


Abstract
Post-transcriptional regulation of gene expression is particularly important in trypanosomatid protozoa. RNA-binding proteins (RBPs) regulate mRNA stability and translation, yet information about how RBPs are able to link environmental cues to post-transcriptional control is scarce. In Trypanosoma brucei, we have previously characterized a short RNA stem-loop cis-element (PuRE, Purine Responsive Element) within the 3′-UTR of the NT8 nucleobase transporter mRNA that is necessary and sufficient to confer a strong repression of gene expression in response to purines. In this study, we have identified a protein complex composed of two RNA-binding proteins (PuREBP1 and PuREBP2) that binds to the PuRE in vitro and to NT8 mRNA in vivo. Depletion of PuREBP1 by RNA interference results in the upregulation of just NT8 and the mRNAs encoding the amino acid transporter AATP6 paralogues. Moreover, we found that the PuREBP1/2 complex is associated with only a handful of mRNAs, and that it is responsible for the observed purine-dependent regulation of NT8 expression.
INTRODUCTION
Orchestrated expression of genes in time and space is essential for a cell. Post-transcriptional processes such as mRNA splicing, turnover, localization and translation are crucial for proper control of protein abundance (1). Key players in these events are RNA-binding proteins (RBPs) that interact with cis-acting RNA elements located usually within the untranslated regions of target mRNAs (2). In eukaryotes, there is increasing evidence that mRNAs encoding functionally-related proteins are often bound to specific RBPs to build dynamic mRNA (or post-transcriptional) regulons, thus providing a molecular scaffold to regulate the fate of multiple mRNAs in a coordinated fashion (3).
Trypanosomatids include many parasitic trypanosomes and Leishmania species, which are responsible for neglected diseases that threaten human health and hinder economic development in impoverished countries around the globe (4). These early-divergent unicellular eukaryotes are considered excellent model organisms to study post-transcriptional regulatory events, since they depend largely on mRNA stability and translation to control gene expression (5,6). This is due to the unconventional organization of their genomes in long polycistronic transcription units that are transcribed in an apparent constitutive manner (7,8). Many trypanosomatids alternate between mammals and insect vectors, and therefore undergo extensive developmental changes in order to adapt to the contrasting extracellular conditions they find within one or the other host (9).
Many RBPs have been identified in trypanosomatids in global surveys following bioinformatics or genetic approaches (10–13), but few have been functionally characterized. Some RBPs have been shown to be important for development and differentiation (14–16), energy metabolism (17,18) or cell cycle progression (19). However, little is known about the specific subsets of mRNAs associated to regulatory RBPs (9), and therefore our knowledge about how RBPs control mRNA fate is rather limited in these pathogens (6).
One of the major challenges trypanosomatids have to face relate to nutrient availability. For example, Trypanosoma brucei, the causative agent of sleeping sickness, depends for energy production exclusively on the bloodstream glucose when it replicates as blood forms in the mammalian host. Upon taken up by a feeding tsetse fly vector, there is a rapid drop in glucose concentration, and bloodstream trypanosomes differentiate to procyclic forms that use the amino acids available within the insect gut as the main energy source (20). Regulatory 3′UTRs have been identified in mRNAs coding for transporters responsible for the uptake of glucose (21), transferrin (22), the prozyme subunit of S-adenosylmethionine decarboxylase (23) and purines (24,25), but the specific regulatory RBPs involved are not known at present.
Trypanosomatids are not able to synthesize the purine ring de novo and thus they need to scavenge purine nucleobases or nucleosides from their hosts or culture environments for viability and growth (26). They express a plethora of transporters, many of them developmentally regulated, to cope with the fluctuations in purine concentrations they likely encounter through their life cycles (27). Purine starvation in trypanosomatids results in the upregulation of nucleoside transport activity and a deep remodelling of purine metabolic pathways (24,28), and there is strong evidence that purines themselves act as powerful signals that trigger the metabolic adaptations necessary for cell survival when extracellular purines are scarce (29). Indeed, in Leishmania purine starvation activates a gene expression program that governs a developmental transition from promastigote to metacyclic cells, which are responsible for the initiation of infection in the mammalian host (30). Even though purine uptake is an essential process considered a promising target for therapeutic intervention, and many components of purine transport and metabolism have been characterized in depth at the biochemical and structural level, very little is known about how purine transport is regulated at the gene expression level in response to changes in the concentration of extracellular purines.
The nucleobase transporter NT8 is mainly expressed in the insect (procyclic) stage of T. brucei (31). A predicted stem–loop RNA regulatory element was discovered within the 3′-UTR of NT8 mRNA that is necessary and sufficient to repress NT8 expression when purines are abundant (25); this element has been termed PuRE in the present work (Purine Responsive Element). Structures equivalent to PuRE have been recently identified in NT8 orthologues from other trypanosomatids (32), but the relevant trans-acting factors interacting with PuREs have remained elusive to date.
In this work, we used the T. brucei NT8 PuRE as a bait to biochemically purify and characterize an RNA-binding protein complex that is responsible for the observed purine-dependent regulation of NT8 expression.
MATERIALS AND METHODS
Trypanosome culture
Trypanosoma brucei 449 procyclic cells (33) were cultured at 27°C in SDM-79 medium supplemented with 10% fetal bovine serum (34) and transfected following standard procedures (35). SDM-79 lacking purines was supplemented with 10% dialyzed fetal bovine serum (Gibco). When needed, guanosine was added at a concentration of 100 μM (25).
RNA affinity chromatograpy
We adapted the method described in (36) as follows:
Extract. 1.5 × 1010 cells were washed in serum-free SDM-79 and frozen at –80°C until use. Cells were resuspended in 3 ml of 10 mM Tris–HCl, pH 7.4, 1 mM DTT, 0.1% Igepal CA-630 and protease inhibitors and lysed by passing through a 27-gauge syringe thrice on ice. The cell extract was centrifuged at 16,000g for 10 min at 4°C, and NaCl and tRNA were added to the supernatant at final concentrations of 150 mM and 100 μg/ml, respectively.
RNA baits. dsDNA corresponding to the 35-mer stem-loop regulatory purine-responsive wild-type element (PuRE) or a structurally synonymous mutated version (25) were cloned into the BamHI site of pGR24 immediately downstream of the sequence corresponding to the T. brucei spliced-leader (SL) sequence (37). The resulting plasmids were linearized at the XbaI site and in vitro transcribed by T7 RNA polymerase. Five microgram of gel-purified RNAs were mixed with 400 pmol of oligodeoxynucleotide AE48 [complementary to SL (37), Supplementary Table S1] in 70 μl of 10 mM Tris–HCl, pH 7.4, 50 mM NaCl, heated for 3 min at 95°C, chilled on ice for 5 min, and hybridized for 30 min at 37°C.
Preparation of beads. Paramagnetic beads coupled with streptavidin (Invitrogen, 11205D) were washed twice with 100 mM NaOH, 50 mM NaCl, once with 100 mM NaCl and once with IPP-150 (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.1% Igepal CA630). Beads (1 mg) were incubated with either wild-type or mutated PuRE:AE48 hybrids for 15 min at room temperature in 0.1 ml of 10 mM Tris–HCl, pH 7.4, 1 M NaCl and then washed twice with IPP-150.
RNA chromatography and identification of RNA-binding proteins. The extract was split into two identical fractions; one was mixed with beads bound to the wild-type PuRE, the other with beads bound to the mutated version. Proteins were allowed to bind for 15 min at room temperature, followed by an incubation of 15 min at 4°C, and washed four times with IPP-150. To release bound RNA-binding proteins, beads were incubated for 30 min at 37°C in 20 μl of 10 mM Tris–HCl, pH 7.4, 2 mM MgCl2 and 50 units of benzonase (Sigma E1014) in a Thermomixer at 800 rpm. Eluates were loaded in a 10% SDS-PAGE gel, and proteins were visualized with Sypro Ruby staining. Those proteins appearing only in the wild-type sample were excised and subjected to MALDI TOF/TOF analysis in a UltrafleXtreme mass spectrometer (Bruker). Protein identification was assigned by peptide mass fingerprinting and confirmed by MS/MS analysis of at least two peptides in each sample.
Epitope tagging and tandem affinity purification (TAP)
Details of the plasmids and oligodeoxynucleotides used for epitope-tagging are available in Supplementary Table S1. Briefly, for N-terminal TAP tagging, fragments of PuREBP1 (Tb927.4.4550) or PuREBP2 (Tb927.3.3060) ORFs were cloned into p2676 (38) to yield pGR333 and pGR336, respectively. PuREBP1 was also TAP-tagged at the C-terminus to yield pGR351. To create a double tagged cell line, PuREBP2 was tagged with a 4xTy epitope at the N-terminus to yield pGR362, which was transfected to trypanosomes expressing PuREBP1-TAP. For tetracycline-inducible overexpression of N-terminal 4xTy-tagged proteins, ORFs were cloned into pGR12, a pGR19-derivative (35) bearing a blasticidin S-resistance gene (Supplementary Table S1).
Detection of TAP-tagged proteins was carried out by western blot analysis using peroxidase anti-peroxidase (PAP) reagent or anti-protein A antiserum (Sigma), whereas 4xTy tagged protein levels were monitored using a BB2 monoclonal antiserum (39). Proteins were visualized using either chemiluminescence or an Odyssey CLx Near-Infrared Fluorescence Imaging System.
Protein complexes were purified from 1–2 × 1010 cells using the TAP method (40) with the modifications described in (41). When stringent conditions were assayed, IgG-sepharose bound material was washed with 100 volumes of 10 mM Tris–HCl, pH 7.4, 500 mM NaCl, 0.5% Igepal, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulphate. After Sypro staining, individual bands were excised and identified by mass spectrometry as indicated above. RNase A+T1 treatment was carried out as in (41).
Size exclusion chromatography
About 1 mg of soluble protein extract (16 000g supernatant, see above) obtained from trypanosomes expressing both PuREBP1-TAP and 4xTy-PuREBP2 was fractionated in a Superdex-200 column coupled to a FPLC Äkta Purifier (GE Healthcare) using 10 mM Tris–HCl, pH 7.4, 150 mM NaCl as running buffer. Fractions of 0.5 ml were collected, and 15 μl of each fraction were loaded in SDS-PAGE gels and analyzed by western blot assays. The Superdex column was previously calibrated using a high molecular weight gel filtration kit (GE Healthcare).
RNA interference and luciferase assays
A fragment of PuREBP1 ORF corresponding to nucleotides 2 to 2017 or a fragment of PuREBP2 ORF corresponding to nucleotides 2–473 were PCR-amplified and cloned into pGR19 (35) to yield pGR335 and pGR338, respectively (Supplementary Table S1). Trypanosomes expressing PuREBP1-TAP or TAP-PuREBP2 were transfected with Not I-linearized pGR335 or pGR338, respectively, and selected in the presence of 50 μg/ml hygromycin. For RNAi induction, tetracycline was added to the culture medium at a concentration of 1 μg/ml. Depletion of tagged proteins was monitored by immunoblot using PAP reagent (Sigma). For luciferase assays, trypanosomes expressing PuREBP1-TAP or TAP-PuREBP2 were transfected with plasmids pGR292 or pGR108 (25), bearing a luciferase gene flanked by an actin 3′-UTR with or without the PuRE element, respectively. The resulting cell lines were further transfected with the RNAi plasmids pGR335 or pGR338. Luciferase assays were carried out as in (25) in at least three biological replicates.
Orthogonal organic phase separation (OOPS)
2 × 108 trypanosomes were washed and resuspended in 0.4 ml of serum-free and hemin-free SDM-79 medium. Half of the cell suspension was transferred to a well of a 24-well cell culture plate on ice and irradiated twice at 400 mJ/cm2 in a Stratalinker UV crosslinker (Stratagen); the other half was kept on ice (non-crosslinked control). After centrifugation, cells were lysed in peqGOLD TriFast (VWR) and processed as described in (42).
Small-scale RNA immunoprecipitation (RIP) and quantitative RT-PCR
1 × 109 cells were washed in serum-free SDM-79, resuspended in 1.4 ml of 10 mM Tris–HCl, pH 7.4, 0.5% Igepal, 1 mM DTT, 2 mM ribonucleoside vanadyl complexes (Invitrogen), 40 units/ml of RNase A inhibitor (RiboLock, Thermo Scientific) and protease inhibitors (Roche) and lysed by passing the cell suspension thrice on ice. Lysates were centrifuged at 16 000g for 10 min at 4°C, and NaCl was added to 1.2 ml of the supernatant to a final concentration of 150 mM. To obtain input RNA, 40 μl of the mixture were extracted with peqGOLD TriFast (VWR), treated with RQ1 DNase I (Promega), extracted with phenol:chloroform (1:1), and ethanol precipitated. The remaining supernatant was incubated for 1.5 h at 4°C with 1 mg of paramagnetic epoxy beads (M-270, Invitrogen) coupled with rabbit IgGs (Sigma) according to the manufacturer's instructions. Beads were washed four times with IPP*-150 buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.5% Igepal) and once with TEV buffer (same as IPP*-150 but containing 0.1% Igepal and 0.5 mM EDTA). TEV protease digestion was carried out in 50 μl of TEV buffer containing 30 units of TEV protease (Invitrogen) and 40 units of RNAseA inhibitor (RiboLock, ThermoFisher) for 1.5 h at 18°C in a Thermomixer at 800 rpm. The eluate was then extracted with phenol:chloroform (1:1), and ethanol precipitated.
Both input and immunoprecipitated RNA were converted to cDNA using 0.5 μg of random hexamers (Invitrogen), 0.25 mM of each dNTP, 20 units of RiboLock (Thermo Scientific) and 200 units of Maxima reverse transcriptase (Thermo Scientific) in a final volume of 20 μl. Reactions were incubated for 30 min at 50°C and then heated at 85°C for 5 min. Quantitative RT-PCR reactions were carried out in 96-well pates (Thermo Scientific) in a BioRad CFX96 thermal cycler. The PCR conditions were: 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s alternating with 55°C for 1 min. Melting curve analyses were performed to confirm a single amplicon for each mRNA tested. Reactions were set up in a final volume of 10 μl containing 0.1–0.5 μl of cDNA, 1× SYBR Green master mix (Thermo Scientific K0251) and 0.5 μM of each oligodeoxynucleotide. The fraction of immmunoprecipitated transcripts with respect to input RNA was calculated as 2ΔCt, where ΔCt = Ct (input) − Ct (IP). The resulting values were then corrected for the dilution factor and multiplied by 100 to obtain a percentage figure. When qRT-PCR was used to assess mRNA abundance upon PuREBP1 depletion, fold-changes in expression were calculated using the 2−ΔΔCT method (43) using actin mRNA as a reference. All qRT-PCR experiments were performed with at least three biological replicates. Oligodeoxynucleotide pairs used are listed in Supplementary Table S1.
High-throughput sequencing
To identify transcripts associated with PuREB1/2 complex (RIP-seq), TAP was performed using 5 × 109 cells expressing PuREBP1-TAP as described above, except that IgG chromatography was carried out using IgG-Sepharose beads (GE) as for TAP purification, and TEV digestion was performed in 0.45 ml of TEV buffer containing 45 units of TEV protease and 100 units of RiboLock. Bound RNA was extracted with phenol:chloroform and precipitated with ethanol. Input RNA was isolated from 50 μl of lysates using peqGOLD TriFast reagent (VWR) as indicated above. For transcriptome analyses of PuREBP1-depleted trypanosomes, RNAi was induced for 48 h, and total RNA was obtained from either uninduced or depleted cells and treated with DNase I. Messenger RNA libraries were obtained using the TruSeq Stranded mRNA sample preparation protocol (Illumina). Biological duplicates were sequenced at the Genomics Unit of the Centre for Genomics and Oncological Research (Genyo, Granada, Spain) and the Genomics Unit of the IPBLN-CSIC (Granada, Spain) using a NextSeq 500 platform (Illumina). The resulting 76-nt paired end sequences were checked for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and mapped to the T. brucei 11 megabase chromosomes (TREU927, version 5.1) using the ‘align’ program of the Subread package (44) with the options nTrim5 = 1, nTrim3 = 1 y tieBreakQS = TRUE. Reads assignment to mRNAs and counting were carried out using the ‘featureCounts’ program of the Subread package (45). Only fragments that had both ends successfully aligned were considered for summarization, and those mRNAs with ≤1 fragments per kilobase and million (FPKMs) were filtered out. Replicates were compared using the edgeR package (46).
RESULTS
Identification of proteins that bind to the purine-responsive regulatory element (PuRE)
To discover proteins that regulate purine-mediated gene expression in T. brucei, we followed an RNase-assisted RNA chromatography approach designed to identify proteins that could bind to the PuRE but not to a mutated version known to be unable to confer regulation [(25) and Figure 1]. We detected two proteins bound only to the wild-type PuRE, with apparent electrophoretic mobilities of ∼105 kDa and ∼42 kDa (Figure 1). Protein bands were subjected to mass spectrometry and identified as Tb927.4.4550 and Tb927.3.3060. Both proteins migrated significantly slower than expected from their theoretical molecular masses (92 kDa for PuREBP1 and 32 kDa for PuREBP2). PuREBP1 seems to have a cytosolic localization (http://tryptag.org/, (47)). We could also identify a protein bound non-specifically to the RNA baits, ZC3H41, which was previously described as an RNA-binding protein that interacted loosely to the spliced-leader RNA (48). We named Tb927.4.4550 and Tb927.3.3060 PuREBP1 and PuREBP2, respectively (for Purine Responsive Element Binding Proteins). Neither protein contained conserved motifs we were able to identify, and neither share apparent similarity to any known protein outside the Trypanosomatidae family.
Figure 1.
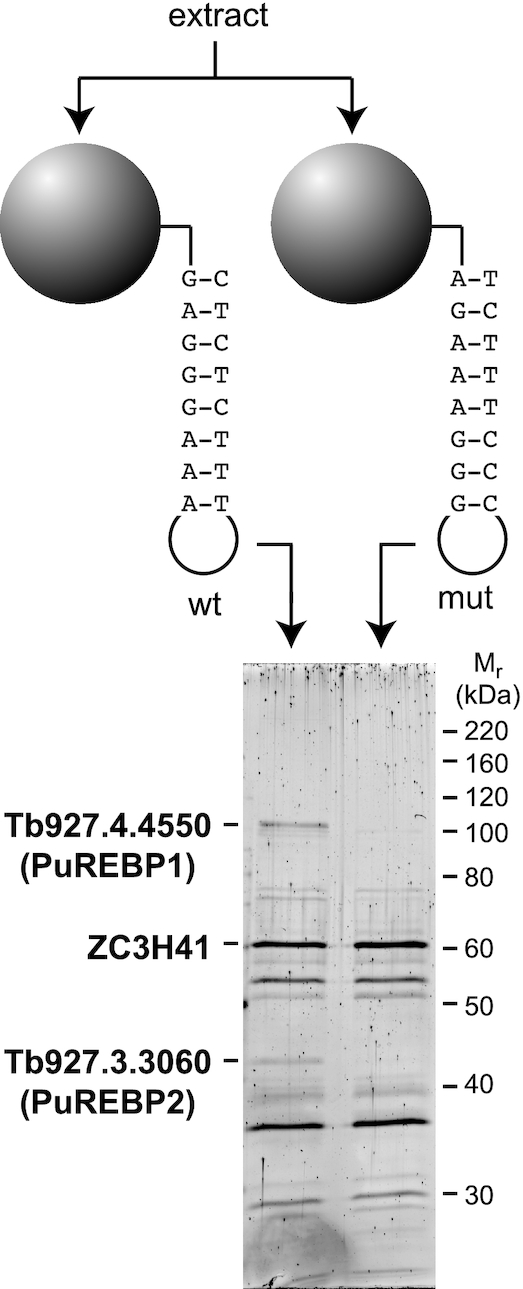
Purification of proteins that bind to the PuRE. An RNA-affinity purification approach was followed to identify proteins that bind to a wild-type PuRE but not to a mutated, non-functional version of the element. Protein bands were identified by mass-spectrometry.
PuREBP1 and PuREBP2 form a stable complex
To study both PuREBPs in more detail, trypanosome cell lines were generated that expressed TAP (tandem-affinity purification) tagged versions from their endogenous loci. PuREBP1 was tagged at either the N- or C-terminus, whereas PuREBP2 was tagged at the N-terminus. Tagged proteins were detected as single bands of the expected sizes in western blot analysis of total cell extracts (Figure 2A). To analyze whether PuREBP1 and PuREBP2 were associated with each other, we performed TAP of either protein tagged at its N-terminus, and identified associated proteins by mass spectrometry of the individual bands. We could detect PuREBP1 upon TAP purification of PuREBP2, but PuREBP2 was not present in TAP-PuREBP1 samples (Figure 2B). However, when the TAP tag was placed at the C-terminus of PuREBP1, PuREBP2 readily co-purified (Figure 2C). PuREBP1-PuREBP2 interaction was not dependent on RNA, and could withstand stringent washes in the presence of detergents (0.5% Igepal, 0.5% deoxycholate and 0.1% SDS) and 0.5M NaCl (Figure 2D).
Figure 2.

PuREBP1 and PuREBP2 form a stable complex. (A) Immunoblot of the cell lines expressing TAP-tagged versions of PuREBP1 and PuREBP2 used in this study. An extract from the parental cell line 449 was included as a negative control. TAP-tagged proteins were detected using peroxidase anti-peroxidase (PAP) reagent, and an antiserum raised against DRBD3 (37) was used to confirm equal loading. (B and C) SDS-PAGE/Sypro staining of tandem-affinity purifications using TAP-PuREBP1, PuREBP1-TAP or TAP-PuREBP2 as baits. ‘CBP’ refers to the calmodulin binding peptide tag that remains after tobacco-etch virus (TEV) protease cleavage in the TAP procedure. (D) SDS-PAGE/Sypro staining of a tandem-affinity purification of TAP-PuREBP2 after treating the protein extract with RNases A and T1, and washing IgG sepharose beads with high salt and detergent concentrations (see main text). (E) Size exclusion chromatography of a protein extract obtained from a cell line expressing both PuREBP1-TAP and 4xTy-PuREBP2. Fractions were subjected to SDS-PAGE, and PuREBP1, PuREBP2 and the exosome component RRP45 (49) were detected by western blot analysis. Elution of different molecular size markers is indicated with arrows.
In addition, we examined the elution behaviour of the PuREBP1/2 complex during size exclusion chromatography. For that purpose we generated a cell line that expressed each PuREBP with a different tag (PuREBP1-TAP and 4xTy-PuREBP2). Extracts from double tagged cells were subjected to size fractionation on Superdex 200, and fractions were analyzed for the presence of PuREBP1, PuREBP2 and the exosome component RRP45 (49) as a control. As seen in Figure 2E, both PuREBPs peaked between thyroglobulin (669 kDa) and ferritin (440 kDa) in fractions corresponding to size ranges larger than their combined monomeric masses. We could also observe an additional peak of PuREBP1 with an estimated mass of ∼160–200 kDa (indicated with an asterisk in Figure 2E). The distribution of the PuREBP1/2 complex was rather broad when compared to that of the exosome. These results suggest that PuREBP1 and PuREBP2 assemble into multimeric complexes that are heterogeneous in size, and that a fraction of PuREBP1 probably exists also as free species. PuREBP2 could also be present as free species at levels below the detection limit of our immunoblot assays.
PuREBP1 and PuREBP2 are RNA-binding proteins that associate with NT8 in vivo
We showed above that PuREBPs are able to bind to RNA in vitro. To see whether they can associate with RNA in vivo, we carried out orthogonal organic phase separation assays [OOPS, (42)]. Briefly, cells were subjected to UV-crosslinking in vivo and extracted with guanidinium–phenol–chloroform. In these conditions, RNA-protein adducts generated by UV-light migrate to the aqueous-organic interface, whereas unbound proteins partition to the organic phase. As seen in Figure 3A, both PuREBP1 and PuREBP2 were detected in the interphase of UV-treated cells, but not in non-irradiated controls. Moreover, when the interphases were treated with a mixture of RNase A and RNase T1, the association of PuREBPs with the interphase was lost. Samples were also analyzed for the presence of the glycolytic enzyme phosphoglycerate kinase (PGK-B), which is not expected to bind RNA and therefore was not detected in the interphase. These results indicate that both PuREBP1 and PuREBP2 are able to bind RNA.
Figure 3.
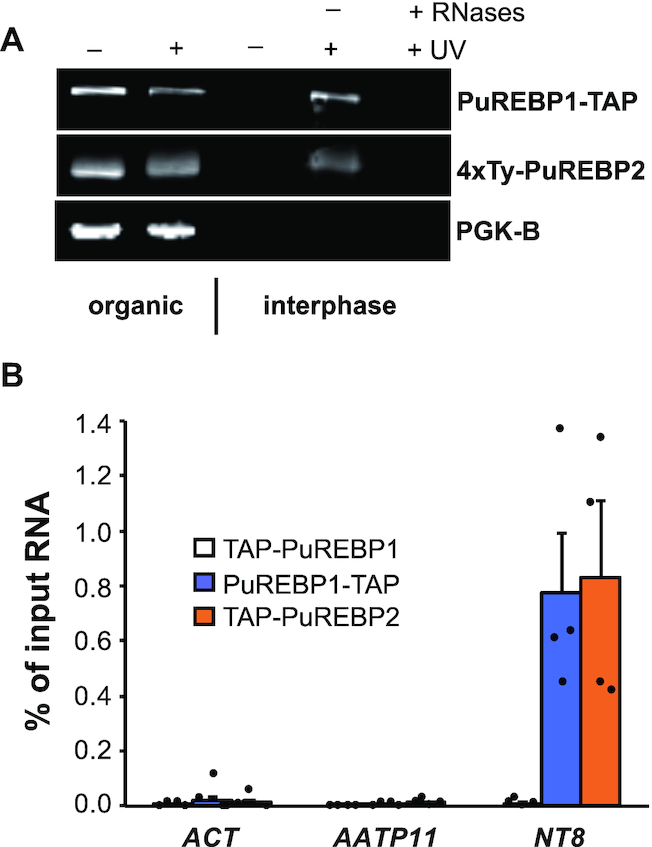
PuREBP1 and PuREBP2 bind to RNA in vivo. (A) Orthogonal Organic Phase Separation (OOPS) assay. Cells expressing PuREBP1-TAP or TAP-PuREBP2 were irradiated with UV light in vivo and lysed in peqGOLD TriFast reagent. Partition of proteins to the organic phase or to the interphase was monitored by immunoblot. Phosphoglycerate kinase B served as a negative control. (B) RNA immunoprecipitation (RIP) followed by quantitative RT-PCR assays. Actin (ACT) and amino-acid transporter 11 (AATP11) transcripts were used as non-bound controls. Percentages of immunoprecipitated RNA relative to input RNA are expressed as the mean ± s.e.m. (n = 4).
Since PuREBPs were purified using as bait the PuRE present in NT8 mRNA, one could expect to detect NT8 associated with the PuREBP protein complex. Indeed, NT8 was highly enriched in PuREBP1-TAP or TAP-PuREBP2 purifications as compared to control transcripts actin (ACT) and amino acid transporter 11 (AATP11), but was not detected above control levels in TAP-PuREBP1 purifications (Figure 3B).
PuREBP1/2 complex is involved in the regulation of the abundance of NT8 and AATP6 transcripts
To gain insight into the function of PuREBPs, the expression of each protein was silenced in vivo using RNA interference (RNAi) in a tetracycline-inducible manner. A marked reduction in protein abundance was observed for both PuREBPs after 48h of RNAi induction (insets in Figure 4A and B). Depletion of PuREBP1 resulted in cell death (Figure 4A), whereas PuREBP2 silencing reduced cell growth rate by ∼50% (Figure 4B). To study whether the depletion of PuREBP1 had an effect on the levels of PuREBP2 and vice versa, we transfected the double tagged cell line described above with plasmids targeting either PuREBP1 or PuREBP2 for RNAi. As shown in Figure 4C, depletion of any PuREBP caused partial codepletion of the other.
Figure 4.

RNAi of PuREBPs and effects on growth and transcriptome. Cell lines were generated that expressed dsRNAs corresponding to PuREBP1 (A) or PuREBP2 (B) in a tetracycline-dependent fashion. Cultures were followed for up 8 days and diluted every two days as needed. Depletion of each protein was monitored by immunoblot after 48 h of tetracycline induction (insets); DRBD3 (37) served as a loading control. (C) Ablation of any PuREBP caused partial co-depletion of the other. A cell line expressing both PuREBP1-TAP and 4xTy-PuREBP2 was transfected with plasmids expressing dsRNA corresponding to either PuREBP1 or PuREBP2. Protein levels were monitored by immunoblot as above. (D) Volcano plot of differential gene expression in PuREBP1-depleted cells compared to uninduced cells. Blue dots correspond to downregulated transcripts, orange blots indicate upregulated transcripts and grey dots represent unregulated mRNAs; dashed lines show thresholds for differential abundance (|log2 fold change| > 1) and false discovery rate (FDR < 0.01). Only one paralogue of NT8 and AATP6 are shown. (E) Quantitative RT-PCR to confirm differential abundance of NT8, AATP6, PuREBP1 and AT-E transcripts upon PuREBP1 depletion by RNAi. Fold-changes are expressed as the mean ± s.e.m. (n = 3).
Next, we analyzed the effect of the depletion of PuREBP1 on the transcriptome of T. brucei by high-throughput sequencing (RNA-seq) analysis after 48h of tetracycline induction, when cell growth was barely affected. A volcano plot for the differentially expressed genes in PuREBP1-silenced cells compared to non-induced controls is shown in Figure 4D. We used cut-offs of 1 for |log2 fold change| (i.e. ± 2-fold linear fold change) and 0.01 for false discovery rate. It could be readily seen that changes in the transcriptome were highly specific, as 99.8% of the mRNAs were inside the threshold limits (Figure 4D and Supplementary Tables S2 and S5). Only those mRNAs corresponding to NT8 and the amino acid transporter 6 (AATP6) paralogues (there are three NT8 and at least five AATP6 genes in tandem in the T. brucei genome) significantly increased in abundance in PuREBP1-depleted cells, whereas just three mRNAs were downregulated: the transcript encoding PuREBP1 (as expected), the adenosine transporter AT-E, and the mRNAs corresponding to paralogues Tb927.2.1460/Tb927.1.3760 (Figure 4D and Supplementary Table S2). Changes in the levels of NT8, AATP6, PuREBP1 and AT-E transcripts were confirmed by quantitative RT-PCR (Figure 4E).
PuREBP1/2 complex binds to few mRNAs
The changes observed in the transcriptome of PuREBP1-depleted trypanosomes suggested that PuREBP1/2 complex is associated with a small set of mRNAs. To test this, we performed TAP followed by RNA-seq (RIP-seq) in order to identify transcripts bound to the complex. We showed already that NT8 was found associated with PuREBP1/2, but not with a N-terminal TAP-tagged version of PuREBP1 (Figure 3B). Therefore, we performed RIP-seq on cells expressing either TAP-PuREBP1 or PuREBP1-TAP, and used TAP-PuREBP1 purifications as ‘mock’ samples to identify transcripts that copurified non-specifically. The most enriched mRNA detected in the mock purification encoded the SET domain-containing protein Tb927.8.6470, with a log2FC value of 1.8 (Supplementary Tables S3 and S6). We therefore considered only those transcripts with a log2FC value >1.8 in PuREBP1-TAP purifications as highly likely to be considered genuine PuREBP1/2 targets (Supplementary Tables S4 and S7). A volcano plot corresponding to the differentially enriched genes in PuREBP1-TAP purifications compared to input RNA is shown in Figure 5A. The ‘threshold transcript’ corresponding to the SET domain-containing protein is indicated as a blue dot. Only transcripts corresponding to NT8 paralogues, AATP6 paralogues and nine other mRNA species were significantly associated with the PuREBP1/2 complex (Figure 5A, orange dots, and Supplementary Table S4). These observations were confirmed by quantitative RT-PCR using either PuREBP1-TAP or TAP-PuREBP2 purifications (Figure 5B). With the exception of NT8, we could not detect PuRE RNA motifs within the 3′UTRs of bound transcripts. The transcript encoding the adenosine transporter AT-E was not found associated with PuREBP1/2 above control levels (Figure 5B), and therefore the decrease in AT-E abundance observed in PuREBP1-depleted cells (Figure 4D and E) was probably due to indirect effects unrelated to PuREBPs binding.
Figure 5.
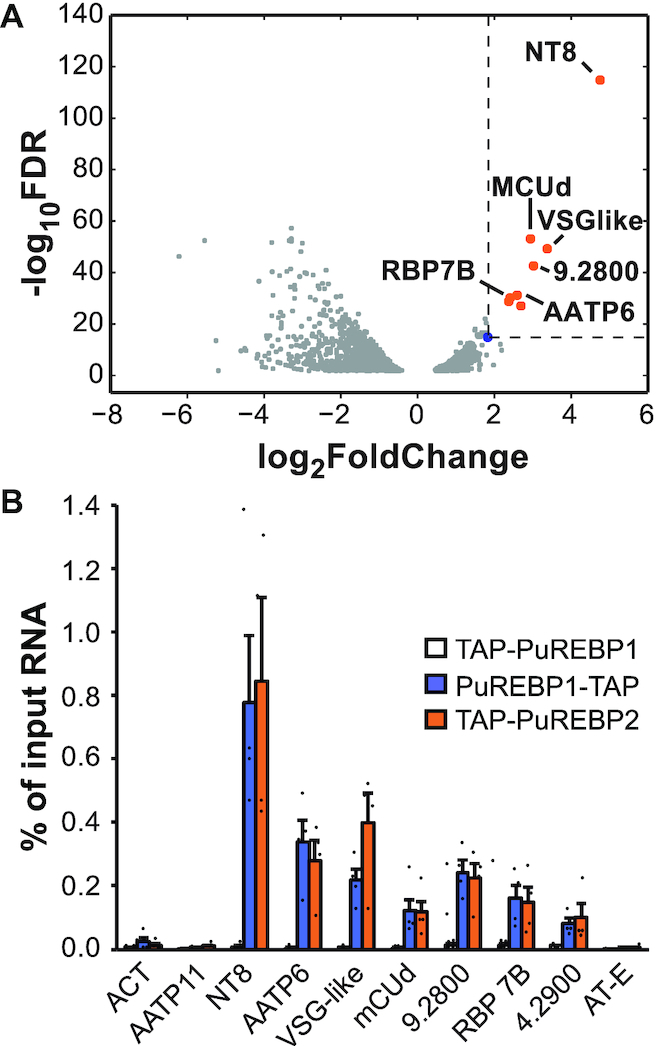
Identification of mRNAs bound to the PuREBP1/2 complex. Purification of PuREBPs-associated mRNAs followed by RNA-seq (RIP-seq) was performed in order to identify transcripts associated to the complex. (A) Volcano plot showing differential fold-abundance of PuREBP1-TAP-purified RNAs compared with total input RNA. The blue dot indicates the most abundant mRNA detected in a mock purification using cell lines expressing a non-functional version of PuREBP1; this transcript was used to set thresholds for log2 fold change and FDR (dashed lines). Only one paralogue of NT8 and AATP6 are shown. (B) RIP followed by quantitative RT-PCR to confirm association of the indicated transcripts to either PuREBP1, PuREBP2 or a non-functional version of PuREBP1 (TAP-PuREBP1, white bars). Actin (ACT) and amino-acid transporter 11 (AATP11) transcripts were used as non-bound controls. Percentages of immunoprecipitated RNA relative to input RNA are expressed as the mean ± s.e.m. (n = 4).
PuREBPs repress NT8 gene expression in a purine-dependent manner
Since the insertion of a PuRE in the 3′UTR of a reporter mRNA results in a substantial decrease in activity in the presence of extracellular purines (25), we reasoned that the PuREBP1/2 complex could be involved in the observed inhibition of gene expression. To test this hypothesis, we depleted PuREBP1 or PuREBP2 by RNAi in cell lines expressing a luciferase transgene under the control of an actin 3′UTR bearing a PuRE [Figure 6A, (25)]. Purine-dependent repression of luciferase expression was completely (PuREBP1) or partially (PuREBP2) abolished upon induction of RNAi. This phenomenon was not due to the slowing in growth and cell death associated with RNAi, as it was not observed when RNAi of PuREBP1 was induced in a cell line expressing the luciferase gene fused to a 3′-UTR lacking the PuRE (Figure 6A, control). These results indicate that PuREBPs act as repressors of gene expression in a purine-dependent fashion. To confirm that the PuREBP1/2 complex binds luciferase mRNA (LUC), we performed RIP + quantitative RT-PCR assays in cell lines expressing both PuREBP1-TAP and luciferase reporter genes bearing or lacking a PuRE (Figure 6B). Cells lines expressing non-functional TAP-PuREBP1 were used as negative controls. As shown in Figure 6B, LUC mRNA was found associated with the complex only in the cell line expressing both a functional PuREBP1 and a LUC reporter bearing a PuRE. NT8 was readily detected in PuREBP1-TAP cell lines expressing both types of reporter mRNAs, which confirms efficient immunoprecipitation. These results further support that PuREBP1/2 complex binds the PuRE in vivo.
Figure 6.

PuREBPs regulate NT8 expression in a purine-dependent manner. (A) A cell line expressing a luciferase reporter gene fused to an actin 3′UTR containing a PuRE was transfected with plasmids expressing dsRNA corresponding to either PuREBP1 or PuREBP2. A control cell line was also constructed that expressed both a luciferase reporter gene fused to an actin 3′UTR lacking a PuRE and dsRNA corresponding to PuREBP1. Luciferase activity was monitored in uninduced versus RNAi-induced cells for 48 h (± tet) in the absence (blue bars) or presence (orange bars) of guanosine. Activity measured in the control cell line in —tet and —gua conditions was set at 100%. Values are expressed as the mean ± s.e.m. (n = 3) (B) Cell lines expressing either TAP-PuREBP1 or PuREBP1-TAP were transfected with luciferase reporter genes bearing or lacking a PuRE, and used to assess binding of the PuREBP1/2 complex to luciferase mRNA by RIP followed by quantitative RT-PCR. Actin (ACT) transcript was used as non-bound control, whereas NT8 served as positive control. Percentages of immunoprecipitated RNA relative to input RNA are expressed as the mean ± s.e.m. (n = 4). (C and D) Effect of PuREBP1 depletion on protein AATP6 and NT8 levels. Proteins were monitored by immunoblot in the presence or absence of guanosine; DRBD3 was used as a loading control. A representative experiment is shown. A quantification of the relative amounts of AATP6 (blue bars) and NT8 (orange bars) relative to —tet/–gua conditions in three independent experiments is shown in (D). Values are expressed as mean ± s.e.m. (n = 3).
We next analyzed the effect of PuREBP1 silencing on NT8 and AATP6 at the protein level. Cell lines were generated that expressed either NT8 or AATP6 fused to 4xTy tags, and were subsequently transfected with a plasmid targeting PuREBP1 for RNAi. As expected, there was a strong repression of NT8 expression in the presence of guanosine, and regulation was lost upon RNAi of PuREBP1 (Figure 6C and D). In the case of AATP6, depletion of PuREBP1 also caused a marked increase in the levels of the transporter, but regulation was purine-independent, even in uninduced cells (Figure 6C and D).
To assess whether overexpression of NT8 or AATP6 was detrimental for growth, we generated cell lines that ectopically produced 4xTy-tagged versions under the control of a tetracycline-inducible promoter, and the amount of expressed proteins was modulated using a range of tetracycline concentrations. We also tested another protein encoded by a PuREBP1/2-bound mRNA, Tb927.9.2800. As shown in Supplementary Figure S1, NT8 overexpression did not have a large effect on cell growth, whereas AATP6 overexpression decreased generation time by ∼ 25% and Tb927.9.2800 ectopic expression had a dramatic effect on cell growth.
DISCUSSION
Cells need to sense and respond to changes in their environment. Trypanosomatid parasites are no exception, and they have to adapt in a fast and precise manner to the different conditions they find within each host. In spite of the importance of responding to changes in the external milieu, very little is known about how these parasites regulate gene expression in order to respond to nutrient availability. We have previously shown in procyclic T. brucei cells that the expression of the nucleobase transporter NT8 is strongly downregulated when extracellular purines are abundant, and that a regulatory 35-mer RNA element (coined ‘PuRE’ in the present work) was necessary and sufficient to confer purine-dependent repression (25). Here, we have followed a biochemical approach involving PuRE affinity purifications to identify trans-acting factors that could act as repressors of NT8 expression. We succeeded in identifying two RBPs, PuREBP1 and PuREBP2, that bind in vitro to NT8 PuRE but not to a mutated, non-functional version of the element. Using tandem-affinity purification, size-exclusion chromatography and RNA immunoprecipitation we have shown that both proteins form a stable complex that binds NT8 mRNA in vivo. PuREBP1/2 complex looks heterogeneous in size, as judged by the wide elution profile observed during size-exclusion chromatography compared to that of the exosome complex. This suggests that PuREBP1/2 heterodimers can associate to form higher-order oligomers, and that individual proteins probably exist as free species as well. In addition, PuREBP1 could also associate to itself, since a band corresponding in size to endogenous, untagged PuREBP1 is observed in PuREBP1-TAP purifications (Figure 2C). Proteolysis seems unlikely in this case, since peptide fingerprints corresponding to the different PuREBP1 species are nearly indistinguishable (Supplementary Figure S2).
Silencing PuREBP1 expression by RNAi resulted in cell death, indicating that this protein is essential for trypanosome viability. Depletion of PuREBP2, on the other hand, reduced growth rate to ∼50% of uninduced cells. Since depletion of PuREBP2 caused partial co-depletion of PuREBP1 (Figure 4C), the observed reduction in growth could be the result of a diminished expression of PuREBP1 rather than to an essential role of PuREBP2 itself. Interestingly, although PuREBP1 orthologues can be easily identified in Leishmania and Trypanosoma species, PuREBP2 gene is only present in the genome of Trypanosoma species (https://tritrypdb.org/tritrypdb/).
We hypothesize that PuREBP2 could alter the RNA-binding potential of the PuREBP1/2 complex in trypanosomes, thus allowing the regulation of a broader or narrower set of mRNAs. Alternatively, it could be involved in the regulation/localization of the complex, or play a more significant role in other developmental stages of the parasite. In any case, both PuREBPs seem to bind RNA individually, according to OOPS experiments (Figure 3A). Both PuREBPs were detected in the T. brucei mRNA-binding proteome (11); although their false-discovery rates were above the significance cut-off, their scores were very similar to that of poly(A)-binding protein 2.
Depletion of PuREBP1 caused the upregulation of NT8 mRNA, indicating that PuREBP1 acts as a repressor of NT8 expression. In agreement with this observation, PuREBP2 was shown to decrease expression of a reporter in genome-wide tethering assays in bloodstream forms (11,50). Whether this is a direct effect or the result of association with PuREBP1 remains to be investigated. PuREBP1 was not detected in tethering screenings; it could be that the whole protein is required for the tethering to occur, or it only takes place in procyclic forms.
In addition, PuREBP1 depletion resulted in an increase in the levels of just another mRNA, encoding the amino acid transporter AATP6, and in the downregulation of the nucleobase transporter AT-E and two paralogues coding for proteins of unknown function. Thus, PuREBP1/2 complex seems to regulate a very specific subset of mRNAs. Indeed, RIP-seq analysis revealed only a handful of transcripts associated with the complex above background levels: NT8 and AATP6 paralogues, as expected, and nine other mRNAs, half of which encode putative membrane proteins (Supplementary Table S4). AT-E transcript, whose levels were reduced upon PuREBP1 ablation, was not found associated with the complex. This suggests that the observed downregulation of AT-E and Tb927.2.1460/Tb927.1.3760 paralogues is probably due to an indirect effect of PuREBP1 depletion, and that the protein acts mainly as a repressor. For some of the other bound mRNAs, the effects of PuREBPs loss might not be observed because of the influence of other RNA-binding proteins. Besides NT8, none of the PuREBP1/2-bound transcripts contain PuREs. This is in agreement with the observation that the only PuRE detected in the Leishmania donovani genome was that of LdNT3, the orthologue of NT8 (32), and that minor changes in the PuRE sequence abolish purine-dependent regulation (25,32). It is possible, therefore, that PuREBP1/2 can bind to several distinct regulatory motifs, as have been shown for other RBPs (51,52).
Purine-dependent repression of a reporter gene bearing a PuRE was completely lost upon ablation of PuREBP1 by RNAi (Figure 6A). Silencing of PuREBP2 resulted in a milder phenotype; as reasoned above, this could be due to partial co-depletion of PuREP1. Tagging a NT8 transporter allowed us to test whether PuREBP1 depletion abrogated purine-dependent NT8 repression as well. Indeed, NT8 transporter was expressed regardless of the presence or absence of purines in PuREBP1-depleted trypanosomes. Taken together, these results indicate that the PuREBP1/2 complex is responsible for the purine-dependent repression of NT8 expression, and represent the first report in trypanosomatids linking a nutritional cue with specific regulatory trans- and cis-acting factors.
How do purines regulate PuREBPs function? The PuRE could act as a riboswitch that adopts different conformations depending on purine levels, which in turn would facilitate or hinder binding of PuREBPs. Alternatively, PuREBPs could bind purines in an allosteric fashion, altering their RNA-binding activity (53). Finally, PuREBPs binding capacity could be modulated by phosphorylation or other post-translational modifications in a purine-dependent manner. In this regard, it is worth mentioning that both PuREBP1 and PuREBP2 migrate slower than expected from their molecular masses, and that they are phosphorylated in multiple residues according to global phosphoproteome surveys [(54) and https://tritrypdb.org/tritrypdb/].
PuREBP1 acts as a repressor of AATP6 expression as well, since the levels of the transporter increased upon PuREBP1 ablation. Interestingly, repression of AATP6 expression was not dependent on available purine levels, and the same is likely to hold true for the remaining target mRNAs, as they seem to lack PuREs. These results open the intriguing possibility that PuREBP1/2 complex is able to integrate several extra- and/or intracellular cues that culminate in the coordinated regulation of target transcripts.
Inducible overexpression of NT8 had little effect on growth rate, whereas AATP6 and especially Tb927.9.2800 overexpression was detrimental for trypanosome growth. This could explain why the PuREBP1/2 complex is essential; additional and critical roles of PuREBPs in other aspects of mRNA metabolism (i.e. mRNA transport/intracellular localization) are also possible. Although overexpressing NT8 did not result in large changes in cell viability, we cannot rule out that high levels of NT8 be toxic in other stages of the parasite's life cycle. Interestingly, NT8 expression seems to be strongly downregulated in bloodstream trypanosomes (31). In addition, PuREBP1/2 complex could play a role in parasite development. The RNA-binding protein RBP7B, whose transcript is associated with PuREBPs, has been shown to be involved in the slender to stumpy differentiation in the bloodstream of the mammalian host (16). Binding of RBP7B transcript to the PuREBP1/2 complex could thus alter mRNA localization and/or translation. Moreover, purine-dependent regulation mediated by PuREBPs is likely to be important as the parasite progresses to colonize different insect niches where purines are likely to be scarce. In this regard, in Leishmania purine starvation triggers the transition toward metacyclics, the insect stage that initiates infection in the vertebrate host (30).
Adaptation to nutrient availability is crucial for parasite survival, yet no signalling pathways regulating the response to nutrients have been described so far in any trypanosomatid species. Our work provides the first steps to understand how these pathogens link nutritional sensing with post-transcriptional regulation of gene expression.
DATA AVAILABILITY
The RNA-seq and RIP-seq data discussed in this publication are accessible through GEO Series accession number GSE145515.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mark Carrington for epitope-tagging plasmids and critical reading, to Paul Michels for anti-PGK antiserum and to Francisco Macías for suggesting using benzonase in the RNA affinity purification procedure. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Contributor Information
Miriam Rico-Jiménez, Instituto de Parasitología y Biomedicina ‘López-Neyra’, IPBLN-CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016 Armilla, Granada, Spain.
Gloria Ceballos-Pérez, Instituto de Parasitología y Biomedicina ‘López-Neyra’, IPBLN-CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016 Armilla, Granada, Spain.
Claudia Gómez-Liñán, Instituto de Parasitología y Biomedicina ‘López-Neyra’, IPBLN-CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016 Armilla, Granada, Spain.
Antonio M Estévez, Instituto de Parasitología y Biomedicina ‘López-Neyra’, IPBLN-CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016 Armilla, Granada, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministerio de Economía, Industria y Competitividad [BFU2014-55193-P to A.M.E.]; Spanish National Research Council (CSIC) [201920E114 to A.M.E.]. Funding for open access charge: CSIC Open Access Publication Support Initiative, Unit of Information Resources for Research (URICI).
Conflict of interest statement. None declared.
REFERENCES
- 1. Schaefke B., Sun W., Li Y.S., Fang L., Chen W.. The evolution of posttranscriptional regulation. Wiley Interdiscipl. Rev. 2018; 9:e1485. [DOI] [PubMed] [Google Scholar]
- 2. Kilchert C., Strasser K., Kunetsky V., Anko M.L.. From parts lists to functional significance-RNA-protein interactions in gene regulation. Wiley Interdiscipl. Rev. 2020; 11:e1582. [DOI] [PubMed] [Google Scholar]
- 3. Bisogno L.S., Keene J.D.. RNA regulons in cancer and inflammation. Curr. Opin. Genet. Dev. 2018; 48:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stuart K., Brun R., Croft S., Fairlamb A., Gurtler R.E., McKerrow J., Reed S., Tarleton R.. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 2008; 118:1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandez-Moya S.M., Estevez A.M.. Posttranscriptional control and the role of RNA-binding proteins in gene regulation in trypanosomatid protozoan parasites. Wiley Interdiscipl. Rev. 2010; 1:34–46. [DOI] [PubMed] [Google Scholar]
- 6. Clayton C.E. Gene expression in kinetoplastids. Curr. Opin. Microbiol. 2016; 32:46–51. [DOI] [PubMed] [Google Scholar]
- 7. Michaeli S. Trans-splicing in trypanosomes: machinery and its impact on the parasite transcriptome. Future Microbiol. 2011; 6:459–474. [DOI] [PubMed] [Google Scholar]
- 8. Martínez-Calvillo S., Yan S., Nguyen D., Fox M., Stuart K., Myler P.J.. Transcription of Leishmania major Friedlin chrosomose 1 initiates in both directions within a single region. Mol. Cell. 2003; 11:1291–1299. [DOI] [PubMed] [Google Scholar]
- 9. Clayton C. Regulation of gene expression in trypanosomatids: living with polycistronic transcription. Open Biol. 2019; 9:190072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Gaudenzi J., Frasch A.C., Clayton C.. RNA-binding domain proteins in kinetoplastids: a comparative analysis. Eukaryot. Cell. 2005; 4:2106–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lueong S., Merce C., Fischer B., Hoheisel J.D., Erben E.D.. Gene expression regulatory networks in Trypanosoma brucei: insights into the role of the mRNA-binding proteome. Mol. Microbiol. 2016; 100:457–471. [DOI] [PubMed] [Google Scholar]
- 12. Kramer S., Carrington M.. Trans-acting proteins regulating mRNA maturation, stability and translation in trypanosomatids. Trends Parasitol. 2011; 27:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kramer S., Kimblin N.C., Carrington M.. Genome-wide in silico screen for CCCH-type zinc finger proteins of Trypanosoma brucei, Trypanosoma cruzi and Leishmania major. BMC Genomics. 2010; 11:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kolev N.G., Ramey-Butler K., Cross G.A., Ullu E., Tschudi C.. Developmental progression to infectivity in Trypanosoma brucei triggered by an RNA-binding protein. Science. 2012; 338:1352–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mugo E., Clayton C.. Expression of the RNA-binding protein RBP10 promotes the bloodstream-form differentiation state in Trypanosoma brucei. PLoS Pathog. 2017; 13:e1006560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mony B.M., MacGregor P., Ivens A., Rojas F., Cowton A., Young J., Horn D., Matthews K.. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature. 2014; 505:681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Das A., Morales R., Banday M., Garcia S., Hao L., Cross G.A., Estevez A.M., Bellofatto V.. The essential polysome-associated RNA-binding protein RBP42 targets mRNAs involved in Trypanosoma brucei energy metabolism. RNA. 2012; 18:1968–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trenaman A., Glover L., Hutchinson S., Horn D.. A post-transcriptional respiratome regulon in trypanosomes. Nucleic Acids Res. 2019; 47:7063–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Archer S.K., Luu V.D., de Queiroz R.A., Brems S., Clayton C.. Trypanosoma brucei PUF9 regulates mRNAs for proteins involved in replicative processes over the cell cycle. PLoS Pathog. 2009; 5:e1000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bringaud F., Riviere L., Coustou V.. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol. Biochem. Parasitol. 2006; 149:1–9. [DOI] [PubMed] [Google Scholar]
- 21. Hotz H.R., Lorenz P., Fischer R., Krieger S., Clayton C.. Role of 3′-untranslated regions in the regulation of hexose transporter mRNAs in Trypanosoma brucei. Mol. Biochem. Parasitol. 1995; 75:1–14. [DOI] [PubMed] [Google Scholar]
- 22. Benz C., Lo W., Fathallah N., Connor-Guscott A., Benns H.J., Urbaniak M.D.. Dynamic regulation of the Trypanosoma brucei transferrin receptor in response to iron starvation is mediated via the 3′UTR. PLoS One. 2018; 13:e0206332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A.et al.. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013; 499:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin J.L., Yates P.A., Soysa R., Alfaro J.F., Yang F., Burnum-Johnson K.E., Petyuk V.A., Weitz K.K., Camp D.G. 2nd, Smith R.D.et al.. Metabolic reprogramming during purine stress in the protozoan pathogen Leishmania donovani. PLoS Pathog. 2014; 10:e1003938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fernandez-Moya S.M., Carrington M., Estevez A.M.. A short RNA stem-loop is necessary and sufficient for repression of gene expression during early logarithmic phase in trypanosomes. Nucleic Acids Res. 2014; 42:7201–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Koning H.P., Bridges D.J., Burchmore R.J.. Purine and pyrimidine transport in pathogenic protozoa: from biology to therapy. FEMS Microbiol. Rev. 2005; 29:987–1020. [DOI] [PubMed] [Google Scholar]
- 27. Landfear S.M., Ullman B., Carter N.S., Sanchez M.A.. Nucleoside and nucleobase transporters in parasitic protozoa. Eukaryot Cell. 2004; 3:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Koning H.P., Watson C.J., Sutcliffe L., Jarvis S.M.. Differential regulation of nucleoside and nucleobase transporters in Crithidia fasciculata and Trypanosoma brucei brucei. Mol. Biochem. Parasitol. 2000; 106:93–107. [DOI] [PubMed] [Google Scholar]
- 29. Martin J.L., Yates P.A., Boitz J.M., Koop D.R., Fulwiler A.L., Cassera M.B., Ullman B., Carter N.S.. A role for adenine nucleotides in the sensing mechanism to purine starvation in Leishmania donovani. Mol. Microbiol. 2016; 101:299–313. [DOI] [PubMed] [Google Scholar]
- 30. Serafim T.D., Figueiredo A.B., Costa P.A., Marques-da-Silva E.A., Goncalves R., de Moura S.A., Gontijo N.F., da Silva S.M., Michalick M.S., Meyer-Fernandes J.R.et al.. Leishmania metacyclogenesis is promoted in the absence of purines. PLoS Negl.Trop. Dis. 2012; 6:e1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Henriques C., Sanchez M.A., Tryon R., Landfear S.M.. Molecular and functional characterization of the first nucleobase transporter gene from African trypanosomes. Mol. Biochem. Parasitol. 2003; 130:101–110. [DOI] [PubMed] [Google Scholar]
- 32. Licon M.H., Yates P.A.. Purine-responsive expression of the Leishmania donovani NT3 purine nucleobase transporter is mediated by a conserved RNA stem-loop. J. Biol. Chem. 2020; 295:8449–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Biebinger S., Wirtz L.E., Lorenz P., C. C.. Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 1997; 85:99–112. [DOI] [PubMed] [Google Scholar]
- 34. Brun R., Schönenberger M.. Cultivation and in vitro cloning of procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979; 36:289–292. [PubMed] [Google Scholar]
- 35. Clayton C., Estevez A.M., Hartmann C., Alibu V.P., Field M.C., Horn D.. Down-regulating gene expression by RNA intereference in Trypanosoma brucei. Methods Mol. Biol. 2005; 309:39–60. [DOI] [PubMed] [Google Scholar]
- 36. Michlewski G., Caceres J.F.. RNase-assisted RNA chromatography. RNA. 2010; 16:1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Estevez A.M. The RNA-binding protein TbDRBD3 regulates the stability of a specific subset of mRNAs in trypanosomes. Nucleic Acids Res. 2008; 36:4573–4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kelly S., Reed J., Kramer S., Ellis L., Webb H., Sunter J., Salje J., Marinsek N., Gull K., Wickstead B.et al.. Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol. Biochem. Parasitol. 2007; 154:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bastin P., Bagherzadeh Z., Matthews K.R., Gull K.. A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Mol. Biochem. Parasitol. 1996; 77:235–239. [DOI] [PubMed] [Google Scholar]
- 40. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B.. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001; 24:218–229. [DOI] [PubMed] [Google Scholar]
- 41. Fernandez-Moya S.M., Garcia-Perez A., Kramer S., Carrington M., Estevez A.M.. Alterations in DRBD3 ribonucleoprotein complexes in response to stress in Trypanosoma brucei. PLoS One. 2012; 7:e48870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Queiroz R.M.L., Smith T., Villanueva E., Marti-Solano M., Monti M., Pizzinga M., Mirea D.M., Ramakrishna M., Harvey R.F., Dezi V.et al.. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 2019; 37:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔ C(T)). Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 44. Liao Y., Smyth G.K., Shi W.. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 46. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dean S., Sunter J.D., Wheeler R.J.. TrypTag.org: a trypanosome genome-wide protein localisation resource. Trends Parasitol. 2017; 33:80–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eliaz D., Kannan S., Shaked H., Arvatz G., Tkacz I.D., Binder L., Waldman Ben-Asher H., Okalang U., Chikne V., Cohen-Chalamish S.et al.. Exosome secretion affects social motility in Trypanosoma brucei. PLoS Pathog. 2017; 13:e1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Estevez A.M., Kempf T., Clayton C.. The exosome of Trypanosoma brucei. EMBO J. 2001; 20:3831–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Erben E.D., Fadda A., Lueong S., Hoheisel J.D., Clayton C.. A genome-wide tethering screen reveals novel potential post-transcriptional regulators in Trypanosoma brucei. PLoS Pathog. 2014; 10:e1004178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dominguez D., Freese P., Alexis M.S., Su A., Hochman M., Palden T., Bazile C., Lambert N.J., Van Nostrand E.L., Pratt G.A.et al.. Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell. 2018; 70:854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jolma A., Zhang J., Mondragón E., Morgunova E., Kivioja T., Laverty K.U., Yin Y., Zhu F., Bourenkov G., Morris Q.et al.. Binding specificities of human RNA binding proteins towards structured and linear RNA sequences. Genome Res. 2020; 30:962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Clingman C.C., Deveau L.M., Hay S.H., Genga R.M., Shandilya S.M.D., Massi F., Ryder S.P.. Allosteric inhibition of a stem cell RNA-binding protein by an intermediary metabolite. Elife. 2014; 16:e02848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Urbaniak M.D., Guther M.L., Ferguson M.A.. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS One. 2012; 7:e36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and RIP-seq data discussed in this publication are accessible through GEO Series accession number GSE145515.