

Abstract
Compound materials, such as transition-metal (TM) carbides, are anticipated to be effective electrocatalysts for the carbon dioxide reduction reaction (CO2RR) to useful chemicals. This expectation is nurtured by density functional theory (DFT) predictions of a break of key adsorption energy scaling relations that limit CO2RR at parent TMs. Here, we evaluate these prospects for hexagonal Mo2C in aqueous electrolytes in a multimethod experiment and theory approach. We find that surface oxide formation completely suppresses the CO2 activation. The oxides are stable down to potentials as low as −1.9 V versus the standard hydrogen electrode, and solely the hydrogen evolution reaction (HER) is found to be active. This generally points to the absolute imperative of recognizing the true interface establishing under operando conditions in computational screening of catalyst materials. When protected from ambient air and used in nonaqueous electrolyte, Mo2C indeed shows CO2RR activity.
Keywords: electrocatalysis, transition-metal carbides, electrochemical CO2 reduction, surface Pourbaix diagram, ab initio thermodynamics, solid/liquid interface, XPS, HER
1. Introduction
Transition-metal compounds are widely applicable as catalyst or catalyst support materials in heterogeneous catalysis. The reason for this is their structural and compositional diversity, which can be adopted to the reaction of interest and which can suppress the correlation of adsorption energies, a phenomenon frequently referred to as scaling relations.1 Such scaling is made responsible for numerous limitations in heterogeneous catalysis on transition metals (TMs),2,3 but can be overcome by compound materials, such as oxides,4 sulfides, or carbides.2,3,5−7 High prospects for usage in thermal catalysis8−19 and electrocatalysis6 are therefore assigned to TM carbides (TMCs) as least explored materials class among these compound catalysts. Their ability to uptake and activate CO2 and thus drive the eminent electrochemical carbon dioxide reduction reaction (CO2RR) has recently been predicted by a number of theoretical works.20−22 The two most severe generic issues in the CO2RR are the competing hydrogen evolution reaction (HER) that often dominates in aqueous electrolytes6 and the adsorption energy scaling of CO and CHO, which are central reaction intermediates.23 And indeed, for a range of single-crystal TMC surfaces, the density functional theory (DFT) calculations, underlying those promising forecasts, indicated a breaking of this key limiting scaling23 due to a more carbophobic and oxophilic nature in comparison to the parent metals.7,20,21 Such guiding predictions often made in the context of larger computational screening studies2,3 are essential for experimentalists because the chemical diversity of compound materials is too vast for empirical approximation.
In particular for Mo2C, an active-site computational screening study performed by some of us investigated the numerous and various active sites offered by this TMC and further confirmed its high suitability for the CO2RR.7 Irritatingly, despite all this praise, only one experimental study of the CO2RR on Mo2C powder and sheets has been reported to date.24 A minuscule conversion to methane (CH4) with a Faradaic efficiency of <0.1% was detected at ≤−0.95 V versus the standard hydrogen electrode (SHE), denoted as VSHE in the following, while the dominant current was related to the competing hydrogen evolution reaction (HER).
Aiming to understand this blatant discrepancy between claim and reality, we here perform a multimethod approach to investigate Mo2C’s CO2 electroreduction capability and the related surface chemistry in aqueous electrolyte. Our experiments confirm an in fact exclusive activity toward the HER, without any formation of CO2 reduction products. In the search for an explanation, we realize that the bulk material and especially the parent metal are prone to oxidation, even at rather negative potentials. Detailed ab initio thermodynamics calculations reveal that the situation at the carbide surface is even worse than expected from the generic bulk Pourbaix (E/pH) diagrams of Mo2C25 and its parent metal.26,27 In the relevant potential range, the carbide surface is always covered by oxygenated species, and it is the HER activity of these oxidic films that is actually seen in the experiments. This understanding is fully confirmed by an experimental surface E/pH diagram recorded with quasi in-situ X-ray photoelectron spectroscopy (XPS)28 under consideration of surface pH effects. To one end, these findings underscore the urgent need to extend computational screening studies from their present evaluation of the bulklike material to an account of the catalyst surface structure and composition that is actually forming operando.29,30 On the other hand, given the highly appealing properties of the carbide motivates us to search for experimental reaction conditions where these properties may indeed be harvested. First experiments correspondingly performed in nonaqueous electrolyte immediately show a high CO2 reduction activity of the then nonoxidized or barely oxidized Mo2C.
2. Results and Discussion
2.1. Electrocatalytic Performance of Planar Mo2C Films
The planar molybdenum carbide (Mo2C) films that we synthesized for our study consist of large, several-micrometer-wide terraces (see Supporting Information Note 1 and Figure S1) that are smooth and chemically homogeneous (see Figure S2). Figure 1a shows a bright-field (BF) transmission electron microscopy (TEM) image of the Mo2C film cross section. The film is very homogeneous in thickness and shows different areas of ordered growth with changes in the crystallite orientation depending on the underlying substrate grains. The high-resolution (HR) TEM image (inset in Figure 1a), together with the selected area electron diffraction (SAED) pattern, reveal the best agreement of our material’s phase with hexagonal Mo2C (see Figures S3 and S4). Details of the evaluation of the structural data are given in Supporting Information Note 1.
Figure 1.

Mo2C film cross section and structure, and its cathodic water reduction activity. (a) Bright-field (BF) TEM image of the Mo2C film in a focused ion beam (FIB) lamella embedded between the polycrystalline Mo substrate and a Pt/C protection layer, and HRTEM image of the Mo2C film (inset). (b) Cathodic scans of Mo2C recorded at 20 mV s–1 in 0.1 M CO2-saturated KHCO3 (orange line) and Ar-saturated 0.1 M NaClO4 (blue line), both at pH 6.9. The dashed blue line shows an independently recorded cyclic voltammogram (CV) featuring anodic oxidation (see text); j is the current density, and E is the electrode potential versus the standard hydrogen electrode (SHE, upper axis) or the reversible hydrogen electrode (RHE, lower axis).
To get a macroscopic picture of the electrocatalytic activity of this planar hexagonal Mo2C film electrode, cyclic voltammograms (CVs) were recorded in 0.1 M KHCO3 with CO2 and 0.1 M NaClO4 without CO2 (both pH 6.9) over the potential (E) region of interest (see Figure 1b and Supporting Information Note 2). These electrolytes were selected to have a minimized interaction of ions with the interface and for the meaningful comparison of the Mo2C electrochemistry with and without CO2 at the same pH. After immersion under potential control at −0.70 VSHE, the potential was cycled between −1.6 and −0.2 VSHE. A current density of ca. −20 mA cm–2 is reached at −1.6 VSHE when CO2 is present. Notably, care has to be taken to not apply potentials more positive than −0.2 VSHE due to the danger of anodically oxidizing the electrodes, as indicated by the positive current peak (independently recorded CV, dashed blue line) visible in Figure 1b. The cathodic current is due to the reduction of neutral water and shows the same shape for both electrolytes. With CO2 present, the onset of the reduction is slightly shifted to less negative potentials, which we attribute to the presence of carbonate and bicarbonate species (see below).
To qualitatively identify the reaction products forming upon cathodic polarization, differential electrochemical mass spectrometry (DEMS, Figure S5) has been employed. No valuable CO2 reduction product, such as methane (CH4) or ethylene (C2H4), but exclusively hydrogen was detected under the conditions applied in this work at pH 6.9. This means that the whole potential range of interest is governed by the HER. To guarantee the detectability of all products of interest, a reference measurement was conducted with a polycrystalline Cu sample, where CH4 and C2H4 were indeed found at potentials <−1.2 VSHE. At a different pH of 3.7, gas chromatography also confirmed the exclusive formation of hydrogen over our Mo2C films in the complete cathodic potential range (see Supporting Information Note 2). The HER is enhanced when buffering CO2/carbonate species are present in the electrolyte, and it is this enhancement, not any CO2 reduction, that stands behind the slight shift of the onset potential seen in Figure 1b.
2.2. Ab Initio Thermodynamics of Mo2C in Aqueous Electrolyte
Unexpectedly, no CO2 electroreduction is thus found upon cathodic polarization of this heralded compound material. One immediate explanation for this could be the material’s known susceptibility to oxidation. However, in full agreement with the experimental Pourbaix diagram of Mo2C,25 bulk oxidation is only clearly remarkable at ∼0.04 VSHE at pH 6.9 (Figure 1b), a potential which we deliberately never applied in the above studies. In principle, one would also expect residing surface oxides or oxygenated adsorbates to be cathodically reduced during the highly negative potentials applied for the CO2 reduction. On the other hand, a sometimes surprisingly large extra thermodynamic stability of surface oxides has been reported in thermal oxidation catalysis.31,32
To assess this for the present case, we performed DFT-based ab initio thermodynamics to determine the stable surface phases of the dominant Mo2C(110) facet in aqueous electrolyte (see Supporting Information Note 3). We computed the surface Pourbaix diagrams for two different bulk phases, each time considering more than 300 candidate structures of pristine Mo2C terminations with or without coincidence and minimally strained MoO2(100) surface oxide films of varying thickness, and with varying degrees of hydrogenation or hydroxylation. The resulting surface Pourbaix diagram for the (more stable) hexagonal close-packed (β)-2 bulk phase is depicted in Figure 2. The surface Pourbaix diagram of the (β)-1 phase is given in Figure S6 and leads to analogue conclusions. Eight different stable surface phases emerge in the relevant E/pH range. At extremely negative potentials and low pH values, a carbon-rich Mo2C termination with a low amount of adsorbed hydrogen prevails (see region ① in Figure 2). Oxygen or hydroxyl surface films dominate in the less reducing regions ② and ③, followed by a slightly hydroxylated monolayer (ML) thick surface oxide in region ④. An oxide bilayer with a varying degree of hydroxylation finally forms in regions ⑤–⑧.
Figure 2.

Ab initio thermodynamics surface Pourbaix (E/pH) diagram for Mo2C(110) and possible MoO2(100) surface oxide formation. The data here are for the more stable hexagonal (β)-2 bulk phase, while analogue results for the (β)-1 phase are given in Figure S6. The generic stability ranges of bulk MoO2 and dissolved MoO42–, as calculated for the parent metal,27 are indicated by black hatched areas. Side views of the stable surface phases ①–⑧ are shown on the right (Mo, C, O, and H atoms are depicted as green, gray, red, and white spheres, respectively). The surface terminations in terms of fractions of monolayers (ML) defined with respect to MoO2(100)-(1 × 1) are also provided. The blue dashed line indicates the thermodynamic onset potential of the hydrogen evolution reaction. Experimentally tested conditions are marked by stars, solid stars for nominal pH conditions, and hollow stars after considering surface pH changes (see text and Figure 4).
Not surprisingly, the surface thus changes with decreasing E and pH from an oxidized to a reduced state with a still partially hydrogenated carbon-rich Mo2C termination. Intriguingly however, this transition of the surface chemistry happens at much more negative potentials than expected from the experimental bulk Pourbaix diagram of Mo2C,25 i.e., higher overpotentials are required to reduce the surface. Comparing the experimental apparent bulk stability and theoretical surface stability lines (Figure S7) suggests that the overpotential to theoretically reach a Mo2C surface free of oxygen-containing adsorbates is in fact almost pH-independent and as high as ∼1.0 V. This implies that the benefit of a possibly suitable reactive carbide surface chemistry for CO2 electroreduction will only become accessible under conditions where the competing HER already proceeds at extremely high overpotentials (see blue dashed line in Figure 2 for the HER onset). Different from the bare Mo2C surface,7 MoO2 is a recognized HER catalyst.33−35 Free-energy calculations for the first protonation step of the HER and CO2RR summarized in Figure S8 indeed indicate a high selectivity toward the HER also for the Mo2C surface covered with a MoO2 bilayer, i.e., structure ⑧ in Figure 2. This rationalizes the dominant hydrogen evolution seen in our electrocatalytic measurements and casts severe doubts on a promising activity toward the CO2RR that could be reached with Mo2C catalysts in aqueous electrolyte.
2.3. Experimental Surface Pourbaix Diagram
While the ab initio thermodynamics results thus provide a sound rationalization for the initially surprising inactivity toward the CO2RR, one has to stress that the absolute position of stability lines in computed Pourbaix diagrams depends sensitively on the employed DFT functional.32 The surface Pourbaix diagram was furthermore only computed for the dominant Mo2C(110) facet, and the formation of a coincidence surface oxide film was assumed. Before reaching a final conclusion regarding a future use of Mo2C as a CO2RR catalyst, we therefore scrutinize the obtained picture with quasi in-situ X-ray photoelectron spectroscopy (XPS).
2.4. Mo2C Film Analysis after Its Synthesis
We started our analysis of the exact chemical composition in terms of existing oxidation states and stoichiometry before and after cathodic polarization with the pristine Mo2C films, directly after their synthesis. Here, we first handled the Mo2C without protection from ambient air. Importantly, due to air exposure, adventitious carbon and oxidized Mo species are always detected at the Mo2C surface; an example of the typical surface chemistry is shown in Figure 3 (top panels). The same behavior has been observed in the case of titanium oxycarbide surfaces and is very likely related to the low intrinsic thermodynamic stability of the material at ambient oxygen pressures.36 The amount of oxide can vary slightly and depends on the exact synthesis conditions (details are given in Table S2). No other undesirable element is found, which confirms the chemical purity of the synthesized Mo2C specimen (see Figure S9).
Figure 3.

Surface chemistry of Mo2C showing MoO3 after air exposure and MoO2 after electrolyte contact at reductive potentials. Deconvoluted X-ray photoelectron C 1s (left) and Mo 3d (right) high-resolution spectra of the freshly synthesized film (top) with native MoO3 after short (15 min) air contact, (middle) without oxide due to handling under hydrogen and in an Ar-filled glovebox at all times, and (bottom) with MoO2 after immersion of oxide-free Mo2C into 0.1 M NaClO4 (pH 3.7) at −0.2 VSHE for 15 min. The fit components are directly given in the figure; the takeoff angle is 0° in all cases.
The fit of the XPS spectra of Mo2C after a 15 min air contact in Figure 3 (top) reveals contributions from MoO3, graphitic, and adventitious carbon species, while the primary components are carbidic Mo and carbidic carbon. Details of the evaluation and fitting of the spectra are given in Supporting Information Note 4. The average thickness determined for the native oxide surface film lies between 0.6 and 0.7 nm (see Figures S10 and S14), which fits nicely to the expected thickness of one double layer of edge-shared MoO6 octahedra constituting layered MoO3.37 Carbon and oxide can be easily removed from the surface through 60 s of Ar sputtering in the XPS (Figure S12). It has to be noted that the synthesis of oxide-free stoichiometric Mo2C is possible in a reactor where the Mo2C specimen can be transported protected under Ar and/or H2 gas (see Figure 3 (middle) and Table S4). Systematic contact to air leads to surface oxide (MoO3) formation within less than 15 min (see Figure S15).
Importantly, exposure of the oxide-free carbide to aqueous electrolyte also leads to the spontaneous formation of an oxide overlayer, even if reducing conditions are applied through immersion at −0.2 VSHE into the most acidic electrolyte (see Figure 3, bottom). After contact of the specimen with electrolyte, the sole detectable oxides are MoO2 and/or nonstoichiometric suboxides (see Figure 3 and Table S5 and explanation in the next paragraph). This insight is central to the application of oxidation-prone carbide materials in electrocatalysis in general, because it reveals the sheer impossibility to start from an oxide- or oxygen-free catalyst/electrolyte interface, which has not been clearly recognized in the past. The only conceivable way to remove the oxide from the surface could therefore be the polarization at extremely negative potentials in regions where massive HER activity is expected. However, in view of the concomitant surface pH effects described below, it might be that electrochemical surface oxide reduction is not practically feasible at all.
2.5. Mo2C Surface Chemistry after Cathodic Polarization
To investigate the surface chemistry of the Mo2C films after electrolyte exposure at specific applied potentials, quasi in-situ XPS studies were carried out with the help of an air-tight transfer vessel for specimen transport from the Ar-filled glovebox, where the electrochemistry was carried out, to the XPS, where a second transfer under Ar was undertaken as described in ref. (38) (see Figure S16). Different E/pH values were selected to monitor changes of the electrode surface after cathodic polarization in 0.1 M NaClO4 solution. The potential steps were individually selected for three specific pH values of 3.7, 7, and 10, based on central points in the theoretical surface Pourbaix diagram. The corresponding CVs are shown in Figure S17. After potentiostatic polarization, XPS spectra (Figure S18) were recorded to calculate the Mo/Ccarbidic ratio and estimate the oxide layer thicknesses shown in Figure 4. The oxide overlayer exclusively consists of MoO2 and/or, as the case may be, suboxides. This suggests that MoO3 gets dissolved upon electrode immersion, which agrees well with the bulk Pourbaix diagram of the Mo/MoOx system.26,27,39 Therefore, for the estimation of the surface oxide layer thickness, it was assumed that the film only consists of MoO2. A detailed description of the data evaluation assuming uniform oxide distribution (complete “wetting”) is given in Supporting Information Note 4 and Figure S19.
Figure 4.
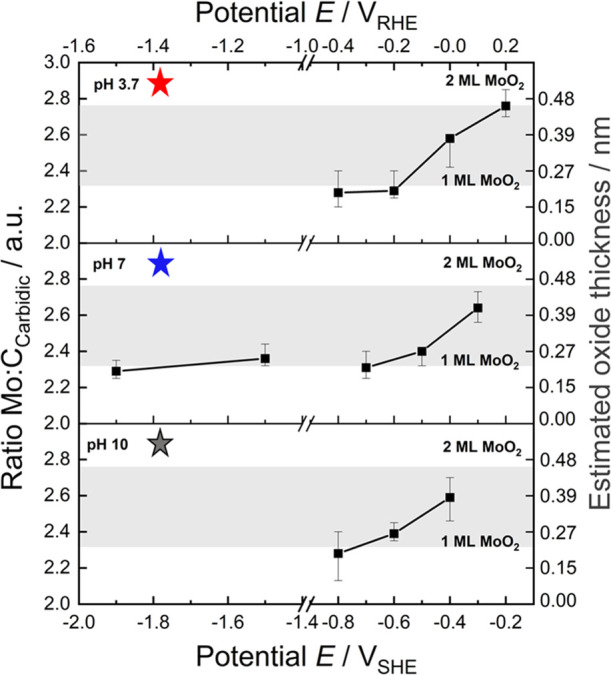
Experimental Pourbaix diagram confirming surface oxide formation under all relevant CO2RR conditions. The correlation between the Mo/Ccarbidic ratio and the estimated oxide layer thickness is given in Figure S19. For the calculation of the error bars, we refer to Supporting Information Note 4. The shaded regions indicate the thickness expected for one or two monolayers (ML) MoO2(100). The probed reaction conditions are indicated by correspondingly colored stars in the theoretical surface Pourbaix diagram in Figure 2.
At pH 3.7, polarization at −0.2 VSHE leads to the formation of an ∼0.46 nm thick film, which corresponds almost perfectly to the thickness of 0.48 nm of a bilayer MoO2(100) predicted to form atop the Mo2C under these conditions in the theoretical surface Pourbaix diagram (region ⑤ in Figure 2). Stepping to more negative potentials at pH 3.7 results in a decrease of the oxide film thickness down to ∼0.20 nm at ≤−0.6 VSHE, which would then correspond to roughly one or below one monolayer coverage, again exactly as expected from the theoretical surface Pourbaix diagram. A similar evolution from about bilayer surface oxide to below monolayer surface oxide is found upon increasing cathodic polarization at the other two probed pH values 7 and 10. In the case of pH 7, this oxide is stable down to potentials as low as −1.9 VSHE. As further discussed below, all of these findings are fully consistent with the theoretical surface Pourbaix diagram; see the stars in Figure 2 marking the location of the probed reaction conditions.
For comparison with realistic CO2RR conditions, additional studies have been carried out in 0.1 M CO2-saturated KHCO3 (pH 6.9). After polarization at −0.4 and −1.5 VSHE, thin surface oxide films with thicknesses in the same range as those determined for 0.1 M NaClO4 (pH 7) are detected with XPS (see Figure S20 and Tables S9–S11).
2.6. Surface pH Correction
The overall consistency of the experimental and theoretical Pourbaix diagram is truly remarkable considering not least the known uncertainty in computed absolute stability regions.32 Nevertheless, it is intriguing to note that at pH 3.7, our experiments still indicate the presence of a surface oxide of about 1 ML thickness even at the most negative potential probed. Under such reducing conditions, the intrinsic state of the surface should have just been reached according to theory. To further analyze this, we extended the measurements to very negative potentials, specifically for pH 7, as this is the sole pH where meaningful CO2 electroreduction is possible in aqueous electrolytes. Surprisingly, these measurements also indicate the stability of about 1 ML thick surface oxide even at a polarization as negative as −1.9 VSHE (see Figure 4), where theory would predict the bare carbide surface beyond doubt (see Figure 2).
This discrepancy can be resolved by recognizing that non-negligible water and/or proton reduction currents flow at these negative potentials (Figure S21 and Table S12). This can give rise to significant changes in the surface pH due to mass transfer limitations, i.e., the actual pH at the carbide/liquid interface could differ strongly from the nominal bulk pH set in the experiments. To assess this, we calculated the approximate surface pH that occurs during the potentiostatic experiments solving a simplified Poisson–Nernst–Planck (PNP) transport model40 (see Supporting Information Note 4, Figures S22–S24, and Table S13 for a detailed account). Indeed, these calculations confirm strong changes, which, for instance, at pH 7 and −1.5 VSHE lead to an effective pH shift to strongly alkaline conditions (pH 13–14) in front of the electrode. The real reaction conditions thus probed in the experiments are indicated by hollow stars in Figure 2 and are now fully consistent with the theoretical predictions within the expected uncertainties. Note that we did not explicitly engage in a calculation of the pH shift for the most negative −1.9 VSHE at pH 7, as bubble formation and a likely ion deposition from the electrolyte under the then highly violent HER is not captured by the simple PNP transport model anymore.
2.7. CO2 Electroreduction in Nonaqueous Electrolyte
The analyses presented so far unfortunately confirm that the TMC’s theoretically predicted promising CO2RR chemistry is not experimentally accessible in aqueous electrolytes due to the persistent formation of a thin oxidic film at the surface. Truly harvesting this chemistry thus requires to move to alternative approaches that prevent such passivation. As shown in Figure 3, it is possible to avoid oxidation after synthesis, to transfer the electrode to the electrochemical cell without any contact to ambient air, and to immerse it under potential control. The last consequent measure to prevent operando oxidation is then to switch to a nonaqueous electrolyte.
Recognizing that CO2RR in nonaqueous electrolytes is a widely unchartered territory, we carried out corresponding proof-of-principle experiments in acetonitrile-based electrolyte that only contains traces of water (see Supporting Information Note 5 and Table S14). As shown in Figure 5, this immediately leads to a pronounced reduction current in the presence of CO2 with an approximate onset at −1.7 V versus the ferrocene/ferrocenium (Fc/Fc+) redox couple (calibration in Figure S25) and thus roughly at −1.08 VSHE.41 Polycrystalline Cu shows an approximate CO2 reduction onset at −1.31 VSHE in the same electrolyte.42 A current density of 0.1 mA cm–2 is reached at a potential of ≤−2.52 VFc/Fc+ (≤−1.9 VSHE). In contrast, no reduction peak is visible in Ar-purged electrolyte, where no CO2 is present. First electrochemical infrared spectroscopy data indicate that CO forms as a product at potentials ≤−2.5 VFc/Fc+ (≤−1.9 VSHE) (see Figure S26). This preliminary insight confirms that under conditions that preserve the Mo2C chemistry, the material indeed shows high CO2 electroreduction activity as had been predicted in the earlier computational screening studies.20,21 This immediate success now motivates follow-up work that analyzes the formed reaction products and systematically explores the activity toward the CO2RR of this and other TMCs in nonaqueous electrolytes.
Figure 5.
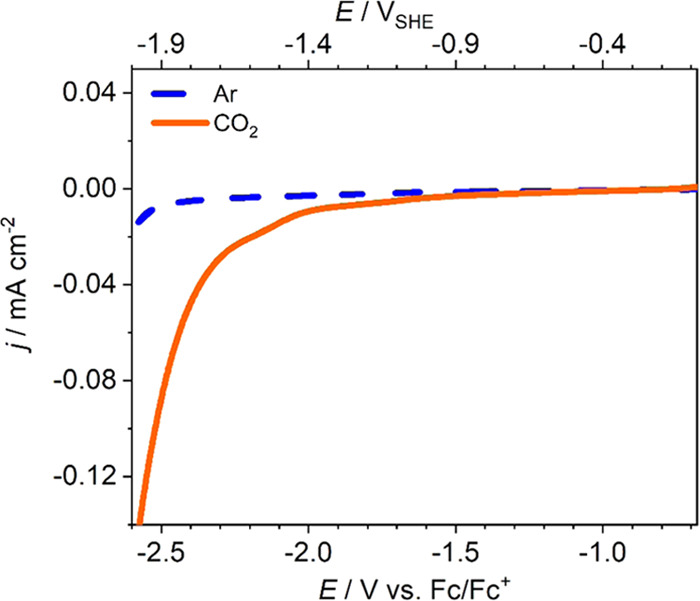
CO2 electroreduction at Mo2C in nonaqueous electrolyte. CVs of Mo2C without (blue) and with (orange) CO2 in acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6). Scan rate: 20 mV s–1.
3. Conclusions
In summary, we have demonstrated that well-defined planar hexagonal Mo2C films exhibit the intrinsic property to passivate at their surface upon air exposure and immersion in aqueous electrolyte, even under reductive conditions and regardless if they are native oxide-free or already passivated before contact with the electrolyte. After immersion in aqueous electrolyte with pHs 3.7, 7, and 10, monolayer-thin MoO2-like oxidic films are present at the surface that are stable down to potentials as negative as <−1.9 VSHE at pH 7. The oxide-covered carbides exclusively show activity toward water reduction, and no CO2 electroreduction products are found. The presence of CO2 in the electrolyte only enhances the HER, likely through changes of the surface pH and/or transport of protons by buffer ions.
This means that the promising CO2RR properties of this TMC heralded by earlier computational studies7,20,21 are experimentally not accessible in aqueous electrolytes. Quite discomfortingly, the bulk Pourbaix diagrams of other prominent carbides like W2C or WC,25 as well as the bulk Pourbaix diagrams of important parent metals like V, Zr, or Nb,27 suggest a similar propensity to oxidation as molybdenum. A possible operando formation of ultrathin surface oxidic films was generally not considered in previous screening work on TMCs. Our results thus dictate to revisit the suitability of this materials class for CO2RR from this perspective. Alternatively, first experiments with acetonitrile indicate that a switch to nonaqueous base electrolytes (with only controlled water addition as a proton source) circumvents this issue and indeed confirms Mo2C as an efficient CO2RR catalyst.
4. Experimental Section
4.1. Synthesis of Mo2C Films
A detailed description of the applied synthesis route is given in Supporting Information Note 6.
4.2. Surface Characterization
4.2.1. Microscopy
The surface morphology of the synthesized Mo2C was investigated with atomic force microscopy (AFM) and scanning electron microscopy coupled with energy-dispersive X-ray spectroscopy (SEM–EDX). Transmission electron microscopy (TEM) was used for cross-sectional characterization. Details are outlined in Supporting Information Note 1.
4.2.2. X-ray Photoelectron Spectroscopy (XPS)
XPS spectra were acquired on a MultiLab 2000 instrument (Thermo Fisher Scientific) comprising a hemispherical sector analyzer (Alpha 110, Thermo Fisher Scientific) and a monochromated X-ray source (Al Kα, 1486.6 eV). A flood gun was used for charge compensation via emission of electrons at a kinetic energy of 6 eV, and charge-induced shifts were corrected with reference to the carbidic carbon component at 283.1 eV. Ar sputtering (3 keV, ion current of 1.0 μA) was used for depth profiling by scanning across an area of (1.5 × 1.5) mm2. Nondestructive depth profiling was carried out by adjusting the angle between the sample and the analyzer (0–60°). Survey scans were obtained at a pass energy of 100 eV with an energy step size of 1 eV. XPS spectra of the binding energy (BE) regions of interest (C 1s, O 1s, Mo 3d) were recorded at a pass energy of 25 eV and an energy step size of 0.1 eV. Peak fitting was processed with CasaXPS software,43 always using a Shirley-type background and mixed Gaussian–Lorentzian (GL30) functions except for the carbide peaks, where asymmetric peak shapes were utilized. The quantification of all spectra was corrected for different escape depths via the Gries44 formula.
4.3. Electrochemistry
All electrochemistry measurements were carried out at room temperature in aqueous 0.1 M Ar-saturated NaClO4 and CO2-saturated KHCO3 or in nonaqueous acetonitrile solutions containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) using an Autolab (Metrohm) or a BioLogic (VMP3, BioLogic) potentiostat. Details are outlined in Supporting Information Notes 2 and 5.
4.3.1. Quasi In-Situ XPS Studies
The Mo2C electrodes were potentiostatically polarized in a glovebox (MB 200B Eco, MBraun) using a commercial three-electrode glass cell with a flame-annealed carbon rod (Ultra Carbon Corporation) counter electrode and an activated carbon quasi-reference electrode.45 Selected potentials were applied for 15 min, followed by emersion under potential control and flushing with deionized water. Afterward, the specimen was transported to the XPS analyzing chamber in a home-built transfer cell without exposure to ambient for characterization of their surface chemistries. The transfer cell is depicted in Figure S16.
4.4. Computational Details
4.4.1. Ab Initio Thermodynamics
The Pourbaix diagram is calculated by evaluating the surface free energy γ for a range of candidate structures and identifying the most stable surface composition or geometry as the one that minimizes γ at any given bias E and pH. In-depth information on these calculations is provided in Supporting Information Note 3.
4.4.2. DFT Calculations
All DFT calculations (see also Supporting Information Note 3) were performed with the plane-wave DFT code QuantumESPRESSO (QE)46 using the van der Waals-corrected BEEF-vdW exchange correlation functional.47 The basis for all surface candidate structures consists of a seven-layer Mo2C slab, to which oxide layers were added at both sides. A vacuum layer of at least 15 Å ensured a decoupling of consecutive slabs in the supercell geometry. A (1 × 2) MoO2(110) surface unit cell allowed to test O, H, and/or OH concentration variations in 1/4 ML steps. The corresponding surface unit-cell areas A = 27.30 Å2 for bulk phase (β)-1 (Figure S6) and 24.88 Å2 for phase (β)-2 (Figure 2) result from the optimized bulk lattice constants of these two phases of hexagonal Mo2C. Using a plane-wave cutoff of 800 eV and a (4 × 4 × 1) k-grid, all structures were fully relaxed until residual forces fell below 0.03 eV/Å, while keeping the middle three Mo2C slab layers in their frozen bulklike positions. Test calculations with higher cutoffs and k-point grids indicate the obtained surface free energies to be converged within 10 and 1 meV/Å2, respectively. Solvation effects were considered within an implicit solvation approach using the Environ module provided with QE.48 The relative permittivity value of the dielectric layer was set to 78.3 to simulate water. The DFT calculations for the bulk and molecular reservoirs were performed as follows: The bulk hexagonal phase Mo2C was calculated with a (1 × 1 × 1) unit cell and a (12 × 12 × 12) k-grid. The gas-phase molecules (H2, H2O, and CO2) were calculated for electronic energies and vibrational frequencies separately in each supercell with a side length of 10 Å.
Acknowledgments
E.-M.W. and J.K.-L. acknowledge funding by the Austrian Science Foundation (FWF) via the grant I-4114. H.L. gratefully acknowledges funding through the Alexander von Humboldt (AvH) foundation. C.G. thanks the Austrian Research Promotion Agency (FFG) for funding by the project number 870523. Ample computing time was provided through the John von Neumann Institute for Computing (NIC) on the GCS Supercomputer JUWELS at Juelich Supercomputing Centre (JSC). D.E., C.S., and K.R. gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy–EXC 2089/1-390776260. T.G. acknowledges funding by the FWF project number J4278. The authors gratefully acknowledge the support of Prof. Sariciftci for providing the experimental facilities of LIOS in this collaboration and for reading the manuscript. They also acknowledge U. Griesser for giving them access to the Karl–Fischer titration apparatus and B. Kindler for help with the sample preparation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c00415.
Additional characterization (AFM, SEM, TEM, XPS); product analysis (DEMS, GC, IR); additional ab initio thermodynamic calculations; fitted XPS spectra; XPS fitting parameters; determination of Mo2C and its native oxide film thicknesses; current transients; pH correction calculations; and Karl–Fischer titration results (PDF)
Author Contributions
C.G., H.L., and E.-M.W. contributed equally. C.G. and E.-M.W. designed and conducted the experiments. H.L. performed the DFT calculations. D.W. and N.S.N. contributed to sample preparation and product detection. T.G., T.S., A.S.-T., and S.P. performed the TEM studies. D.W. and N.S.S. performed the GC studies. D.E. and C.S. performed calculations of the surface pH changes. J.K.-L. and K.R. supervised and coordinated the project. E.-M.W., C.G., K.R., and J.K.-L. contributed to the manuscript writing. All authors discussed and revised the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Nørskov J. K.; Bligaard T.; Rossmeisl J.; Christensen C. H. Towards the Computational Design of Solid Catalysts. Nat. Chem. 2009, 1, 37–46. 10.1038/nchem.121. [DOI] [PubMed] [Google Scholar]
- Greeley J. Theoretical Heterogeneous Catalysis: Scaling Relationships and Computational Catalyst Design. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 605–635. 10.1146/annurev-chembioeng-080615-034413. [DOI] [PubMed] [Google Scholar]
- Zhao Z.-J.; Liu S.; Zha S.; Cheng D.; Studt F.; Henkelman G.; Gong J. Theory-Guided Design of Catalytic Materials Using Scaling Relationships and Reactivity Descriptors. Nat. Rev. Mater. 2019, 4, 792–804. 10.1038/s41578-019-0152-x. [DOI] [Google Scholar]
- Védrine J. Heterogeneous Catalysis on Metal Oxides. Catalysts 2017, 7, 341 10.3390/catal7110341. [DOI] [Google Scholar]
- Li Z.; Wu Y. 2D Early Transition Metal Carbides (MXenes) for Catalysis. Small 2019, 15, 1804736 10.1002/smll.201804736. [DOI] [PubMed] [Google Scholar]
- Wan W.; Tackett B. M.; Chen J. G. Reactions of Water and C1 Molecules on Carbide and Metal-Modified Carbide Surfaces. Chem. Soc. Rev. 2017, 46, 1807–1823. 10.1039/C6CS00862C. [DOI] [PubMed] [Google Scholar]
- Li H.; Reuter K. Active-Site Computational Screening: Role of Structural and Compositional Diversity for the Electrochemical CO2 Reduction at Mo Carbide Catalysts. ACS Catal. 2020, 10, 11814–11821. [Google Scholar]
- Levy R. B.; Boudart M. Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis. Science 1973, 181, 547–549. 10.1126/science.181.4099.547. [DOI] [PubMed] [Google Scholar]
- Weigert E. C.; Stottlemyer A. L.; Zellner M. B.; Chen J. G. Tungsten Monocarbide as Potential Replacement of Platinum for Methanol Electrooxidation. J. Phys. Chem. C 2007, 111, 14617–14620. 10.1021/jp075504z. [DOI] [Google Scholar]
- Rodriguez J. A.; Illas F. Activation of Noble Metals on Metal-Carbide Surfaces: Novel Catalysts for CO Oxidation, Desulfurization and Hydrogenation Reactions. Phys. Chem. Chem. Phys. 2012, 14, 427–438. 10.1039/C1CP22738F. [DOI] [PubMed] [Google Scholar]
- Hunt S. T.; Nimmanwudipong T.; Román-Leshkov Y. Engineering Non-Sintered, Metal-Terminated Tungsten Carbide Nanoparticles for Catalysis. Angew. Chem., Int. Ed. 2014, 53, 5131–5136. 10.1002/anie.201400294. [DOI] [PubMed] [Google Scholar]
- Huo C.-F.; Li Y.-W.; Wang J.; Jiao H. Insight into CH4 Formation in Iron-Catalyzed Fischer–Tropsch Synthesis. J. Am. Chem. Soc. 2009, 131, 14713–14721. 10.1021/ja9021864. [DOI] [PubMed] [Google Scholar]
- Liu P.; Rodriguez J. A. Water-Gas-Shift Reaction on Molybdenum Carbide Surfaces: Essential Role of the Oxycarbide. J. Phys. Chem. B 2006, 110, 19418–19425. 10.1021/jp0621629. [DOI] [PubMed] [Google Scholar]
- Dubois J.-L.; Sayama K.; Arakawa H. CO2 Hydrogenation over Carbide Catalysts. Chem. Lett. 1992, 1, 5–8. 10.1246/cl.1992.5. [DOI] [Google Scholar]
- Medford A. J.; Vojvodic A.; Studt F.; Abild-Pedersen F.; Nørskov J. K. Elementary Steps of Syngas Reactions on Mo2C(001): Adsorption Thermochemistry and Bond Dissociation. J. Catal. 2012, 290, 108–117. 10.1016/j.jcat.2012.03.007. [DOI] [Google Scholar]
- Gracia J. M.; Prinsloo F. F.; Niemantsverdriet J. W. Mars-van Krevelen-like Mechanism of CO Hydrogenation on an Iron Carbide Surface. Catal. Lett. 2009, 133, 257–261. 10.1007/s10562-009-0179-5. [DOI] [Google Scholar]
- Dhandapani B.; St. Clair T.; Oyama S. T. Simultaneous Hydrodesulfurization, Hydrodeoxygenation, and Hydrogenation with Molybdenum Carbide. Appl. Catal., A 1998, 168, 219–228. 10.1016/S0926-860X(97)00342-6. [DOI] [Google Scholar]
- Ren H.; Hansgen D. A.; Stottlemyer A. L.; Kelly T. G.; Chen J. G. Replacing Platinum with Tungsten Carbide (WC) for Reforming Reactions: Similarities in Ethanol Decomposition on Ni/Pt and Ni/WC Surfaces. ACS Catal. 2011, 1, 390–398. 10.1021/cs200057y. [DOI] [Google Scholar]
- Vojvodic A. Steam Reforming on Transition-Metal Carbides from Density-Functional Theory. Catal. Lett. 2012, 142, 728–735. 10.1007/s10562-012-0820-6. [DOI] [Google Scholar]
- Michalsky R.; Zhang Y.-J.; Medford A. J.; Peterson A. A. Departures from the Adsorption Energy Scaling Relations for Metal Carbide Catalysts. J. Phys. Chem. C 2014, 118, 13026–13034. 10.1021/jp503756g. [DOI] [Google Scholar]
- Peterson A. A.; Nørskov J. K. Activity Descriptors for CO2 Electroreduction to Methane on Transition Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. 10.1021/jz201461p. [DOI] [Google Scholar]
- Li N.; Chen X.; Ong W.-J.; MacFarlane D. R.; Zhao X.; Cheetham A. K.; Sun C. Understanding of Electrochemical Mechanisms for CO2 Capture and Conversion into Hydrocarbon Fuels in Transition-Metal Carbides (MXenes). ACS Nano 2017, 11, 10825–10833. 10.1021/acsnano.7b03738. [DOI] [PubMed] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Kim S. K.; Zhang Y.-J.; Bergstrom H.; Michalsky R.; Peterson A. Understanding the Low-Overpotential Production of CH4 from CO2 on Mo2C Catalysts. ACS Catal. 2016, 2003–2013. 10.1021/acscatal.5b02424. [DOI] [Google Scholar]
- Weidman M. C.; Esposito D. V.; Hsu Y.-C.; Chen J. G. Comparison of Electrochemical Stability of Transition Metal Carbides (WC, W2C, Mo2C) over a Wide pH Range. J. Power Sources 2012, 202, 11–17. 10.1016/j.jpowsour.2011.10.093. [DOI] [Google Scholar]
- Deltombe E.; Pourbaix M.. Atlas of Electrochemical Equilibria in Aqueous Solutions; National Association of Corrosion Engineers, 1974; p 272. [Google Scholar]
- Takeno N.Atlas of E-pH Diagrams: Intercomparison of Thermodynamic Databases, Geological Survey of Japan Open File Report No. 419; Research Center for Deep Geological Environments, 2005.
- Foelske-Schmitz A.X-ray Photoelectron Spectroscopy in Electrochemistry Research. In Encyclopedia of Interfacial Chemistry; Elsevier, 2018; pp 591–606. [Google Scholar]
- Auer A.; Andersen M.; Wernig E.; Hörmann N. G.; Buller N.; Reuter K.; Kunze-Liebhäuser J. Self-Activation of Copper Electrodes during CO Electro-Oxidation in Alkaline Electrolyte. Nat. Catal. 2020, 3, 797 10.1038/s41929-020-00505-w. [DOI] [Google Scholar]
- Bruix A.; Margraf J. T.; Andersen M.; Reuter K. First-Principles-Based Multiscale Modelling of Heterogeneous Catalysis. Nat. Catal. 2019, 2, 659–670. 10.1038/s41929-019-0298-3. [DOI] [Google Scholar]
- Campbell C. T. Transition Metal Oxides: Extra Thermodynamic Stability as Thin Films. Phys. Rev. Lett. 2006, 96, 066106 10.1103/PhysRevLett.96.066106. [DOI] [PubMed] [Google Scholar]
- Reuter K. Ab Initio Thermodynamics and First-Principles Microkinetics for Surface Catalysis. Catal. Lett. 2016, 146, 541–563. 10.1007/s10562-015-1684-3. [DOI] [Google Scholar]
- Chen X.; Liu G.; Zheng W.; Feng W.; Cao W.; Hu W.; Hu P. Vertical 2D MoO2/MoSe2 Core-Shell Nanosheet Arrays as High-Performance Electrocatalysts for Hydrogen Evolution Reaction. Adv. Funct. Mater. 2016, 26, 8537–8544. 10.1002/adfm.201603674. [DOI] [Google Scholar]
- Jian C.; Cai Q.; Hong W.; Li J.; Liu W. Edge-Riched MoSe2/MoO2 Hybrid Electrocatalyst for Efficient Hydrogen Evolution Reaction. Small 2018, 14, 1703798 10.1002/smll.201703798. [DOI] [PubMed] [Google Scholar]
- Zeng H.; Chen S.; Jin Y. Q.; Li J.; Song J.; Le Z.; Liang G.; Zhang H.; Xie F.; Chen J.; Jin Y.; Chen X.; Meng H. Excellent Hydrogen Evolution and Oxidation Activities in Acid. ACS Energy Lett. 2020, 5, 1908–1915. 10.1021/acsenergylett.0c00642. [DOI] [Google Scholar]
- Calvillo L.; Fittipaldi D.; Rüdiger C.; Agnoli S.; Favaro M.; Valero-Vidal C.; Di Valentin C.; Vittadini A.; Bozzolo N.; Jacomet S.; Gregoratti L.; Kunze-Liebhäuser J.; Pacchioni G.; Granozzi G. Carbothermal Transformation of TiO2 into TiOxCy in UHV: Tracking Intrinsic Chemical Stabilities. J. Phys. Chem. C 2014, 118, 22601–22610. 10.1021/jp506728w. [DOI] [Google Scholar]
- Floquet N.; Bertrand O.; Heizmann J. J. Structural and Morphological Studies of the Growth of MoO3 Scales during High-Temperature Oxidation of Molybdenum. Oxid. Met. 1992, 37, 253–280. 10.1007/BF00665191. [DOI] [Google Scholar]
- Watschinger M.; Ploner K.; Winkler D.; Kunze-Liebhäuser J.; Klötzer B.; Penner S. Operando Fourier-Transform Infrared–Mass Spectrometry Reactor Cell Setup for Heterogeneous Catalysis with Glovebox Transfer Process to Surface-Chemical Characterization. Rev. Sci. Instrum. 2021, 92, 024105 10.1063/5.0041437. [DOI] [PubMed] [Google Scholar]
- Saji V. S.; Lee C.-W. Molybdenum, Molybdenum Oxides, and Their Electrochemistry. ChemSusChem 2012, 5, 1146–1161. 10.1002/cssc.201100660. [DOI] [PubMed] [Google Scholar]
- Bazant M. Z.; Chu K. T.; Bayly B. J. Current-Voltage Relations for Electrochemical Thin Films. SIAM J. Appl. Math. 2005, 65, 1463–1484. 10.1137/040609938. [DOI] [Google Scholar]
- Pavlishchuk V. V.; Addison A. W. Conversion Constants for Redox Potentials Measured versus Different Reference Electrodes in Acetonitrile Solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. 10.1016/S0020-1693(99)00407-7. [DOI] [Google Scholar]
- Figueiredo M. C.; Ledezma-Yanez I.; Koper M. T. M. In Situ Spectroscopic Study of CO2 Electroreduction at Copper Electrodes in Acetonitrile. ACS Catal. 2016, 6, 2382–2392. 10.1021/acscatal.5b02543. [DOI] [Google Scholar]
- Walton J.; Wincott P.; Fairley N.; Carrick A.. Peak Fitting with CasaXPS: A Casa Pocket Book; Accolyte Science: Knutsford, UK, 2010. http://www.casaxps.com.
- Gries W. H. A Universal Predictive Equation for the Inelastic Mean Free Pathlengths of X-Ray Photoelectrons and Auger Electrons. Surf. Interface Anal. 1996, 24, 38–50. . [DOI] [Google Scholar]
- Auer A.; Kunze-Liebhäuser J. A Universal Quasi-Reference Electrode for in situ EC-STM. Electrochem. Commun. 2019, 98, 15–18. 10.1016/j.elecom.2018.11.015. [DOI] [Google Scholar]
- Giannozzi P.; Baroni S.; Bonini N.; Calandra M.; Car R.; Cavazzoni C.; Ceresoli D.; Chiarotti G. L.; Cococcioni M.; Dabo I.; Dal Corso A.; De Gironcoli S.; Fabris S.; Fratesi G.; Gebauer R.; Gerstmann U.; Gougoussis C.; Kokalj A.; Lazzeri M.; Martin-Samos L.; Marzari N.; Mauri F.; Mazzarello R.; Paolini S.; Pasquarello A.; Paulatto L.; Sbraccia C.; Scandolo S.; Sclauzero G.; Seitsonen A. P.; Smogunov A.; Umari P.; Wentzcovitch R. M. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials Related Content QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys.: Condens. Matter 2009, 21, 395502 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- Wellendorff J.; Lundgaard K. T.; Møgelhøj A.; Petzold V.; Landis D. D.; Nørskov J. K.; Bligaard T.; Jacobsen K. W. Density Functionals for Surface Science: Exchange-Correlation Model Development with Bayesian Error Estimation. Phys. Rev. B 2012, 85, 235149 10.1103/PhysRevB.85.235149. [DOI] [Google Scholar]
- Andreussi O.; Hörmann N. G.; Nattino F.; Fisicaro G.; Goedecker S.; Marzari N. Solvent-Aware Interfaces in Continuum Solvation. J. Chem. Theory Comput. 2019, 15, 1996–2009. 10.1021/acs.jctc.8b01174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.