

Abstract
The apolipoprotein E (APOE) gene associates with Alzheimer's disease (AD) and cholesterol levels. Upstream transcription factor 1 (USF1) regulates lipid metabolism genes, including APOE, and the AD Aβ‐precursor protein. We investigated associations between 6 haplotype‐tagging USF1 single‐nucleotide polymorphisms (and haplotypes) and AD‐related neuropathological lesions [senile plaques (SP), neurofibrillary tangles (NFT) ] in an autopsy series comprising 603 cases (ages 0–97, mean 62 years, 215 women) that died out‐of‐hospital. In age‐ and APOE‐adjusted analyses, the minor G‐allele of rs2774276, previously linked to elevated cholesterol, associated with late‐stage burnt out SP among women and early non‐neuritic SP among men. The G‐allele of the previously unreported rs10908821 showed significant risk of having SP, especially neuritic and burnt out SP, among women but not men. USF1 haplotype GCGCAC carriers (risk alleles of rs2774276 and rs10908821) associated with SP risk, especially neuritic and late‐stage burnt out SP, among women but not men. Younger CCGCAC carriers (risk allele of rs2774276 and protective of rs10908821) were more likely to have non‐neuritic and diffuse SP. Conversely, USF1 CCGCAC haplotype carriers had lower NFT prevalence among 65+ year‐olds. These results suggest USF1 has an independent but gender‐ and age‐associated effect on AD‐related brain lesion development.
Keywords: Alzheimer's disease, neurofibrillary tangles, senile plaques, upstream transcription factor 1
INTRODUCTION
Alzheimer's disease (AD) is the most common form of neurodegenerative disease, affecting millions of people and estimated to affect 81.1 million by the year 2040 (10). Although the prevalence of AD is high, the underlying pathogenesis has not been unraveled. Extracellular senile plaques (SP) formed from amyloid beta (Aβ) and intracellular neurofibrillary tangles (NFT) consisting of hyperphosphorylated tau protein are considered hallmarks of AD (37). It is thought that these brain lesions, by interrupting neuronal signaling and instigating oxidation and inflammation, gradually lead to loss of memory, orientation, judgment and reasoning, the typical clinical manifestations of AD. Definite AD diagnosis involves postmortem detection of SP and NFT in the brain (8). Because the pathogenesis of this disease is still largely unknown, much interest has focused on possible genetic and environmental factors responsible for this progressive disorder. Up to 80% of an individuals' risk for AD is thought to be genetic (13). The apolipoprotein E (APOE) ε4 allele is the most commonly accepted AD risk gene, increasing the risk for the disease and significantly lowering the age of onset 4, 18, 28, although other risk genes have been recently identified 15, 25.
AD and coronary artery disease (CAD) share many risk factors, such as elevated cholesterol levels and carriership of the APOEε4 allele 29, 47. The APOEε4 allele may affect AD risk both directly through Aβ‐modifying mechanisms (18) and/or through cholesterol metabolism (32), although the exact mechanisms have yet to be established.
A gene that ties cholesterol metabolism and AD together would be useful in elucidating the pathogenesis of AD. The upstream transcription factor 1 (USF1) gene located on chromosome 1q22‐q23 (39) encodes a ubiquitously expressed important and general transcription factor with multiple roles in transcriptional regulation of several genes involved in glucose and lipid metabolism (27), including APOE (36). Polymorphisms of the USF1 gene associate with familial combined hyperlipidemia (FCHL) (33), increased risk for cardiovascular disease in women (21), high plasma triglyceride and low‐density lipoprotein (LDL) levels (3), early markers of atherosclerosis (2), and coronary artery calcifications and atherosclerotic lesions (23), and lowered APOE gene expression in fat tissue (31). USF1 also represses the gene encoding ATP‐binding cassette A1 (ABCA1) transporter protein, which has a key role in the cellular efflux of cholesterol and phospholipids (48). Additionally, USF1 modulates genes involved in the immune response and cell cycle control (6), and up‐regulates the expression of the APP gene encoding amyloid precursor protein (APP) (22). USFs also associate with genes regulating synaptic plasticity (41), neuronal survival and differentiation (44).
In a previous case‐control study of living AD patients, no association between USF1 gene polymorphisms and disease onset was found (38). We have previously found that the accumulation of SP is strongly dependent on the APOEε4 allele and this association begins in middle age 20, 34. We chose to investigate the possible associations between USF1 polymorphisms and the prevalence of neuropathologically confirmed AD‐related lesions in the brains of a sample representing that of a normal, non‐demented population, consisting of all ages in the Tampere Autopsy Study (TASTY).
MATERIALS AND METHODS
The Tampere Autopsy Study (TASTY) comprises a prospective autopsy series of 603 (aged 0–97, mean 62 years, 215 women) cases, who underwent medicolegal autopsy because of sudden or unexpected death as described in detail in our earlier article (20). The study was approved by the Board of Medicolegal Affairs of Finland. The cohort is thought to represent the best available sample of a general non‐institutionalized population. None of the cases died of AD, but 22 (3.7%) had some form of dementia, of which six (1.0%) were diagnosed with AD prior to death. Ten cases (1.7%) had memory problems and one had been diagnosed with Parkinson's disease. These data were obtained according to available hospital records and next‐of‐kin reports.
The salt precipitation method was used on frozen blood samples for DNA isolation. Six reported USF1 haplotype‐tagging single‐nucleotide polymorphisms (SNPs) were genotyped (rs10908821, rs2073658, rs2774276, rs2516839, rs1556259 and rs2774279) to capture common allelic variants of the gene using the commercially available TaqMan® assays with the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) (21). APOE genotyping was performed as described elsewhere (16). Bielschowsky silver staining was used to determine the SP (cortex) and NFT (hippocampus) assessments under the supervision of an experienced neuropathologist (HH) in brain tissue samples and Aβ‐staining was used to confirm Bielschowsky staining as previously described (20).
Estimation of USF1 haplotype frequencies was performed using PHASE v2.1.1 42, 43. SNP order was rs10908821, rs2073658, rs2774276, rs2516839, rs1556259 and rs2774279 and created the haplotypes CCCTAC (haplotype 1), CCCTAT (2), CCCCGC (3), CCGCAC (5), CTCTAC (4), CTCTAT (6) and GCGCAC (7). Haplotypes 1 and 6 were excluded from analyses based on their low prevalence (0.001% and 0.1%, respectively).
Statistical analyses used SPSS v16.0 for Windows (SPSS Inc., Chicago, IL, USA). The variables used were: USF1 SNPs (reported genotypes vs. the common homozygous genotype, and also carriership of the rare allele vs. the common homozygous genotype), haplotypes (yes/no carriership), SP (yes/no) and NFT (yes/no). SP were further grouped into non‐neuritic (diffuse and primitive) and neuritic (classic and burnt out). We also used a variable (converted from the SP variable) with percentage of SP cortex coverage, based on the CERAD protocol, which semi‐quantitatively assessed the categories “no SP”, and equal‐sized groups “≤1.05% SP coverage” and “>1.05% SP coverage”, in order to create the strongest statistical power assessments. Logistic regression analyses used age and APOEε4 carriership as covariates. Based on an “a priori” hypothesis and reported differences in age and gender on the effect of SP (5) and AD 1, 12 prevalence, we also chose to investigate the cohort in a stratified manner. The analyses were performed splitting the entire data series by gender and into two age groups at the median value: 0–64 (49.4%) and ≥65 (50.6%) years.
False discovery rate (FDR) multiple correction calculations were performed assuming there were 11 independent tests (6 SNPs and 5 haplotypes), using the calculation shown later and assuming an FDR value of <0.05 was acceptable.
RESULTS
Demographics of the TASTY cohort can be seen in Table 1. Of 603 individuals SP frequency was available for 548 (90.9%), NFT counts for 484 (80.3%), APOE genotypes for 601 (99.7%). USF1 SNPs had a mean genotyping success rate of 87.75% (rs10908821–87.6%, rs2073658–87.7%, rs2774276–88.4%, rs2516839–87.7%, rs1556259–87.7% and rs2774279–87.4%). The haplotype frequencies of USF1 can be seen in Table 2. All SNPs were in Hardy–Weinberg equilibrium. There were significant interactions between SNPs, and both age and gender on SP prevalence, as seen in Table 3. Based on this, we did the analyses separately among those below and above 65 years of age, and among men and women. Women were more likely to have SP compared with men [odds ratio (OR) 2.15, confidence interval (CI) 1.49–3.11, P < 0.0001], although on average, women were 10 years older than men.
Table 1.
Characteristics of the TASTY Cohort.
| Valid N | All | Male | Female | P‐value |
|---|---|---|---|---|
| 603 | 388 (64.3%) | 215 (35.7%) | ||
| Age, years ± SD | 62.7 ± 19.1 | 59.6 ± 18.8 | 68.2 ± 18.6 | <0.0001 |
| <65 years | 298 | 219 (73.5%) | 79 (26.5%) | <0.0001 |
| ≥65 years | 305 | 169 (55.4%) | 136 (44.6%) | NS |
| BMI, kg/m2 ± SD | 27.4 ± 6.15 | 27.1 ± 5.86 | 27.8 ± 6.65 | NS |
| Cause of death | ||||
| Disease | 59.1% | 59.6% | 58.1% | NS |
| Accident | 27.0% | 26.2% | 28.4% | NS |
| Suicide | 12.0% | 12.4% | 11.2% | NS |
| Homicide | 0.5% | 0.8% | 0% | NS |
| Unknown | 1.5% | 1.0% | 2.3% | NS |
| Dementia Status | ||||
| AD | 1.0% | 0.5% | 1.9% | NS |
| Dementia | 2.7% | 1.8% | 4.2% | NS |
| Memory problems | 1.7% | 1.3% | 2.3% | NS |
| Parkinson's disease | 0.2% | 0.3% | 0% | . |
| Neuropathological lesions | ||||
| SP | 31.1% | 24.9% | 41.5% | <0.0001 |
| Non neuritic | 9.9% | 8.7% | 12.1% | NS |
| Neuritic | 19.1% | 15.1% | 26.1% | 0.001 |
| Diffuse | 3.7% | 3.9% | 3.4% | NS |
| Primitive | 6.2% | 4.8% | 8.7% | 0.02 |
| Classic | 14.7% | 11.2% | 20.8% | 0.001 |
| Burnt out | 4.4% | 3.9% | 5.3% | NS |
| SP coverage ≤ 1.05% | 15.5% | 13.6% | 18.6% | 0.02 |
| SP coverage > 1.05% | 15.5% | 11.0% | 23.0% | <0.0001 |
| NFT | 42.1% | 35.3% | 54.3% | <0.0001 |
AD = Alzheimer's disease; BMI = body mass index; NFT = neurofibrillary tangles; NS = nonsignificant; N = number of cases SP = senile plaques; SD = standard deviation; TASTY = Tampere Autopsy Study.
Table 2.
Upstream transcription factor 1 haplotype and allele frequencies.
| rs10908821 | rs2073658 | rs2774276 | rs2516839 | rs1556259 | rs2774279 | ||
|---|---|---|---|---|---|---|---|
| Haplotype 1 | C | C | C | T | A | C | 0.001% |
| Haplotype 2 | C | C | C | T | A | T | 24.6% |
| Haplotype 3 | C | C | C | C | G | C | 13.2% |
| Haplotype 4 | C | C | G | C | A | C | 12.2% |
| Haplotype 5 | C | T | C | T | A | C | 36.6% |
| Haplotype 6 | C | T | C | T | A | T | 0.1% |
| Haplotype 7 | G | C | G | C | A | C | 13.3% |
| (Major > minor allele) Frequency in percent % | (C > G) 79.9/20.1 | (C > T) 59.1/40.9 | (C > G) 67.9/32.1 | (T > C) 57.8/42.2 | (A > G) 79.9/20.1 | (C > T) 68.2/31.8 |
Table 3.
Upstream transcription factor 1 single‐nucleotide polymorphism interaction terms.
| P‐value | OR | 95% CI | |
|---|---|---|---|
| Age * rs10908821 G carriers vs. CC | 0.001 | 1.0 | 1.01–1.02 |
| Age * rs2073658 T carriers vs. CC | 0.002 | 1.0 | 1.00–1.01 |
| Age * rs2774276 G carriers vs. CC | 0.008 | 1.0 | 1.00–1.01 |
| Age * rs2516839 C carriers vs. TT | 0.001 | 1.0 | 1.00–1.02 |
| Age * rs1556259 G carriers vs. AA | 0.359 | 1.0 | 0.99–1.01 |
| Age * rs2774279 T carriers vs. CC | 0.077 | 1.0 | 0.99–1.01 |
| Gender * rs10908821 G carriers vs. CC | 0.542 | 0.9 | 0.51–1.42 |
| Gender * rs2073658 T carriers vs. CC | 0.025 | 0.6 | 0.43–0.95 |
| Gender * rs2774276 G carriers vs. CC | 0.101 | 0.7 | 0.46–1.07 |
| Gender * rs2516839 C carriers vs. TT | 0.011 | 0.6 | 0.41–0.89 |
| Gender * rs1556259 G carriers vs. AA | 0.110 | 0.7 | 0.38–1.10 |
| Gender * rs2774279 T carriers vs. CC | 0.006 | 0.5 | 0.33–0.83 |
CI = confidence interval; OR = odds ratio.
In general, age‐ and APOE‐adjusted analyses did not provide statistically significant associations between the SNPs or haplotypes, and the neuropathological lesions. In contrast, division by gender and age division into younger (0–64 years) and older (≥65 years) individuals revealed a number of statistically significant associations between the USF1 SNPs and haplotypes, and the neuropathological lesions. The supplementary tables show complete results for the cohort, both unstratified and stratified.
The previously unreported SNP rs10908821 female G‐allele carriers were found to associate with SP in the simplest analysis (OR 2.95, CI 1.25–6.95, P = 0.014), as shown in Figure 1A, compared with the common CC genotype. Additionally, the female minor G‐allele carriers were statistically more likely to have neuritic SP (OR 3.27, CI 1.29‐8.29, P = 0.013) and burnt out (OR 10.79, CI 2.34–49.71, P = 0.002) SP. In a CERAD‐like analysis, female carriers of the G‐allele were more likely to have increasing levels of SP coverage (≤1.05% OR 3.20, CI 1.20–8.51, P = 0.020; >1.05% OR 2.66, CI 0.98–7.22, P = 0.055). In analyses investigating NFT prevalence within gender, there were no statistically significant values. There were no significant associations seen in men.
Figure 1.
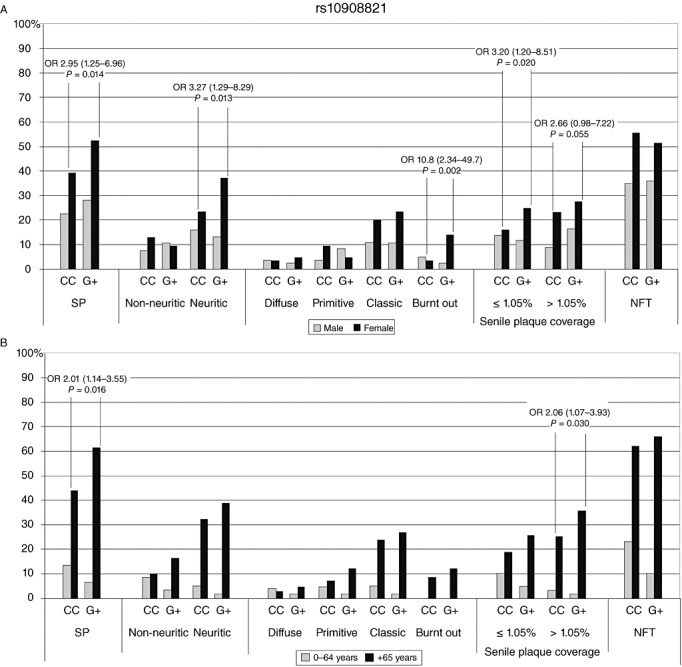
SNP rs10908821 prevalence (%) of SP and NFT against genotypes. (A) Grey indicates men and black for women; (B) grey indicates 0–64‐year‐old individuals and black refers to those over 65 years. NFT = neurofibrillary tangles; OR = odds ratio; SNP = single‐nucleotide polymorphism; SP = senile plaques.
With age division analyses, rs10908821 G‐allele carriers in the older age group (≥65 years) were more likely to have SP (OR 2.01, CI 1.14–3.55, P = 0.016) and higher SP coverage (>1.05% OR 2.06, CI 1.07–3.93, P = 0.030), compared with the CC genotype (see Figure 1B). There were no significant associations seen in younger individuals.
Female carriers of SNP rs2073658 showed no statistical associations with the neuropathological findings. Conversely, male rs2073658 TT genotype carriers were more likely to have neuritic SP vs. CC carriers (OR 2.82, CI 0.99–7.98, P = 0.050). No statistically significant values were discovered when comparing the minor allele carriers against the common homozygote carriers in analyses with age divisions.
Among women, the minor G‐allele of rs2774276 increased the risk of late stage burnt out SP (Fig. 2A), compared with CC homozygotes (OR 6.56, CI 1.36–31.5, P = 0.019), while male carriers of the G‐allele were more likely to have early stage non‐neuritic SP (OR 2.53, CI 1.09–5.88, P = 0.030). Analyses with age division (Fig. 2B) revealed that younger (0–64 years) individuals were more likely to have diffuse SP if they had a G‐allele, compared with CC homozygotes (OR 11.1, CI 1.37–89.7, P = 0.024). Older individuals showed no statistically significant associations.
Figure 2.
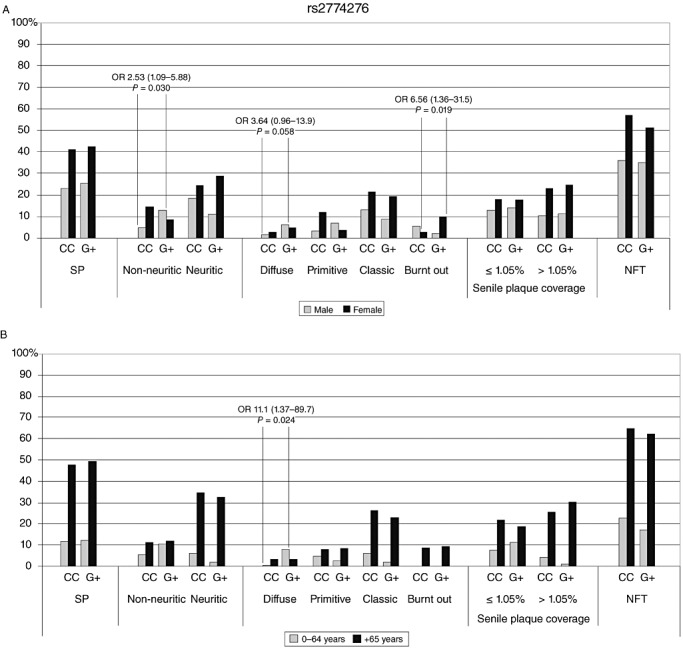
SNP rs2774276 prevalence (%) of SP and NFT against genotypes. (A) Grey indicates men and black for women; (B) grey indicates 0–64‐year‐old individuals and black refers to those over 65 years. NFT = neurofibrillary tangles; OR = odds ratio; SNP = single‐nucleotide polymorphism; SP = senile plaques.
Female carriers of the rs2516839 C‐allele were more likely to have late stage burnt out SP (OR 9.20, CI 1.04–81.6, P = 0.046), while we did not observe this in men (Fig. 3A). Age division analyses did not reveal statistically significant associations (Fig. 3B); however, a trend toward protection could be seen in C‐carriers for neuritic (OR 0.31, CI 0.08–1.12, P = 0.074) and classic (OR 0.31, CI 0.09–1.14, P = 0.078) SP in younger individuals. Age or gender divisions did not reveal statistically significant results between the SNPs rs1556259 or rs2774279 and SP or NFT variables (see Supporting Information Tables S1–S5).
Figure 3.
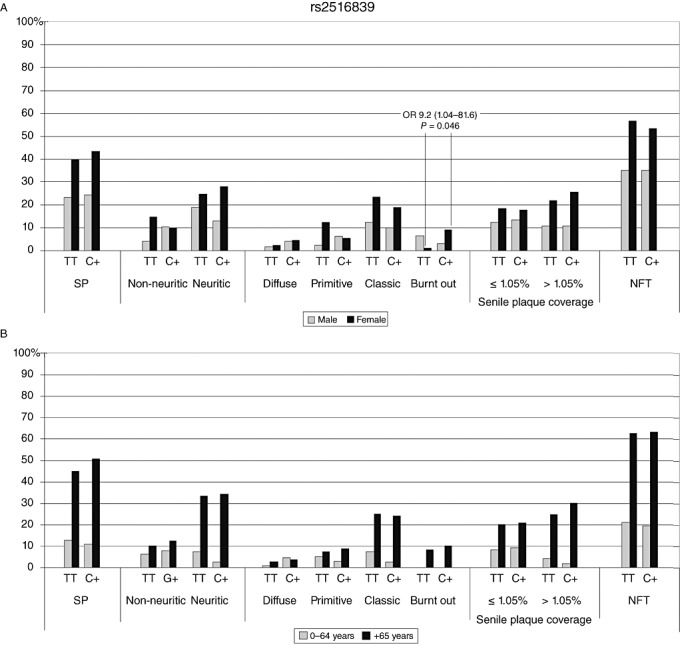
SNP rs2516839 prevalence (%) of SP and NFT against genotypes. (A) Grey indicates men and black for women; (B) grey indicates 0–64‐year‐old individuals and black refers to those over 65 years. NFT = neurofibrillary tangles; OR = odds ratio; SNP = single‐nucleotide polymorphism; SP = senile plaques.
Haplotypes 2, 3, 5 and 6 did not reveal statistically significant associations with the SP or NFT variables in simple analyses, or with gender or age divisions in our cohort. Gender division analyses of haplotype CCGCAC (haplotype 4) carriers did not reveal any associations with the neuropathological lesions (Fig. 4A), whereas older (≥65 years) carriers were less likely to have NFT (OR 0.50, CI 0.28–0.91, P = 0.024). Conversely, younger individuals carrying CCGCAC were more likely to have non‐neuritic (OR 2.98, CI 1.17–7.59, P = 0.022) and diffuse (OR 9.21, CI 2.26–37.5, P = 0.002) SP (Fig. 4B).
Figure 4.
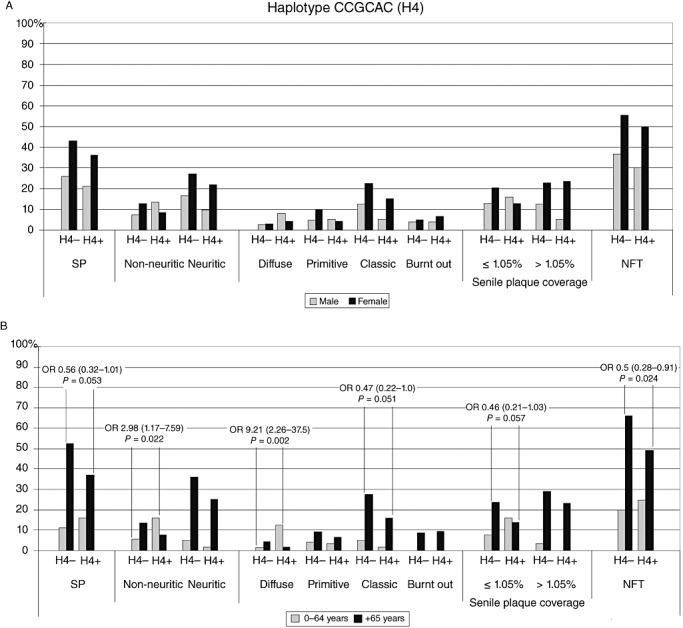
Haplotype CCGCAC (H4) prevalence (%) of SP and NFT against haplotype carriership. (A) Grey indicates men and black for women; (B) grey indicates 0–64‐year‐old individuals and black refers to those over 65 years. NFT = neurofibrillary tangles; OR = odds ratio; SP = senile plaques.
The strongest associations between the SP and haplotypes were seen for female carriers of GCGCAC (haplotype 7). These carriers (Fig. 5A) were more likely to have SP (OR 2.93, CI 1.25–6.85, P = 0.013), neuritic (OR 3.35, CI 1.32–8.47, P = 0.011) and late stage burnt out SP (OR 12.4, CI 2.67–57.1, P = 0.001), as well as increased risk for increasing SP cortical coverage (≤1.05% OR 2.94, CI 1.12–7.73, P = 0.029; > 1.05% OR 2.75, CI 1.01–7.44, P = 0.047). When concerning age division (Fig. 5B), younger individuals were not associated with the neuropathological findings; however, older (≥65 years) individuals with the GCGCAC haplotype were more likely to have SP (OR 1.97, CI 1.13–3.43, P = 0.017) and higher SP coverage (>1.05% OR 2.13, CI 1.12–4.04, P = 0.020).
Figure 5.
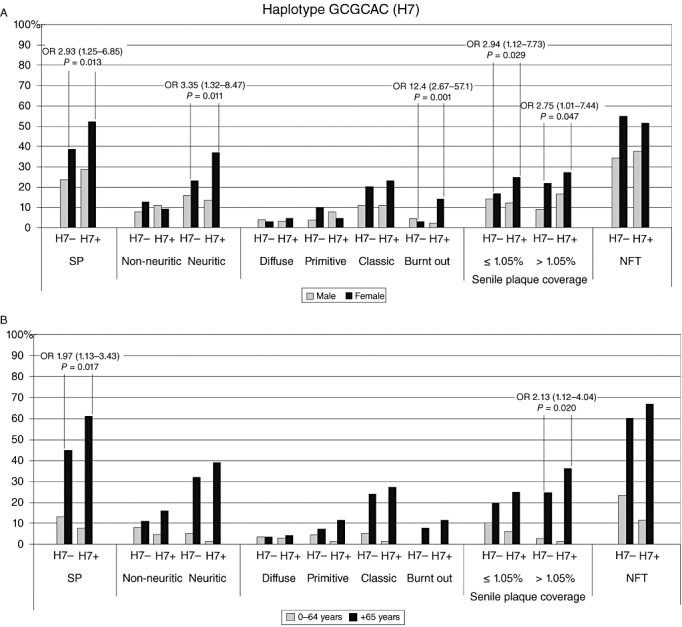
Haplotype GCGCAC (H7) prevalence (%) of SP and NFT against haplotype carriership. (A) Grey indicates men and black for women; (B) grey indicates 0–64‐year‐old individuals and black refers to those over 65 years. NFT = neurofibrillary tangles; OR = odds ratio; SP = senile plaques.
We performed FDR calculations on our results in order to correct for multiple testing. These showed that with an FDR < 0.05 (5% false positives), all of our results survived multiple testing (see Supporting Information Tables S1–S5 for full cohort results). The SNPs and haplotypes of the USF1 gene seen most often in statistical analyses were rs10908821 (G carriers), rs2774276 (G carriers), and haplotypes CCGCAC (5) and GCGCAC (7).
DISCUSSION
USF1 has been previously identified as a crucial and general transcription factor with multiple roles in the transcription regulation of several genes involved in lipid and glucose metabolism (27), stress and immune responses, cell cycle and proliferation (6). It is ubiquitously expressed, its E‐box motif is broadly distributed across the genome and the number of USF1‐dependent genes is extremely high (27). We discovered gender‐ and age‐dependent associations between the USF1 SNPs and haplotypes with both SP and NFT, which are the characteristic neuropathological lesions in AD.
A substantial gender difference with relation to age was seen in our cohort and is most likely caused by a bias toward the longevity of women and the higher number of young men seen in accidental deaths. For this reason, we included age as a covariate when investigating gender differences, in order to counteract this effect. Additionally, there were differential effects related to age and SP type among our results, possibly caused by small sample sizes, the younger ages of men in general in our cohort, or simply that different effects come into play at different stages of SP development.
The rs10908821 G‐carrier genotype appeared as a risk factor for SP changes in women and older individuals. This SNP has not been previously linked to disturbances in lipid metabolism. The rs2774276 has previously been linked to higher total serum cholesterol and LDL‐levels (GG genotype), and the G‐allele with higher waist‐to‐hip ratios (21). In our study, the G‐allele of rs2774276 associated with increased risk for both early‐stage (men and younger individuals) and late‐stage SP (women).
The rs2516839 C‐allele has been previously linked to higher high‐density lipoprotein (HDL) levels, lower triglyceride levels (26) and also identified as a protective factor for calcification of coronary arteries and less severe coronary atherosclerosis (23). In our study, a trend toward the C‐allele being protective for neuritic and classic SP could be seen in the younger individuals. In contrast, the C‐allele appeared as a risk factor for late‐stage SP in women, which is not in correlation with previous findings concerning lipid metabolism, although this could be a confounding factor because of small sample numbers. The rs2073658 T‐allele has been linked to an increased risk of cardiovascular diseases (CVD) and among female carriers to a higher risk for mortality (21). In accordance with these previous results, we found that rs2073658 TT genotype associated with later‐stage SP in men.
Previous studies investigating USF1 haplotypes (21, 23) did not find any significant associations with dyslipidemia for haplotypes CCGCAC and GCGCAC (haplotypes 4 and 7, respectively) that in our study associated with AD‐related neuropathological lesions. Although dyslipidemia was found to correlate with the haplotypes CCCTAT and CTCTAC (haplotypes 2 and 5, respectively) (21), these did not indicate any increased or decreased risks with neuropathological lesion prevalence in our study. In our study, younger carriers (<65 years) of CCGCAC were more likely to have non‐neuritic SP. On the other hand, a trend toward a lower tendency for SP and NFT was seen for older carriers of the same haplotype, possibly indicating that different effects come into play at different ages. Carriers of GCGCAC associated with SP prevalence, especially neuritic, and higher SP cortical coverage in women. Although this risk haplotype contains the low‐risk allele of rs2073658, it contains the high‐risk G‐alleles of rs2774276 and rs10908821, indicating their strong effects. Both of these haplotypes appeared to increase the risk of having SP, while differing only with regards to rs10908821 alleles, suggesting a differential effect for this particular SNP, depending on gender and age.
USF1 protein, like the tau protein of NFT, is also regulated through phosphorylation (7). It has also been suggested that increased intracellular cholesterol levels may accelerate phosphorylation of tau (47), and possibly also USF1. The reverse has also been shown, where reduction of cholesterol can cause the hyperphosphorylation of tau (9), suggesting signaling pathways affected by cholesterol levels may be the true activators. This might indicate that risk alleles—either in the USF1 gene, or other as yet unknown genes—could be activated by cholesterol levels.
Our current results suggest that USF1 SNPs and haplotypes associate with neuropathological lesions. Because previous studies on USF1 have suggested its involvement in lipid metabolism disorders 3, 21, 23, 26, 27, 31, 33, 35 and elevated cholesterol associates with AD 17, 19, 29, 32, our findings suggest that USF1 may affect the formation of these lesions through alterations in lipid metabolism. Cholesterol is actively turned over among neurons and glial cells, with the help of apolipoproteins and their receptors, and cholesterol has an essential role in synaptic plasticity in the central nervous system (47). A significant increase in the levels of LDL cholesterol, as well as a significant decrease in the levels of HDL cholesterol was found post‐mortem in AD patients (24). Suppression of de novo synthesis of cholesterol and decreased generation of Aβ has also been suggested to play a role in AD (40). The use of statins (3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors) has been suggested to potentially suppress the development of AD (46), but this observed effect might be caused by other beneficial effects they have such as anti‐inflammatory actions, immunosuppression and blockage of macrophage activation (45).
In addition to the hypothesis of disturbances in lipid metabolism, there are also several other possible mechanisms by which USF1 might affect the development of AD‐related lesions. The accumulation of these lesions might depend upon differential expression of USF1 target genes such as ABCA1 and APP. USF1 represses the gene encoding the ABCA1 transporter protein (48), which has an essential role in the cellular efflux of cholesterol and phospholipids. Disturbances in its production might cause diminished cellular efflux of lipids resulting in disturbances in cell function, possibly leading to cell death. Another possible mechanism might be the overproduction of Aβ, as USF1 up‐regulates transcription of the APP gene (22).
It has been previously hypothesized that inflammation might play a part in the pathogenesis of AD. A major transcription factor for controlling inflammation, NF‐kappaB (NFκB), is activated in the brains of AD patients (14), and the APOEε4 allele is related to the hyperactivation of NFκB and enhanced brain inflammation (11). USF1 has been found to regulate the transcription of APOE (31). The T‐allele of USF1 SNP rs2073658 has also been associated with higher plasma C‐reactive protein (CRP) and interleukin‐6 levels (IL‐6) (35). As USF1 regulates genes involved in immune responses (6), this offers yet another possible mechanism by which USF1 contributes to AD.
One of the drawbacks of our study is that although we show that polymorphisms of USF1 gene significantly associate with AD‐related lesions, we do not have many cases with clinically defined AD in our series. Additionally, we did not measure cholesterol levels or cerebrospinal fluid amyloid and tau metabolites. Our study probably also underestimates the SP and NFT load, as the lesions were examined only in three sections of brain tissue. The selected locations are, however, thought to be informative enough according to the CERAD protocol (30). Our cohort also comprised mostly of non‐demented individuals, and not clinically diagnosed AD patients, although these are most probably underdiagnosed, but our unique study thus offers an interesting insight into the brain pathology of a series that is the best available sample of a community‐dwelling non‐demented population.
The only previous study to our knowledge that looked at the possible associations between USF1 genotypes and clinical AD cases did not find any associations (38). The SNPs studied differed from the ones in our study, however, and the AD cases were not neuropathologically confirmed. To our knowledge, this is the first study of its kind that investigated the association with USF1 genotypes at a neuropathological level.
Our interesting findings will need to be investigated further in AD patient cohorts and replicated in larger epidemiological studies to determine which and how USF1 polymorphisms may contribute to the development of AD‐related lesions. Our unique study revealed associations between USF1 SNPs and haplotypes, and AD‐related lesions, supporting evidence for the hypothesis that disturbances in lipid metabolism might play a part in the formation of AD‐related lesions and therefore maybe the disease itself.
Supporting information
Table S1. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership and age as covariates).
Table S2. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership and age as covariates) in males.
Table S3. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership and age as covariates) in females.
Table S4. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership as a covariate) in under 65‐year‐old.
Table S5. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership as a covariate) in over 65‐year‐old.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
This study has been supported by the Medical Research Fund of Tampere University Hospital, the Pirkanmaa Regional Fund of the Finnish Cultural Foundation and the Finnish Foundation for Cardiovascular Research.
REFERENCES
- 1. Candore G, Balistreri CR, Grimaldi MP, Vasto S, Listi F, Chiappelli M et al (2006) Age‐related inflammatory diseases: role of genetics and gender in the pathophysiology of Alzheimer's disease. Ann N Y Acad Sci 1089:472–486. [DOI] [PubMed] [Google Scholar]
- 2. Collings A, Hoyssa S, Fan M, Kahonen M, Hutri‐Kahonen N, Marniemi J et al (2008) Allelic variants of upstream transcription factor 1 associate with carotid artery intima‐media thickness: the Cardiovascular Risk in Young Finns study. Circ J 72:1158–1164. [DOI] [PubMed] [Google Scholar]
- 3. Coon H, Xin Y, Hopkins PN, Cawthon RM, Hasstedt SJ, Hunt SC (2005) Upstream stimulatory factor 1 associated with familial combined hyperlipidemia, LDL cholesterol, and triglycerides. Hum Genet 117:444–451. [DOI] [PubMed] [Google Scholar]
- 4. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921–923. [DOI] [PubMed] [Google Scholar]
- 5. Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H (2004) The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci 1019:24–28. [DOI] [PubMed] [Google Scholar]
- 6. Corre S, Galibert MD (2005) Upstream stimulating factors: highly versatile stress‐responsive transcription factors. Pigment Cell Res 18:337–348. [DOI] [PubMed] [Google Scholar]
- 7. Corre S, Primot A, Baron Y, Le Seyec J, Goding C, Galibert M (2009) Target gene specificity of USF‐1 is directed via p38‐mediated phosphorylation‐dependent acetylation. J Biol Chem 284:18851–18862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger‐Gateau P, Cummings J et al (2007) Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS‐ADRDA criteria. Lancet Neurol 6:734–746. [DOI] [PubMed] [Google Scholar]
- 9. Fan QW, Yu W, Senda T, Yanagisawa K, Michikawa M (2001) Cholesterol‐dependent modulation of tau phosphorylation in cultured neurons. J Neurochem 76:391–400. [DOI] [PubMed] [Google Scholar]
- 10. Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M et al, Alzheimer's Disease International (2005) Global prevalence of dementia: a Delphi consensus study. Lancet 366:2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fontalba A, Gutierrez O, Llorca J, Mateo I, Berciano J, Fernandez‐Luna JL, Combarros O (2008) Deficiency of CARD8 is associated with increased Alzheimer's disease risk in women. Dement Geriatr Cogn Disord 26:247–250. [DOI] [PubMed] [Google Scholar]
- 12. Gao S, Hendrie HC, Hall KS, Hui S (1998) The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta‐analysis. Arch Gen Psychiatry 55:809–815. [DOI] [PubMed] [Google Scholar]
- 13. Gatz M, Reynolds C, Fratiglioni L, Johansson B, Mortimer J, Berg S et al (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63:168–174. [DOI] [PubMed] [Google Scholar]
- 14. Granic I, Dolga A, Nijholt I, van Dijk G, Eisel U (2009) Inflammation and NF‐kappaB in Alzheimer's disease and diabetes. J Alzheimers Dis 16:809–821. [DOI] [PubMed] [Google Scholar]
- 15. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML et al (2009) Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hixson JE, Vernier DT (1990) Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 31:545–548. [PubMed] [Google Scholar]
- 17. Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB (1995) Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer's disease: a case‐control study. Neurology 45:1092–1096. [DOI] [PubMed] [Google Scholar]
- 18. Kim J, Basak J, Holtzman D (2009) The role of apolipoprotein E in Alzheimer's disease. Neuron 63:287–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kivipelto M, Helkala E, Laakso M, Hanninen T, Hallikainen M, Alhainen K et al (2002) Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late‐life Alzheimer disease. Ann Intern Med 137:149–155. [DOI] [PubMed] [Google Scholar]
- 20. Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ (2009) Apolipoprotein E‐dependent accumulation of Alzheimer disease‐related lesions begins in middle age. Ann Neurol 65:650–657. [DOI] [PubMed] [Google Scholar]
- 21. Komulainen K, Alanne M, Auro K, Kilpikari R, Pajukanta P, Saarela J et al (2006) Risk alleles of USF1 gene predict cardiovascular disease of women in two prospective studies. PLoS Genet 2:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kovacs DM, Wasco W, Witherby J, Felsenstein KM, Brunel F, Roeder RG, Tanzi RE (1995) The upstream stimulatory factor functionally interacts with the Alzheimer amyloid beta‐protein precursor gene. Hum Mol Genet 4:1527–1533. [DOI] [PubMed] [Google Scholar]
- 23. Kristiansson K, Ilveskoski E, Lehtimaki T, Peltonen L, Perola M, Karhunen PJ (2008) Association analysis of allelic variants of USF1 in coronary atherosclerosis. Arterioscler Thromb Vasc Biol 28:983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, Drumm D, Roher AE (1998) Elevated low‐density lipoprotein in Alzheimer's disease correlates with brain abeta 1–42 levels. Biochem Biophys Res Commun 252:711–715. [DOI] [PubMed] [Google Scholar]
- 25. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M et al (2009) Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 26. Laurila PP, Naukkarinen J, Kristiansson K, Ripatti S, Kauttu T, Silander K et al (2010) Genetic association and interaction analysis of USF1 and APOA5 on lipid levels and atherosclerosis. Arterioscler Thromb Vasc Biol 30:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee JC, Lusis AJ, Pajukanta P (2006) Familial combined hyperlipidemia: upstream transcription factor 1 and beyond. Curr Opin Lipidol 17:101–109. [DOI] [PubMed] [Google Scholar]
- 28. Lehtimäki T, Pirttilä T, Mehta PD, Wisniewski HM, Frey H, Nikkari T (1995) Apolipoprotein E (apoE) polymorphism and its influence on ApoE concentrations in the cerebrospinal fluid in Finnish patients with Alzheimer's disease. Hum Genet 95:39–42. [DOI] [PubMed] [Google Scholar]
- 29. Martins IJ, Berger T, Sharman MJ, Verdile G, Fuller SJ, Martins RN (2009) Cholesterol metabolism and transport in the pathogenesis of Alzheimer's disease. J Neurochem 111:1275–1308. [DOI] [PubMed] [Google Scholar]
- 30. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 31. Naukkarinen J, Gentile M, Soro‐Paavonen A, Saarela J, Koistinen HA, Pajukanta P et al (2005) USF1 and dyslipidemias: converging evidence for a functional intronic variant. Hum Mol Genet 14:2595–2605. [DOI] [PubMed] [Google Scholar]
- 32. Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P et al (1998) Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology 17:14–20. [DOI] [PubMed] [Google Scholar]
- 33. Pajukanta P, Lilja HE, Sinsheimer JS, Cantor RM, Lusis AJ, Gentile M et al (2004) Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1). Nat Genet 36:371–376. [DOI] [PubMed] [Google Scholar]
- 34. Pirttilä T, Soininen H, Mehta PD, Heinonen O, Lehtimäki T, Bogdanovic N et al (1997) Apolipoprotein E genotype and amyloid load in Alzheimer disease and control brains. Neurobiol Aging 18:121–127. [DOI] [PubMed] [Google Scholar]
- 35. Reiner AP, Carlson CS, Jenny NS, Durda JP, Siscovick DS, Nickerson DA, Tracy RP (2007) USF1 gene variants, cardiovascular risk, and mortality in European Americans: analysis of two US cohort studies. Arterioscler Thromb Vasc Biol 27:2736–2742. [DOI] [PubMed] [Google Scholar]
- 36. Salero E, Gimenez C, Zafra F (2003) Identification of a non‐canonical E‐box motif as a regulatory element in the proximal promoter region of the apolipoprotein E gene. Biochem J 370:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Savioz A, Leuba G, Vallet PG, Walzer C (2009) Contribution of neural networks to Alzheimer disease's progression. Brain Res Bull 80:309–314. [DOI] [PubMed] [Google Scholar]
- 38. Shibata N, Ohnuma T, Higashi S, Higashi M, Usui C, Ohkubo T et al (2006) Genetic association between USF 1 and USF 2 gene polymorphisms and Japanese Alzheimer's disease. J Gerontol A Biol Sci Med Sci 61:660–662. [DOI] [PubMed] [Google Scholar]
- 39. Shieh BH, Sparkes RS, Gaynor RB, Lusis AJ (1993) Localization of the gene‐encoding upstream stimulatory factor (USF) to human chromosome 1q22‐q23. Genomics 16:266–268. [DOI] [PubMed] [Google Scholar]
- 40. Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K (1998) Cholesterol depletion inhibits the generation of beta‐amyloid in hippocampal neurons. Proc Natl Acad Sci U S A 95:6460–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steiger JL, Bandyopadhyay S, Farb DH, Russek SJ (2004) cAMP response element‐binding protein, activating transcription factor‐4, and upstream stimulatory factor differentially control hippocampal GABABR1a and GABABR1b subunit gene expression through alternative promoters. J Neurosci 24:6115–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stephens M, Donnelly P (2003) A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tabuchi A, Sakaya H, Kisukeda T, Fushiki H, Tsuda M (2002) Involvement of an upstream stimulatory factor as well as cAMP‐responsive element‐binding protein in the activation of brain‐derived neurotrophic factor gene promoter I. J Biol Chem 277:35920–35931. [DOI] [PubMed] [Google Scholar]
- 45. Tan ZS, Seshadri S, Beiser A, Wilson PW, Kiel DP, Tocco M et al (2003) Plasma total cholesterol level as a risk factor for Alzheimer disease: the Framingham Study. Arch Intern Med 163:1053–1057. [DOI] [PubMed] [Google Scholar]
- 46. Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G (2000) Decreased prevalence of Alzheimer disease associated with 3‐hydroxy‐3‐methyglutaryl coenzyme a reductase inhibitors. Arch Neurol 57:1439–1443. [DOI] [PubMed] [Google Scholar]
- 47. Yanagisawa K (2002) Cholesterol and pathological processes in Alzheimer's disease. J Neurosci Res 70:361–366. [DOI] [PubMed] [Google Scholar]
- 48. Yang XP, Freeman LA, Knapper CL, Amar MJ, Remaley A, Brewer HB Jr, Santamarina‐Fojo S (2002) The E‐box motif in the proximal ABCA1 promoter mediates transcriptional repression of the ABCA1 gene. J Lipid Res 43:297–306. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership and age as covariates).
Table S2. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership and age as covariates) in males.
Table S3. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership and age as covariates) in females.
Table S4. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership as a covariate) in under 65‐year‐old.
Table S5. USF1 SNPs and haplotypes and their association with SP type and NFT prevalence (APOE4 carriership as a covariate) in over 65‐year‐old.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item