTS
We thank Drs. Grzegorz Ira, Jim Haber, Sarit Smolikove, and Adam Dupuy for helpful discussions and comments on this manuscript. This work was supported by NIH grants R21ES030307 to A.M. and J.M.C., and R35GM127006 to A.M.
Footnotes
DECLARATION OF INTERESTS
The Authors have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anand RP, Tsaponina O, Greenwell PW, Lee CS, Du W, Petes TD, and Haber JE (2014). Chromosome rearrangements via template switching between diverged repeated sequences. Gene Dev 28, 2394–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, and Reddel RR (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11, 319–330. [DOI] [PubMed] [Google Scholar]
- Chang M, Arneric M, and Lingner J (2007). Telomerase repeat addition processivity is increased at critically short telomeres in a Tel1-dependent manner in Saccharomyces cerevisiae. Genes Dev 21, 2485–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Dittmar JC, and Rothstein R (2011). Long telomeres are preferentially extended during recombination-mediated telomere maintenance. Nat Struct Mol Biol 18, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Ijpma A, and Greider CW (2001). Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol Cell Biol 21, 1819–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churikov D, Charifi F, Simon MN, and Geli V (2014). Rad59-facilitated acquisition of Y’ elements by short telomeres delays the onset of senescence. Plos Genet 10, e1004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, and Lydall D (2012). Similarities and differences between “uncapped” telomeres and DNA double-strand breaks. Chromosoma 121, 117–130. [DOI] [PubMed] [Google Scholar]
- Dilley RL, and Greenberg RA (2015). ALTernative Telomere Maintenance and Cancer. Trends Cancer 1, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango R, Kockler Z, Liu L, and Malkova A (2018). Investigation of Break-Induced Replication in Yeast. Methods Enzymol 601, 161–203. [DOI] [PubMed] [Google Scholar]
- Elango R, Sheng Z, Jackson J, DeCata J, Ibrahim Y, Pham NT, Liang DH, Sakofsky CJ, Vindigni A, Lobachev KS, et al. (2017). Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun 8, 1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallet E, Jolivet P, Soudet J, Lisby M, Gilson E, and Teixeira MT (2014). Length-dependent processing of telomeres in the absence of telomerase. Nucleic Acids Res 42, 3648–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf M, Bonetti D, Lockhart A, Serhal K, Kellner V, Maicher A, Jolivet P, Teixeira MT, and Luke B (2017). Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell 170, 72–85 e14. [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, and Hegemann JH (2002). A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JC, and Haber JE (2006). Surviving the breakup: The DNA damage checkpoint. Annual Review of Genetics 40, 209–235. [DOI] [PubMed] [Google Scholar]
- Heyer WD (2015). Regulation of recombination and genomic maintenance. Cold Spring Harb Perspect Biol 7, a016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, Ding Z, Ying H, Boutin AT, Zhang H, et al. (2012). Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148, 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Tang HB, Liu NN, Tong XJ, Dang W, Duan YM, Fu XH, Zhang Y, Peng J, Meng FL, et al. (2013). Telomerase-null survivor screening identifies novel telomere recombination regulators. Plos Genet 9, e1003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PH, Pryde FE, Lester D, Maddison RL, Borts RH, Hickson ID, and Louis EJ (2001). SGS1 is required for telomere elongation in the absence of telomerase. Curr Biol 11, 125–129. [DOI] [PubMed] [Google Scholar]
- Ira G, and Haber JE (2002). Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Molecular and Cellular Biology 22, 6384–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Marciniak RA, McVey M, Stewart SA, Hahn WC, and Guarente L (2001). The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. Embo Journal 20, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, and Phillippy AM (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, and Tamura K (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 35, 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivee M, and Wellinger RJ (2006). Telomerase- and capping-independent yeast survivors with alternate telomere states. Nat Cell Biol 8, 741–747. [DOI] [PubMed] [Google Scholar]
- Le S, Moore JK, Haber JE, and Greider CW (1999). RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvay TS, Morris DK, Sah J, Balasubramanian B, and Lundblad V (1996). Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics 144, 1399–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DL, and Blackburn EH (2004). Counting of Rif1p and Rif2p on Saccharomyces cerevisiae telomeres regulates telomere length. Mol Cell Biol 24, 10857–10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, and Heyer WD (2011). Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 479, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis EJ, and Haber JE (1990a). Mitotic Recombination among Subtelomeric Y’ Repeats in Saccharomyces-Cerevisiae. Genetics 124, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis EJ, and Haber JE (1990b). The Subtelomeric Y’ Repeat Family in Saccharomyces-Cerevisiae - an Experimental System for Repeated Sequence Evolution. Genetics 124, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, and Blackburn EH (1993). An Alternative Pathway for Yeast Telomere Maintenance Rescues Est1- Senescence. Cell 73, 347–360. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, and Haber JE (2007). Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448, 820–823. [DOI] [PubMed] [Google Scholar]
- Malkova A, Swanson J, German M, McCusker JH, Housworth EA, Stahl FW, and Haber JE (2004). Gene conversion and crossing over along the 405-kb left arm of Saccharomyces cerevisiae chromosome VII. Genetics 168, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcand S, Brevet V, and Gilson E (1999). Progressive cis-inhibition of telomerase upon telomere elongation. Embo Journal 18, 3509–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, and Haber JE (2006). Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75, 111–135. [DOI] [PubMed] [Google Scholar]
- Neumann AA, and Reddel RR (2002). Telomere maintenance and cancer -- look, no telomerase. Nat Rev Cancer 2, 879–884. [DOI] [PubMed] [Google Scholar]
- Niu HY, and Klein HL (2017). Multifunctional roles of Saccharomyces cerevisiae Srs2 protein in replication, recombination and repair. Fems Yeast Res 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeyre C, and Shore D (2012). Anticheckpoint pathways at telomeres in yeast. Nat Struct Mol Biol 19, 307–313. [DOI] [PubMed] [Google Scholar]
- Shampay J, Szostak JW, and Blackburn EH (1984). DNA sequences of telomeres maintained in yeast. Nature 310, 154–157. [DOI] [PubMed] [Google Scholar]
- Shore D, and Bianchi A (2009). Telomere length regulation: coupling DNA end processing to feedback regulation of telomerase. Embo J 28, 2309–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer MS, and Gottschling DE (1994). TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science 266, 404–409. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Ira G, and Haber JE (2000). DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol 20, 5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MT, Arneric M, Sperisen P, and Lingner J (2004). Telomere length homeostasis is achieved via a switch between telomerase- extendible and -nonextendible states. Cell 117, 323–335. [DOI] [PubMed] [Google Scholar]
- Teng SC, Chang J, McCowan B, and Zakian VA (2000). Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol Cell 6, 947–952. [DOI] [PubMed] [Google Scholar]
- Teng SC, and Zakian VA (1999). Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19, 8083–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaponina O, and Haber JE (2014). Frequent Interchromosomal Template Switches during Gene Conversion in S. cerevisiae. Molecular Cell 55, 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, and Haber JE (2002). Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10, 373–385. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, and Philippsen P (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Wellinger RJ, and Zakian VA (2012). Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics 191, 1073–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Haber JE (1995). MATa donor preference in yeast mating-type switching: activation of a large chromosomal region for recombination. Genes Dev 9, 1922–1932. [DOI] [PubMed] [Google Scholar]
- Xu Z, Fallet E, Paoletti C, Fehrmann S, Charvin G, and Teixeira MT (2015). Two routes to senescence revealed by real-time analysis of telomerase-negative single lineages. Nat Commun 6, 7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Frequency of ALT measured by passaging yeast in liquid cultures. Related to Figure 1.
Table S2: Frequency of ALT measured by plating senescent cells. Related to Figure 1.
Table S3: Chromosome end structure analysis by Oxford Nanopore Technology (ONT). Related to Figures 5 and 6.
Data Availability Statement
All sequencing reads from ONT and PacBio studies reported in this study are deposited in the NCBI SRA PRJNA630168.
All raw data for Figures and Tables are available at http://dx.doi.org/10.17632/m6xzygyvit.1