

SUMMARY
Acetylation of lysine 16 on histone H4 (H4K16ac) is catalyzed by histone acetyltransferase KAT8 and can prevent chromatin compaction in vitro. Although extensively studied in Drosophila, the functions of H4K16ac and two KAT8-containing protein complexes (NSL and MSL) are not well understood in mammals. Here, we demonstrate a surprising complex-dependent activity of KAT8: it catalyzes H4K5ac and H4K8ac as part of the NSL complex, whereas it catalyzes the bulk of H4K16ac as part of the MSL complex. Furthermore, we show that MSL complex proteins and H4K16ac are not required for cell proliferation and chromatin accessibility, whereas the NSL complex is essential for cell survival, as it stimulates transcription initiation at the promoters of housekeeping genes. In summary, we show that KAT8 switches catalytic activity and function depending on its associated proteins and that, when in the NSL complex, it catalyzes H4K5ac and H4K8ac required for the expression of essential genes.
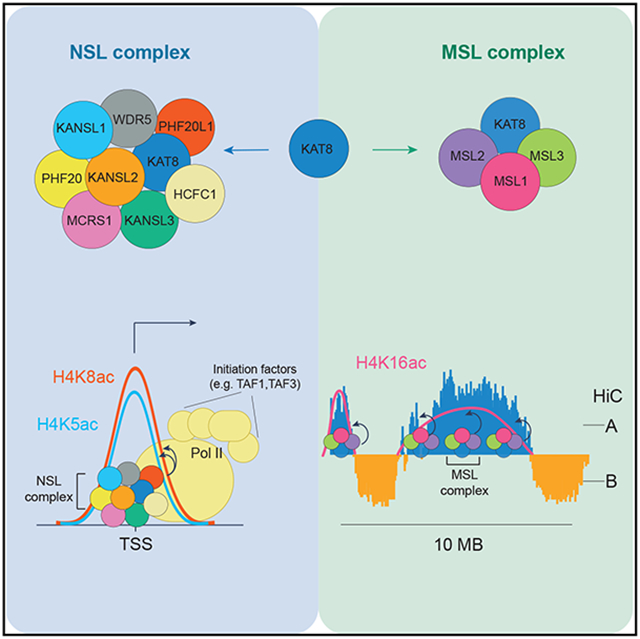
Graphical Abstract
In brief
Radzisheuskaya et al. report that histone acetyltransferase KAT8 switches functions depending on associated proteins. It catalyzes promoter-associated H4K5 and H4K8 acetylation as part of the NSL complex, which is required for the activation of essential genes. As part of the MSL complex, it places H4K16 acetylation marking all open chromatin.
INTRODUCTION
Histone acetylation is well known for its association with transcriptionally active chromatin. It is assumed to contribute to open chromatin directly, by introducing structural barriers to compaction, and indirectly, by providing a platform for binding of proteins with specific reader domains. Most evidence for direct regulation of chromatin compaction stems from research on lysine 16 acetylation of histone H4. Specifically, H4K16ac was shown to prevent nucleosome array assembly in vitro (Allahverdi et al., 2011; Liu et al., 2011; Robinson et al., 2008; Shogren-Knaak et al., 2006), potentially by increasing disorder of the H4 tail basic patch, which, in turn, decreases H4K16 interactions with adjacent nucleosomes (Zhang et al., 2017; Zhou et al., 2012). Despite strong in vitro evidence, the role of H4K16ac in regulating chromatin accessibility in vivo has not been demonstrated.
H4K16ac is catalyzed by the MYST-family histone acetyltransferase KAT8, also known as MOF or MYST1 (Akhtar and Becker, 2000; Hilfiker et al., 1997). KAT8 serves as a catalytic subunit of two independent protein complexes conserved from Drosophila to mammals: MSL (male-specific lethal) and NSL (non-specific lethal). In Drosophila, the MSL complex is required for sex chromosome dosage compensation in males and, thereby, their viability (Belote and Lucchesi, 1980; Hilfiker et al., 1997; Tanaka et al., 1976). It places widespread H4K16ac on the male X chromosome, which increases expression of its genes 2-fold (Larschan et al., 2011; Lucchesi and Kuroda, 2015). In contrast, depletion of the NSL complex is lethal in both Drosophila sexes (Mendjan et al., 2006). The NSL complex was found to bind to promoters of housekeeping genes in Drosophila cells and to stimulate expression of a subset of these genes (Feller et al., 2012; Lam et al., 2012; Raja et al., 2010). Particularly, NSL complex depletion was shown to reduce RNA polymerase II (Pol II) (Gaub et al., 2020; Lam et al., 2012), TBP (Lam et al., 2012), TFIIB (Lam et al., 2012), and BRD4 (Gaub et al., 2020) binding and to induce loss of nucleosome-free regions (Lam et al., 2019) at target promoters. Although it is currently unknown how the NSL complex directly contributes to this regulation, many attribute these phenotypes to H4K16 acetylation, as the main catalytic activity of KAT8 (Gaub et al., 2020; Sheikh et al., 2019).
Studies in mammalian cells have shown that loss of KAT8 and consequent global loss of H4K16ac lead to a number of phenotypes, including a deficient DNA damage response (Sharma et al., 2010), reduced fatty acid oxidation and mitochondrial respiration (Khoa et al., 2020), cell death and apoptosis (Li et al., 2012; Sheikh et al., 2016; Yin et al., 2017), and autophagy (Füllgrabe et al., 2013). Although Kat8−/− mouse embryos exhibit a global loss of H4K16ac and die at implantation (Gupta et al., 2008; Thomas et al., 2008), the precise regulation of these processes by different KAT8-associated complexes is not well understood. Previous results in mouse embryonic stem cells (ESCs) show that, similarly to Drosophila, the NSL and MSL complexes have different genomic localizations and affect different sets of genes: the NSL complex is enriched at promoter regions, while the MSL complex is associated with gene bodies and intergenic regions (Ravens et al., 2014). Additionally, the bulk of H4K16ac in mammalian cells is produced by the MSL complex, but depletion of the NSL complex has a stronger effect on ESC proliferation (Chelmicki et al., 2014; Ravens et al., 2014). If both KAT8-associated complexes function through H4K16ac, a critical question is how one specific modification exerts different cellular effects depending on genome localization. It could be that members of the NSL and MSL complexes have additional, non-overlapping functions, for instance recruiting different interacting partners. Alternatively, KAT8-containing complexes could have distinct, previously uncharacterized, substrate specificities. In fact, in vitro histone acetylation studies have shown that KAT8 can catalyze H4K16ac as part of both complexes, while the NSL complex can also acetylate H4K5 and H4K8 (Cai et al., 2010; Zhao et al., 2013). However, evidence that the NSL complex catalyzes those modifications in vivo is thus far lacking (Karoutas et al., 2019; Ravens et al., 2014).
In this study, we investigated the chromatin functions of KAT8 and provide insights into how they are specified by the associated protein complexes in mammalian cells.
RESULTS
The NSL but not the MSL complex is essential for proliferation of human cells
To determine the functions of KAT8 and its associated complexes in human cells, we used the human acute myeloid leukemia cell line THP-1 as a main model because of the ability to study chromatin changes in both steady state and during cell differentiation into macrophages. To confirm the existence of two distinct KAT8-containing chromatin complexes, we generated THP-1 cells expressing either 3xFLAG-tagged mammalian NSL complex members KANSL2 and KANSL3 or KAT8 and performed immunoprecipitation followed by mass spectrometry-based identification of associated proteins. We identified all the reported human NSL complex subunits (KANSL1, KANSL2, KANSL3, KAT8, MCRS1, PHF20, PHF20L1, HCFC1, and WDR5) (Figures 1A and 1B; Table S1). Moreover, KAT8 also co-purified with all the reported subunits of the human MSL complex (MSL1, MSL2, and MSL3) (Figures 1A and 1B; Table S1). We also identified three unique interactors of KANSL3 (OGT, HSPA5, and PPM1G), suggesting it has functions outside the NSL complex (Figure 1A).
Figure 1. The NSL complex is essential for cell survival and predominantly localizes to transcription start sites.

(A) Venn diagram of KANSL2, KANSL3, and KAT8 interactors (log2 fold enrichment over untagged cell line > 2, minimum two peptides identified per protein, and q ≤ 0.05). KAT8 did not reach the selected enrichment threshold in the KANSL2 and KANSL3 immunoprecipitations but was significantly enriched over the untagged cell line (bar blot; values are shown as mean ± SD). *q ≤ 0.05.
(B) NSL and MSL complex composition in human cells.
(C) CRISPR KO competition assays testing essentiality of the NSL and MSL complex subunits. Cutting efficiency of all sgRNAs was verified using Sanger sequencing with subsequent sequence trace decomposition.
(D) Boxplot describing distribution of the DepMap dependency scores for the NSL and MSL complex subunits. Genes with dependency scores ≤ −0.5 are considered essential (pink dashed line).
(E) Heatmaps of normalized KANSL3 ChIP-seq signal at 5,000 bp regions surrounding all significant KANSL3 peaks.
(F) Representative tracks of normalized KANSL3 and H3K4me3 ChIP-seq signals.
(G) Distribution of distances between KANSL3 peaks and the nearest H3K4me3 ChIP-seq peaks.
(H) Average normalized KANSL3 ChIP-seq signal across the genes nearest to KANSL3 peaks. TSS, transcription start site, TES, transcription end site.
To determine whether the two KAT8-containing complexes are required for growth of human cells, we performed CRISPR-mediated knockout (KO) of each subunit followed by a competition assay with wild-type cells. THP-1 cells expressing wild-type Cas9 were transduced with a lentivirus expressing BFP (blue fluorescent protein) and a single guide RNA (sgRNA) against the gene of interest. The percentage of BFP-positive (sgRNA-expressing) cells was then monitored over time. Depletion of six of eight subunits of the NSL complex led to cell death (KANSL1, KANSL2, KANSL3, MCRS1, WDR5, and HCFC1), while the MSL complex depletion did not (Figure 1C). We also compared essentiality scores for all complex subunits in the DepMap CRISPR database (https://depmap.org/portal/) and observed similar growth dependency patterns in a set of 739 human cell lines (Figure 1D). As PHF20 and PHF20L1 are the only non-essential subunits of the NSL complex and are close orthologs, we knocked out both genes to test redundancy. As shown in Figures S1A and S1B, both genes are dispensable for proliferation.
In summary, we confirmed that KAT8 exists in two distinct protein complexes in human cells, NSL and MSL, and found that only the function of the NSL complex is essential for cell survival.
The NSL complex is required for expression of a subset of essential genes in human cells
To determine genomic occupancy of the NSL complex in human cells, we performed KANSL3 chromatin immunoprecipitation sequencing (ChIP-seq) in wild-type THP-1 cells or cells with CRISPR interference (CRISPRi)-mediated KANSL3 knockdown (KD) 5 days after sgRNA transduction (Figure S1C). We chose CRISPRi over the CRISPR KO approach to avoid truncated or in-frame-modified protein products, which might be recognized by the anti-KANSL3 antibody. KANSL3 ChIP-seq identified 373 KANSL3-specific peaks (Figure 1E; Table S1), the majority of which (77.2%) co-localized with H3K4me3 (Figures 1F and 1G) and were positioned at transcription start sites (TSSs) (Figure 1H; Figure S1D). This localization is consistent with previous reports in Drosophila (Feller et al., 2012; Lam et al., 2012; Raja et al., 2010) and mouse ESCs (Chelmicki et al., 2014; Ravens et al., 2014). KANSL3 peaks were frequently found with ETS family transcription factor motifs (Figure S1E), potentially suggesting a role for these factors in NSL complex recruitment.
To identify genes regulated by the NSL complex, we performed global transcriptomics (RNA sequencing [RNA-seq]) and proteomics profiling following CRISPRi KD of KANSL2, KANSL3, and KAT8 (Figures 2A and 2B). We used the milder CRISPRi assay over CRISPR KO because of the essentiality of KANSL2, KANSL3, and KAT8. CRISPRi-mediated KD of the three genes induced growth defects (Figure 2C) due to cell-cycle arrest in G1 and G2/M phases (Figure 2D) and apoptosis (Figure 2E) starting about 7 days after sgRNA transduction. Thus, we chose a time point of 3 days after transduction to capture changes in gene expression, prior to the onset of cell death. Principal-component analysis of both global transcriptomic and proteomic data revealed that KANSL2- and KANSL3-KD samples clustered together and clustered apart from KAT8-KD (Figures 2F and 2G; Figures S2A and S2B; Table S2), indicating additional KAT8 functions, consistent with its also being a part of the MSL complex. Interestingly, KANSL2 KD led to decreased protein but not transcript levels of KANSL1, KANSL3, MCRS1, and PHF20L1 (Figure S2C), indicating its importance for complex integrity.
Figure 2. The NSL complex is required for expression of a subset of essential genes in human cells.

(A and B) qRT-PCR (A) and western blotting (B) analysis of KANSL2, KANSL3, and KAT8 expression 3 days after transduction of THP-1/cdCas9-KRAB cells with corresponding sgRNAs. In (A), values are normalized to RPLP0 and shown as mean ± SD.
(C) Growth curves of THP-1/cdCas9-KRAB cells transduced with a NegCtrl sgRNA or sgRNAs against KANSL2, KANSL3, or KAT8.
(D and E) Cell cycle (E) and apoptosis (F) analysis 7 days after KANSL2, KANSL3, or KAT8 KD. The values are shown as mean ± SD.
(F and G) Principal-component and non-hierarchical clustering analysis of the RNA-seq (F) and proteomics (G) expression data from NSL complex KD series.
(H) Volcano plots describing expression changes for all genes with a nearby KANSL3 ChIP-seq peak (KANSL3 targets) in NSL complex KD series. Differentially expressed genes are indicated in light blue (q < 0.05).
(I) Venn diagram indicating overlap between downregulated KANSL3 target genes and the list of pan-essential genes from the DepMap project.
(J) The gene set enrichment analysis (GSEA) enrichment plot for GO:0070125 gene set (mitochondrial translation elongation) on the basis of differential protein expression after KANSL2 KD.
(K) CRISPR KO competition assays for the selected NSL complex target genes.
Of the genes with KANSL3 enrichment at their promoters, 48.2% and 49.5% are downregulated by KD of KANSL2 and KANSL3, respectively, whereas relatively few genes had increased expression (5.8% and 4.5%, respectively) (Figure 2H; Figure S2D). These data suggest that the NSL complex facilitates gene expression in human cells, similar to what has been observed in Drosophila (Feller et al., 2012; Lam et al., 2012) and mouse ESCs (Chelmicki et al., 2014). Unexpectedly, KD of KAT8 led to fewer changes in NSL target gene expression than KANSL2 and KANSL3 KD, at both the mRNA and protein levels (Figure 2H; Figures S2D-S2F), and induced a weaker effect on cell proliferation (Figures 2C-2E). This is likely due to incomplete KD of KAT8 and is discussed in detail below.
To understand the essential nature of the NSL complex, we compared the genes directly activated by the NSL complex with the genes identified as pan-essential in the DepMap project. This identified 27 essential genes downregulated by KD of KANSL2 and KANSL3 (Figure 2I). Several of these encode proteins involved in mitochondrial translation and other aspects of mitochondrion biology (LARS2, TTC27, POP4, TARS2, MRPL9, MRPS31, QRSL1, SLC25A26, PMPCB, MALSU1, and TEFM). Consistently, Gene Ontology analysis of the downregulated proteins after KANSL2 KD uncovered mitochondrial translation as the top category (Figure 2J; Figure S2G). In addition, essential proteins involved in transcription and RNA splicing (RPAP1, GTF3C3, DDX21, ISY1, and MAGOH), translation (RRP15), telomere elongation (RTEL1), and vacuole homeostasis (TMEM199) were among the direct targets of the NSL complex. To ensure that the identified genes are essential in THP-1 cells, several of these were independently validated (Figure 2K; Figure S2H).
In summary, the NSL complex binds to several essential genes and is required for maintaining their expression and, therefore, for cell survival.
Depletion of KAT8, but not KANSL2 and KANSL3, abrogates global H4K16ac levels
As KAT8 is the main acetyltransferase for H4K16ac, it is widely believed that both KAT8-containing complexes, MSL and NSL, exert their activity through H4K16ac (Ravens et al., 2014; Sheikh et al., 2019; Yang et al., 2014). Therefore, we tested whether H4K16ac is involved in maintaining NSL complex target gene expression in mammalian cells. First, we validated the specificity of a commercial H4K16ac antibody by ELISA with an in-house library of 158 histone peptides (Figure S3A; Table S3) and by a Luminex-based approach with a panel of 24 designer nucleosomes (Figure S3B; Table S3). Next, we defined the optimal antibody/chromatin ratio (Figure S3C) and performed H4K16ac ChIP-seq in six human cell lines (THP-1, K562, MOLM13, U2OS, HeLa, and HEK293). We found that H4K16ac is an abundant histone modification with extensive presence throughout the genome (Figure 3A). H4K16ac was particularly enriched at gene bodies, positively correlating with gene expression, and depleted at TSSs in all cell lines tested (Figures 3A-3C; Figure S3D). This was consistent with ChIP results using another anti-H4K16ac antibody (Figure S3E). Furthermore, we confirmed the high abundance of H4K16ac (about 30% of all histone H4) in THP-1 and mouse ESCs using mass spectrometry (Figure 3D).
Figure 3. Depletion of KAT8 but not KANSL2 and KANSL3 abrogates global H4K16ac levels.

(A) Heatmaps of H4K16ac ChIP-seq signal at all genes in six human cell lines (gene length is normalized). The signal is normalized to the total number of reads. TSS, transcription start site; TES, transcription end site.
(B) Density plot of normalized THP-1 H4K16ac ChIP-seq signal across different chromatin states defined by the Broad Institute ChromHMM project in K562 cells. Similar results were obtained using K562 H4K16ac ChIP-seq data. Txn, transcription. Color in the density plot conveys shape of the signal distribution normalized to maximum within one lane. The amount of H4K16ac is shown on the y axis.
(C) Boxplots demonstrating statistical summary of H4K16ac ChIP-seq signal in gene bodies of genes binned according to quantile (Q) distribution of expression in THP-1 cells. All groups are significantly different (p < 2.2e-16, two-sided Wilcoxon rank-sum test).
(D) Bar plot indicating abundances of individual H4 peptides (amino acids 4–17) with different acetylation combinations in mouse ESCs and human THP-1 cells measured using mass spectrometry.
(E) Heatmaps of H4K16ac ChIP-seq signal at all genes in NSL complex KD series. The signal is normalized to the number of Drosophila reads (see STAR methods).
(F) Average normalized H4K16ac ChIP-seq profiles across NSL target genes or all genes in NSL complex KD series.
(G) qRT-PCR quantitation of H4K16ac ChIP signal at selected loci after KDs of KANSL2, KANSL3, or KAT8. TSS, transcription start site; GB, gene body. Values are shown as mean ± SD.
(H) Example H4K16ac ChIP-seq tracks in NSL complex KD series.
(I) Western blotting analysis of H4K16ac and H3 levels after KANSL2, KANSL3, or KAT8 KDs.
H4K16ac was previously reported to be enriched at gene promoters and enhancers in mouse ESCs (Taylor et al., 2013). Because this disagrees with our mapping data in human cells, we performed H4K16ac ChIP-seq in mouse ESCs and mouse MLL-AF9-rearranged acute myeloid leukemia cells. Both mouse cell types demonstrated widespread H4K16ac with enrichment at gene bodies (Figure S3F), but H4K16ac was only present at TSSs in mouse ESCs (Figure S3F), suggesting a specialized regulation in this cell type.
To analyze changes in H4K16ac upon downregulation of the NSL complex, we performed H4K16ac ChIP-seq following CRISPRi KD of KANSL2, KANSL3, and KAT8 in THP-1 cells. KAT8 KD led to a widespread genome-wide loss of H4K16ac, with the exception of TSSs, where a low signal was mostly maintained (Figures 3E-3H). This remaining signal could potentially represent “phantom” ChIP-seq peaks commonly occurring at promoter regions (Jain et al., 2015). Alternatively, it could represent the activity of TIP60, also known to acetylate H4K16 (Kimura and Horikoshi, 1998). Surprisingly, KANSL2 and KANSL3 KD did not lead to changes in H4K16ac at their target genes or globally (Figures 3E-3H). Similar results were observed by measuring global levels of H4K16ac using western blotting (Figure 3I). Furthermore, detailed analysis of the abundances of 125 histone PTM combinations using mass spectrometry showed that whereas KANSL2 and KANSL3 KD had no discernible impact, KAT8 KD led to a significant decrease in the abundance of multiple H4K16ac-containing peptides (H4K16ac, H4K8acK16ac, H4K5acK16ac, H4K12acK16ac, H4K5acK12acK16ac, and H4K8acK12acK16ac) (Figure S3G; Table S4).
Taken together, these results demonstrate that KAT8 catalyzes widespread and abundant H4K16ac outside TSS regions in the cell lines tested (with the exception of mouse ESCs, where H4K16ac is also present at TSSs). Importantly, KD of KANSL2 and KANSL3 did not affect H4K16ac levels, suggesting that the NSL complex uses alternative mechanisms for regulating gene expression.
The NSL complex catalyzes H4K5ac and H4K8ac and is required for transcription initiation
Previous reports demonstrated that, in addition to catalyzing H4K16ac, the NSL complex can catalyze H4K5 and H4K8 acetylation when incubated with nucleosomes in vitro (Cai et al., 2010; Zhao et al., 2013). These modifications can also be catalyzed in vitro by histone acetyltransferases from other families (GNAT and p300/CBP) (Anamika et al., 2010), but the contribution of individual enzymes to the in vivo patterns of these marks is unknown. H4K5ac and H4K8ac were shown to localize to gene promoters in human cells (Zhang et al., 2020) though their involvement in transcriptional regulation is unclear. To test whether the NSL complex catalyzes these modifications in vivo, we profiled H4K5 and H4K8 acetylation using ChIP-seq after depletion of NSL complex members. It proved challenging to identify a specific H4K5ac antibody, because of high similarity of the epitope with H2AK5ac (Figure S4A). From a set of five commercial antibodies, we chose the two showing the best results in histone peptide array ELISA and designer nucleosomes assays (Figures S4B-S4E). We also confirmed high specificity of the selected H4K8ac antibody in both antibody specificity assays (Figures S4F-S4H). Both H4K5ac and H4K8ac demonstrated specific enrichment at TSSs, and their global levels were not affected by KD of KANSL2, KANSL3, or KAT8 (Figures 4A and 4B; Figures S4I-S4K). Importantly, KANSL2, KANSL3, or KAT8 KD specifically reduced both modifications at promoters of NSL target genes (Figures 4C-4H; Figures S5A-S5D). These results were verified by ChIP experiments with another H4K5ac antibody (Figures S5E-S5H). This suggests that the NSL complex regulates transcription by placing H4K5ac and H4K8ac at TSSs of target genes. Consistently, RNA Pol II binding is lost at NSL target promoters upon KANSL2 or KANSL3 KD (Figure S5G). Similar to the modest target gene expression changes after KAT8 KD (Figure 2C), it also had milder effect on H4K5ac and H4K8ac in comparison with KANSL2 or KANSL3 KD.
Figure 4. The NSL complex catalyzes H4K5 and H4K8 acetylation and is required for transcription initiation.

(A) Heatmap of normalized H4K5ac antibody #1 and H4K8ac ChIP-seq signal at all genes in wild-type THP-1 cells.
(B) Average normalized H4K5ac antibody #1 and H4K8ac ChIP-seq profiles across NSL target genes or all genes in NSL complex KD series.
(C) Examples of H4K5ac antibody #1 and H4K8ac ChIP-seq tracks at a representative NSL complex target gene.
(D) qRT-PCR quantitation of H4K5ac and H4K8ac ChIP signal at selected loci after KD of KANSL2, KANSL3, or KAT8. TSS, transcription start site; GB, gene body; NC, NegCtrl; K2, K3, and K8 respectively stand for KANSL2, KANSL3, and KAT8 KD. Values are shown as mean ± SD.
(E) Schematic representation of the dTAG Kansl3 degron system. FKBPF36V is indicated as FKBPV.
(F) Western blot of HA-tagged KANSL3 and GAPDH in Kansl3 degron-knockin cell line at different times after dTAG-13 addition.
(G) Bar plots representing fold cell expansion and percentage of Trypan blue-positive cells after 4 days of dTAG-13 treatment.
(H) qRT-PCR analysis of HA-tagged KANSL3 binding at selected target genes and control regions in Kansl3 degron-knockin cell line at 0 and 2 h after dTAG-13 treatment. Values are shown as mean ± SD.
(I) qRT-PCR analysis of total and nascent mRNA expression of NSL complex target genes in Kansl3 degron-knockin cell line at different times after dTAG-13 treatment. Values are normalized to Rplp0 and shown as mean ± SD.
(J) qRT-PCR quantitation of H4K5ac and H4K8ac ChIP signal at selected loci in Kansl3 degron-knockin cell line at different times of dTAG-13 treatment. Values are mean ± SD.
(K) H4K5ac and H4K8ac ChIP-seq tracks at Sumf1 locus (NSL complex target gene) in Kansl3 degron-knockin cell line at 0 and 2 h after dTAG-13 treatment. Arrow indicates Sumf1 TSS.
See also Figures S4-S6 and Tables S3 and S5.
To test whether H4K5ac and H4K8ac are directly regulated by the NSL complex, we generated a mouse ESC line with a biallelic FKBPF36V degradation tag (dTAG) knockin (Nabet et al., 2018) at the Kansl3 locus. In this system, addition of dTAG-13 ligand to culture medium targets KANSL3-FKBPF36V fusion protein for CRBN-mediated protein degradation (Figures 4E and 4F). Consistent with an essential role of the NSL complex in human cells (Figures 1C and 1D), dTAG-13 treatment of degron-knockin ESCs induced cell-cycle arrest and cell death (Figure 4G; Figures S5H and S5I). Because the NSL complex is common essential (and, therefore, presumably binds to the same genes across cell lines), we selected several ubiquitously expressed genes previously identified as targets of the NSL complex in THP-1 cells and showed that KANSL3 is also bound to their promoters in ESCs and that KANSL3 binding was completely lost after dTAG-13 treatment (Figure 4H). Excitingly, total and nascent expression of KANSL3 target genes was markedly reduced just 2 h after dTAG-13 treatment, suggesting that the NSL complex directly regulates their expression (Figure 4I; Figure S5J). Importantly, dTAG-13 treatment also led to the loss of promoter-associated H4K5ac and H4K8ac (Figures 4J and 4K; Figure S5K) as well as RNA Pol II binding (Figure S5L) at NSL complex target genes concomitantly with downregulation of nascent mRNA. Altogether, these results strongly suggest that the NSL complex catalyzes H4K5ac and H4K8ac at its target genes, which is required for recruitment of RNA Pol II and transcriptional initiation.
BET bromodomain proteins, particularly BRD4, are known to have high affinity to H4K5ac and H4K8ac in vitro (Filippakopoulos et al., 2012; Zaware and Zhou, 2019). However, BRD4 recruitment cannot provide a downstream mechanism for transcription activation by these modifications, as it facilitates formation of a functional elongation complex, downstream of Pol II recruitment (Muhar et al., 2018; Winter et al., 2017). Among the Pol II initiation factors, TAF1 has been shown to bind acetylated H4 tails through its bromodomains in vitro (Jacobson et al., 2000). Strikingly, TAF1 binding was decreased at NSL target genes after KANSL2 or KANLS3 KD (Figure S5M), highlighting a potential mechanism for regulation of transcription initiation by H4K5ac and H4K8ac.
To test whether the essential function of the NSL complex depends on the catalytic activity of KAT8, we expressed wild-type KAT8 (wtKAT8) or catalytic dead (E350Q) KAT8 (cdKAT8) in THP-1 cells. In contrast to wtKAT8, it was not possible to achieve high levels of cdKAT8 overexpression (Figures S6A and S6B), which may suggest a dominant-negative effect on cell proliferation. Moreover, cdKAT8 expression did not rescue cell survival after KAT8 KD (Figure S6C), suggesting that the catalytic activity of KAT8 is essential for its function.
We hypothesized that the milder effect of KAT8 KD could be due to incomplete KAT8 loss and that the remaining amount of KAT8 (Figure 2B) could be associated with the NSL complex and sufficient to partially preserve its function, despite loss of the bulk of H4K16ac. Consistent with this hypothesis, the effects of KANSL2 and KAT8 CRISPR KO were comparable (Figures S6D-S6G). Moreover, we observed higher enrichment of KAT8 at KANSL3 binding sites than at other KAT8 binding sites, and the overlapping KANSL3 and KAT8 sites were less sensitive to KAT8 loss after KAT8 KD (Figures S6H-S6J; Table S5), further suggesting a high affinity of KAT8 for the NSL complex. Interestingly, we did not observe KAT8 binding in gene body regions (Figures S6I and S6J), where it catalyzes broad patterns of H4K16ac. This suggests that KAT8 is more transiently associated with chromatin when not in the NSL complex.
Taken together, we show that the catalytic activity of KAT8 is required for its function and that the NSL complex facilitates transcriptional initiation of its target genes potentially through acetylation of H4K5 and H4K8 at their TSSs.
H4K16ac broadly correlates with open chromatin and does not regulate chromatin accessibility in vivo
As global levels of H4K16ac were affected by the loss of KAT8 but not the NSL complex, we tested whether the bulk of H4K16ac is established by the MSL complex. We performed CRISPR KO of individual MSL complex members and found that loss of MSL1 and MSL3, but not MSL2, causes global decrease in H4K16ac, with MSL1 KO having the strongest effect (Figure 5A; Figure S7A). H4K16ac ChIP-seq in MSL1-KO cells revealed dramatic signal loss throughout the genome, similar to KAT8-KD cells (Figure 5B; Figure S7B). Consistent with our competition assays (Figure 1B), MSL1-KO cells did not exhibit any proliferation defects (Figure 5C) and underwent efficient monocyte-to-macrophage differentiation (data not shown).
Figure 5. H4K16ac is associated with open chromatin but does not regulate chromatin accessibility in vivo.

(A) Western blot analysis of H4K16ac and H4 in THP-1/Cas9 cells transduced with sgRNAs targeting MSL complex members.
(B) Heatmaps of H4K16ac ChIP-seq signal at all genes in wild-type (NegCtrl) or MSL1-KO cells. The signal is normalized to the number of Drosophila reads (see STAR methods).
(C) Growth curves of THP-1/Cas9 cells transduced with a NegCtrl sgRNA or two independent sgRNAs against MSL1.
(D) Agarose gel electrophoresis image comparing chromatin digestion profiles of wild-type and MSL1-KO cells at different times after MNase addition.
(E) Volcano plot illustrating changes in ATAC-seq peaks after MSL1 KO in THP-1 cells.
(F) Boxplot summarizing H4K16ac ChIP-seq signal intensity in HiC-defined compartments A and B. p value was calculated using Wilcoxon rank-sum test.
(G) Example tracks demonstrating correlation between H4K16ac and open chromatin (ATAC-seq peaks and HiC-defined compartments A) in THP-1 cells.
As in vitro experiments suggest that H4K16ac facilitates chromatin openness, we tested whether MSL1-KO cells exhibit altered chromatin accessibility patterns. MNase digestion of extracted nuclei did not reveal any difference in chromatin accessibility between MSL1-KO and wild-type cells (Figure 5D). assay for transposase-accessible chromatin sequencing (ATAC-seq) revealed some accessibility changes induced by MSL1 KO (161 significantly changed peaks, 1.5-fold change, p < 0.05) but no global effect on chromatin accessibility (Figure 5E; Figure S7C; Table S6). Consistently, we did not find changes in global levels of any of the 125 histone PTM combinations in KAT8-KD cells assessed using mass spectrometry, except for those containing H4K16ac (Figure S3G; Table S4).
Interestingly, we observed that H4K16ac forms large megabase-scale domains conserved between different cell lines (Figure S7D). These domains largely coincided with genomic compartment A, defined by HiC data in THP-1 cells and representing open chromatin with high transcriptional activity and high density of ATAC-seq peaks (Figures 5F and 5G; Figure S7E). Thus, although not regulating chromatin compaction globally, H4K16ac represents a hallmark of open chromatin and transcription.
KAT8-low cancers retain NSL complex function but lose H4K16ac globally
Multiple studies reported decreased levels of KAT8 and H4K16ac in different types of cancer cells (Cai et al., 2015; Cao et al., 2014; Fraga et al., 2005; Liu et al., 2013; Pfister et al., 2008; Zhang et al., 2014), but the significance of this observation is unknown. As we found that the NSL complex is essential for cell survival, and that KAT8 is strongly associated with it, we hypothesized that KAT8-low cancers would have decreased MSL, but not NSL, complex activity. To test this, we selected several cancer cell lines with low KAT8 expression (SKMEL5, SKHEP1, and Caki1) on the basis of the DepMap project data (Figure 6A). In those cells, we confirmed several NSL complex target genes identified in THP-1 cells (Figure 6B) and found comparable expression of these genes between the KAT8-high (THP-1) and KAT8-low (SKMEL5, SKHEP1, Caki1) cell lines (Figure 6C), as well as comparable levels of H4K5ac and H4K8ac at their promoters (Figures 6D and 6E). Excitingly, KAT8-low cancer cells had lower H4K16ac at gene body regions (Figure 6F). Next, we analyzed a large RNA-seq dataset from The Cancer Genome Atlas (n = 10,433) and found that both NSL and MSL complexes are highly expressed in different cancer subtypes (Figure 6G). At the same time, in these cancer samples, we observed either very weak or no correlation between the levels of KAT8 expression and expression levels of NSL complex target genes defined in THP-1 cells (Figures 6H and 6I). Taken together, these results indicate that KAT8-low cancer cells exhibit mostly intact activity of the NSL complex, while their H4K16ac-associated MSL complex function is markedly decreased. This could suggest that KAT8-low cancers are more susceptible to further KAT8 loss, as their remaining KAT8 is associated with the essential NSL complex. On the basis of the data from the DepMap project, there is weak correlation between KAT8 expression and cell sensitivity to KAT8 KO (Figure 6J), suggesting that complete KAT8 loss is detrimental independently of the starting KAT8 expression level. At the same time, there was a correlation between KAT8 expression and cell sensitivity to KAT8 RNAi-mediated KD (Figure 6K), which was even more pronounced for certain cancer lineages (Figure 6L).
Figure 6. KAT8-low cancers retain NSL complex function but lose H4K16ac globally.

(A) Western blot analysis and quantitation of KAT8, H4K16ac, and beta-actin in THP-1, Caki1, SKHEP1, SKMEL28, and SKMEL5 cells.
(B) qRT-PCR quantitation of KANSL3 ChIP signal at a subset of NSL complex target TSSs in SKMEL5, SKHEP1, and Caki1 cells. Values are shown as mean ± SD.
(C) qRT-PCR gene expression analysis of a subset of NSL complex target genes in THP-1, Caki1, SKHEP1, and SKMEL5 cells. Values are normalized to RPLP0 and shown as mean ± SD.
(D–F) qRT-PCR analysis of H4K5ac (D), H4K8ac (E), and H4K16ac (F) ChIP signal at gene bodies (GBs) and TSS regions of a subset of NSL complex target genes in THP-1, SKMEL5, SKHEP1, and Caki1 cells. Values are shown as mean ± SD.
(G) Expression of the NSL and MSL complex members in different types of human cancer cells. Data are from The Cancer Genome Atlas.
(H) Vioplots demonstrating distribution of Pearson’s correlation coefficients (r) for KAT8 expression versus expression of 20,501 genes measured using RNA-seq in 10,433 cancer samples in The Cancer Genome Atlas for NSL complex target genes (as defined in THP-1 cells) and non-target genes.
(I) Volcano plots illustrating gene expression differences between the KAT8high and KAT8low human cancer samples (defined by median KAT8 expression) in the selected dataset from The Cancer Genome Atlas datasets.
(J and K) Scatterplots describing relationship between KAT8 expression level and cell sensitivity to KAT8 KO (J) defined using CRISPR KO (DepMap CERES dependency score) in 721 cancer cell lines or KAT8 KD (K) defined using RNAi (DepMap DEMETER2 dependency score) in 640 cancer cell lines. The robust Pearson’s correlation coefficient was calculated by bootstrapping 10% of the data.
(L) Scatterplot describing relationship between KAT8 expression level and cell sensitivity to KAT8 KD in selected cancer lineages.
DISCUSSION
One of our main findings is that KAT8 catalyzes H4K5 and H5K8 acetylation as part of the NSL complex, while it catalyzes H4K16 acetylation as part of the MSL complex. Although the mechanism leading to this difference in activity remains to be determined, the NSL complex and, therefore, potentially H4K5 and H4K8 acetylation are essential for cell survival, whereas the MSL complex and H4K16 acetylation are not (Figure 7).
Figure 7. Model for the function of the NSL and MSL complexes.

As part of the NSL complex, KAT8 binds to a subset of active gene promoters, where it stimulates TAF1 binding, through H4K5 and H4K8 acetylation. This facilitates Pol II recruitment and transcription initiation. As part of the MSL complex, KAT8 catalyzes the bulk of H4K16ac in open chromatin regions and, especially, in gene bodies. K1, KANSL1; K2, KANSL2; K3, KANSL3; K8, KAT8; M1, MSL1; M2, MSL2; M3, MSL3.
Investigation of how the NSL complex regulates transcription revealed that it stimulates RNA Pol II recruitment and, thereby, transcriptional initiation of a subset of essential genes, potentially through acetylation of H4K5 and H4K8. Acetylated H4 is known to be recognized by TAF1, a subunit of TFIID transcription initiation complex (Jacobson et al., 2000). Consistently, TAF1 binding at NSL complex target genes was reduced after KANSL2 or KANSL3 KD. Previous studies suggest that H3K4me3 recognition by TAF3, another TFIID subunit, aids pre-initiation complex binding and stabilization (Lauberth et al., 2013). Thus, TAF1 recognition of promoter-associated H4K5ac and H4K8ac could represent an additional mechanism facilitating transcription initiation. The dual H4K5ac/H4K8ac mark is also known to be recognized by the BET bromodomain family members in vitro (Filippakopoulos et al., 2012; Zaware and Zhou, 2019). In fact, BRD4 was recently reported to colocalize with the NSL complex in Drosophila (Gaub et al., 2020). However, BRD4 functions after RNA Pol II recruitment (Muhar et al., 2018; Winter et al., 2017) and is unlikely to act downstream of the NSL complex. Indeed, although Nsl1 KD in Drosophila led to RNA Pol II loss at promoter regions, BRD4 inhibition decreased RNA Pol II occupancy only at gene body regions (Gaub et al., 2020). Interestingly, depletion of the NSL complex does not lead to complete removal of H4K5ac and H4K8ac at target promoters or complete gene repression, which may suggest that other histone acetyltransferases can deposit these modifications and sustain basal transcription levels.
We found that H4K16ac is catalyzed by the MSL complex and is highly abundant (i.e., present on about 30% of all histone H4). It forms megabase-scale genomic domains, which highly correlate with transcriptionally active genome compartment A. This indicates that H4K16ac is not a mere consequence of transcription but rather a hallmark of all euchromatin in cells. Despite this strong association with open chromatin, depletion of H4K16ac did not lead to cell proliferation and differentiation phenotypes or changes in chromatin accessibility patterns. This is particularly surprising, as the strong phenotypes of KAT8 loss observed in different cell types are widely believed to be caused by a global loss of H4K16ac. Our study decouples the two and allows a more direct assessment of H4K16ac function. Interestingly, two recent reports (Armstrong et al., 2018; Copur et al., 2018) demonstrated that female Drosophila flies in which H4K16 was substituted with arginine are viable. Another report (Taylor et al., 2013) demonstrated that changes in H4K16ac levels did not lead to significant differences in distances between fluorescence in situ hybridization (FISH) probes at several genomic loci. Collectively, these studies support our observation that H4K16ac does not have a key role regulating global chromatin structure. Chromatin compaction is known to be regulated by several other mechanisms (e.g., nucleosome spacing and binding of linker histone H1 and HMG proteins) (Gibson et al., 2019; Postnikov and Bustin, 2016). Thus, further studies are warranted to determine if there is a redundancy between these mechanisms and H4K16ac.
Our results also suggest that KAT8 is more stable when associated with the NSL complex, which may have an impact in cancer biology. Specifically, we showed that KAT8-low cancer cells have diminished MSL complex function but retained activity of the NSL complex. These results may suggest that there is a “therapeutic window” for inhibition of KAT8 activity in KAT8-low tumors, as the remaining KAT8 in these cells is associated with the essential NSL complex.
In summary, this study uncovers a dichotomy of KAT8 functions depending on the associated proteins. As part of the NSL complex, KAT8 binds to a subset of active gene promoters, where it stimulates initiation of transcription, potentially via H4K5ac and H4K8ac. As part of the MSL complex, KAT8 catalyzes the majority of H4K16ac at euchromatic regions, but loss of this function does not affect cell proliferation or chromatin accessibility in vivo.
Limitations
Our results suggest that the NSL complex is essential because of its role in activating a set of housekeeping genes. However, it should be noted that the complex also has been reported to regulate mitochondrial respiration (Chatterjee et al., 2016), nuclear lamina integrity (Karoutas et al., 2019), and p53 activity (Li et al., 2009). Thus, the phenotypic effects induced by NSL complex loss may to some degree be attributed to defects in non-transcriptional NSL complex functions.
We also propose that H4K16ac does not regulate chromatin compaction in vivo. It should be noted, that although MSL1 KO causes 70%–80% loss of H4K16ac, we cannot exclude that remaining H4K16ac is sufficient to maintain wild-type patterns of chromatin accessibility.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kristian Helin (helink@mskcc.org).
Materials availability
Plasmids and cell lines generated in this study will be made available upon request.
Data and code availability
Next-generation sequencing data were deposited to GEO and can be accessed as follows: ATAC-sequencing (GSE157583), RNA-sequencing (GSE158521), ChIP-sequencing (GSE158736). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaíno et al., 2013) with the dataset identifiers: PXD022267, PXD021943, PXD023865. Uncropped western blots are available at Mendeley (https://doi.org/10.17632/v7mbmfxxy5.1). GitHub project with the RNA-sequencing analysis code is available at: https://github.com/VGrinev/TranscriptomicFeatures.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293FT packaging cells (female, RRID: CVCL_6911), HEK293 (female, RRID: CVCL_0045), HeLa (female, RRID: CVCL_0030) and U2OS (female, RRID: CVCL_0042) cells were cultured in DMEM, high glucose, GlutaMAX supplement, sodium pyruvate medium (Thermo Fisher SCIENTIFIC) containing 10% heat-inactivated FBS (HyClone) and 1x Penicillin/Streptomycin (Thermo Fisher SCIENTIFIC). THP-1 (male, RRID: CVCL_0006), K562 (female, RRID: CVCL_0004), MOLM13 (male, RRID: CVCL_2119), Caki1 (male, RRID: CVCL_0234), SKHEP1 (male, RRID: CVCL_0525), SKMEL28 (male, RRID: CVCL_0526) and SKMEL5 (female, RRID: CVCL_0527) cells were cultured in RPMI 1640, GlutaMAX supplement, sodium pyruvate medium (Thermo Fisher SCIENTIFIC) containing 10% heat-inactivated FBS (HyClone) and 1x Penicillin/Streptomycin (Thermo Fisher SCIENTIFIC). Mouse MLL-AF9 secondary leukemia cells (female) were cultured in RPMI 1640, GlutaMAX supplement, sodium puryvate medium (Thermo Fisher SCIENTIFIC) containing 20% heat-inactivated FBS (HyClone), 1x Penicillin/Streptomycin (Thermo Fisher SCIENTIFIC) and 20% of culture media supernatant from the IL-3-secreting cell line (homemade). E14 mouse ES cells (male, RRID: CVCL_C320) were cultured in the serum/LIF medium containing Glasgow Minimum Essential Media (Sigma), 10% heat-inactivated FBS (HyClone), 1x Penicillin/Streptomycin (Thermo Fisher SCIENTIFIC), 2 mM GlutaMAX (Thermo Fisher SCIENTIFIC), 100 μM β-Mercaptoethanol (Thermo Fisher SCIENTIFIC), 1 mM sodium pyruvate (Thermo Fisher SCIENTIFIC), 0.1 mM MEM Non-Essential Amino Acids Solution (Thermo Fisher SCIENTIFIC) and Leukemia Inhibitory Factor (homemade). Where indicated the cells were selected using the following antibiotic concentrations: puromycin 2 ug/ml, hygromycin 200 ug/ml, blasticidin 10ug/ml. dTAG-13 was used at a final concentration of 500 nM. PMA was used at a final concentration of 100 nM. All the cell lines were cultured at 37°C. All the cell lines were routinely tested for mycoplasma, but were not authenticated.
METHOD DETAILS
Plasmid and sgRNA cloning
The U6-sgRNA-SFFV-puro-P2A-EGFP vector was generated by substituting Cas9 open reading frame with a puromycin resistance cassette in the pL-CRISPR.SFFV.GFP plasmid (Addgene cat. no 57827). For sgRNA cloning into pU6-sgRNA-EF1α-puro-T2A-BFP, oligos were annealed in annealing buffer (200 mM potassium acetate, 60 mM HEPES-KOH pH 7.4, 4 mM magnesium acetate) and ligated into BstXI+BlpI (NEB) digested pU6-sgRNA-EF1α-puro-T2A-BFP. For sgRNA cloning into U6-sgRNA-SFFV-puro-P2A-EGFP, the oligos were phosphorylated by T4 PNK (NEB) and annealed in the T4 ligation buffer (NEB). The oligos and plasmid mixture was then subjected to digestion by BsmBI (NEB) and ligation by T4 ligase (NEB) (4 cycles of 42°C – 5 min and 16°C – 5 min, inactivation 55°C – 15 min). 3xFLAG-KANSL2, 3x-FLAG-KANSL3 and 3xFLAG-KAT8 open reading frames were expressed using the pLenti PGK Hygro plasmid. The FKBPF36V cassette was amplified from pCRIS-PITChv2-BSD-dTAG (BRD4) plasmid (Addgene cat. no 91792). The eSpCas9(1.1)-T2A-mCherry plasmid used for degron knock-in generation.
sgRNA design
CRISPRko sgRNAs were designed using the sgRNA Designer: CRISPRko – Broad Institute (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). CRISPRi sgRNAs were designed as previously (Radzisheuskaya et al., 2016). Table S7 contains sequences of all the sgRNAs used in this study.
Generation of the KANSL3 degron knock-in cell line
The targeting construct was assembled from the PCR products or synthetic DNA blocks (IDT) using the In-Fusion cloning kit (Takara) and contained the following elements: 500 bp left homology arm, blasticidin resistance gene, P2A, 2x HA tags, FKBPF36V, 3x GGGGS linkers, 500 bp right homology arm. The complete sequence can be found in the supplementary methods. The targeting construct and the eSpCas9(1.1)-T2A-mCherry plasmid with the sgRNA targeting Kansl3 start codon were co-transfected into the wild-type ES cells using Lipofectamine 3000 (Thermo Fisher SCIENTIFIC) according to the manufacturer’s protocol. The transfected cells were selected with blasticidin, single cell sorted to obtain clonal cell lines and screened for correct bi-allelic integration. The sequence of the sgRNAs used for knock-in generation can be found in Table S7.
Flow cytometry
Flow cytometry was performed using a BD LSR II flow cytometer, BD FACSAria III Cell Sorter and Beckman Coulter CytoFlex.
Virus production and lentiviral transduction
HEK293FT cells were co-transfected with the construct of interest and pAX8 and pCMV-VSV using a standard calcium phosphate protocol. The viral supernatant was collected 72 hours after HEK293FT transfection and used for transduction. Leukemia cells were transduced using a RetroNectin Bound Virus Infection Method (TaKaRa) according to the manufacturer’s instructions. Transduction was performed in presence of polybrene at 8 μg/ml. Antibiotic selection was added 24 hours after transduction.
RNA extraction, cDNA synthesis and qRT-PCR analysis
Total RNA was extracted using RNeasy Plus Mini Kit (QIAGEN) in accordance with the manufacturer’s protocol. One microgram of total RNA was subjected to reverse transcription using Transcriptor Universal cDNA Master (Roche). Quantitative PCR with reverse transcription (qRT-PCR) reactions were set up in triplicate using LightCycler 480 SYBR Green I Master (Roche) or PowerUp SYBR Green Master Mix (Thermo Fisher SCIENTIFIC) and primers listed in Table S7. qRT-PCR experiments were performed on a LightCycler 480 Instrument II (Roche) or Quant Studio 6 FLEX Real-Time PCR (Thermo Fisher Scientific) system. Relative quantitation was performed to a housekeeping gene either using a standard curve or a delta delta CT method, as indicated in the corresponding figure legends.
Growth curves and competition assays
For the growth curve experiments, cells were plated at 500,000 per well in a 6-well plate. The cells were counted every 72 hours and 500,000 cells used for replating. For competition assays, cells were collected 2 days after the addition of puromycin (3 days posttransduction) and mixed with untransduced cells at approximately 8:1 [transduced to untransduced cells]. The percentage of BFP+ or GFP+ (transduced) cells was then recorded on a flow cytometer at the specified time points.
RNA-seq
RNA was extracted using a QIAGEN RNeasy Kit. Libraries were prepared using an Illumina TruSeq v2 kit and samples sequenced on a NextSeq 500.
RNA-seq data analysis
We used 11 samples for RNA-seq analysis (3 replicates of cells transduced with the non-targeting sgRNA or sgRNAs against KANSL3 and KAT8 and 2 replicates with the sgRNA against KANSL2, the third KANSL2 replicate was a strong outlier in the set and was excluded from the analysis).
A comprehensive RNA-seq data quality assessment and pre-processing of the raw data was performed using the R/Bioconductor package ShortRead v.1.36.1 (Morgan et al., 2009) and the standard R infrastructure. GRCh38/hg38 reference assembly of the human genome was downloaded as a twoBit file from the FTP server of the UCSC Genome Browser. It was then converted to a standard FASTA format with twoBitToFa utility (Speir et al., 2016). A hash table for the reference genome was built with the function buildindex from the R/Bioconductor package Rsubread v.1.32.2 (Liao et al., 2019). At this step, 16-mers subreads were extracted in every 3 bases from the reference genome and the threshold 24 was used to exclude highly repetitive subreads from the created hash table.
A global alignment of RNA-seq reads against the reference genome was carried out with the function subjunc from the R/Bioconductor package Rsubread v.1.32.2 (Liao et al., 2019) and the above created hash table. This function implements a seed-and-vote mapping paradigm for a fast and accurate alignment (Liao et al., 2013). At this step, we used the default settings of the function subjunc and collected only uniquely mapped reads with the maximum of 3 mismatched bases in the alignment. The resulting BAM files were sorted with the function sortBam and indexed with the function indexBam, both from the R/Bioconductor package Rsamtools v.2.3.3 (Morgan et al., 2016).
For each gene, the genomic coordinates of exons were retrieved from Ensembl annotations (Ensembl release 85, GRCh38.p7 reference assembly of human genome) (Zerbino et al., 2018). These coordinates were intersected and joined into overlapping groups called exon clusters. The exon clusters shorter than 10 nucleotides were removed, and the final list of genomic intervals was sorted, assigned with gene information and converted into an object of a class GRanges. We used the function featureCounts from the R/Bioconductor package Rsubread v.1.32.2 (Liao et al., 2013; 2019; 2014) to assign the mapped RNA-seq reads to the exon clusters and summarize to the genomic meta-features (genes). Each read was counted in an unstranded mode with the minimum 1 base overlapping an exon cluster, and only uniquely mapped reads were collected for subsequence calculations.
To identify differentially expressed genes with DESeq2, the mapped RNA-seq reads were assigned to the genomic meta-features (genes) as described above. Next, the resulting count matrix was subjected to filtering against too low sequencing depth and it was wrapped (together with the sample information) into a DESeq DataSet object (Love et al., 2018). Then, differentially expressed genes were identified using the functions DESeq and results from the R/Bioconductor package DESeq2 v.1.20.0 (Love et al., 2018). These functions were run in a default mode and according to the standard DESeq2 pipeline (Anders et al., 2013; Love et al., 2018). Finally, the results were parsed, and genes with the q-value below 0.01 were annotated as differentially expressed.
To identify differentially expressed genes with edgeR/limma, the mapped RNA-seq reads were assigned to the genomic meta-features as described above. Second, the resulting count matrix was subjected to filtering against too low sequencing depth and it was wrapped (together with the sample information) into a DGEList object (Robinson et al., 2010). Third, to calculate an effective size of each RNA-seq library, the scaling factors were estimated using the “trimmed mean of M-values” method (Ritchie et al., 2015). Fourth, by applying the calculated scaling factors, the count data were converted into counts per million (CPM) and logarithmically transformed, the mean-variance relationship was estimated, and the appropriate observational-level weights were calculated using the voom algorithm (Law et al., 2014). Fifth, the multiple simple linear models were fitted to the normalized count matrix by least-squares method using the function lmFit from the R/Bioconductor package limma v.3.42.2 (Ritchie et al., 2015). Sixth, fold changes (log2 FC) and empirical Bayes statistics were calculated using respective functions from R/Bioconductor package edgeR v.3.28.0 (Robinson et al., 2010) and limma v.3.42.2 (Ritchie et al., 2015). Finally, results were parsed and genes with the q-value below 0.01 were annotated as differentially expressed.
To perform RNA-seq clustering analysis, the mapped RNA-seq reads were summarized as described above. Then, the resulting count matrix was normalized, logarithmically transformed and standardized (by scaling and centering of data). The optimal number of sample clusters was determined by comparing the outcomes from the with-in-sum-of-squares, “silhouette” and gap statistics methods. Non-hierarchical clustering of samples was performed using the method of k-means. For better visualization of cluster composition and proximity, the conventional principal component analysis was used.
Enrichment tests
We used up-to-date ODO and GAF files from the Gene Ontology Consortium (Ashburner et al., 2000) to develop a comprehensive list of the reference functional gene sets. As above, from this list, we selected the gene sets containing ten or more members for downstream analysis. Next, two-sided Fisher’s exact test was used to identify the under- and/or over-represented query gene set(-s) among the reference gene sets. Query results were parsed and under- or over-represented gene sets that passed members size > = 10 and FDR adjusted p-value threshold of 0.05 were collected. If indicated, differential gene expression was determined before gene enrichment analysis. According to the sign of log2 FC, all differentially expressed genes were grouped into up- and downregulated and significantly enriched gene sets were inferred by enrichment test separately for each group. Finally, Cytoscape plug-in EnrichmentMap19 was used to handle gene-set redundancy and hierarchical visualization of the enrichment results. In enrichment map, nodes represent significantly enriched gene sets, node size is proportional to the number of members in a gene set and color intensity depends on the q-value. Edges indicate gene overlap between the nodes and the thickness of edges is equivalent to the degree of gene overlap between the nodes. Functionally related gene sets were clustered and named.
Chromatin immunoprecipitation (ChIP) and sequencing
The cells were fixed with 1% formaldehyde for 10 min, after which the reaction was quenched by the addition of glycine to the final concentration of 0.125 M. The fixed cells were washed with PBS and resuspended in SDS buffer (100 mM NaCl, 50 mM Tris-HCl pH 8.0, 5 mM EDTA, 0.5% SDS, 1x protease inhibitor cocktail from Roche). The resulting nuclei were spun down, resuspended in the immunoprecipitation buffer at 1 mL per 16 million cells (SDS buffer and Triton Dilution buffer (100 mM NaCl, 100 mM TrisHCl pH 8.0, 5 mM EDTA, 5% Triton X-100) mixed in 2:1 ratio with the addition of 1x protease inhibitor cocktail from Roche) and processed on a Bioruptor Plus Sonicator (Diagenode) to achieve an average fragment length of 200-300 bps. Chromatin concentrations were estimated using Bradford (Bio-Rad) or BCA assays (Thermo Fisher SCIENTIFIC) according to manufacturer’s protocols. The immunoprecipitation reactions were set up in 1 mL of the immunoprecipitation buffer as indicated below and incubated overnight at 4°C. The next day, BSA-blocked Fast-flow Protein G Sepharose beads (GE Healthcare) or Protein G Dynabeads (Thermo Fisher SCIENTIFIC) were added to the reactions and incubated for 3 hours at 4°C. The beads were then washed three times with low-salt washing buffer (150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA, 20 mM TrisHCl pH8.0) and two times with high-salt washing buffer (500 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA, 20 mM TrisHCl pH8.0). The samples were then reverse-crosslinked overnight in the elution buffer (1% SDS, 0.1 M NaHCO3) and purified using QIAQuick PCR purification kit (QIAGEN). The following chromatin/antibody amounts were used: KANSL3 ChIP – 250ug/5ug, H3K4me3 ChIP – 5ug/2ul, H4K16ac ChIP – 10ug/2ug, H4K5ac ChIP – 20ug/2ug, H4K8ac ChIP – 20ug/2ug, PolII ChIP – 300ug/2ul, KAT8 ChIP – 700ug/3ug, HA ChIP – 300ug/2ul, TAF1 ChIP – 100ug/1ug. Inhibitors used in the ChIP immunoprecipitation buffer where specified in the figure legend: sodium butyrate (5 mM) and trichostatin A (250 nM). ChIP-seq libraries were prepared using NEBNext® Ultra II DNA Library Prep Kit for Illumina® according to manufacturer’s protocol and samples were sequenced on a NextSeq 500. All the primers used for ChIP-qPCR are in Table S7. For histone acetylation ChIP-seq, 5% of the crosslinked Drosophila chromatin (homemade) was added prior to immunoprecipitation step. For all H4K16ac ChIP-seq reactions, normalization factors were calculated as follows: a) the number of the unique human reads was divided by the number of all the Drosophila reads for each sample including inputs; b) the obtained ratios for each sample were then divided by the corresponding ratios of the inputs. The resulting normalization factors are available at the GEO database together with the raw and processed ChIP-seq data (see Data Availability). Since no global changes in levels of H4K5ac and H4K8ac were observed after depletion of NSL complex members, spike-in normalization was not used for analysis of these modifications.
MNase assay
30 million cells were washed with PBS and resuspended in 3 mL of buffer containing PBS, 1x protease inhibitor cocktail (Roche) and 0.2% Triton X-100 and divided into the required number of aliquots. The samples were rotated for 10 min at 4°C and spun down for 5 min at 4°C and 3300 g. The resulting pellets were resuspended in 50 ul of digestion buffer (10 mM HEPES pH 7.6, 100 mM NaCl, 1.5 mM MgCl2, 0.5 mM EDTA, 10% v/v glycerol, 1 mM DTT, 2 mM CaCl2, 100 ug/ml BSA, 0.2 ul MNase from NEB) and incubated at 37°C for various times (30 s, 1 min, 2 min, 5 min, 15 min, 30 min). Reactions were stopped by the addition of EDTA to the final concentration of 10 mM. The samples were purified using QIAQuick PCR purification kit (QIAGEN) and run on an agarose gel.
ATAC-seq
ATAC-seq was performed using the Omni-ATAC protocol (Corces et al., 2017). 50,000 cells per condition were washed in PBS, resuspended in 50 ul of the ice-cold ATAC-RSB buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2) with the addition of 0.1% NP-40, 0.1% Tween-20 and 0.01% Digitonin and incubated on ice for 3 min. Then, 1 mL of ice-cold ATAC-RSB buffer with the addition of 0.1% Tween-20 was added to each sample and gently mixed by inverting the tubes. The nuclei were then pelleted at 500 g for 10 min at 4°C, resuspended in 50 ul of the transposition mix (25ul 2x TD buffer from Illumina, 2.5 ul Tn5 from Illumina, 16.5 ul PBS, 0.5 ul 1% Digitonin, 0.5 ul 10% Tween-20, 5 ul water) and incubated for 30 min at 37°C with shaking. DNA fragments were isolated using QIAQuick PCR purification kit (QIAGEN), subjected to PCR amplification, size selection and sequencing using NextSeq 500.
ATAC- and ChIP-seq data analysis
ATAC and ChIP sequencing reads were 3′ trimmed and filtered for quality and adaptor content using TrimGalore (v0.4.5), with a quality setting of 15, and running version 1.15 of cutadapt and version 0.11.5 of FastQC. Reads were aligned to human assembly hg38 or mouse assembly mm10 with version 2.3.4.1 of bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) and were deduplicated using MarkDuplicates in Picard Tools (v2.16.0). For KANSL3 and KAT8 ChIP-seq analysis, to ascertain enriched regions, MACS2 (https://github.com/macs3-project/MACS) was used with a p-value setting of 0.001 with KANSL3 KD cells as background controls or input, respectively. The BEDTools suite (https://bedtools.readthedocs.io) was used to create normalized read density profiles, using a read extension of 200 bp for ChIP samples and 0 bp for ATAC samples. Subsequent ChIP-seq data visualization and analysis were performed using EaSeq software (Lerdrup et al., 2016). To analyze H4K16ac distribution across different chromatin states, Chromatin State Segmentation by HMM data for K562 cells from ENCODE/Broad (Ernst and Kellis, 2010; Ernst et al., 2011) was downloaded from the UCSC Genome Browser and converted to the GRCh38/hg38 genome assembly using the liftOver tool available through the UCSC Genome Browser. Motif signatures were obtained using Homer v4.5 (http://homer.ucsd.edu).
For ATAC-seq, a global peak atlas was created by first removing blacklisted regions (http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg38-human/hg38.blacklist.bed.gz) then merging all peaks within 500 bp and counting reads with version 1.6.1 of featureCounts (http://subread.sourceforge.net). Comparison of intra versus inter-group clustering in principle component analysis was used to determine normalization strategy, using either the median ratio method of DESeq2 or a sequencing depth-based factor normalized to ten million uniquely mapped fragments. Differential enrichment was scored using DESeq2 for all pairwise group contrasts. Peak-gene associations were created using linear genomic distance to transcription start site.
Histone PTM analysis
Histones were extracted from 4 million cells as described (Sidoli et al., 2016). TCA precipitation was substituted with desalting using disposable SEC PD-10 columns (GE healthcare, 17085101), and the samples were subsequently concentrated by speedvac to 100 ul. Histones were quantified by BCA assay (each sample produced 20-30 ug), and 10 ug were digested and derivatized in 30 ul of buffer, as previously described (Maile et al., 2015). Histones were desalted using 3 mg of HLB material (Waters), as recommended by the manufacturer. The samples were analyzed using DIA strategy with high resolution orbitrap MS1 and low resolution ion trap MS2 scans (Sidoli et al., 2015) on Orbitrap Velos coupled to nanoAcquity UPLC system (Waters). Peptides (600 ng per injection) were directly loaded on 30 cm long column with 150 uM inner diameter packed with intertsil 2 um sorbent using flashPack method (Kovalchuk et al., 2019). The flow rate was kept at 1 ul/min and column temperature was 30°C during the analysis. The aqueous mobile phase A was 0.1% formic acid, the organic mobile phase B was acetonitrile with 0.1% formic acid. The gradient was 4%–25% B in 49 min, 25%–40% in 13 minutes, 40%–80% in 4 minutes. Peptides were electrosprayed through etched emitters (Kelly et al., 2006). MS1 scans were performed at 15K resolution in Orbitrap analyzer, MS2 peptides were isolated in ten 80 Da windows and fragmented using CID with 35NCE. To improve peak definitions, every scan was acquired with two microscans. The results were analyzed by epiProfile (Yuan et al., 2015), the code was modified to specify phenyl isocyanate (119.037114) as N-terminal modification.
NSL complex interactomics analysis
For each of the four replicates, 10 million cells were lysed in 50 mM EPPS, pH 7.4,150 mM NaCl, 1 mM EDTA, 1% Triton X-100, complete protease inhibitors and 25 U/mL of Benzonase. The lysate was kept on ice for 5 minutes to allow DNA digestion and cleared by centrifugation at 20.000 g for 5 minutes and filtering through acropep advance 96-well 2 ml, 1 μm glass fiber filter plate. Immunopreciptation was performed for 1 hour at 4°C with ANTI-FLAG® M2 Affinity Gel (Sigma, A2220) in Eppendorf Deepwell Plates 96 with shaking at 1300 rpm. After immunoprecipitation, the beads were washed five times in wash buffer (50 mM EPPS, pH 7.4, 150 mM NaCl) and subjected to trypsin digestion for two hours at 37°C with 15 μl of 10 ng/μl LysC and 20 ng/μl Trypsin in 10 mM EPPS pH 8.5 on Orochem OF1100 plates. The digest was labeled with 4 μl 20 g/L TMTPro tags, as recommended by the manufacturer, and the material was fractionated by Pierce High pH Reversed-Phase Peptide Fractionation Kit concatenating 2 fractions into a superfraction (e.g., 1 and 5). After fractionation, the samples were evaporated using vacuum centrifuge, resuspended in 20 μl 0.1% TFA and 4.5 μl was analyzed by Waters nanoAcquity M Class UPLC on 2 μm particle size, 75 μm x 500 mm easyspray column in direct injection mode. The samples were separated using the following gradient of buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in acetonitrile): 0%–7% in 5 min, 7%–30% in 90 min, 30%–50% in 20 min. Eluting peptides were analyzed on Orbitrap Fusion Lumos instrument using MS3 SPS method with the settings recommended by the instrument manufacturer with the following modifications: 1. CID NCE for MS2 was set at 32; 2. HCD NCE for MS3 was set at 45; 3. C series exclusion was disabled, since TMTPro reagent was not enabled in C-series exclusion. Data were analyzed in Proteome Discoverer 2.4 software. A database search was performed with Sequest HT search engine using Homo sapiens UniProt database containing only reviewed entries and canonical isoforms (retrieved on 14/06/2019). Oxidation (M) was set as a variable modification, while TMTPro was set as fixed modification. A maximum of two missed cleavages were permitted. The precursor and fragment mass tolerances were 10 ppm and 0.6 Da, respectively. PSMs were validated by percolator with a 0.01 posterior error probability (PEP) threshold. Only PSMs with isolation interference < 25% and at least 5 MS2 fragments selected for MS3 matched to peptide sequence were considered. The quantification results of peptide spectrum matches were combined into protein-level quantitation using MSstatsTMT R package (Choi et al., 2014) with at least 2 peptides per protein.
Global proteomics profiling
Data acquisition and analysis were performed as previously described (Radzisheuskaya et al., 2019) with sample pre-fractionated into 30 high pH fractions. We used TMT11-plex for sample multiplexing (3 replicates of the cells transduced with NegCtrl sgRNA, 3 replicates with sgRNAs against KANSL2 and KAT8 and 2 replicates with the sgRNA against KANSL3, total 11 samples).
Checking antibody specificity by ELISA using the histone peptide array library
Streptavidin-coated plates (Thermo Fisher SCIENTIFIC) were rinsed three times with wash buffer (0.1% BSA, 0.05% Tween-20 in PBS), and 50 ul containing 20 ng of biotinylated histone tail peptide (Table S3, tab 1) was added to individual wells and incubated for two hours at room temperature with shaking (1000 rpm). Each well was washed three times with wash buffer, and 50 ul of primary antibody (1: 10,000 in wash buffer) added and incubated for one hour at room temperature with shaking (1000 rpm). Wells were washed three times with wash buffer, and 50 ul of peroxidase-labeled anti-rabbit IgG secondary antibody (Vector Laboratories; 1: 5,000 in wash buffer) was added to each well and incubated for 30 min at room temperature with shaking (1000 rpm). Wells were washed three times with wash buffer and once with water. 50 ul of the TMB ELISA substrate was added per well, left for 10-30 min (until bright color was observed) and 50 ul of H2SO4 added to each well to stop the reaction. Results were read using a spectrophotometer at 450 nm.
Luminex
To examine antibody specificity in a nucleosome context by Luminex, we assembled a diversity set to mirror the related DNA-barcoded SNAP-ChIP K-AcylStat panel (EpiCypher #19-3001) (Table S3, tab 2). In brief, designer nucleosomes [a PTM-defined human histone octamer assembled on 147 bp 5′ biotinylated 601 DNA; EpiCypher] were individually coupled to saturation on different MagPlex avidin bead regions (Luminex), multiplexed at equivalence (1000 beads/region/well) in flat black-bottom 96-well plates (GreinerBio; VWR 89089-582), and used to query the binding specificity of each antibody at three different concentrations (1:250, 1:1000, 1:4000). 50 μl of each antibody dilution in TB_NaCl (25mM Tris pH 7.5, 0.1% Tween 20; 0.1% BSA, 200mM NaCl) was added per well, the plate covered in a dark seal, and incubated for one hour at room temperature with shaking (800 rpm). Beads were then captured on a plate-base magnet, the liquid in each well discarded, and the magnet removed. 100 μl of ChIP buffer #2 (25mM Tris pH 7.5, 5mM MgCl2, 300mM KCl, 10% Glycerol, 0.1% NP-40) was added per well, the plate covered in a dark seal, and incubated for two minutes at room temperature with shaking (800 rpm). Beads were then captured and successively washed as above with ChIP buffer #3 (25mM Tris pH 7.5, 240mM LiCl, 1mM EDTA, 0.5% sodium deoxycholate, 0.5% NP-40), ChIP buffer #1 (25mM Tris pH 7.5, 5mM MgCl2, 100mM KCl, 10% Glycerol, 0.1% NP-40) and TB-NaCl. Beads were then resuspended in 100 ul anti-rabbit IgG*PE (1:1000 in TB-NaCl), the plate covered in a dark seal, incubated for 30 minutes at room temperature with shaking (800 rpm), and successively washed three times with TB (25mM Tris pH 7.5, 0.1% Tween 20; 0.1% BSA). Beads were then resuspended in 50 ul TE, read on a FlexMap-3D (Luminex), and data presented as % on-target signal for each antibody.
Annexin V staining
To assess cell death, 150,000 cells per replicate were washed in AnnexinV binding buffer (BD) and incubated for 30 min with FITC-Annexin V dye (BD). After two more washing steps, stained cells were resuspended in Annexin V binding buffer containing 10 ng/ml DAPI and analyzed using flow cytometry. Early and late apoptotic cells were determined by single Annexin V positivity or double positivity with AnnexinV and DAPI, respectively.
Cell cycle analysis
20,000 ES cells were seeded in 6 well plates and treated for 4 days with 500 nM dTAG-13 or DMSO. To label cells undergoing DNA synthesis, 20 uM EdU was added for 10 min before harvest. 500,000 sgRNA-expressing THP-1/cdCas9-KRAB cells were seeded in triplicates the day before and labeled with 10 uM EdU for 60 min before harvest.
For both ES and THP-1 cells, 250,000 cells per replicate were fixed for 15 min with Cytofix/Cytoperm solution (BD), followed by washes with PBS and Perm/Wash buffer (BD). EdU labeling reaction was performed for 30 min in the dark in 150 ul reaction buffer (2 mM CuSO4, 20 mg/ml L-ascorbic acid, 8 uM Cy5-azide in PBS). After 3 washes, cells were resuspended in Perm/wash buffer containing 1ug/ml DAPI and incubated for 1 hour before acquisition on a flow cytometer.
HiC analysis
HiC data for THP-1 cells was generated as a part of another project (unpublished). HiC was performed using the Arima HiC Kit according to manufacturer’s protocol. The samples were sequenced using Novaseq. The HiC data were processed with HiC-Pro version 2.11.0 (Servant et al., 2015). Replicates were pooled to obtain 431 million validate interactions in wild-type THP-1 cells. Iterative correction (ICE) (Imakaev et al., 2012) was applied to the pooled validate interactions using HiC-Pro with default settings. Then the samples were further normalized together to the smallest read using HiCExplorer (3.4.3) (Ramírez et al., 2018). The final HiC matrices were binned at 25 kb and the median counts per bin is ~2700 in the range of ~2600 – 2850. A/B compartments were then computed using hicPCA function provided by HiCExplorer (3.4.3) with ‘lieberman’ algorithm (Lieberman-Aiden et al., 2009) and the signs were corrected by the gene density.
For the integrated representation of the complex ChIP-Seq and HiC data, an implementation of Circos plot in R/Bioconductor package circlize v.0.4.8 was used (Krzywinski et al., 2009).
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of the experiments can be found in the figure legends and the method details. Boxplot summary: outliers (points), minimum (lower whisker), first quartile (lower bound of box), median (horizontal line inside box), third quartile (upper bound of box), interquartile range (box), and maximum (upper whisker).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-KANSL2 | Sigma | Cat# HPA038497, RRID: AB_10674685 |
| Anti-KANSL3 | Sigma | Cat# HPA035018, RRID: AB_10601763 |
| Anti-KAT8 | Abcam | Cat# ab200660 |
| Anti-H4K5ac (#1) | Abcam | Cat# ab51997, RRID: AB_2264109 |
| Anti-H4K5ac (#2) | Abcam | Cat# ab114146 |
| Anti-H4K5ac (#3) | Diagenode | Cat# C1541005 |
| Anti-H4K16ac (#1) | Abcam | Cat# ab109463, RRID: AB_10858987 |
| Anti-H4K16ac (#2) | Millipore | Cat# 07-329, RRID: AB_310525 |
| Anti-H4K8ac | Abcam | Cat# ab45166, RRID: AB_732937 |
| Anti-Pol II | Cell Signaling | Cat# 14958, RRID: AB_2687876 |
| Anti-H3K4me3 | Cell Signaling | Cat# 9751, RRID: AB_2616028 |
| Anti-H3 | Abcam | Cat# ab24834, RRID: AB_470335 |
| Anti-H4 | Abcam | Cat# ab17036, RRID: AB_1209245 |
| Anti-HA | Cell Signaling | Cat# 3724S, RRID: AB_1549585 |
| Anti-FLAG | Sigma | Cat# F1804, RRID: AB_262044 |
| Anti-Vinculin | Sigma | Cat# SAB4200080, RRID: AB_10604160 |
| Anti-GAPDH | Abcam | Cat# ab181602, RRID: AB_2630358 |
| Anti-b-Actin | Abcam | Cat# ab6276, RRID: AB_2223210 |
| Anti-MSL2 | Cell Signaling | Cat# 44006, RRID: AB_2799256 |
| Anti-TAF1 | Cell Signaling | Cat# 12781, RRID: AB_2798025 |
| Goat Anti-Rabbit IgG Antibody (H+L), Peroxidase | Vector Laboratories | Cat# PI-1000, RRID: AB_2336198 |
| Horse Anti-Mouse IgG Antibody (H+L), Peroxidase | Vector Laboratories | Cat# PI-2000, RRID: AB_2336177 |
| IRDye® 800CW Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 925-32211, RRID: AB_2651127 |
| IRDye® 680RD Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926-68070, RRID: AB_10956588 |
| Bacterial and Virus Strains | ||
| DH5alpha Chemically Competent Cells | Lab prep | N/A |
| Stbl3 Chemically Competent Cells | Lab prep | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Agencourt Ampure XP beads | Beckman Coulter | A63881 |
| LIF | Lab prep | N/A |
| Polybrene | Fisher | NC9200896 |
| RetroNectin | Fisher | 50444032 |
| Protein G Sepharose 4 Fast Flow | GE Healthcare | 17061802 |
| Protein G Dynabeads | Thermo Fisher Scientific | 10004D |
| ANTI-FLAG® M2 Affinity Gel | Sigma | A2220 |
| TMTpro™ 16plex Label Reagent Set | Thermo Fisher Scientific | A44520 |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent | Thermo Fisher Scientific | A34808 |
| EdU (5-ethynyl-2'-deoxyuridine) | Lumiprobe | 10540 |
| Sulfo-Cyanine5 azide | Lumiprobe | A3330 |
| FITC Annexin V | BD | 556420 |
| Digitonin | Promega | G9441 |
| Critical Commercial Assays | ||
| SNAP-ChIP K-AcylStat panel | EpiCypher | 193001 |
| Illumina Tagment DNA Enzyme and Buffer | Illumina | 20034197 |
| NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® | NEB | E7645L |
| Illumina TruSeq v2 kit | Illumina | RS1222001 |
| Deposited Data | ||
| Raw and analyzed RNA-seq data | GEO | GSE158521 |
| Raw and analyzed ChIP-seq data | GEO | GSE158736 |
| Raw and analyzed ATAC-seq data | GEO | GSE157583 |
| Uncropped western blots | Mendeley | DOI: 10.17632/v7mbmfxxy5.1 |
| Raw and analyzed proteomics data | ProteomeXchange | PXD022267, PXD021943, PXD023865 |
| Experimental Models: Cell Lines | ||
| HEK293FT | Kristian Helin lab | RRID: CVCL_6911 |
| HEK293 | Kristian Helin lab | RRID: CVCL_0045 |
| HeLa | Xuejun Jiang Lab | RRID: CVCL_0030 |
| U2OS | Xuejun Jiang Lab | RRID: CVCL_0042 |
| THP-1 | Kristian Helin lab | RRID: CVCL_0006 |
| K562 | Kristian Helin lab | RRID: CVCL_0004 |
| MOLM13 | Kristian Helin lab | RRID: CVCL_2119 |
| Caki1 | MSKCC cell line core | RRID: CVCL_0234 |
| SKHEP1 | MSKCC cell line core | RRID: CVCL_0525 |
| SKMEL28 | Jedd Wolchok lab | RRID: CVCL_0526 |
| SKMEL5 | MSKCC cell line core | RRID: CVCL_0527 |
| Mouse MLL-AF9 secondary leukemia cells | Lab prep | N/A |
| E14 mouse embryonic stem cells | Joshua Brickman lab | RRID: CVCL_C320 |
| See Table S7 for primer and sgRNA sequences | ||
| Recombinant DNA | ||
| pLentiCas9-blast | Addgene | 60955 |
| pU6-sgRNA-EF1α-puro-T2A-BFP | Addgene | 52962 |
| pHR-SFFV-KRAB-dCas9-2A-CHERRY | Addgene | 60954 |
| pLenti-PGK-Hygro dest | Addgene | 19066 |
| U6-sgRNA-SFFV-puro-P2A-EGFP | This study | N/A |
| eSpCas9(1.1)-T2A-mCherry | Ian Chambers lab | N/A |
| pCRIS-PITChv2-BSD-dTAG (BRD4) | Addgene | 91792 |
| pUC19_FKBPF36V-KANSL3-targeting | This study | N/A |
| Software and Algorithms | ||
| sgRNA Designer: CRISPRko | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design | N/A |
| CRISPRi sgRNA design | Radzisheuskaya et al., 2016 | N/A |
| Image Studio Lite software | LI-COR Biosciences | N/A |
| R/Bioconductor package ShortRead v.1.36.1 | Morgan et al., 2009 | N/A |
| twoBitToFa utility | Speir et al., 2016 | N/A |
| R/Bioconductor package Rsubread v.1.32.2 | Liao et al., 2019 | N/A |
| R/Bioconductor package Rsamtools v.2.3.3 | Morgan et al., 2016 | N/A |
| R/Bioconductor package DESeq2 v.1.20.0 | Love et al., 2018 | N/A |
| R/Bioconductor package limma v.3.42.2 | Ritchie et al., 2015 | N/A |
| R/Bioconductor package edgeR v.3.28.0 | Robinson et al., 2010 | N/A |
| bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | N/A |
| MACS2 | https://github.com/taoliu/MACS | N/A |
| BEDTools suite | http://bedtools.readthedocs.io | N/A |
| EaSeq | Lerdrup et al., 2016 | N/A |
| Homer v4.5 | http://homer.ucsd.edu | N/A |
| HiC-Pro version 2.11.0 | Servant et al., 2015 | N/A |
| HiCExplorer 3.4.3 | Ramírez et al., 2018 | N/A |
| R/Bioconductor package circlize v.0.4.8 | Krzywinski et al., 2009 | N/A |
Highlights.
The NSL but not the MSL complex is essential for the proliferation of human cells
The NSL complex places H4K5ac and H4K8ac at TSSs, promoting transcription initiation
H4K16ac is a highly abundant modification placed by KAT8 as part of the MSL complex
H4K16ac marks open chromatin but does not affect global chromatin accessibility
ACKNOWLEDGMENTS
We thank members of the Helin laboratory for discussions; Robin Armstrong for critical reading of the manuscript; Sarah Teed, Abbas Nazir, Eugenia Lorenzini, Charlotte Holck, and Zhifan Yang for technical assistance; Zuo-Fei Yuan for epiProfile software support; Jedd Wolchok’s lab for SKMEL28 cells; Joshua Brickman’s lab for E14 mouse ESCs; and Xuejun Jiang’s lab for HeLa and U2OS cells. A.R. and D.S. were in part funded by the European Union’s Horizon 2020 research and innovation program under Marie Skłodowska-Curie grant agreements 659171 and 749362, respectively. The work in the Helin laboratory was supported by the Danish Cancer Society (R167-A10877), through a center grant from the Novo Nordisk Foundation (NNF) to the NNF Center for Stem Cell Biology (NNF17CC0027852), and through the Memorial Sloan Kettering Cancer Center Support Grant (NIH P30 CA008748). Studies in EpiCypher were funded by the NIH (R44 CA212733, R44 GM136172, R44 HG008907, and R44 GM116584). Experimental and computational proteomics work at the University of Southern Denmark (SDU) (P.V.S., V.G., S.K., and O.N.J.) was supported by the research infrastructure provided by the Danish National Mass Spectrometry Platform for Functional Proteomics (PRO-MS; 5072-00007B) and the VILLUM Center for Bioanalytical Sciences (7292). P.V.S. was supported by a postdoctoral fellowship from the Lundbeck Foundation (R231-2016-3093). S.K. was supported by a research grant from Independent Research Fund Denmark (to O.N.J., 4181-00172B). Research in the V.V.G. laboratory was supported in part by the Ministry of Education of the Republic of Belarus (grant 3.08.3 [469/54]).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.02.012.
DECLARATION OF INTERESTS
EpiCypher is a commercial developer and supplier of reagents (recombinant semi-synthetic modified nucleosomes) and the antibody characterization platforms used in this study. K.H. is a consultant for Inthera Bioscience AG and a scientific advisor for Hannibal Health Innovation.
REFERENCES
- Akhtar A, and Becker PB (2000). Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 5, 367–375. [DOI] [PubMed] [Google Scholar]
- Allahverdi A, Yang R, Korolev N, Fan Y, Davey CA, Liu C-F, and Nordenskiöld L (2011). The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 39, 1680–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anamika K, Krebs AR, Thompson J, Poch O, Devys D, and Tora L (2010). Lessons from genome-wide studies: an integrated definition of the coactivator function of histone acetyl transferases. Epigenetics Chromatin 3, 18, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, and Robinson MD (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- Armstrong RL, Penke TJR, Strahl BD, Matera AG, McKay DJ, MacAlpine DM, and Duronio RJ (2018). Chromatin conformation and transcriptional activity are permissive regulators of DNA replication initiation in Drosophila. Genome Res. 28, 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. ; The Gene Ontology Consortium (2000). Gene Ontology: tool for the unification of biology. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote JM, and Lucchesi JC (1980). Male-specific lethal mutations of Drosophila melanogaster. Genetics 96, 165–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Jin J, Swanson SK, Cole MD, Choi SH, Florens L, Washburn MP, Conaway JW, and Conaway RC (2010). Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J. Biol. Chem 285, 4268–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Hu Z, Liu J, Gao J, Tan M, Zhang D, Zhu L, Liu S, Hou R, and Lin B (2015). Expression of hMOF in different ovarian tissues and its effects on ovarian cancer prognosis. Oncol. Rep 33, 685–692. [DOI] [PubMed] [Google Scholar]
- Cao L, Zhu L, Yang J, Su J, Ni J, Du Y, Liu D, Wang Y, Wang F, Jin J, and Cai Y (2014). Correlation of low expression of hMOF with clinicopathological features of colorectal carcinoma, gastric cancer and renal cell carcinoma. Int. J. Oncol 44, 1207–1214. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Seyfferth J, Lucci J, Gilsbach R, Preissl S, Böttinger L, Mårtensson CU, Panhale A, Stehle T, Kretz O, et al. (2016). MOF acetyl transferase regulates transcription and respiration in mitochondria. Cell 167, 722–738.e23. [DOI] [PubMed] [Google Scholar]
- Chelmicki T, Dündar F, Turley MJ, Khanam T, Aktas T, Ramírez F, Gendrel A-V, Wright PR, Videm P, Backofen R, et al. (2014). MOF-associated complexes ensure stem cell identity and Xist repression. eLife 3, e02024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Chang C-Y, Clough T, Broudy D, Killeen T, MacLean B, and Vitek O (2014). MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. [DOI] [PubMed] [Google Scholar]
- Copur Ö, Gorchakov A, Finkl K, Kuroda MI, and Müller J (2018). Sex-specific phenotypes of histone H4 point mutants establish dosage compensation as the critical function of H4K16 acetylation in Drosophila. Proc. Natl. Acad. Sci. U S A 115, 13336–13341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2010). Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol 28, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller C, Prestel M, Hartmann H, Straub T, Söding J, and Becker PB (2012). The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Res. 40, 1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al. (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400. [DOI] [PubMed] [Google Scholar]
- Füllgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, and Joseph B (2013). The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500, 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaub A, Sheikh BN, Basilicata MF, Vincent M, Nizon M, Colson C, Bird MJ, Bradner JE, Thevenon J, Boutros M, and Akhtar A (2020). Evolutionary conserved NSL complex/BRD4 axis controls transcription activation via histone acetylation. Nat. Commun 11, 2243, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Guerin-Peyrou TG, Sharma GG, Park C, Agarwal M, Ganju RK, Pandita S, Choi K, Sukumar S, Pandita RK, et al. (2008). The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell. Biol 28, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker A, Hilfiker-Kleiner D, Pannuti A, and Lucchesi JC (1997). mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 16, 2054–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, and Tjian R (2000). Structure and function of a human TAFII250 double bromodomain module. Science 288, 1422–1425. [DOI] [PubMed] [Google Scholar]
- Jain D, Baldi S, Zabel A, Straub T, and Becker PB (2015). Active promoters give rise to false positive ‘phantom peaks’ in ChIP-seq experiments. Nucleic Acids Res. 43, 6959–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoutas A, Szymanski W, Rausch T, Guhathakurta S, Rog-Zielinska EA, Peyronnet R, Seyfferth J, Chen H-R, de Leeuw R, Herquel B, et al. (2019). The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nat. Cell Biol 21, 1248–1260. [DOI] [PubMed] [Google Scholar]
- Kelly RT, Page JS, Luo Q, Moore RJ, Orton DJ, Tang K, and Smith RD (2006). Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal. Chem. 78, 7796–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoa LTP, Tsan Y-C, Mao F, Kremer DM, Sajjakulnukit P, Zhang L, Zhou B, Tong X, Bhanu NV, Choudhary C, et al. (2020). Histone acetyltransferase MOF blocks acquisition of quiescence in ground-state ESCs through activating fatty acid oxidation. Cell Stem Cell 27, 441–458.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, and Horikoshi M (1998). Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells 3, 789–800. [DOI] [PubMed] [Google Scholar]
- Kovalchuk SI, Jensen ON, and Rogowska-Wrzesinska A (2019). FlashPack: fast and simple preparation of ultrahigh-performance capillary columns for LC-MS. Mol. Cell. Proteomics 18, 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KC, Mühlpfordt F, Vaquerizas JM, Raja SJ, Holz H, Luscombe NM, Manke T, and Akhtar A (2012). The NSL complex regulates housekeeping genes in Drosophila. PLoS Genet. 8, e1002736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KC, Chung H-R, Semplicio G, Iyer SS, Gaub A, Bhardwaj V, Holz H, Georgiev P, and Akhtar A (2019). The NSL complex-mediated nucleosome landscape is required to maintain transcription fidelity and suppression of transcription noise. Genes Dev. 33, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, and Kuroda MI (2011). X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, and Roeder RG (2013). H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152, 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerdrup M, Johansen JV, Agrawal-Singh S, and Hansen K (2016). An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat. Struct. Mol. Biol 23, 349–357. [DOI] [PubMed] [Google Scholar]
- Li X, Wu L, Corsa CAS, Kunkel S, and Dou Y (2009). Two mammalian MOF complexes regulate transcription activation by distinct mechanisms. Mol. Cell 36, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li L, Pandey R, Byun JS, Gardner K, Qin Z, and Dou Y (2012). The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell 11, 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu C, Yang Y, Fan Y, Yang R, Liu C-F, Korolev N, and Nordenskiöld L (2011). Influence of histone tails and H4 tail acetylations on nucleosome-nucleosome interactions. J. Mol. Biol 414, 749–764. [DOI] [PubMed] [Google Scholar]
- Liu N, Zhang R, Zhao X, Su J, Bian X, Ni J, Yue Y, Cai Y, and Jin J (2013). A potential diagnostic marker for ovarian cancer: Involvement of the histone acetyltransferase, human males absent on the first. Oncol. Lett 6, 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Anders S, and Huber W (2018). Differential gene expression analysis based on the negative binomial distribution. https://bioconductor.org/packages/release/bioc/html/DESeq2.html.
- Lucchesi JC, and Kuroda MI (2015). Dosage compensation in Drosophila. Cold Spring Harb. Perspect. Biol 7, a019398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maile TM, Izrael-Tomasevic A, Cheung T, Guler GD, Tindell C, Masselot A, Liang J, Zhao F, Trojer P, Classon M, and Arnott D (2015). Mass spectrometric quantification of histone post-translational modifications by a hybrid chemical labeling method. Mol. Cell. Proteomics 14, 1148–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, Vermeulen M, Buscaino A, Duncan K, Mueller J, et al. (2006). Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol. Cell 21, 811–823. [DOI] [PubMed] [Google Scholar]
- Morgan M, Anders S, Lawrence M, Aboyoun P, Pagès H, and Gentleman R (2009). ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25, 2607–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M, Pages H, Obenchain V, and Hayden N (2016). Rsamtools: binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. https://bioconductor.org/packages/release/bioc/html/Rsamtools.html.
- Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B, et al. (2018). SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister S, Rea S, Taipale M, Mendrzyk F, Straub B, Ittrich C, Thuerigen O, Sinn HP, Akhtar A, and Lichter P (2008). The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int. J. Cancer 122, 1207–1213. [DOI] [PubMed] [Google Scholar]
- Postnikov YV, and Bustin M (2016). Functional interplay between histone H1 and HMG proteins in chromatin. Biochim. Biophys. Acta 1859, 462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzisheuskaya A, Shlyueva D, Müller I, and Helin K (2016). Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res. 44, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzisheuskaya A, Shliaha PV, Grinev V, Lorenzini E, Kovalchuk S, Shlyueva D, Gorshkov V, Hendrickson RC, Jensen ON, and Helin K (2019). PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat. Struct. Mol. Biol 26, 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja SJ, Charapitsa I, Conrad T, Vaquerizas JM, Gebhardt P, Holz H, Kadlec J, Fraterman S, Luscombe NM, and Akhtar A (2010). The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol. Cell 38, 827–841. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, and Manke T (2018). High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun 9, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravens S, Fournier M, Ye T, Stierle M, Dembele D, Chavant V, and Tora L (2014). Mof-associated complexes have overlapping and unique roles in regulating pluripotency in embryonic stem cells and during differentiation. eLife 3, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PJJ, An W, Routh A, Martino F, Chapman L, Roeder RG, and Rhodes D (2008). 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J. Mol. Biol 381, 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, et al. (2010). MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol 30, 3582–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh BN, Bechtel-Walz W, Lucci J, Karpiuk O, Hild I, Hartleben B, Vornweg J, Helmstädter M, Sahyoun AH, Bhardwaj V, et al. (2016). MOF maintains transcriptional programs regulating cellular stress response. Oncogene 35, 2698–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh BN, Guhathakurta S, and Akhtar A (2019). The non-specific lethal (NSL) complex at the crossroads of transcriptional control and cellular homeostasis. EMBO Rep. 20, e47630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, and Peterson CL (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- Sidoli S, Simithy J, Karch KR, Kulej K, and Garcia BA (2015). Low resolution data-independent acquisition in an LTQ-orbitrap allows for simplified and fully untargeted analysis of histone modifications. Anal. Chem 87, 11448–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. J. Vis. Exp (111), 54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. (2016). The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 44 (D1), D717–D725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Fukunaga A, and Oishi K (1976). Studies on the sex-specific lethals of Drosophila melanogaster. II. Further studies on a male-specific lethal gene, maleless. Genetics 84, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GCA, Eskeland R, Hekimoglu-Balkan B, Pradeepa MM, and Bickmore WA (2013). H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 23, 2053–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Dixon MP, Kueh AJ, and Voss AK (2008). Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol. Cell. Biol 28, 5093–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno JA, Côté RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, et al. (2013). The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Han X, Guan J, and Li X (2014). Regulation and function of histone acetyltransferase MOF. Front. Med 8, 79–83. [DOI] [PubMed] [Google Scholar]
- Yin S, Jiang X, Jiang H, Gao Q, Wang F, Fan S, Khan T, Jabeen N, Khan M, Ali A, et al. (2017). Histone acetyltransferase KAT8 is essential for mouse oocyte development by regulating reactive oxygen species levels. Development 144, 2165–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z-F, Lin S, Molden RC, Cao X-J, Bhanu NV, Wang X, Sidoli S, Liu S, and Garcia BA (2015). EpiProfile quantifies histone peptides with modifications by extracting retention time and intensity in high-resolution mass spectra. Mol. Cell. Proteomics 14, 1696–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaware N, and Zhou M-M (2019). Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol 26, 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et al. (2018). Ensembl 2018. Nucleic Acids Res. 46 (D1), D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu H, Pan H, Yang Y, Huang G, Yang Y, Zhou W-P, and Pan Z-Y (2014). The histone acetyltransferase hMOF suppresses hepatocellular carcinoma growth. Biochem. Biophys. Res. Commun 452, 575–580. [DOI] [PubMed] [Google Scholar]
- Zhang R, Erler J, and Langowski J (2017). Histone acetylation regulates chromatin accessibility: role of H4K16 in inter-nucleosome interaction. Biophys. J 112, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lee D, Dhiman V, Jiang P, Xu J, McGillivray P, Yang H, Liu J, Meyerson W, Clarke D, et al. (2020). An integrative ENCODE resource for cancer genomics. Nat. Commun 11, 3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Su J, Wang F, Liu D, Ding J, Yang Y, Conaway JW, Conaway RC, Cao L, Wu D, et al. (2013). Crosstalk between NSL histone acetyltransferase and MLL/SET complexes: NSL complex functions in promoting histone H3K4 di-methylation activity by MLL/SET complexes. PLoS Genet. 9, e1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B-R, Feng H, Ghirlando R, Kato H, Gruschus J, and Bai Y (2012). Histone H4 K16Q mutation, an acetylation mimic, causes structural disorder of its N-terminal basic patch in the nucleosome. J. Mol. Biol 421, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next-generation sequencing data were deposited to GEO and can be accessed as follows: ATAC-sequencing (GSE157583), RNA-sequencing (GSE158521), ChIP-sequencing (GSE158736). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaíno et al., 2013) with the dataset identifiers: PXD022267, PXD021943, PXD023865. Uncropped western blots are available at Mendeley (https://doi.org/10.17632/v7mbmfxxy5.1). GitHub project with the RNA-sequencing analysis code is available at: https://github.com/VGrinev/TranscriptomicFeatures.