

Abstract
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is an ubiquitous DNA repair enzyme present in yeast, plants and animals. It removes a broad range of blocking lesions at the ends of DNA breaks. The catalytic core of TDP1 consists in a pair of conserved histidine-lysine-asparagine (HKN) motifs. Analysis of the human TDP1 (hTDP1) crystal structure reveals potential involvement of additional residues that shape the substrate binding site. In this biochemical study, we analyzed four such conserved residues, tyrosine 204 (Y204), phenylalanine 259 (F259), serine 400 (S400) and tryptophan 590 (W590). We show that the F259 residue of hTDP1 is critical for both 3′- and 5′-phosphodiesterase catalysis. We propose that the double π-π interactions of the F259 residue with the −2 and −3 nucleobases serve to position the nucleopeptide substrate in phase with the active site histidines of hTDP1. Mutating Y204 of hTDP1 to phenylalanine (Y204F), as in fly and yeast TDP1 enzymes, had minor impact on TDP1 activity. In constrast, we find that S400 enhances 3′-processing activity while it suppresses 5′-processing activity, thereby promoting specificity for 3′-substrates. W590 is selectively important for 5′-processing. These results reveal the impact of conserved amino acid residues that participate in defining the DNA binding groove around the dual HKN catalytic core motif of TDP1, and their differential roles in facilitating the 3′- vs 5′-end processing activities of hTDP1.
Keywords: DNA damage and repair, 3′-End blocking DNA lesions, 5′-End blocking DNA lesions, Topoisomerases, Camptothecin, Indenoisoquinoline, Oxidative DNA damage, Bleomycin
1. Introduction
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a DNA repair enzyme ubiquitously present in eukaryotes including yeast, plants and metazoans, which is capable of cleansing 3′-DNA blocking ends both in the nuclear and mitochondrial genomes [1–8]. For example, phosphotyrosyl linkages resulting from aborted topoisomerase I-DNA cleavage complexes (TOP1cc) are repaired by TDP1 [9,10]. Other blockages removed by TDP1 include 3′-phosphoglycolate ends generated by the anticancer drugs bleomycin, alkylating agents and oxygen radicals [11,12]. TDP1 also acts as nucleosidase, though less efficiently, by removing 3′-nucleoside [4], chain terminating anticancer and antiviral nucleosides (cytosine arabinoside, acyclovir, abacavir, AZT) [13–16]. Additionally, both human and yeast TDP1 enzymes are capable of excising the 5′-phosphotyrosyl bonds resulting from aborted topoisomerase II-DNA cleavage complexes (TOP2cc) [11,17], an activity normally displayed by the structurally and mechanistically unrelated TDP2 enzyme [2,18]. The yeast TDP1 has a more pronounced 5′-diesterase activity than human TDP1, compensating for the lack of yeast TDP2 orthologue [2,17,19]. The importance of TDP1 in resolving the TOP1-associated DNA lesions and 3′-blocking lesions has prompted the search for inhibitors particularly to be used in conjunction with TOP1 inhibitors and other anticancer agents that introduce DNA breaks [20].
The biological and medical importance of TDP1 was brought to light by the discovery of a functionally disruptive active site His493Arg mutation in patients with spinocerebellar ataxia with axonal neuropathy (SCAN1) [21]. A neurological phenotype has also been described in flies with genetic inactivation of TDP1 (Glaikit) [22] and in mice with double-inactivation of Tdp1 and Atm [23]. The neuroprotective role of TDP1 has been ascribed to the critical function of TDP1 in removing 3′-blocking lesions that form as a result of DNA oxidative lesions and abortive TOP1cc in neurons [6,7,22,23].
TDP1 catalyzes phosphodiester hydrolysis at 3′-ends of DNA at least 4-bases long [24], which is consistent with co-crystal structure of TDP1 with DNA [25]. TDP1 also efficiently acts on double-stranded substrates with blunt ends or gaps [13,26]. It catalyzes the hydrolysis of phosphotyrosyl linkages in cofactor-independent manner (no metal or nucleotide cofactor as energy source). Sequence analysis and subsequent evaluation of mutations at conserved residues have placed TDP1 within the phospholipase D superfamily of enzymes, pointing to the organization of its catalytic site and its molecular mechanism of action [27]. TDP1-catalyzed hydrolysis can be summarized in two phosphoryl transfer steps through two HKN motifs, which form the enzyme catalytic pocket: H263-K495-N516 and H493-K265-N283 [2,25,28] (see Figs. 1–5 of the current study). In the first step, H263 acts as a nucleophile attacking the 3′-phosphate, displacing the TOP1 tyrosine while forming a covalent TDP1-DNA adduct. In the second step, H493 of the second HKN motif performs base-catalyzed hydrolysis of the phosphoramide intermediate, releasing the competent TDP1 and free 3′-phosphate DNA [20]. This mechanism is substantiated by crystal structures of TDP1 [25,28] and accounts for the molecular mechanism of the H493R SCAN1 mutation [21,29].
Fig. 1.
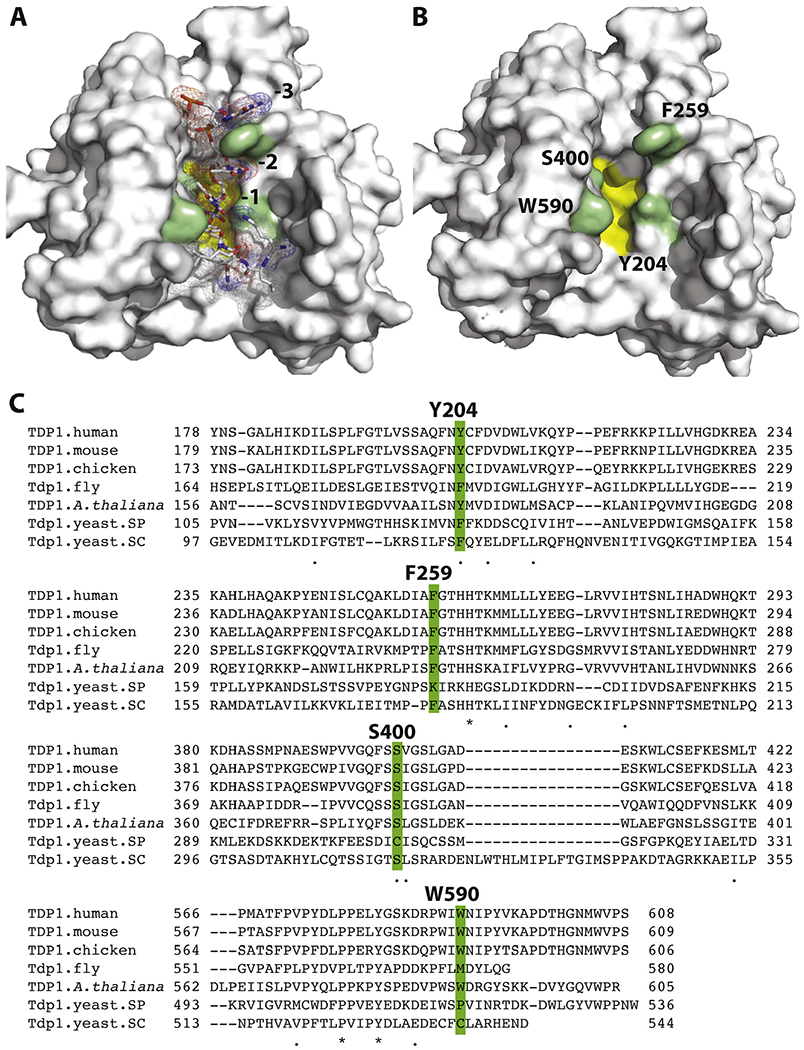
Structure of TDP1 (surface representation). A: with the substrate (sticks and mesh representation) (PDB ID 1NOP); B: with the substrate removed to expose the corresponding binding cavity.The catalytic HKN motifs are shown as yellow surface. Residues examined in this study and shaping the catalytic cavity are shown in green; C: sequence alignment highlighting the four residues examined in the present study, and high conservation of the Y204, F259 and S400 amino acid residues of TDP1.
Fig. 5.
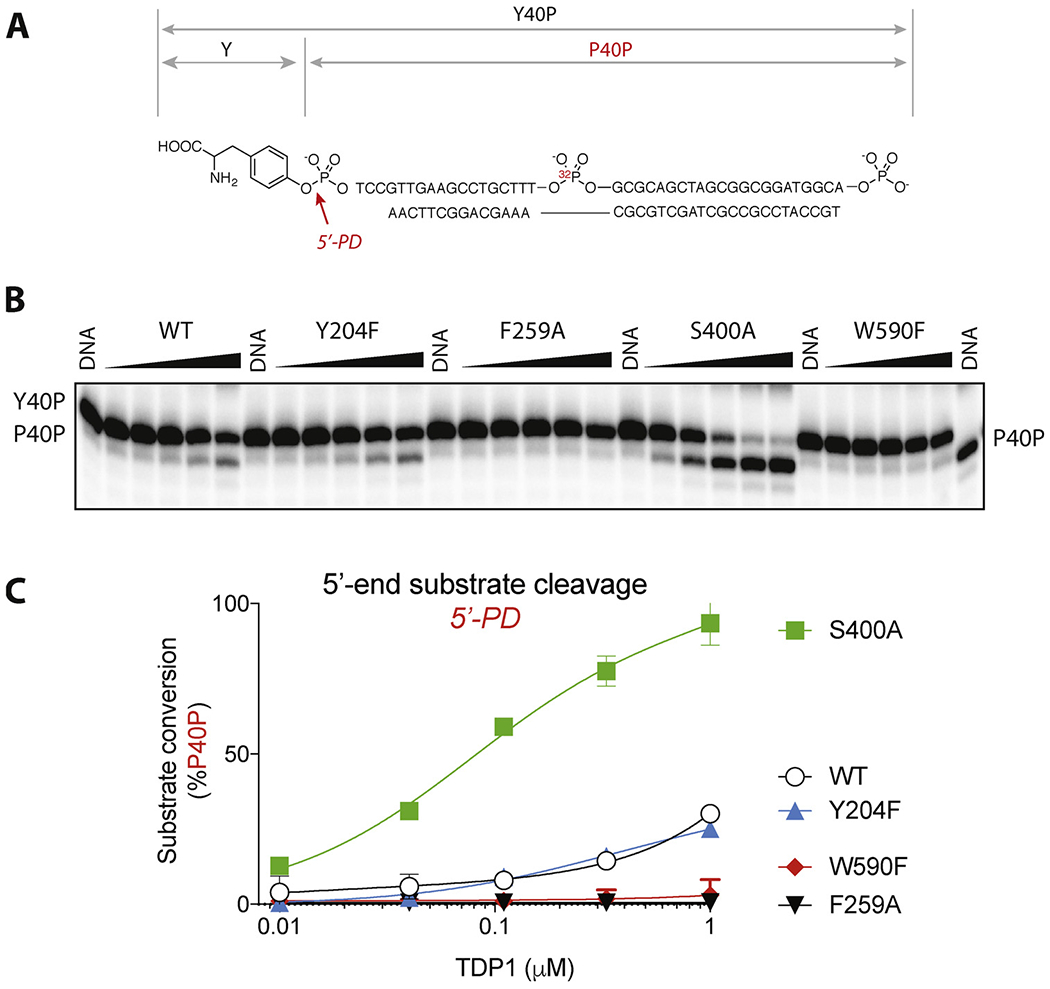
Processing of the 5′-tyrosyl substrate by hTDP1 WT, Y204F, F259A, S400A and W590F.A: Scheme of the hydrolyzable bond, structure and labeling of the 5′ substrate and product. B: Representative gel of the substrate processing. The substrate was incubated for 30 min at 25 °C with each TDP1 mutant in a series of three-fold dilutions, starting from the highest concentration of 1 μM. C: Quantification of the product formation by hTDP1 WT and the mutants. Each point on the graph represents mean (n = 3) ± standard deviation. Invisible bars for standard deviations are within symbol size.
In addition to elucidating the details of the catalytic site, the crystal structures of hTDP1 point to the potential roles of other conserved residues in substrate binding [25,28,30,31] (Fig. 1). Among those residues, phenyl-Cα dihedral of F259 changes from 64° (PDBID 1QZQ) to 108° (PDBID 1NOP) upon substrate binding; this rotation allows for phenyl group to stack with the nucleobases at −2 and −3 positions [25,30] (see Fig. 3D and E). Other potentially important residues revealed by the crystallography studies include S400, which participates in substrate binding in the proximity of the cleavage site by engaging the substrate phosphodiester groups, as well as Y204 and W590, which form a narrow passage separating the DNA and TOP1-derived peptide binding regions (Fig. 1A and B) [25]. All three residues are conserved across multiple species (Fig. 1C).
Fig. 3.
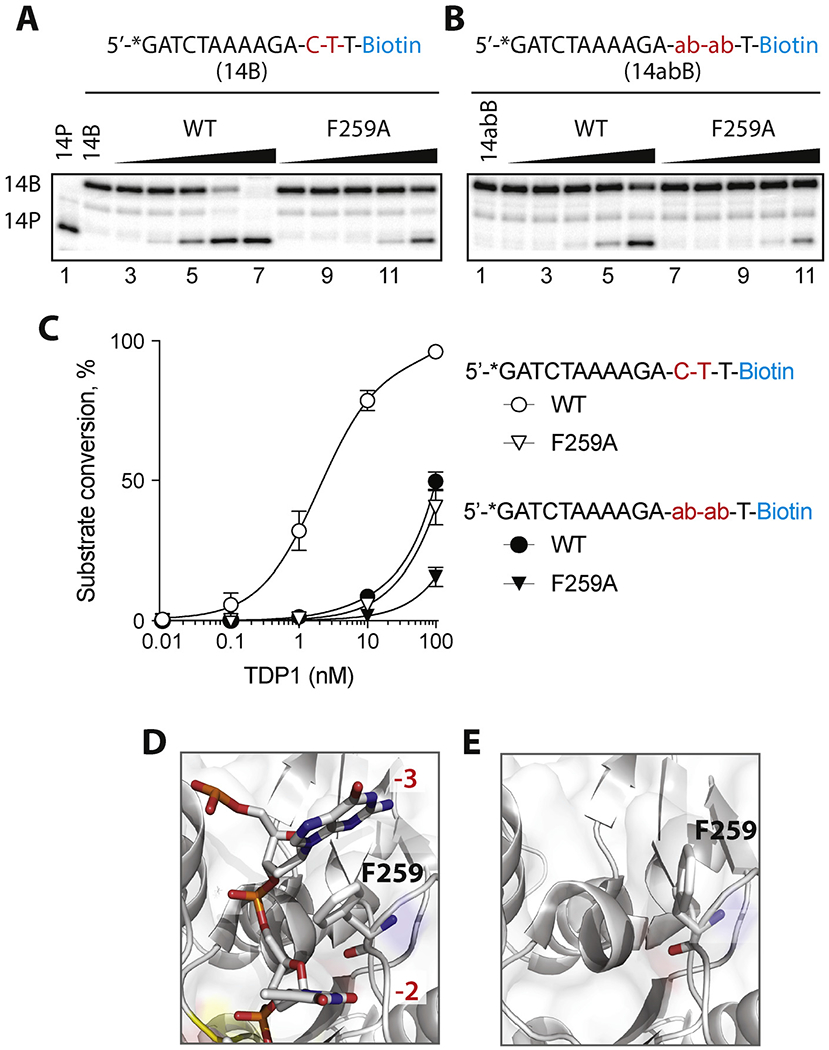
Enzymatic hydrolysis of 3′-phosphotyrosyl substrates containing abasic site at positions −2 and −3 by hTDP1 WT and F259A. A-B: Schematic representation of the substrates above representative gels showing hydrolysis of normal substrate (14B) (A) and substrate with abasic sites at −2 and −3 positions (14abB) (B);C: Quantification of substrate processing by the WT and F259A TDP1 enzyme. Each point on the graph represents the mean (n = 3) ± standard deviation. D-E: Structure of TDP1 (grey surface) in the vicinity of F259 (green surface) in presence (PDBID: 1NOP) and absence (PDBID: 1QZQ) of DNA oligonucleotide (wire mesh).
In this study, we analyze the impact of the above-mentioned four residues that shape the substrate binding site: Y204, F259, S400 and W590. We examined the biochemical activities of each hTDP1 mutant in an attempt to discern their importance and role in the cleavage of different substrates with 3′- and 5′-blocking ends. This work represents a biochemical approach to probe the enzymatic function of previously untested conserved TDP1 amino acid residues and the mechanisms of substrate recognition and conversion. Our conclusions regarding the mechanistic role of the F259 residue are consistent with an independent study [31] published while ours was under review.
2. Materials and methods
Oligomeric DNA substrates were obtained from Midland Certified Reagent Co. (Midland, TX): 14B, 5′-GATCTAAAAGACTT-pBiotin-3′; 14abB, 5′-GATCTAAAAGA-ab-ab-T-pBiotin-3′, where ‘ab’ is abasic site; 14Y, 5′-GATCTAAAAGACTT-pY-3′; Y18dA, 5′-Yp-TCCGTTGAAGCCTGCTTT-3′dA-3′. The 5′-phosphotyrosine substrate DNA with internal 32P label (Y40P, 5′-Yp-TCCGTTGAAGCCTGCTTT-32P-GCGCAGCTAGCGGCGGATGGCA) was prepared as previously reported [11] and annealed to complementary oligonucleotide TGCCATCCGCCGCTAGCTGCGCAAAGCAGGCTTCAA allowing for 4 nucleotide overhang on the 5′-end of the labeled strand, Y40P was used as a double-stranded substrate [11]. TDP1 mutants were expressed in BL21 cells and purified as described [13,32].
TDP1 cleavage reactions were conducted as previously described [11,20]. 5′-32P-labeled DNA substrate (1 nM) was incubated with TDP1 (five 10-fold dilutions from 100 nm to 10 pM for reactions with N14 B and N14abB; nine 2-fold dilutions from 2 nM to 8 pM for reactions with 14Y; and five 3-fold dilutions from 1 μM to 10 nM for reactions with Y18dA and Y40P) for 15 min (for reactions with 14B, 14abB, 14Y) or 30 min (for reactions with Y18dA and Y40P) at room temperature in a buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2mM EDTA, 1 mM DTT, 40 μg/ml BSA and 0.01% Tween-20. Reactions were terminated by the addition of 1 vol of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE and gels were exposed after drying to a PhosphoImager screen (GE Healthcare). Gel images were scanned using a Typhoon 9500 (GE Healthcare) and densitometry analyses were performed using the ImageQuant software (GE Healthcare).
3. Results
3.1. Impact of residues Y204, F259, S400 and W590 on TDP1 3′-phosphodiesterase activity
We selected four residues Y204, F259, S400 and W590 based on previous structural studies which showed their contribution in shaping the walls of the substrate binding site (Fig. 1A and B) and their conservation across species (Fig. 1C). Y204 is conserved as tyrosine or phenylalanine across all species; F259 and S400 are highly conserved except in yeast saccharomyces cerevisiae, and the W590 residue is conserved in vertebrates and plants (Fig. 1C).
Based on the published TDP1 co-crystal structures [25,28], we posited that the Y204F mutation would eliminate the possibility of hydrogen bond formation between the substrate −1 nucleobase and the Y204 tyrosine hydroxyl group, and that mutating the hTDP1 tryptophan 590 residue to phenylalanine (W590F) would broaden the channel between the peptide and DNA portions of the substrate binding site, allowing for greater mobility and flexibility of the substrate at the site of cleavage (Fig. 1B and see Fig. 6). Enzymatic assays with 3′-phosphotyrosyl DNA were performed to test the impact of these residues on substrate conversion.
Fig. 6.
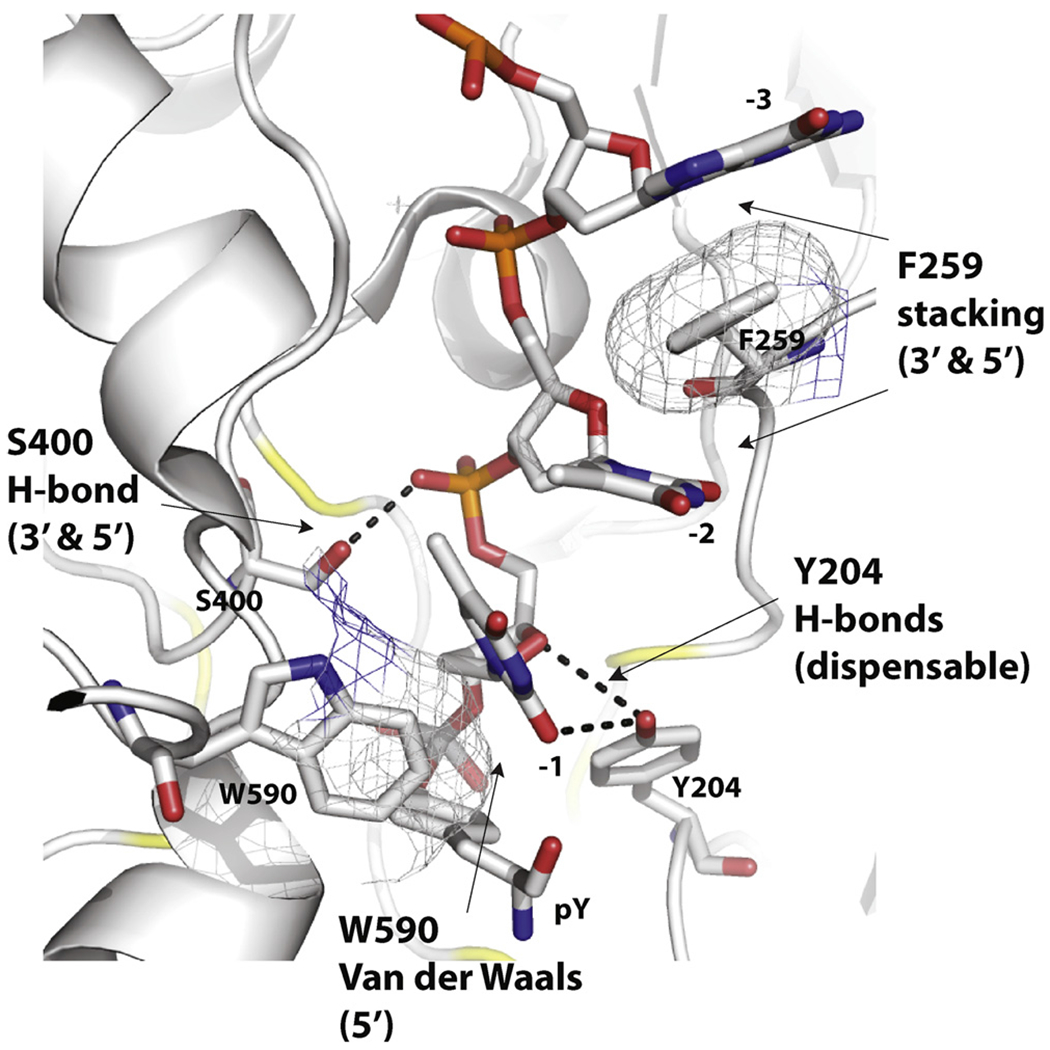
Summary of the atomic interactions and functional importance for the four residues examined in the present study: Y204, F259, S400 and W590. Specific interactions and role on 3′- vs. 5′-processing are noted in parenthesis for each residue (see text for details).
TDP1-catalyzed 3′-end processing was not affected by either the Y204F or W590F mutations (Fig. 2), indicating that any potential interactions formed by Y204 with the substrate nucleobases are dispensable, and spatial restriction placed by bulky W590 residue is not a necessary prerequisite for efficient 3′-phosphodiesterase catalysis. These results indicate that despite conservation (Fig. 1C), mutating Y204 and W590 to phenylalanine has limited or no impact on 3′-end substrate recognition and binding. This observation is in line with the fact that TDP1 is highly efficient at removing chemically diverse lesions from 3′-phosphate ends [4,11,13,33].
Fig. 2.
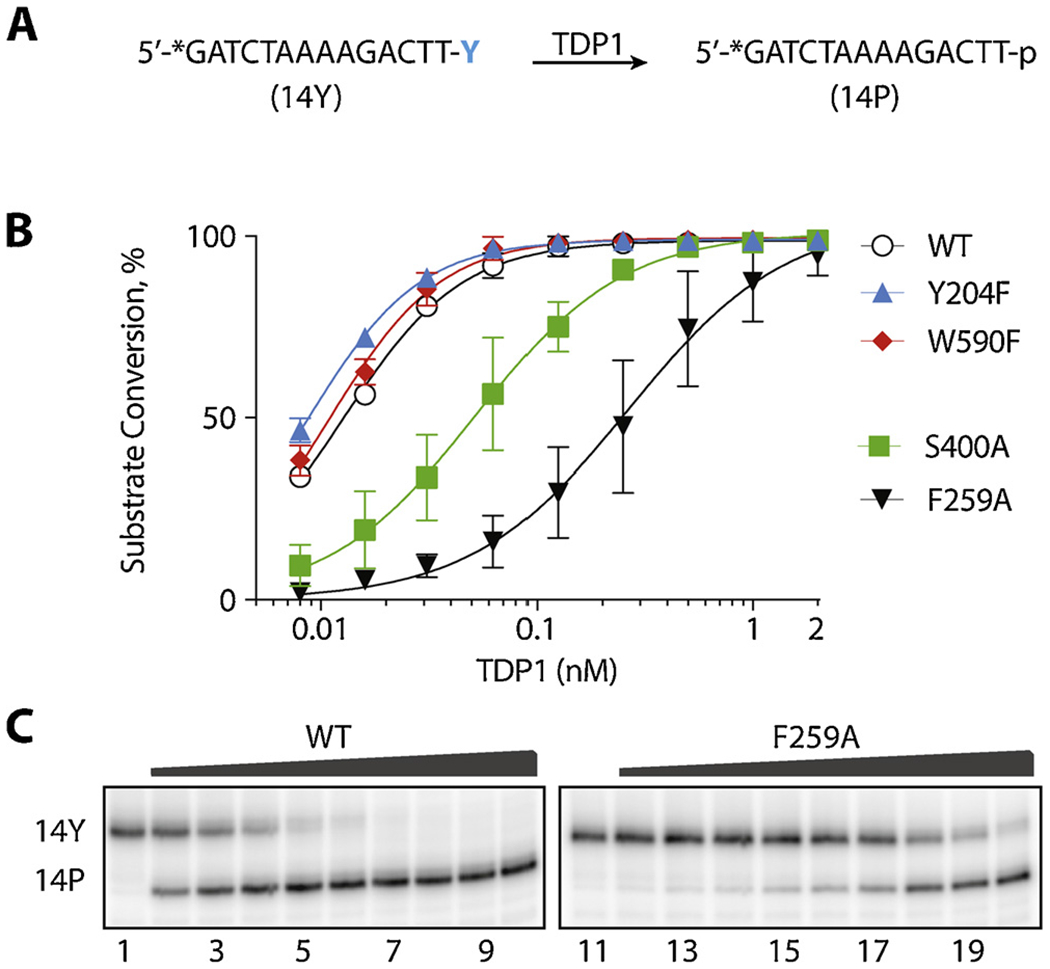
Enzymatic hydrolysis of 3′-phosphotyrosyl substrates by hTDP1. A: Schematic representation of substrate (14Y) conversion into product (14P). B: Quantification of the processing of the 3′-tyrosyl substrate by hTDP1 WT, Y204F, F259A, S400A, and W590F. Each point on the graph represents the mean (n = 3) ± standard deviation. C: Representative gel showing hydrolysis of 14Y by TDP1 F259A mutant.
The aromatic side chain of F259 is relatively distant (circa 13 Å) from the pair of HKN catalytic sites of TDP1 (highlighted in yellow in Fig. 1A and B) [25], but is conserved across species (Fig. 1C). As revealed in the crystal structure of TDP1 in complex with the substrate GTT segment (PDB ID 1NOP) [25], the F259 aromatic ring is sandwiched between the −3 and −2 nucleobases of the substrate (Figs. Figure 1A and Figure 3D). These interactions pack tightly against each other as all the atoms of the F259 aromatic ring are within 5 Å of the atoms of the −3 and −2 nucleobases. As shown in Figs. Figure 1A and Figure 3D, the −3 and −2 nucleotides clamp around the F259 side chain and form a π-π stacking network. On the other hand, in the TDP1 structure without substrate (PDB ID 1QZQ) [28], the aromatic ring of the F259 residue rotates to minimize the exposed hydrophobic area (Fig. 3E). To determine whether TDP1 catalytic competency could be adversely affected by compromising the π-π stacking between F259 and the −3 and −2 nucleobases, we tested the activity of TDP1 after replacing the F259 aromatic ring with an alanine residue (F259A mutant). Fig. 2 (panels A & C) shows that the catalytic efficiency of hTDP1 is negatively affected by the F259A mutation as over 10-fold higher concentration of the F259A mutant enzyme was required to produce the same amount of 3′-tyrosyl substrate hydrolisys as WT. Together with a parallel independent study [31], these results demonstrate the importance of the F259 residue for optimum 3′-processing activity.
The fourth residue tested here is the conserved S400 residue, which is placed deeper in the DNA binding groove (Fig. 1B), interacting with the phosphodiester group bridging the −1 and −2 nucleobases (Fig. 1A) [25]. Fig. 2 shows that mutating S400 to alanine (S400A) led to a decrease in 3′-hydrolysis, reflecting the importance of this residue, as suggested by crystallographic studies of TDP1 [25] and conservation of S400 among different organisms (see Fig. 1C).
Together, these experiments show that, in the context of 3′-end DNA processing, the catalytic competency of hTDP1 is highly dependent on the residues responsible in holding the DNA portion of the substrate in place, i.e. F259 and S400. It is worth noting that the interactions formed by F259 and S400 do not entail any sequence specificity; S400 interacts with the phosphate of the DNA backbone and F259 stacks against nucleobases without forming any other interactions to specific nucleobase features. By contrast, residues Y204 and W590, which shape the substrate channel around the 3′-phosphotyrosyl junction, do not act as critical determinants for the 3′-processing activity of hTDP1.
3.2. Critical role of DNA stacking interactions of the F259 residue for positioning the DNA substrate in the catalyic site of TDP1
To further establish the importance of the π-π stacking of F259 between the −3 and −2 bases of the substrate (Fig. 3D) [31], we introduced two abasic sites at positions −3 and −2 of the 3′-tyrosyl substrate (Fig. 3B). This alteration, which eliminates π-π stacking of the F259 residue, decreased the catalytic activity of hTDP1 almost as much as mutating F259 of hTDP1 to alanine (F259A) in the context of the normal DNA substrate (Fig. 3A and C). Introducing the abasic site-containing substrate in addition to the F259A mutation slightly further reduced the catalytic hydrolysis (Fig. 3C). It should also be noted that no additional cleavage by TDP1 at the abasic sites was observed, contrary to a previous report [34].
These experiments demonstrate the critical role F259 plays in positioning the DNA substrate in the TDP1 catalytic cleft through π–π interactions with the −2 and −3 bases of the substrate. They also highlight the conformational change in the side chain of F259 observed in crystallographic studies, in which the F259 side chain was observed to rotate to minimize the exposed hydrophobic area in the crystal structure without DNA substrate [28] (Fig. 3, compare panels D & E).
3.3. Selective impact of residues Y204, S400 and W590 on the 5′-phosphodiesterase activity of hTDP1
In addition to cleansing 3′-end blockages, TDP1 is capable of hydrolyzing 5′-phosphotyrosyl conjugates arising from stalled TOP2cc [11,17]. To assess the 5′-phosphodiesterase activity of our four TDP1 mutants, we first performed experiments with a previously described DNA substrate [11] containing a 5′-phosphotyrosyl group in addition to 3′-end linked to the 3′-adenosine analog, cordycepin, Y18dA (Fig. 4A). This substrate has the advantage of allowing simultaneous assessment of 3′- and 5′-phosphodiesterase activities of TDP1 (Fig. 4B and C). The previously reported [11] cleavage scheme of such substrate (5′-Y-p-18nt-p-3′dA = Y18dA) taking into account both 3′- and 5′-end processing abilities is shown in Fig. 4B. Hydrolysis of the 3′-cordycepin linkage results in a 3′-phosphate product, Y18P, and cleavage of the 5′-tyrosine yields the 5′-phosphate, P18P product. Products Y18P or P18dA migrate similarly on sequencing gels [11] and appear as a merged band P18dA/Y18P (Fig. 4C). Subsequent hydrolysis produces the same oligonucleotide product with phosphates at both DNA ends (P18P). Hence, both TDP1 3′- and 5′-phosphodiesterase activities (3′-PD and 5′-PD; Fig. 4A) are required for the generation of P18P.
Fig. 4.
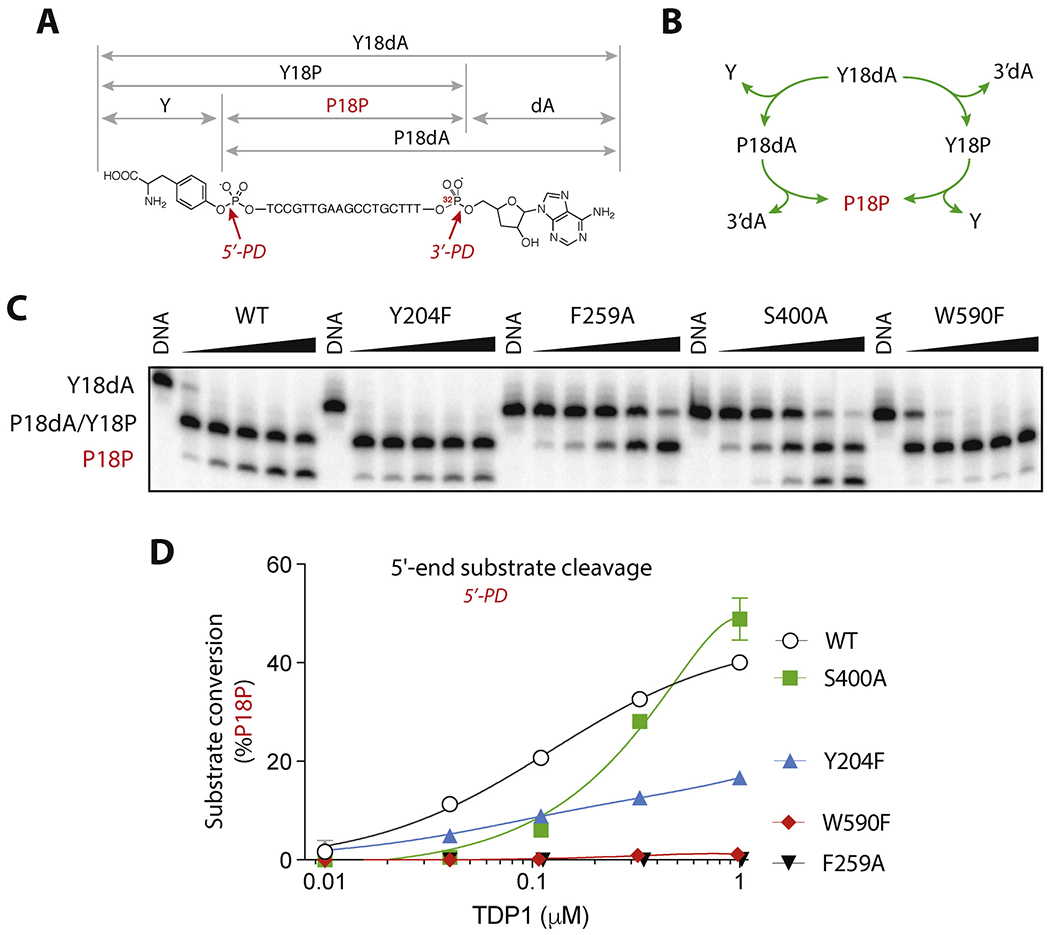
Processing of the 5′-tyrosyl/3′-cordycepin substrate by hTDP1 WT, Y204F, F259A, S400A, and W590F.A: Scheme of the hydrolyzable bonds, structure and labeling convention for the dual substrate and products. B: Stepwise mechanism of the 5′- and 3′-phophodiesterase (PD) activities of TDP1. Products P18dA and Y18P have indistinguishable electrophoretic migrations and appear in the same band on the gel (see panel C). C: Representative gel of the dual substrate processing. The substrate was incubated for 30 min at 25 °C with each TDP1 mutant in a series of three-fold dilutions, starting from the highest concentration of 1 μM. D: Quantification of the 5′-product formation by hTDP1 WT and the mutants, each point on the graph represents the mean (n = 3) ± standard deviation. Invisible bars for standard deviations are within symbol size.
Formation of the doubly hydrolyzed product, P18P was used to assess the 5′-end processing ability of our 4 mutant hTDP1 enzymes [11]. The representative experiment shown in Fig. 4C demonstrates that the F259A mutant is unable to catalyze 5′-hydrolysis, and therefore catalytically deficient for both 3′- and 5′-DNA phosphodiesterase activities (Fig. 4C). The partial deficiency of the S400A mutant (Fig. 4C and D) is consistent with its defective 3′-processing activity (see Fig. 2B).
Mutations of the two residues (Y204F and W590F) forming the groove of the substrate binding site next to the phosphotyrosyl residue (see Fig. 1A and B) gave notable results. The Y204F mutant despite being somewhat deficient in generating the P18P product (Fig. 4C and D) was nethetheless capable of efficiently clearing the starting substrate (Y18dA) (Fig. 4C and D). Mutation of W590 to phenylalanine while inconsequential for 3′-processing (see Fig. 2B) abolished processing of the 5′-end by TDP1 (Fig. 4C and D). These results show the importance of interactions beyond the known HKN motifs and DNA binding groove for selective processing of 5′-tyrosyl-DNA substrates by TDP1.
To unambiguously delineate the 3′- from 5′-phosphodiesterase activities of the mutant hTDP1, experiments were conducted with an internally-labeled 5′-phosphotyrosyl substrate (Y40P) containing 3′-phosphate end, which is resistant to TDP1 end-processing activity [33] [11] (Fig. 5A). Such substrate is subject to 5′-phosphodiesterase activity only and produces only one product upon 5′-tyrosine cleavage (P40P) [11].
Consistent with conclusions made from the experiments with the Y18dA experiments (see Fig. 4), WT and Y204F TDP1 hydroziled Y40P to the single P40P product by removing tyrosine from the 5′-end (Fig. 5B). Also, incubation of F259A and W590F with Y40P did not yield any product (Fig. 5B and C), once again demonstrating the lack of 5′-phosphodiesterase activity of the W590F and F259A mutants.
Notably, S400A TDP1 was capable hydrolyzing the single 5′-substrate Y40P to a much greater extent then the WT, completely processing the entire pool of substrate at highest concentrations of S400A TDP1 tested (Fig. 5B and C). These results demonstrate a previously unanticipated role of the S400 residue in selectively promoting the 3′-phosphodiesterase activity of hTDP1 over its 5′-phosphodiesterase activity.
4. Discussion
By mutating conserved residues that shape the substrate binding groove of human TDP1, we reveals the importance of specific interactions between TDP1 and its DNA substrates beyond the two canonical HKN catalytic motifs (see Fig. 1A and B). The interactions of the four residues probed in the present study, Y204, F259, S400 and W590 are detailed and annotated in Fig. 6. Notably, none of the interactions involve the nucleobases, which is consistent with the broad range of activity of TDP1 for different substrates [1–4,17,24].
Here, we propose that the π-π interactions between the phenylalanine residue F259 and the −3 and −2 nucleotides act as a locking mechanism to pair the TDP1 catalytic histidines with the substrates for both 3′- and 5′-processing. Coincidentally, during the preparation of the present report, an independent study confirmed the importance of the π-π stacking interactions of F259 with the −2 and −3 bases of the substrate for optimum 3′-phosphodiesterase activity of hTDP1 [31]. In that report, the authors show selective photocrosslinking of TDP1 to its substrate by incorporating 5-iodouracil (5IdU) at different positions of the DNA substrate. For both the −2 or −3 incorporated substrates, the substrate was found to form covalent crosslink to F259, highlighting the close interactions of F259 with the −2 and −3 nucleobases. Consistent with this result, we show here that removing the −2 and −3 nucleobases reduces 3′-processing to the same extent as removing the aromatic residue F259 of TDP1 (see Fig. 3). It is plausible that this molecular recognition mechanism is a key determinant of the selectivity of TDP1 for DNA substrates rather than phosphodiesterase activity against non-nucleic acid substrates (as is the case of the related phospholipase D enzymes). This type of interaction is reminiscent of the DNA locking mechanism used by the TATA-binding protein (TBP), which utilizes stacking interactions of two pairs of phenylalanines to rig the DNA conformation and stabilize the TBP-TATA box complex [35]. Analogous substrate positioning control also applies to protein substrates, such as the 19S proteasome catalytic system, in which the cleavable peptide bond is placed at the heart of the catalytic site by positioning substrate amino acid side chains in specificity pockets [36].
The reduction in 3′-end processing by mutating the serine 400 residue of hTDP1 to alanine (S400A) (see Figs. 2 and 4) is consistent with the presence of a hydrogen bond between S400 and the non-bridging oxygen atom of the phosphate joining −1 and −2 nucleobases of the substrates [25] (Fig. 6). Hence, TDP1 engages its substrates by a network of interactions involving hydrogen bonds beyond the HKN catalytic motifs. Serine 400, which is a previously untested residue and highly conserved across species (see Fig. 1C), appears to participate in the critical network of interactions, anchoring the phosphodiester backbone of TDP1 substrates for processing blocking lesions at the 3′-end of DNA breaks. Such anchoring is essential with respect to 3′-phosphodiestrease activity, selectively promoting the 3′-phosphodiestrease activity over 5′-phosphodiesterase activity of hTDP1. This could be explained by the fact that S400 binds and coordinates the substrate through hydrogen bond to the phosphodiester group between −1 and −2 nucleotides. The coordination of the phosphate by S400 likely restrics the conformation of the DNA substrate backbone. While such anchorage is optimal for 3′-phosphodiesterase activity, it has the opposite effect for the 5′-phosphodiesterase activity of hTDP1. Removal of this phosphodiester coordination and relaxation of the backbone restriction in the S400A mutant potentially enhances the binding of substrate for 5′-end hydrolysis. Hence, we conclude that the S400 residue drives the selectivity of hTDP1 for 3′- over 5′-phosphodiesterase activity.
The selective defect in 5′-end processing activity of the W590F mutants is notable. The W490F aromatic residue is situated across the Y204 aromatic residue, and both form a groove above the HKN catalytic residues near the target residues to be removed by TDP1 (see Fig. 1, yellow shading, and Fig. 6). Y204 forms two hydrogen bonds with the terminal (−1) nucleobase while W590 forms Van der Waals interactions with that same terminal nucleobase (Fig. 6). Hence, the Y204F and W590F mutations were predicted to increase the size of the peptidic groove where TDP1 accommodates its nucleoprotein substrates. The lack of impact of the Y204F and W590F mutations on the 3′-processing activity of hTDP1 may be due to the many other interactions between TDP1 and its substrate with 3′-end lesions, which suffice for TDP1 3′-processing activity. The selective impact of the W590F mutations on the 5′-processing activity of hTDP1 might reflect the fact that hTDP1 is markedly less active against 5′-tyrosyl vs. 3′-tyrosyl substrates [11], and that the 5′-processing reaction has to accommodate a substrate with opposite polarity with respect to the target phosphodiester bond. We speculate that minimizing the substrate interactions around the Y204F and W590F substrate groove at the junction between the DNA product has a greater impact on enzyme activity toward its less preferred 5′-end substrates.
While it seems that the hydrogen bond between S400 and phosphate needs to be disrupted for 5′-end cleavage to proceed efficiently, the Van der Waals interactions and spatial restriction provided by bulky W590 become crucial for retention of the 5′-phosphotyrosyl linkage within the catalytic site. Co-crystallization studies are warranted to elucidate the specific interactions of TDP1 with 5′-end substrates.
Acknowledgements
We thank all members of the Developmental Therapeutics Branch and Laboratory of Molecular Pharmacology, CCR-NCI for insights and discussion during the course of our studies.
Funding
Our studies are supported by the Center for Cancer Research, the Intramural Research Program of the National Cancer Institute (Z01 BC 006150), NIH.
Abbreviations:
- hTDP1
human tyrosyl-DNA phosphodiesterase 1
- TOP1
topoisomerase I
- TOP2
topoisomerase II
- TOP1cc
TOP1 cleavage complex
- TOP2cc
TOP2 cleavage complex
Footnotes
Conflict of interest statement
The authors declare no conflict of interest.
References
- [1].Menon V, Povirk LF, End-processing nucleases and phosphodiesterases: an elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair, DNA Repair (Amst.) 43 (2016) 57–68. [DOI] [PubMed] [Google Scholar]
- [2].Pommier Y, Huang SY, Gao R, Das BB, Murai J, Marchand C, Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2), DNA Repair (Amst.) 19 (2014) 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yang S-W, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA, A. eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases, Proc. Natl. Acad. Sci. U. S. A 93 (1996) 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Interthal H, Chen HJ, Champoux JJ, Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages, J. Biol. Chem 280 (2005) 36518–36528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Das BB, Dexheimer TS, Maddali K, Pommier Y, Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 19790–19795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chiang S-C, Meagher M, Kassouf N, Hafezparast M, McKinnon PJ, Haywood R, El-Khamisy SF, Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical–induced DNA damage, Sci. Adv 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ashour ME, Atteya R, El-Khamisy SF, Topoisomerase-mediated chromosomal break repair: an emerging player in many games, Nat. Rev. Cancer 15 (2015) 137–151. [DOI] [PubMed] [Google Scholar]
- [8].Lee SY, Kim H, Hwang HJ, Jeong YM, Na SH, Woo JC, Kim SG, Identification of tyrosyl-DNA phosphodiesterase as a novel DNA damage repair enzyme in Arabidopsis, Plant Physiol. 154 (2010) 1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pommier Y, Sun Y, Huang SN, Nitiss JL, Roles of eukaryotic topoisomerases in transcription, replication and genomic stability, Nat. Rev. Mol. Cell Biol 17 (2016) 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D, TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II, J. Biol. Chem 279 (2004) 55618–55625. [DOI] [PubMed] [Google Scholar]
- [11].Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y, Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells, J. Biol. Chem 287 (2012) 12848–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, Povirk LF, Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1), Nucleic Acids Res. 33 (2005) 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang SY, Murai J, Dalla Rosa I, Dexheimer TS, Naumova A, Gmeiner WH, Pommier Y, TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs, Nucleic Acids Res. 41 (2013) 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Abo MA, Sasanuma H, Liu X, Rajapakse VN, Huang S.-y., Kiselev E, Takeda S, Plunkett W, Pommier Y, TDP1 is critical for the repair of DNA breaks induced by sapacitabine, a nucleoside also targeting ATM- and BRCA-deficient tumors, Mol. Cancer Ther 16 (2017) 2543–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Takiuchi Y, Kobayashi M, Tada K, Iwai F, Sakurada M, Hirabayashi S, Nagata K, Shirakawa K, Shindo K, Yasunaga JI, Murakawa Y, Rajapakse V, Pommier Y, Matsuoka M, Takaori-Kondo A, HTLV-1 bZIP factor suppresses TDP1 expression through inhibition of NRF-1 in adult T-cell leukemia, Sci. Rep 7 (2017) 12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tada K, Kobayashi M, Takiuchi Y, Iwai F, Sakamoto T, Nagata K, Shinohara M, Io K, Shirakawa K, Hishizawa M, Shindo K, Kadowaki N, Hirota K, Yamamoto J, Iwai S, Sasanuma H, Takeda S, Takaori-Kondo A, Abacavir, an anti-HIV-1 drug, targets TDP1-deficient adult T cell leukemia, Sci. Adv 1 (2015) e1400203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nitiss KC, Malik M, He X, White SW, Nitiss JL, Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 8953–8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW, A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage, Nature 461 (2009) 674–678. [DOI] [PubMed] [Google Scholar]
- [19].Gajewski S, Comeaux EQ, Jafari N, Bharatham N, Bashford D, White SW, van Waardenburg RC, Analysis of the active-site mechanism of tyrosyl-DNA phosphodiesterase I: a member of the phospholipase D superfamily, J. Mol. Biol 415 (2012) 741–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Marchand C, Huang SY, Dexheimer TS, Lea WA, Mott BT, Chergui A, Naumova A, Stephen AG, Rosenthal AS, Rai G, Murai J, Gao R, Maloney DJ, Jadhav A, Jorgensen WL, Simeonov A, Pommier Y, Biochemical assays for the discovery of TDP1 inhibitors, Mol. Cancer Ther 13 (2014) 2116–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR, Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy, Nat. Genet 32 (2002) 267–272. [DOI] [PubMed] [Google Scholar]
- [22].Guo D, Dexheimer TS, Pommier Y, Nash HA, Neuroprotection and repair of 3′-blocking DNA ends by glaikit (gkt) encoding Drosophila tyrosyl-DNA phosphordiesterase 1 (TDP1), Proc. Natl. Acad. Sci. U. S. A 111 (2014) 15816–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ, TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo, EMBO J. 26 (2007) 4720–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Debethune L, Kohlhagen G, Grandas A, Pommier Y, Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase, Nucleic Acids Res. 30 (2002) 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Davies DR, Interthal H, Champoux JJ, Hol WG, Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide, Chem. Biol 10 (2003) 139–147. [DOI] [PubMed] [Google Scholar]
- [26].Pouliot JJ, Robertson CA, Nash HA, Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae, Genes Cells 6 (2001) 677–687. [DOI] [PubMed] [Google Scholar]
- [27].Interthal H, Pouliot JJ, Champoux JJ, The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Raymond AC, Rideout MC, Staker B, Hjerrild K, Burgin AB Jr., Analysis of human tyrosyl-DNA phosphodiesterase I catalytic residues, J. Mol. Biol 338 (2004) 895–906. [DOI] [PubMed] [Google Scholar]
- [29].Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ, SCAN1 mutant Tdp1 accumulates the enzyme-DNA intermediate and causes camptothecin hypersensitivity, EMBO J. 24 (2005) 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He X, van Waardenburg RC, Babaoglu K, Price AC, Nitiss KC, Nitiss JL, Bjornsti MA, White SW, Mutation of a conserved active site residue converts tyrosyl-DNA phosphodiesterase i into a DNA topoisomerase I-dependent poison, J. Mol. Biol 372 (2007) 1070–1081. [DOI] [PubMed] [Google Scholar]
- [31].Flett FJ, Ruksenaite E, Armstrong LA, Bharati S, Carloni R, Morris ER, Mackay CL, Interthal H, Richardson JM, Structural basis for DNA 3′-end processing by human tyrosyl-DNA phosphodiesterase 1, Nat. Commun 9 (2018) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Antony S, Marchand C, Stephen AG, Thibaut L, Agama KK, Fisher RJ, Pommier Y, Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1, Nucleic Acids Res. 35 (2007) 4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dexheimer TS, Stephen AG, Fivash MJ, Fisher RJ, Pommier Y, The DNA binding and 3′-end preferential activity of human tyrosyl-DNA phosphodiesterase, Nucleic Acids Res. 38 (2010) 2444–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lebedeva NA, Rechkunova NI, Lavrik OI, AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1, FEBS Lett. 585 (2011) 683–686. [DOI] [PubMed] [Google Scholar]
- [35].Juo ZS, Chiu TK, Leiberman PM, Baikalov I, Berk AJ, Dickerson RE, How proteins recognize the TATA box, J. Mol. Biol 261 (1996) 239–254. [DOI] [PubMed] [Google Scholar]
- [36].Groll M, Heinemeyer W, Jager S, Ullrich T, Bochtler M, Wolf DH, Huber R, The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 10976–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]