

Abstract
Hypercalcemia and hyperphosphatemia associate with an elevated risk of cardiovascular events, yet the pathophysiological basis of this association is unclear. Disturbed mineral homeostasis and the associated hypercalcemia and hyperphosphatemia may result in the formation of circulating calciprotein particles (CPPs) that aggregate the excessive calcium and phosphate ions. If not counteracted, the initially formed harmless amorphous spherical complexes (primary CPPs) may mature into damaging crystalline complexes (secondary CPPs). Secondary CPPs are internalized by vascular cells, causing a massive influx of calcium ions into the cytosol, leading to a proinflammatory response, cellular dysfunction, and cell death. Although the pathophysiological effects induced by CPPs in vascular cells receive increasing attention, a complete picture of how these particles contribute to the development of atherosclerosis and vascular calcification remains elusive. We here discuss existing knowledge on CPP formation and function in atherosclerosis and vascular calcification, techniques for investigating CPPs, and models currently applied to assess CPP-induced cardiovascular pathogenesis. Lastly, we evaluate the potential diagnostic value of serum CPP measurements and the therapeutic potential of anti-CPP therapies currently under development.
Keywords: atherosclerosis, calcium, homeostasis, hypercalcemia, hyperphosphatemia, vascular calcification
Highlights
This review discusses the contribution of calciprotein particles to the pathogenesis of atherosclerosis and vascular calcifications. The important determinants of calciprotein particle formation and the pathogenic processes wherein calciprotein particles are involved are highlighted.
Calciprotein particles are internalized by vascular cells, causing a massive influx of calcium ions into the cytosol, leading to a proinflammatory response, cellular dysfunction, and cell death.
Calciprotein particles are a modifiable risk factor for the development of cardiovascular events.
Pioneering anti-calciprotein particle therapies reduce the risk of cardiovascular events.
Calciprotein particles (CPPs) are blood-borne circulating particles formed of a combination of calcium phosphate and protein.1,2 Their clinical importance stems from the observation that circulating CPP levels are elevated in patients with chronic kidney disease3,4 where vascular calcification develops earlier compared to healthy subjects.5,6 Indeed, increased circulating CPP levels associate with arterial stiffness4 and the development and progression of calcific uremic arteriopathy,3 atherosclerosis,7 and vascular calcification.8 Moreover, the propensity of serum to form CPPs is associated with the occurrence of cardiovascular events and mortality.9–15 Albeit the pathophysiological effects of CPPs receive increasing attention, mechanistic insight into how these particles contribute to the development of atherosclerosis and vascular calcification remains elusive. In this review, we discuss existing knowledge on CPP formation and function in atherosclerosis and vascular calcification, the techniques to investigate CPPs, and models currently applied to assess CPP-induced cardiovascular pathogenesis.
Calcium and Phosphate Homeostasis and the Generation of CPPs
Serum calcium and phosphate levels are tightly regulated in the human body. Calcium and phosphate metabolism includes their intestinal absorption, deposition and resorption from the bone, and renal reabsorption, regulated by calciotropic and phosphotropic factors (reviewed in Renkema et al,16 Peacock,17 Peacock,18 Blaine et al19). Mechanisms maintaining calcium and phosphate homeostasis are redundant and interconnected,16 and their dysregulation may result in hypercalcemia and hyperphosphatemia as well as extraskeletal calcifications, including vascular calcifications.17,18
A network of endogenous inhibitors, with distinct mechanisms of action, prevents and inhibits the formation of extraskeletal calcifications.20 First, the prevention of bone resorption, the decrease in calcium and phosphate reabsorption by the kidneys, and the inhibition of calcium phosphate crystal growth all inhibit extraskeletal calcification. Osteoprotegerin is a decoy RANKL (receptor for the receptor activator of NFκB [nuclear factor κB] ligand)21 precluding osteoclastic differentiation, activation, and bone resorption.22,23 Osteopontin inhibits osteoclastic differentiation and bone resorption, but its vascular expression promotes mineral resorption via unknown mechanisms.24–26 Klotho is a coreceptor for fibroblast growth factor 23 that abates phosphate reabsorption in kidney proximal tubules and biosynthesis of calcitriol, thereby reducing renal tubular calcium reabsorption and intestinal calcium and phosphate absorption.27 Furthermore, inorganic pyrophosphate hinders the nucleation and crystallization of amorphous calcium and inhibits the growth of mature hydroxyapatite crystals.20
Second, circulating calcium scavengers buffer the amount of free calcium available for extraskeletal calcification. Albumin binds ionized calcium (Ca2+) via its negatively charged amino acids distributed on the surface of the tertiary protein structure, scavenging Ca2+ from the microenvironment.1 Similarly, osteonectin scavenges Ca2+ via multiple negatively charged amino acids focused on specific domains, for example, EF-hand (helix-loop-helix) domain.28
Third, CPPs scavenge both free Ca2+ and phosphate (PO43−) ions and sequestering minerals available for extraskeletal calcification. CPPs are blood-borne spongeous carbonate-hydroxyapatite particles, 50 to 500 nm in diameter29,30 that adsorb proteins from their environment.31,32 Fetuin-A, MGP (matrix γ-carboxylated glutamate protein) and GRP (γ-carboxylated glutamate–rich protein) scavenge Ca2+ and PO43− ions from the serum and complex these into clusters of protein and amorphous calcium phosphate (Ca3[PO4]2).1,2,33,34 Fetuin-A scavenges serum Ca2+ and PO43− via its negatively charged extended β-sheet within the amino-terminal cystatin-like D1 domain1,33 and stabilizes nascent clusters of calcium phosphate in its monomeric form33 (Figure 1A). MGP and GRP contain negatively charged γ-carboxylated glutamate residues34,35 which bind both Ca2+ and calcium-containing compounds (Figure 1A).36–39 The interaction between fetuin-A and MGP integrates calcium and phosphate clusters into amorphous proteinaceous spherical particles called primary CPPs (Figure 1B). In physiology, these initially formed primary CPPs are generally considered harmless and facilitate clearance of calcium and phosphate. However, in conditions of hypercalcemia or hyperphosphatemia, primary CPPs ripe into harmful needle-shaped crystalline secondary CPPs containing calcium hydroxyapatite (Ca10[PO4]6[OH]2) by a process called amorphous-to-crystalline transition31,40 (Figure 1C). Serum fetuin-A levels inversely associate with secondary CPP formation,13,41 implying that fetuin-A may act as an inhibitor of amorphous-to-crystalline transition.31 The key determinants of amorphous-to-crystalline transition need further investigation.
Figure 1.
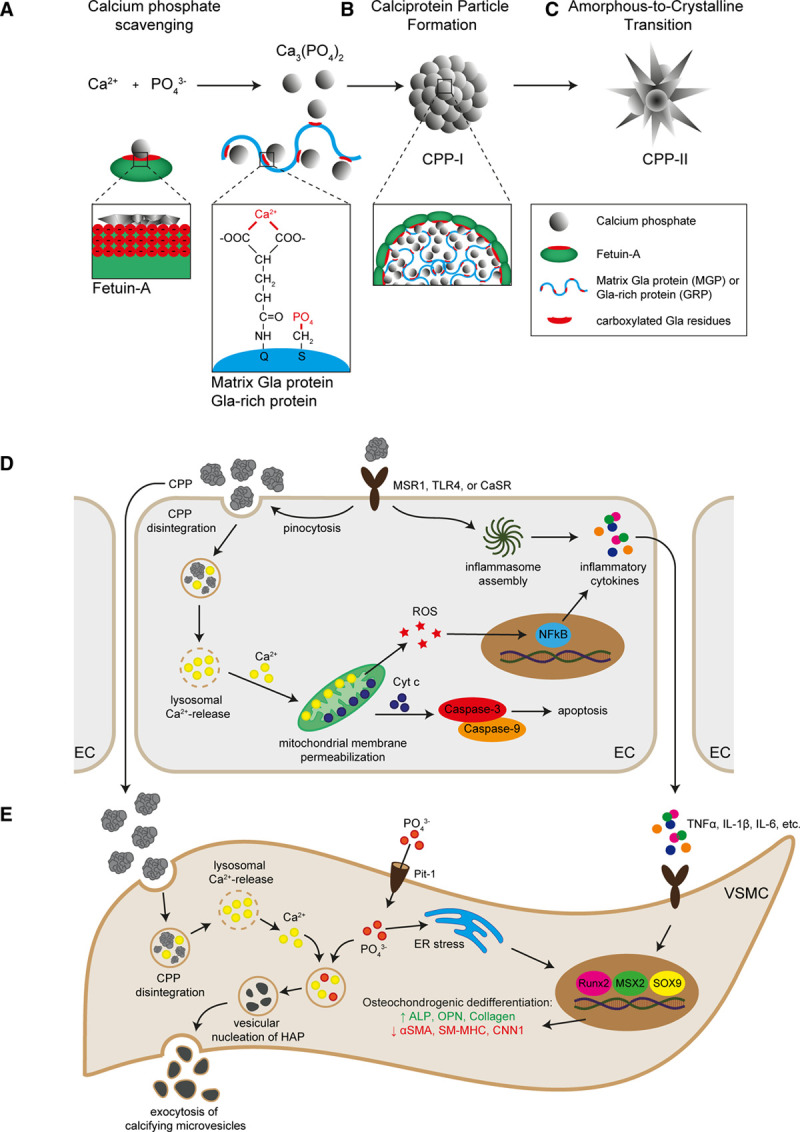
Calciprotein particle (CPP) formation and pathophysiological mechanisms. In the blood, Ca2+ and PO43− form complexes of calcium phosphate that can be scavenged by fetuin-A via the β-sheet of the amino-terminal cystatin-like D1 domain, which contains multiple negatively charged amino acids. MGP (matrix γ-carboxylated glutamate protein) and GRP (γ-carboxylated glutamate–rich protein) scavenge calcium phosphate via their negatively charged amino acids in the γ-carboxylated glutamate residues. Additionally, MGP and GRP scavenge PO43− via the phosphorylation of serine residues (A). The interaction between fetuin-A and MGP integrates calcium phosphate into amorphous spherical particles named primary CPP (B). These primary CPP may ripe into highly crystalline CPP (secondary CPP) under conditions of hypercalcemia and hyperphosphatemia (C). Endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) can internalize CPP via receptor-mediated pinocytosis. In ECs, CPP internalization induces a rise in intracellular Ca2+ level, which results in the inflammatory activation of the ECs, characterized by increased transcellular permeability, oxidative stress, and inflammatory cytokine production (D). In VSMCs, CPP internalization results in a rise in intracellular Ca2+ and PO43− levels that evoke osteochondrogenic dedifferentiation via various mechanisms including inflammatory signaling and oxidative stress. An important molecular consequence of osteochondrogenic dedifferentiation of VSMCs is the production and excretion of calcifying microvesicles, which facilitate vascular calcification (E). α-SMA indicates alpha smooth muscle actin; ALP, alkaline phosphatase; CaSR, calcium-sensing receptor; CNN, calponin; ER, endoplasmatic reticulum; HAP, hydroxyapatite; IL, interleukin; MSR, macrophage scavenge receptor; MSX, homeobox transcription factor muscle segment homeobox; NF, nuclear factor kappa B; OPN, osteopontin; Pit, phosphate transporter; ROS, reactive oxygen species; Runx, runt-related transcription factor; SM-MHC, smooth muscle myosin heavy chain; SOX, sex-determining region Y-box; TLR, toll-like receptor; and TNF, tumor necrosis factor.
MGP, GRP, and fetuin-A are essential to calcium and phosphate homeostasis as mice lacking either protein spontaneously develop extraskeletal calcifications in soft tissues. MGP- and GRP-deficient mice develop medial arterial calcifications34,42,43 and may prematurely die from blood vessel rupture.34 Fetuin-A–deficient mice develop numerous calcified thrombi in the microvasculature44,45 and intimal arterial calcifications on atherosclerosis-prone genetic backgrounds.46 Exogenous fetuin-A supplementation inhibits the development of calcified thrombi in fetuin-A–deficient mice, confirming its relevance to vasculopathy.44 Expectedly, serum Ca2+, PO43−, low fetuin-A, and high CPP levels all associate with the development of vascular pathology.47–49
Hereinafter, it must be noted that proteinaceous CPPs should be clearly distinguished from inorganic calcium phosphate crystals, although an identical mineral composition of these entities may evoke similar downstream events.
CPPs in Cardiovascular Pathophysiology
Internalization, Cell Death and Proinflammatory Signaling
CPPs exert considerable cytotoxic effects on multiple vascular and valvular cell types, including vascular endothelial cells (ECs),32 vascular smooth muscle cells (VSMCs),50 adventitial fibroblasts,51 valve interstitial cells, and valvular ECs.52
Internalization of CPPs is an active process that may occur via clathrin-mediated endocytosis, involving MSR (macrophage scavenge receptor) 1 scavenger receptors and actin polymerization53–55(Figure 1D). CPP shape and crystallinity greatly impact internalization,54 and different cell types have distinct internalization efficacies. Macrophages preferentially internalize secondary CPPs, whereas ECs preferentially internalize primary CPPs.54 The molecular basis behind these distinct internalization patterns is currently unknown but may reflect distinct receptors for primary and secondary CPPs. Indeed, knockdown of the MSR1 gene or blockade of the MSR1 receptor in macrophages diminishes the internalization of secondary CPPs without affecting the internalization of primary CPPs.53,54 Furthermore, the CaSR (calcium-sensing receptor) is expressed on a variety of vascular cells, including ECs, smooth muscle cells, and monocytes56,57 and offers an alternative route for CPP internalization. Blood monocytes internalize secondary CPPs via the CaSR in a Ca2+ concentration-dependent manner, but independently of PO43−.56 Of note, the internalization of inorganic calcium phosphate crystals is also accomplished by clatherin-mediated endocytosis and macropinocytosis,58 suggesting that CPPs and calcium phosphate crystals use similar internalization routes (Figure 1D).
Cytochalasin D, chlorpromazine, and polyinosinic acid lower CPP internalization rates regardless of their physical or chemical properties, indicating that although different surface receptors are responsible for the CPP binding, the downstream mechanism of internalization is similar.53,54 Nevertheless, it should be noted that the mechanisms of CPP internalization have received limited attention to date and need further investigation and independent confirmation.
Inorganic calcium phosphate crystals induce cell death via Ca2+-dependent mitochondrial outer membrane permeabilization.59 Controversy exists as to the exact mechanism of the cytosolic calcium influx; some experimental results indicate mild lysosome membrane permeabilization59,60; other studies report severe lysosomal rupture due to the osmotic difference between the crystal-carrying lysosomes and the cytosol.61 CPPs also induce cell death in a variety of vascular cells, albeit to a lesser extent,32,62,63 and it is tempting to speculate that CPP-induced cell death occurs via similar mechanisms. Of note, the incorporation of fetuin-A into calcium phosphate crystals—effectively generating secondary CPPs—dose-dependently decreases cytotoxicity by limiting particle-induced intracellular Ca2+ elevations.63 The exact mechanism by which CPPs induce cell death remains unclear and may differ between primary and secondary CPPs, as these have distinct crystallinity and therefore solubility in lysosomes.54 Nonetheless, cleavage of caspase-3 and caspase-9 following CPP internalization by vascular cells implies a central role for intrinsic apoptosis (Figure 1D).32,64
CPPs induce expression and secretion of proinflammatory cytokines, including IL (interleukin)-1β, IL-6, IL-8, and TNF (tumor necrosis factor)-α,50,54,55,65,66 potentially via the Ca2+-reactive oxygen species-NFκB-axis or inflammasome activation.56,67–69 Knockdown of the toll-like receptor 4 (TLR4), RANKL, or CaSR gene abrogates secretion of TNF-α and IL-1β after CPP exposure, indicating a paramount role for TLR4, RANKL, and CaSR in CPP-induced cytokine responses.54,56,65 Primary CPPs promote the release of IL-1β, whereas secondary CPPs induce TNF-α secretion,54 suggesting that primary and secondary CPP have distinct receptor binding affinities and evoke distinct signaling cascades. Nonetheless, inflammasome activation is required for CPP-induced cytokine expression, as blocking inflammasome assembly abrogates overall cytokine expression (Figure 1D).70
Endothelial Dysfunction
The endothelium represents a barrier between circulating CPPs and underlying vascular tissue and are the first cell population exposed to CPPs upon their formation. Endothelial inflammatory activation and endothelial dysfunction are triggered by proatherogenic and proinflammatory signaling molecules and key in the development of atherosclerosis and vascular calcification (reviewed in Gimbrone and García-Cardeña,71 Davignon and Ganz,72 Karwowski,73 and Boström74). Understanding how CPPs affect EC behavior75 may partly explain how CPPs contribute to these and possibly other vascular pathologies.
Endothelial dysfunction is defined as the pathological state wherein vasoconstriction occurs as a consequence of an imbalance in the relative contribution of endothelium-derived relaxing and contracting factors.76 It is well established that proatherogenic signaling molecules, including oxidized lipids, evoke endothelial dysfunction,72 which may culminate in hypertensive responses.77,78 CPP number and serum calcification propensity both associate with blood pressure,9,10,79,80 implying CPP may also induce endothelial dysfunction. Moreover, endothelial dysfunction associates with serum fetuin-A levels81 and sevelamer—a calcium binder that reduces circulating CPPs82—preserves endothelial-dependent vasorelaxation and maintains endothelial integrity in mice with chronic kidney disease.83 One possible mechanism by which CPP may induce endothelial dysfunction is by reducing NO bioavailability, either by repressing the expression or activity of eNOS (endothelial NO synthase),84,85 or by the ROS-mediated scavenging of NO.86 Alternatively, CPPs might increase levels of asymmetrical dimethylarginine, an endogenous inhibitor of NO.87 The exact mechanism by which CPPs induce endothelial dysfunction is unknown and warrants further investigation.
Osteochondrogenic dedifferentiation
Vascular calcification is associated with the osteochondrogenic dedifferentiation of VSMCs,88,89 induced by the proatherogenic and proinflammatory milieu.90–92 The osteochondrogenic dedifferentiation of VSMCs is controlled by distinct transcription factors like Runx2 (runt-related transcription factor 2), Osterix, MSX2 (homeobox transcription factor muscle segment homeobox 2), and SOX9 (sex-determining region Y-box 9; reviewed in Durham et al93). Activation of the osteochondrogenic transcription machinery culminates in decreased expression of contractile proteins (eg, α-smooth muscle actin, smooth muscle myosin heavy chain, smoothelin, calponin) and increased expression of osteogenic markers (osteopontin, osteocalcin, alkaline phosphatase, and collagens).94
Another sequel of the osteochondrogenic dedifferentiation of VSMCs is excessive production of core matrisome components (ie, collagens, proteoglycans, and glycoproteins) and extracellular matrix regulators (ie, matrix metalloproteinases and metalloproteases) that contribute to blood vessel remodeling.95,96 This further potentiates the osteochondrogenic dedifferentiation process, aggravating impairment of vascular homeostasis and resulting in a stable proatherogenic microenvironment and increased vascular stiffness.96
VSMC osteochondrogenic dedifferentiation may be induced by a plethora of factors, including oxidized lipids97 and oxidative stress,98 inflammatory cytokines,99 growth factors,100 hormones,101 vitamin D,102 and calcium phosphate crystals.103 Hence, the use of HMG-CoA (β-hydroxy β-methylglutaryl-CoA) reductase inhibitors—more commonly known as statins—has received high interest as potential therapeutic in vascular calcification because of their lipid-lowering and anti-inflammatory effects.104 The inhibition of cholesterol synthesis diminishes cAMP-dependent matrix calcification by VSMC105 and mitigates inflammation-induced artery calcification in rodents106 via mechanisms including the lowering of plasma Ca2+ levels,107 the suppression of autophagy,108 the prevention of phosphate-induced VSMC apoptosis,109,110 and microarchitectural changes in calcium deposits.111 Yet, clinical studies on the use of statin therapy in vascular calcification have been discordant: statins are reported to promote,112,113 suppress,114,115 or have no effect on vascular calcification.116 These discrepancies may be explained by the interaction between statins and BMP (bone morphogenic protein)-2 signaling in VSMC.117,118 The activation of BMP-2 signaling is a key event in vascular calcification as it evokes the expression of the osteochondrogenic transcription factors Runx2 and Osterix.119,120 Indeed, the loss of the BMP-2 inhibitory molecule Smad6 culminates in the aggravation of vascular calcification.92,121 Statins induce the expression of BMP-2117 and BMP receptor II118 in VSMC, which may change the calcification process. Indeed, statins promote macrocalcification of atherosclerotic plaques, irrespective of their plaque-regressing effects.122,123 As macrocalcifications associate with plaque stability,124 these observations may explain why statins decrease cardiovascular risk, despite increasing vascular calcification.124 Thus, a deeper understanding of the mechanisms underlying vascular calcification is warranted and the clinical need for new treatments remains.
It is well accepted that CPPs promote calcification by VSMCs.2,50,62,125 However, controversy exists on the induction of osteochondrogenic dedifferentiation by CPPs. To illustrate, some studies report reduced osteochondrogenic dedifferentiation when the formation of secondary CPPs is blocked125 or CPPs are removed from serum,2 whereas others fail to identify osteochondrogenic gene signatures in the calcified lesions.45
Mechanistic insight on the interference of CPPs on the osteochondrogenic dedifferentiation of VSMC is limited, yet the elimination of CPPs from the serum of patients with end-stage renal disease (ESRD) reduces the serum capacity to induce osteochondrogenic dedifferentiation and abrogates its procalcific capacity.2 Likewise, the addition of CPPs derived from ESRD patients to the serum of healthy blood donors promotes the osteochondrogenic dedifferentiation of VSMCs.2 CPP-induced osteochondrogenic dedifferentiation appears restricted to secondary CPPs, as inhibiting amorphous-to-crystalline transition prevents VSMC calcification.125 In VSMCs, CPPs provoke an increase in cell-bound calcium50,126 and may induce osteochondrogenic differentiation via a multitude of mechanisms (Figure 1E). First, CPPs induce the expression and secretion of TNFα by VSMC,50 which can trigger osteochondrogenic dedifferentiation via the MSX2127 and AP-1 (activator protein 1)128 transcriptional regulators augmenting the expression of Runx2. Second, CPPs may provoke the expression and secretion of BMP-2 by VSMC,103 which induces osteochondrogenic dedifferentiation via increased phosphate transport,129 resulting in endoplasmic reticulum stress and the activation of osteogenic transcription factor XBP1 (x-box binding protein 1).130 Third, CPPs induce VSMC oxidative stress50 which activates a multitude of downstream signaling cascades (eg, Akt [Ak-strain transforming], p38 MAPK [mitogen-activated protein kinase], and NFκB) enhancing the transcriptional activation of the osteochondrogenic differentiation program.131–134 Alternatively, CPPs promote the secretion of IL-6 from EC,64 which may drive the osteochondrogenic differentiation of VSMC in a STAT3 (signal transducer and activator of transcription 3)-dependent manner.135
Calcifying Microvesicles
Vascular calcification occurs in the extracellular space136,137 and is initiated by the secretion of calcifying microvesicles (CMVs) from VSMC138 and plaque macrophages,139 which represent nucleation sites for matrix calcification.140 Cell-derived CMVs are distinct from blood-borne CPPs. CMVs and CPPs differ in origin, size, the presence of membranous proteins and lipids, and crystallinity (Table). CMVs are a heterogeneous group of secreted vesicles, including matrix vesicles and exosomes,157,164,165 which function to maintain mineral homeostasis. Under physiological conditions, CMVs contain inhibitors of calcification, whereas under pathogenic conditions, promoters of calcification are present.158,159,166,167 Once released in the extracellular space, CMV aggregate by annexin-dependent tethering158,160 and bind to matrix collagens161 to form nucleation sites for calcification, culminating in microcalcifications,140 which may fuse to form macrocalcifications within the vessel wall.168
Table 1.
Characteristics of the Various Procalcifying Particles: CaP, CPPs, and CMVs
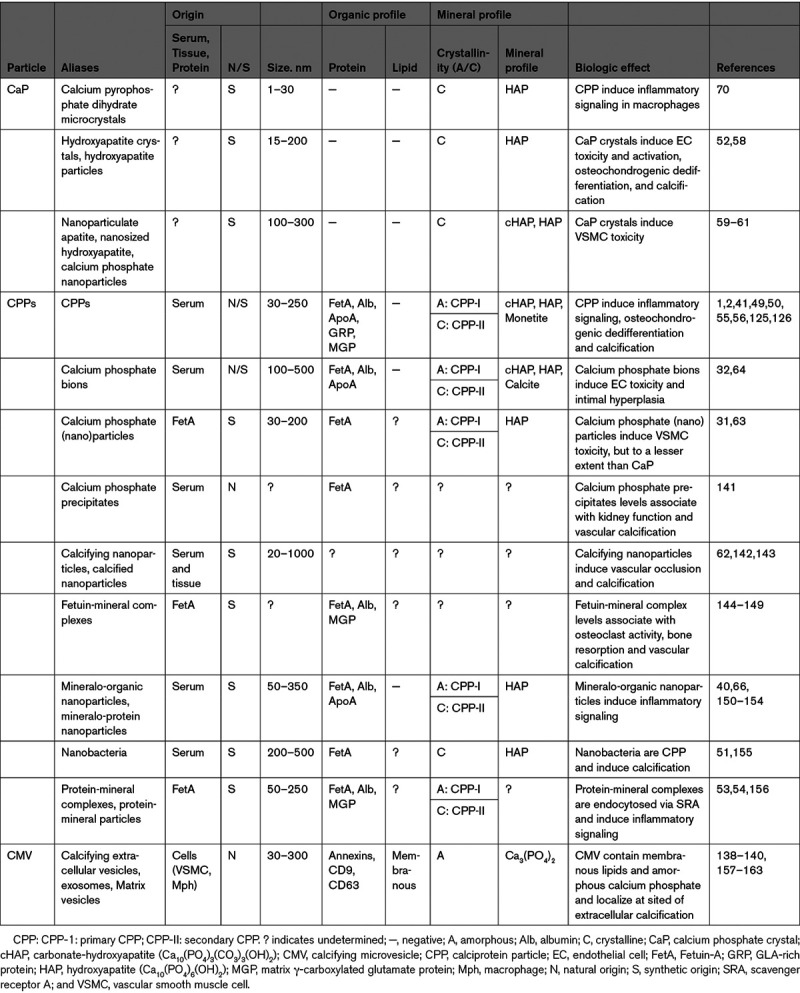
CPPs may influence CMV-mediated calcification in several ways. First, CPPs induce apoptosis of VSMC59 and apoptotic bodies form a nidus for calcification.169,170 Second, CPPs cause a rise in cytoplasmic Ca2+,59 and high cytosolic Ca2+ levels in VSMC result in the formation of procalcifying CMVs158 (Figure 1E). Third, CPPs can be isolated from calcified atherogenic lesions32 wherein CPPs may fuse to and integrate into the developing microcalcifications. How CPPs interfere with CMV-mediated calcification is understudied and a complete picture is lacking. Nonetheless, serum calcification propensity and CPP maturity associate with calcified lesion size,8,171 suggesting an interaction that deserves further evaluation.
Perivascular Adipocytes and Adventitial Fibroblasts
It is increasingly recognized that the perivascular adipose tissue actively contributes to atherogenesis172,173 and vascular calcification.174,175 The perivascular adipose tissue, wherein perivascular adipocytes reside, is a highly metabolic tissue, which secretes a plethora of paracrine signaling molecules, including vasoactive and immunomodulatory factors.176–178 Proatherogenic actions of perivascular adipocytes include the secretion of proinflammatory cytokines,179 the recruitment of inflammatory cells into the vessel wall,180 the induction of smooth muscle cell proliferation in the neointima,181 and the activation of adventitial fibroblasts,182 all facilitating atherogenesis. Moreover, inflammatory activation of the perivascular adipose tissue is associated with decreased plaque stability, vascular calcification, and an increased cardiovascular risk score.174
Adventitial fibroblasts also contribute to atherogenesis183 and vascular calcification.184 Stimulated by atherogenic and proinflammatory signaling molecules, adventitial fibroblasts acquire a motile myofibroblastic phenotype185,186 and migrate into the forming neointima.187,188 Myofibroblast are professional extracellular matrix producing cells, that facilitate neointimal growth by the secretion of collagens and other matrix components.189 Moreover, myofibroblasts secrete a variety of proinflammatory cytokines,190 which enhance endothelial dysfunction, inflammatory cell recruitment into the neointima,191–193 and smooth muscle cell proliferation.186 Notably, vascular calcification may not only occur in the intima or media but also occurs in the adventitia,194 where—under conditions of hypercalcemia and hyperphosphatemia—adventitial myofibroblasts actively contribute to calcium deposition.19
Thus, perivascular adipocytes and adventitial fibroblasts actively contribute to atherogenesis and calcification. Hitherto, it is obscure if, and how CPPs might alter the behavior of these cells, and thus if CPPs mediate vascular pathogenesis via the perivascular adipose tissue or adventitia is unknown.
Dynamics of CPPs In Vivo
Serum CPPs can be isolated from a variety of (pre)clinical animal models32 and patient samples by (ultra)centrifugation,49,141,144,145,150,195 allowing analysis of their quantity, morphology, constituents, and subsequent study of their pathogenicity in in vitro or in vivo models. Alternatively, CPP formation can be replicated in vitro by the supersaturation of serum-supplemented culture medium with calcium salts and phosphates.32,151 Primary and secondary CPPs are, respectively, synthesized by moderate and severe calcium/phosphate supersaturation of the culture medium66,152 or short- and long-term incubation.54 Notably, plaque-derived and synthesized CPPs show morphological and chemical resemblance.32
Intravenous administration of CPPs into normolipidemic rats leads to aortic neointimal lesions in 30% to 40% of rats.64 Such preatherosclerotic niches are characterized by endothelial activation and the osteochondrogenic dedifferentiation of VSMCs, which produce abundant extracellular matrix,64 resembling that in human atherosclerotic plaque development.93,196 Combining CPP administration with balloon-induced vascular injury provokes development of intimal hyperplasia in 50% to 90% of animals,32,142,143 which vary in the presence of calcium phosphate deposits,32,64,142,143 suggesting a secondary hit (eg, dyslipidemia or a chronic low-grade inflammation) as prerequisite for vascular calcification. Intravenous CPP administration has to date only been performed in normolipidemic animals, and it remains unclear whether CPPs are involved in the transition of developing plaques to calcified plaques. Administration of CPPs into atherosclerosis-prone apoE-deficient or low-density lipoprotein receptor–deficient mice with preestablished plaques could clearly answer this question and provide new insights into how CPPs affect atherosclerotic plaque calcification.
Despite the differences between the actions of primary and secondary CPPs in vitro, administration of either CPP type culminates in a similar outcome in vivo; that is, the prevalence of intimal hyperplasia and features of neointima formation by these 2 particle types is similar.32,64 It is tempting to speculate that the administered primary CPPs would mature into secondary CPPs in vivo, but evidence for this is lacking. Alternatively, the shape factor of toxicity of secondary CPPs may become negligible in vivo because of the adsorption of numerous serum proteins that smooth out the otherwise sharp particles.54 In keeping with this hypothesis, mass spectrometry analysis documented a similar protein composition for primary and secondary CPPs derived from various biofluids like serum and ascites, suggestive of an identical adsorption pattern.150
The ability to fluorescently label CPPs by tagging fetuin-A or albumin with fluorescent dyes or generating a fluorescent-fusion fetuin-A/albumin and subsequently incorporating it into synthesized CPPs allows for their pharmacokinetic and pharmacodynamic evaluation (eg, serum half-life, biodistribution, and clearance characteristics) as well as their cellular localization at sites of vascular injury. Alternatively, fluorescent bisphosphonate labeling of calcium phosphate offers a similar strategy to track CPPs in vivo. To illustrate, the intravenous administration of fluorescently labeled CPPs in healthy normolipidemic mice suggests that CPPs have a relatively short serum half-life and are rapidly cleared by the liver and spleen.53,54 In mice deficient in the macrophage scavenger receptor class A/macrophage receptor with a collagenous structure, administered CPPs did not accumulate in liver Kupffer cells or spleen macrophages, suggesting that clearance of CPPs is largely dependent on macrophage uptake.53 Furthermore, in a mouse model of calcified atherosclerosis, fluorescently labeled CPPs accumulate in the vessel lumen and plaque area and colocalize to the endothelium and macrophages.53 No CPPs were found in the arterial wall, suggesting that CPPs did not associate with VSMCs. Noteworthy, however, is that the fluorescence intensity of CPPs critically depends on the maturity of the particles and the extent of crystallinity54 and may not provide a sufficiently strong signal for complete in vivo imaging.
Although investigations on the in vivo effects of CPPs on the vasculature are in their infancy, development of in vivo imaging tools to assess the dynamics of CPPs, their distribution, and detection of the cell types they associate with, will undoubtedly increase insight into the pathophysiological role of CPPs in the cardiovascular system. Advances in CPP imaging enable investigation of key questions about the identity of cell types affected by CPPs in vivo or whether the detrimental effects of CPPs are limited to the cardiovascular system. These developments could culminate in the development of specific therapies targeting CPPs.
Clinical Relevance of CPPs: a Biomarker and Modifiable Risk Factor for Cardiovascular Pathology
The serum of patients with ESRD, coronary artery disease, or arterial hypertension has a greater propensity to CPP formation than serum from healthy blood donors.79 Increased propensity to generate CPPs is associated with adverse cardiovascular outcomes (ie, all-cause and cardiovascular death, myocardial infarction, and peripheral artery disease) in patients with predialysis chronic kidney disease (CKD)9 and ESRD, including kidney transplant recipients.12,15 Moreover, the augmented propensity to form CPPs associates with the occurrence and progression of severe coronary artery calcifications and atherosclerotic cardiovascular events in patients with CKD stages 2 to 4.14,171 These observations were partially verified by findings of a recent study that patients with acute coronary syndrome have higher CPP serum levels than patients with stable angina (without predialysis CKD or ESRD) and serum CPP levels correlate with the total and lipid plaque volumes.7 Hence, serum CPP levels may be considered a surrogate marker of coronary atherosclerosis and coronary artery calcification. Meta-analyses demonstrating a link between reduced serum fetuin-A and albumin and a higher risk of coronary artery disease, additionally testify to the potential importance of elevated calcification propensity in the pathogenesis of atherosclerosis.197,198
A method to determine calcification propensity has been developed which may be used for diagnostic approaches; CPP formation in patient serum is induced by supersaturating the serum with calcium and phosphate and measuring the optical density after incubation (Figure 2A). Other methods to quantify CPPs in serum and biofluids include microplate-based dynamic light-scattering and electron or atomic force microscopy. Microplate-based dynamic light scattering is both a high-throughput and precise method for estimating the hydrodynamic radius of nanoparticles and can be modified to detect CPPs.8 Alternatively, electron or atomic force microscopy are low-throughput but demonstrative methods for CPP visualization2,49 (Figure 2B). Alternatively, one-half maximal transition time has been established as a measure of primary-to-secondary CPP transition, and a prognostic biomarker in various patient cohorts (Figure 2C).9–15,79 Although this method provides a surrogate marker suggesting elevated CPP formation in disease, it remains unclear if all types of CPPs are equally detected, what their composition is, and whether the actual concentration of circulating CPPs is indeed elevated. Nonetheless, validation by independent groups of the association between a decreased one-half maximal transition time and the occurrence of pathology are appearing in literature.199,200
Figure 2.
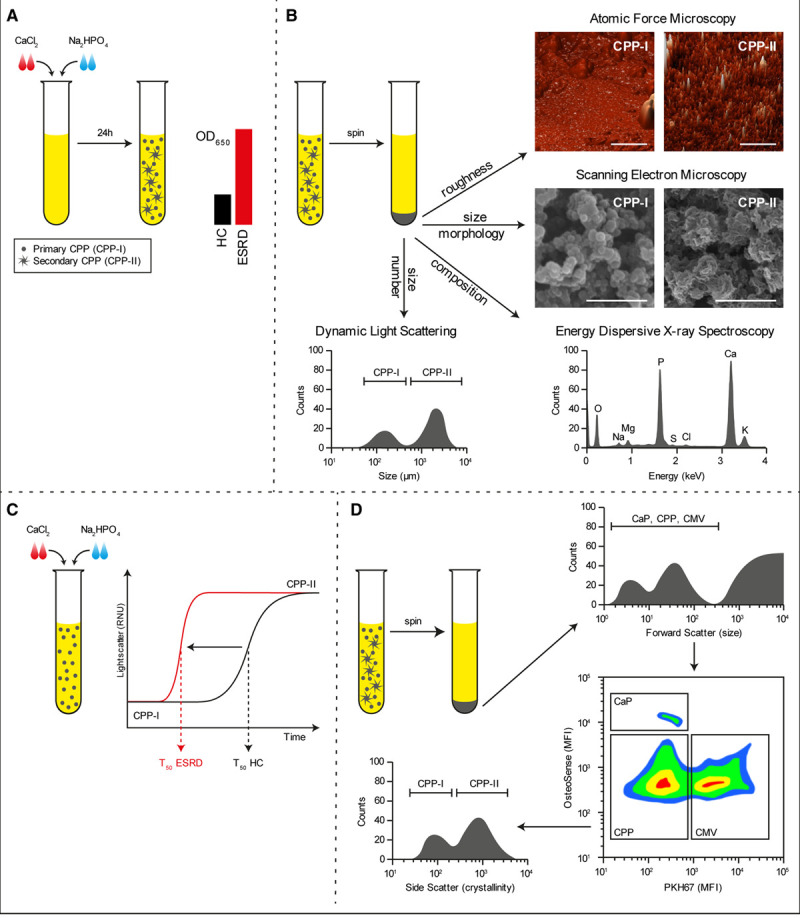
Methods to detect calciprotein particles (CPPs) in clinical samples. Supersaturation of serum with calcium chloride (CaCl2) and sodium diphosphate (Na2HPO4) followed by incubation under culture conditions for 24 h causes the formation of CPPs that can be measured by absorbance at 650 nm. In disease conditions wherein CPP levels are increased, the OD650 readings increase (A). Alternatively, CPPs can be pelleted by centrifugation and investigated by dynamic light scattering to assess particle size, electron and atomic force microscopy to assess morphology, or elemental analysis (EDX) to assess mineral constituent (B). Supersaturation of serum is also used to measure the one-half maximal transition time needed for amorphous-to-crystalline transition (T50). An increased serum propensity for secondary CPP formation is observed as a reduction in T50 (C). A novel flow cytometry-based technique allows for the direct quantification of CPP levels in serum. Here, serum precipitates are labeled with a combination of a fluorescent bisphosphonate (osteoSense) and a fluorescent membrane-intercalating dye (PKH67) and separated based on size, calcium phosphate content, and the presence of membranous lipids. CPPs are observed as OsteoSense+/PKH67− events that fluoresce dim compared to calcium phosphate crystal (CaP) crystals. CPPs are further characterized as primary- or secondary CPPs based on crystallinity (D). CMVs indicates calcifying microvesicles; ESRD, end-stage renal disease sample; HC, healthy control sample; MFI, mean fluorescence intensity; and OD, optical density.
A recently introduced flow cytometry-based technique allows for direct quantification of CPPs in serum and other biofluids (Figure 2D), which may be translated into routine clinical diagnostics. In this protocol, CPP and membranous extracellular vesicles are separated from other cellular particulates by size-exclusion or ultracentrifugation and further characterized by a combination of a fluorescently labeled bisphosphonate (OsteoSense 680EX) that labels mineral deposits and a green fluorescent membrane-intercalating dye (PKH67) that labels membranous structures. Using this technique, CPPs are detected as OsteoSense+/PKH67− events, whereas calcifying extracellular vesicles appear as OsteoSense+/PKH67+ events.7,162 Moreover, CPPs can be further discriminated on basis of their light-scattering properties, allowing for the separate quantification of primary and secondary CPPs162 (Figure 2D).
The clinical significance of serum CPPs is highlighted by the recent TACT (Trial to Assess Chelation Therapy; https://www.clinicaltrials.gov; Unique identifier: NCT00044213). Serum CPPs can be routinely decalcified using EDTA disodium salt in vitro, and infusion of EDTA culminates in reduced cardiovascular risk in patients. In TACT, the EDTA treatment regimen was associated with 1.22-fold lower risk of a primary composite end point (death from any cause, repeated myocardial infarction, stroke, coronary revascularization, or hospitalization for angina pectoris).201 Notably, in subgroups of patients with diabetes,202 and those having diabetes mellitus and peripheral artery disease—2 conditions whereby patients have elevated serum CPP levels—the reduction in risk scores was even greater (1.69- and 1.92-fold, respectively).203 Although EDTA therapy is relatively safe,204 its limited bioavailability (≈5%) when taken orally205 limits its clinical use. Follow-up trials (TACT2 [Trial to Assess Chelation Therapy-2; https://www.clinicaltrials.gov; Unique identifier: NCT02733185] and TACT3a [Trial to Assess Chelation Therapy-3a; https://www.clinicaltrials.gov; Unique identifier: NCT03982693] trials) are ongoing, focused on the efficacy of chelation therapy specifically in diabetic patients with prior myocardial infarctions and individuals with diabetes and critical limb ischemia resulting from severe peripheral atherosclerosis, respectively. Besides chelation therapy, new clinical studies are starting that specifically aim to reduce the serum calcification propensity or the number of circulating CPPs.82,206,207 Albeit their initial data indicates a successful reduction in CPP formation, their effects on long-term cardiovascular risk have yet to become apparent.
Future Perspectives and Therapeutic Implications for CPPs in Cardiovascular Pathology
The clinical relevance of elevated circulating CPP levels is illustrated by a significant correlation between an augmented calcification propensity or increased number of circulating CPPs and a higher risk of adverse outcomes, including major cardiovascular events and mortality.9–14 As CPPs represent a modifiable risk factor for cardiovascular diseases, pioneering clinical trials aimed at reducing the level of circulating CPPs are ongoing.82,206,207 Despite current advances in CPP research, revealing their clinical relevance to cardiovascular morbidity and primary modes of action, many questions remain unanswered.
First, we propose that the methods for obtaining CPPs require standardization, as their current nomenclature (Table), isolation techniques, and synthesis methods are diverse. CPP extraction from biological fluids is currently limited to the serum of etidronate-, vitamin D–treated, or uremic rats,141,144–148,156 with only few studies reporting the isolation of CPPs from human blood or tissue.2,32 Moreover, the pathogenic capacity of CPPs may depend on the health of the serum donor. Although CPPs can be synthesized in vitro by combining serum, Ca2+, and PO43−, a systematic and detailed comparison of CPPs synthesized using serum from cardiovascular patients and CPPs produced using serum from healthy human volunteers is lacking. We recommend performing in-depth characterization of CPPs’ physicochemical properties (eg, Ca2+, phosphate and protein content, particle size, and crystallinity) and comparing them to native CPPs isolated from patient sera, before using in vitro synthesized CPPs for mechanistic studies. Moreover, rather than the current multitude of protocols used to synthesize CPP in vitro, the research field would benefit from standardization.
Second, the current classification of CPPs into either primary (amorphous) or secondary (crystalline) particles may be oversimplified. CPPs can adsorb macromolecules from the ambient fluid and undergo dissolution-reprecipitation and ion exchange reactions.150,153,154,208 This leads to formation of a variety of different particles, not limited to certain sets hitherto defined as primary or secondary CPPs. Moreover, the exact shape, crystallinity, and chemical composition of CPPs within tissues are affected by several local factors including pH, amount, and relative proportion of available mineral ions,209 and the conformation of CPPs present in the vascular tissues they affect remains unclear. We strongly recommend comprehensive mineral and organic profiling as CPP effects, and their molecular mechanisms are defined by these physical and chemical features. This profiling would preferentially include the visualization of CPP size, structure, shape, crystallinity, and chemical composition combined with mass spectrometry approaches to determine the protein composition.
Third, it remains unclear whether particle formation under conditions of hyperphosphatemia is restricted to Ca2+ and whether alternative protein-mineral particles have pathophysiological properties like those of CPPs. Comparing the pathogenic effects of magnesium phosphate particles with the same size, shape, and organic profile as CPPs, we found that, unlike CPPs, these particles lack pathogenic capacity, suggesting that the pathogenic potential of CPPs is defined by its mineral component and possibly its crystallinity and not its proteinaceous constituents.64 Moreover, administration of CPPs produced using pyrophosphate—a phosphate substitute that does not allow for hydroxyapatite crystal formation—causes no pathogenic effects, suggesting that the specific crystals, and not the Ca2+ or phosphate, possess pathogenic capacity.210
Fourth, current understanding of the signaling mechanisms evoked by CPP exposure is inadequate. Valuable information on the signaling mechanisms underlying CPP-mediated pathogenesis has been obtained from in vitro experiments (discussed in this review), but the observation that CPPs induce massive cell death in vitro but not in vivo suggests that CPP may evoke different signaling events in vitro and in vivo and may explain why current methodologies have been unable to identify clear alterations in signaling pathways. This illustrates the need to develop in vitro systems that mimic pathophysiology more closely. Furthermore, recent advances in high-throughput “-omics” approaches (RNA-sequencing, ribosome profiling, and mass spectrometry) will in the future provide a better insight into CPP-mediated signaling in primary vascular cells, as the lack of such data currently inhibits our understanding of cell-specific effects of CPPs and their involvement in pathogenesis. We propose that using single-cell RNA-sequencing can separate the process of cell death and other signaling events after exposure of vascular cell populations to CPPs. This approach can be complemented by combining CPP exposure with established cardiovascular risk factors (hypoxia, oxidized low-density lipoprotein cholesterol, advanced glycation end-products).
Regarding the in vivo studies reported to date, CPPs display different pathogenic behavior in animals and humans. In humans, elevated levels of CPPs have been primarily associated with increased vascular calcification,3,9,149 whereas in rodents CPP administration is associated with intimal hyperplasia and atherosclerosis64 and a highly variable frequency of vessel calcification.32,64,142,143 It should, however, be noted that the animal models currently used for CPP administration are normolipidemic, without a renal phenotype. Performing further studies to investigate the ability of CPPs to induce or aggravate vascular calcification would best be conducted in animal models that are predisposed to vascular calcification, such as partially nephrectomized rodents, or animals with dyslipidemia or inherently disturbed mineral homeostasis.
From clinical perspective, the elevation of circulating CPPs levels in patients with acute coronary syndrome compared with those with stable angina suggest possible importance of this parameter to prognosticate ischemic heart disease. Circulating CPP levels may also have prognostic value in other patient cohorts, including individuals with osteopenia/osteoporosis, primary hyperparathyroidism, or CKD, as these conditions are characterized by hypercalcemia and hyperphosphatemia, and the concentration of CPPs in the blood is closely reflected by patients’ mineralization status. As such, investigations into circulating CPP levels may explain the relationship between elevated bone turnover and the increased risk of cardiovascular disorders observed in these patients. Also, noteworthy, however, is that current investigations have focused primarily on measurement of calcification propensity rather than on direct detection of CPPs in the blood. The number of circulating CPPs may better predict cardiovascular outcomes in these patients and would be a valuable addition to measuring calcification propensity.
From a translational perspective, pioneering studies using chelation therapy have established that circulating CPPs indeed represent a modifiable risk factor for cardiovascular outcome, although generalized chelation therapy has its limitations. Future research should focus on identifying Ca2+ chelators with a superior pharmacokinetic profile, or medicaments to facilitate the hepatic clearance of CPPs in patients at risk of developing cardiovascular events. For instance, Mg2+ has been recently suggested as a promising new therapeutic intervention in the development of CPP-induced vascular calcifications, as it dose-dependently delays maturation from primary to secondary CPPs and prevents VSMC calcification in vitro.125 Mg2+-supplementation prevents and reverses the development of vascular calcifications in mice,211 making it a promising therapeutic intervention for patients with increased CPP levels.212 Replacement of calcium carbonate with lanthanum carbonate lowers serum CPP levels in patients with ESRD,213 which may explain its beneficial effect on the attenuation of aortic calcification.214 A recent study proposed 4,6-di-O-(methoxy-diethyleneglycol)-myo-inositol-1,2,3,5-tetrakis(phosphate)—an inositol phosphate analog—as an agent limiting primary-to-secondary CPP transition and preventing vascular calcification.215 These results suggest avenues for future clinical trials of crystallization inhibitors specifically targeting the formation of harmful secondary CPPs, at least in high-risk patients with CKD.
Conclusions
CPPs may be proposed as a relatively novel potential culprit of vascular disease which can be particularly important in patients with a concomitant chronic kidney disease. Yet, exactly how CPPs influence vascular cells and cardiovascular pathology in vitro and vivo remains obscure. Upcoming research may uncover additional detrimental effects of CPPs, or pathways mediating the underlying pathophysiological mechanisms, whereas clinical investigations aim at direct identification of CPPs in the serum to evaluate their association with various cardiovascular pathologies. New insights into CPP-induced cardiovascular pathology will certainly lead to improved therapeutic interventions and possibly benefit cardiovascular outcome.
Sources of Funding
This study was performed as a collaboration between the Research Institute for Complex Issues of Cardiovascular Diseases (Kemerovo, Russia) and the University Medical Center Groningen (The Netherlands), and funded by the Russian Science Foundation (project no. 19-15-00032), the Netherlands Organization for Health Research and Development (project no. 917.16.446) and the Graduate School of Medical Sciences of the University of Groningen. J.-L. Hillebrands is principal investigator within the NIGRAM2+ (NIer Gerichte Research van Arterie tot Mens: centrale rol voor Magnesium++) consortium, funded by Health Holland (LSHM17034) and the Dutch Kidney Foundation (16TKI02).
Disclosures
G. Krenning is Chief Scientific Officer of Sulfateq B.V. (Groningen, The Netherlands), a company that develops small molecule therapeutics. Sulfateq B.V. has no small molecule in development for anti–circulating calciprotein particle (CPP) therapy at present and had no influence on the content of this article. The other authors report no conflicts.
Nonstandard Abbreviations and Acronyms
- BMP
- bone morphogenic protein
- Ca2+
- ionized calcium
- CaSR
- calcium-sensing receptor
- CKD
- chronic kidney disease
- CMVs
- calcifying microvesicles
- CPPs
- calciprotein particles
- ECs
- endothelial cells
- eNOS
- endothelial nitric oxide synthase
- ESRD
- end-stage renal disease
- GRP
- γ-carboxylated glutamate–rich protein
- HAP
- hydroxyapatite
- IL
- interleukin
- MGP
- matrix γ-carboxylated glutamate protein
- MSR
- macrophage scavenge receptor
- MSX
- homeobox transcription factor muscle segment homeobox
- NF-κB
- nuclear factor kappa B
- RANKL
- receptor activator of nuclear factor κB ligand
- RUNX
- runt-related transcription factor
- SOX
- sex-determining region Y-box
- TACT
- trial to assess chelation therapy
- TLR
- toll-like receptor
- TNF
- tumor necrosis factor
- VSMCs
- vascular smooth muscle cells
A.G. Kutikhin and L. Feenstra contributed equally.
For Sources of Funding and Disclosures, see page 1618.
Contributor Information
Lian Feenstra, Email: l.feenstra@umcg.nl.
Alexander E. Kostyunin, Email: kostae@kemcardio.ru.
Arseniy E. Yuzhalin, Email: yuzhalin@gmail.com.
Jan-Luuk Hillebrands, Email: j.l.hillebrands@umcg.nl.
References
- 1.Heiss A, DuChesne A, Denecke B, Grötzinger J, Yamamoto K, Renné T, Jahnen-Dechent W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J Biol Chem. 2003;278:13333–13341. doi: 10.1074/jbc.M210868200 [DOI] [PubMed] [Google Scholar]
- 2.Viegas CSB, Santos L, Macedo AL, Matos AA, Silva AP, Neves PL, Staes A, Gevaert K, Morais R, Vermeer C, et al. Chronic kidney disease circulating calciprotein particles and extracellular vesicles promote vascular calcification: a role for GRP (Gla-Rich Protein). Arterioscler Thromb Vasc Biol. 2018;38:575–587. doi: 10.1161/ATVBAHA.117.310578 [DOI] [PubMed] [Google Scholar]
- 3.Smith ER, Cai MM, McMahon LP, Pedagogos E, Toussaint ND, Brumby C, Holt SG. Serum fetuin-A concentration and fetuin-A-containing calciprotein particles in patients with chronic inflammatory disease and renal failure. Nephrology (Carlton). 2013;18:215–221. doi: 10.1111/nep.12021 [DOI] [PubMed] [Google Scholar]
- 4.Smith ER, Ford ML, Tomlinson LA, Rajkumar C, McMahon LP, Holt SG. Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol Dial Transplant. 2012;27:1957–1966. doi: 10.1093/ndt/gfr609 [DOI] [PubMed] [Google Scholar]
- 5.Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–1483. doi: 10.1056/NEJM200005183422003 [DOI] [PubMed] [Google Scholar]
- 6.Moe SM, O’Neill KD, Fineberg N, Persohn S, Ahmed S, Garrett P, Meyer CA. Assessment of vascular calcification in ESRD patients using spiral CT. Nephrol Dial Transplant. 2003;18:1152–1158. doi: 10.1093/ndt/gfg093 [DOI] [PubMed] [Google Scholar]
- 7.Nakazato J, Hoshide S, Wake M, Miura Y, Kuro-O M, Kario K. Association of calciprotein particles measured by a new method with coronary artery plaque in patients with coronary artery disease: a cross-sectional study. J Cardiol. 2019;74:428–435. doi: 10.1016/j.jjcc.2019.04.008 [DOI] [PubMed] [Google Scholar]
- 8.Chen W, Anokhina V, Dieudonne G, Abramowitz MK, Kashyap R, Yan C, Wu TT, de Mesy Bentley KL, Miller BL, Bushinsky DA. Patients with advanced chronic kidney disease and vascular calcification have a large hydrodynamic radius of secondary calciprotein particles. Nephrol Dial Transplant. 2019;34:992–1000. doi: 10.1093/ndt/gfy117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith ER, Ford ML, Tomlinson LA, Bodenham E, McMahon LP, Farese S, Rajkumar C, Holt SG, Pasch A. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J Am Soc Nephrol. 2014;25:339–348. doi: 10.1681/ASN.2013060635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahle DO, Åsberg A, Hartmann A, Holdaas H, Bachtler M, Jenssen TG, Dionisi M, Pasch A. Serum calcification propensity is a strong and independent determinant of cardiac and all-cause mortality in kidney transplant recipients. Am J Transplant. 2016;16:204–212. doi: 10.1111/ajt.13443 [DOI] [PubMed] [Google Scholar]
- 11.Lorenz G, Steubl D, Kemmner S, Pasch A, Koch-Sembdner W, Pham D, Haller B, Bachmann Q, Mayer CC, Wassertheurer S, et al. Worsening calcification propensity precedes all-cause and cardiovascular mortality in haemodialyzed patients. Sci Rep. 2017;7:13368. doi: 10.1038/s41598-017-12859-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasch A, Block GA, Bachtler M, Smith ER, Jahnen-Dechent W, Arampatzis S, Chertow GM, Parfrey P, Ma X, Floege J. Blood calcification propensity, cardiovascular events, and survival in patients receiving hemodialysis in the EVOLVE trial. Clin J Am Soc Nephrol. 2017;12:315–322. doi: 10.2215/CJN.04720416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bostom A, Pasch A, Madsen T, Roberts MB, Franceschini N, Steubl D, Garimella PS, Ix JH, Tuttle KR, Ivanova A, et al. Serum calcification propensity and Fetuin-A: biomarkers of cardiovascular disease in kidney transplant recipients. Am J Nephrol. 2018;48:21–31. doi: 10.1159/000491025 [DOI] [PubMed] [Google Scholar]
- 14.Bundy JD, Cai X, Mehta RC, Scialla JJ, de Boer IH, Hsu CY, Go AS, Dobre MA, Chen J, Rao PS, et al. ; CRIC Study Investigators. Serum calcification propensity and clinical events in CKD. Clin J Am Soc Nephrol. 2019;14:1562–1571. doi: 10.2215/CJN.04710419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keyzer CA, de Borst MH, van den Berg E, Jahnen-Dechent W, Arampatzis S, Farese S, Bergmann IP, Floege J, Navis G, Bakker SJ, et al. Calcification propensity and survival among renal transplant recipients. J Am Soc Nephrol. 2016;27:239–248. doi: 10.1681/ASN.2014070670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renkema KY, Alexander RT, Bindels RJ, Hoenderop JG. Calcium and phosphate homeostasis: concerted interplay of new regulators. Ann Med. 2008;40:82–91. doi: 10.1080/07853890701689645 [DOI] [PubMed] [Google Scholar]
- 17.Peacock M. Calcium metabolism in health and disease. Clin J Am Soc Nephrol. 2010;5Suppl 1S23–S30. doi: 10.2215/CJN.05910809 [DOI] [PubMed] [Google Scholar]
- 18.Peacock M. Phosphate metabolism in health and disease. Calcif Tissue Int. 2021;108:3–15. doi: 10.1007/s00223-020-00686-3 [DOI] [PubMed] [Google Scholar]
- 19.Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. 2015;10:1257–1272. doi: 10.2215/CJN.09750913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bäck M, Aranyi T, Cancela ML, Carracedo M, Conceição N, Leftheriotis G, Macrae V, Martin L, Nitschke Y, Pasch A, et al. Endogenous calcification inhibitors in the prevention of vascular calcification: a consensus statement from the COST action EuroSoftCalcNet. Front Cardiovasc Med. 2018;5:196. doi: 10.3389/fcvm.2018.00196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collin-Osdoby P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res. 2004;95:1046–1057. doi: 10.1161/01.RES.0000149165.99974.12 [DOI] [PubMed] [Google Scholar]
- 22.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min H, Morony S, Sarosi I, Dunstan CR, Capparelli C, Scully S, Van G, Kaufman S, Kostenuik PJ, Lacey DL, et al. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med. 2000;192:463–474. doi: 10.1084/jem.192.4.463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Speer MY, McKee MD, Guldberg RE, Liaw L, Yang HY, Tung E, Karsenty G, Giachelli CM. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J Exp Med. 2002;196:1047–1055. doi: 10.1084/jem.20020911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H, Giachelli CM. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol. 2002;161:2035–2046. doi: 10.1016/S0002-9440(10)64482-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paloian NJ, Leaf EM, Giachelli CM. Osteopontin protects against high phosphate-induced nephrocalcinosis and vascular calcification. Kidney Int. 2016;89:1027–1036. doi: 10.1016/j.kint.2015.12.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mencke R, Hillebrands JL; NIGRAM consortium. The role of the anti-ageing protein Klotho in vascular physiology and pathophysiology. Ageing Res Rev. 2017;35:124–146. doi: 10.1016/j.arr.2016.09.001 [DOI] [PubMed] [Google Scholar]
- 28.Busch E, Hohenester E, Timpl R, Paulsson M, Maurer P. Calcium affinity, cooperativity, and domain interactions of extracellular EF-hands present in BM-40. J Biol Chem. 2000;275:25508–25515. doi: 10.1074/jbc.M001770200 [DOI] [PubMed] [Google Scholar]
- 29.Heiss A, Jahnen-Dechent W, Endo H, Schwahn D. Structural dynamics of a colloidal protein-mineral complex bestowing on calcium phosphate a high solubility in biological fluids. Biointerphases. 2007;2:16–20. doi: 10.1116/1.2714924 [DOI] [PubMed] [Google Scholar]
- 30.Rochette CN, Rosenfeldt S, Heiss A, Narayanan T, Ballauff M, Jahnen-Dechent W. A shielding topology stabilizes the early stage protein-mineral complexes of fetuin-A and calcium phosphate: a time-resolved small-angle X-ray study. Chembiochem. 2009;10:735–740. doi: 10.1002/cbic.200800719 [DOI] [PubMed] [Google Scholar]
- 31.Heiss A, Eckert T, Aretz A, Richtering W, van Dorp W, Schäfer C, Jahnen-Dechent W. Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J Biol Chem. 2008;283:14815–14825. doi: 10.1074/jbc.M709938200 [DOI] [PubMed] [Google Scholar]
- 32.Kutikhin AG, Velikanova EA, Mukhamadiyarov RA, Glushkova TV, Borisov VV, Matveeva VG, Antonova LV, Filip’ev DE, Golovkin AS, Shishkova DK, et al. Apoptosis-mediated endothelial toxicity but not direct calcification or functional changes in anti-calcification proteins defines pathogenic effects of calcium phosphate bions. Sci Rep. 2016;6:27255. doi: 10.1038/srep27255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heiss A, Pipich V, Jahnen-Dechent W, Schwahn D. Fetuin-A is a mineral carrier protein: small angle neutron scattering provides new insight on Fetuin-A controlled calcification inhibition. Biophys J. 2010;99:3986–3995. doi: 10.1016/j.bpj.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0 [DOI] [PubMed] [Google Scholar]
- 35.Viegas CS, Rafael MS, Enriquez JL, Teixeira A, Vitorino R, Luís IM, Costa RM, Santos S, Cavaco S, Neves J, et al. Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler Thromb Vasc Biol. 2015;35:399–408. doi: 10.1161/ATVBAHA.114.304823 [DOI] [PubMed] [Google Scholar]
- 36.Berkner KL. Vitamin K-dependent carboxylation. Vitam Horm. 2008;78:131–156. doi: 10.1016/S0083-6729(07)00007-6 [DOI] [PubMed] [Google Scholar]
- 37.Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med. 2013;19:217–226. doi: 10.1016/j.molmed.2012.12.008 [DOI] [PubMed] [Google Scholar]
- 38.Rafael MS, Cavaco S, Viegas CS, Santos S, Ramos A, Willems BA, Herfs M, Theuwissen E, Vermeer C, Simes DC. Insights into the association of Gla-rich protein and osteoarthritis, novel splice variants and γ-carboxylation status. Mol Nutr Food Res. 2014;58:1636–1646. doi: 10.1002/mnfr.201300941 [DOI] [PubMed] [Google Scholar]
- 39.Tesfamariam B. Involvement of vitamin K-dependent proteins in vascular calcification. J Cardiovasc Pharmacol Ther. 2019;24:323–333. doi: 10.1177/1074248419838501 [DOI] [PubMed] [Google Scholar]
- 40.Wong TY, Peng HH, Wu CY, Martel J, Ojcius DM, Hsu FY, Young JD. Nanoparticle conversion to biofilms: in vitro demonstration using serum-derived mineralo-organic nanoparticles. Nanomedicine (Lond). 2015;10:3519–3535. doi: 10.2217/nnm.15.171 [DOI] [PubMed] [Google Scholar]
- 41.Pasch A, Farese S, Gräber S, Wald J, Richtering W, Floege J, Jahnen-Dechent W. Nanoparticle-based test measures overall propensity for calcification in serum. J Am Soc Nephrol. 2012;23:1744–1752. doi: 10.1681/ASN.2012030240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao Y, Bennett BJ, Wang X, Rosenfeld ME, Giachelli C, Lusis AJ, Boström KI. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ Res. 2010;107:485–494. doi: 10.1161/CIRCRESAHA.110.219071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willems BA, Furmanik M, Caron MMJ, Chatrou MLL, Kusters DHM, Welting TJM, Stock M, Rafael MS, Viegas CSB, Simes DC, et al. Ucma/GRP inhibits phosphate-induced vascular smooth muscle cell calcification via SMAD-dependent BMP signalling. Sci Rep. 2018;8:4961. doi: 10.1038/s41598-018-23353-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Babler A, Schmitz C, Buescher A, Herrmann M, Gremse F, Gorgels T, Floege J, Jahnen-Dechent W. Microvasculopathy and soft tissue calcification in mice are governed by fetuin-A, magnesium and pyrophosphate. PLoS One. 2020;15:e0228938. doi: 10.1371/journal.pone.0228938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrmann M, Babler A, Moshkova I, Gremse F, Kiessling F, Kusebauch U, Nelea V, Kramann R, Moritz RL, McKee MD, et al. Lumenal calcification and microvasculopathy in fetuin-A-deficient mice lead to multiple organ morbidity. PLoS One. 2020;15:e0228503. doi: 10.1371/journal.pone.0228503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westenfeld R, Schäfer C, Krüger T, Haarmann C, Schurgers LJ, Reutelingsperger C, Ivanovski O, Drueke T, Massy ZA, Ketteler M, et al. Fetuin-A protects against atherosclerotic calcification in CKD. J Am Soc Nephrol. 2009;20:1264–1274. doi: 10.1681/ASN.2008060572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foley RN, Collins AJ, Ishani A, Kalra PA. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 2008;156:556–563. doi: 10.1016/j.ahj.2008.05.016 [DOI] [PubMed] [Google Scholar]
- 48.Larsson TE, Olauson H, Hagström E, Ingelsson E, Arnlöv J, Lind L, Sundström J. Conjoint effects of serum calcium and phosphate on risk of total, cardiovascular, and noncardiovascular mortality in the community. Arterioscler Thromb Vasc Biol. 2010;30:333–339. doi: 10.1161/ATVBAHA.109.196675 [DOI] [PubMed] [Google Scholar]
- 49.Smith ER, Hewitson TD, Hanssen E, Holt SG. Biochemical transformation of calciprotein particles in uraemia. Bone. 2018;110:355–367. doi: 10.1016/j.bone.2018.02.023 [DOI] [PubMed] [Google Scholar]
- 50.Aghagolzadeh P, Bachtler M, Bijarnia R, Jackson C, Smith ER, Odermatt A, Radpour R, Pasch A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis. 2016;251:404–414. doi: 10.1016/j.atherosclerosis.2016.05.044 [DOI] [PubMed] [Google Scholar]
- 51.Kajander EO, Ciftçioglu N. Nanobacteria: an alternative mechanism for pathogenic intra- and extracellular calcification and stone formation. Proc Natl Acad Sci U S A. 1998;95:8274–8279. doi: 10.1073/pnas.95.14.8274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richards JM, Kunitake JAMR, Hunt HB, Wnorowski AN, Lin DW, Boskey AL, Donnelly E, Estroff LA, Butcher JT. Crystallinity of hydroxyapatite drives myofibroblastic activation and calcification in aortic valves. Acta Biomater. 2018;71:24–36. doi: 10.1016/j.actbio.2018.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herrmann M, Schäfer C, Heiss A, Gräber S, Kinkeldey A, Büscher A, Schmitt MM, Bornemann J, Nimmerjahn F, Herrmann M, et al. Clearance of fetuin-A–containing calciprotein particles is mediated by scavenger receptor-A. Circ Res. 2012;111:575–584. doi: 10.1161/CIRCRESAHA.111.261479 [DOI] [PubMed] [Google Scholar]
- 54.Köppert S, Büscher A, Babler A, Ghallab A, Buhl EM, Latz E, Hengstler JG, Smith ER, Jahnen-Dechent W. Cellular clearance and biological activity of calciprotein particles depend on their maturation state and crystallinity. Front Immunol. 2018;9:1991. doi: 10.3389/fimmu.2018.01991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith ER, Hanssen E, McMahon LP, Holt SG. Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PLoS One. 2013;8:e60904. doi: 10.1371/journal.pone.0060904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jäger E, Murthy S, Schmidt C, Hahn M, Strobel S, Peters A, Stäubert C, Sungur P, Venus T, Geisler M, et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat Commun. 2020;11:4243. doi: 10.1038/s41467-020-17749-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schreckenberg R, Schlüter KD. Calcium sensing receptor expression and signalling in cardiovascular physiology and disease. Vasc Pharmacol. 2018;107:35–42. [DOI] [PubMed] [Google Scholar]
- 58.Huang LH, Sun XY, Ouyang JM. Shape-dependent toxicity and mineralization of hydroxyapatite nanoparticles in A7R5 aortic smooth muscle cells. Sci Rep. 2019;9:18979. doi: 10.1038/s41598-019-55428-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ewence AE, Bootman M, Roderick HL, Skepper JN, McCarthy G, Epple M, Neumann M, Shanahan CM, Proudfoot D. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ Res. 2008;103:e28–e34. doi: 10.1161/CIRCRESAHA.108.181305 [DOI] [PubMed] [Google Scholar]
- 60.Zhang CY, Sun XY, Ouyang JM, Gui BS. Diethyl citrate and sodium citrate reduce the cytotoxic effects of nanosized hydroxyapatite crystals on mouse vascular smooth muscle cells. Int J Nanomedicine. 2017;12:8511–8525. doi: 10.2147/IJN.S145386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Z, Xiao Y, Chen W, Wang Y, Wang B, Wang G, Xu X, Tang R. Calcium phosphate nanoparticles primarily induce cell necrosis through lysosomal rupture: the origination of material cytotoxicity. J Mater Chem B. 2014;2:3480–3489. doi: 10.1039/c4tb00056k [DOI] [PubMed] [Google Scholar]
- 62.Hunter LW, Charlesworth JE, Yu S, Lieske JC, Miller VM. Calcifying nanoparticles promote mineralization in vascular smooth muscle cells: implications for atherosclerosis. Int J Nanomedicine. 2014;9:2689–2698. doi: 10.2147/IJN.S63189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dautova Y, Kozlova D, Skepper JN, Epple M, Bootman MD, Proudfoot D. Fetuin-A and albumin alter cytotoxic effects of calcium phosphate nanoparticles on human vascular smooth muscle cells. PLoS One. 2014;9:e97565. doi: 10.1371/journal.pone.0097565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shishkova D, Velikanova E, Sinitsky M, Tsepokina A, Gruzdeva O, Bogdanov L, Kutikhin A. Calcium phosphate bions cause intimal hyperplasia in intact aortas of normolipidemic rats through endothelial injury. Int J Mol Sci. 2019;20:5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Callegari A, Coons ML, Ricks JL, Rosenfeld ME, Scatena M. Increased calcification in osteoprotegerin-deficient smooth muscle cells: dependence on receptor activator of NF-κB ligand and interleukin 6. J Vasc Res. 2014;51:118–131. doi: 10.1159/000358920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peng HH, Wu CY, Young D, Martel J, Young A, Ojcius DM, Lee YH, Young JD. Physicochemical and biological properties of biomimetic mineralo-protein nanoparticles formed spontaneously in biological fluids. Small. 2013;9:2297–2307. doi: 10.1002/smll.201202270 [DOI] [PubMed] [Google Scholar]
- 67.Dada LA, Sznajder JI. Mitochondrial Ca2+ and ROS take center stage to orchestrate TNF-α-mediated inflammatory responses. J Clin Invest. 2011;121:1683–1685. doi: 10.1172/JCI57748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alevriadou BR, Shanmughapriya S, Patel A, Stathopulos PB, Madesh M. Mitochondrial Ca(2+) transport in the endothelium: regulation by ions, redox signalling and mechanical forces. J R Soc Interface. 2017;14:20170672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Campillo-Gimenez L, Renaudin F, Jalabert M, Gras P, Gosset M, Rey C, Sarda S, Collet C, Cohen-Solal M, Combes C, et al. Inflammatory potential of four different phases of calcium pyrophosphate relies on NF-κB activation and MAPK pathways. Front Immunol. 2018;9:2248. doi: 10.3389/fimmu.2018.02248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gimbrone MA, Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–32. [DOI] [PubMed] [Google Scholar]
- 73.Karwowski W, Naumnik B, Szczepański M, Myśliwiec M. The mechanism of vascular calcification - a systematic review. Med Sci Monit. 2012;18:RA1–R11. doi: 10.12659/msm.882181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boström KI. Where do we stand on vascular calcification? Vascul Pharmacol. 2016;84:8–14. doi: 10.1016/j.vph.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shishkova D, Markova V, Sinitsky M, Tsepokina A, Velikanova E, Bogdanov L, Glushkova T, Kutikhin A. Calciprotein particles cause endothelial dysfunction under flow. Int J Mol Sci. 2020;21:8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–1992. doi: 10.1097/01.ASN.0000132474.50966.DA [DOI] [PubMed] [Google Scholar]
- 77.Hayakawa H, Raij L. Relationship between hypercholesterolaemia, endothelial dysfunction and hypertension. J Hypertens. 1999;17:611–619. doi: 10.1097/00004872-199917050-00004 [DOI] [PubMed] [Google Scholar]
- 78.Konukoglu D, Uzun H. Endothelial dysfunction and hypertension. Adv Exp Med Biol. 2017;956:511–540. doi: 10.1007/5584_2016_90 [DOI] [PubMed] [Google Scholar]
- 79.Pruijm M, Lu Y, Megdiche F, Piskunowicz M, Milani B, Stuber M, Bachtler M, Vogt B, Burnier M, Pasch A. Serum calcification propensity is associated with renal tissue oxygenation and resistive index in patients with arterial hypertension or chronic kidney disease. J Hypertens. 2017;35:2044–2052. doi: 10.1097/HJH.0000000000001406 [DOI] [PubMed] [Google Scholar]
- 80.Eelderink C, Te Velde-Keyzer CA, Frenay AS, Vermeulen EA, Bachtler M, Aghagolzadeh P, van Dijk PR, Gansevoort RT, Vervloet MG, Hillebrands JL, et al. ; NIGRAM2+ consortium. Serum calcification propensity and the risk of cardiovascular and all-cause mortality in the general population: the PREVEND study. Arterioscler Thromb Vasc Biol. 2020;40:1942–1951. doi: 10.1161/ATVBAHA.120.314187 [DOI] [PubMed] [Google Scholar]
- 81.Karabakan M, Bozkurt A, Gunay M, Aktas BK, Hirik E, Aydin M, Nuhoglu B. Association between serum fetuin-A level and erectile function. Andrologia. 2016;48:787–792. doi: 10.1111/and.12513 [DOI] [PubMed] [Google Scholar]
- 82.Smith ER, Pan FFM, Hewitson TD, Toussaint ND, Holt SG. Effect of sevelamer on calciprotein particles in hemodialysis patients: the sevelamer versus calcium to reduce Fetuin-A-containing calciprotein particles in dialysis (SCaRF) randomized controlled trial. Kidney Int Rep. 2020;5:1432–1447. doi: 10.1016/j.ekir.2020.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Six I, Maizel J, Barreto FC, Rangrez AY, Dupont S, Slama M, Tribouilloy C, Choukroun G, Mazière JC, Bode-Boeger S, et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc Res. 2012;96:130–139. doi: 10.1093/cvr/cvs240 [DOI] [PubMed] [Google Scholar]
- 84.Peng A, Wu T, Zeng C, Rakheja D, Zhu J, Ye T, Hutcheson J, Vaziri ND, Liu Z, Mohan C, et al. Adverse effects of simulated hyper- and hypo-phosphatemia on endothelial cell function and viability. PLoS One. 2011;6:e23268. doi: 10.1371/journal.pone.0023268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stevens KK, Denby L, Patel RK, Mark PB, Kettlewell S, Smith GL, Clancy MJ, Delles C, Jardine AG. Deleterious effects of phosphate on vascular and endothelial function via disruption to the nitric oxide pathway. Nephrol Dial Transplant. 2017;32:1617–1627. doi: 10.1093/ndt/gfw252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hong Q, Qi K, Feng Z, Huang Z, Cui S, Wang L, Fu B, Ding R, Yang J, Chen X, et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium. 2012;51:402–410. doi: 10.1016/j.ceca.2012.01.003 [DOI] [PubMed] [Google Scholar]
- 87.D’Marco L, Bellasi A, Raggi P. Cardiovascular biomarkers in chronic kidney disease: state of current research and clinical applicability. Dis Markers. 2015;2015:586569. doi: 10.1155/2015/586569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D, Giachelli CM. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. 2009;104:733–741. doi: 10.1161/CIRCRESAHA.108.183053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Voelkl J, Luong TT, Tuffaha R, Musculus K, Auer T, Lian X, Daniel C, Zickler D, Boehme B, Sacherer M, et al. SGK1 induces vascular smooth muscle cell calcification through NF-κB signaling. J Clin Invest. 2018;128:3024–3040. doi: 10.1172/JCI96477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867 [DOI] [PubMed] [Google Scholar]
- 91.Nadra I, Mason JC, Philippidis P, Florey O, Smythe CD, McCarthy GM, Landis RC, Haskard DO. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ Res. 2005;96:1248–1256. doi: 10.1161/01.RES.0000171451.88616.c2 [DOI] [PubMed] [Google Scholar]
- 92.Li X, Lim J, Lu J, Pedego TM, Demer L, Tintut Y. Protective role of Smad6 in inflammation-induced valvular cell calcification. J Cell Biochem. 2015;116:2354–2364. doi: 10.1002/jcb.25186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114:590–600. doi: 10.1093/cvr/cvy010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tintut Y, Parhami F, Boström K, Jackson SM, Demer LL. cAMP stimulates osteoblast-like differentiation of calcifying vascular cells. Potential signaling pathway for vascular calcification. J Biol Chem. 1998;273:7547–7553. doi: 10.1074/jbc.273.13.7547 [DOI] [PubMed] [Google Scholar]
- 95.Hortells L, Sur S, St Hilaire C. Cell phenotype transitions in cardiovascular calcification. Front Cardiovasc Med. 2018;5:27. doi: 10.3389/fcvm.2018.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jaminon A, Reesink K, Kroon A, Schurgers L. The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci. 2019;20:5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Proudfoot D, Davies JD, Skepper JN, Weissberg PL, Shanahan CM. Acetylated low-density lipoprotein stimulates human vascular smooth muscle cell calcification by promoting osteoblastic differentiation and inhibiting phagocytosis. Circulation. 2002;106:3044–3050. doi: 10.1161/01.cir.0000041429.83465.41 [DOI] [PubMed] [Google Scholar]
- 98.Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med. 2001;31:509–519. doi: 10.1016/s0891-5849(01)00610-4 [DOI] [PubMed] [Google Scholar]
- 99.Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler Thromb Vasc Biol. 2007;27:2589–2596. doi: 10.1161/ATVBAHA.107.153668 [DOI] [PubMed] [Google Scholar]
- 100.Nakagawa Y, Ikeda K, Akakabe Y, Koide M, Uraoka M, Yutaka KT, Kurimoto-Nakano R, Takahashi T, Matoba S, Yamada H, et al. Paracrine osteogenic signals via bone morphogenetic protein-2 accelerate the atherosclerotic intimal calcification in vivo. Arterioscler Thromb Vasc Biol. 2010;30:1908–1915. doi: 10.1161/ATVBAHA.110.206185 [DOI] [PubMed] [Google Scholar]
- 101.Carrillo-López N, Panizo S, Alonso-Montes C, Martínez-Arias L, Avello N, Sosa P, Dusso AS, Cannata-Andía JB, Naves-Díaz M. High-serum phosphate and parathyroid hormone distinctly regulate bone loss and vascular calcification in experimental chronic kidney disease. Nephrol Dial Transplant. 2019;34:934–941. doi: 10.1093/ndt/gfy287 [DOI] [PubMed] [Google Scholar]
- 102.Han MS, Che X, Cho GH, Park HR, Lim KE, Park NR, Jin JS, Jung YK, Jeong JH, Lee IK, et al. Functional cooperation between vitamin D receptor and Runx2 in vitamin D-induced vascular calcification. PLoS One. 2013;8:e83584. doi: 10.1371/journal.pone.0083584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sage AP, Lu J, Tintut Y, Demer LL. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;79:414–422. doi: 10.1038/ki.2010.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taqueti VR, Ridker PM. Lipid-lowering and anti-inflammatory benefits of statin therapy: more than meets the plaque. Circ Cardiovasc Imaging. 2017;10:e006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Geng Y, Hsu JJ, Lu J, Ting TC, Miyazaki M, Demer LL, Tintut Y. Role of cellular cholesterol metabolism in vascular cell calcification. J Biol Chem. 2011;286:33701–33706. doi: 10.1074/jbc.M111.269639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin CP, Huang PH, Lai CF, Chen JW, Lin SJ, Chen JS. Simvastatin attenuates oxidative stress, NF-κB activation, and artery calcification in LDLR-/- Mice Fed with high fat diet via down-regulation of tumor necrosis factor-α and TNF receptor 1. PLoS One. 2015;10:e0143686. doi: 10.1371/journal.pone.0143686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li H, Tao HR, Hu T, Fan YH, Zhang RQ, Jia G, Wang HC. Atorvastatin reduces calcification in rat arteries and vascular smooth muscle cells. Basic Clin Pharmacol Toxicol. 2010;107:798–802. doi: 10.1111/j.1742-7843.2010.00580.x [DOI] [PubMed] [Google Scholar]
- 108.Liu D, Cui W, Liu B, Hu H, Liu J, Xie R, Yang X, Gu G, Zhang J, Zheng H. Atorvastatin protects vascular smooth muscle cells from TGF-β1-stimulated calcification by inducing autophagy via suppression of the β-catenin pathway. Cell Physiol Biochem. 2014;33:129–141. doi: 10.1159/000356656 [DOI] [PubMed] [Google Scholar]
- 109.Iijima K, Ito Y, Son BK, Akishita M, Ouchi Y. Pravastatin and olmesartan synergistically ameliorate renal failure-induced vascular calcification. J Atheroscler Thromb. 2014;21:917–929. doi: 10.5551/jat.23218 [DOI] [PubMed] [Google Scholar]
- 110.Son BK, Kozaki K, Iijima K, Eto M, Kojima T, Ota H, Senda Y, Maemura K, Nakano T, Akishita M, et al. Statins protect human aortic smooth muscle cells from inorganic phosphate-induced calcification by restoring Gas6-Axl survival pathway. Circ Res. 2006;98:1024–1031. doi: 10.1161/01.RES.0000218859.90970.8d [DOI] [PubMed] [Google Scholar]
- 111.Xian JZ, Lu M, Fong F, Qiao R, Patel NR, Abeydeera D, Iriana S, Demer LL, Tintut Y. Statin effects on vascular calcification: microarchitectural changes in aortic calcium deposits in aged hyperlipidemic mice. Arterioscler ThrombVasc Biol. 2021;41:e185–e192. doi: 10.1161/ATVBAHA.120.315737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Henein M, Granåsen G, Wiklund U, Schmermund A, Guerci A, Erbel R, Raggi P. High dose and long-term statin therapy accelerate coronary artery calcification. Int J Cardiol. 2015;184:581–586. doi: 10.1016/j.ijcard.2015.02.072 [DOI] [PubMed] [Google Scholar]
- 113.Saremi A, Bahn G, Reaven PD; VADT Investigators. Progression of vascular calcification is increased with statin use in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care. 2012;35:2390–2392. doi: 10.2337/dc12-0464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ikegami Y, Inoue I, Inoue K, Shinoda Y, Iida S, Goto S, Nakano T, Shimada A, Noda M. The annual rate of coronary artery calcification with combination therapy with a PCSK9 inhibitor and a statin is lower than that with statin monotherapy. NPJ Aging Mech Dis. 2018;4:7. doi: 10.1038/s41514-018-0026-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Achenbach S, Ropers D, Pohle K, Leber A, Thilo C, Knez A, Menendez T, Maeffert R, Kusus M, Regenfus M, et al. Influence of lipid-lowering therapy on the progression of coronary artery calcification: a prospective evaluation. Circulation. 2002;106:1077–1082. doi: 10.1161/01.cir.0000027567.49283.ff [DOI] [PubMed] [Google Scholar]
- 116.Rifai MA, Blaha MJ, Patel J, Xiaoming J, Cainzos-Achirica M, Greenland P, Budoff M, Yeboah J, Nasir K, Al-Mallah MH, et al. Coronary artery calcification, statin use and long-term risk of atherosclerotic cardiovascular disease events (from the Multi-Ethnic Study of Atherosclerosis). Am J Cardiol. 2020;125:835–839. doi: 10.1016/j.amjcard.2019.12.031 [DOI] [PubMed] [Google Scholar]
- 117.Emmanuele L, Ortmann J, Doerflinger T, Traupe T, Barton M. Lovastatin stimulates human vascular smooth muscle cell expression of bone morphogenetic protein-2, a potent inhibitor of low-density lipoprotein-stimulated cell growth. Biochem Biophys Res Commun. 2003;302:67–72. doi: 10.1016/s0006-291x(03)00109-8 [DOI] [PubMed] [Google Scholar]
- 118.Hu H, Sung A, Zhao G, Shi L, Qiu D, Nishimura T, Kao PN. Simvastatin enhances bone morphogenetic protein receptor type II expression. Biochem Biophys Res Commun. 2006;339:59–64. doi: 10.1016/j.bbrc.2005.10.187 [DOI] [PubMed] [Google Scholar]
- 119.Matsubara T, Kida K, Yamaguchi A, Hata K, Ichida F, Meguro H, Aburatani H, Nishimura R, Yoneda T. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J Biol Chem. 2008;283:29119–29125. doi: 10.1074/jbc.M801774200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Balderman JA, Lee HY, Mahoney CE, Handy DE, White K, Annis S, Lebeche D, Hajjar RJ, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 decreases microRNA-30b and microRNA-30c to promote vascular smooth muscle cell calcification. J Am Heart Assoc. 2012;1:e003905. doi: 10.1161/JAHA.112.003905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lim J, Ehsanipour A, Hsu JJ, Lu J, Pedego T, Wu A, Walthers CM, Demer LL, Seidlits SK, Tintut Y. Inflammation drives retraction, stiffening, and nodule formation via cytoskeletal machinery in a three-dimensional culture model of aortic stenosis. Am J Pathol. 2016;186:2378–2389. doi: 10.1016/j.ajpath.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Puri R, Nicholls SJ, Shao M, Kataoka Y, Uno K, Kapadia SR, Tuzcu EM, Nissen SE. Impact of statins on serial coronary calcification during atheroma progression and regression. J Am Coll Cardiol. 2015;65:1273–1282. doi: 10.1016/j.jacc.2015.01.036 [DOI] [PubMed] [Google Scholar]
- 123.Lee SE, Sung JM, Andreini D, Budoff MJ, Cademartiri F, Chinnaiyan K, Choi JH, Chun EJ, Conte E, Gottlieb I, et al. Differential association between the progression of coronary artery calcium score and coronary plaque volume progression according to statins: the Progression of AtheRosclerotic PlAque DetermIned by Computed TomoGraphic Angiography Imaging (PARADIGM) study. Eur Heart J Cardiovasc Imaging. 2019;20:1307–1314. doi: 10.1093/ehjci/jez022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Barrett HE, Van der Heiden K, Farrell E, Gijsen FJH, Akyildiz AC. Calcifications in atherosclerotic plaques and impact on plaque biomechanics. J Biomech. 2019;87:1–12. doi: 10.1016/j.jbiomech.2019.03.005 [DOI] [PubMed] [Google Scholar]
- 125.Ter Braake AD, Eelderink C, Zeper LW, Pasch A, Bakker SJL, de Borst MH, Hoenderop JGJ, de Baaij JHF. Calciprotein particle inhibition explains magnesium-mediated protection against vascular calcification. Nephrol Dial Transplant. 2020;35:765–773. doi: 10.1093/ndt/gfz190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Aghagolzadeh P, Radpour R, Bachtler M, van Goor H, Smith ER, Lister A, Odermatt A, Feelisch M, Pasch A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis. 2017;265:78–86. doi: 10.1016/j.atherosclerosis.2017.08.012 [DOI] [PubMed] [Google Scholar]
- 127.Lee HL, Woo KM, Ryoo HM, Baek JH. Tumor necrosis factor-alpha increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem Biophys Res Commun. 2010;391:1087–1092. doi: 10.1016/j.bbrc.2009.12.027 [DOI] [PubMed] [Google Scholar]
- 128.Zickler D, Luecht C, Willy K, Chen L, Witowski J, Girndt M, Fiedler R, Storr M, Kamhieh-Milz J, Schoon J, et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol Dial Transplant. 2018;33:574–585. doi: 10.1093/ndt/gfx316 [DOI] [PubMed] [Google Scholar]
- 129.Li X, Yang HY, Giachelli CM. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis. 2008;199:271–277. doi: 10.1016/j.atherosclerosis.2007.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liberman M, Johnson RC, Handy DE, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun. 2011;413:436–441. doi: 10.1016/j.bbrc.2011.08.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu H, Li X, Qin F, Huang K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J Biol Inorg Chem. 2014;19:375–388. doi: 10.1007/s00775-013-1078-1 [DOI] [PubMed] [Google Scholar]
- 133.Blanc A, Pandey NR, Srivastava AK. Distinct roles of Ca2+, calmodulin, and protein kinase C in H2O2-induced activation of ERK1/2, p38 MAPK, and protein kinase B signaling in vascular smooth muscle cells. Antioxid Redox Signal. 2004;6:353–366. doi: 10.1089/152308604322899422 [DOI] [PubMed] [Google Scholar]
- 134.Zhao MM, Xu MJ, Cai Y, Zhao G, Guan Y, Kong W, Tang C, Wang X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79:1071–1079. doi: 10.1038/ki.2011.18 [DOI] [PubMed] [Google Scholar]
- 135.Kurozumi A, Nakano K, Yamagata K, Okada Y, Nakayamada S, Tanaka Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone. 2019;124:53–61. doi: 10.1016/j.bone.2019.04.006 [DOI] [PubMed] [Google Scholar]
- 136.New SE, Aikawa E. Role of extracellular vesicles in de novo mineralization: an additional novel mechanism of cardiovascular calcification. Arterioscler Thromb Vasc Biol. 2013;33:1753–1758. doi: 10.1161/ATVBAHA.112.300128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schurgers LJ, Akbulut AC, Kaczor DM, Halder M, Koenen RR, Kramann R. Initiation and propagation of vascular calcification is regulated by a concert of platelet- and smooth muscle cell-derived extracellular vesicles. Front Cardiovasc Med. 2018;5:36. doi: 10.3389/fcvm.2018.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116:1312–1323. doi: 10.1161/CIRCRESAHA.116.305012 [DOI] [PubMed] [Google Scholar]
- 139.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113:72–77. doi: 10.1161/CIRCRESAHA.113.301036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. 2016;15:335–343. doi: 10.1038/nmat4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yamada S, Tokumoto M, Tatsumoto N, Tsuruya K, Kitazono T, Ooboshi H. Very low protein diet enhances inflammation, malnutrition, and vascular calcification in uremic rats. Life Sci. 2016;146:117–123. doi: 10.1016/j.lfs.2015.12.050 [DOI] [PubMed] [Google Scholar]
- 142.Schwartz MA, Lieske JC, Kumar V, Farell-Baril G, Miller VM. Human-derived nanoparticles and vascular response to injury in rabbit carotid arteries: proof of principle. Int J Nanomedicine. 2008;3:243–248. doi: 10.2147/ijn.s2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cenizo Revuelta N, Gonzalez-Fajardo JA, Bratos MA, Alvarez-Gago T, Aguirre B, Vaquero C. Role of calcifying nanoparticle in the development of hyperplasia and vascular calcification in an animal model. Eur J Vasc Endovasc Surg. 2014;47:640–646. doi: 10.1016/j.ejvs.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 144.Price PA, Nguyen TM, Williamson MK. Biochemical characterization of the serum fetuin-mineral complex. J Biol Chem. 2003;278:22153–22160. doi: 10.1074/jbc.M300739200 [DOI] [PubMed] [Google Scholar]
- 145.Matsui I, Hamano T, Mikami S, Fujii N, Takabatake Y, Nagasawa Y, Kawada N, Ito T, Rakugi H, Imai E, et al. Fully phosphorylated fetuin-A forms a mineral complex in the serum of rats with adenine-induced renal failure. Kidney Int. 2009;75:915–928. doi: 10.1038/ki.2008.700 [DOI] [PubMed] [Google Scholar]
- 146.Price PA, Caputo JM, Williamson MK. Bone origin of the serum complex of calcium, phosphate, fetuin, and matrix Gla protein: biochemical evidence for the cancellous bone-remodeling compartment. J Bone Miner Res. 2002;17:1171–1179. doi: 10.1359/jbmr.2002.17.7.1171 [DOI] [PubMed] [Google Scholar]
- 147.Price PA, Williamson MK, Nguyen TM, Than TN. Serum levels of the fetuin-mineral complex correlate with artery calcification in the rat. J Biol Chem. 2004;279:1594–1600. doi: 10.1074/jbc.M305199200 [DOI] [PubMed] [Google Scholar]
- 148.Price PA, Lim JE. The inhibition of calcium phosphate precipitation by fetuin is accompanied by the formation of a fetuin-mineral complex. J Biol Chem. 2003;278:22144–22152. doi: 10.1074/jbc.M300744200 [DOI] [PubMed] [Google Scholar]
- 149.Hamano T, Matsui I, Mikami S, Tomida K, Fujii N, Imai E, Rakugi H, Isaka Y. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. 2010;21:1998–2007. doi: 10.1681/ASN.2009090944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Martel J, Young D, Young A, Wu CY, Chen CD, Yu JS, Young JD. Comprehensive proteomic analysis of mineral nanoparticles derived from human body fluids and analyzed by liquid chromatography-tandem mass spectrometry. Anal Biochem. 2011;418:111–125. doi: 10.1016/j.ab.2011.06.018 [DOI] [PubMed] [Google Scholar]
- 151.Wu CY, Young L, Young D, Martel J, Young JD. Bions: a family of biomimetic mineralo-organic complexes derived from biological fluids. PLoS One. 2013;8:e75501. doi: 10.1371/journal.pone.0075501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Young JD, Martel J, Young D, Young A, Hung CM, Young L, Chao YJ, Young J, Wu CY. Characterization of granulations of calcium and apatite in serum as pleomorphic mineralo-protein complexes and as precursors of putative nanobacteria. PLoS One. 2009;4:e5421. doi: 10.1371/journal.pone.0005421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wu CY, Young D, Martel J, Young JD. A story told by a single nanoparticle in the body fluid: demonstration of dissolution-reprecipitation of nanocrystals in a biological system. Nanomedicine (Lond). 2015;10:2659–2676. doi: 10.2217/nnm.15.88 [DOI] [PubMed] [Google Scholar]
- 154.Martel J, Wu CY, Hung CY, Wong TY, Cheng AJ, Cheng ML, Shiao MS, Young JD. Fatty acids and small organic compounds bind to mineralo-organic nanoparticles derived from human body fluids as revealed by metabolomic analysis. Nanoscale. 2016;8:5537–5545. doi: 10.1039/c5nr08116e [DOI] [PubMed] [Google Scholar]
- 155.Raoult D, Drancourt M, Azza S, Nappez C, Guieu R, Rolain JM, Fourquet P, Campagna B, La Scola B, Mege JL, et al. Nanobacteria are mineralo fetuin complexes. PLoS Pathog. 2008;4:e41. doi: 10.1371/journal.ppat.0040041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Price PA, Thomas GR, Pardini AW, Figueira WF, Caputo JM, Williamson MK. Discovery of a high molecular weight complex of calcium, phosphate, fetuin, and matrix gamma-carboxyglutamic acid protein in the serum of etidronate-treated rats. J Biol Chem. 2002;277:3926–3934. doi: 10.1074/jbc.M106366200 [DOI] [PubMed] [Google Scholar]
- 157.Hutcheson JD, Goettsch C, Pham T, Iwashita M, Aikawa M, Singh SA, Aikawa E. Enrichment of calcifying extracellular vesicles using density-based ultracentrifugation protocol. J Extracell Vesicles. 2014;3:25129. doi: 10.3402/jev.v3.25129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808 [DOI] [PubMed] [Google Scholar]
- 159.Chen NX, O’Neill K, Chen X, Kiattisunthorn K, Gattone VH, Moe SM. Transglutaminase 2 accelerates vascular calcification in chronic kidney disease. Am J Nephrol. 2013;37:191–198. doi: 10.1159/000347031 [DOI] [PubMed] [Google Scholar]
- 160.Rogers MA, Buffolo F, Schlotter F, Atkins SK, Lee LH, Halu A, Blaser MC, Tsolaki E, Higashi H, Luther K, et al. Annexin a1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci Adv. 2020;6:eabb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chen NX, O’Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res. 2008;23:1798–1805. doi: 10.1359/jbmr.080604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Smith ER, Hewitson TD, Cai MMX, Aghagolzadeh P, Bachtler M, Pasch A, Holt SG. A novel fluorescent probe-based flow cytometric assay for mineral-containing nanoparticles in serum. Sci Rep. 2017;7:5686. doi: 10.1038/s41598-017-05474-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schlieper G, Schurgers L, Brandenburg V, Reutelingsperger C, Floege J. Vascular calcification in chronic kidney disease: an update. Nephrol Dial Transplant. 2016;31:31–39. doi: 10.1093/ndt/gfv111 [DOI] [PubMed] [Google Scholar]
- 164.Aikawa E. Extracellular vesicles in cardiovascular disease: focus on vascular calcification. J Physiol. 2016;594:2877–2880. doi: 10.1113/JP272112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol. 2017;14:259–272. doi: 10.1038/nrcardio.2017.7 [DOI] [PubMed] [Google Scholar]
- 166.Cui L, Houston DA, Farquharson C, MacRae VE. Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone. 2016;87:147–158. doi: 10.1016/j.bone.2016.04.007 [DOI] [PubMed] [Google Scholar]
- 167.Dusso A, Colombo MI, Shanahan CM. Not all vascular smooth muscle cell exosomes calcify equally in chronic kidney disease. Kidney Int. 2018;93:298–301. doi: 10.1016/j.kint.2017.08.036 [DOI] [PubMed] [Google Scholar]
- 168.Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34:724–736. doi: 10.1161/ATVBAHA.113.302642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062. doi: 10.1161/01.res.87.11.1055 [DOI] [PubMed] [Google Scholar]
- 170.Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28 [DOI] [PubMed] [Google Scholar]
- 171.Bundy JD, Cai X, Scialla JJ, Dobre MA, Chen J, Hsu CY, Leonard MB, Go AS, Rao PS, Lash JP, et al. ; CRIC Study Investigators. Serum calcification propensity and coronary artery calcification among patients with CKD: the CRIC (Chronic Renal Insufficiency Cohort) Study. Am J Kidney Dis. 2019;73:806–814. doi: 10.1053/j.ajkd.2019.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ahmadieh S, Kim HW, Weintraub NL. Potential role of perivascular adipose tissue in modulating atherosclerosis. Clin Sci (Lond). 2020;134:3–13. doi: 10.1042/CS20190577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Qi XY, Qu SL, Xiong WH, Rom O, Chang L, Jiang ZS. Perivascular adipose tissue (PVAT) in atherosclerosis: a double-edged sword. Cardiovasc Diabetol. 2018;17:134. doi: 10.1186/s12933-018-0777-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Oikonomou EK, Thomas S, Mancio J, Antonopoulos AS, Sabharwal N, Kelion A, Neubauer S, Channon KM, Antoniades C. Computed tomography-based perivascular fat phenotyping identifies unstable coronary lesions and active vascular calcification. Eur Heart J. 2018;39:ehy565.1182. doi: 10.1093/eurheartj/ehy565.1182 [Google Scholar]
- 175.Shields KJ, El Khoudary SR, Ahearn JM, Manzi S. Association of aortic perivascular adipose tissue density with aortic calcification in women with systemic lupus erythematosus. Atherosclerosis. 2017;262:55–61. doi: 10.1016/j.atherosclerosis.2017.04.021 [DOI] [PubMed] [Google Scholar]
- 176.Gao YJ, Lu C, Su LY, Sharma AM, Lee RM. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151:323–331. doi: 10.1038/sj.bjp.0707228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mancio J, Oikonomou EK, Antoniades C. Perivascular adipose tissue and coronary atherosclerosis. Heart. 2018;104:1654–1662. doi: 10.1136/heartjnl-2017-312324 [DOI] [PubMed] [Google Scholar]
- 178.Oikonomou EK, Antoniades C. The role of adipose tissue in cardiovascular health and disease. Nat Rev Cardiol. 2019;16:83–99. doi: 10.1038/s41569-018-0097-6 [DOI] [PubMed] [Google Scholar]
- 179.Henrichot E, Juge-Aubry CE, Pernin A, Pache JC, Velebit V, Dayer JM, Meda P, Chizzolini C, Meier CA. Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol. 2005;25:2594–2599. doi: 10.1161/01.ATV.0000188508.40052.35 [DOI] [PubMed] [Google Scholar]
- 180.Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol. 2017;174:3496–3513. doi: 10.1111/bph.13705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A. 1991;13:277–296. doi: 10.3109/10641969109042063 [DOI] [PubMed] [Google Scholar]
- 182.Ruan CC, Zhu DL, Chen QZ, Chen J, Guo SJ, Li XD, Gao PJ. Perivascular adipose tissue-derived complement 3 is required for adventitial fibroblast functions and adventitial remodeling in deoxycorticosterone acetate-salt hypertensive rats. Arterioscler Thromb Vasc Biol. 2010;30:2568–2574. doi: 10.1161/ATVBAHA.110.215525 [DOI] [PubMed] [Google Scholar]
- 183.Tillie RJHA, van Kuijk K, Sluimer JC. Fibroblasts in atherosclerosis: heterogeneous and plastic participants. Curr Opin Lipidol. 2020;31:273–278. doi: 10.1097/MOL.0000000000000700 [DOI] [PubMed] [Google Scholar]
- 184.Li W, Su SA, Chen J, Ma H, Xiang M. Emerging roles of fibroblasts in cardiovascular calcification. J Cell Mol Med. 2021;25:1808–1816. doi: 10.1111/jcmm.16150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Singh S, Torzewski M. Fibroblasts and their pathological functions in the fibrosis of aortic valve sclerosis and atherosclerosis. Biomolecules. 2019;9:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dutzmann J, Koch A, Weisheit S, Sonnenschein K, Korte L, Haertlé M, Thum T, Bauersachs J, Sedding DG, Daniel JM. Sonic hedgehog-dependent activation of adventitial fibroblasts promotes neointima formation. Cardiovasc Res. 2017;113:1653–1663. doi: 10.1093/cvr/cvx158 [DOI] [PubMed] [Google Scholar]
- 187.Zhu Z, Hao X, Li T, Guo J. Homocysteine promotes migration of adventitial fibroblasts via angiotensin ii type 1 receptor activation to aggravate atherosclerosis. BMC Cardiovasc Disord. Preprint posted online December 17, 2020. doi: 10.21203/rs.3.rs-32632/v3. https://www.researchsquare.com/article/rs-32632/v3 [DOI] [PubMed] [Google Scholar]
- 188.Yuan F, Wang D, Xu K, Wang J, Zhang Z, Yang L, Yang GY, Li S. Contribution of vascular cells to neointimal formation. PLoS One. 2017;12:e0168914. doi: 10.1371/journal.pone.0168914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Krishnan P, Purushothaman KR, Purushothaman M, Turnbull IC, Tarricone A, Vasquez M, Jain S, Baber U, Lascano RA, Kini AS, et al. Enhanced neointimal fibroblast, myofibroblast content and altered extracellular matrix composition: implications in the progression of human peripheral artery restenosis. Atherosclerosis. 2016;251:226–233. doi: 10.1016/j.atherosclerosis.2016.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jabs A, Okamoto E, Vinten-Johansen J, Bauriedel G, Wilcox JN. Sequential patterns of chemokine- and chemokine receptor-synthesis following vessel wall injury in porcine coronary arteries. Atherosclerosis. 2007;192:75–84. doi: 10.1016/j.atherosclerosis.2006.05.050 [DOI] [PubMed] [Google Scholar]
- 191.Milutinović A, Šuput D, Zorc-Pleskovič R. Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: an updated review. Bosn J Basic Med Sci. 2020;20:21–30. doi: 10.17305/bjbms.2019.4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. 2001;89:1111–1121. doi: 10.1161/hh2401.100844 [DOI] [PubMed] [Google Scholar]
- 193.Xu F, Ji J, Li L, Chen R, Hu W. Activation of adventitial fibroblasts contributes to the early development of atherosclerosis: a novel hypothesis that complements the “Response-to-Injury Hypothesis” and the “Inflammation Hypothesis”. Med Hypotheses. 2007;69:908–912. doi: 10.1016/j.mehy.2007.01.062 [DOI] [PubMed] [Google Scholar]
- 194.Li N, Cheng W, Huang T, Yuan J, Wang X, Song M. Vascular adventitia calcification and its underlying mechanism. PLoS One. 2015;10:e0132506. doi: 10.1371/journal.pone.0132506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cai MMX, Smith ER, Kent A, Huang L, Hewitson TD, McMahon LP, Holt SG. Calciprotein particle formation in peritoneal dialysis effluent is dependent on dialysate calcium concentration. Perit Dial Int. 2018;38:286–292. doi: 10.3747/pdi.2017.00163 [DOI] [PubMed] [Google Scholar]
- 196.Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018;114:540–550. doi: 10.1093/cvr/cvy022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Danesh J, Whincup P, Walker M, Lennon L, Thomson A, Appleby P, Gallimore JR, Pepys MB. Low grade inflammation and coronary heart disease: prospective study and updated meta-analyses. BMJ. 2000;321:199–204. doi: 10.1136/bmj.321.7255.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sun ZL, Xie QY, Guo GL, Ma K, Huang YY. Serum fetuin-A levels in patients with cardiovascular disease: a meta-analysis. Biomed Res Int. 2014;2014:691540. doi: 10.1155/2014/691540 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 199.Bielesz B, Reiter T, Marculescu R, Gleiss A, Bojic M, Kieweg H, Cejka D. Calcification propensity of serum is independent of excretory renal function. Sci Rep. 2017;7:17941. doi: 10.1038/s41598-017-18336-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Aigner C, Cejka D, Sliber C, Fraunschiel M, Sunder-Plassmann G, Gaggl M. Oral sodium bicarbonate supplementation does not affect serum calcification propensity in patients with chronic kidney disease and chronic metabolic acidosis. Kidney Blood Press Res. 2019;44:188–199. doi: 10.1159/000498975 [DOI] [PubMed] [Google Scholar]
- 201.Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Lindblad L, Lewis EF, Drisko J, Lee KL; TACT Investigators. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA. 2013;309:1241–1250. doi: 10.1001/jama.2013.2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Escolar E, Lamas GA, Mark DB, Boineau R, Goertz C, Rosenberg Y, Nahin RL, Ouyang P, Rozema T, Magaziner A, et al. The effect of an EDTA-based chelation regimen on patients with diabetes mellitus and prior myocardial infarction in the Trial to Assess Chelation Therapy (TACT). Circ Cardiovasc Qual Outcomes. 2014;7:15–24. doi: 10.1161/CIRCOUTCOMES.113.000663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ujueta F, Arenas IA, Escolar E, Diaz D, Boineau R, Mark DB, Golden P, Lindblad L, Kim H, Lee KL, et al. The effect of EDTA-based chelation on patients with diabetes and peripheral artery disease in the Trial to Assess Chelation Therapy (TACT). J Diabetes Complications. 2019;33:490–494. doi: 10.1016/j.jdiacomp.2019.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Avila MD, Escolar E, Lamas GA. Chelation therapy after the trial to assess chelation therapy: results of a unique trial. Curr Opin Cardiol. 2014;29:481–488. doi: 10.1097/HCO.0000000000000096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Foreman H, Trujillo TT. The metabolism of C14 labeled ethylenediaminetetraacetic acid in human beings. J Lab Clin Med. 1954;43:566–571. [PubMed] [Google Scholar]
- 206.Quiñones H, Hamdi T, Sakhaee K, Pasch A, Moe OW, Pak CYC. Control of metabolic predisposition to cardiovascular complications of chronic kidney disease by effervescent calcium magnesium citrate: a feasibility study. J Nephrol. 2019;32:93–100. doi: 10.1007/s40620-018-0559-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bressendorff I, Hansen D, Schou M, Silver B, Pasch A, Bouchelouche P, Pedersen L, Rasmussen LM, Brandi L. Oral magnesium supplementation in chronic kidney disease stages 3 and 4: efficacy, safety, and effect on serum calcification propensity-A prospective randomized double-blinded placebo-controlled clinical trial. Kidney Int Rep. 2017;2:380–389. doi: 10.1016/j.ekir.2016.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wu CY, Martel J, Young JD. Comprehensive organic profiling of biological particles derived from blood. Sci Rep. 2018;8:11310. doi: 10.1038/s41598-018-29573-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cottignoli V, Relucenti M, Agrosì G, Cavarretta E, Familiari G, Salvador L, Maras A. Biological niches within human calcified aortic valves: towards understanding of the pathological biomineralization process. Biomed Res Int. 2015;2015:542687. doi: 10.1155/2015/542687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Holt SG, Smith ER. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol Dial Transplant. 2016;31:1583–1587. doi: 10.1093/ndt/gfw048 [DOI] [PubMed] [Google Scholar]
- 211.Diaz-Tocados JM, Peralta-Ramirez A, Rodríguez-Ortiz ME, Raya AI, Lopez I, Pineda C, Herencia C, Montes de Oca A, Vergara N, Steppan S, et al. Dietary magnesium supplementation prevents and reverses vascular and soft tissue calcifications in uremic rats. Kidney Int. 2017;92:1084–1099. doi: 10.1016/j.kint.2017.04.011 [DOI] [PubMed] [Google Scholar]
- 212.Bressendorff I, Hansen D, Pasch A, Holt SG, Schou M, Brandi L, Smith ER. The effect of increasing dialysate magnesium on calciprotein particles, inflammation and bone markers: post hoc analysis from a randomized controlled clinical trial. Nephrol Dial Transplant. 2019;34:gfz234. doi: 10.1093/ndt/gfz234 [DOI] [PubMed] [Google Scholar]
- 213.Nakamura K, Nagata Y, Hiroyoshi T, Isoyama N, Fujikawa K, Miura Y, Matsuyama H, Kuro-O M. The effect of lanthanum carbonate on calciprotein particles in hemodialysis patients. Clin Exp Nephrol. 2020;24:323–329. doi: 10.1007/s10157-019-01832-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Toussaint ND, Lau KK, Polkinghorne KR, Kerr PG. Attenuation of aortic calcification with lanthanum carbonate versus calcium-based phosphate binders in haemodialysis: a pilot randomized controlled trial. Nephrology (Carlton). 2011;16:290–298. doi: 10.1111/j.1440-1797.2010.01412.x [DOI] [PubMed] [Google Scholar]
- 215.Schantl AE, Verhulst A, Neven E, Behets GJ, D’Haese PC, Maillard M, Mordasini D, Phan O, Burnier M, Spaggiari D, et al. Inhibition of vascular calcification by inositol phosphates derivatized with ethylene glycol oligomers. Nat Commun. 2020;11:721. doi: 10.1038/s41467-019-14091-4 [DOI] [PMC free article] [PubMed] [Google Scholar]