

SUMMARY
Post-transcriptional mechanisms regulate the stability and hence expression of coding and noncoding RNAs. Sequence-specific features within the 3’-untranslated region (3’UTR) often direct mRNAs for decay. Here we characterize a genome-wide RNA decay pathway that reduces the half-lives of mRNAs based on overall 3’UTR structure formed by base-pairing. The decay pathway is independent of specific single-stranded sequences, as regulation is maintained in both the original and reverse complement orientation. Regulation can be compromised by reducing the overall structure by fusing the 3’UTR with an unstructured sequence. Mutating base-paired RNA regions can also compromise this structure-mediated regulation, which can be restored by re-introducing base-paired structures of different sequences. The decay pathway requires the RNA-binding proteins UPF1 and its associated protein G3BP1. Depletion of either protein increased steady-state levels of mRNAs with highly-structured 3’UTRs as well as highly-structured circular RNAs. This structure-dependent mechanism therefore enables cells to selectively regulate coding and noncoding RNAs.
Keywords: RNA decay, 3’-untranslated region, RNA structure, RNA base-pairing, UPF1, G3BP1, circular RNA
eTOC BLURB
Fischer et al report an RNA decay that selectively degrades highly-structured RNAs. This pathway senses overall RNA structuredness, but not defined sequences or structures. The pathway is predicted to regulate up to one-quarter of the 3’UTRs of protein-coding mRNAs and one-third of circular RNAs in humans.
Graphical abstract
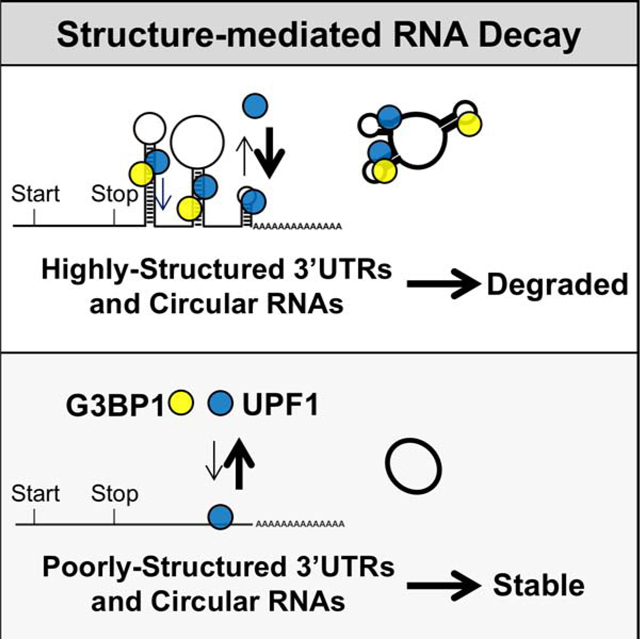
INTRODUCTION
Modulating RNA turnover is critical for controlling many cellular processes (Mitchell and Parker, 2014). mRNA decay is often regulated by proteins that recognize specific single-stranded sequences within the 3’UTR of mRNAs, such as the presence of AU-rich elements (Mayr, 2019). These elements recruit sequence-specific proteins that, in turn, facilitate deadenylation and exoribonuclease degradation (Mayr, 2019). mRNA decay can also be regulated by proteins that recognize discrete RNA structures within the 3’UTR, such as iron response elements (Binder et al., 1994) and stem-loop structures (Mino et al., 2015). These RNA structures are formed through intramolecular base-pairing and subsequently recognized and targeted for degradation by specific RNA binding proteins.
The overall structure of RNA is extrinsically determined by protein binding and local cation concentrations as well as intrinsically determined by its GC content, length and nucleotide order. Based on these intrinsic parameters, computational methods have been developed to predict RNA structures by optimizing the base-pairing probability of individual nucleotides and/or minimizing the thermodynamic free energy (−ΔG) of the resultant folded structures (Bellaousov et al., 2013; Lorenz et al., 2011; Zuker, 2003). Genome-wide RNA structure profiling revealed that a sizeable fraction of RNA is highly-structured in vivo, which is prevalent across untranslated mRNAs or at 3’UTRs of translating mRNAs (Beaudoin et al., 2018). Highly-structured mRNAs in plants have a lower expression (Su et al., 2018), and mammalian mRNAs with more structure generally have shorter half-lives (Sun et al., 2019). However, it is unclear how highly-structured mRNAs are targeted for degradation. Here we have characterized a decay pathway that targets mRNAs with highly-structured 3’UTRs for degradation mediated by two RNA-binding proteins, UPF1 and G3BP1.
RESULTS
UPF1 binds and regulates mRNAs with highly-structured 3’UTRs
Given that UPF1 unwinds highly-structured RNA (Fiorini et al., 2015) and most UPF1-mediated pathways target 3’UTRs (Kim and Maquat, 2019), we postulated that 3’UTR RNA structure could play a role in UPF1-mediated degradation. Using the RNA secondary structure prediction program ViennaRNA (Lorenz et al., 2011), we assessed the overall 3’UTR structure by calculating the length-normalized, minimum thermodynamic free energy (−ΔG/nt) of all human 3’UTRs (Figure 1A; Table S1). The −ΔG/nt value calculated by in silico prediction is not drastically different from the value calculated with additional constraints placed by the genome-wide profile of RNA structures based on their reactivity to dimethylsulfate (DMS) (Zubradt et al., 2017) (R2 = 0.883; Figure S1A; Table S2), so we used the predicted −ΔG/nt and, wherever available, the DMS-guided values as proxies for overall structure in our analyses.
Figure 1: UPF1 regulates transcripts with highly-structured 3’UTRs.

(A) Global analysis of predicted RNA structure (−ΔG/nt) for all 3’UTRs (n = 47,760; NCBI RefSeq database [O’Leary et al., 2016]) (Table S1). (B) Cumulative distribution function of −ΔG/nt for all 3’UTRs (n = 47,760) and helicase-dependent UPF1-bound 3’UTRs (Lee et al., 2015) (n = 3,839) (Table S1), with the maximal separation between these two populations indicated with a dashed line. (C) DLD-1 and SH-SY5Y cells stably expressing shScrambled (negative control) or 2 shRNAs targeting UPF1 (average of 3 colonies per shRNA; individual rows) were analyzed for the expression of transcripts with either highly-structured (HSU) or poorly-structured (PSU) 3’UTRs (individual columns) by RT-qPCR in triplicate. Colors represent fold change in expression compared to control (Ctrl) KD. (D) Candidate mRNA half-life changes in UPF1 KD cells (compared to Ctrl KD) treated with actinomycin D and analyzed by RT-qPCR. The two rows represent biological replicates, with additional data in Fig. S1G. (E) UPF1 KD cells stably integrated with GFP-tagged UPF1 WT, R615A, or DEAA mutants. Three colonies from each were analyzed by RT-qPCR and normalized to GFP vector alone. (F) Simplified diagram of dual luciferase construct. Analysis of Renilla luciferase expression with no 3’UTR (None) or two candidate HSUs and PSUs normalized to the control firefly luciferase in UPF1 DOX-inducible DLD-1 cells (performed in triplicate; see Fig. S1I for induced protein expression level). (G-H) Renilla and Firefly luciferase mRNA half-lives for UPF1 KD DLD-1 cells stably reintegrated with DOX-inducible UPF1. Experiments were performed as in Panel D for (G) WT vs. low levels of WT UPF1 and (H) WT vs. DEAA mutant of UPF1. (I) DLD-1 cells stably expressing shRNAs targeting UPF1-associated genes were analyzed for changes in candidate gene expression as in Panel C. (J) Positive control analysis for genes targeted by NMD (Mendell et al., 2004), Staufen-mediated decay (Cho et al., 2012; Kim et al., 2014; Park et al., 2013), and Regnase1-mediated decay (Mino et al., 2015). Significant differences (p-value <0.05) were determined by Student’s t-tests and denoted with * in panels E and F. See also Figure S1, and the statistics of all RT-qPCR data is documented in Table S5.
UPF1 ATP-dependent helicase activity is required for target mRNA discrimination (Lee et al., 2015) and unwinding of highly-structured RNAs (Fiorini et al., 2015). To determine if UPF1 preferentially binds to transcripts with more overall structure (larger - ΔG/nt), we examined a CLIP-seq dataset (Lee et al., 2015) that determined the RNA-binding sites of wild-type (WT) UPF1 and the ATP hydrolysis-deficient helicase mutant D637A/E638A (DEAA; Figure S1B; Table S1). Using the peak calling function MACS2 (Feng et al., 2012), we defined helicase-dependent UPF1-binding regions as those enriched for wild-type (WT) CLIP-seq reads relative to the DEAA mutant (Figure S1C). Though WT UPF1 associates with 3’UTRs irrespective of structure based on multiple CLIP-seq datasets (Colombo et al., 2017; Imamachi et al., 2017; Lee et al., 2015; Zünd et al., 2013) (Figure S1D), 3’UTRs containing helicase-dependent UPF1-binding regions are more structured based on in silico prediction and genome-wide profile data (Figure 1B and S1B; Tables S1 and S2), indicating that the UPF1 helicase activity is required for association with highly-structured 3’UTRs in cells.
Nearly all mRNAs containing UPF1 helicase-dependent binding regions at 3’UTR have −ΔG/nt ≥ 0.2, and they maximally separate from all 3’UTRs at −ΔG/nt = 0.295 (Figure 1B). To test whether the overall 3’UTR structure could serve as a signal for UPF1-dependent mRNA decay, transcripts were classified based on these thresholds as either containing a highly-structured 3’UTR (HSU; −ΔG/nt ≥ 0.3) or poorly-structured 3’UTR (PSU; ≤ 0.2). For each class, we chose seven candidate transcripts of different degrees of structure that have been predicted by multiple folding algorithms and supported by DMS-based sequencing data with similar coverage (Zubradt et al., 2017) (Figure S1E). To avoid the confounding factor of 3’UTR length, which may affect UPF1-dependent regulation (Hogg and Goff, 2010; Kim and Maquat, 2019), we also chose these candidates because they have similar 3’UTR lengths for analyses (Figure S1E). Analysis of the HSU and PSU candidates in control and UPF1-knockdown (KD) DLD-1 colon cancer cells and SH-SY5Y neuroblastoma cells revealed that all seven HSU candidates were upregulated (mean = 2.0–2.5 fold) in UPF1 KD cells whereas all seven PSU candidates were unaffected (Figure 1C and S1F; Table S5).
To determine if these expression changes are due to post-transcriptional regulation, we performed half-life analyses using the transcription inhibitor actinomycin D. Half-lives of HSU candidates in UPF1 KD cells were significantly increased relative to control whereas the half-lives of PSU candidates remained unchanged (Figure 1D and S1G; Table S5). To confirm that the observed mRNA upregulation is UPF1-specific, we reintroduced WT or mutant UPF1 to the KD cells in either a constitutive or doxycycline (DOX)-inducible manner (Figure 1E and S1H–I; Table S5). Expression of WT UPF1 in the UPF1 KD cells selectively reduced the level of all HSU candidates, while neither HSU nor PSU candidates were reduced upon reintroduction of the RNA-binding (R615A) or helicase (DEAA) mutants, indicating that both RNA-binding and helicase activity are critical for regulating mRNA with HSUs.
To test if 3’UTRs are sufficient for the observed mRNA decay, we characterized two pairs of HSUs and PSUs (EIF3B/SDHAF3 and TMED3/ZC3H15) with closely matched 3’UTR length using a dual reporter luciferase construct. Individual 3’UTRs were cloned downstream of the Renilla luciferase gene, with firefly luciferase as a control (Figure 1F). Transient transfection into UPF1 KD cells, with inducible expression of WT UPF1, revealed that mRNA levels of the HSU reporters were higher in cells with low levels of UPF1 whereas PSU reporters were unaffected (Figure 1F and S1J; Table S5). Overexpression of WT UPF1, but not the RNA-binding mutant, in UPF1 KD cells reduced the levels of HSU reporters (Figure S1K), indicating that 3’UTR structure-dependent regulation depends on RNA-binding ability of UPF1.
As with the endogenous transcripts (Figure 1D), HSU reporters had a shorter half-life in cells expressing WT levels of UPF1 compared to KD cells (Figure 1G and S1L; Table S5). The half-lives of both the unmodified and PSU reporters were unaffected by UPF1 expression. The shorter half-life of the HSU reporter in cells expressing WT UPF1 was not observed in cells expressing the helicase mutant (DEAA; Figure 1H and S1M; Table S5). Thus, 3’UTR structures dictate the half-lives of mRNAs through the post-transcriptional regulation by the helicase activity of UPF1.
UPF1 associates with several factors in various RNA decay pathways (Kim and Maquat, 2019). We tested whether the regulation of mRNAs with HSUs is mediated by these factors. Knockdown of nonsense-mediated decay (NMD) factors UPF2, UPF3A, UPF3B or SMG6 resulted in robust upregulation of NMD targets (Mendell et al., 2004), but only modest, if any, upregulation of HSU candidates (mean = 0.9–1.4 fold; Figure 1I–J; Table S5). We observed similar negative results upon knockdown of components of Staufen-mediated decay (SMD; Kim et al., 2005) and Regnase1-mediated decay (Mino et al., 2015) (Figure 1I–J; Table S5). Because we observed the upregulation of previously characterized targets upon knockdown of individual proteins for all three decay pathways (Figure 1J), we conclude that the UPF1-mediated regulation of mRNAs with HSUs involves a previously uncharacterized decay pathway.
G3BP1 binds and regulates mRNAs with highly-structured 3’UTRs
We hypothesized that this structure-dependent mRNA regulation may be mediated through other UPF1-associated proteins. Analyzing three proteomic studies (Flury et al., 2014; Schweingruber et al., 2016; Youn et al., 2018), we identified two UPF1-associated proteins that do not associate with NMD factors, but are implicated in mRNA decay (Tourrière et al., 2001): G3BP1 and its homolog, G3BP2. G3BP1 binds double-stranded (ds)RNA (Kim et al., 2019); analyzing CLIP-seq data (ENCODE Project Consortium et al., 2012) revealed that G3BP1 also preferentially bound 3’UTRs that are more structured (Figure 2A). Approximately 37% of the 3’UTRs bound by helicase-dependent UPF1 were also bound by G3BP1, including our seven candidate HSUs (Figure 2A and S2A; Tables S1–2). Consistent with the CLIP-seq data (Colombo et al., 2017; ENCODE Project Consortium et al., 2012; Imamachi et al., 2017; Zünd et al., 2013), RNA immunoprecipitation confirmed that WT UPF1 bound to both mRNAs with HSUs or PSUs while G3BP1 preferentially bound to mRNAs with HSUs (Figure 2B and S2B; Table S5).
Figure 2: G3BP1 regulates transcripts with highly-structured 3’UTRs.

(A) Cumulative distribution function of −ΔG/nt for all 3’UTRs (n = 47,760), G3BP1-bound 3’UTRs (Dunham et al., 2012) (n = 6,903) and G3BP1- and helicase-dependent UPF1-bound 3’UTRs (Lee et al., 2015) (n = 1,473; Table S1). (B) Enrichment of candidate mRNAs in WT pulldowns relative to RNA binding mutants from RNA immunoprecipitations (RIP) using GFP antibodies from lysates of DLD-1 cells stably expressing GFP-tagged UPF1 (WT or R615A) or G3BP1 (WT or ΔRBP). (C) Expression of candidate transcripts in G3BP1 KD cells compared to control KD cells as in Fig. 1C. (D) Expression of candidate transcripts in G3BP1/G3BP2 knockout (KO) cells. (E) Changes in RNA half-lives from actinomycin D-treated G3BP1 KO cells as in Fig. 1D, with additional data in Fig. S2E. (F) Analysis of Renilla luciferase expression with candidate 3’UTRs in G3BP1 DOX-inducible DLD-1 cells as in Fig. 1F. (G) Changes in luciferase RNA half-lives from actinomycin D-treated G3BP1 WT and KO cells as in Fig. 1G. (H) Candidate mRNA expression in G3BP1 KD cells with WT, ΔRBP, or S149A GFP-tagged G3BP1 stably integrated and analyzed as in Fig. 1E. Significant differences (p-value <0.05) were determined by Student’s t-tests and denoted with * in panels B, F and H. See also Figure S2, and the statistics of all RT-qPCR data is documented in Table S5.
To determine if G3BP1, like UPF1, plays a role in regulating mRNAs with HSUs, we stably knocked down G3BP1 in DLD-1 and SH-SY5Y cells (Figure S2C). G3BP1 KD significantly increased the expression of all seven HSU candidates (mean = 2.5–2.7 fold), but no PSU candidates (Figure 2C; Table S5). The specific upregulation of HSU candidates by G3BP1 was also observed in DLD-1 and U2OS osteosarcoma cells where G3BP1, its homolog G3BP2, or both were knocked out (Figure 2D and S2D; Table S5). Compared to WT cells, the half-lives of the HSU candidates were longer in G3BP1 knockout (KO) cells (Figure 2E and S2E; Table S5), suggesting that G3BP1 also destabilizes mRNAs with HSUs post-transcriptionally. Luciferase assays also indicated that the 3’UTR is sufficient for the selective reduction in half-lives of HSU reporters (Figure 2F–G and S2F–I; Table S5). Thus, G3BP1, like UPF1, post-transcriptionally regulates the half-lives of mRNAs with HSUs.
To identify specific features of G3BP1 essential for the regulation of transcripts with HSUs, we mutated the G3BP1 RNA binding domains (ΔRBP) and the phosphorylation site (S149A) that is implicated in G3BP1 endoribonuclease activity (Tourrière et al., 2001). Reintroduction of WT G3BP1, but neither mutant, selectively reduced the expression of the HSU candidates in G3BP1 KD cells (Figure 2H and S2J–K; Table S5). Additionally, there were no significant changes in expression upon knockdown of the G3BP1-associated proteins CAPRIN1 and USP10 (Kedersha et al., 2016) (Figure S2L; Table S5). Thus, the structure-dependent mRNA regulation by G3BP1 does not require the associated proteins CAPRIN1 and USP10, but it requires the RNA binding and the phosphorylation-competent S149 residue of G3BP1.
G3BP1 and UPF1 work cooperatively to regulate highly-structured 3’UTRs
To determine if UPF1, G3BP1, and G3BP2 work cooperatively or independently to regulate mRNAs with HSUs, DLD-1 G3BP1 KO cells were further stably modified with control KD, UPF1 KD, or G3BP2 KO. Neither the additional UPF1 KD nor G3BP2 KO in G3BP1 KO cells led to significant changes in expression compared to control KD (Figure 3A and S3A; Table S5). These combinatorial genetic depletion assays revealed that UPF1 cannot regulate mRNAs on the basis of HSUs without G3BP1.
Figure 3: UPF1 and G3BP1 regulate transcripts with highly-structured 3’UTRs through a mechanism independent of other UPF1-associated pathways.

(A) G3BP1 KO DLD-1 cell lines were modified by the knockout of G3BP2 with CRISPR-Cas9 or stable expression of shScrambled (Ctrl KD) or shUPF1. Isolated colonies were analyzed for changes in candidate transcript expression in relation to G3BP1 KO + Ctrl KD cells. (B) Pulldown of WT and DEAA UPF1 as in Fig. 2B with an additional WT UPF1 pulldown treated with RNaseA to degrade RNA. Enriched proteins were analyzed by western blots and enriched candidate mRNAs were analyzed by RT-qPCR. (C) Simplified diagram of the tethering assay with λN-tagged UPF1 and MS2BP-tagged G3BP1 using the Renilla luciferase reporter with three alternating MS2 and BoxB hairpins on its 3’UTR. Panel right summarizes the analysis of Renilla luciferase reporters in G3BP1 KO + UPF1 KD cells from Panel A. Additionally, combinations of WT and DEAA (DE) λN-UPF1 was transfected with WT and S149A (S) MS2BP-G3BP1 and analyzed as in Fig. 1F. (D) Cumulative distribution function of −ΔG/nt for all 3’UTRs (n = 47,760), G3BP1-bound (Dunham et al., 2012) (n = 6,903), G3BP1- and helicase-dependent UPF1-bound (Lee et al., 2015) (n = 1,473), STAU1-bound (Sugimoto et al. 2015) (n = 5,629), and STAU2-bound 3’UTRs (Dunham et al., 2012) (n = 6,324). (E) Previously characterized SMD targets determined by RNA-seq (Lucas et al. 2018) and CLIP-seq (Sugimoto et al. 2015) that possessed either an HSU or PSU were analyzed in KD cell lines as in Fig. 1I. (F) Analysis of Renilla luciferase expression with candidate 3’UTRs in STAU1 and STAU2 KD DLD-1 cells as in Fig. 1F. Significant differences (p-value <0.05) were determined by Student’s t-tests and denoted with * in panels B and C. See also Figure S3, and the statistics of all RT-qPCR data is documented in Table S5.
To evaluate the interaction between UPF1 and G3BP1 biochemically, GFP-tagged WT UPF1 was immunoprecipitated from transfected cells and probed for endogenous G3BP1. A weak association with G3BP1 was detected in WT UPF1 immunoprecipitation, but the association was diminished when the helicase mutant DEAA was immunoprecipitated (Figure 3B). This UPF1–G3BP1 association is also sensitive to RNase A treatment (Figure 3B). Notably, as in CLIP analyses (Figure 1B and S1B), UPF1 binds selectively to HSUs in a helicase-dependent manner (Figure 3B and S3B; Table S5). Given that the selective helicase-dependent UPF1 binding to HSUs is similar to G3BP1, these data indicate that the RNase-sensitive interaction between UPF1 and G3BP1 is likely mediated through mRNAs with HSUs.
To further evaluate the effect of the interaction between UPF1 and G3BP1 on mRNA regulation, we utilized the MS2BP/MS2 and λN/BoxB tethering systems (Bos et al., 2016) to enhance the binding of UPF1 and G3BP1 on the same 3’UTR. Three MS2 and three BoxB RNA hairpins were inserted into the Renilla luciferase 3’UTR in an alternating fashion (Figure 3C). Upon expressing WT G3BP1 (MS2BP-tagged) and WT UPF1 (λN-tagged) in G3BP1 KO + UPF1 KD DLD-1 cells (Figure S3C), the expression of Renilla luciferase was diminished (Figure 3C). Expression of a tagged WT UPF1 or WT G3BP1 alone was unable to alter mRNA expression. Additionally, expression of UPF1 helicase DEAA mutant with WT G3BP1 or WT UPF1 with S149A G3BP1 did not affect luciferase reporter expression (Figure 3C), indicating the importance of UPF1 helicase activity and the S149 residue of G3BP1. These findings further confirm that the interaction between UPF1 and G3BP1 is required to modulate the expression of mRNAs through the 3’UTR.
UPF1–G3BP1 regulation of mRNAs with highly-structured 3’UTR is distinct from SMD that recognizes defined double-stranded 3’UTR structures
Although helicase-dependent UPF1 and G3BP1 both selectively bind to HSUs (Figure 2B and 3B), it is possible that these HSUs can also be regulated by other UPF1-associated factors such as STAU1 and STAU2, since SMD is known to recognize and degrade mRNA targets with defined 3’UTR double-stranded structures (Kim et al., 2005, 2007). CLIP-seq analyses (Sugimoto et al. 2015; Dunham et al., 2012) revealed that these SMD factors, similar to G3BP1 and helicase-dependent UPF1, tend to bind mRNAs with highly structured 3’UTRs (Figure 3D). STAU1 peaks did not significantly overlap with G3BP1 peaks, compared to the overlap between UPF1 with G3BP1 or between UPF1 with STAU1 (Figure S3D), whereas UPF1, G3BP1, and STAU2 peaks overlap (Figure S3E). Therefore, G3BP1 and STAU2 may bind to overlapping and distinct regions of 3’UTRs that could facilitate their interactions or utilize the same dsRNA structures to mediate distinct degradative pathways.
To clarify the role of SMD and this UPF1–G3BP1 decay pathway in regulating mRNAs with HSUs, we tested the regulation of six SMD targets (Lucas et al., 2018; Sugimoto et al., 2015) possessing either HSUs (REQ, KLF2, UNG) or PSUs (NUP155, BEX1, ABCC4) in STAU1, STAU2 and G3BP1 KD cells. KD of either STAU1 or STAU2 increased all six SMD targets irrespective of their overall structure in 3’UTRs (Figure 3E; Table S5). For G3BP1 KD cells, we observed only a minimal upregulation of one of the three SMD targets with an HSU and no change for all PSUs tested (Figure 3E). Conversely, mRNA targets with HSUs regulated by UPF1 and G3BP1, as shown earlier, are not sensitive to KD of SMD factors (Figure 1I–J). To further confirm that these HSUs are not regulated by SMD, we repeated the luciferase assay with the two candidate PSUs and two HSUs (Figure 1F) in both STAU1 and STAU2 KD cells. We found no change in luciferase expression upon depleting either component essential for SMD (Figure 3F). These data altogether suggest that STAU1/2 and G3BP1 regulate different transcripts.
G3BP1 regulates genes with highly-structured 3’UTRs genome-wide
Given that both CLIP-seq and candidate mRNA analyses demonstrate that G3BP1 can bind and regulate mRNAs with HSUs and that UPF1 cannot regulate these mRNAs without G3BP1, we used RNA-seq to globally analyze whether the steady-state expression level and decay of mRNAs with HSUs change in the absence of G3BP1. Utilizing External RNA Controls Consortium (ERCC) spike-in RNAs, we found that ERCC transcript counts were highly correlated with the known concentrations (R2 = 0.969; Figure S4A). Based on the concentration range where ERCC spike-in RNAs were readily detectable and correlated with counts linearly, we analyzed genes with 10–2,000 counts, resulting in a list of 8,742 genes for analyses. To confirm the RNA-seq results, we examined changes in RNA expression using RT-qPCR. We chose 10 mRNAs (5 HSUs and 5 PSUs) that were either significantly upregulated or downregulated for validation. As observed in the RNA-seq data, RT-qPCR results confirmed the differential expression of these genes (Table S5).
Overall, we observed 1.8-fold more upregulated genes than downregulated genes in G3BP1 KO cells, which may be due to this UPF1-G3BP1 decay pathway or other effects (Figure S4B). Because many genes code for both HSU- and PSU-containing transcripts, we narrowed our analyses to genes that code for exclusively HSUs or PSUs (Figure S4B). Among PSUs, we observed no significant enrichment of upregulated genes over downregulated genes in KO cells compared with all genes [1.5 fold; χ2(2, N = 1502) = 0.5, p = 0.48]. In contrast, among HSUs, we observed statistically significantly more upregulated genes than downregulated genes in KO cells [2.8 fold; χ2(2, N = 1913) = 15.9, p = 0.00007]. These data indicate that G3BP1 indeed regulates mRNAs with HSUs.
To identify genes regulated by G3BP1 through RNA decay, we performed RNA-seq on DLD-1 WT and KO cells treated with the transcriptional inhibitor actinomycin D for 18 h. We chose this time point because our previously characterized candidate mRNAs have relatively long half-lives (Figure S1G, S2E; Table S5). Consistent with our candidate 3’UTRs, genes that decayed 2-fold slower in G3BP1 KO vs WT cells contained more structured 3’UTRs (Figure 4A and S4C).
Figure 4: G3BP1 preferentially regulates the expression and decay of genes with HSUs globally.

(A) Cumulative distribution function of the average −ΔG/nt for all analyzed genes with significantly slower decay (>2 fold) in G3BP1 KO cells. (B) Analysis of the enrichment of genes that were differentially expressed and/or decreased decay (rows) based on different 3’UTR features (columns) compared to all genes analyzed in the RNA-seq experiment between G3BP1 WT and KO DLD-1 cells. High GC% and long 3’UTRs represent the top 50% of all RNA-seq genes for their respective 3’UTR feature. Corresponding statistics are detailed in Table S3. (C) Cumulative distribution function of the change in decay in G3BP1 KO cells for genes enriched or depleted in arsenite-induced stress granules from Khong et al. (2017). (D) Simplified splicing diagram of mRNA isoform pairs with structurally distinct 3’UTRs. Boxes represent exons, dark grey boxes represent PSUs and red boxes represent HSUs. Introns are represented by a black line. Analyses of mRNA isoform pairs for predicated RNA structure and length. Expression of mRNA isoform pairs in UPF1 and G3BP1 KD compared to control KD in DLD-1 cells. See also Figure S4, and the statistics of all RT-qPCR data is documented in Table S5.
Analysis of genes that were significantly upregulated (>2.0-fold) with decreased decay (>2.0-fold) in KO cells identified significant enrichment of HSUs [1.7 fold; χ2(2, N = 88) = 6.1, p = 0.048], whereas PSUs were unaffected [0.9 fold; χ2(2, N = 60) = 0.05, p = 0.98] compared to all genes (Figure 4B; Table S3). A similar enrichment pattern was observed for genes upregulated by >1.5-fold with decay decreased by >1.5-fold (Figure S4D; Table S3), suggesting that G3BP1 depletion leads to the upregulation of genes with HSUs globally through decreased rates of RNA decay. For other 3’UTR features, a weaker enrichment was observed in G3BP1 KO cells for genes with GC-rich 3’UTRs and no enrichment for long 3’UTRs, when compared to all genes (Figure 4B; Table S3). Cross-comparison with CLIP-seq data revealed that G3BP1- and helicase-dependent UPF1-bound mRNAs that are upregulated with slower decay rates in G3BP1 KO cells have more structured 3’UTRs (Figure S4E). Thus, G3BP1 regulates mRNA expression through targeting genes with HSUs for degradation. However, the rules of how and when these transcripts are regulated by G3BP1 are unclear.
G3BP1 is a key protein involved in the assembly of stress granules, a class of biomolecular condensates induced by stress in the cytoplasm that are enriched with RNAs and proteins (Kedersha et al., 2016; Tourrière et al., 2003). To determine if this G3BP1-mediated mRNA decay pathway is involved in any stress granule regulation, we compared our RNA-seq data with the transcripts identified to be enriched or depleted in stress granules (Khong et al., 2017). We found that the decay rates for transcripts enriched in stress granules to be mostly unaffected in G3BP1 KO cells (Figure 4C). In contrast, transcripts depleted from stress granules decayed more slowly [χ2(2, N = 2,624) = 243.6, p < 0.0001], and the genes with HSUs were most affected in G3BP1 KO cells [χ2(2, N = 862) = 10.7, p = 0.001; Table S3]. Analyzing transcriptome data from an independent study that used a different method to isolate stress granules (Namkoong et al., 2018) also showed that only HSUs depleted from granules have moderately slower decay in G3BP1 KO cells for each of the three stresses tested: arsenite, ER-stress, and heat-shock (Figure S4F; Table S3). While our RNA-seq was performed in unstressed conditions, it is interesting that G3BP1 may preferentially modulate the decay of mRNAs that are excluded from stress granules, which warrant future investigations.
One limitation of the RNA-seq experiment was that insufficient sequencing depth hampered our ability to accurately determine which mRNA isoform was differentially regulated by G3BP1. Of 19,337 human genes, 177 genes encode both an HSU and a PSU isoform (Table S1). Given that 3’UTR overall structure dictates regulation of mRNA decay, we tested whether UPF1 and G3BP1 selectively regulate 3’UTR isoforms of the same gene. We chose five isoform pairs with similar 3’UTR length but different overall structure for further analyses (Figure 4D). We found that UPF1 and G3BP1 KD selectively increased expression of the HSU isoforms, but none of the PSU isoforms (Figure 4D). These data further emphasize that this mRNA regulation mediated by UPF1 and G3BP1 depends on the overall 3’UTR structures.
UPF1 and G3BP1 recognizes overall 3’UTR structure independent of a defined sequence or structure
Based on these findings, we aimed to better characterize how highly-structured 3’UTRs are recognized and targeted for decay. We specifically asked (1) whether the decay mechanism is sequence-dependent, (2) what are the minimum structure requirements, and (3) what is the role of non-structured sequence. Given that G3BP1 is the determining factor of this new UPF1-dependent mRNA regulation, we focused on evaluating how G3BP1 drives this novel mode of 3’UTR regulation.
To test if G3BP1-mediated regulation depends on a specific sequence or secondary structure, we inserted either an HSU or a PSU downstream of the Renilla reporter in the reverse complement orientation. We reasoned that this reverse orientation will change the single-stranded sequence but preserve overall RNA structure (R2 = 0.915; Figure 5A). The reverse complements of the HSUs led to an upregulation of luciferase expression and a longer half-life after depletion of G3BP1, while no differences were observed for PSU (Figure 5B–C and S5A–B; Table S5). Therefore, G3BP1 likely mediates mRNA decay through the recognition of base-paired secondary structures in the 3’UTR rather than specific single-stranded sequences.
Figure 5: SRD regulation is dependent on overall 3’UTR structure.

(A) Positive correlation with the coefficient of determination (R2) calculated between the overall structure of the original vs. the reverse complement orientation of all 3’UTRs. (B) Reverse complements of candidate 3’UTRs were inserted into the Renilla luciferase construct and analyzed as in Fig. 1F using G3BP1 WT vs. KO DLD-1 cells. (C) Change in RNA half-lives of luciferase reporter fused to reverse complement EIF3B 3’UTR from actinomycin D-treated G3BP1 WT and KO cells as in Fig. 2G. (D) Simplified diagram of the RNA secondary structure of the EIF3B 3’UTR (Fig. S5C) that was fragmented and inserted as the 3’UTR for the Renilla luciferase gene and analyzed for expression. (E) An artificial 200-nt sequence with little defined structure (−ΔG/nt = 0.08) was inserted into the Renilla luciferase 3’UTR either 5’ or 3’ of full length or truncated (465–552) EIF3B 3’UTR and analyzed for expression in G3BP1 WT vs KO DLD-1 cells as well as WT vs DEAA UPF1 DOX-inducible UPF1 KD DLD-1 cells from Fig. 1H. Significant differences (p-value <0.05) were determined by Student’s t-tests and denoted with * in panels B, D and E. See also Figure S5, and the statistics of all RT-qPCR data is documented in Table S5.
To isolate elements in 3’UTRs required for this structure-dependent mRNA decay, the HSU EIF3B was divided into three fragments based on DMS-guided structural predictions (Figure 5D and S5C). The fragments, analyzed using luciferase reporters, were all expressed at significantly higher levels in G3BP1 KO compared to WT cells (Figure 5D), suggesting that different regions of the 3’UTR all contribute to its regulation. While fragmentation of the EIF3B 3’UTR reduced the overall structure, even the least-structured fragment (465–552) had a −ΔG/nt = 0.28 close to the HSU threshold (−ΔG/nt ≥ 0.3). The structured nature of this fragment was confirmed by DMS analyses on in vitro transcribed EIF3B 3’UTR, which revealed an extensive reduction in DMS modification between the denatured and folded RNA compared to the PSU candidate (SDHAF3; Figure S5D). Thus, G3BP1 can also regulate the highly structured fragments derived from the original EIF3B 3’UTR.
To test if G3BP1-mediated regulation can be lost by reducing the overall 3’UTR structure, we generated a 200-nt artificial 3’UTR sequence containing minimal structure (−ΔG/nt = 0.08) with no difference in DMS reactivity between the denatured and folded states (Figure S5E). This unstructured 3’UTR was not regulated by G3BP1 (Figure 5E). Insertion of this sequence either 5’ or 3’ to the full-length EIF3B 3’UTR (552-nt) in the luciferase reporter maintained its relatively high overall structure (−ΔG/nt = 0.31) and its regulation by G3BP1 (Figure 3E), suggesting that the artificial sequence was not inhibitory. Addition of the artificial sequence to the 465–552 fragment (88-nt) compromised G3BP1-mediated regulation, consistent with a significant reduction in overall structure (−ΔG/nt = 0.16) to a value below the cutoff for PSU (−ΔG/nt ≤ 0.2). Consistently, we observed a reduced change in DMS reactivity between the denatured and folded state for the 88-nt fusion construct compared to the 88-nt alone construct (Figure S5E), suggesting a reduction in the overall structure. Therefore, non-structured sequence can dilute the overall structure of the 3’UTR resulting in differential regulation by G3BP1.
UPF1 helicase activity also plays a critical role in regulating these fusion constructs with different degrees of the overall structure. Compared with the helicase DEAA mutant, WT UPF1 preferentially regulated the fusion constructs that had a high overall structure but was unable to regulate the constructs where the overall structure was diluted (Figure 5E). Taken together, we conclude that UPF1 and G3BP1 require a high overall structure in the 3’UTR to regulate transcripts, and we term this novel pathway as structure-mediated RNA decay (SRD).
Delineating parameters underlying overall 3’UTR structure that are critical for SRD
While SRD regulation is highly correlated with overall structure (−ΔG/nt) of the 3’UTR, these values are determined by several factors including length, GC content, and nucleotide order (Bellaousov et al., 2013; Lorenz et al., 2011; Zuker, 2003). We decided to individually examine these three factors in relation to SRD regulation.
Length and overall structure (−ΔG/nt) are not correlated linearly (Figure 6A), although longer 3’UTRs (>1,000 nt) are generally more capable of forming HSUs than short 3’UTRs (<100 nt). Given that we observed variable regulation of the candidate mRNAs with similar 3’UTR lengths by SRD (Figure S1E) and no enrichment of differentially regulated genes with long 3’UTRs in WT vs. G3BP1 KO cells (Figure 4B and S4D, Table S3), SRD regulation is not dependent on 3’UTR length.
Figure 6: SRD regulates highly base-paired structures.

(A) Correlation of predicted overall structure and length for all 3’UTRs with the coefficient of determination (R2) calculated. (B) Correlation of predicted overall structure and GC content for all 3’UTRs and analysis of GC content for 7 HSU and 7 PSU candidates. (C) Analysis of GC content for EIF3B structure dilution experiments from Fig. 5E. (D) Cumulative distribution function of z-scores derived by comparing −ΔG/nt of the native 3’UTR to the −ΔG/nt of one hundred permutations of the same sequence for all unique 3’UTRs (n = 14,169), HSUs (n = 4,801), and PSUs (n = 2,290). Large negative z-scores represent 3’UTRs that have more overall structure than the permuted population. (E) Cumulative distribution function of the fraction of base-paired nucleotides (≥90% probability) for all 3’UTRs, HSUs, and PSUs. (F-G) Cumulative distribution function of the (F) fraction of base-paired nucleotides (≥90% probability) and (G) DMS reactivity for G3BP1 + UPF1-bound regions (peak and its surrounding 0, 100, and 200 nts with the peak as the mid-point). (H) Schematic diagrams demonstrating the modifications to the 88-nt EIF3B 3’UTR (465–552) for luciferase assays. Analysis of Renilla luciferase expression with the mutated 88-nt 3’UTRs in G3BP1 WT and KO cells as in Fig. 5E. Experiment was performed in two batches, and each 88-nt mutation was plotted with their respective controls (None and WT). Significant differences (p-value <0.05) were determined by Student’s t-tests and denoted with * in panel H. See also Figure S6, and the statistics of all RT-qPCR data is documented in Table S5.
In contrast to length, GC content is highly correlated with −ΔG/nt (Figure 6B). GC-rich 3’UTRs were enriched in the genes upregulated in G3BP1 KO cells based on RNA-seq analyses (Figure 4B and S4D, Table S3). Given that GC-rich sequences will have a higher frequency of GC base-pairing and a GC-bond is thermodynamically more stable than an AU-bond, GC-rich sequences likely yield more overall structure. Within the candidate mRNAs (7 PSUs and 7 HSUs), we were unable to determine if the specific regulation by UPF1 and G3BP1 was due to overall structure or GC content (Figure 6B). However, the fusion of the artificial sequence to the luciferase constructs altered SRD regulation in a −ΔG/nt-dependent manner but did not change the GC content (Figure 5E and 6C). Thus, GC content is likely not the critical determinant of SRD.
Another factor utilized in RNA structure prediction programs is nucleotide order. To test whether the nucleotide order, rather than nucleotide content, is critical for establishing the large −ΔG/nt observed for HSUs, we permutated each 3’UTR in the genome (n = 14,169) 100 times and calculated the −ΔG/nt for each sequence. We compared the −ΔG/nt of the native 3’UTR to this population of theoretical −ΔG/nt values and derived a z-score for each 3’UTR. A large negative z-score indicates that a 3’UTR is substantially more structured than would be expected by chance given its nucleotide content. We observe that HSUs have large negative z-scores (95% CI −5.034±0.128) while PSUs have small negative z-scores (95% CI - 0.083±0.064) (Figure 6D). To confirm that 100 permutations were sufficient for robust z-score analysis, we permutated the seven candidate HSUs and seven PSUs an additional 4,900 times. The derived z-scores were highly correlated with the corresponding value after only 100 permutations (R2 = 0.834; Figure S6A). Based on these permutation analyses, we conclude that the native sequence plays an important role in determining whether mRNAs can be assigned as HSUs or PSUs for SRD regulation, even though G3BP1 does not recognize a specific sequence for binding.
UPF1 and G3BP1 recognize experimentally-verified base-paired structures within 3’UTR
One possibility is that the nucleotide order of the native sequence dictates the maximum probability of local base-pairing within 3’UTRs regulated by SRD. We therefore used the partition function for “RNAfold” (Lorenz et al., 2011) and calculated the base-pairing probability within the 3’UTR at single-nucleotide level. We first established a threshold for 3’UTRs to be considered base-paired (Figure S6B). The base-paired fraction of each 3’UTR was, on average, higher in HSUs than PSUs, with an increasing difference between HSUs and PSUs as the base-pairing probability threshold approached 100% (Figure S6C). The absolute difference maximized in the 85–90% base-pairing probability range (Figure S6D), and therefore we considered nucleotides above the 90% probability threshold base-paired (Figure 6E).
Utilizing these single-nucleotide analyses, we analyzed the CLIP-seq data to determine if UPF1 and G3BP1 specifically bind to regions that are predicted to be highly base-paired. Analyses revealed that although WT UPF1-bound regions were not enriched for base-paired nucleotides, the helicase-dependent UPF1-bound and G3BP1-bound regions were highly base-paired (Figure S6E). The regions co-bound by both helicase-dependent UPF1 and G3BP1 contained an even higher density of base-paired nucleotides (Figure 6F and S6E), indicating that these two proteins preferentially bind to or have access to dsRNA regions.
To verify these in silico structural analyses, we also utilized DMS-based sequencing data (Zubradt et al., 2017) to determine if the predicted base-paired nucleotides were unavailable for DMS modification in cells. Analysis of 3’UTR with DMS coverage revealed that DMS-modified bases tended to be single-stranded while unmodified bases were double-stranded (Figure S6F). By analyzing DMS-modified bases within UPF1 and G3BP1 CLIP-seq peaks, we found significantly reduced DMS modification within the UPF1 helicase-dependent and G3BP1 CLIP-seq peaks (Kolmogorov-Smirnov test [one-sided, p-value = 0]), further confirming that these proteins bind to regions of structured RNA in cells (Figure 6G and S6G). We also found that increasingly distal regions surrounding the UPF1 and G3BP1 peaks had progressively increased DMS reactivity (Figure 6G and S6G), which coincides with the reduction in predicted base-pairing probabilities (Figure 6F and S6E).
Besides utilizing DMS to probe RNA structures in vivo, SHAPE (selective 2′-hydroxyl acylation and primer extension) is another chemical that can probe RNA structures by modifying all four RNA bases that are structurally unconstrained. We analyzed two genome-wide SHAPE datasets (Lu et al., 2016; Sun et al., 2019) and found that regions with G3BP1 and helicase-dependent UPF1 binding had significantly lower SHAPE reactivity in vivo (Kolmogorov-Smirnov test [one-sided, p-values < 0.0005]; Figure S6H), which was similar to what we observed with DMS reactivity (Figure 6G). Based on SHAPE probing in vivo and in vitro folded RNA, we observed that nucleotides with a high probability of base-pairing had low SHAPE reactivity, whereas nucleotides predicted to be single-stranded had higher SHAPE reactivity (Figure S6I). Based on these analyses, we conclude that the regulation of mRNAs with HSUs is due to the ability of UPF1 and G3BP1 to access or bind dsRNA regions, consistent with previous findings (Fiorini et al., 2015; Kim et al., 2019).
Disruption of base-pairing within 3’UTR structures prevents SRD regulation
To experimentally demonstrate the importance of RNA base-pairing, we further modified the short EIF3B 88-nt fragment (Figure 5E), which are folded into three hairpins based on DMS-guided structure prediction (Figure S5C). To better characterize how the SRD determining factor G3BP1 regulates this 88-nt fragment, we created additional mutations and deletions (Figure 6H), which changed the overall structure and base-pairing, while minimally changing the GC content (Figure S6J). We identified that the first hairpin and particularly a double-stranded region of that hairpin were essential for SRD regulation (Figure 6H, Δhairpin and 1st hairpin).
We mutated a double-stranded region of the first hairpin to prevent RNA base-pairing and reduce the overall structure, which greatly reduced regulation of the construct by G3BP1 (Figure 6H; disrupted hairpin). To confirm that G3BP1-mediated regulation is dependent on base-paired structure, we introduced a compensatory mutation on the hairpin to restore base-pairing and overall structure, which restored G3BP1 regulation (Figure 6H; restored hairpin).
To further examine whether the regulation of HSUs by G3BP1 is independent of GC content, we mutated the G:C base-pairs to A:U base-pairs in the first hairpin (Figure 6H). Even though the GC content was reduced, G3BP1 was able to regulate the A:U rich 3’UTR (Figure 6H, A:U hairpin). This series of luciferase assays strongly indicates that SRD regulation is sequence-independent and instead relies on the formation of RNA base-paired structures within the 3’UTR.
UPF1 and G3BP1 differentially regulate highly-structured circular RNAs
Because 3’UTRs are by definition untranslated regions in mRNAs, we wondered whether UPF1 and G3BP1 could also regulate the expression of non-coding RNAs, such as circular RNAs (circRNAs) (Jeck et al., 2013; Kramer et al., 2015; Nigro et al., 1991). CircRNAs are generated during RNA splicing and form covalently closed molecules that lack both a 5’ and 3’ end, which make circRNAs highly stable and resistance to major RNA decay pathways that are often initiated via exoribonucleases (Łabno et al., 2016). However, circRNA expression can dynamically change during development (Venø et al., 2015)—yet, the mechanism of this dynamic regulation has not been identified.
The most likely mechanism to degrade circRNAs is through endoribonucleases. Endoribonucleases are enzymes that cleave RNA internally and do not require a free 5’ or 3’ end (Li et al., 2010). Select circRNAs can be degraded through perfect complementary binding with microRNAs and subsequent Ago2‐mediated endoribonuclease cleavage (Hansen et al., 2011). CircRNAs can also be globally degraded through the endoribonuclease RNase L upon virus infection (Liu et al., 2019). As G3BP1 has been implicated for endoribonuclease activity (Tourrière et al., 2001), we were interested to determine if UPF1 and G3BP1 selectively act on highly-structured circRNAs.
To test whether UPF1 and G3BP1 regulate circRNAs in a structure-dependent manner, we analyzed the overall structure of all exonic circRNAs from circBase (Glažar et al., 2014) (Figure 7A; Table S4). We screened over 100 circRNAs of varying degrees of structure, with 37 circRNAs sufficiently expressed in DLD-1 cells for further investigation. Similar to what we observed with 3’UTRs, highly-structured circRNAs were upregulated in UPF1 and G3BP1 KD cells compared to control, while poorly-structured circRNAs remained unchanged (Figure 7B).
Figure 7: UPF1 and G3BP1 differentially regulate circRNAs.

(A) Global analysis of the predicted overall structure of circRNAs from non-terminal exons (n = 96,123; Table S4). (B) Change in expression of 37 circRNAs in UPF1 and G3BP1 KD cells compared to control KD cells grouped into bins of ΔG/nt = 0.05. CircRNAs significantly upregulated in at least 3 of the 4 KD cell lines are indicated with *. (C) The RNA immunoprecipitations performed in Fig. 2B were analyzed for candidate circRNA association. (D) DLD-1, SH-SY5Y, and U2OS cells in Fig. 1C and 2C–D were analyzed for changes in expression for candidate circRNAs. (E) Generated G3BP1 KO cells with a second genetic manipulation were further analyzed for changes in circRNA expression as in Fig. 3A. (F) Analyses of circRNA expression in DLD-1 cells stably expressing shRNAs targeting UPF1-associated genes in DLD-1 cells as in Fig. 1I. (G) Constitutive expression of UPF1 and G3BP1 using random integration in respective KD cells were analyzed for changes in circRNA expression as in Fig. 1E and 2H. (H) Helicase-dependent UPF1 and G3BP1 preferentially associate with highly-structured 3’UTRs through their ability to bind to dsRNA. The helicase activity of UPF1 may bring the proteins in close proximity on highly-structured RNA and facilitate its degradation. Significant differences (p-value <0.05) were determined by Student’s t-tests and denoted with * in panels B, C and G. See also Figure S7, and the statistics of all RT-qPCR data is documented in Table S5.
To further characterize the regulation of circRNAs, we decided to interrogate seven highly-structured and seven poorly-structured circRNAs with confirmed splice junctions (Figure S7A), comparable −ΔG/nt estimated by three prediction algorithms (Bellaousov et al., 2013; Zuker, 2003) and similar circRNA size range (Figure S7B). While UPF1 was bound to both highly-structured and poorly-structured circRNAs, G3BP1 was preferentially bound to highly-structured circRNAs (Figure 7C and S7C). We also found that highly-structured circRNAs were selectively upregulated upon depletion of G3BP1, G3BP2 or UPF1 in multiple cell lines (Figure 7D). Similar to 3’UTRs, the additional KD of UPF1 or KO of G3BP2 in G3BP1 KO cells had no additional effect, suggesting the proteins work cooperatively to regulate highly-structured circRNAs and G3BP1 is the determining factor (Figure 7E).
Notably, candidate circRNAs were not strongly regulated by known factors of the UPF1-mediated decay pathways, including the NMD-associated endoribonuclease SMG6 as well as SMD-associated STAU1 and STAU2 (Figure 7F). Given that SMD is translation- and decapping-dependent (Kim et al., 2005; Park et al., 2013) whereas SRD targets circRNAs (which are non-coding and do not have a 5’ cap), our findings further indicate that SRD is a unique pathway for regulating highly-structured RNAs. Consistent with mRNAs with HSUs, the regulation of these highly-structured circRNAs requires the RNA-binding and helicase activity of UPF1 as well as the RNA-binding and S149 phosphorylation site of G3BP1 (Figure 7G and S7D). Taken together, these data suggest that UPF1 and G3BP1 regulate highly-structured circRNAs in a manner similar to mRNAs with HSUs.
DISCUSSION
Here we report that the overall RNA structure of the 3’UTR can serve as a signal for genome-wide mRNA degradation. This structure-mediated RNA decay (SRD) pathway requires G3BP1 and UPF1 but is independent of other UPF1-mediated decay pathways. Combinatorial genetic assays revealed that UPF1 cannot regulate SRD targets without G3BP1. Biochemically, UPF1 and G3BP1 associate in an RNA-dependent manner. Informatics analyses of CLIP-seq and genome-wide RNA structure profiling data revealed that UPF1 and G3BP1 bind at highly base-paired regions. While G3BP1 preferentially binds to mRNAs with HSUs, UPF1 binds both HSUs and PSUs. The specificity of UPF1 for HSUs relies on its helicase activity. We propose that the unwinding of dsRNA regions by UPF1 facilitates the enrichment of G3BP1 in proximity to UPF1. Therefore, RNA with a higher density of base-pairing will have an increased chance of having these two RNA-binding proteins in proximity, which could be the key signal required to initiate SRD (Figure 7H).
Comparing our RNA-seq data with the published stress granule transcriptomes, we discovered that SRD targets are preferentially excluded from stress granules. Intriguingly, both G3BP1 and UPF1 localize in stress granules (Brown et al., 2011; Tourrière et al., 2003). Therefore, G3BP1 and UPF1 may be sequestered from their targets in stress granules but able to act on highly-structured mRNAs in normal conditions when these proteins are diffuse in the cytoplasm. Alternatively, UPF1 and G3BP1 may be post-translationally modified at stress granules to inactivate their SRD activity locally. The inability for SRD to act on stress granule transcripts may be the reason why they are enriched.
In this study, we have also identified two biological functions of SRD. First, SRD enables cells to differentially regulate specific isoforms of mRNAs, which may have defined functions. For example, the SRD-targeted ARAF HSU isoform, unlike the PSU isoform, inhibits apoptosis (Rauch et al., 2011). Second, although circRNAs are generally stable, their expression dynamically changes during development (Venø et al., 2015), suggesting an undefined global pathway for selective degradation. Here, we demonstrate that SRD regulates circRNAs in a structure-dependent manner. Given that UPF1 and G3BP1 regulate mRNAs and circRNAs similarly and that degradation of circRNAs requires internal cleavage, we deduce that SRD likely involves an endoribonuclease.
STAR METHODS
Lead Contact and Materials Availability
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Anthony Leung (anthony.leung@jhu.edu). Plasmids generated in this study have been deposited to Addgene (IDs: 135994–136061). All stable cell lines generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details Cell lines and cell culture conditions
DLD-1, SH-SY5Y, U2OS, and 293T cells were grown in DMEM (Thermo) containing 10% heat-inactivated FBS (Thermo) at 37 °C with 5% CO2.
Method Details
Sequence and Coordinate Retrieval
mRNAs from NCBI RefSeq database (O’Leary et al., 2016) were downloaded and 3’UTR sequences as defined by NCBI were extracted. hg19 coordinates were retrieved by aligning 3’UTR sequences using HISAT2 (Kim et al., 2015). Only 3’UTRs with mappable coordinates were included in Table S1.
Circular RNAs (circRNAs) were downloaded from circBase (Glažar et al., 2014). To isolate only exonic circRNAs, circRNA sequences were aligned using HISAT2 (Kim et al., 2015) to the NCBI RefSeq mRNA sequences missing the first and last exons. The first exon does not have an intron upstream and the last exon does not have an intron downstream, which would be required for a circRNA generated by the canonical splicing machinery.
RNA Folding
RNA sequences were folded using the “RNAfold” function from ViennaRNA package (Lorenz et al., 2011) to calculate the minimum thermodynamic free energy (−ΔG/nt). ΔG is defined as the minimum free energy of the RNA secondary structure as determined by the default RNAfold function within the ViennaRNA Package 2.0 (Lorenz et al., 2011). For each 3’UTR, the minimum free energy value is divided by the length of the 3’UTR to calculate the −ΔG/nt value. The “shape” modifier was used to guide the folding with DMS-MaPseq data. The “circ” modifier was used to fold circular RNAs. To generate DMS-guided RNA structure images, the RNAstructure package (Bellaousov et al., 2013) was used to fold the RNA using DMS and DMS-MaPseq data as guides. RNA structure of candidate transcripts and circular RNAs was also determined using mFOLD (Zuker, 2003) and RNAstructure (Bellaousov et al., 2013). RNA base-pairing probability for every nucleotide was determined using the partition function (-p) in RNAfold. The number of nucleotides within the 3’UTR that was base-paired (0–99% probability thresholds) were counted and normalized to the length to determine the fraction of nucleotides base-paired. UPF1 and G3BP1-bound regions were calculated for base-paired nucleotides (≥90% probability) within the CLIP-seq peaks. Due to the small peak size for G3BP1 CLIP-seq data, we also analyzed base-paired nucleotides 50, 100, and 200-nt around the center of the peak for G3BP1 and UPF1+G3BP1-bound regions.
RT-qPCR
Total RNA was extracted by Trizol (Thermo), phase-separated with chloroform, precipitated with isopropyl alcohol, washed with 75% ethanol and re-dissolved in water. Reverse transcription was performed with 1 μg RNA and random hexamers using the SuperScript VILO cDNA Synthesis Kit (Thermo). CircRNA primers (Table S6) were designed to cross the back-splice junction and RT-qPCR products were sent for Sanger sequencing to confirm specific amplification.
Western Blotting
Cells were lysed in 1% SDS lysis buffer and ran on a 10% SDS-PAGE gels. Protein was semi-dry-transferred to a 0.45 μm nitrocellulose membrane (BioRad), blocked in Tris-buffered saline (TBS; Sigma) at pH 7.6 with Tween-20 (TBS-T; Sigma) containing 3% non-fat milk (Giant Eagle), and probed with primary antibodies in TBS overnight at 4 °C. Membranes were washed 3 times with TBS-T, probed for eithr IRDye 800 anti-mouse or IRDye 680 anti-rabbit (LiCor) secondary antibodies, washed an additional 3 times with TBS-T for 5 min each and imaged using the Odyssey Imaging System (Licor). UPF1 (1:1000; Santa Cruz, sc-393594), G3BP1 (1:1000; Santa Cruz, sc-365338), G3BP2 (1:1000; Bethyl Laboratories, A302–040A-M), ACTB (1:3000; Sigma, A1978), and GFP (1:1000; Roche, 11814460001) primary antibodies were used.
Immunoprecipitations
For immunoprecipitations, ~2.0 × 107 DLD-1 cells stably expressing GFP-tagged genes were washed in 1X PBS, trypsinized and pelleted by centrifugation (1,000 xg for 5 min). The cell pellet was washed in 1X PBS and 1/10 of the pellet was used for RNA extraction (input). Cells were then lysed in 500 μl of lysis buffer [HEPES pH 7.4 (50 mM), NaCl (150 mM), MgCl2 (1 mM), EGTA (1 mM), Triton-X-100 (1%), DTT (1 mM), SUPERase RNase inhibitor (Thermo; 100 U/ml), SigmaFast Protease inhibitor cocktail (Sigma, 0.01%)], and cell debris was pelleted (21,100 xg for 10 min) and 1/10 of the lysate was used for western blot analysis (input). For each sample, antibody-conjugated beads were generated using 0.1 μg of anti-GFP antibody (Invitrogen, A-11120) and 50 μl of Dynabeads Protein G (Thermo, 10004D) and rotated constantly for 10 min at room temperature and subsequently washed once in lysis buffer. GFP-tagged proteins from lysate were immunoprecipitated with the antibody-conjugated beads at 4 °C for 2 h with constant rotation. Beads were then washed twice in lysis buffer and twice in high salt lysis buffer (300 mM NaCl) buffers for 5 min each. A fifth of the pulldown was used for western blot analysis, while the rest was used for RNA isolation using Trizol, reverse-transcribed, and analyzed by RT-qPCR in triplicate.
Cell line transfections
For plasmid transfections, cells were transfected with lipofectamine 2000 (Thermo) or polyethylenimine (Polysciences).
Actinomycin D treatment
DLD-1 cells were treated with actinomycin D (ACTD; Cell Signaling, 10 μg/ml) to block transcription. Cells were harvested every 6 h over a 24 h time course. RNA expression was analyzed by RT-qPCR.
Stable cell line generation
For the generation of stable shRNA knockdown cells, lentiviruses were generated in 293T using the PLKO.1 lentivirus packing system with gene-specific shRNAs (Table S6). Following 48 h of lentivirus production, media was collected, and cell debris was removed by centrifugation. Lentivirus was immediately added with 8 μg/ml polybrene (Santa Cruz) to cells in a 6-well dish. Cells were diluted 1/20 and transferred to a 15 cm2 plate plated 24 h after transduction and treated with puromycin (4 μg/ml for DLD-1 and 1 μg/ml for SH-SY5Y; Sigma) until single-cell colonies could be isolated and analyzed by RT-qPCR to confirm knockdown. Three colonies for each knockdown (2 knockdowns per gene) were isolated and analyzed.
For the generation of stable CRISPR-Cas9 knockout cells, DLD-1 cells were transfected with the pSpCas9(BB)-2A-GFP plasmid that expresses both Cas9 and a gRNA targeting the gene of interest. Following transfection, single cells were isolated and cultured. Individual colonies were analyzed by western blot to confirm gene knockout.
For the generation of stable constitutively expressing cells, stable knockdown cells with the shRNA targeting the 3’UTR were transfected with a GFP-tagged gene of interest that lacked the 3’UTR. Cells were treated with Geneticin (0.6 mg/ml; Thermo) and grown until single colonies could be analyzed for GFP expression by microscopy. Three GFP-positive colonies were isolated per integration.
For the generation of DOX-inducible cells, the Flp-In T-Rex system (Thermo) was utilized. DLD-1 cells had the initial integration site already inserted (Andrew Holland Lab, Johns Hopkins University). Stable knockdown DLD-1 cells with the shRNA targeting the 3’UTR were transfected with a GFP-tagged gene of interest inserted into the Flp-In plasmid system. Successfully inserted colonies were isolated after Hygromycin treatment (0.5 mg/ml; Gibco). Gene induction was achieved through treatment with Doxycycline hyclate (10 μg/ml; Sigma)
Plasmid Construction
Plasmids expressing GFP-tagged UPF1 and G3BP1 were modified using site-directed mutagenesis (Table S6). In brief, overlapping primers containing the desired mutation were used to amplify the entire plasmid. PCR products were treated with DpnI (NEB) for 2 h at 37 °C, PCR purified using the QIAquick PCR Purification Kit (Qiagen) and transformed into DH5α competent cells with antibiotic selection. Mutated genes were sequenced to confirm no additional mutations.
For luciferase assay, the psiCHECK-2 plasmid (Promega) was further modified so both luciferase genes within the plasmid contained an intron (psiCHECK-3). This construct allows RT-qPCR reactions to have one primer spanning the intron in both luciferase genes. To clone 3’UTRs into luciferase assay, cDNA from DLD-1 cells were amplified using 3’UTR-specific primers (Table S6) or synthesized (Thermo; Table S6). Sequences were cloned into the firefly luciferase 3’UTR with XhoI (5’ end) and PmeI (3’ end). The unstructured artificial sequence was designed using NUPACK (Zadeh et al., 2011).
Luciferase Assay and RT-qPCR
DLD-1 cells were seeded in 12-well plates and transfected with 100 ng of plasmid when cells reached 60–80% confluency. After 24 h transfection, RNA was extracted, cDNA generated as described earlier, and RT-qPCR was performed in triplicate. 3’UTRs from candidate transcripts and modified sequences were inserted as the 3’UTR for the Renilla luciferase (Table S6). Cells were transfected with luciferase plasmids and harvested 24 hours after transfection for RT-qPCR analysis in triplicate. Renilla luciferase expression was normalized to the control firefly luciferase gene within the same plasmid. For all steady-state luciferase-based experiments, the assays were repeated three separate times. When using the DOX-inducible DLD-1 cells, cells were treated with or without DOX-induction (10 μg/ml) for 24 h prior to transfection. For ACTD luciferase assays, the experiment was repeated twice and values reported (Table S5).
The MS2BP/MS2 and λN/BoxB tethering system (Bos et al., 2016) was utilized with the luciferase assay. UPF1 (WT and DEAA) was tagged with λN and G3BP1 (WT and S149A) with MS2BP on the N-terminus. The MS2 and BoxB sequences were inserted into the 3’UTR of the Renilla luciferase gene in an alternating fashion to generate 3 repeats of MS2/BoxB sequences (Table S6). The luciferase assay was performed as before, but the cells were transfected with the luciferase system and proteins at the same time and harvested 48 h later for RNA analysis.
RNA-sequencing
RNA-sequencing was performed by the Experimental and Computational Genomics Core at Johns Hopkins University. In brief, 1 μg of RNA extracted from G3BP1 WT or KO DLD-1 cells treated with or without actinomycin D (ACTD; 10 μg/ml for 18 h) were used for library preparation. To better normalize changes in RNA expression between 0 h and 18 h of ACTD treatment, 1 μl of 1:100 diluted ERCC RNA Spike-In Mix (Thermo) was added to each sample. Libraries were prepared following the manufacturer’s instructions using the TruSeq Stranded Total RNA Kit (Illumina) with barcoding and sequenced on the HiSeq2500 (Illumina) with 151×151 paired-end reads. Illumina’s CASAVA 1.8.4 was used to convert BCL files to FASTQ files using default parameters.
in vitro DMS analysis
DMS analysis was performed on in vitro-transcribed RNA. In brief, the EIF3B 3’UTR was PCR amplified from the luciferase plasmids using a forward primer containing a T7 promoter sequence. RNA was in vitro-transcribed using the AmpliScribe T7 High Yield Transcription Kit (Epicentre) and purified by Trizol isolation. RNA (1 μg) in 25 μl of 10 mM MOPS pH 7 was heated to 95 °C for 1 min and slowly cooled to 4 °C using a thermocycler. MgCl2 (0.1 mM final) was added to RNA and incubated at 37 °C for 30 min. Denatured controls underwent the same protocol but did not receive MgCl2, and following the 37 °C incubation, the RNA was denatured at 95 °C for 2 min and then moved directly to ice.
Folded RNA was subjected to 5% DMS treatment (Sigma, 1:5 diluted DMS:EtOH) for 3 min at room temperature. DMS reactions were stopped with 475 μl quench solution (30% β-mercaptoethanol [Sigma], 0.3 M sodium acetate), mixed with 1 ml of EtOH and incubated at −20 °C for 20 min. RNA was pelleted by centrifugation and resuspended in 100 μl of 0.3 M sodium acetate. RNA was extracted with Trizol.
10 pmol reverse transcription primer was radio-labeled with P32-γ ATP (Perkin Elmer) and purified with a G-25 column (GE). 0.25 pmol of radio-labeled primer was used for each reaction. DMS-labeled RNA was reverse transcribed using SuperScript IV Reverse Transcriptase (Thermo) following the manufacturer’s protocol. In brief, 32P-labeled primers, dNTPs, and RNA was denatured at 65 °C for 5 min and moved directly to ice. The kit-provided SSIV buffer, DTT, RNase inhibitor, and SuperScript IV Reverse Transcriptase was added to each tube. Samples were incubated at 50 °C for 10 min, 80 °C for 10 min and cooled to 4 °C. RNA was degraded with RNaseH, prior to loading on gel. Sanger sequencing reactions were also performed using ddTTP and ddGTP (GE) at a 1:1 molar ratio with the dNTPs. The reactions were then run on a 12% denaturing urea-acrylamide sequencing gels using the UreaGel System (National Diagnostics). Gels were then exposed to Imaging Plates (Fujifilm) for 72 h at −80 °C to prevent band diffusion and imaged with the FLA-7000 Fujifilm imager. Images were normalized to the “No DMS” full-length reverse transcription band.
Quantification and Statistical Analysis
RNA Folding
RNA folding was performed as described above and the average difference in the fraction of base-paired nucleotides for HSUs and PSUs and z-scores representing the significance of the difference between the two populations were determined
RNA Permutations and Folding
14,169 different 3’UTR sequences were randomly permuted one hundred times, and the −ΔG/nt for the new sequences were calculated by “RNAfold” from ViennaRNA (Lorenz et al., 2011). Additionally, the 14 candidate 3’UTRs were permuted an additional 5,000 times to compare the difference in z-scores between the two permutations. The −ΔG/nt mean and standard deviation for each permuted 3’UTR population were used to calculate the z-score of the actual 3’UTR −ΔG/nt. Negative z-scores represent 3’UTRs that have a larger −ΔG/nt and therefore more structure than the folding of the permuted population would predict.
DMS-MaPseq Analysis
DMS-MaPseq data (Zubradt et al., 2017) (NCBI Gene Expression Omnibus, accession number GSE84537, runs SRR3929619 and SRR3929620 for control and DMS-treated HEK293T cells, respectively) was downloaded and uploaded to Galaxy (Afgan et al., 2018). Reads were clipped [Trimmomatic (Bolger et al., 2014), adaptor sequence “TruSeq3 (single-end for MiSeq and HiSeq)”; all settings were otherwise default] and aligned to hg19 with Bowtie2 (Langmead and Salzberg, 2012); all settings were otherwise set as default “simple run”). The resulting indexed BAM files were downloaded and input with a dataset of 3’UTR coordinates into the mismatch frequency analysis pysam (https://github.com/pysam-developers/pysam), a python wrapper around htslib and the samtools package (Li et al., 2009).
Analysis of the pysam output, which listed the read depth and called nucleotide for every 3’UTR position, and all further data manipulation was performed through base R (Team, 2018) and the tidyverse package (Wickham, 2017) unless otherwise specified. Control sample 3’UTR positions were filtered for a minimum read depth of 10x, and the identity of each position was called if all nucleotides at the position were concordant to ensure that positions prone to the incorporation of mutated bases were excluded. DMS-treated sample 3’UTR positions were filtered for a minimum read depth of 30x to provide a balance between adequate signal resolution and site retention (44% of DMS-treated 3’UTR positions met this cut-off). DMS modification frequency was calculated by determining the percent of non-consensus nucleotides for each position that had a corresponding control consensus base.
DMS modifications were normalized using the 2–8% rule (Low and Weeks, 2010) and 3’UTRs were filtered for regions of completeness. 3’UTRs with sufficient coverage were defined as having DMS modification information for at least 80% of all positions or for a stretch of at least 200 nucleotides that have no wider than a four-nucleotide gap.
The sequence and DMS modification information for 3’UTRs with sufficient coverage were inputted to ViennaRNA package’s RNAfold function (Lorenz et al., 2011), which allows the supplementation of RNA sequences with the normalized rate of each nucleotide DMS modification to refine in silico-predicted RNA folding structures by constraining which nucleotides are bound in the predicted secondary structures. There was a moderate correlation (R2 = 0.66) between the percent of DMS modification coverage for each 3’UTR and the difference between DMS modification-informed and -uninformed ΔG per nucleotide predictions for folded 3’UTRs, suggesting that the DMS constraints systematically drove the UTRs to seem more structured than RNAfold on its own predicted. This overestimation was adjusted for in the final DMS modification-informed predicted −ΔG per nucleotide values based on the slope of the linear regression. DMS reactivity for individual 3’UTR nucleotides was compared to their respective base-pairing probabilities. Nucleotides with DMS reactivity within CLIP-seq peaks were also analyzed to determine RNA structure within protein binding regions.
icSHAPE Analysis
Analyzed icSHAPE reactivity datasets were downloaded from NCBI GEO [GSE74353 (Lu et al., 2016) and GSE117840 (Sun et al., 2019)]. Coordinates for the transcripts from the datasets were retrieved from their respective Ensembl Builds (GRch37.75 (Lu et al., 2016) and GRch38.83 (Sun et al., 2019) and single nucleotides were assigned their icSHAPE reactivities. NCBI 3’UTRs (Table S1) were aligned to the respective Ensembl transcripts using HISAT (Kim et al., 2015) to identify identical transcripts between databases. NCBI 3’UTRs aligned to the Ensemble transcripts were utilized to determine icSHAPE reactivity within the NCBI 3’UTRs. Nucleotides with icSHAPE reactivity were analyzed for predicted base-pairing probabilities as well as CLIP-seq peaks were analyzed for icSHAPE reactivity.
CLIP-seq Analysis
Publicly available data was downloaded and entered the following workflow based on file type available using the online Galaxy server (Afgan et al., 2018). For fastq sequencing files, sequences were trimmed with Trimmomatic (Bolger et al., 2014), and aligned using HISAT (Kim et al., 2015). BAM files were counted using StringTie (Pertea et al., 2015). Bed files were intersected with all 3’UTR coordinates to retrieve bound 3’UTRs. To compare the enrichment of WT vs DEAA UPF1 bound 3’UTRs, MACS2 “callpeak” (Feng et al., 2012) was used on the Galaxy servers. The UPF1 mutants were used as the control file to identify regions where WT UPF1 was enriched compared to the helicase mutant DEAA. Composite broad regions were called to identify large regions of UPF1 enrichment. The rest of the settings were left as default, which define the cutoff as peaks that have an FDR (q-value) >0.05. Based on this FDR cutoff, the lowest fold-change observed in the UPF1-helicase dependent enriched peaks was 2.3 fold. Enriched regions were intersected with all 3’UTR coordinates to retrieve bound 3’UTRs and visually inspected using the Integrative Genomics Viewer (Robinson et al., 2011). Cumulative distribution functions were calculated using the ggplot2 (Wickham, 2011) package in R (Team, 2018).
RT-qPCR
RT-qPCR was performed as described above. RNA levels were measured using gene-specific primers (Table S6) in triplicate with PowerUp SYBR Green (Thermo) using the 7500 Fast Real-Time PCR Machine (Applied Biosystems). RNA expression was normalized to the internal control GAPDH. All RT-qPCR data were included in Table S5 including the standard deviation and Student’s t-tests.
For immunoprecipitations performed as described above, RNA enrichment was determined by calculating the fold difference between WT pulldown and the RNA binding mutant pulldown by RT-qPCR. Student’s t-tests were performed between the WT and mutant RT-qPCR pulldown values for all RNAs within the specified groups. RNase A treatment (15 μg/ml; Thermo) was performed during the 2 h pulldown and RNA digestion was confirmed by RT-qPCR of candidate transcripts (data not shown).
For actinomycin D analysis of RNA half-lives performed as described above, changes in RNA half-lives were calculated by the linear fitting of the log-transformed exponential decay function and normalizing the half-life of the gene KD/KO cells to the control cells.
RNA-sequencing
RNA-seq libraries were constructed as described above and rsem-1.3.0 was used for running the alignments as well as generating gene expression levels. The ‘rsem-calculate-expression’ module was used with the following options: --star, --star-gzipped-read-file, --calc-ci, --star-output-genome-bam, --paired-end, --forward-prob 0. The data were aligned to “hg19” reference genome with ERCC spike-in RNA sequences.
To determine differences in gene expression and decay between G3BP1 WT and KO DLD-1 cells, the “remove unwanted variation (RUV)” function was used with default settings to normalize gene counts by the upper quartile and the ERCC spike-in counts (Risso et al., 2014). For changes in steady-state levels between G3BP1 WT and KO DLD-1 cells, normalized counts were analyzed for differential expression by edgeR (Robinson et al., 2010) to identify significantly differentially expressed genes (Fold change >1.5; p-value < 0.05).
For changes in RNA decay between G3BP1 WT and KO cells, the normalized gene counts were converted to absolute concentrations based on the slope of the ERCC spike-in counts compared to their known concentrations. Genes with RNA-seq coverage within the range of the ERCC spike-in counts were utilized for further analyses (10–2,000 counts). To determine RNA decay, replicates were averaged and the 16 h gene counts were normalized to the 0 h time point to determine the fraction of RNA remaining at 16 h. The slope was determined for the natural log of the two time points and the fold difference in decay between WT and KO cells was determined.
Data and Code Availability
All sequencing data generated as a part of this study have been deposited to NCBI BioProject under PRJNA591294.
Supplementary Material
Length, GC content and secondary structure, defined by the lowest thermodynamic free energy using the nearest neighbor energy model in the ViennaRNA Package (Lorenz et al., 2011), was normalized to RNA length (−ΔG/Nucleotide) were determined. CLIP-seq datasets were analyzed for 3’UTR peaks. A (+) indicates that the 3’UTR contained a CLIP-seq peak and a (−) indicates that the 3’UTR did not contain a peak.
DMS sequencing data (Zubradt et al., 2017) was used to guide the folding of 3’UTRs. DMS coverage of modified A and C nucleotides is also denoted for each 3’UTR. CLIP-seq datasets were analyzed for 3’UTR peaks. A (+) indicates that the 3’UTR contained a CLIP-seq peak and a (−) indicates that the 3’UTR did not contain a peak.
G3BP1 WT and KO DLD-1 cells were treated with or without actinomycin-D (10 μg/ml) for 18 h and analyzed by RNA-seq for changes in steady-state expression and decay. RNA-sequencing coverage and alignment are included. For each gene, the average overall structure, GC content, and length of all mRNA isoforms were included. HSUs and PSUs are defined as genes in which all 3’UTRs are highly-structured (−ΔG/nt ≥ 0.3) or poorly-structured (−ΔG/nt ≤ 0.2) respectively. UPF1 and G3BP1 binding are defined as genes in which at least 1 3’UTR isoform was bound. Enrichment and Chi-square (χ2) of 3’UTR features were determined.
CircRNAs that contained introns or contained the first or last exon of an mRNA transcript were excluded. CircRNAs were analyzed for length, GC content and predicted structure normalized to RNA length (−ΔG/nt).
Each analysis also includes the standard deviation (SD) and p-value. Fold changes are indicated by color coding (blue = upregulated and orange = downregulated) using the Excel “Conditional Formatting” function.
Sequences used in this study are included with additional information included (NCBI/circBase, etc.)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RENT1 (UPF1) | Santa Cruz | sc-393594 |
| G3BP1 | Santa Cruz | sc-365338 |
| G3BP2 | Bethyl Laboratories | A302-040A-M |
| ACTB | Sigma | A1978 |
| GFP | Roche | 11814460001 |
| Critical Commercial Assays | ||
| SuperScript VILO cDNA Synthesis Kit | Thermo | 11754–250 |
| PowerUp SYBR Green | Thermo | A25777 |
| AmpliScribe T7 High Yield Transcription Kit |
Epicentre | AS3107 |
| Deposited Data | ||
| DMS-MaPseq | NCBI GEO, this paper | GSE84537, Table S2 |
| UPF1 CLIP-seq | NCBI GEO, DDBJ Sequence Read, this Paper |
GSE69586, GSE86148, GSE47976, DRA003675, Tables S1 and S2 |
| G3BP1 CLIP-seq | ENCODE, this paper | ENCSR721HPX Tables S1 and S2 |
| STAU1 CLIP-seq | ArrayStar | E-MTAB-2937 |
| STAU2 CLIP-seq | ENCODE | ENCSR979EWD |
| icSHAPE | NCBI GEO |
GSE74353, GSE117840 |
| Stress Granule Transcriptome | NCBI GEO | GSE99304, GSE90869 |
| Experimental Models: Cell Lines | ||
| Human: DLD-1 cells | Andrew Holland, ATCC | CCL-221 |
| Human: SH-SY5Y cells | Ted Dawson, ATCC | CRL-2266 |
| Human: U2OS cells | Nancy Kedersha, ATCC |
HTB-96 |
| Oligonucleotides | ||
| List of sequences and primers (Table S6) | ||
| Recombinant DNA | ||
| PLKO.1 Lentivirus expression system | Addgene | Plasmid #8453 |
| psiCHECK2 Luciferase expression system | Promega | C8021 |
| Flp-In T-Rex Core Kit | Andrew Holland, Thermo |
K650001 |
| pEGFP-C1 | Clontech | GenBank #U55763 |
| Software and Algorithms | ||
| ViennaRNA v2.4.12 | (Lorenz et al., 2011) | RRID: SCR_008550 |
| HISAT2 v2.1 | (Kim et al., 2015) | RRID: SCR_015530 |
| RNAstructure v6.1 | (Bellaousov et al., 2013) | RRID: SCR_017216 |
| mFOLD v3.6 | (Zuker, 2003) | RRID: SCR_008543 |
| Trimmomatic v0.36 | (Bolger et al., 2014) | RRID: SCR_011848 |
| Bowtie2 v2.3.4.2 | (Wickham, 2018) | RRID: SCR_005476 |
| pysam v0.15.0 | pysam | https://github.com/pysam-developers/pysam |
| R v3.5.1 | (Team, 2018) | https://www.r-project.org/ |
| Tidyverse v1.2.1 | (Wickham, 2017) | https://www.tidyverse.org/ |
| Galaxy v19.05 | (Afgan et al., 2018) | RRID: SCR_006281 |
| StringTie v1.3.4 | (Pertea et al., 2015) | RRID: SCR_016323 |
| MACS2 v2.1.1.20160309.6 | (Feng et al., 2012) | RRID: SCR_013291 |
| Integrative Genomics Viewer v2.5 | (Robinson et al., 2011) | RRID: SCR_011793 |
| ggplot2 v3.1.1 | (Wickham, 2011) | RRID: SCR_014601 |
HIGHLIGHTS.
UPF1 and G3BP1 globally regulate the decay of mRNAs with highly-structured 3’UTRs.
This structure-mediated RNA decay (SRD) is independent of defined linear sequences.
SRD is dependent on the overall base-pairing density of 3’UTR.
SRD selectively regulates mRNA isoforms and circular RNAs.
ACKNOWLEDGEMENTS
We thank the Leung laboratory, C. Fischer, P. Sharp, R. Green, B. Zinshteyn, H. Dietz, J. Kim, S. Bailey, S. Woodson, G. Seydoux, M. Matunis, S. Myong and J. Wilusz for discussion and reading the manuscript. We thank the ENCODE Consortium and the G. Yeo laboratory for the G3BP1 eCLIP dataset. We thank N. Kedersha for U2OS G3BP1/2 KO cells and G3BP1-GFP plasmid; L. Maquat for UPF1-FLAG plasmid; A. Holland for DLD-1 cells with integrated Flip-In site; and T. Dawson for SH-SY5Y cells. We thank A. Jaffe for providing the ERCC RNA spike-in reagent and J. Ellenberg for lambdaN constructs. This work was supported by Johns Hopkins Bloomberg School of Public Health Start-up fund (AKLL), Core Coin Pilot grant from Experimental and Computational Core, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (JWF, VFB, AKLL), and Brian D. Crawford and Family Research Scholarship (JWF), and NIH grants F31GM125109 (JWF), T32CA009110 (JWF), and T32GM007814 (JWF).
Footnotes
DECLARATION OF INTERESTS
Authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin J-D, Novoa EM, Vejnar CE, Yartseva V, Takacs CM, Kellis M, and Giraldez AJ (2018). Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol 25, 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellaousov S, Reuter JS, Seetin MG, and Mathews DH (2013). RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 41, W471–W474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder R, Horowitz JA, Basilion JP, Koeller DM, Klausner RD, and Harford JB (1994). Evidence that the pathway of transferrin receptor mRNA degradation involves an endonucleolytic cleavage within the 3’ UTR and does not involve poly(A) tail shortening. EMBO J. 13, 1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos TJ, Nussbacher JK, Aigner S, and Yeo GW (2016). Tethered Function Assays as Tools to Elucidate the Molecular Roles of RNA-Binding Proteins. Adv. Exp. Med. Biol 907, 61–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JAL, Roberts TL, Richards R, Woods R, Birrell G, Lim YC, Ohno S, Yamashita A, Abraham RT, Gueven N, et al. (2011). A novel role for hSMG-1 in stress granule formation. Mol. Cell. Biol 31, 4417–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, and Mühlemann O. (2017). Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA 23, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, and Snyder M. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini F, Bagchi D, Le Hir H, and Croquette V. (2015). Human Upf1 is a highly processive RNA helicase and translocase with RNP remodelling activities. Nat. Commun 6, 7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flury V, Restuccia U, Bachi A, and Mühlemann O. (2014). Characterization of phosphorylation- and RNA-dependent UPF1 interactors by quantitative proteomics. J. Proteome Res 13, 3038–3053. [DOI] [PubMed] [Google Scholar]
- Glažar P, Papavasileiou P, and Rajewsky N. (2014). circBase: a database for circular RNAs. RNA 20, 1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, and Kjems J. (2011). miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 30, 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JR, and Goff SP (2010). Upf1 senses 3′ UTR length to potentiate mRNA decay. Cell 143, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamachi N, Salam KA, Suzuki Y, and Akimitsu N. (2017). A GC-rich sequence feature in the 3’ UTR directs UPF1-dependent mRNA decay in mammalian cells. Genome Res. 27, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, and Sharpless NE (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, et al. (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol 212, 845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, and Parker R. (2017). The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol. Cell 68, 808–820.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, and Maquat LE (2019). UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 25, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SS-Y, Sze L, Liu C, and Lam K-P (2019). The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-β response. J. Biol. Chem 294, 6430–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Furic L, Desgroseillers L, and Maquat LE (2005). Mammalian Staufen1 recruits Upf1 to specific mRNA 3’UTRs so as to elicit mRNA decay. Cell 120, 195–208. [DOI] [PubMed] [Google Scholar]
- Kim YK, Furic L, Parisien M, Major F, DesGroseillers L, and Maquat LE (2007). Staufen1 regulates diverse classes of mammalian transcripts. EMBO J. 26, 2670–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, and Wilusz JE (2015). Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 29, 2168–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łabno A, Tomecki R, and Dziembowski A. (2016). Cytoplasmic RNA decay pathways - Enzymes and mechanisms. Biochim. Biophys. Acta 1863, 3125–3147. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Pratt GA, Martinez FJ, Yeo GW, and Lykke-Andersen J. (2015). Target Discrimination in Nonsense-Mediated mRNA Decay Requires Upf1 ATPase Activity. Mol. Cell 59, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WM, Barnes T, and Lee CH (2010). Endoribonucleases--enzymes gaining spotlight in mRNA metabolism. FEBS J. 277, 627–641. [DOI] [PubMed] [Google Scholar]
- Liu C-X, Li X, Nan F, Jiang S, Gao X, Guo S-K, Xue W, Cui Y, Dong K, Ding H, et al. (2019). Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 177, 865–880.e21. [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol. Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JT, and Weeks KM (2010). SHAPE-directed RNA secondary structure prediction. Methods 52, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, et al. (2016). RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas BA, Lavi E, Shiue L, Cho H, Katzman S, Miyoshi K, Siomi MC, Carmel L, Ares M Jr, and Maquat LE (2018). Evidence for convergent evolution of SINE-directed Staufen-mediated mRNA decay. Proc. Natl. Acad. Sci. U. S. A 115, 968–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C. (2019). What are 3′ UTRs doing? Cold Spring Harb. Perspect. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, and Dietz HC (2004). Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet 36, 1073–1078. [DOI] [PubMed] [Google Scholar]
- Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels H-H, Ori D, Uehata T, Tartey S, Akira S, Suzuki Y, et al. (2015). Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 161, 1058–1073. [DOI] [PubMed] [Google Scholar]
- Mitchell SF, and Parker R. (2014). Principles and properties of eukaryotic mRNPs. Mol. Cell 54, 547–558. [DOI] [PubMed] [Google Scholar]
- Namkoong S, Ho A, Woo YM, Kwak H, and Lee JH (2018). Systematic Characterization of Stress-Induced RNA Granulation. Mol. Cell 70, 175–187.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, and Vogelstein B. (1991). Scrambled exons. Cell 64, 607–613. [DOI] [PubMed] [Google Scholar]
- O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J, Moran-Jones K, Albrecht V, Schwarzl T, Hunter K, Gires O, and Kolch W. (2011). c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res. 71, 4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S. (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweingruber C, Soffientini P, Ruepp M-D, Bachi A, and Mühlemann O. (2016). Identification of Interactions in the NMD Complex Using Proximity-Dependent Biotinylation (BioID). PLoS One 11, e0150239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Tang Y, Ritchey LE, Tack DC, Zhu M, Bevilacqua PC, and Assmann SM (2018). Genome-wide RNA structurome reprogramming by acute heat shock globally regulates mRNA abundance. Proc. Natl. Acad. Sci. U. S. A 115, 12170–12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, D’Ambrogio A, Luscombe NM, and Ule J. (2015). hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, Huang W, Kool ET, Chang HY, and Zhang QC (2019). RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol 26, 322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.C. (2018). R: A language and environment for statistical computing; 2015. [Google Scholar]
- Tourrière H, Gallouzi IE, Chebli K, Capony JP, Mouaikel J, van der Geer P, and Tazi J. (2001). RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Mol. Cell. Biol 21, 7747–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, and Tazi J. (2003). The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol 160, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Venø MT, Hansen TB, Venø ST, Clausen BH, Grebing M, Finsen B, Holm IE, and Kjems J. (2015). Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 16, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2011). ggplot2. Wiley Interdisciplinary Reviews: Computational Statistics 3, 180–185. [Google Scholar]
- Wickham H. (2017). The tidyverse. R Package Ver. 1. 1 1. [Google Scholar]
- Youn J-Y, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM, et al. (2018). High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 69, 517–532.e11. [DOI] [PubMed] [Google Scholar]
- Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM, and Pierce NA (2011). NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem 32, 170–173. [DOI] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, and Rouskin S. (2017). DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zünd D, Gruber AR, Zavolan M, and Mühlemann O. (2013). Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3’ UTRs. Nat. Struct. Mol. Biol 20, 936–943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Length, GC content and secondary structure, defined by the lowest thermodynamic free energy using the nearest neighbor energy model in the ViennaRNA Package (Lorenz et al., 2011), was normalized to RNA length (−ΔG/Nucleotide) were determined. CLIP-seq datasets were analyzed for 3’UTR peaks. A (+) indicates that the 3’UTR contained a CLIP-seq peak and a (−) indicates that the 3’UTR did not contain a peak.
DMS sequencing data (Zubradt et al., 2017) was used to guide the folding of 3’UTRs. DMS coverage of modified A and C nucleotides is also denoted for each 3’UTR. CLIP-seq datasets were analyzed for 3’UTR peaks. A (+) indicates that the 3’UTR contained a CLIP-seq peak and a (−) indicates that the 3’UTR did not contain a peak.
G3BP1 WT and KO DLD-1 cells were treated with or without actinomycin-D (10 μg/ml) for 18 h and analyzed by RNA-seq for changes in steady-state expression and decay. RNA-sequencing coverage and alignment are included. For each gene, the average overall structure, GC content, and length of all mRNA isoforms were included. HSUs and PSUs are defined as genes in which all 3’UTRs are highly-structured (−ΔG/nt ≥ 0.3) or poorly-structured (−ΔG/nt ≤ 0.2) respectively. UPF1 and G3BP1 binding are defined as genes in which at least 1 3’UTR isoform was bound. Enrichment and Chi-square (χ2) of 3’UTR features were determined.
CircRNAs that contained introns or contained the first or last exon of an mRNA transcript were excluded. CircRNAs were analyzed for length, GC content and predicted structure normalized to RNA length (−ΔG/nt).
Each analysis also includes the standard deviation (SD) and p-value. Fold changes are indicated by color coding (blue = upregulated and orange = downregulated) using the Excel “Conditional Formatting” function.
Sequences used in this study are included with additional information included (NCBI/circBase, etc.)
Data Availability Statement
All sequencing data generated as a part of this study have been deposited to NCBI BioProject under PRJNA591294.