


Abstract
Palindromic sequences are a potent source of chromosomal instability in many organisms and are implicated in the pathogenesis of human diseases. In this study, we investigate which nucleases are responsible for cleavage of the hairpin and cruciform structures and generation of double-strand breaks at inverted repeats in Saccharomyces cerevisiae. We demonstrate that the involvement of structure-specific nucleases in palindrome fragility depends on the distance between inverted repeats and their transcriptional status. The attack by the Mre11 complex is constrained to hairpins with loops <9 nucleotides. This restriction is alleviated upon RPA depletion, indicating that RPA controls the stability and/or formation of secondary structures otherwise responsible for replication fork stalling and DSB formation. Mus81-Mms4 cleavage of cruciforms occurs at divergently but not convergently transcribed or nontranscribed repeats. Our study also reveals the third pathway for fragility at perfect and quasi-palindromes, which involves cruciform resolution during the G2 phase of the cell cycle.
INTRODUCTION
Palindromic sequences or inverted repeats (IRs) are a potent source of chromosomal breakage and rearrangements in bacteria and many eukaryotic organisms because they can adopt hairpin and cruciform structures. In bacteria, perfect and quasi-palindromes (IRs separated by nonrepeated spacer) in plasmid or phage DNA are frequently deleted with the length of the spacer being the major factor for their instability (reviewed in (1)). When present on the Escherichia coli chromosome, IRs cause DSB formation and large inverted duplications (2). In budding and fission yeast and in mice, IRs have been shown to strongly induce ectopic and allelic recombination, crossing-over, and a variety of gross chromosomal rearrangements (GCRs) including deletions, translocations and gene amplification (e.g. (3–10)). In humans, palindromic sequences have been found at breakpoints of chromosomal translocations and are implicated in the pathogenesis of diseases. For example, palindromic AT-rich repeats (PATRRs) are responsible for the most frequent, recurrent non-Robertsonian translocation t(11;22)(q23;q11), which causes Emanuel syndrome, as well as nonrecurrent translocations (11). Palindrome-mediated large deletions, interchromosomal insertions and translocations also cause several types of ϵγΔβ thalassemia (12), X-linked congenital hypertrichosis syndrome (13) and hereditary renal cell carcinoma (14). Finally, palindromes promote oncogene amplification (15–22).
Several nucleases have been implicated in the attack of hairpin or cruciform structures leading to the genetic instability of IRs. One major player in the metabolism of both palindromes and quasi-palindromes is the prokaryotic SbcCD nuclease and its eukaryotic homolog Mre11-Rad50, part of the Mre11 complex (Mre11-Rad50-Xrs2/Nbs1) (23). In E. coli, SbcCD induces breakage at IRs in a replication-dependent manner. SbcCD targets hairpins formed by quasi-palindromes on the lagging strand template leading to a two-ended DSB that can be repaired by recombination with the sister chromatid (24,25). In contrast, perfect palindrome cleavage by SbcCD leads to cell death presumably because of hairpin formation on both the leading and lagging strands, thus eliminating the templates amenable to repair (26). In fission yeast, the recombinogenic effect of a 160 bp palindrome is dependent on the Mre11 complex. It has been postulated that the recombinogenic DSB is generated by the nuclease activity of Mre11 (3,4). However, in Saccharomyces cerevisiae, the Mre11 complex is not involved in breakage at a large IR consisting of two Ty1 elements with a ∼280 bp spacer in strains where DNA polymerase δ was down-regulated (27). Previously, we demonstrated that in S. cerevisiae the Mre11 complex does not generate DSBs at a closely spaced Alu quasi-palindrome (Alu-QP), but it is required, along with Sae2, for opening and resection of breaks that have hairpin-capped ends (6,28). This disparity on the effect of the Mre11 complex on DSB generation at IRs might be attributed to the differences in the formation or stability of hairpins with different loop sizes during replication and their availability for nuclease attack. This surmise is experimentally addressed in this study.
Several structure-specific endonucleases (SSEs) were described as a responsible for generating DSBs at IRs in eukaryotes. A genetic-based assay in budding yeast identified Rad1/Rad10 as a contributor to chromosomal fragility at a short IR (29). Mus81/Mms4 acting together with Rad1/Rad10 and Slx1/Slx4 was found to be involved in causing fragility at the AT-rich Flex1 sequence motif located on a yeast artificial chromosome (30). However, Nag et al. showed that Rad1/Rad10 is neither required for generating nor for processing DSBs at a 140 bp IR in budding yeast (31). Mus81/Mms4 was also shown to cleave plasmid-born cruciform structures formed by PATTRs in budding yeast (32). In cancer cell lines, MUS81/EME1 attacks secondary structures formed by AT-rich microsatellites upon depletion of the WRN helicase (33). Inagaki et al. found that GEN1 (homologue of yeast Yen1) can cleave cruciforms formed by PATTRs in a plasmid transfection assay in human cells (34). However, Mus81/Mms4 is not involved in recombination induced by either quasi-palindromes (28) or palindromes (35). Yen1 is not required for fragility at Flex1 and is not responsible for palindrome-induced intra-chromosomal recombination (30,35). This rather diverse spectrum of effects could be explained by the fact that (i) repeats used in these studies had different structural characteristics; (ii) IRs were located on plasmids versus chromosomes; (iii) whether the conclusions about the effect of the SSEs were based only on biological assays or were accompanied by direct detection of DSBs.
In this study, we examined the effect of SSEs on IR-mediated chromosomal DSB formation in budding yeast using a set of structurally different palindromic sequences. The strength of our study is the use of a combination of experimental approaches that include: (i) a sensitive assay for GCR induction and (ii) direct physical detection of chromosomal breaks and replication intermediates. We found that perfect and quasi-palindromes with spacers <9 bp can efficiently block DNA replication and are targets for MRX/Sae2 attack, a mechanism reminiscent of SbcCD attack on hairpins in bacteria. The specificity of MRX/Sae2 activity depends on the formation or stability of hairpins since RPA depletion leads to replication arrest and Sae2-dependent breaks in strains containing Alu-QP with spacers >8 bp. Besides the spacer length, transcription is another important factor affecting palindrome fragility and specificity of the involved nucleases. We observed that Mus81/Mms4 contributes to DSB formation at divergently transcribed URA3 or HIS4 IRs. However, Mus81/Mms4 and other known SSEs (Yen1, Slx1/Slx4, Rad1/Rad10 and Mlh1/Mlh3) do not generate breaks at the nontranscribed Alu and IS50 palindromes. Interestingly, we found that MRX/Sae2- and Mus81/Mms4-independent breakage at palindromes is not replication-dependent which points towards cruciform formation and resolution by an unknown nuclease during the G2 phase. These results suggest that chromosomal fragility at palindromic sequences is caused by the interplay of multiple nucleases at different stages of the cell cycle and is dependent on the type of secondary structure formed. Our data define parameters of palindrome attack by nucleases and extend our understanding of modalities of DSBs formation at IRs to elucidate the poorly defined mechanisms underlying palindrome-associated chromosomal aberrations and human diseases.
MATERIALS AND METHODS
Strains, plasmids and oligonucleotides
Yeast strains and oligonucleotides used in this study are listed in Supplementary Tables S3 and S4, respectively. The strains used in this study are isogenic and based on MATα bar1Δ his7–2 trp1Δ ura3Δ leu2–3, 112 ade2Δ lys2Δ cup1Δ yhr054cΔ cup2Δ V34205::ADE2lys2::IR V29616::CUP1. The GCR cassette composed of the counter selectable markers CUP1, CAN1 and ADE2 (in this order from telomere to centromere) is located on the left arm of chromosome V. IRs are located within lys2 that is telomere-distal to the GCR cassette. For all experiments, freshly thawed yeast strains were used. All strains were grown at 30°C and manipulated using standard yeast genetics methods. The media used in this study are YPD, YPR (1% yeast extract, 2% peptone and 2% dextrose of raffinose), uracil-drop out synthetic medium and histidine-drop out synthetic medium supplemented with 2 mM 3-AT. Target nonessential genes were disrupted using one of the following drug-resistance or prototrophic markers: kanMX (kanamycin), hphMX (hygromycin), natMX (nourseothricin) or TRP1 (tryptophan). The essential gene RFA2 was placed under control of a tetO7 repressible promoter to create the strain TET-RFA2 and expression was downregulated by adding doxycycline to the media (6). All IRs were inserted into LYS2 which had been placed telomere-distal to the GCR cassette (V34205). Strains containing Alu perfect palindrome (Alu-PAL) and Alu palindromes interrupted by spacers (Alu-QPs) were generated by the delitto perfetto approach (36). A CORE cassette containing the markers kanMX and URA3 was inserted at the center of symmetry of the Alu inverted repeat. The CORE cassette was then replaced by transformation with an oligomer (Supplementary Table S4) containing homology to the flanking Alu repeats separated by the SwaI palindromic restriction site to create Alu-PAL. Similarly, Alu-QPs were generated by transformation with oligomers (Supplementary Table S4) containing asymmetrical spacers ranging from 5 to 12 bp flanked by homology to the Alu repeats. Transformants were selected on synthetic media lacking adenine and containing 1 g/l of 5-fluoroorotic acid (5-FOA). Uracil auxotroph and kanamycin-sensitive clones were selected and tested for the presence of the expected Alu-PAL or Alu-QP by Southern blot. Genomic DNA was extracted and digested with BstEII to reveal replacement of the CORE cassette and with BstEII and SfaNI (or BsmI, whichever enzyme corresponded to the restriction site in the asymmetrical spacer) to reveal the presence of the spacer. LYS2-specific probes located upstream and downstream of the Alu repeats were used to reveal the resulting fragments. Strains containing IS50-PAL (37) URA3- and HIS4-PAL (38) were created as previously described. pRSafe_CRE-EBD (gift from Gartenberg’s lab) was integrated into LEU2 to build conditional quasi-palindrome strains.
GCR and mutagenesis rate estimation by fluctuation test
Strains were grown on YPD agar media for 3 days at 30°C for most of the experiments. URA3-PAL strains were grown on YPD or uracil-drop out medium. HIS4-PAL strains were grown on YPD and single colonies were cultured overnight in histidine-drop out medium supplemented with 2 mM of 3-AT. TET-RFA2 strains were grown on YPD with 62.5 μg/l of doxycycline. For each strain 12–24 independent colonies were selected for a fluctuation test (39). Appropriate dilutions of cells were plated on YPD and canavanine-containing plates (arginine-drop out medium containing a low amount of adenine (4 mg/l) and 60 mg/l of L-canavanine). White and red colonies grown on canavanine-containing media reflect CAN1 mutagenesis and GCR events, respectively. GCR and mutation rates were calculated as previously described (28).
DSB detection
Cells from overnight cultures were embedded in 0.75% low-melting agarose plugs at a concentration of 25 × 108 cells/ml. Each plug (∼4 × 108 cells/plug) was treated with 0.5 mg of zymolyase and 1 mg of proteinase K. Around one-fourth of a plug (∼40 μl) was used per sample for DSB detection either by separating chromosomes with contour-clamped homogeneous electric field (CHEF) electrophoresis or restriction digestion prior to standard gel electrophoresis. Each plug was equilibrated in the electrophoresis buffer prior to gel casting. The broken left arm of chromosome V (∼43 kb) was separated from intact chromosome V (∼580 kb) by CHEF for 26 h (or 28 h, Figure 4A) in a 1% PFGE certified agarose gel in 0.5× TBE at 14°C and 6 V/cm with an included angle of 120°, an initial switching time 3.14 s and final switching time of 7.68 s (or 12.56 and 17.53 s, respectively, Figure 4A). For restriction digestion of DNA, the plugs were washed twice in 1× TE (10 mM Tris-Cl, pH 8.0, 0.1 mM EDTA), once in 1× TE containing 1 mM of PMSF, once in water and equilibrated in 2× and then 1× restriction buffer. Each plug was digested with 50 units of AflII, BglII or BsrGI overnight. Digested plugs containing DNA were loaded in 1% (AflII digestion) or 0.8% (BglII and BsrGI digestions) agarose gels and run in 1× TBE for 18 and 20 h, respectively. The agarose gels containing the separated DNA fragments were treated consecutively with 0.25 N HCl, alkaline buffer (1.5 M NaCl, 0.5 M NaOH) and neutralization buffer (1.5 M NaCl, 1 M Tris, pH7.5). DNA was then transferred to a charged nylon membrane in 10× SSC for 2 h at 70 mmHg via Posiblotter (Stratagene). Southern hybridization was performed using a 32P-radiolabeled HPA3- or LYS2-specific probe in PerfectHyb Plus Hybridization Buffer at 68°C overnight. The membranes were washed twice in a buffer containing 0.1× SSC and 0.1% SDS at 68°C and were exposed to a phosphor storage screen. Densitometry analysis of the broken fragments was performed using ImageJ software (NIH).
Figure 4.

Role of Mus81/Mms4 on DSB formation at URA3-PAL. (A) Representative DSB detection at URA3-PAL in strains deleted for the indicated SSE. Detection has been carried out as described in Figure 1A and B. Chromosomes were separated by CHEF for 28 h. (B) Quantification of DSBs presented in (A). Values are the mean of at least three independent biological replicates ± standard deviation (SD); * P< 0.001. (C) Representative DSB detection at 5′ and 3′ URA3-PAL in sae2Δ and sae2Δ mus81Δ in presence (+) and absence (-) of uracil in the growth media. Detection has been carried out as described in Figure 1A and B. Chromosomes were separated by CHEF for 26 h. (D) Quantification of DSBs presented in (C). Values are the mean of at least three independent biological replicates ± standard deviation (SD); * P< 0.0001.
Replication intermediate analysis by 2DGE
Strains were inoculated in 400 ml of YPD and grown overnight. When OD600 reached 0.8, the cells were synchronized in G1 for 3 h with 0.1 μg/ml of alpha factor. For TET-RFA2 strains, 2 μg/ml of doxycycline was added to the medium to downregulate RPA. After washing the cells twice with water, cells were released into fresh YPD containing 12.5 μg/ml of pronase E. Fifty minutes post release, sodium azide was added to a final concentration of 0.1% and the cells were collected in the presence of 0.2 M frozen EDTA pH8 (20 mM final). Genomic DNA was extracted by the standard cesium chloride method using the protocol ‘Joel Huberman’s DNA isolation procedure with modifications by Bonny Brewer’ (https://fangman-brewer.genetics.washington.edu/DNA_prep.html) and DNA was processed as described in (40). DNA was digested with 50 units of AflII for several hours and precipitated in ethanol. For the first dimension, restriction digestion fragments were separated in a 0.4% agarose gel and run in 1× TBE at 1.7 V/cm for 22 h. For the second dimension, gel slices from the first dimension containing the fragment of interest, were loaded into a 1.2% agarose gel containing 0.3 mg/l of ethidium bromide. Gel electrophoresis was performed in 1X TBE containing 0.3 mg/ml of ethidium bromide at 6 V/cm for 10 h at 4°C. The gels were processed for Southern hybridization as described in the previous section. A LYS2-specific probe, corresponding to the ARS-proximal side of the IR, was used to highlight replication intermediates.
DSB formation across the cell cycle (conditional IR)
Cells were grown overnight in YPR supplemented with 300 mg/l G418. At an OD600 of 0.8, the cultures were either synchronized in G1 with 0.1 μg/ml of alpha factor or in G2/M with 15 μM nocodazole or 500 nM 1-NM-PP-1. For each time point, the volume of culture necessary to make a plug was transferred into a falcon tube containing sodium azide (0.1% final) and frozen 0.2 M EDTA pH8 (20 mM final). The first aliquot was taken after 3 h of synchronization and represents the condition (-) for Cre induction in the figures. Galactose (2% final) and estradiol (4 μM final) were added to the remaining cultures to induce Cre expression and additional alpha-factor was added to the G1 arrested cells. After 3 h of induction, an aliquot of cells was collected for FACS analysis and to make plugs. This time point is represented by the condition (+) for Cre induction for the G2/M arrested cells (with nocodazole or 1-NM-PP-1) and 0 min for the G1 arrested cells. To release G1 arrested cells into S phase, the remainder of the culture was washed twice with water and resuspended in fresh YPD containing 12.5 μg/ml of pronase E. Samples were taken 20, 40, 50, 70 and 90 min after release. Plugs were made and DSBs were revealed as described in the section ‘DSB detection.’ Plugs were digested either with AflII or BsrGI to detect the ARS-proximal or telomere-proximal side of the break, respectively, using the corresponding LYS2-specific probe.
Flow cytometry
For each time point, 1 ml of cells was harvested and fixed in cold 70% ethanol. An aliquot was collected by centrifugation and resuspended in 1 ml of 50 mM Tris, pH 8.0, 15 mM NaCl containing 2 mg/ml of RNase A. After 2 h of RNase treatment at 37°C, 25 μl of 20 mg/ml Proteinase K was added and the samples were incubated at 55°C for an additional hour. DNA was stained with 1× SYTOX green in 50 mM Tris pH 7.5. The samples were sonicated briefly and DNA content was measured using an Accuri C6 (Bio-Rad).
RT-qPCR
At least three independent colonies from each strain were inoculated in YPD and grown overnight. Cells were washed once with water and were diluted to an OD600 of 0.1 in the appropriate medium. URA3-PAL strains were cultured in YPD (+ Uracil condition) and uracil-drop out medium (- Uracil condition) and HIS4-PAL strains were cultured in YPD (+ Histidine condition) and histidine-drop out medium supplemented with 2 mM of 3-AT (- Histidine + 3-AT condition). After 4 h of growth, RNA was isolated using an Aurum Total RNA Kit (BIO-RAD) with the DNaseI treatment step lengthened to 45 min at 37°C. The absence of gDNA was confirmed by PCR using the extracted RNA as a template. Reverse transcription and RT-qPCR were performed using a Luna Universal One-Step RT-qPCR Kit (New England BioLabs) on a QuantStudio 6 Flex Real-Time PCR system (Applied Biosystems). Primers used are listed in Supplementary Table S4. mRNA levels were normalized to ACT1 to determine relative abundance (41). Significance was estimated by two sample t tests.
Quantification and statistical analysis
GCR and mutagenesis rate: All data presented are the median of the rates for at least 12 biological replicates and a 95% confidence interval is indicted (39).
DSB detection: For each graph, the nature of the values and errors bars is mentioned in the figure legends. All data presented are the average of at least three biological replicates. Bands were visualized by phosphorimaging (Typhoon, GE Healthcare) and quantified with ImageJ software. Percent of DSB corresponds to the DSB signal normalized to the corresponding unbroken fragment/chromosome V. Data were analyzed and graphed with Prism8. Statistical analysis was performed using a t test. When error bars correspond to the 95% confidence interval, statistics are not indicated.
RESULTS
Experimental system to study DSB formation at palindromic repeats
The experimental system to monitor DSB formation at IRs is based on the sensitive GCR assay described in (10) (Figure 1A). Briefly, the 43 kb region between telomere and CAN1 on chromosome V does not contain any essential genes and can be lost without affecting cell viability. ADE2 is placed between CAN1 and the IR to differentiate between DSB-mediated arm loss (leading to GCRs) and CAN1 mutations. GCR isolates are canavanine-resistant red (Ade-) colonies, while mutations in CAN1 give rise to canavanine-resistant white colonies. The spontaneous rate of arm loss in control strains without repeats, is extremely low: 3 × 10–9/division. Four different repeats were inserted in inverted orientation into LYS2: 320 bp Alu, 1.3 kb IS50, and actively transcribed 1.1 kb URA3 and 2.7 kb HIS4. Repeats are either separated by a spacer (abbreviated as -QP) or have no spacer (abbreviated as -PAL). Overall, insertion of interrupted or perfect palindromes next to CAN1 causes a dramatic (>10 000-fold) increase in the GCR rate (Table 1). The deduced mechanism for IR-induced GCRs is the formation of hairpin-capped breaks, that after DNA synthesis, result in dicentric dimers, followed by breakage in anaphase and repair involving break-induced replication using a non-homologous chromosome as a template (10).
Figure 1.

DSBs at perfect but not quasi palindromes are partly induced by Sae2/MRX. (A) Scheme of IRs located on chromosome V. Different palindromes cloned into LYS2 have been inserted on the left arm of chromosome V, ∼26 kb away from ARS507. Secondary structures at IRs lead to the formation of hairpin-capped breaks (∼43 kb), duplication of which leads to the formation of an inverted dimer twice the break size (∼86 kb). DSBs can be revealed using Southern blot and hybridization after chromosome separation by CHEF. HPA3 probe (gray rectangle) is used to highlight the different chromosomal fragments. IRs with the corresponding size of the repeat are indicated. (B) Representative image of DSB detection at different IRs in WT and sae2Δ. Genomic DNA embedded in agarose plugs was separated by CHEF. Chromosomal fragments (indicated by black arrows) correspond to unbroken chromosome V (585 kb), dimer (86 kb) and break (43 kb). Due to a lower DSB frequency at QP compared to perfect palindromes, a higher exposure of Alu-QP is presented on the left. (C) Representative image of DSB detection at Alu-PAL in WT and in the absence of MRX-Sae2 complex. Detection and analysis were performed as in (B). (D and E) Quantification of DSBs presented in (B) and (C), respectively. Values are the mean of at least three independent biological replicates ± standard deviation (SD); * P< 0.02.
Table 1.
Effect of palindromic sequences on GCRs in WT and sae2Δ
| GCR rate (×10−7) | ||
|---|---|---|
| lys2-inserted repeat | WT | sae2Δ |
| No repeat a,b | 0.03 (0.02–0.04)c | 0.4 (0.3–0.6) |
| Alu-QP b | 617 (559–691) | 1343 (1134–1717) |
| Alu-PAL b | 7000 (5017–9943) | 7829 (6560–9459) |
| IS50-PAL | 8600 (4190–8620) | 7200 (5840–8060) |
| URA3-PAL b | 850 (680–960) | 13460 (11090–15570) |
| HIS4-PAL | 8620 (8200–9710) | 9750 (8700–11220) |
aRates previously published (Saini et al., 2013).
bCell plating efficiency was assayed in these strains and was ∼ 95–100%.
cNumbers in parenthesis correspond to the 95% confidence interval.
MRX/Sae2 attacks secondary structures at perfect palindromes but not quasi-palindromes separated by a 12 bp spacer
We have previously shown that Alu-QP with a 12 bp spacer located on chromosome II induces hairpin-capped DSBs. We have also found that the Mre11 complex does not trigger DSBs at Alu-QP but is required along with Sae2 to process hairpin termini (28). This is consitent with Sae2 being a regulator of the Mre11 endonuclease activity (42). A defect in hairpin opening results in replication of broken fragments and formation of inverted dimers, including a dicentric chromosome. Accordingly, Alu-QP-induced GCRs are increased in mre11Δ, rad50Δ (6,10) and sae2Δ (Table 1). The frequency of breakage on chromosome V was similar (∼1%) in WT and sae2Δ (Figure 1B), consistent with previous studies of Alu-QP-induced DSBs on chromosome II (28). IR-induced DSB formation is always accompanied by the appearance of inverted dimers. In WT, resected broken arms run with delay and show a spread signal compared to unresected broken arms in sae2Δ strains (43,44). This explains why DSBs may seem more visible in sae2Δ albeit DSB quantification is similar to WT. In strains containing Alu-PAL, GCR rates and chromosomal breakage were greatly increased, compared to Alu-QP (Table 1 and Figure 1B). When SAE2 or MRE11 were disrupted, we observed 50–70% reduction in the Alu-PAL-mediated breaks (Figure 1B–E). In these mutants, broken molecules migrated as a discrete band and faster than resected breaks in WT, indicating the presence of unprocessed hairpin termini (43,44). Similarly, the breakage level at IS50-PAL, URA3-PAL and HIS4-PAL ranged from 8 to 30%, and sae2Δ caused a 2–3-fold reduction in DSB formation (Figure 1B–D). Remarkably, URA3-PAL exhibited the strongest level of fragility: approximately one-third of the WT cells carried broken chromosome V. Nevertheless, viability of these strains was not affected and URA3-PAL was stably maintained suggesting recurring breakage and restoration of the palindrome (see ‘Discussion’ section). Notably, in sae2Δ strains, high levels of URA3-PAL-induced breaks correlated with high levels of GCRs, in contrast to WT (Table 1)
From these observations we can draw several conclusions. First, MRX/Sae2 has a dual role in IR-induced DSBs metabolism: it is responsible for ∼30 to 50% of breaks formed at perfect but not at quasi-palindromes and is required for processing hairpin-capped breaks induced by both types of IR. Notably, despite the strong effect on DSB formation, disruption of SAE2 in strains with perfect palindromes did not lead to a decrease in GCRs (Table 1), indicating that MRX/Sae2-mediated breaks are not channeled into GCRs in contrast to hairpin-capped breaks. Second, there are at least two pathways for DSBs formation at IRs: an MRX/Sae2-dependent pathway operating only at perfect palindromes and an MRX/Sae2-independent pathway that operates at both types of IR, yet has a stronger effect at perfect palindromes.
Spacer length determines Alu-IR DSB potential, susceptibility to the MRX/Sae2 attack and ability to block replication forks
To identify structural parameters governing IR stability and MRX/Sae2 attack, we generated strains with Alu-QPs separated by spacers ranging from 5 to 12 bp. Since even in the case of perfect palindromes 4–5 nt remain unpaired in the hairpin loop due to stereochemical constraints (45,46), Alu-QP with 1–4 bp spacers were omitted from the analysis. The spacers contained the nonpalindromic SfaNI restriction site and random nucleotides (Supplementary Figure S1A). We refer to each quasi-palindrome as Alu-QP, followed by the spacer length. The impact of each spacer on GCRs and DSBs was assessed in WT and sae2Δ.
In WT, DSB levels at Alu-QP5 and 6 were only slightly decreased compared to Alu-PAL. Alu-QP7 and 8 showed a drop of ∼50% in the frequency of breaks (∼5%) whereas Alu-QP9 showed a drop of 80% (Figure 2A and B). The length of the spacer had a similar impact in sae2Δ. However, the DSB level was reduced in sae2Δ, compared to WT, at Alu-PAL and Alu-QPs5–8 but not at Alu-QPs with longer spacers (Figure 2A and B). This indicates that MRX/Sae2 attack occurs at hairpins with loops <9 nt.
Figure 2.

Analysis of the ability of IRs to induce DSBs and replication fork block according to the spacer length. (A) Scheme of Alu-PAL (0 bp) and Alu-IRs separated by 5 to 12 bp asymmetrical spacers (top panel) and representative image of DSB detection at these IRs (bottom panel). Detection was performed as described in Figure 1A and B. (B) Quantification of DSBs in (A). Values are the mean of at least six independent biological replicates ± standard deviation (SD); * P< 0.02. (C) 2D gels analysis of replication intermediates (RIs) in strains containing the indicated Alu-IRs. Illustration of restriction digestion with AflII and the position of the probe used to reveal RIs (black rectangle) is depicted on the top. Bottom panel: Representative RIs analysis in the indicated Alu-IRs. Scheme of RIs analysis is depicted on the left. (D) DSB detection in WT (TET-RFA2) and sae2Δ (TET-RFA2 sae2Δ). Detection was carried out as described in Figure 1A and B. (E) Quantification of DSBs in TET-RFA2 strains (WT and sae2Δ); * P< 0.04. (F) Replication intermediate analysis in WT and sae2Δ strains containing Alu-PAL and in TET-RFA2 strains with Alu-QP9 and 10.
GCRs analysis showed the same inverse correlation between the spacer length and fragility, in both WT and sae2Δ (Supplementary Table S1). While disruption of SAE2 in strains with Alu-PAL or Alu-QPs5–8 did not lead to a significant decrease in GCRs, Alu-QPs with spacers ≥9 bp exhibited higher GCR rates in sae2Δ compared to WT. We confirmed this observation using Alu-QPs with a different intervening sequence containing the nonpalindromic BsmI restriction site (Supplementary Figure S1B), thus excluding an effect due to the sequence composition (Supplementary Table S2). We concluded that MRX/Sae2 plays a role in DSB formation at Alu-QP separated by up to 8 bp whereas IRs with longer spacers are insensitive to MRX/Sae2 attack.
Stable secondary structures formed by IRs cause replication fork pausing (6,47). Therefore, we analyzed the impact of spacer length on replication fork progression using 2D gel electrophoresis of replication intermediates. Strains with Alu-PAL and Alu-QP8 showed a robust fork arrest at the location of the repeats, as evidenced by the accumulation of molecules at one point along the replication arc (Figure 2C; Supplementary Figure S2C and D). Similar to Alu-QP12 (6), we did not detect fork pausing in strains harboring Alu-QP9 or 10. This shows that MRX/Sae2 attack on secondary structures and their capacity to hinder fork progression are correlated. We propose that stable hairpins on the lagging strand template are formed when IRs are separated by <9 bp. Overall, our data suggest that the length of the spacer is the main structural feature that determines MRX/Sae2 attack and predisposition to strongly block replication, with an 8 bp spacer being the transition point.
Rfa2 downregulation destabilizes Alu-IRs and mitigates spacer effect
Based on the data presented above, we reasoned that Alu-PAL and Alu-QP may form hairpins in single-stranded DNA (ssDNA) on the lagging strand template with a comparable efficiency, but hairpin loops >8 nt may be targeted by proteins involved in secondary structure dissolution. Replication protein A (RPA) is one of the possible players in this process. RPA is an essential eukaryotic ssDNA-binding protein playing pivotal roles in DNA metabolism. RPA can efficiently bind ssDNA stretches with an 8 nt initial binding site (48,49) and is known to be involved in destabilizing secondary structures such as hairpins (50,51). We previously showed that downregulation of one RPA subunit, Rfa2, increases Alu-QP12 fragility (6). The natural RFA2 promoter was replaced by a tetO-repressible promoter to create the strain referred to as TET-RFA2. Downregulation was induced by adding doxycycline in the media (6). In TET-RFA2 strains, that is, upon Rfa2 downregulation, all IRs showed the same high level of DSBs (35–40%) regardless of the spacer length (Figure 2D–E). Surprisingly, DSBs were decreased not only at Alu-PAL but also at all Alu-QPs in sae2Δ TET-RFA2 (Figure 2D–E). Similar frequencies of breakage were observed in Alu-PAL and Alu-QP7–9 (18–23%) in sae2Δ strains, whereas break level was affected to a greater extent at Alu-QP10 (12%).
We noticed that Rfa2 downregulation affected DSB levels in Alu-PAL and Alu-QP9 and 10 in WT strains to different extents. TET-RFA2 strains exhibited a 3.6-fold increase in breakage at Alu-PAL compared to RFA2 proficient strains, whereas a ∼20-fold increase was observed at Alu-QP9 and 10 (Supplementary Figure S2A). This difference was not as pronounced in sae2Δ strains (Supplementary Figure S2B). These data indicate that RPA both stabilizes Alu-IRs in general and prevents MRX/Sae2 attack at Alu-IRs separated by >8 bp.
We then tested if Alu-QP9 and 10 acquire the ability to arrest replication forks when RPA is downregulated. In contrast to WT strains, a strong replication fork arrest was observed at Alu-QP9 and 10 in TET-RFA2 strains (Figure 2F; Supplementary Figure S2C and D).
Altogether, these data suggest that in the absence of RPA, Alu-QPs with spacers longer than 8 bp form stable secondary structures that can both arrest replication forks and become targets for MRX/Sae2.
Breaks occurring at Alu-PAL are symmetrical and not affected by known SSEs
Accumulation of DSBs in sae2Δ (Figure 1B and C) demonstrates the existence of another pathway for break formation at perfect palindromes. We analyzed the effect of different SSEs on chromosomal fragility induced by Alu-PAL in sae2Δ strains. We assayed the involvement of SSEs capable of processing Holliday junctions or other branched DNA structures: Mus81/Mms4, Yen1, Slx4/Slx1, Rad1/Rad10 and Mlh1/Mlh3 (reviewed in (52,53)). GCR analysis revealed that the deletion of MUS81, YEN1, SLX4, RAD1 or MLH1 did not significantly affect GCR rates (Supplementary Figure S3A). This indicates that none of these nucleases is solely responsible for MRX/Sae2-independent DSBs induced at Alu-PAL. Consistent with the GCR analysis, DSB detection did not reveal involvement of the above SSEs in Alu-PAL fragility. DSBs were detected using an ARS- and a telomere-proximal probe upon digestion of agarose embedded chromosomal DNA with AflII and BglII, respectively (Figure 3A and B, top panels). The size of the fragment corresponding to DSBs occurring at Alu-PAL was expected to be 1.3 kb upon AflII digestion and detection using the ARS-proximal probe. A 3.2 kb fragment was expected upon BglII digestion and detection using the telomere-proximal probe. Replication of hairpin-capped breaks leads to inverted dimers twice the size of the breaks, which are manifested by the appearance of a 2.6 or 6.4 kb fragment upon AflII or BglII digestion, respectively. The results presented in Figure 3 revealed that DSBs induced at Alu-PAL can be detected from both sides of the repeat. Deletion of MUS81, YEN1, SLX4, RAD1 and MLH1 did not significantly affect the accumulation of breaks.
Figure 3.

DSBs at Alu-PAL are symmetrical and not affected by known SSEs. (A and B) Detection of the ARS- and telomere proximal DSBs, respectively, formed at Alu-PAL in sae2Δ strains deleted for the indicated SSE. Genomic DNA embedded in agarose plugs was digested with AflII (A) or with BglII (B). The relative position of the repeat and the restriction sites are indicated. The solid bar indicates the position of the LYS2-specific probes. The bands corresponding to the unbroken fragment, the dimer and the DSB are indicated on the representative images for DSB detection. The DNA ladder, highlighted with ethidium bromide prior Southern blot hybridization, with indicated sizes is shown on the left. (C and D) Quantification of DSBs presented in (A and B), respectively, relative to unbroken chromosome V. Values are the mean of at least three independent biological replicates ± 95% confidence intervals.
The simultaneous presence of broken molecules from both sides of Alu-PAL indicates a cleavage of the cruciform at or near its axis of symmetry. This conclusion is strongly supported by analyzing DSB formation at IRs specifically induced in the G2 phase (see below).
Mus81/Mms4 is involved in breakage at convergently transcribed palindromes
Among all perfect palindromes analyzed, URA3-PAL exhibited the strongest fragility in both WT and sae2Δ strains (Figure 1B). GCR rates and DSB frequencies remained unchanged upon YEN1, SLX4, RAD1 or MLH1 deletion in sae2Δ strains (Figure 4A and Supplementary Figure S3B). However, deficiency in Mus81 led to a modest, ∼2-fold, but reproducible reduction in GCRs (Supplementary Figure S3B) and DSBs induced by URA3-PAL (Figure 4B). Similarly, the GCR rate was ∼2-fold lower in sae2Δ mus81Δ than in sae2Δ at HIS4-PAL (Supplementary Figure S4C). This effect was specific to URA3- and HIS4-PAL since chromosomal fragility in Alu- and IS50-PAL strains was not affected in Δmus81 (Supplementary Figure S3A and C). The notable difference between these palindromes is that URA3- and HIS4-PAL consist of genes with active promoters located at the center of the IRs (from here on, we refer to this orientation by 5′URA3-PAL and 5′HIS4-PAL). This suggests that transcription can be a modifier of palindrome fragility and susceptibility for Mus81/Mms4 attack. Interestingly, the breakage potential of these two transcriptionally active palindromes is different when cells are propagated in YPD media: 30% (WT) and 10% (sae2Δ) at 5′URA3-PAL vs 8% (WT) and 4% (sae2Δ) at 5′HIS4-PAL (Figure 1B and C). Since breakage and repair might interfere with gene expression (54), we quantified the relative amount of mRNA expressed by the pairs of URA3 and HIS4 genes in sae2Δ mus81Δ. We found that the expression level of URA3 pair is 3.5-fold higher than HIS4 pair (Supplementary Figure S4A). This reinforces the idea that transcription at URA3-PAL fosters DSBs formation.
To assess the role of transcription on 5′URA3-PAL fragility, we analyzed the GCR rate and DSB level upon uracil starvation. It is known that in this condition URA3 expression is induced (55). RT-qPCR analysis confirmed that the transcription level of URA3 IRs was stimulated by a factor of 2.5 in the absence of uracil (Supplementary Figure S4A). The GCR rate in sae2Δ showed a ∼7.5-fold increase upon uracil starvation and deletion of MUS81 in sae2Δ led to a significant ∼5-fold decrease (Supplementary Figure S4B). Consistently, DSB detection at 5′URA3-PAL in sae2Δ showed a significant increase in breakage level when the strains were grown in the absence of uracil (Figure 4C and D). In sae2Δ mus81Δ, DSB levels were similar in presence or absence of uracil (Figure 4C and D). Taken together, these results show that transcription-related DSBs are strongly dependent on Mus81/Mms4 and indicate that the complex attacks a structure whose formation is driven by transcription.
To gain insight into transcriptionally driven secondary structure formation, we analyzed the impact of transcription orientation at URA3-PAL. We flipped the orientation of the URA3 genes so that transcription is directed towards the center of symmetry to create the palindrome referred to as 3′URA3-PAL. In this construct, sae2Δ strains exhibited a much lower DSB level (∼5%) compared to 5′URA3-PAL (∼10%). In contrast to 5′URA3-PAL, neither transcription stimulation nor MUS81 deletion had a profound impact on DSB levels at 3′URA3-PAL (Figure 4C and D). Uracil starvation induced a small but significant, 3-fold increase in GCRs in both sae2Δ and sae2Δ mus81Δ (2.9 and 2.7-fold, respectively) at 3′URA3-PAL. This increase did not, however, translate into an increase in DSB level. Deletion of MUS81 reduced the GCR rates by only 1.5- and 1.7-fold in the presence and the absence of uracil, respectively (Supplementary Figure S4B).
To confirm that substrate formation for Mus81/Mms4 cleavage at divergently transcribed palindromes is not restricted to URA3-PAL, we performed a similar analysis using the other transcribed palindrome, HIS4-PAL. HIS4 induction was achieved by growing cells in medium lacking histidine and supplemented with 3-amino-1,2,4-triazole (3-AT). 3-AT is a competitive inhibitor of an enzyme involved in histidine production and is known to increase HIS4 expression (56). We confirmed that growing cells in this medium stimulated the expression of the two HIS4 genes (Supplementary Figure S4A). Similar to 5′URA3-PAL, in sae2Δ strains, HIS4 induction led to a 4-fold increase in GCRs induced by 5′HIS4-PAL. Transcription-related GCRs were significantly decreased, by 2-fold, in sae2Δ mus81Δ compared to sae2Δ. GCRs rates were not impacted by HIS4 induction at 3′HIS4-PAL in sae2Δ and sae2Δ mus81Δ (1.3- and 1.7-fold increase, respectively). Deletion of MUS81 reduced the GCR rates by only 1.7- and 1.4-fold in the presence and the absence of histidine, respectively (Supplementary Figure S4C). Overall, these results are highly consistent with the fragility observed at URA3-PAL.
We concluded that the fragility potential at transcribed palindromes resulting from Mus81/Mms4 attack is mainly governed by both the level and the orientation of transcription.
Cruciform extrusion and subsequent DSB formation can occur independently of replication
Here we aimed to determine at which stage of the cell cycle MRX/Sae2-independent and Mus81/Mms4-mediated DSBs at IRs occur. Because DSB formation at a given phase of the cell cycle can be concealed by the existence of pre-formed DSBs, we built a URA3-based Cre-loxP conditional quasi-palindrome system. The strains contain 1 kb 5′URA3-IRs separated by a 1.5 kb kanMX gene flanked by loxP sites (URA3-loxP-kanMX-loxP-URA3, Figure 5A and Supplementary Figure S5A). A long spacer between repeats greatly compromises the potential to adopt a secondary structure and generate DSBs. Hence, the advantage of this system is that the pre-existing populations of DSBs and dimer intermediates are absent prior to the creation of the quasi-palindrome. The second component of the system is Cre recombinase fused in frame with the estradiol-binding domain (EBD) under the control of a GAL1 promoter. The fusion construct is integrated into the LEU2 locus on chromosome III (57). Upon induction with galactose and estradiol, the functional Cre recombinase is expressed, which then excises the kanMX leaving a single, 34 bp, palindromic loxP site with an 8 bp spacer (Supplementary Figure S5A). Thus, expression of the Cre recombinase and subsequent kanMX cassette loss lead to the creation of a URA3-QP8 (URA3-loxP-URA3). By inducing Cre expression, and thus URA3-QP8 formation, we followed the appearance of DSB intermediates in G1 (cells arrested with α-factor), S (cells released from G1 arrest) and G2/M (cells arrested with nocodazole) phases in sae2Δ and sae2Δ mus81Δ strains. The cell cycle stage was determined by monitoring cellular morphology and by flow cytometry (Supplementary Figure S5B).
Figure 5.

Analysis of DSB formation through the cell cycle in the conditional URA3-quasi palindrome. (A) Diagram of the conditional URA3-QP system. URA3 genes (black arrows) are separated by the kanMX gene (gray arrow) flanked by loxP sites (red double arrowhead and gray triangle). This construct, URA3-loxP-kanMX-loxP-URA3, is located on chromosome V. The construct, pGAL1-Cre-EBD, is integrated into the LEU2 locus on chromosome III. kanMX excision leaves a 34 bp single quasi-palindromic loxP site with an AT-rich spacer. The resulting sequence, URA3-loxP-URA3, is a palindrome separated by 8 bp (gray bar). (B) DSB formation during the cell cycle in sae2Δ and sae2Δ mus81Δ. Cells were arrested in G1 (with α-factor) or G2/M (with nocodazole) prior to Cre-EBD expression. After 3h of incubation in media containing galactose, G1 arrested cells were released in fresh media and cells were collected at different timepoints, indicated by minutes after G1 release, for DSB analysis. G2/M arrested cells were collected after Cre expression. Genomic DNA embedded in agarose plugs was digested with AflII. The position of the resulting quasi-palindrome (URA3-loxP-URA3) relative to AflII restriction site is represented. Southern hybridization was performed using a LYS2-specific probe located on the ARS-proximal side of the IR (black bar). The DNA ladder, highlighted with ethidium bromide prior Southern blot hybridization, with indicated sizes is shown on the left. (C) Analysis of DSB formation in G2 phase in sae2Δ and sae2Δ mus81Δ upon Cdc28 inactivation. DSB analysis was performed like in (B). Cells were arrested in G2/M either with nocodazole in CDC28 or cdc28-as1 or with 1-NM-PP-1 inhibitor in cdc28-as1. The DNA ladder, highlighted with ethidium bromide prior Southern blot hybridization, with indicated sizes is shown on the right.
DSBs were revealed using an ARS-proximal probe upon digestion of agarose embedded chromosomal DNA with AflII (Figure 5B, top panel). The size of the fragment corresponding to DSBs occurring at URA3–8QP was expected to be 2.4 kb. In addition, a dimer of 4.8 kb was expected because of the hairpin-capped nature of the break. In uninduced conditions (-), a 7.6 kb fragment corresponding to URA3-loxP-kanMX-loxP-URA3 was the predominant species highlighted and no DSBs were observed. Upon Cre induction (+), we observed the predominance of a 6.1 kb fragment devoid of kanMX cassette that corresponds URA3-loxP-URA3.
DSB analysis in sae2Δ showed that DSBs started to accumulate between 40 and 50 min (late S phase) after the cells were released from G1. DSB levels were even more pronounced at 70 and 90 min post-release (G2/M). In sae2Δ mus81Δ, DSBs appeared with the same dynamics, though the DSB level was lower upon MUS81 deletion (Figure 5B bottom panel and Supplementary Figure S5C). These data show that DSBs at URA3-QP8 are generated by Mus81/Mms4 and another SSE after the bulk of DNA replication and suggest the DSBs accumulate in G2. Recently, Ivanova et al., 2020 demonstrated that hard-to-replicate sequences such as G4 structures can be replicated when chromosome segregation has begun, in anaphase. This can explain accumulation of DSBs 90 min after α-factor release (58). However, breaks are also induced in nocodazole-arrested cells, excluding the contribution of replication. The fact that a high level of DSBs is observed in nocodazole-arrested cells strongly suggests that the breaks occur during the G2 phase. Interestingly, inverted dimers were observed in G2-arrested cells suggesting that they can result from DNA synthesis of the broken molecule outside of S-phase.
We also analyzed the symmetry of the breaks occurring at URA3-QP8 by cutting DNA with BsrGI and probing for the telomere-proximal side of the break (Supplementary Figure S5D). This revealed accumulation of DSBs at 50, 70 and 90 min after G1 release and also in nocodazole-arrested cells. This confirms the two-ended nature of the breaks at URA3-QP8 and proves that cruciform resolution is the mechanism by which DSBs are generated. These data provide direct evidence that DSBs are driven by cruciform formation and resolution in G2.
Cdk regulates Mus81-mediated breaks in G2 but not the third pathway for the cruciform resolution
To further support a replication-independent break formation at IRs, we used another way to arrest cells in G2, prior to Cre induction. We used the conditional hypomorphic cdc28-as1 allele that encodes a form of Cdc28Cdk1 that can bind the nonhydrolyzable ATP analog 1-NM-PP1. Treatment of cells harboring the cdc28-as1 allele with low doses of 1-NM-PP1 results in a specific inhibition of Cdc28 and causes cells to arrest in G2 (59).
DSB detection revealed that breaks are formed in sae2Δ in both nocodazole- and 1-NM-PP1-arrested cells (Figure 5C, top panel), confirming that breaks indeed occur in G2 phase. However, 1-NM-PP1-treated cdc28-as1 cells showed a decrease in DSB level, compared to the nocodazole-treated cells. Since Cdc28 potentiates Mus81/Mms4 (60,61), the decrease in break level reflects the inactivity of Mus81/Mms4 in 1-NM-PP1-treated cdc28-as1 cells. Indeed, DSB analysis in sae2Δ mus81Δ revealed that inactivation of Cdc28 did not further decrease DSB formation (Figure 5C, bottom panel and Supplementary Figure S5E). Strikingly, the dimer formation in 1-NM-PP1-treated cdc28-as1 cells was strongly decreased, if not entirely absent, in both sae2Δ and sae2Δ mus81Δ (Figure 5C). This suggests that one or several factors involved in hairpin-capped break duplication are under the control of Cdc28.
Overall, these data allow three main conclusions to be drawn: (i) breaks at URA3-QP8 are the result of a symmetrical cleavage of a cruciform structure by Mus81/Mms4 and a putative SSE, (ii) this cleavage occurs independently of DNA replication and (iii) MRX/Sae2- and Mus81/Mms4-independent breaks are perpetrated by a yet unknown nuclease whose activity is upregulated in G2, but is not under control of the Cdc28 kinase.
DISCUSSION
In this study, we have investigated the role of SSEs in generating chromosome breaks at palindromic sequences in yeast. We uncovered three distinct pathways by which secondary structures can initiate DSBs and promote chromosomal instability. Very specific structural characteristics of inverted repeats define the nuclease attack mediated by MRX/Sae2 and Mus81/Mms4. Moreover, we obtained direct evidence for cruciform formation and resolution, a pathway that operates for all types of inverted repeats and which involves an unknown nuclease. A model for DSB formation at perfect and interrupted palindromes is presented in Figure 6.
Figure 6.
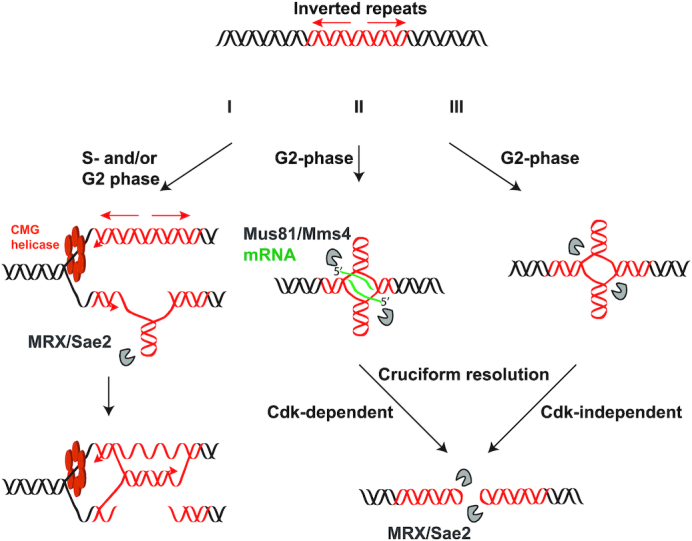
Model for DSB formation at palindromic sequences during the cell cycle. See ′Discussion’ for details.
MRX/Sae2 plays a dual role in the instability of palindromes
We revealed a specific role for MRX/Sae2 in DSB generation at IRs with spacers from 0 to 8 bp. Remarkably, strong replication arrest occurs at IRs prone to MRX/Sae2 cleavage. Therefore, it is likely that stable hairpins formed on ssDNA-containing lagging strand template are attacked by the complex (Figure 6I). These breaks are repaired using the sister chromatid as a template and do not generate GCRs, thus explaining the absence of decrease in GCR rates upon SAE2 deletion (Table 1). We also noticed that the WT strain with the most fragile palindrome (URA3-PAL) exhibited pink color when propagated on YPD, which indicates partial inactivation of ADE2 expression, likely due to ongoing breakage and repair. These observations are in favor of recurrent DSB formation and restoration of palindromic sequence since palindromes are stably maintained and viability is not affected in these strains.
Even though MRX/Sae2 cleaves replication-born hairpins that stall fork progression, SAE2 deletion does not lead to a stronger fork arrest (Figure 2F). This suggests that (i) fork pausing imposed by secondary structures at palindromes is a surmountable barrier and that (ii) MRX/Sae2 attack on hairpins occurs later, thus generating a two-ended DSB, which is consistent with the mode of action of SbcCD (25). Overall, the mechanism of MRX/Sae2-mediated breaks at palindromic sequences demonstrated in this study is very similar to the mechanism of palindrome breakage by SbcCD in bacteria described in pioneering studies of Leach and colleagues (1). MRN has also been implicated in DSB formation at palindromes in fission yeast (3,4). Thus, MRX/Sae2 DNA-structure processing properties at IRs are evolutionary conserved and conceivably, MRN/CtIP in mammals possesses the same avidity for cleaving secondary structures.
MRX/Sae2 is responsible for ∼50% of total DSBs occurring at palindromes. MRX-Sae2-independent breaks occur symmetrically and yield dimers, which is indicative of cruciform resolution and the hairpin-capped nature of the break. Thus, similar to the breaks induced at Alu-QP12 (28), hairpin-capped DSBs triggered at perfect palindromes require the Mre11 endonuclease activity for opening and resection.
RPA safeguards IR stability according to the spacer length
We showed that DSB level decreases as the spacer length increases. However, DSB levels remained steady as the spacer length increased from 9 to 12 bp (Figure 2A and B). Why MRX/Sae2 does not target secondary structures formed at Alu-QP with >8 bp spacer is intriguing since Mre11 has the ability to cleave loops as long as 30 nt in vitro (62). Upon downregulation of RPA, Alu-QPs9–10 behaved like perfect palindromes in terms of DSB frequency, MRX/Sae2 attack, and the ability to arrest replication forks (Figure 2D–F). Hence, the apparent absence of MRX/Sae2 effect on DSB formation at Alu-QPs9–10 is likely due to the decreased stability and/or probability of formation of secondary structures with loops >8 nt. We propose that RPA affects both processes. When the loop size reaches 9 nt, RPA might directly destabilize hairpin structures or participate in recruiting proteins involved in DNA unwinding. For example, RPA stimulates Sgs1 unwinding activity (63) and sgs1 mutants show an increase in both GCRs and DSB formation at Alu-QP12 (6) as well as an increase in recombination induced by a quasi-palindrome with 10 bp spacer (35). The scenario where RPA plays a specific role in preserving stability of quasi-palindromes is strengthened by the fact that RPA depletion augmented MRX-Sae2-dependent DSB formation by up to 20-fold at quasi-palindromes versus only 4-fold at the perfect palindrome. Binding of RPA to ssDNA loops ≤ 8 nt seems unlikely. However, a defect in RPA binding during DNA replication may lead to uncoupling between the lagging and leading DNA strand synthesis and result in a greater ssDNA exposure, thereby producing favorable conditions for hairpin formation. Indeed, instability of Alu-QP12 and Alu-QP100 in replication defective mutants prone to ssDNA accumulation, such as polymerase δ, is dramatically increased (6,28). The fact that DSB levels were similar between the perfect and quasi-palindromes in TET-RFA2 (∼40%) argues in favor of the same mechanism increasing secondary structure formation and DSB potential, i.e. presence of unprotected ssDNA leading to hairpin formation. This is in agreement with RPA having a protective role against hairpin formation.
Mus81/Mms4 targets preferentially transcribed palindromes
We demonstrated that breakage at perfect palindromes composed of actively transcribed URA3 or HIS4 genes is partially dependent on Mus81/Mms4. Transcription initiated near the center of symmetry, but not at the extremities, enhances susceptibility of palindromes to breakage, either by stimulating cruciform extrusion by creating local, negative DNA supercoiling and/or by generating a substrate targetable by Mus81/Mms4. It is well-established that in vitro Mus81/Mms4 prefers DNA substrates that have a free 5′ end over closed substrates such as perfect 4-way junctions (53). It is possible that the presence of an RNA molecule (specifically providing an unannealed 5′ end) at the DNA secondary structure promotes cleavage by Mus81/Mms4 (Figure 6II). The fact that breakage levels in sae2Δ mus81Δ at 5′URA3-PAL and in sae2Δ at nontranscribed palindromes (Alu- and IS50-PAL) are approximately the same, favors the idea that Mus81/Mms4 recognizes a branched DNA/RNA structure instead of transcription simply driving cruciform extrusion. Using a conditional quasi-palindrome system, we also demonstrated that Mus81/Mms4 attacks cruciform structures, likely comprising RNA molecules, in G2 stage.
Several groups reported an involvement of Mus81/Mms4 in generation of breaks at hairpin/cruciform-forming repeats (30,32,33). It is worth noting that the common feature of the repeats investigated in these studies is AT-richness. Poly-AT sequences are poorly bound by histones (64–66), which generates nucleosome-depleted regions where transcription is often initiated (67–69). In light of our data, we suggest that transcription taking place at AT-rich repeats signals Mus81/Mms4 attack.
Cruciform extrusion and resolution are G2-mediated events
Cruciform extrusion in vivo in E. coli on plasmids has been reported in several studies (70,71) and cruciform extrusion has been proposed to occur in eukaryotic cells. For example, a replication-independent model for cruciform formation and cleavage was proposed to explain mechanism of t(11;22) translocation (72,73). However, experimental support for cruciform extrusion occurring in a chromosomal context was lacking. Using a novel conditional quasi-palindrome assay, we provide here direct evidence that in vivo conditions are sufficient to support cruciform extrusion in a chromosomal context in budding yeast (Figure 5). We show that susceptibility of palindromic sequences to breakage emanates in a timely manner, with cruciform extrusion and resolution occurring independently of replication, in G2 phase. It remains to be determined if cruciform resolution in G2 happens because the activity of the SSEs targeting the cruciform structure peaks in G2 and/or conditions of torsional strain leading to cruciform extrusion are met specifically in G2.
Mus81/Mms4-independent breaks appear with the same kinetic as the Mus81-induced DSBs, suggesting that the putative enzyme instigating the breaks is subject to the same regulation but is not subject to Cdc28 control. Strikingly, when we addressed the genetic dependency of DSBs at Alu-PAL, none of the known SSEs emerged as solely responsible for generating hairpin-capped breaks. Importantly, our conclusion is unequivocal since it is based on the physical detection of the chromosomal breaks, which is the most stringent criteria to assess the involvement of SSEs in DSB formation. We propose that a discrete pathway involving a putative nuclease is responsible for generating the MRX/Sae2 and Mus81/Mms4-independent DSBs (Figure 6III). Although we cannot completely exclude it, we do not favor the scenario where known SSEs play a redundant role in cruciform resolution. This premise is supported by the fact that inactivation of Cdc28, which is involved in the positive (Mus81, Slx4 (74,75)) and negative (Yen1 (75,76)) regulation of several nucleases, did not affect DSB formation. We also noticed that triple nuclease knockout strains grow poorly and quickly accumulate suppressors. Therefore, to address redundancy in cruciform cleavage adequately, special approaches, such as the use of auxin-regulatable conditional alleles (77), are required. Experiments are underway to identify the factor(s) involved in the proposed third pathway.
DATA AVAILABILITY
Flow cytometry data were deposited with Flow Repository under ID FR-FCM-Z39K.
Supplementary Material
ACKNOWLEDGEMENTS
We are very thankful to Dr Natalya P. Degtyareva and Dr Dmitry A. Gordenin, for the critical reading of the manuscript and constructive comments and to Dr M. Gartenberg for providing the pRSafe_GAL-CRE-EBD vector.
Author contribution: Conceptualization: A.A.S., A.B.C. and K.S.L.; Methodology: A.A.S., A.B.C., J.E.H. and K.S.L.; Investigation: A.A.S., A.B.C., Z.S. and W.G.; Writing—Original Draft: A.A.S. and K.S.L.; Writing—review and editing: A.A.S., A.B.C, J.E.H. and K.S.L.; Funding Acquisition: K.S.L.; Resources: K.S.L.; Supervision: K.S.L.; Project Administration: K.S.L.
Contributor Information
Anissia Ait Saada, School of Biological Sciences and Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GE 30332, USA.
Alex B Costa, School of Biological Sciences and Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GE 30332, USA.
Ziwei Sheng, School of Biological Sciences and Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GE 30332, USA.
Wenying Guo, School of Biological Sciences and Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GE 30332, USA.
James E Haber, Department of Biology and Rosenstiel Basic Medical Sciences Research Center, Waltham, MA 02454-9110, USA.
Kirill S Lobachev, School of Biological Sciences and Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GE 30332, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH[R01GM129119to K.S.L.; R35GM127029to J.E.H.]. Funding for open access charge: NIH [R01GM129119].
Conflict of interest statement. None declared.
REFERENCES
- 1. Leach D.R. Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. Bioessays. 1994; 16:893–900. [DOI] [PubMed] [Google Scholar]
- 2. Darmon E., Eykelenboom J.K., Lincker F., Jones L.H., White M., Okely E., Blackwood J.K., Leach D.R.. E. coli SbcCD and RecA control chromosomal rearrangement induced by an interrupted palindrome. Mol. Cell. 2010; 39:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Farah J.A., Cromie G., Steiner W.W., Smith G.R.. A novel recombination pathway initiated by the Mre11/Rad50/Nbs1 complex eliminates palindromes during meiosis in Schizosaccharomyces pombe. Genetics. 2005; 169:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farah J.A., Hartsuiker E., Mizuno K., Ohta K., Smith G.R.. A 160-bp palindrome is a Rad50.Rad32-dependent mitotic recombination hotspot in Schizosaccharomyces pombe. Genetics. 2002; 161:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. St Charles J., Petes T.D.. High-resolution mapping of spontaneous mitotic recombination hotspots on the 1.1 Mb arm of yeast chromosome IV. PLos Genet. 2013; 9:e1003434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y., Saini N., Sheng Z., Lobachev K.S.. Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLos Genet. 2013; 9:e1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collick A., Drew J., Penberth J., Bois P., Luckett J., Scaerou F., Jeffreys A., Reik W.. Instability of long inverted repeats within mouse transgenes. EMBO J. 1996; 15:1163–1171. [PMC free article] [PubMed] [Google Scholar]
- 8. Akgün E., Zahn J., Baumes S., Brown G., Liang F., Romanienko P.J., Lewis S., Jasin M.. Palindrome resolution and recombination in the mammalian germ line. Mol. Cell. Biol. 1997; 17:5559–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waldman A.S., Tran H., Goldsmith E.C., Resnick M.A.. Long inverted repeats are an at-risk motif for recombination in mammalian cells. Genetics. 1999; 153:1873–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Narayanan V., Mieczkowski P.A., Kim H.M., Petes T.D., Lobachev K.S.. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 2006; 125:1283–1296. [DOI] [PubMed] [Google Scholar]
- 11. Kato T., Kurahashi H., Emanuel B.S.. Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev. 2012; 22:221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rooks H., Clark B., Best S., Rushton P., Oakley M., Thein O.S., Cuthbert A.C., Britland A., Ruf A., Thein S.L.. A novel 506kb deletion causing ϵγδβ thalassemia. Blood Cells Mol. Dis. 2012; 49:121–127. [DOI] [PubMed] [Google Scholar]
- 13. Zhu H., Shang D., Sun M., Choi S., Liu Q., Hao J., Figuera L.E., Zhang F., Choy K.W., Ao Y.et al.. X-linked congenital hypertrichosis syndrome is associated with interchromosomal insertions mediated by a human-specific palindrome near SOX3. Am. J. Hum. Genet. 2011; 88:819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato T., Franconi C.P., Sheridan M.B., Hacker A.M., Inagakai H., Glover T.W., Arlt M.F., Drabkin H.A., Gemmill R.M., Kurahashi H.et al.. Analysis of the t(3;8) of hereditary renal cell carcinoma: a palindrome-mediated translocation. Cancer Genet. 2014; 207:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guenthoer J., Diede S.J., Tanaka H., Chai X., Hsu L., Tapscott S.J., Porter P.L.. Assessment of palindromes as platforms for DNA amplification in breast cancer. Genome Res. 2012; 22:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka H., Bergstrom D.A., Yao M.C., Tapscott S.J.. Large DNA palindromes as a common form of structural chromosome aberrations in human cancers. Hum. Cell. 2006; 19:17–23. [DOI] [PubMed] [Google Scholar]
- 17. Tanaka H., Bergstrom D.A., Yao M.C., Tapscott S.J.. Widespread and nonrandom distribution of DNA palindromes in cancer cells provides a structural platform for subsequent gene amplification. Nat. Genet. 2005; 37:320–327. [DOI] [PubMed] [Google Scholar]
- 18. Tanaka H., Cao Y., Bergstrom D.A., Kooperberg C., Tapscott S.J., Yao M.C.. Intrastrand annealing leads to the formation of a large DNA palindrome and determines the boundaries of genomic amplification in human cancer. Mol. Cell. Biol. 2007; 27:1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tanaka H., Yao M.C.. Palindromic gene amplification–an evolutionarily conserved role for DNA inverted repeats in the genome. Nat. Rev. Cancer. 2009; 9:216–224. [DOI] [PubMed] [Google Scholar]
- 20. Neiman P.E., Elsaesser K., Loring G., Kimmel R.. Myc oncogene-induced genomic instability: DNA palindromes in bursal lymphomagenesis. PLoS Genet. 2008; 4:e1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neiman P.E., Kimmel R., Icreverzi A., Elsaesser K., Bowers S.J., Burnside J., Delrow J.. Genomic instability during Myc-induced lymphomagenesis in the bursa of Fabricius. Oncogene. 2006; 25:6325–6335. [DOI] [PubMed] [Google Scholar]
- 22. Mangano R., Piddini E., Carramusa L., Duhig T., Feo S., Fried M.. Chimeric amplicons containing the c-myc gene in HL60 cells. Oncogene. 1998; 17:2771–2777. [DOI] [PubMed] [Google Scholar]
- 23. Sharples G.J., Leach D.R.. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol. Microbiol. 1995; 17:1215–1217. [DOI] [PubMed] [Google Scholar]
- 24. Connelly J.C., Kirkham L.A., Leach D.R.. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:7969–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eykelenboom J.K., Blackwood J.K., Okely E., Leach D.R.. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol. Cell. 2008; 29:644–651. [DOI] [PubMed] [Google Scholar]
- 26. Azeroglu B., Lincker F., White M.A., Jain D., Leach D.R.. A perfect palindrome in the Escherichia coli chromosome forms DNA hairpins on both leading- and lagging-strands. Nucleic Acids Res. 2014; 42:13206–13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Casper A.M., Greenwell P.W., Tang W., Petes T.D.. Chromosome aberrations resulting from double-strand DNA breaks at a naturally occurring yeast fragile site composed of inverted ty elements are independent of Mre11p and Sae2p. Genetics. 2009; 183:423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lobachev K.S., Gordenin D.A., Resnick M.A.. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002; 108:183–193. [DOI] [PubMed] [Google Scholar]
- 29. Lu S., Wang G., Bacolla A., Zhao J., Spitser S., Vasquez K.M.. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 2015; 10:1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaushal S., Wollmuth C.E., Das K., Hile S.E., Regan S.B., Barnes R.P., Haouzi A., Lee S.M., House N.C.M., Guyumdzhyan M.et al.. Sequence and nuclease requirements for breakage and healing of a structure-forming (AT)n sequence within fragile site FRA16D. Cell Rep. 2019; 27:1151–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nag D.K., Fasullo M., Dong Z., Tronnes A.. Inverted repeat-stimulated sister-chromatid exchange events are RAD1-independent but reduced in a msh2 mutant. Nucleic Acids Res. 2005; 33:5243–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coté A.G., Lewis S.M.. Mus81-dependent double-strand DNA breaks at in vivo-generated cruciform structures in S. cerevisiae. Mol. Cell. 2008; 31:800–812. [DOI] [PubMed] [Google Scholar]
- 33. van Wietmarschen N., Sridharan S., Nathan W.J., Tubbs A., Chan E.M., Callen E., Wu W., Belinky F., Tripathi V., Wong N.et al.. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature. 2020; 586:292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inagaki H., Ohye T., Kogo H., Tsutsumi M., Kato T., Tong M., Emanuel B.S., Kurahashi H.. Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat. Commun. 2013; 4:1592. [DOI] [PubMed] [Google Scholar]
- 35. Svetec Miklenić M., Gatalica N., Matanović A., Žunar B., Štafa A., Lisnić B., Svetec I.K.. Size-dependent antirecombinogenic effect of short spacers on palindrome recombinogenicity. DNA Repair (Amst.). 2020; 90:102848. [DOI] [PubMed] [Google Scholar]
- 36. Storici F., Resnick M.A.. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 2006; 409:329–345. [DOI] [PubMed] [Google Scholar]
- 37. Saini N., Zhang Y., Nishida Y., Sheng Z., Choudhury S., Mieczkowski P., Lobachev K.S.. Fragile DNA motifs trigger mutagenesis at distant chromosomal loci in saccharomyces cerevisiae. PLos Genet. 2013; 9:e1003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lobachev K.S., Shor B.M., Tran H.T., Taylor W., Keen J.D., Resnick M.A., Gordenin D.A.. Factors affecting inverted repeat stimulation of recombination and deletion in Saccharomyces cerevisiae. Genetics. 1998; 148:1507–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Drake J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Friedman K.L., Brewer B.J.. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 1995; 262:613–627. [DOI] [PubMed] [Google Scholar]
- 41. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 42. Cannavo E., Cejka P.. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014; 514:122–125. [DOI] [PubMed] [Google Scholar]
- 43. Ait Saada A., Costa A.B., Lobachev K.S.. Genetic and molecular approaches to study chromosomal breakage at secondary structure-forming repeats. Methods Mol. Biol. 2021; 2153:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Westmoreland J., Ma W., Yan Y., Van Hulle K., Malkova A., Resnick M.A.. RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma-induced double-strand break ends. PLos Genet. 2009; 5:e1000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gough G.W., Sullivan K.M., Lilley D.M.. The structure of cruciforms in supercoiled DNA: probing the single-stranded character of nucleotide bases with bisulphite. EMBO J. 1986; 5:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hilbers C.W., Haasnoot C.A., de Bruin S.H., Joordens J.J., van der Marel G.A., van Boom J.H.. Hairpin formation in synthetic oligonucleotides. Biochimie. 1985; 67:685–695. [DOI] [PubMed] [Google Scholar]
- 47. Voineagu I., Narayanan V., Lobachev K.S., Mirkin S.M.. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brosey C.A., Yan C., Tsutakawa S.E., Heller W.T., Rambo R.P., Tainer J.A., Ivanov I., Chazin W.J.. A new structural framework for integrating replication protein A into DNA processing machinery. Nucleic Acids Res. 2013; 41:2313–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang X., Klimovich V., Arunkumar A.I., Hysinger E.B., Wang Y., Ott R.D., Guler G.D., Weiner B., Chazin W.J., Fanning E.. Structural mechanism of RPA loading on DNA during activation of a simple pre-replication complex. EMBO J. 2006; 25:5516–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen H., Lisby M., Symington L.S.. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell. 2013; 50:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deng S.K., Chen H., Symington L.S.. Replication protein A prevents promiscuous annealing between short sequence homologies: Implications for genome integrity. Bioessays. 2015; 37:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dehé P.M., Gaillard P.H.L.. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 2017; 18:315–330. [DOI] [PubMed] [Google Scholar]
- 53. Schwartz E.K., Heyer W.D.. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma. 2011; 120:109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Manfrini N., Clerici M., Wery M., Colombo C.V., Descrimes M., Morillon A., d’Adda di Fagagna F., Longhese M.P.. Resection is responsible for loss of transcription around a double-strand break in Saccharomyces cerevisiae. Elife. 2015; 4:e08942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lacroute F. Regulation of pyrimidine biosynthesis in Saccharomyces cerevisiae. J. Bacteriol. 1968; 95:824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Apone L.M., Virbasius C.A., Holstege F.C., Wang J., Young R.A., Green M.R.. Broad, but not universal, transcriptional requirement for yTAFII17, a histone H3-like TAFII present in TFIID and SAGA. Mol. Cell. 1998; 2:653–661. [DOI] [PubMed] [Google Scholar]
- 57. Cheng T.H., Chang C.R., Joy P., Yablok S., Gartenberg M.R.. Controlling gene expression in yeast by inducible site-specific recombination. Nucleic Acids Res. 2000; 28:E108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ivanova T., Maier M., Missarova A., Ziegler-Birling C., Dam M., Gomar-Alba M., Carey L.B., Mendoza M.. Budding yeast complete DNA synthesis after chromosome segregation begins. Nat. Commun. 2020; 11:2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bishop A.C., Ubersax J.A., Petsch D.T., Matheos D.P., Gray N.S., Blethrow J., Shimizu E., Tsien J.Z., Schultz P.G., Rose M.D.et al.. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000; 407:395–401. [DOI] [PubMed] [Google Scholar]
- 60. Gallo-Fernández M., Saugar I., Ortiz-Bazán M., Vázquez M.V., Tercero J.A.. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 2012; 40:8325–8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Szakal B., Branzei D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013; 32:1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trujillo K.M., Sung P.. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J. Biol. Chem. 2001; 276:35458–35464. [DOI] [PubMed] [Google Scholar]
- 63. Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J.L., Kowalczykowski S.C.. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010; 467:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruan H., Wang Y.H.. Friedreich's ataxia GAA.TTC duplex and GAA.GAA.TTC triplex structures exclude nucleosome assembly. J. Mol. Biol. 2008; 383:292–300. [DOI] [PubMed] [Google Scholar]
- 65. Russell D.W., Smith M., Cox D., Williamson V.M., Young E.T.. DNA sequences of two yeast promoter-up mutants. Nature. 1983; 304:652–654. [DOI] [PubMed] [Google Scholar]
- 66. Struhl K. Naturally occurring poly(dA-dT) sequences are upstream promoter elements for constitutive transcription in yeast. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:8419–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M.. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008; 320:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Munster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M.. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009; 457:1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tubbs A., Sridharan S., van Wietmarschen N., Maman Y., Callen E., Stanlie A., Wu W., Wu X., Day A., Wong N.et al.. Dual Roles of Poly(dA:dT) tracts in replication initiation and fork collapse. Cell. 2018; 174:1127–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sinden R.R., Zheng G.X., Brankamp R.G., Allen K.N.. On the deletion of inverted repeated DNA in Escherichia coli: effects of length, thermal stability, and cruciform formation in vivo. Genetics. 1991; 129:991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dayn A., Malkhosyan S., Duzhy D., Lyamichev V., Panchenko Y., Mirkin S.. Formation of (dA-dT)n cruciforms in Escherichia coli cells under different environmental conditions. J. Bacteriol. 1991; 173:2658–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Inagaki H., Kato T., Tsutsumi M., Ouchi Y., Ohye T., Kurahashi H.. Palindrome-Mediated translocations in humans: A new mechanistic model for gross chromosomal rearrangements. Front Genet. 2016; 7:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kurahashi H., Inagaki H., Ohye T., Kogo H., Tsutsumi M., Kato T., Tong M., Emanuel B.S.. The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangements. Clin. Genet. 2010; 78:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gritenaite D., Princz L.N., Szakal B., Bantele S.C., Wendeler L., Schilbach S., Habermann B.H., Matos J., Lisby M., Branzei D.et al.. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014; 28:1604–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Matos J., Blanco M.G., Maslen S., Skehel J.M., West S.C.. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011; 147:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Blanco M.G., Matos J., West S.C.. Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol. Cell. 2014; 54:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., Kanemaki M.. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 2009; 6:917–922. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Flow cytometry data were deposited with Flow Repository under ID FR-FCM-Z39K.