

Abstract
Ageing is a major risk factor for the development of cardiovascular disease (CVD) and cancer. Whilst the cumulative effect of exposure to conventional cardiovascular risk factors is important, recent evidence highlights clonal haematopoiesis of indeterminant potential (CHIP) as a further key risk factor. CHIP reflects the accumulation of somatic, potentially pro-leukaemic gene mutations within haematopoietic stem cells over time. The most common mutations associated with CHIP and CVD occur in genes that also play central roles in the regulation of inflammation. While CHIP carriers have a low risk of haematological malignant transformation (<1% per year), their relative risk of mortality is increased by 40% and this reflects an excess of cardiovascular events. Evidence linking CHIP, inflammation and atherosclerotic disease has recently become better defined. However, there is a paucity of information about the role of CHIP in the development and progression of heart failure, particularly heart failure with preserved ejection fraction (HFpEF). While systemic inflammation plays a role in the pathophysiology of both heart failure with reduced and preserved ejection fraction (EF), it may be of greater relevance in the pathophysiology of HFpEF, which is also strongly associated with ageing. This review describes CHIP and its pathogenetic links with ageing, inflammation and CVD, while providing insight into its putative role in HFpEF.
Keywords: ageing, atherosclerosis, cardiovascular disease, clonal haematopoiesis of indeterminate potential, heart failure
Introduction
Cardiovascular disease (CVD) and cancer are the two leading causes of deaths worldwide and ageing is a major risk factor for the development of both of these major disease processes [1,2]. To a large extent, these age-associated risks reflect the cumulative effects of exposure to ‘conventional’ shared risk factors such as smoking and obesity. However, clonal haematopoiesis of indeterminate potential (CHIP) may provide a further important link, particularly in the pathogenesis of CVD [3]. CHIP, also known as age-related clonal haematopoiesis (ARCH), reflects the accumulation of potentially pre-leukaemic, somatic mutations in haematopoietic stem cells (HSCs) over time [3,4]. However, whilst the risk of malignant transformation of CHIP is low, its presence confers a substantially greater risk of CVD (Figure 1) [3,5–9].
Figure 1. Development of clonal haematopoiesis, associated risk factors and its role in heart failure (murine and humans).

Abbreviations: HSC, haematopoietic stem cells; EF, ejection fraction; FS, fractional shortening; HFH, heart failure hospitalisation; HFrEF, heart failure with reduced ejection fraction; KO, knockout; LAD, left anterior descending artery; NT-proBNP, B-type natriuretic peptide; TAC, transverse aortic constriction.
In this review, we will provide a primer on CHIP and its pathogenetic links with ageing, inflammation and CVD. In particular, we provide a framework to inform further investigation of the role of CHIP as a risk factor and pathogenetic mediator in patients with heart failure, especially in heart failure with preserved ejection fraction (HFpEF).
CHIP: a primer
Definition and overview of CHIP
Current diagnostic criteria for clonal haematopoiesis of indeterminate potential (CHIP) include: (1) the absence of overt haematological malignancy; (2) a normal peripheral blood count and (3) mutant cells bearing relevant driver mutations in ≥2% of peripheral white blood cells (variant allele frequency [VAF] ≥ 2%) [10]. By the age of 70 years, 10–20% of the otherwise healthy population have a peripheral blood leucocyte clone with a VAF of at least 2% and meet the criteria for CHIP [5,9,11–13]. Conversely, CHIP is found in fewer than 1% of patients under the age of 50 years [3,9,13].
CHIP can be detected via DNA sequencing of peripheral blood, saliva and tumour samples and, while deep-sequencing methods may detect a VAF of less than 2%, the clinical consequences of these smaller clonal populations are unknown [14–16]. The majority of these age-associated mutations are cytosine (C) to thymine (T) transitions, consistent with the signature of mutations seen across many different types of cancer [3,9]. The most frequently encountered somatic mutations are within the driver genes ten-eleven-translocation-2 (TET2), DNA methyltransferase 3 [DNMT3]), Janus kinase 2 (Jak2) and additional sex comb-like 1 (ASXL1). CHIP-associated mutations are also found, albeit less frequently, in other driver genes outlined in (Table 1) [17].
Table 1. Most frequent somatic mutations in CHIP.
| Gene | Name | Description |
|---|---|---|
| TET2 | Ten-eleven-translocation-2 | A methylcytosine dioxygenase that catalyses the conversion of 5-methylcytosine into 5-hydroxymethylcytosine. An epigenetic regulator that can activate or repress transcription. |
| DNMT3A | DNA methyltransferase 3A | A de novo DNA methyltransferase. |
| Jak2 | Janus kinase 2 | Receptor tyrosine kinase involved in haematopoietic cytokine signalling and myelopoiesis. |
| ASXL1 | Additional sex comb-like 1 | Polycomb chromatin-binding protein that is involved in the transcriptional regulation of Hox genes. |
| PPMD1 | Protein phosphatase, magnesium/manganese- dependent 1D | Protein phosphatase involved in dephosphorylation and inactivation of proteins in the DNA damage response pathway. |
| SF3B1 | Splicing factor 3B, subunit 1 | A component of the U2 small nuclear riboprotein that binds to the 3′ branch site in pre-mRNA splicing and processing. |
| SRSF2 | Serine/Arginine-rich splicing factor 2 | Required for 5′ and 3′ spliceosome assembly, splice-site selection, U1 and U2 snRNP interactions with pre-mRNA, and alternative splicing. |
| TP53 | Transformation-related protein 53 | Tumour suppressor transcription factor that responds to cellular stress and DNA damage. |
Adapted from [11]. Abbreviations: DNA, deoxyribonucleic acid; mRNA, messenger ribonucleic acid; snRNP, small nuclear ribonucleioprotein.
Triggers and risk factors for CHIP
Little is known about the triggers to clone initiation and expansion. The natural process of ageing results in an increased likelihood of retaining somatic mutations [18,19]. At the molecular level, DNA damage, telomere shortening and autophagy appear to be central mechanisms underlying age-related functional impairment and decline in the durability of HSCs [20,21]. Chronic low-grade inflammation occurs with ageing (recently described as inflammageing) and may also be partly responsible [22]. Indeed, exposure of mice to the pro-inflammatory mediator, tumour necrosis factor-α (TNF-α), promotes the expansion of TET2 mutant clones and exposure to inflammatory stress in myeloid cells results in the rapid increase in frequency and absolute number of TET2-mutated myeloid cells [23,24]. Exogenous stressors that directly provoke inflammation, DNA damage, telomere shortening and production of reactive oxygen species may lead to the premature exhaustion of HSCs and an increased likelihood of retaining somatic mutations at a younger age [25]. Consistent with this hypothesis, prior chemotherapy and radiotherapy are associated with an increased susceptibility to the retention of these somatic mutations in humans [26,27]. To date, little attention has been paid to the environmental factors that may influence the development of CHIP but smoking, diet and diabetes have been associated with risk for clonal expansion in humans [3,13,28] (Table 2). While there has been speculation about a potentially heritable risk of CHIP, this was not confirmed in studies of mono- and di-zygotic twins [29,30].
Table 2. Risk factors for CHIP.
| Degree of risk | References | |
|---|---|---|
| Non-modifiable risk factors | ||
| Age | ↑ | [3,9,13] |
| Male sex | ↑ | [3] |
| Race | ||
| Hispanic ancestry | ↓ | [3] |
| Asian ancestry | ↓ | [34] |
| Modifiable risk factors | ||
| Smoking | ↑ | [13] |
| Diabetes | ↑ | [3] |
| Unhealthy diet | ↑ | [28] |
| Radiation exposure | ↑ | [26] |
| Chemotherapy exposure | ||
| Platinum agents (cisplatin, carboplatin and oxaliplatin) | ↑ | [26,157] |
| Topoisomerase inhibitors (e.g. etoposide) | ↑ |
CHIP and risk for haematological malignancy and CVD
CHIP belongs to a spectrum of haematological pre-malignant states and is associated with the development of various haematological malignancies including leukaemia, lymphoma and myeloma [3,26]. However, most carriers will not develop malignancy and the progression rate is approximately 0.5–1% per year [31]. Malignant transformation or progression generally requires the acquisition of multiple mutations and directly correlates with the mean VAF [3,10]. It is notable that patients found to have CHIP at the time of autologous stem cell transplantation are at an increased risk for the subsequent development of therapy-related myeloid neoplasm (myelodysplastic syndrome and acute myeloid leukaemia) [32].
Despite the low risk of progression to haematologically important diagnoses, all-comers with CHIP have a 40% higher mortality than those without CHIP, and this striking excess is a reflection of cardiovascular events [3]. The presence of CHIP confers a substantially increased risk for CVD independent of traditional risk factors including diabetes and hypercholesterolaemia [3,5].
CHIP and inflammation
The effects of specific CHIP-associated mutations are yet to be fully described, but a core feature appears to be the establishment of a pro-inflammatory state. Compared with those without CHIP, people with evidence of clonal haematopoiesis have higher circulating concentrations of pro-inflammatory markers including interleukin-6 (IL-6), TNF-α and monocyte chemoattractant protein 1 (MCP-1) [33,34]. Driver gene-specific analysis of a large cohort of individuals with CHIP highlighted the association of TET2 mutations with increased IL-1β, whereas Jak2 and SF3B1 mutations were associated with higher circulating IL-18 [34]. Other, potentially less sensitive markers of inflammation such as white blood count (WBC), neutrophil count, C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) are not normally elevated in people with CHIP [33,34]. It has been proposed that the role of inflammation in CHIP is bidirectional, whereby inflammation initially predisposes to the development of CHIP, with consequent unregulated pro-inflammatory cytokine release via a feedback loop [35]. Of the mutations associated with CHIP, TET2, DNMT3A, Jak2V617F and ASXL1 are the most frequent. To date, it is unknown whether specific mutations in the TET2, DNMT3A and ASXL1 genes have different clinical consequences. Several different mutations have been reported to occur in each gene and the pathophysiologic effects of these have not yet been individually characterised [3,10].
TET2
Mutation of TET2 was the first somatic genetic abnormality to be reported in blood cells from individuals with clonal haematopoiesis without overt haematological malignancy [36]. TET2 is a member of a family of enzymes located on chromosome 4q23 and is an epigenetic regulator of DNA methylation. It catalyses the oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcystosine (5hmC) as the first step in cytosine demethylation [37]. This activity is critical for maintaining the normal development of HSCs. TET2 mutations are loss-of-function mutations associated with a decrease in 5hmC availability and consequently this has been proposed as a potential diagnostic and prognostic biomarker in haematological malignancy [38]. Whether it holds the same potential utility in the prediction of CHIP/TET2 mutation-associated CVD remains to be tested. TET2 also plays an important role in the regulation of the immune system, and evidence suggests that TET2-mediated clonal haematopoiesis contributes to the pathophysiology and progression of CVD through its induction of a pro-inflammatory state. TET2 controls the secretion of pro-inflammatory cytokines through modulation of histone acetylation [39,40]. Lipopolysaccharide (LPS) and interferon-γ (IFN-γ) stimulation of macrophages from TET2 deficient mice induces the hyperactivation of pro-inflammatory cytokines and chemokines including IL-1β and IL-6 [39]. Furthermore, loss of TET2 in myeloid-derived cells results in a higher expression of IL-6 in mice [40]. In an unselected cohort of patients without CVD, the presence of TET2 mutation was associated with over two-fold higher circulating concentrations of IL-8 than in those without this mutation [5].
DNMT3A
DNMT3A modulates gene transcription via the catalysis of DNA methylation and is the most frequently mutated gene in people with CHIP. DNMT3A mutations are thought to be loss-of-function mutations although there are reports that some mutations may lead to gain-of-function, conferring increased HSC self-renewal and subsequent clonal expansion [41,42]. DNMT3A also has multiple roles in the regulation of inflammation. In particular, it controls cytokine expression through the regulation of the scaffold protein IQ motif containing GTPase Activating Protein 2 (IQGAP2) in mast cells [43]. In patients with osteoarthritis, IL-6 gene activity is associated with the expression of DNMT3A and significantly lower levels of IL-6 secretion are found in those with DNMT3A overexpression [44]. Furthermore, in patients with severe aortic stenosis, the presence of DNMT3A mutations has been associated with significantly elevated T helper 17 cell (TH17): regulatory T cells (Tregs) ratio, representing pro-inflammatory T-cell polarisation [8].
Jak2V617F
Of CHIP-associated genetic abnormalities, Jak2V617F gain-of-function mutation has been linked most clearly to inflammatory processes. In humans, it serves as a signal transmitter downstream of major cytokine receptors resulting in activation of granulocytes, T cells, enhanced inflammation in macrophages and activation of neutrophil extracellular traps [45]. V617F somatic mutation of the Jak2 gene reflects substitution of phenylalanine for valine at position 617. Jak2V617F mutations are commonly associated with myeloproliferative neoplasms including essential thrombocythaemia (ET) and polycythaemia vera (PV) [46]. These conditions are associated with an increased risk of stroke, myocardial infarction and deep vein thrombosis, primarily as a result of increased blood viscosity and a pro-coagulant state. However, Jak2V617F mutations are increasingly recognised in individuals with normal peripheral blood counts, and remain associated with increased cardiovascular mortality [13,47–49].
ASXL1
ASXL1 encodes an epigenetic regulator which binds to chromatin. It is one of the most frequently mutated genes in myeloid neoplasms and its presence is associated with poor prognosis [50–52]. The majority of mutations are frameshift or nonsense mutations and frequently coexist with TET2, IDH1 and IDH2 mutations [52–55]. However, whether these truncations of the protein lead to loss- or gain-of-function remains controversial [56–58]. Mutation of ASXL1 is common in patients with atherosclerosis and chronic ischaemic heart failure but the mechanisms contributing to this increased CV risk are not defined [5,7].
CHIP and vascular disease
Atherosclerosis is an inflammatory disease, predominantly of the macro-vasculature. Almost 60% of elderly patients with atherosclerosis have either no conventional risk factors (e.g. hypertension or hypercholesterolaemia) or have only one risk factor, thus implying the presence of otherwise unidentified predisposing conditions [59]. CHIP has been identified as a potential factor closely linked to the initiation and progression of atherosclerosis [5]. Microvascular disease involves a complex interplay between upstream atherosclerosis, inflammation and endothelial dysfunction. Of the CHIP-related mutations, the role of TET2 has been most clearly defined in relation to vascular disease and normal TET2 function has been implicated in several important regulatory processes in both the macro- and microcirculation [60–64]. These include suppression of vascular smooth muscle cell (VSMC) phenotypic transformation, protective effects upon endothelial cells as well as anti-inflammatory and anti-atherogenic effects [60–64].
CHIP and human atherosclerosis
Nested case–control analyses of prospective cohorts, that together enrolled 4726 participants with coronary artery disease and 3529 controls, revealed that carriers of CHIP (DNMT3A, TET2 and ASXL1 mutations) have a risk of coronary artery disease that is substantially greater than controls (Table 3). Indeed, patients with CHIP were twice as likely to have a history of myocardial infarction or coronary revascularisation than people without CHIP [5]. CHIP-associated DNMT3A mutation was associated with a hazard ratio of 1.7 for coronary artery disease while TET2 mutation conferred a hazard ratio of 1.9. Those with JakV617F mutation had the highest increased risk of coronary artery disease, which was 12-times greater than people with no mutation. In younger patients, the association between CHIP and atherosclerotic risk was even stronger than in older individuals [5]. In the same study, people with CHIP without a prior diagnosis of coronary artery disease were three times more likely to have a computed tomography (CT) coronary artery calcification (CAC) score of at least 615 Agatston units [5], the empirical cutoff for the identification of older patients at high risk of coronary events [65]. This coronary artery calcification score correlated positively with percentage VAF implying a ‘dose effect’ of the accumulation of mutated cells. Patients with large mutant clone populations (VAF > 10%) without a prior diagnosis of coronary artery disease were 12-times more likely to have a CAC score over 615 Agatston units [5]. In a large genome-wide association study, the presence of CHIP-associated Jak2 mutation was associated with increased risk of coronary artery disease despite lower levels of triglycerides and low-density lipoprotein (LDL) cholesterol [66].
Table 3. CHIP mutations and associated cardiovascular risk.
| Cohort | Mutation | Age | Cardiovascular risk | HR | Ref |
|---|---|---|---|---|---|
| US population based | Any CHIP mutation | Median 58 years | Incident coronary artery disease | 2.0 (1.2–3.5) | [3] |
| Ischaemic stroke | 2.6 (1.3–4.8) | ||||
| PROMIS | Any CHIP mutation | <50 years | Early onset myocardial infarction (before the age of 50) | 4.0 (2.4–6.7) | [5] |
| ATVB | 5.4 (2.3–13.0) | ||||
| U.K. Biobank | Any CHIP mutation | Mean 61 years | Myocardial infarction, coronary artery revascularisation, stroke or death | 1.27 (1.04–1.56) | [6] |
| Chronic ischaemic HFrEF | TET2 or DNMT3A | Median 69 years | Heart failure hospitalisation or all-cause death | 2.1 (1.1–4.0) | [7] |
| Severe aortic stenosis undergoing transcatheter aortic valve replacement | TET2 or DNMT3A | Median 83 years | Risk of death following transcatheter aortic valve replacement | 3.1 (1.17–8.08) | [8] |
Abbreviations: ATVB, Atherosclerosis, Thrombosis, and Vascular Biology Italian Study Group; HFrEF, heart failure with reduced ejection fraction; HR, hazard ratio; PROMIS, Pakistan Risk of Myocardial Infarction Study.
Endothelial dysfunction is the earliest feature in the development of atherosclerosis. Patients with coronary endothelial dysfunction (assessed via vasomotor responses to intra-coronary acetylcholine infusion) have significantly higher prevalence of CHIP-associated mutations in comparison with people with normal coronary endothelial function (9.2 versus 1.5%, respectively) [67]. Furthermore, somatic mutations in ASXL1, DNMT3A and TET2 are associated with higher levels of IL-6 and IL-8 in this group [67].
The potential association between CHIP, inflammation and CVD was assessed in 35416 people included in the U.K. Biobank (Table 3) [6]. Participants did not have a history of CVD at inclusion but those with DNMT3A or TET2 mutation had a 27% higher risk of CVD over 6.9 years of follow-up when compared with those without these CHIP mutations [6]. This risk was larger in those with larger clones denoted by VAF >10% (hazard ratio 1.59 [95% CI: 1.21–2.09]) [6]. Furthermore, to examine the potential interaction with inflammation, the effect of carrying a genetic proxy of IL-6 inhibition (IL6R p.Asp358Ala) and simultaneous CHIP was also assessed [6]. In people with large CHIP clones (VAF > 10%), the presence of this genetic proxy was associated with a 54% lower risk of CVD events and was without effect upon CVD event risk in individuals without CHIP [6]. In those aged over 50 years with a history of prior myocardial infarction and CHIP, each additional IL6R p.Asp358Ala allele attenuated the risk of CVD events [6]. Not only do these genetic data provide further mechanistic insight concerning interactions between CHIP, inflammation and CVD, they also give weight to the hypothesis that therapeutic inhibition of IL-6 signalling may prove to be beneficial in patients with large CHIP clones and CVD.
TET2 – preclinical vascular models
The first murine model to implicate the role of CHIP in atherosclerosis aimed to mimic human clonal haematopoiesis by initially introducing a small number of mutant TET2 cells. This model used a competitive bone marrow transplantation strategy to generate atherosclerotic prone, LDL receptor deficient (Ldlr−/−) chimeric mice with a small proportion of TET2-deficient HSC (10% TET2−/− bone marrow) [68]. Importantly, when compared with control mice, there was no difference in body weight, plasma cholesterol levels, glucose and systemic insulin sensitivity. Following nine weeks of a high fat/high cholesterol diet, TET-2 deficient mice (10% knockout [KO]-BMT) developed aortic root plaques that were 60% larger than those of control animals. This increased atherogenesis in 10% KO-BMT mice was paralleled by an increase in total macrophage content in the intima, and these TET2-deficient macrophages exhibited markedly increased expression of pro-inflammatory cytokines. In particular, the transcription of aortic arch IL-1β in macrophages was doubled and treatment with the nucleotide-binding domain leucine-rich repeat containing receptor 3 (NLRP3) inflammasome inhibitor, MCC950, reduced the atherosclerotic plaque burden. Furthermore, IL-1β secretion was completely abrogated in macrophages following treatment with MCC950, suggesting that TET2 deficiency affects NLRP3-mediated IL-1β secretion. Subsequently, these findings have been replicated in other murine models of TET2 deficiency, confirming the association of TET2 deficiency in accelerated atherosclerosis through induction of a proinflammatory state [5]. There has been a suggestion from a small cohort of TET2 deficient atherosclerotic-prone mice (n=30) that the response to IL-1β inhibition may be sex-dependent although this needs further exploration [69].
VSMC-derived cells in mouse atherosclerotic plaques are generated by clonal expansion of cells within the vessel wall [70–72]. TET2 is highly expressed in human coronary artery SMCs and, in response to arterial injury, TET2 loss-of-function exacerbates intimal hyperplasia after injury [73]. It has previously been demonstrated that rapamycin induces contractile protein expression in human VSMCs [74]. Rapamycin-induced VSMC differentiation is prevented by TET2 coronary arterial SMC knockdown, whereas TET2 overexpression induces a contractile phenotype suggesting that TET2 acts a regulator of VSMC phenotypic transformation [73].
The endothelium exerts substantial vasoprotective effects. Abnormalities of autophagy homoeostasis, the natural process regulating the removal of unnecessary or damaged cellular components, has been implicated in endothelial cell dysfunction and the development of atherosclerosis, microvascular dysfunction and heart failure. TET2 is an important regulator of autophagy and, following low shear stress, endothelial cell autophagy is reduced via the down-regulation of TET2 [63]. Furthermore, in the ApoE−/− murine model, autophagy is up-regulated by TET2 overexpression and decreased by TET2 silencing [63].
JakV617F – preclinical vascular models
The JakV617F mutation has also been examined in a mouse model of atherosclerosis. Irradiated Ldlr−/− mice were transplanted with bone marrow from either wild type or Jak2VF617 mutant mice and subsequently fed a high fat/high cholesterol diet. Despite lower plasma cholesterol levels, the aortic root atherosclerotic lesion size was 1.6-fold higher in Jak2VF617F mice in comparison with WT [75]. Furthermore, Jak2VF617F macrophages had greater expression of pro-inflammatory cytokines and chemokines including, IL-1β, IL-6, IL-18, TNF-α and MCP-1 following challenge with LPS [75]. Even in the absence of LPS stimulation, Jak2V617F mice had higher plasma levels of IL-18 compared with WT controls [75]. However, these Jak2V617F mice developed marked erythrocytosis, thrombocytosis and neutrophilia which is more consistent with a myeloproliferative neoplastic phenotype than CHIP and these confounding effects limit further interpretation. A subsequent experiment examined endothelial function in the common carotid artery of LDLr−/− mice transplanted with Jak2V617F bone marrow cells following constrictive cuff placement across the artery [76]. The carotid arteries of these Jak2V617F mice displayed increased endothelial permeability, reduced endothelial continuity, increased intimal neutrophil extracellular trap accumulation with a subsequent increase in thrombus formation [76]. Treatment with ruxolitinib, a Jak1/2 inhibitor, reduced endothelial cell apoptosis and improved endothelial continuity in Jak2V617F mice [76].
Heart failure
While heart failure with reduced ejection fraction (HFrEF) is a consequence of impaired left ventricular systolic function (left ventricular ejection fraction [LVEF] <40%), patients with heart failure and preserved ejection fraction (HFpEF; LVEF >50%) reflect a less well understood group in whom ageing and inflammation may play a much larger relative role [77]. Unlike HFrEF, no evidence-based therapies currently exist for the treatment of patients with HFpEF which is more commonly associated with multi-morbidity, myocardial stiffening and macro- and micro-vascular endothelial dysfunction [78–81].
Human HFrEF and CHIP
The prevalence of CHIP in patients with HF has been assessed in 200 patients with chronic ischaemic HFrEF enrolled in clinical trials of autologous stem cell therapy. In this relatively young cohort (median age 65 years) with a mean LVEF of 31%, CHIP was present in 18.5%. [7]. DNMT3A mutations were observed in 30% of patients and 18% of patients had mutations in TET2. These CHIP mutations were independently associated with heart failure hospitalisation and death (HR 2.1; 95% CI 1.1–4.0) (Table 3) [7]. Notably, the majority of this mortality was attributable to progressive heart failure with only one death occurring as a result of subsequent MI. There was a significant association between clinical outcome and %VAF, further implying a ‘dose effect’ of CHIP [7]. VAF cut-off values of ≥0.73% and ≥1.15% for TET2 and DNMT3A mutations, respectively, were predictive of poorer prognosis [82]. Circulating inflammatory cytokines were not measured in this group but, in a separate very small cohort of six patients with heart failure, the presence of DNMT3A mutation was associated with higher transcription of IL-1β and IL-6 when compared with patients with HF and no DNMT3A mutation [83].
Human HFpEF and CHIP
HFpEF now accounts for more than 50% of patients with HF, the incidence of which rises substantially with age [81,84]. The pathophysiology of HFpEF remains incompletely understood although structural and functional abnormalities are becoming better defined. Cardiac biopsies obtained from patients with HFpEF reveal structural alterations including cardiomyocyte hypertrophy [85,86] and interstitial fibrosis [85,87–89], while functional changes include impaired myocardial relaxation [90] and increased myocardial stiffness [85,87,88]. Cardiac biopsies also reveal higher levels of myocardial inflammatory cells in patients with HFpEF [91]. Post mortem findings from patients with HFpEF reveal more extensive coronary artery disease, a greater burden of myocardial fibrosis and reduced microvascular density compared with controls without heart failure [92]. Large vessel stiffening is also a feature of vascular ageing and inflammation may, at least in part, contribute to the pathophysiology of HFpEF [93].
Inflammation appears to be more important in the pathophysiology of HFpEF than HFrEF [80]. Circulating concentrations of inflammatory biomarkers including IL-1, CRP and growth differentiation factor 15 are high in HFpEF [94–97] and more so in HFpEF than in HFrEF [98–100]. Network analysis of circulating biomarkers obtained from patients with HFrEF and HFpEF in the BIOlogy Study to TAilored Treatment in Chronic Heart Failure (BIOSTAT-CHF) cohort revealed important differences between the two heart failure phenotypes. In patients with HFrEF, pathways related to cellular growth and metabolism were specifically up-regulated [80] while inflammatory pathways were specifically up-regulated in those with HFpEF [80]. In addition to the inflammatory hypothesis for the aetiology of HFpEF, micro- and macro-vascular disease involving the cardiac, pulmonary and peripheral circulation are highly prevalent in patients with HFpEF [101].
Non-cardiac comorbidities are common in HFpEF, particularly obesity, diabetes, chronic kidney disease and hypertension [79] and the systemic inflammatory state induced by these conditions has recently been shown to be predictive of incident HFpEF but not HFrEF [102]. A novel paradigm to explain the underlying pathogenesis of HFpEF proposes that the systemic inflammatory state induced by these comorbidities induces coronary microvascular endothelial dysfunction. The production of inflammation-induced reactive oxygen species limits the bioavailability of nitric oxide with consequent impairment of cardiomyocyte protein kinase G activity, microvascular ischaemia, fibrosis and left ventricular concentric remodelling [78,98].
Given the associations between HFpEF, vascular dysfunction, inflammation and ageing, we propose that CHIP may be a particularly potent risk factor for the development, progression and potentiation of HFpEF. This hypothesis is yet to be tested directly in humans.
Preclinical models – heart failure and CHIP
HSC-specific TET2 mutation is associated with the accelerated development of heart failure in murine models of heart failure as a result of left ventricular pressure overload induced by transverse aortic constriction (TAC) and as a consequence of chronic ischaemia induced by ligation of the left anterior descending (LAD) coronary artery [103]. While TAC has been employed as a murine model of HFpEF, after 2–3 weeks TAC results in a reduction in systolic function and progression to HFrEF [104–106]. Following permanent ligation of the LAD, 10% TET2 KO mice had significantly reduced ejection fraction (EF) and this was associated with increased transcription of pro-inflammatory mediators including IL-1β, IL-18, Chemokine (C–X–C motif) ligand 2 (Cxcl2), Chemokine (C–C motif) ligand 2 (Ccl2) and 5 (Ccl5) [103]. Myeloid-specific TET2-deficient mice also had worse cardiac remodelling following LAD ligation with lower LVEF and increased fibrotic area when compared with control mice. Ten percent TET2 KO mice subjected to TAC exhibit marked left ventricular hypertrophy with greater posterior wall thickness and cardiac fibrosis when compared with WT mice. These structural changes were also associated with higher concentrations of circulating IL-1β when compared with control mice [103]. IL-1β cleavage is mediated by the NLRP3 inflammasome, a complex intracellular protein which upon activation, cleaves procaspase-1 protein to functional caspase-1. The primary function of caspase-1 is the conversion of the inactive pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into their active, potently pro-inflammatory states. Over time, TET2 KO mice subjected to TAC also developed systolic impairment. Importantly, administration of MCC950, an NLRP3 inflammasome inhibitor, was associated with significant protection from adverse cardiac remodelling in both models [103].
Bone marrow-specific deletion of TET2 or DNMT3A is associated with cardiac hypertrophy, fibrosis and impaired LV fractional shortening after infusion of angiotensin II in comparison with WT controls [107]. TET2 deletion promoted the expression of IL-1β and IL-6, whereas DNMT3 deletion significantly increased the expression of IL-6 with a trend towards increased IL-1β [107]. Importantly, DNMT3A has been demonstrated to have both direct and indirect roles in maintaining overall cardiomyocyte homeostasis and function [108]. Specifically, DNMT3A−/− engineered human induced pluripotent stem cell-derived cardiomyocytes have up-regulation of pathways involved in cardiac hypertrophy and cardiac proliferation pathways when compared with WT [108]. DNMT3A knock-out also affected contraction kinetics, cell diameter was greater and intracellular lipid accumulation was greater in comparison with the WT [108].
Myeloid-specific Jak2V617F mutation in mice is not associated with abnormalities of peripheral blood count, as would be expected in human CHIP. These animals also do not appear to have abnormalities of cardiac structure or function in the unstressed state [109]. However, following LAD ligation or TAC these mice have greater myocardial macrophage infiltration and concentrations of IL-6 and IL-1β are greater than WT. It has been proposed that Jak2V617F activates the IFN-γ receptor 1 Jak2 signalling transduction pathway (IFNGR1-Jak2-STAT1) resulting in the release of pro-inflammatory cytokines [109]. In the myeloid-specific Jak2V617F model, this mutation was associated with a more substantial deterioration in cardiac function, larger infarct size and increased cardiac fibrosis following TAC/LAD ligation [109]. Furthermore, the adoptive transfer of Jak2V617F bone marrow cells into mice exposed to chronic hypoxia was associated with increased right ventricular systolic pressure and increased muscularisation of pulmonary vessels when compared with control chronically hypoxic mice [110].
While these models have focused upon the investigation of the effects of an exogenous injury or stressor, a recent investigation has attempted to replicate the effects of CHIP in the otherwise ‘unstressed’ state. By transferring TET2-mutant bone marrow cells into mice without prior myeloablative irradiation preconditioning, an attempt was made to replicate the accumulation of somatically abnormal cells over time [111]. In this model, TET2-deficient cardiac macrophages had an overrepresentation of immune response effectors, with specific increases in IL-1β, Ccl17 and IL1-receptor antagonist gene [111]. Concentrations of brain natriuretic peptide (released in response to cardiac pressure overload) were significantly higher in TET2 mutant mice and these animals had greater posterior wall dimension, left ventricular end systolic volume, heart weight and cardiac fibrosis in comparison to control. While LVEF declined slightly, all mice had an LVEF of ≥40% providing evidence that CHIP may be important in the development of HFpEF [111].
Interplay between CHIP, ageing, inflammation and HFpEF
As outlined, both CHIP and HFpEF are considered to be diseases of the ageing population and both are associated with a systemic pro-inflammatory state (Figure 2). The incidence and prevalence of HFpEF increases sharply with age [112–116], and the mean age of patients with HFpEF in recent cohorts is 72 years [113–115,117–138]. In the context of findings describing the prevalence of CHIP in all-comers, it is reasonable to expect that CHIP is found in at least 10–20% of patients with HFpEF. However, this may be a substantial underestimate. CHIP was found in 27% of patients with chronic ischaemic HFrEF aged between 70 and 79 years [7] and in an elderly population (median age 83 years) with severe aortic stenosis undergoing transcatheter aortic valve implantation (TAVI), the prevalence of CHIP was 33% [8]. In this cohort of patients with severe aortic stenosis, the presence of TET2 or DNMT3A was also associated with an elevated pro-inflammatory subset of circulating leucocytes and conferred a profound increased in mortality even after successful correction of the aortic valve stenosis (HR 3.1 [95% CI: 1.17–8.08]) [8].
Figure 2. Potential mechanistic links between CHIP and HFpEF.
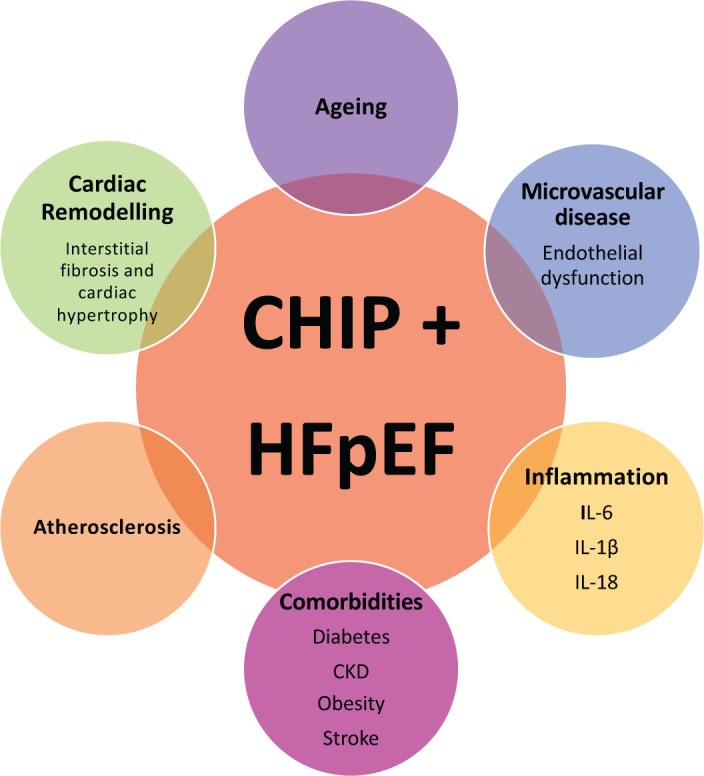
Abbreviations: CKD, chronic kidney disease; HFpEF, heart failure with preserved ejection fraction; IL-6, interleukin-6; IL-18, interleukin 18; IL-1β, interleukin-1β.
In tandem with ageing, the prevalence of comorbidity increases in patients with chronic heart failure [139]. Indeed, nearly half of patients with HFpEF have five or more comorbidities [140]. Many of these comorbidities are associated with a pro-inflammatory state and, furthermore, circulating markers of inflammation are predictive of incident HFpEF [102]. Diabetes occurs in approximately 40% of male patients with HFpEF and 30% of female patients with HFpEF [140]. Diabetes is associated with a two-fold increased risk of developing CHIP and individuals with both diabetes and CHIP have a higher burden of cardiovascular comorbidities than those with diabetes alone [6,141]. It is unclear to what extent these pro-inflammatory comorbidities, considered to be central to the concept of inflammageing, are the cause or effect of CHIP, but it is likely that a positive feedback loop is established between them [35].
Younger patients with HFpEF are more likely to be male and have a history of obesity and diabetes [142–144], both of which are strongly associated with chronic low-grade inflammation [142–144]. The presence of CHIP may be of even greater relevance in these younger patients as an indicator of increased epigenetic age. Indeed, the presence of any CHIP mutation confers a 4-year increase in epigenetic age, while CHIP-related TET2 mutation confers a 6-year increase [145]. Deviations from chronological age towards an increased epigenetic age are associated with increased risk of earlier mortality and age related morbidities [146,147].
In 5214 postmenopausal women included in the Womens Health Initiative dataset, the presence of any of the top three CHIP-associated mutations (TET2, DNMT3A and ASXL1) was associated with incident HFpEF but not HFrEF [148]. Women with premature menopause have increased risk of heart failure, stroke, coronary and peripheral arterial disease [149]. Furthermore, systemic markers of inflammation, including CRP, are higher in post-menopausal women than they are in those who are pre-menopausal [150,151]. It is of note that, in women included in the U.K. Biobank and Womens Health Initiative, the prevalence of CHIP was 60% higher in women with premature menopause compared with those without and the presence of CHIP was independently associated with incident coronary artery disease [152]. Whether or not the presence of CHIP and early-onset menopause increases the risk of developing HFpEF is unknown.
CHIP and personalised cardiovascular management
Historical trials of anti-inflammatory therapy for the treatment of CVD have mainly been disappointing. However, Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) has reinvigorated this area and highlights CHIP as a potential biomarker to inform personalised therapy. CANTOS examined the effects of canakinumab, a monoclonal antibody directed against IL-1β, in patients with a history of prior myocardial infarction and elevated CRP. Canakinumab reduced CRP and the incidence of atherosclerotic cardiovascular events was decreased by 15% versus control [153]. Notably, canakinumab also reduced heart failure hospitalisation and heart failure-related mortality by 23% in patients who achieved a CRP level of <2 mg/l [154]. Given the association of CHIP with inflammation and, in particular, the secretion of IL-1β (the immediate upstream precursor to IL-6), CHIP has been proposed as a potential biomarker for personalised therapy with canakinumab and potentially other anti-inflammatory therapies. Indeed, in an exploratory analysis of CANTOS, canakinumab reduced the relative risk of major adverse cardiovascular events by 64% in those with TET2 mutations and by 15% in the treatment overall [155]. Whether or not this impressive effect will also be seen in patients with HF is unknown.
Inzomelid, a novel small-molecule inhibitor of the NLRP3 inflammasome, is currently under clinical investigation for its safety and tolerability in humans (NCT04015076). Whether any potential effect is amplified in patients with CHIP may be a logical future step in its assessment. Recent intriguing data reveal that the sodium-glucose co-transporter 2 (SGLT-2) inhibitor, dapagliflozin, reduces IL-1β via up-regulation of serum β-hydroxybutyrate [156]. Again, the potential benefits of personalisation of SGLT2 inhibitor therapy on the basis of CHIP status is an intriguing but untested hypothesis.
Conclusion
While early attention has been paid to the potential role of CHIP in progression to haematological malignancy, it has rapidly become clear that its association with CVD is much stronger. The mechanistic basis to its role in the pathogenesis of atherosclerosis and vascular dysfunction is becoming clearer and further highlights the central role of inflammation in these processes. Preliminary clinical data have highlighted the prevalence of CHIP and its association with poorer outcome in patients with chronic ischaemic HFrEF, while animal models have provided further insight. Given the important intersections among ageing, inflammation and vascular disease in the pathogenesis of HFpEF we believe that CHIP reflects a ripe target for further assessment in this growing group of patients who currently lack evidence-based therapy. Whether CHIP status will allow personalisation of therapy for these patients and others remains an open avenue for future work, with the optimistic aim of harnessing the potential of anti-inflammatory treatments for heart failure (Figure 3).
Figure 3. CHIP and future research.

Abbreviations
- ASXL1
additional sex comb-like 1
- CAC
coronary artery calcification
- CANTOS
Canakinumab Anti-Inflammatory Thrombosis Outcome Study
- CHIP
clonal haematopoiesis of indeterminant potential
- CRP
C-reactive protein
- CVD
cardiovascular disease
- DNMT3
DNA methyltransferase 3
- EF
ejection fraction
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- HSC
haematopoietic stem cell
- IFN-γ
interferon-γ
- IL
interleukin
- KO
knockout
- LAD
left anterior descending artery
- LDL
low-density lipoprotein
- LVEF
left ventricular ejection fraction
- MCP-1
monocyte chemoattractant protein 1
- NLRP3
nucleotide-binding domain leucine-rich repeat containing receptor 3
- TAC
transverse aortic constriction
- TET2
ten-eleven-translocation-2
- TNF-α
tumour necrosis factor-α
- VAF
variant allele frequency
- VSMC
vascular smooth muscle cell
- 5hmC
5-hydroxymethylcystosine
Competing Interests
C.G. has received research funding from AstraZeneca, Bristol-Myers Squibb, ISTESSO, Eli-Lilly, MedAnnex, Pfizer and UCB. C.G. is/has been an advisory board member for Bristol-Myers Squibb, MedAnnex, Medincell and Pfizer and has received honoraria from Abbvie and Bristol-Myers Squibb. M.C.P. has received research funding from Novartis, Bristol-Myers Squibb, Cyclacel and Takeda/Incyte, is/has been an advisory board member for Bristol-Myers Squibb, Novartis, Incyte, Daiichi Sankyo, Jazz and Pfizer and has received honoraria from Astellas, Bristol-Myers Squibb, Novartis, Incyte, Pfizer and Gilead. M.C.P. has received research grants or consultancy fees from SQ Innovations, AstraZeneca, Roche, Boehringer Ingelheim, Eli Lilly, Napp Pharmaceuticals, Novartis, and Novo Nordisk and has served on clinical events committees for AbbVie, Alnylam, Astra Zeneca, Bayer, Boehringer Ingelheim, GlaxoSmithKline, Resverlogix, and Novo Nordisk. N.N.L. has received research funding from Roche Diagnostics, Bristol-Myers Squibb, is/has been an advisory board member for Vifor Pharma, Pharmacosmos and has received honoraria from Roche Diagnostics, Takeda, Pfizer and Novartis.
Funding
This work was supported by the British Heart Foundation Centre of Research Excellence Award [grant number RE/18/6/34217 (to L.M., M.C.P. and N.N.L.)]; T.C. is funded by the Chancellor’s Fellowship held at the University of Edinburgh; K.K. is funded by the John Goldman Fellowship sponsored by Leukaemia U.K. [grant number 2019/JCF/003 (to K.K.)].
References
- 1.Department of Health (2017) Health Survey for Engand: Cardiovascualar Diseases. pp. 1–25, http://healthsurvey.hscic.gov.uk/media/78646/HSE17-CVD-rep.pdf [Google Scholar]
- 2.Cancer Research U.K. (2015-2017) Cancer incidence by age. Data is for UK, pp. 1–52, https://www.cancerresearchuk.org/health-professional/cancer-statistics/incidence/age#heading-Zero [Google Scholar]
- 3.Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G.et al. (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 10.1056/NEJMoa1408617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKerrell T., Park N., Moreno T., Grove C.S., Ponstingl H., Stephens J.et al. (2015) Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 10, 1239–1245 10.1016/j.celrep.2015.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaiswal S., Natarajan P., Silver A.J., Gibson C.J., Bick A.G., Shvartz E.et al. (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 10.1056/NEJMoa1701719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bick A.G., Pirruccello J.P., Griffin G.K., Gupta N., Gabriel S., Saleheen D.et al. (2020) Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 141, 124–131 10.1161/CIRCULATIONAHA.119.044362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorsheimer L., Assmus B., Rasper T., Ortmann C.A., Ecke A., Abou-El-Ardat K.et al. (2019) Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 4, 25–33 10.1001/jamacardio.2018.3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mas-Peiro S., Hoffmann J., Fichtlscherer S., Dorsheimer L., Rieger M.A., Dimmeler S.et al. (2019) Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 41, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genovese G., Kähler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F.et al. (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 10.1056/NEJMoa1409405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeZern A.E., Malcovati L. and Ebert B.L. (2019) CHIP, CCUS, and other acronyms: definition, implications, and impact on practice. Am. Soc. Clin. Oncol. Educ. B. 39, 400–410 10.1200/EDBK_239083 [DOI] [PubMed] [Google Scholar]
- 11.Libby P., Sidlow R., Lin A.E., Gupta D., Jones L.W., Moslehi J.et al. (2019) Clonal hematopoiesis: crossroads of aging, cardiovascular disease, and cancer: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 74, 567–577 10.1016/j.jacc.2019.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busque L., Buscarlet M., Mollica L. and Levine R. (2018) Age-related clonal hematopoiesis: stem cells tempting the devil. Stem Cells 36, 1287–1294 10.1002/stem.2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zink F., Stacey S.N., Norddahl G.L., Frigge M.L., Magnusson O.T., Jonsdottir I.et al. (2017) Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 10.1182/blood-2017-02-769869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gondek L. and DeZern A. (2020) Assessing clonal haematopoiesis: clinical burdens and benefits of diagnosing myelodysplastic syndrome precursor states. Lancet Haematol. 7, 73–81, https://pubmed.ncbi.nlm.nih.gov/31810765/ 10.1016/S2352-3026(19)30211-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young A.L., Challen G.A., Birmann B.M. and Druley T.E. (2016) Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 7, 1–7 10.1038/ncomms12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razavi P., Li B.T., Brown D.N., Jung B., Hubbell E., Shen R.et al. (2019) High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 25, 1928–1937 10.1038/s41591-019-0652-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen S. and Liu Y. (2019) P53 involvement in clonal hematopoiesis of indeterminate potential. Curr. Opin. Hematol. 26, 235–240 10.1097/MOH.0000000000000509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.López-otín C., Blasco M.A., Partridge L., Serrano M. and Kroemer G. (2013) The hallmarks of aging longevity. Cell 153, 1194–1217 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiskopf K., Schnorr P., Pang W., Chao M., Chhabra A., Seita J.et al. (2016) Myeloid cell origins, differentiation, and clinical implications. Microbiol. Spectr. 4, 1–23 10.1128/microbiolspec.MCHD-0031-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossi D.J., Bryder D., Seita J., Nussenzweig A., Hoeijmakers J. and Weissman I.L. (2007) Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725–729 10.1038/nature05862 [DOI] [PubMed] [Google Scholar]
- 21.Ho T., Warr M., Adelman E., Lansinger O., Flach J., Verovkaya E.et al. (2017) Autophagy maintains the metabolism and function of young and old (hematopoietic) stem cells. Nature 543, 205–210 10.1038/nature21388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovtonyuk L.V., Fritsch K., Feng X., Manz M.G. and Takizawa H. (2016) Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol. 7, 1–13 10.3389/fimmu.2016.00502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abegunde S.O., Buckstein R., Wells R.A. and Rauh M.J. (2018) An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 59, 60–65 10.1016/j.exphem.2017.11.002 [DOI] [PubMed] [Google Scholar]
- 24.Cai Z., Kotzin J.J., Ramdas B., Chen S., Nelanuthala S., Palam L.R.et al. (2018) Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell 23, 833.e5–849.e5 10.1016/j.stem.2018.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beerman I. (2017) Accumulation of DNA damage in the aged hematopoietic stem cell compartment. Semin. Hematol. 54, 12–18 10.1053/j.seminhematol.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coombs C.C., Zehir A., Devlin S.M., Kishtagari A., Jonsson P., Hyman D.M.et al. (2017) Therapy-related clonal hematopoiesis in patients with non- hematologic cancers is common and impacts clinical outcome. Cell Stem Cell 21, 374–382 10.1016/j.stem.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren J.T. and Link D.C. (2020) Clonal hematopoiesis and risk for hematologic malignancy. Blood 136, 1599–1605 10.1182/blood.2019000991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhattacharya R., Zekavat S.M., Pirruccello J., Griffin G.K., Bick A.G. and Natarajan P. (2020) Abstract 16686 : Improved diet quality is associated with lower prevalence of clonal hematopoiesis of indeterminate potential. Circulation 142 [Google Scholar]
- 29.Hansen J.W., Pedersen D.A., Larsen L.A., Husby S., Clemmensen S.B., Hjelmborg J.et al. (2020) Clonal hematopoiesis in elderly twins: concordance, discordance, and mortality. Blood 135, 261–268 10.1182/blood.2019001793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fabre M.A., McKerrell T., Zwiebel M., Vijayabaskar M.S., Park N., Wells P.M.et al. (2020) Concordance for clonal hematopoiesis is limited in elderly twins. Blood 135, 269–273 10.1182/blood.2019001807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steensma D.P. (2018) Clinical consequences of clonal hematopoiesis of indeterminate potential. Hematology 2018, 264–269 10.1182/asheducation-2018.1.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson C.J., Lindsley R.C., Tchekmedyian V., Mar B.G., Shi J., Jaiswal S.et al. (2017) Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J. Clin. Oncol. 35, 1598–1605 10.1200/JCO.2016.71.6712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook E.K., Izukawa T., Young S., Rosen G., Jamali M., Zhang L.et al. (2019) Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 3, 2482–2486 10.1182/bloodadvances.2018024729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bick A.G., Weinstock J.S., Nandakumar S.K., Fulco C.P., Bao E.L., Zekavat S.M.et al. (2020) Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768 10.1038/s41586-020-2819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandarakov O. and Belyavsky A. (2020) Clonal hematopoiesis, cardiovascular diseases and hematopoietic stem cells. Int. J. Mol. Sci. 21, 1–15 10.3390/ijms21217902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Busque L., Patel J.P., Figueroa M., Vasanthakumar A., Provost S., Hamilou Z.et al. (2012) Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 44, 1179–1181 10.1038/ng.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A.et al. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (80-) 333, 1300–1303 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran-Crusio K., Reavie L., Shih A., Abdel-wahab O., Ndiaye-lobry D., Lobry C.et al. (2011) Tet2 loss leads to increased hematopoietic stem cell self- renewal and myeloid transformation. Cancer Cell 20, 11–24 10.1016/j.ccr.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cull A.H., Snetsinger B., Buckstein R., Wells R.A. and Rauh M.J. (2017) Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 55, 56.e13–70.e13 10.1016/j.exphem.2017.08.001 [DOI] [PubMed] [Google Scholar]
- 40.Zhang Q., Zhao K., Shen Q., Han Y., Gu Y., Li X.et al. (2015) Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 10.1038/nature15252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okano M., Bell D.W., Haber D.A. and Li E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 10.1016/S0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- 42.Sandoval J.E., Huang Y.H., Muise A., Goodell M.A. and Reich N.O. (2019) Mutations in the DNMT3A DNA methyltransferase in acute myeloid leukemia patients cause both loss and gain of function and differential regulation by protein partners. J. Biol. Chem. 294, 4898–4910 10.1074/jbc.RA118.006795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leoni C., Montagner S., Rinaldi A., Bertoni F., Polletti S., Balestrieri C.et al. (2017) Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. U.S.A. 114, E1490–E1499 10.1073/pnas.1616420114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang F., Zhou S., Wang C., Huang Y., Li H., Wang Y.et al. (2017) Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci. Rep. 7, 1–11 10.1038/srep43592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Shea J.J., Kontzias A., Yamaoka K., Tanaka Y. and Laurence A. (2013) Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 72, 111–115 10.1136/annrheumdis-2012-202576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levine R.L., Wadleigh M., Cools J., Ebert B.L., Wernig G., Huntly B.J.P.et al. (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397 10.1016/j.ccr.2005.03.023 [DOI] [PubMed] [Google Scholar]
- 47.Nielsen C., Birgens H.S., Nordestgaard B.G. and Bojesen S.E. (2013) Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49 488 individuals from the general population. Br. J. Haematol. 160, 70–79 10.1111/bjh.12099 [DOI] [PubMed] [Google Scholar]
- 48.Nielsen C., Birgens H.S., Nordestgaard B.G., Kjær L. and Bojesen S.E. (2011) The JAK2 V617F somatic mutation, mortality and cancer risk in the general population. Haematologica 96, 450–453 10.3324/haematol.2010.033191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sidon P., El Housni H., Dessars B. and Heimann P. (2006) The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 20, 1622 10.1038/sj.leu.2404292 [DOI] [PubMed] [Google Scholar]
- 50.Thol F., Friesen I., Damm F., Yun H., Weissinger E.M., Krauter J.et al. (2011) Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J. Clin. Oncol. 29, 2499–2506 10.1200/JCO.2010.33.4938 [DOI] [PubMed] [Google Scholar]
- 51.Itzykson R., Kosmider O., Renneville A., Gelsi-Boyer V., Meggendorfer M., Morabito M.et al. (2013) Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 31, 2428–2436 10.1200/jco.2012.47.3314 [DOI] [PubMed] [Google Scholar]
- 52.Bejar R., Stevenson K., Abdel-Wahab O., Galili N., Nilsson B., Garcia-Manero G.et al. (2011) Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 364, 2496–2506 10.1056/NEJMoa1013343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin C.C., Hou H.A., Chou W.C., Kuo Y.Y., Liu C.Y., Chen C.Y.et al. (2014) IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am. J. Hematol. 89, 137–144 10.1002/ajh.23596 [DOI] [PubMed] [Google Scholar]
- 54.Paschka P., Schlenk R.F., Gaidzik V.I., Herzig J.K., Aulitzky T., Bullinger L.et al. (2015) ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian acute myeloid leukemia study group. Haematologica 100, 324–330 10.3324/haematol.2014.114157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin C.C., Hou H.A., Chou W.C., Kuo Y.Y., Wu S.J., Liu C.Y.et al. (2014) SF3B1 mutations in patients with myelodysplastic syndromes: the mutation is stable during disease evolution. Am. J. Hematol. 89, 109–115 10.1002/ajh.23734 [DOI] [PubMed] [Google Scholar]
- 56.Balasubramani A., Larjo A., Bassein J.A., Chang X., Hastie R.B., Togher S.M.et al. (2015) Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 6, 1–15 10.1038/ncomms8307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abdel-Wahab O., Adil M., LaFave L., Gao J., Hricik T., Shih A.et al. (2012) ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193 10.1016/j.ccr.2012.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang H., Kurtenbach S., Guo Y., Lohse I., Durante M.A., Li J.et al. (2018) Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 131, 328–341 10.1182/blood-2017-06-789669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rinkuniene E., Petrulioniene Ž., Laucevičius A., Ringailaite E. and Laučyte A. (2009) Prevalence of conventional risk factors in patients with coronary heart disease. Medicina 45, 140–146 10.3390/medicina45020018 [DOI] [PubMed] [Google Scholar]
- 60.Li G., Peng J., Liu Y., Li X., Yang Q., Li Y.et al. (2015) Oxidized low-density lipoprotein inhibits THP-1-derived macrophage autophagy via TET2 down-regulation. Lipids 50, 177–183 10.1007/s11745-014-3977-5 [DOI] [PubMed] [Google Scholar]
- 61.Liu R., Jin Y., Tang W., Qin L., Zhange X., Tellider G.et al. (2013) TET2 is a master regulator of smooth muscle cell plasticity. Circulation 128, 2047–2057, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf 10.1161/CIRCULATIONAHA.113.002887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peng J., Yang Q., Li A.F., Li R.Q., Wang Z., Liu L.S.et al. (2016) Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE-/- mice. Oncotarget 7, 76423–76436 10.18632/oncotarget.13121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Q., Li X., Li R., Peng J., Wang Z., Jiang Z.et al. (2016) Low shear stress inhibited endothelial cell autophagy through TET2 downregulation. Ann. Biomed. Eng. 44, 2218–2227 10.1007/s10439-015-1491-4 [DOI] [PubMed] [Google Scholar]
- 64.Fuster J.J., MacLauchlan S., Zuriaga M.A., Polackal M.N., Ostriker A.C., Chakraborty R.et al. (2017) Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (80-) 355, 842–847 10.1126/science.aag1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elias-Smale S.E., Proença R.V., Koller M.T., Kavousi M., Van Rooij F.J.A., Hunink M.G.et al. (2010) Coronary calcium score improves classification of coronary heart disease risk in the elderly: The Rotterdam study. J. Am. Coll. Cardiol. 56, 1407–1414 10.1016/j.jacc.2010.06.029 [DOI] [PubMed] [Google Scholar]
- 66.Liu D.J., Peloso G.M., Yu H., Butterworth A.S., Wang X., Mahajan A.et al. (2017) Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet. 49, 1758–1766 10.1038/ng.3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ganji M., Lasho T., Toya T., Ahmed A., Corban M., Lerman L.O.et al.(2020) Abstract 17182 : Coronary endothelial dysfunction in humans is associated with elevated expression of clonal hematopoiesis of indeterminate potential. Circulation 142 10.1161/circ.142.suppl_3.17182 [DOI] [Google Scholar]
- 68.Fuster J.J., MacLauchlan S., Zuriaga M.A., Polackal M.N., Ostriker A.C., Chakraborty R.et al. (2017) Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (80-) 335, 842–847 10.1126/science.aag1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vromman A., Vellarikkal S., Gomes M.J., Ruvkun V., Shvartz E., Kritikou E.et al. (2020)Interleukin-1beta inhibition attenuates accelerated atherosclerosis in mice with Tet2 loss of function in a sex-dependent fashion. Circulation 142, Abstract 15355 [Google Scholar]
- 70.Feil S., Fehrenbacher B., Lukowski R., Essmann F., Schulze-Osthoff K., Schaller M.et al. (2014) Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 115, 662–667 10.1161/CIRCRESAHA.115.304634 [DOI] [PubMed] [Google Scholar]
- 71.Chappell J., Harman J.L., Narasimhan V.M., Yu H., Foote K., Simons B.D.et al. (2016) Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ. Res. 119, 1313–1323 10.1161/CIRCRESAHA.116.309799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jacobsen K., Lund M.B., Shim J., Gunnersen S., Füchtbauer E.-M., Kjolby M.et al. (2017) Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2, 1–13 10.1172/jci.insight.95890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu R., Jin Y., Tang W.H., Qin L., Zhang X., Tellides G.et al. (2013) Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 128, 2047–2057 10.1161/CIRCULATIONAHA.113.002887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martin K.A., Merenick B.L., Ding M., Fetalvero K.M., Rzucidlo E.M., Kozul C.D.et al. (2007) Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J. Biol. Chem. 282, 36112–36120 10.1074/jbc.M703914200 [DOI] [PubMed] [Google Scholar]
- 75.Wang W., Lui Q., Fidler T., Wang Y., Tang Y., Woods B.et al. (2018) Macrophage inflammation, erythrophagocytosis and accelerated atherosclerosis in Jak2V617F mice. Circ. Res. 123, 34–47 10.1161/CIRCRESAHA.118.313283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Molinaro R., Sellar R.S., Sausen G., Vromman A., Folco E., Sukhova G.K.et al. (2020)The clonal hematopoiesis mutation Jak2 V617F aggravates NETosis and endothelial injury in mouse arteries with erosion-like intimas. Circulation 142, Abstract 15575 [Google Scholar]
- 77.Ponikowski P., Voors A.A., Anker S.D., Bueno H., Cleland J.G.F., Coats A.J.S.et al. (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 37, 2129–2200 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 78.Paulus W.J. and Tschöpe C. (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271 10.1016/j.jacc.2013.02.092 [DOI] [PubMed] [Google Scholar]
- 79.Streng K.W., Nauta J.F., Hillege H.L., Anker S.D., Cleland J.G., Dickstein K.et al. (2018) Non-cardiac comorbidities in heart failure with reduced, mid-range and preserved ejection fraction. Int. J. Cardiol. 271, 132–139 10.1016/j.ijcard.2018.04.001 [DOI] [PubMed] [Google Scholar]
- 80.Tromp J., Westenbrink B.D., Ouwerkerk W., van Veldhuisen D.J., Samani N.J., Ponikowski P.et al. (2018) Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J. Am. Coll. Cardiol. 72, 1081–1090 10.1016/j.jacc.2018.06.050 [DOI] [PubMed] [Google Scholar]
- 81.Pfeffer M.A., Shah A.M. and Borlaug B.A. (2019) Heart failure with preserved ejection fraction in perspective. Circ. Res. 124, 1598–1617 10.1161/CIRCRESAHA.119.313572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Assmus B., Cremer S., Kirschbaum K., Culmann D., Kiefer K., Dorsheimer L.et al. (2021) Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A-and TET2-driver genemutations. Eur. Heart J. 42, 257–265 [DOI] [PubMed] [Google Scholar]
- 83.Abplanalp W., Cremer C., Cremer S., Hoffmann J., Rieger M., Vasa-Nicotera M.et al. (2020) DNMT3A clonal hematopoiesis-driver mutations are associated with profound changes in monocyte and T cell signatures in humans with heart failure. Eur. Heart J.42:257-265 10.1093/ehjci/ehaa946.362632293672 [DOI] [Google Scholar]
- 84.Conrad N., Judge A., Tran J., Mohseni H., Hedgecott D., Crespillo A.P.et al. (2018) Temporal trends and patterns in heart failure incidence: a population-based study of 4 million individuals. Lancet 391, 572–580 10.1016/S0140-6736(17)32520-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Van Heerebeek L., Borbély A., Niessen H.W.M., Bronzwaer J.G.F., Van Der Velden J., Stienen G.J.M.et al. (2006) Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 113, 1966–1973 10.1161/CIRCULATIONAHA.105.587519 [DOI] [PubMed] [Google Scholar]
- 86.Hahn V.S., Ying W., Lee Y.Z., Yanek L.R., Vaidya D., Subramanya V.et al. (2019) Tissue is the issue: endomyocardial biopsy characterization of heart failure with preserved ejection fraction and incident cardiac amyloidosis. J. Card. Fail. 25, S53 10.1016/j.cardfail.2019.07.150 [DOI] [Google Scholar]
- 87.Borbély A., Van Der Velden J., Papp Z., Bronzwaer J.G.F., Edes I., Stienen G.J.M.et al. (2005) Cardiomyocyte stiffness in diastolic heart failure. Circulation 111, 774–781 10.1161/01.CIR.0000155257.33485.6D [DOI] [PubMed] [Google Scholar]
- 88.Van Heerebeek L., Hamdani N., Falcão-Pires I., Leite-Moreira A.F., Begieneman M.P.V., Bronzwaer J.G.F.et al. (2012) Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126, 830–839 10.1161/CIRCULATIONAHA.111.076075 [DOI] [PubMed] [Google Scholar]
- 89.Kasner M., Westermann D., Lopez B., Gaub R., Escher F., Kühl U.et al. (2011) Diastolic tissue doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J. Am. Coll. Cardiol. 57, 977–985 10.1016/j.jacc.2010.10.024 [DOI] [PubMed] [Google Scholar]
- 90.Chaturvedi R.R., Herron T., Simmons R., Shore D., Kumar P., Sethia B.et al. (2010) Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation 121, 979–988 10.1161/CIRCULATIONAHA.109.850677 [DOI] [PubMed] [Google Scholar]
- 91.Westermann D., Lindner D., Kasner M., Zietsch C., Savvatis K., Escher F.et al. (2011) Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 4, 44–52 10.1161/CIRCHEARTFAILURE.109.931451 [DOI] [PubMed] [Google Scholar]
- 92.Mohammed S.F., Hussain S., Mirzoyev S.A., Edwards W.D., Maleszewski J.J. and Redfield M.M. (2015) Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 131, 550–559 10.1161/CIRCULATIONAHA.114.009625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Desai A.S., Mitchell G.F., Fang J.C. and Creager M.A. (2009) Central aortic stiffness is increased in patients with heart failure and preserved ejection fraction. J. Card. Fail. 15, 658–664 10.1016/j.cardfail.2009.03.006 [DOI] [PubMed] [Google Scholar]
- 94.Cheng J.M., Akkerhuis K.M., Battes L.C., Van Vark L.C., Hillege H.L., Paulus W.J.et al. (2013) Biomarkers of heart failure with normal ejection fraction: a systematic review. Eur. J. Heart Fail. 15, 1350–1362 10.1093/eurjhf/hft106 [DOI] [PubMed] [Google Scholar]
- 95.D'Elia E., Vaduganathan M., Gori M., Gavazzi A., Butler J. and Senni M. (2015) Role of biomarkers in cardiac structure phenotyping in heart failure with preserved ejection fraction: critical appraisal and practical use. Eur. J. Heart Fail. 17, 1231–1239 10.1002/ejhf.430 [DOI] [PubMed] [Google Scholar]
- 96.Sanders-Van Wijk S., Van Empel V., Davarzani N., Maeder M.T., Handschin R., Pfisterer M.E.et al. (2015) Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur. J. Heart Fail. 17, 1006–1014 10.1002/ejhf.414 [DOI] [PubMed] [Google Scholar]
- 97.Santhanakrishnan R., Chong J.P.C., Ng T.P., Ling L.H., Sim D., Toh G.et al. (2012) Growth differentiation factor 15, ST2, high-sensitivity troponin T, and N-terminal pro brain natriuretic peptide in heart failure with preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 14, 1338–1347 10.1093/eurjhf/hfs130 [DOI] [PubMed] [Google Scholar]
- 98.Lam C.S.P., Voors A.A., De Boer R.A., Solomon S.D. and Van Veldhuisen D.J. (2018) Heart failure with preserved ejection fraction: From mechanisms to therapies. Eur. Heart J. 39, 2780–2792 10.1093/eurheartj/ehy301 [DOI] [PubMed] [Google Scholar]
- 99.Shah S.J., Marcus G.M., Gerber I.L., McKeown B.H., Vessey J.C., Jordan M.V.et al. (2006) High-sensitivity C-reactive protein and parameters of left ventricular dysfunction. J. Card. Fail. 12, 61–65 10.1016/j.cardfail.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 100.Sciarretta S., Ferrucci A., Ciavarella G.M., De Paolis P., Venturelli V., Tocci G.et al. (2007) Markers of inflammation and fibrosis are related to cardiovascular damage in hypertensive patients with metabolic syndrome. Am. J. Hypertens. 20, 784–791 10.1016/j.amjhyper.2007.01.023 [DOI] [PubMed] [Google Scholar]
- 101.Shah S.J., Lam C.S.P., Svedlund S., Saraste A., Hage C., Tan R.S.et al. (2018) Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 39, 3439–3450 10.1093/eurheartj/ehy531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kalogeropoulos A., Georgiopoulous V., Psaty B., Rodondi N., Smith A., Harrison D.et al. (2010) Inflammatory markers and incident heart failure risk in older adults: The Health, Aging, and Body Composition Study. JAMA Cardiol. 55, 2129–2137 10.1016/j.jacc.2009.12.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sano S., Oshima K., Wang Y., MacLauchlan S., Katanasaka Y., Sano M.et al. (2018) Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol. 71, 875–886 10.1016/j.jacc.2017.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Richards D.A., Aronovitz M.J., Calamaras T.D., Tam K., Martin G.L., Liu P.et al. (2019) Distinct phenotypes induced by three degrees of transverse aortic constriction in mice. Sci. Rep. 9, 1–15 10.1038/s41598-019-42209-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Valero-Munõz M., Li S., Wilson R.M., Hulsmans M., Aprahamian T., Fuster J.J.et al. (2016) Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ. Heart Fail. 9, 1–10 10.1161/CIRCHEARTFAILURE.115.002724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ma J., Luo T., Zeng Z., Fu H., Asano Y., Liao Y.et al. (2016) Histone deacetylase inhibitor phenylbutyrate exaggerates heart failure in pressure overloaded mice independently of HDAC inhibition. Sci. Rep. 6, 1–12 10.1038/srep34036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sano S., Oshima K., Wang Y., Katanasaka Y., Sano M. and Walsh K. (2018) CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res. 123, 335–341 10.1161/CIRCRESAHA.118.313225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Madsen A., Höppner G., Krause J., Hirt M.N., Laufer S.D., Schweizer M.et al. (2020) An important role for DNMT3A-mediated DNA methylation in cardiomyocyte metabolism and contractility. Circulation 10.1161/CIRCULATIONAHA.119.044444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sano S., Wang Y., Yura Y., Sano M., Oshima K., Yang Y.et al. (2019) JAK2V617F-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl. Sci. 4, 684–697 10.1016/j.jacbts.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kimishima Y., Misaka T., Yokokawa T., Wada K., Ueda K., Sugimoto K.et al. (2020) Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension through ALK1. Circulation 142, Abstract 12873 10.1161/circ.142.suppl_3.12873 [DOI] [Google Scholar]
- 111.Wang Y., Pietras E.M. and Walsh K. (2020) Tet2-mediated clonal hematopoiesis in non-conditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5, e135204 10.1172/jci.insight.135204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dunlay S.M., Roger V.L. and Redfield M.M. (2017) Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 14, 591–602 10.1038/nrcardio.2017.65 [DOI] [PubMed] [Google Scholar]
- 113.Bursi F., Weston S.A., Redfield M.M., Jacobsen S.J., Pakhomov S., Nkomo V.T.et al. (2006) Systolic and diastolic heart failure in the community. J. Am. Med. Assoc. 296, 2209–2216 10.1001/jama.296.18.2209 [DOI] [PubMed] [Google Scholar]
- 114.Gottdiener J.S., Mcclelland R.L., Marshall R., Shemanski L., Furberg C., Kitzman D.et al. (2002) Outcome of congestive heart failure in elderly persons : influence of left ventricular systolic function : The Cardiovascular Health Study. Ann. Intern. Med. 137, 631–639 10.7326/0003-4819-137-8-200210150-00006 [DOI] [PubMed] [Google Scholar]
- 115.Devereux R.B., Roman M.J., Liu J.E., Welty T.K., Lee E.T., Rodeheffer R.et al. (2000) Congestive heart failure despite normal left ventricular systolic function in a population-based sample: The Strong Heart Study. Am. J. Cardiol. 86, 1090–1096 10.1016/S0002-9149(00)01165-6 [DOI] [PubMed] [Google Scholar]
- 116.Fonarow G.C., Stough W.G., Abraham W.T., Albert N.M., Gheorghiade M., Greenberg B.H.et al. (2007) Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure. A Report From the OPTIMIZE-HF Registry. J. Am. Coll. Cardiol. 50, 768–777 10.1016/j.jacc.2007.04.064 [DOI] [PubMed] [Google Scholar]
- 117.Lenzen M.J., Scholte Op Reimer W.J.M., Boersma E., Vantrimpont P.J.M.J., Follath F., Swedberg K.et al. (2004) Differences between patients with a preserved and a depressed left ventricular function: A report from the EuroHeart Failure Survey. Eur. Heart J. 25, 1214–1220 10.1016/j.ehj.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 118.Yancy C.W., Lopatin M., Stevenson L.W., De Marco T. and Fonarow G.C. (2006) Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: A report from the Acute Decompensated Heart Failure National Registry (ADHERE) database. J. Am. Coll. Cardiol. 47, 76–84 10.1016/j.jacc.2005.09.022 [DOI] [PubMed] [Google Scholar]
- 119.Owan T.E., Hodge D.O., Herges R.M., Jacobsen S.J., Roger V.L. and Redfield M.M. (2006) Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 355, 251–259 10.1056/NEJMoa052256 [DOI] [PubMed] [Google Scholar]
- 120.Senni M., Tribouilloy C.M., Rodeheffer R.J., Jacobsen S.J., Evans J.M., Bailey K.R.et al. (1998) Congestive heart failure in the community: a study of all incident cases in Olmsted county, Minnesota, in 1991. Circulation 98, 2282–2289 10.1161/01.CIR.98.21.2282 [DOI] [PubMed] [Google Scholar]
- 121.Klapholz M., Maurer M., Lowe A.M., Messineo F., Meisner J.S., Mitchell J.et al. (2004) Hospitalization for heart failure in the presence of a normal left ventricular ejection fraction: Results of the New York heart failure registry. J. Am. Coll. Cardiol. 43, 1432–1438 10.1016/j.jacc.2003.11.040 [DOI] [PubMed] [Google Scholar]
- 122.MacCarthy P.A., Kearney M.T., Nolan J., Lee A.J., Prescott R.J., Shah A.M.et al. (2003) Prognosis in heart failure with preserved left ventricular systolic function: prospective cohort study. Br. Med. J. 327, 78–79 10.1136/bmj.327.7406.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.van Veldhuisen D.J., Cohen-Solal A., Böhm M., Anker S.D., Babalis D., Roughton M.et al. (2009) Beta-blockade with nebivolol in elderly heart failure patients with impaired and preserved left ventricular ejection fraction. Data From SENIORS (Study of Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors With Heart Failure). J. Am. Coll. Cardiol. 53, 2150–2158 10.1016/j.jacc.2009.02.046 [DOI] [PubMed] [Google Scholar]
- 124.Massie B.M., Carson P.E., McMurray J.J., Komajda M., McKelvie R., Zile M.R.et al. (2008) Irbesartan in patients with heart failure and preserved ejection fraction. N. Engl. J. Med. 359, 2456–2467 10.1056/NEJMoa0805450 [DOI] [PubMed] [Google Scholar]
- 125.Yip G.W.K., Wang M., Wang T., Chan S., Fung J.W.H., Yeung L.et al. (2008) The Hong Kong diastolic heart failure study: a randomised controlled trial of diuretics, irbesartan and ramipril on quality of life, exercise capacity, left ventricular global and regional function in heart failure with a normal ejection fraction. Heart 94, 573–580 10.1136/hrt.2007.117978 [DOI] [PubMed] [Google Scholar]
- 126.Cleland J.G.F., Tendera M., Adamus J., Freemantle N., Polonski L. and Taylor J. (2006) The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur. Heart J. 27, 2338–2345 10.1093/eurheartj/ehl250 [DOI] [PubMed] [Google Scholar]
- 127.Ahmed A., Rich M.W., Fleg J.L., Zile M.R., Young J.B., Kitzman D.W.et al. (2006) Effects of digoxin on morbidity and mortality in diastolic heart failure: The Ancillary Digitalis Investigation Group Trial. Circulation 114, 397–403, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf 10.1161/CIRCULATIONAHA.106.628347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bergström A., Andersson B., Edner M., Nylander E., Persson H. and Dahlström U. (2004) Effect of carvedilol on diastolic function in patients with diastolic heart failure and preserved systolic function. Results of the Swedish Doppler-echocardiographic study (SWEDIC). Eur. J. Heart Fail. 6, 453–461 10.1016/j.ejheart.2004.02.003 [DOI] [PubMed] [Google Scholar]
- 129.Yusuf S., Pfeffer M.A., Swedberg K., Granger C.B., Held P., McMurray J.J.V.et al. (2003) Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-preserved trial. Lancet 362, 777–781 10.1016/S0140-6736(03)14285-7 [DOI] [PubMed] [Google Scholar]
- 130.Shah S.J., Heitner K.F., Sweitzer N.K., Anand I.S., Kim Y.H., Harty B.et al. (2013) Baseline characteristics of patients in the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) Trial. Circ. Heart Fail. 6, 184–192 10.1161/CIRCHEARTFAILURE.112.972794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Solomon S.D., Rizkala A.R., Lefkowitz M.P., Shi V.C., Gong J., Anavekar N.et al. (2018) Baseline characteristics of patients with heart failure and preserved ejection fraction in the PARAGON-HF trial. Circ. Heart Fail. 11, 1–10 10.1161/CIRCHEARTFAILURE.118.004962 [DOI] [PubMed] [Google Scholar]
- 132.Gerber Y., Weston S.A., Redfield M.M., Chamberlain A.M., Manemann S.M., Jiang R.et al. (2015) A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern. Med. 175, 996–1004 10.1001/jamainternmed.2015.0924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee D.S., Gona P., Vasan R.S., Larson M.G., Benjamin E.J., Wang T.J.et al. (2009) Relation of disease pathogenesis and risk factors to heart failure with preserved or reduced ejection fraction: insights from the framingham heart study of the national heart, lung, and blood institute. Circulation 119, 3070–3077 10.1161/CIRCULATIONAHA.108.815944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bhatia R.S., Tu J.V., Lee D.S., Austin P.C., Fang J., Haouzi A.et al. (2006) Outcome of heart failure with preserved ejection fraction in a population-based study. N. Engl. J. Med. 355, 260–269 10.1056/NEJMoa051530 [DOI] [PubMed] [Google Scholar]
- 135.Philbin E.F., Rocco T.A., Lindenmuth N.W., Ulrich K. and Jenkins P.L. (2000) Systolic versus diastolic heart failure in community practice: Clinical features, outcomes, and the use of angiotensin-converting enzyme inhibitors. Am. J. Med. 109, 605–613 10.1016/S0002-9343(00)00601-X [DOI] [PubMed] [Google Scholar]
- 136.Brouwers F.P., De Boer R.A., Van Der Harst P., Voors A.A., Gansevoort R.T., Bakker S.J.et al. (2013) Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur. Heart J. 34, 1424–1431 10.1093/eurheartj/eht066 [DOI] [PubMed] [Google Scholar]
- 137.Gurwitz J.H., Madid D.J., Smith D.H., Goldberg R.J., McManus D.D., Allen L.A.et al. (2014) Contemporary prevalence and correlates of incident heart failure with preserved ejection fraction. Clin. Med. 14, s22–s28 10.7861/clinmedicine.14-6-s22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gustafsson F., Torp-Pedersen C., Brendorp B., Seibæk M., Burchardt H. and Køber L. (2003) Long-term survival in patients hospitalized with congestive heart failure: relation to preserved and reduced left ventricular systolic function. Eur. Heart J. 24, 863–870 10.1016/S0195-668X(02)00845-X [DOI] [PubMed] [Google Scholar]
- 139.Triposkiadis F., Giamouzis G., Parissis J., Starling R.C., Boudoulas H., Skoularigis J.et al. (2016) Reframing the association and significance of co-morbidities in heart failure. Eur. J. Heart Fail. 18, 744–758 10.1002/ejhf.600 [DOI] [PubMed] [Google Scholar]
- 140.Chamberlain A.M., Sauver J.L.S., Gerber Y., Manemann S.M., Boyd C.M., Dunlay S.M.et al. (2015) Multimorbidity in heart failure: a community perspective. Am. J. Med. 128, 38–45 10.1016/j.amjmed.2014.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bonnefond A., Skrobek B., Lobbens S., Eury E., Thuillier D., Cauchi S.et al. (2013) Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat. Genet. 45, 1040–1043 10.1038/ng.2700 [DOI] [PubMed] [Google Scholar]
- 142.Tromp J., MacDonald M.R., Ting Tay W., Teng T.H.K., Hung C.L., Narasimhan C.et al. (2018) Heart failure with preserved ejection fraction in the young. Circulation 138, 2763–2773 10.1161/CIRCULATIONAHA.118.034720 [DOI] [PubMed] [Google Scholar]
- 143.Matsushita K., Harada K., Miyazaki T., Miyamoto T., Kohsaka S., Iida K.et al. (2019) Younger- vs older-old patients with heart failure with preserved ejection fraction. J. Am. Geriatr. Soc. 67, 2123–2128 10.1111/jgs.16050 [DOI] [PubMed] [Google Scholar]
- 144.Zacharias M., Joffe S., Konadu E., Meyer T., Kiernan M., Lessard D.et al. (2016) Clinical epidemiology of heart failure with preserved ejection fraction (HFpEF) in comparatively young hospitalized patients. Int. J. Cardiol. 202, 918–921 10.1016/j.ijcard.2015.09.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Robertson N., Deary I., Kirschner K., Marioni R.E. and Chandra T. (2019) Age-related clonal haemopoiesis is associated with increased epigenetic age. Curr. Biol. 29, 86–87 10.1016/j.cub.2019.07.011 [DOI] [PubMed] [Google Scholar]
- 146.Marioni R.E., Shah S., McRae A.F., Ritchie S.J., Muniz-Terrera G., Harris S.E.et al. (2015) The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 44, 1388–1396 10.1093/ije/dyu277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen B.H., Marioni R.E., Colicino E., Peters M.J., Ward-Caviness C.K., Tsai P.C.et al. (2016) DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany N.Y.) 8, 1844–1865 10.18632/aging.101020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Eaton C., Raffield L.M., Bick A., Roberts M., Manson J.E. and Reiner A. (2020) Abstract 11: Prospective association of Tet2 mediated clonal hematoopoiesis and heart failure and its subtypes in postmenopausal women. Circulation 141, 10.1161/circ.141.suppl_1.11 [DOI] [Google Scholar]
- 149.Honigberg M.C., Zekavat S.M., Aragam K., Finneran P., Klarin D., Bhatt D.L.et al. (2019) Association of premature natural and surgical menopause with incident cardiovascular disease. JAMA 322, 2411–2421 10.1001/jama.2019.19191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee C.G., Carr M.C., Murdoch S.J., Mitchell E., Woods N.F., Wener M.H.et al. (2009) Adipokines, inflammation, and visceral adiposity across the menopausal transition: a prospective study. J. Clin. Endocrinol. Metab. 94, 1104–1110 10.1210/jc.2008-0701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Neilson H.K., Conroy S.M. and Friedenreich C.M. (2014) The influence of energetic factors on biomarkers of postmenopausal breast cancer risk. Curr. Nutr. Rep. 3, 22–34 10.1007/s13668-013-0069-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Honigberg M.Zekavat, SM. Niroula, A. Griffi, GK. Bick, AG. Pirruccello, JP et al (2021) Premature Menopause, Clonal Hematopoiesis, and Coronary Artery disease in Post Menopausal Women. Circ 143, 410-423, 10.1161/CIRCULATIONAHA.120.051775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ridker P.M., Everett B.M., Thuren T., MacFadyen J.G., Chang W.H., Ballantyne C.et al. (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 154.Everett B.M., Cornel J.H., Lainscak M., Anker S.D., Abbate A., Thuren T.et al. (2019) Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 139, 1289–1299 10.1161/CIRCULATIONAHA.118.038010 [DOI] [PubMed] [Google Scholar]
- 155.Svensson E., Madar A., Campbell C., He Y., Sultan M., Healey M.et al. (2019) TET2-driven clonal hematopoiesis predicts enhanced response to canakinumab in the CANTOS Trial: an exploratory analysis. Circ. Genet Genomics 138, Abstract 15111 [Google Scholar]
- 156.Lee Y.-H., Kim S.R., Bae J., Lee B.-W., Kang E.S., Ah C.W.et al. (2018) SGLT2 inhibitors suppress NLRP3 inflammasome activity via changes in ketones and insulin in type 2 diabetes and cardiovascular diseases. Diabetes 67, Supplement 1 10.2337/db18-164-OR [DOI] [Google Scholar]
- 157.Hsu J.I., Dayaram T., Tovy A., De Braekeleer E., Jeong M., Wang F.et al. (2018) PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell 23, 700.e6–713.e6 10.1016/j.stem.2018.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]