

SUMMARY
Ibrutinib, a bruton’s tyrosine kinase (BTK) inhibitor, provokes robust clinical responses in aggressive mantle cell lymphoma (MCL), yet many patients relapse with lethal Ibrutinib-resistant (IR) disease. Here, using genomic, chemical proteomic, and drug screen profiling, we report that enhancer remodeling-mediated transcriptional activation and adaptive signaling changes drive the aggressive phenotypes of IR. Accordingly, IR MCL cells are vulnerable to inhibitors of the transcriptional machinery and especially so to inhibitors of cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the positive transcription elongation factor b (P-TEFb) of RNA polymerase II (RNAPII). Further, CDK9 inhibition disables reprogrammed signaling circuits and prevents the emergence of IR in MCL. Finally, and importantly, we find that a robust and facile ex vivo image-based functional drug screening platform can predict clinical therapeutic responses of IR MCL and identify vulnerabilities that can be targeted to disable the evolution of IR.
In brief
Zhao et al. conduct unbiased proteomic, enhancer, and transcriptional profiling in combination with high-throughput drug screening to identify super-enhancer and kinome remodeling as an Ibrutinib resistance mechanism. CDK9 and BRD4 are vulnerabilities for Ibrutinib resistance necessary to sustain super-enhancers, and inhibition of CDK9 or BRD4 prevents the emergence of resistance.
Graphical Abstract
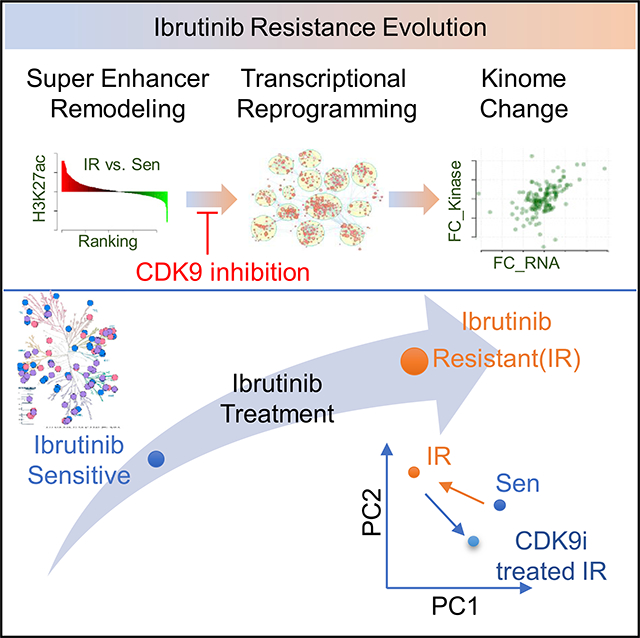
INTRODUCTION
Mantle cell lymphoma (MCL) demonstrates a poor prognosis due to the emergence of drug resistance and the rapid progression of relapsed disease (Jares et al., 2012). B cell receptor (BCR) signaling inhibitors have emerged as promising therapeutic agents for various B cell lymphomas. Ibrutinib, a bruton’s tyrosine kinase (BTK) inhibitor, showed high response rates in patients with MCL (Zucca and Bertoni, 2013; Wang et al., 2013). Unfortunately, despite dramatic clinical responses to Ibrutinib, resistance inevitably develops with treatment (Colomer and Campo, 2014). Once Ibrutinib-treated patients relapse, the 1-year survival rate is only 22% (Cheah et al., 2015; Wang et al., 2015). Thus, new therapeutics that augment the initial response and, more importantly, sustain remission are desperately needed.
Acquired drug resistance emerges following an initial period of drug responsiveness via evolution of drug-tolerant cancer cell populations, and this process is facilitated by the induction of a complex network of survival and proliferative pathways following exposure to therapy (Diaz et al., 2012; Russo et al., 2016). Acquired resistance can involve bypass mutations, loss of the original target, activation of key downstream effectors of the targeted pathway, and/or activation of alternative signaling pathways, all of which can render the malignant cell indifferent to the original therapy (Kobayashi et al., 2005; Pao et al., 2005). We modeled acquired resistance to Ibrutinib by generating Ibrutinib-resistant (IR) MCL cell lines and performing activity-based (ATP-binding) protein profiling (ABPP) (Zhao et al., 2017). These studies revealed that adaptive kinome reprogramming drives IR phenotypes in MCL, such as unrestrained proliferation (Zhao et al., 2017) and increased sensitivity to the BCL-2 inhibitor ABT-199 (Jiang et al., 2019). Collectively, our studies established that IR evolves via a series of responses in both MCL and stromal cells, creating a positive signaling feedback loop that amplifies the pro-survival and growth signals, ultimately leading to the acquired IR phenotype.
Adaptive changes in the kinome also manifest in ABT-199-resistant MCL and large B cell lymphoma. We revealed that drug-resistance evolution was associated with reprogramming of super-enhancers (SEs) that activate the transcription of select genes (Zhao et al., 2019). CDK7 inhibition was shown to disable SE-dependent transcription and kinome programs of ABT-199-resistant lymphoma, leading to cell death ex vivo and tumor regression in vivo (Zhao et al., 2019). Importantly, CDK7 inhibitor also blocked the evolution of ABT-199 resistance, suggesting a combination strategy that could have a significant impact in the clinic (Zhao et al., 2019). Ser2 of RNA polymerase is phosphorylated by P-TEFb (positive transcription elongation factor b), which comprises cyclin-dependent kinase 9 (CDK9) and one of several cyclin subunits and associates with other factors such as BRD4 in a large complex of proteins coined the SE complex (Yang et al., 2005; Smith et al., 2011; He et al., 2010). Notably, BRD4 binding to P-TEFb provokes recruitment of the complex to SEs and productive transcriptional elongation (Yang et al., 2005; Gargano et al., 2007; Guo and Price, 2013; Peterlin and Price, 2006).
Given our findings that ABT-199 resistance in lymphoma was associated with CDK7-dependent SEs and kinome remodeling, and the established roles of CDK9 in controlling P-TEBb function, we reasoned that transcriptional targeting might also disable IR MCL. Using unbiased proteomic, enhancer, and transcriptional profiling as well as drug screening of a broad range of transcriptional inhibitors against IR MCL primary patient samples on an ex vivo imaged-based platform that mimics the tumor microenvironment (TME), we report that SEs and kinome remodeling also manifest in IR MCL; CDK9 and BRD4 are selective vulnerabilities for IR MCL that are necessary to sustain IR SEs; and loss or inhibition of CDK9 or BRD4 disables and prevents the emergence of IR. These studies support the need for the development of chromatin-targeting therapeutic strategies to improve MCL patient outcomes in response to Ibrutinib. Finally, we demonstrated that evaluating drug response in primary MCL patient samples with a robust and facile ex vivo image-based functional platform predicted clinical therapeutic responses of IR MCL and identified vulnerabilities that can be targeted to disable the evolution of IR.
RESULTS
Transcriptome reprograming rewires kinome signaling in IR MCL
IR in MCL occurs via adaptive kinome reprogramming that leads to constitutive activation of the PI3K/AKT/mTOR pathway and increased levels of BCL-2 (Zhao et al., 2017; Jiang et al., 2019). To gain insights into mechanisms that might drive kinome reprogramming in IR MCL cells, we performed RNA sequencing (RNA-seq) analyses on two independent MCL IR derivatives (SP49 versus SP49-IR, Jeko-1 versus Jeko-1-IR) (Zhao et al., 2017). Analysis of significantly differentially expressed genes revealed both shared and selective transcriptomic changes in IR MCL, with 2,412 upregulated and 1,328 downregulated genes in SP49-IR cells and 1,619 upregulated and 1,832 downregulated genes in Jeko-1-IR cells (Figures 1A and S1A). Common differentially expressed genes including upregulated and downregulated genes between SP49 and Jeko-1 (IR versus Sen) were used as inputs for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) term (molecular function) analyses through Enrichr (Table S1). As shown in Figures 1B and S1B, the BCR signaling pathway and mTOR signaling pathway, as well as the RNA polymerase II (RNAPII) core promoter proximal region sequence-specific binding, were on top of the significantly enriched pathways and molecular function terms. GO terms enriched in Jeko-1-IR cells were then mapped as a network of gene sets (nodes) related by mutual overlap (edges), where the color indicates the significance of the gene set (false discovery rate [FDR] q value). Notably, RNAPII, kinase cascade, and cell adhesion regulation are highly enriched in IR compared to parental MCL (Figures 1C and S1C), which were pathways previously identified by ABPP (Zhao et al., 2017). Further, integrating ABPP and RNA-seq data revealed a positive correlation between gene transcript changes and kinase alterations in IR cells (Figure S1C; Table S2). Thus, transcription reprogramming manifestation in IR MCL at least partially contributes to kinome remodeling.
Figure 1. Transcriptome reprograming rewires kinome signaling in IR MCL.

(A) Volcano plots highlighting genes differentially expressed in IR versus parental sensitive (Sen) Jeko-1 (left) and SP49 (right) cells. The numbers of significant differentially expressed upregulated genes (red) or downregulated genes (black) are included in parentheses. Log2(FC): log2 FC cut-off of log2(1.5), p value cut-off of 0.05. n = 3 biologically independent samples.
(B) KEGG pathway analysis of common differentially expressed genes expressed in IR cells compared to Sen cells between SP49 and Jeko-1 using Enrichr. Bar length and top axis represent −log10(p value). Color bar intensity also represents −log10(p value), where the darker colors are indicative of higher significance (lower p value). p value is calculated by hypergeometric test (Chen et al., 2013; Kuleshov et al., 2016).
(C) Enrichment map of IR-associated genes in Jeko-1-IR cells. The map displays the enriched gene sets in Jeko-1-IR cells. Nodes represent gene sets, and edges represent overlap between gene sets. Gene sets that did not pass the enrichment significance threshold are not shown. Clusters of functionally related gene sets were assigned a label using “AutoAnnotate” add-in in Cytoscape; node color intensity is proportional to enrichment significance, and clusters of biological and functional interest for their roles in IR are highlighted in red.
Increased RNAPII activity and dependence on CDK9 are a hallmark of IR MCL
Recent studies have shown that activation of the transcriptional machinery underlies kinome reprogramming in tumors (Zawistowski et al., 2017; Flaherty et al., 2012; Eroglu and Ribas, 2016; Ascierto et al., 2013; Long et al., 2014). To address this, we implemented our automated first-in-class combination of in silico ex vivo drug response assay, termed EMMA (ex vivo multiple myeloma [MM] and MCL advisor), which is capable of screening primary hematologic malignant cells against a panel of targeted and chemotherapeutic agents (Zhao et al., 2017, 2019; Silva et al., 2015, 2017; Ren et al., 2018). Briefly, this drug response assay is capable of screening primary lymphoma cells directly from biopsies against a panel of drugs in an ex vivo reconstruction of the bone marrow TME, including extracellular matrix, patient-derived soluble factors, and patient-derived stroma. Using this platform, we tested the relative sensitivity of MCL cell lines and patient specimens against inhibitors of CDK9 (NVP-2), CDK7 (THZ1), CDK12 (THZ531), and BRD4 (INCB054329 or JQ-1), along with other targeted drugs (Figures 2A and S2A; Table S3). This assay showed drugs targeting the transcriptional kinases CDK9, CDK7, CDK12, and BRD4 have potent and selective activity in IR MCL cell lines and primary MCL (Figures 2A–2C and S2B). The most potent of these agents in primary MCL samples was the CDK9 inhibitor NVP-2. Additionally, NVP-2 sensitivity was shown to be negatively correlated with Ibrutinib sensitivity in MCL samples, though NVP-2 is effective against both Ibrutinib-sensitive and IR MCL cell lines and primary samples (Figures 2C and 2D; Table S4).
Figure 2. IR MCL cells are highly sensitive to CDK9 inhibition.

(A) Z scores of normalized LD50s of drug response curves from drug screening performed in paired IR and Sen cells for both Jeko-1 and SP49 cells. Compounds that have higher potency in IR cells are highlighted and labeled in red.
(B) Drug response assessment of NVP-2 potency in primary MCL specimens using EMMA platform.
(C) Heatmap of LD50s calculated from EMMA drug response assays in primary MCL patient specimens. Red represents low LD50 (sensitive), and blue represents high LD50 (resistant). Patients that are defined as IR by LD50 are highlighted in red font.
(D) Pearson correlation between Ibrutinib sensitivity (AUC) and NVP-2 sensitivity (LD50) from EMMA experiments performed on primary IR MCL samples (n = 19) defined as in (C). Dotted line represents boundary for 95% confidence interval (CI).
(E) NVP-2 treatment suppresses RNA polymerase II (RNAPII) CTD pSer2, pAKT, MYC, and MCL-1 levels and augments PARP cleavage in primary MCL patient samples (Pt0448, Pt1888) in a dose-dependent fashion.
Data shown in (A) and (E) are representatives of three independent experiments.
Next, western blot revealed higher levels of RNAPII phosphorylation in IR compared to parental cells (Figure S2C). Further, treatment with a half maximal effective concentration (EC50) dose of NVP-2 led to more marked reductions in the phosphorylation of Ser2 on RNAPII and AKT, as well as the level of MCL-1, in IR versus parental MCL lines (Figure S2C). NVP-2 treatment triggered apoptosis in IR cells and MCL primary patient samples, as judged by poly (ADP-ribose) polymerase (PARP) cleavage (Figure 2E). Given that BRD4 binds CDK9 as a transcriptional elongation complex to coordinately regulate transcription activities, BRD4 inhibitor was employed in parallel to validate the function of transcriptional activation in driving IR. Indeed, similar selective effects manifested following treatment of primary MCL patient samples with the BDR4 inhibitor (Figure S2D). Thus, transcriptional machinery key regulators CDK9 and BRD4 are vulnerabilities for IR MCL.
Targeting CDK9 or BRD4 compromises transcriptome and kinome reprogramming of IR MCL
Next, chemical proteomic ABPP was performed to test if targeting the transcriptional machinery with CDK9 and BRD4 inhibitors affected the kinome of IR MCL cells. Parental and IR MCL cells were treated with low and high doses of NVP-2 for 24 h and BRD4 inhibitor for 24 and 48 h. Then, lysates were processed for ABPP, and the abundancies of enriched peptides were determined by mass spectrometry. We compared peptide abundancies between treated IR cells and untreated IR cells for each inhibitor and defined inhibitor-regulated kinases as those that had a 1.5-fold change or higher in peptide abundance upon treatment. These analyses identified rather profound effects on kinome activity, where shared kinases whose activity was dependent on CDK9 were upregulated in IR MCL (Figures 3A and S3A; Table S5). KEGG pathway analysis indicated that the NVP-2 or BRD4i-regulated kinases were involved in AKT-mTOR, extracellular-signal-regulated kinase (ERK), chemokine, and BCR pathways that were enriched in the IR MCL cells (Figures 3B and S3B). Thus, targeting CDK9 and BRD4 perturbs the IR-associated kinome.
Figure 3. CDK9 is required to sustain transcriptional and kinase reprogramming in IR MCL cells.

(A) FC in activity of kinases in IR versus Sen MCL cells (x axes) and kinase upregulation in IR cells after treatment with NVP-2 (y axes) (left panel, 10 nM; right panel, 50 nM) in the SP49 cell line. Venn diagram shows overlap in shared protein kinases that are increased in SP49-IR cells versus Sen cells and that are significantly decreased by NVP-2 treatment in IR cells. These overlapped kinases are highlighted by the red box in each scatterplot. Cut-off, FC = 1.5. n = 3 biological replicates.
(B) KEGG pathway enrichment of kinases that are both increased in SP49-IR cells versus Sen cells and significantly decreased by NVP-2 treatment in IR cells. Top axis represents −log10(p value). P values were calculated as in Figure 1B.
(C) RNA-seq heatmap showing genes that are increased in IR versus Sen cells and that are decreased by NVP-2 treatment in Jeko-1 and SP49 IR cells. n = 3 biological replicates.
(D) Pie charts showing the percentage of gene expression changes (increase/conserved/decrease) in IR compared to Sen cells for NVP-2 decreased genes in IR cells. Around half (49.0% for Jeko-1 and 48.8% for SP49) of the NVP-2 decreased genes are increased in IR cells compared to sensitive cells.
(E) Molecular function enrichment results by Enrichr for genes that are commonly upregulated by IR cells compared to Sen cells and downregulated by NVP-2 in IR cells between Jeko-1 and SP49. Top axis represents −log10(p value). P values were calculated as in Figure 1B.
(F) Principal component analysis (PCA) of RNA-seq showing that IR cells are distinct from Sen cells and that Ibrutinib-treated IR cells cluster with IR cells treated with vehicle (DMSO). In contrast, NVP-2 treatment renders IR-associated gene signatures toward those of the Sen cell clusters. n = 3 biological replicates.
To evaluate the possible transcriptome influences on the IR MCL kinome changes resulting from CDK9 and BRD4 targeting, RNA-seq studies were performed in paired parental and IR MCL lines treated with NVP-2, BRD4i, or Ibrutinib. Using 2-fold change as the cut-off, we identified 290 genes in Jeko-1-IR cells and 328 genes in SP49-IR cells that were both significantly upregulated in IR MCL and downregulated following NVP-2 treatment (for BRD4i treatment, the numbers of significantly regulated genes were 426 and 523) (Figures 3C and S3C; Table S5). Interestingly, nearly half of the NVP2- and BRD4i-suppressed genes were upregulated in IR MCL cells when compared with parental MCL cells (Figures 3D, S3D, and S3E). Further, GO analysis for molecular function terms revealed that these genes are consistent with top IR-associated genes, such as RNAPII-activation-associated genes and pathways, and are negatively enriched by NVP-2 and BRD4i treatment (Figures 3E and S3F). Accordingly, principal-component analysis (PCA) revealed that NVP-2 treatment drives and reverses the IR MCL gene profile phenotypes toward those in Ibrutinib-sensitive MCL cells (Figures 3F and S3G). In contrast, Ibrutinib treatment failed to shift and reverse the gene phenotypes of IR lines to those of Ibrutinib-sensitive cells (Figures 3F and S3G). Together, these results support the notion that transcriptome and associated kinome reprogramming characteristics of IR MCL are disabled by inhibition of the transcription machinery.
SE remodeling contributes to transcriptional changes in IR MCL
To assess if the rewired signaling and transcriptional programs in IR MCL reflected remodeling by SE, chromatin immunoprecipitation sequencing (ChIP-seq) analysis of lysine-27 acetylated histone H3 (H3K27Ac), a mark of SEs and actively transcribed genes, was performed on paired parental and IR MCL cells (Bradner et al., 2017; Hnisz et al., 2013). ChIP-seq and ranking of enhancers by the amplitude and density of H3K27ac marks revealed distinct upregulated (gained) and downregulated (lost) SEs in IR cells (Figures 4A and S4A). As expected, increased expression of genes found in IR cells was associated with gained SEs, whereas SEs lost in IR cells were associated with decreased gene expression in IR cells (Figure 4B) (Zhao et al., 2017; Hnisz et al., 2013). Notably, gained SEs in IR MCL cells were associated with increased expression of known drivers of lymphoma, specifically BCL2, CXCR4, PLCG, MYC, TRAF4, MiR-17, and CCND1, among others (Figure 4A). Gene set enrichment analysis (GSEA) confirmed that genes regulated by IR-associated SEs are positively enriched in IR cells relative to Ibrutinib-sensitive parental cells (Figure 4C), and enrichment analysis using the Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al., 2010) established that IR-associated H3K27ac profiles were upregulated in several pathways that were significantly increased in IR MCL cells as determined by ABPP and RNA-seq profiling (Figures 4D and S4B).
Figure 4. SE remodeling drives transcriptional programming and drug sensitivity to CDK9 inhibition in IR MCL lines and primary samples.

(A) ChIP-seq hockey-stick plot ranking enhancers by H3K27ac signal density. Number of SEs in each sample is labeled in plot. SE-associated genes with H3K27ac signal densities that surpass the inflection point by are indicated in red font.
(B) Boxplots showing log2 FC of gene expression in IR compared to Sen cells that are regulated by gained (Gain), lost (Loss) or conserved (Cons) SEs. Gained or lost SE-associated genes are genes with H3K27ac signal enrichment exhibiting a greater or less than 2-fold enrichment in IR versus parental Sen cells. Significance was determined using the Kruskal-Walllis test; SP49 p = 6.5E−16; Jeko-1 p < 2.2E−16.
(C) GSEA shows that SEs defined by H3K27ac signals and transcript levels in both IR and parental MCL cells are highly correlated. Top panels: IR SE-associated genes are enriched in IR cells. Bottom panels: Sen SE-associated genes are enriched in parental sensitive cells. Genes were ranked according to their expression FC between resistant and sensitive cells.
(D) GREAT (Genomic Regions Enrichment of Annotations Tool) analysis of H3K27ac enrichment in SP49-IR cells.
(E) GSEA shows gained SE-regulated genes are decreased by NVP-2 treatment in Jeko-1 and SP49 IR cells.
(F) Boxplots showing log2 FC of gene expression following treatment with NVP-2 versus vehicle (DMSO) in Jeko-1-IR and SP49 IR cells. Typical enhancer (TE) or SE-associated genes are defined by H3K27ac ChIP-seq. Significance was determined using the Kruskal-Walllis test; SP49 p < 2.2E−16; Jeko-1 p = 7.33E−11.
NES, normalized enrichment score; FDR, false discovery rate.
See also Figure S4.
Strikingly, the increased expression of genes associated with gained SEs in IR MCL was compromised by NVP-2 treatment (Figures 4E and S4C). Further, the suppressive effects of NVP-2 or BRD4i treatment on SE-regulated genes were more profound than on genes driven by typical enhancers (TEs) (Figures 4F and S4D). Finally, ChIP-seq and RNA-seq studies performed on primary MCL patient samples revealed that the increased expression of genes associated with SEs was suppressed following inhibition of CDK9 or BRD4 (Figure S4E). Thus, SE-associated transcriptome reprogramming in IR can be disabled by targeting CDK9 or BRD4.
Targeting CDK9 prevents emergence and overcomes IR in MCL
Given that IR in MCL is associated with increased RNAPII phosphorylation and a heightened dependence on CDK9 and BRD4, we next tested the ex vivo efficacy of CDK9i and BRD4i against IR lines and primary IR MCL samples on our EMMA platform and combined these results with matched RNA-seq in these samples. RNA-seq analyses of primary IR MCL samples treated with and without NVP-2 or BRD4i revealed that IR-associated genes and pathways (e.g., mTORC1) are dramatically suppressed by these inhibitors (Figures 5A and S5A). Further, combined treatment by NVP-2 or BRD4i with Ibrutinib demonstrated increased compound effects, measured as decreased cell viability, when compared to any single agent alone (Figure 5B). Importantly, synergy and enhanced effect of the combined treatment with NVP-2 with Ibrutinib was also observed in primary MCL samples, where the combination induced more cell death and suppression of cell viability than did single agents alone (Figures 5C and S5B).
Figure 5. Targeting CDK9 prevents emergence and overcomes IR in MCL ex vivo and in vivo.

(A) GSEA of HALLMARK_MTORC1_SIGNALING pathway for NVP-2 treatment in the indicated IR MCL patient (Pt) specimens.
(B) Cell-based imaging analysis for drug response in SP49-IR cells treated with NVP-2, Ibrutinib, or the NVP-2/Ibrutinib combination (left panel) or with BRD4i (INCB054329), Ibrutinib, or the BRD4i/Ibrutinib combination (right panel).
(C) Drug response assay of primary MCL patient samples (n = 8) treated with Ibrutinib or NVP-2 alone or Ibrutinib/NVP-2 combination. AUC values calculated from cell-based imaging analysis. p values were calculated by one-way ANOVA.
(D) Colony formation analysis revealed the Ibrutinib/NVP-2 and Ibrutinib/BRD4i combination markedly impairs colony formation in HBL-2 MCL cells.
(E) Tumor volume in NSG recipient mice bearing Jeko-1 Sen tumors that were treated daily with Ibrutinib (25 mg/kg, i.p.) or biweekly with NVP-2 (2.5 mg/kg, i.p.) or the Ibrutinib/NVP-2 combination. First arrow, start of drug application; second arrow, onset of drug resistance evolution. p values were calculated by Student’s t test.
(F) Tumor volume in NSG recipient mice bearing Jeko-1-IR tumors that were treated daily with Ibrutinib or biweekly NVP-2, or the Ibrutinib/NVP-2 combination. P values were calculated by Student’s t test.
(G). Kaplan-Meier survival analysis of mice in (F). Mantel-Cox test was used for statistical analysis. For (E) and (F), dosages of the treatment are indicated in the figures. Black arrows indicate treatment start time, and red arrow in (E) indicates the time that Ibrutinib group tumor sizes are larger than vehicle group.
(H), Representative images taken 6 weeks after transplant of NSG mice bearing IR MCL PDX tumors that were treated with vehicle, Ibrutinib daily, biweekly NVP-2, or NVP-2/Ibrutinib combination. n is at least 4 for each group. Dosages of the treatment are same as (F).
For (E) and (F), data are shown as mean ± SD. For (E)–(G), n = 4 mice per treatment group. Data in (B) and (D) are representative of three independent experiments run in triplicate.
We next applied colony formation assays to monitor the emergence of drug resistance as described previously (Zhao et al., 2019) and to test if NVP-2 or BRD4 inhibition could impair the onset of IR ex vivo. As expected, following an initial and profound decrease of cell viability, IR emerged in both cell line models, as reflected by accelerated colonogenic growth following treatment with 1 μM Ibrutinib (Figure 5D). In contrast, co-treatment of these models with the combination of Ibrutinib and NVP-2 blocked this accelerated growth phase and nearly abolished colony formation (Figure 5D). To assess the efficacy of these combinations in vivo, immunocompromised NOD severe combined murine immune deficiency (scid) gamma (NSG) mice bearing parental Jeko-1 MCL cells were used. After tumors reached 100 mm3, mice were treated daily interperitoneally (i.p.) with Ibrutinib (25 mg/kg, daily), low-dose NVP-2 (2.5 mg/kg, twice a week), or both and followed for disease progression. Mice treated with Ibrutinib only experienced an initial transient response to the drug in the first 2 weeks of treatment. However, tumor volumes in mice treated with Ibrutinib alone exceeded those of mice in the vehicle group by week 3 after inoculation (Figure 5E). Notably, the NVP-2 treatment was much more effective than the Ibrutinib treatment at preventing disease progression, and the combination was superior to both Ibrutinib and NVP-2 treatment alone (Figure 5E); thus, these data support that CDK9 inhibition also impairs the development of IR in vivo. CDK9 activity is also necessary for the emergence of IR in this model.
To further test the in vivo efficacy of the NVP-2/Ibrutinib combination, a four-armed study was performed using Jeko-1-IR cells. Again, there was also obvious synergy of the NVP-2 + Ibrutinib combination in NSG mice bearing Jeko-1-IR tumors in delaying tumor progression and in improving overall survival (Figures 5F and 5G). Thus, CDK9 inhibition can resensitize IR MCL to inhibitors of BCR signaling. Finally, in a luciferase-expressing patient-derived xenograft (PDX) IR MCL xenograft model, the NVP-2/Ibrutinib combination was again superior at preventing tumor progression (Figure 5H). Indeed, the NVP-2/Ibrutinib combination provoked nearly complete tumor regression (Figure 5H). No significant weight loss or movement disorders were observed after drug treatment (single or combined treatment). A complete blood count (CBC) evaluation revealed no significant changes of total red blood cell (RBC) count, hemoglobin, neutrophil count, platelet count, and reticulocyte cell numbers with any monotherapy (Ibrutinib or NVP-2) or combination treatments when compared with the vehicle control treatments in these NSG mice (Table S6). Overall, these results provide reassurance that the lower dose of NVP-2 that we chose to use to treat these mice was not toxic, though the potentially toxic off-target effects of using a larger dose of NVP-2 should not be ignored. Collectively, these studies provide proof-of-concept that co-targeting CDK9 or BRD4 with Ibrutinib is a rational therapeutic strategy that can prevent the development of IR and can re-sensitize IR MCL to Ibrutinib.
The EMMA platform as a tool to predict clinical responses and inform vulnerabilities in primary MCL samples
A daunting therapeutic challenge in treating MCL patients once IR develops is inter- and intra-tumoral heterogeneity and plasticity that limits the efficacy and duration of response. We again implemented the EMMA platform and examined 60 MCL patient specimens for their response to Ibrutinib, which were ranked by calculating the area under curve (AUC) of five serial diluted dose response curves, the EC50, and the maximal efficacy at 96 h after treatment. Primary MCL samples displayed variable responses to Ibrutinib, where, for example, Pt61 was sensitive, Pt13 was resistant, and Pt31 was intermediately sensitive (Figure 6A).
Figure 6. The EMMA platform predicts clinical responses and informs vulnerabilities in primary and IR MCL.

(A) Drug response assays of Ibrutinib sensitivity in primary patient samples with cell-based imaging analysis showing representative dose responses of sensitive, intermediate, and resistant primary MCL patients.
(B) Heatmap showing top differential genes between primary IR and Sen MCL samples separated by maximum Ibrutinib effect measured by cell-based imaging analysis. n = 32 primary samples.
(C) GSEA shows MTORC1_SIGNALING is positively enriched in primary IR MCL samples compared to Sen primary MCL samples. NES, normalized enrichment score; FDR, false discovery rate.
(D) Median NES of single-sample GSEA (ssGSEA) reveals the correlation of Moffitt and MD Andersen gene signatures of IR and Sen primary MCL samples.
(E) Shared positively enriched HALLMARK pathways of IR compared to Ibrutinib-sensitive primary MCL samples of Moffitt and MD Andersen datasets.
(F) Receiver operating characteristic (ROC) curves of AUC and maximum effect from cell-based imaging analysis compared to clinical response of patients. AUC and Youden cut-off indexes for maximum effect ROC curve are shown.
To identify a gene expression signature that discriminated Ibrutinib-sensitive from IR samples, RNA-seq was performed on 32 of these patient specimens. Supervised hierarchical clustering of the gene expression of these 32 samples based on the 96-h maximal efficacy to Ibrutinib was performed. These analyses revealed a significant enrichment and correlation of differentially expressed genes between clinical IR and EMMA-defined IR (Figure 6B; Table S4). Pathway analysis and GSEA were performed on genes that were differentially expressed between Ibrutinib responders and non-responders. Consistent with the findings from IR MCL lines (Figure 2), IR primary samples exhibited significant positive enrichment for the mTORC1 HALLMARK signature (Figures 6C and S6A). These findings are in accord with the dependencies of primary IR to kinase and transcriptional inhibitors present in primary IR MCL samples.
To determine and compare the relative significance of the HALLMARK signatures identified in Ibrutinib-sensitive and IR primary MCL patient samples, single-sample GSEA (ssGSEA) was performed, and the resulting enrichment scores were ranked based on the responder and non-responder groups defined by our drug screen. In accordance with our MCL cell line studies, there were higher enrichment scores for MYC, E2F, nuclear factor κB (NF-κB), and mTOR pathways in IR MCL patient samples, while higher enrichment scores for wingless-related integration site (WNT) pathways were observed in Ibrutinib-sensitive MCL patient samples (Figure S6B). Notably, we validated these signatures by querying an independent dataset of Ibrutinib-treated MCL samples with clinically defined responses (Zhang et al., 2019a) and demonstrated that enriched HALLMARK signatures from IR patients as determined by the EMMA platform positively correlated with the findings from this MD Anderson study (Figures 6D and S6C). Thus, as in myeloma patients (Silva et al., 2017), the EMMA platform can predict drug response in primary MCL.
To determine how sensitive and specific our ex-vivo-based drug screen is at predicting clinical Ibrutinib responses, we compared the receiver operating characteristic (ROC) curve for AUC and maximum effect (Figure 6F). ROC curves for clinical response status over the maximum effect were the most predictive of clinical Ibrutinib responses (Figure 6F), where areas under the ROC curve and the 95% confidence intervals (CIs) were 0.830 (0.653–1.007). To assess the potential clinical usefulness of the EMMA maximum effect to predict clinical responses to Ibrutinib, the Youden-index-based cut-off value for this metric was determined and was calculated to be 63.2. Further, the associated positive predictive value (PPV; response rate above the cut-off) was 66.7%, and the negative predictive value (NPV; non-response rate below the cut-off) was 92.9%. This suggests a potential for a high discriminatory value of maximum effect from EMMA that enriches for response rates to Ibrutinib (i.e., increasing PPV) while preserving a high NPV. Having said this, a formal evaluation of clinical utility depends on cut-offs selected for implementation of a clinical-grade diagnostic device in developmental trials that is dependent on the goals of those trials, and such trials are needed for the EMMA platform. Nonetheless, these findings suggest that maximum effect from EMMA can be used to determine clinical responses to Ibrutinib, thus providing a means to stratify and tailor treatment of MCL patients.
DISCUSSION
Our findings are in accord with recent studies that have shown that adaptive kinome reprogramming and altered cancer cell states are due to transcriptional programs coordinated by chromatin and transcriptional regulators (e.g., BRD4, CDK7, CDK9, and RNAPII) that bind to and activate distinct SE sites (Zawistowski et al., 2017; Bradner et al., 2017; Franco et al., 2016). Our findings that such SE-dependent adaptive signaling contributes to the evolution of drug resistance support the notion that clinical resistance to Ibrutinib can arise in the absence of new genetic mutations, as we and others have failed to identify recurrent mutations in IR MCL patients (Zhao et al., 2017; Zhang et al., 2020), and similar observations have been reported in other tumor types such as AML and solid tumors (Zawistowski et al., 2017; Rusan et al., 2018; Agarwal et al., 2019). Indeed, as we have shown, numerous kinase signaling networks are rewired and activated in MCL cells as they become IR (Zhao et al., 2017). Thus, combinations of two or more kinase inhibitors are unlikely to be sufficient to provide durable therapeutic responses. Given inter-tumor heterogeneity, especially in primary MCL samples, comparing gene expression between sensitive and resistant samples failed to detect any single gene or gene pair that clearly defined IR, suggesting more complex molecular processes. Genetic changes, such as mutations in BTK, NOTCH1, and KMT2D, and alternative NF-κB pathway mutations have been shown to be associated with primary and acquired IR in only a minority of cases (Chiron et al., 2014; Martin et al., 2016; Nomie et al., 2020; Rahal et al., 2014). Recent studies have approached overcoming IR by developing treatment strategies that combine Ibrutinib with additional inhibitors targeting proteins such as HSP90, ROR1, and XO1 as well as with inhibitors that target the oxidative phosphorylation pathway, with some of these strategies moving into early-phase clinical trials (Jacobson et al., 2016; Zhang et al., 2019a, 2019b; Hing et al., 2015). However, our global pharmacogenomic approaches instead identified common pathways associated with IR evolution and lead us to focus specifically on transcriptional machinery to disable global signaling reprogramming. Thus, the inhibitions of the transcriptional mechanisms that control kinase remodeling are a very attractive alternative strategy to overcoming drug resistance. Our translational results provide a foundation for CDK9 inhibitors, such as the recently developed agent AZD4573, which is currently in a phase of clinical trials, to be used in the treatment of aggressive and drug-resistant lymphomas (Cidado et al., 2020). Importantly, our studies strongly indicate that CDK9 or BRD4 targeting can be effective against MCL and can prevent the development of IR in initially Ibrutinib-sensitive tumors and can re-sensitize IR MCL to Ibrutinib, addressing two major clinical hurdles in the treatment of MCL patients. Thus, the findings presented fully support such combinations as strategies for the treatment of IR MCL patients that have few therapeutic options and dismal outcomes and as regimens that may prevent the emergence of resistance to improve outcomes of treatment-naive MCL patients.
Finally, we submit that platforms such as EMMA could be powerful clinical tools, as treatment of individual MCL patients with either conventional chemotherapy or an ever-increasing number of targeted agents remains highly empiric, and inter- and intra-tumoral heterogeneity and tumor plasticity limit their efficacy and durability. The EMMA platform is a considerable improvement for deciding drug choices over cell line studies, where by recapitulating the 3D lymphoma TME, it is possible to temporally and robustly assess the dynamics and magnitude of the response of primary lymphoma cells to single agents and drug combinations over a short interval to inform patient therapies. Most importantly, our studies showed a strong correlation of Ibrutinib response detected by EMMA with patient clinical Ibrutinib response. This first-in-kind automated in silico platform is an accurate predictor of the clinical responses of MCL patients and allows for patient-specific approaches for tailored therapies that are effective at eradicating their drug-resistant disease and at preventing the emergence of drug resistance in treatment-naive patients across a broad spectrum of hematological malignancies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jianguo Tao (jianguo.tao@moffitt.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for the RNAseq and CHIPseq data reported in this paper is GEO: GSE141336.
The accession number for the ABPP proteomics data reported in this paper is ProteomeXchange: PXD005734.
Computational code used for ChIP-seq analyses can be obtained from the Jun Qi Laboratory github page. Namely, Bamliquidator was used to calculate read density which can be found at https://github.com/BradnerLab/pipeline/wiki/bamliquidator; and ROSE2, was used to identify enhancers and can be found at https://github.com/BradnerLab/pipeline/(ROSE2_main.py).
All algorithms and codes used to process images and perform downstream analyses for the EMMA platform were provided directly by the Silva Lab (Silva et al., 2015).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and tumor specimens
The primary samples from MCL patients were obtained from fresh biopsy-derived lymphoma tissues (lymph nodes) and from peripheral blood following informed consent from patients and approval by the Moffitt Cancer Center/University of South Florida Institutional Review Board. For preparation of viable, sterile, single cell suspensions, the lymph node tissue was diced and forced through a cell strainer into RPMI-1640 tissue culture medium. Cells, obtained after low-speed centrifugation, were resuspended in media. Lymphoma cells from peripheral blood were isolated by Ficoll-Plaque purification, and only lymphoma samples that had greater than 80% tumor cells were used for experiments. Age and gender of patients from which primary samples were obtained are not included in this publication as this information is not provided to research laboratories by the institution.
The human specimen studies presented were approved by the Moffitt/University of South Florida Institutional Review Board and patients provided signed informed consent forms.
Mice
Six- to eight-week-old male NOD/SCID mice were purchased from the Jackson Laboratory and used for xenograft experiments as described (Zhao et al., 2017).
Four- to eight-week-old male NOD/SCID mice were purchased from the Jackson Laboratory and used for PDX experiments as described (Zhao et al., 2017).
All animal studies were conducted in accordance with the NIH guidelines for animal care. All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of the Moffitt Cancer Center and the University of South Florida.
Cell lines
Mantle cell lymphoma cell line Jeko-1 was purchased from ATCC. SP49 was provided by Dr. M. Wasik from the University of Pennsylvania. Stromal cell line HK was purchased from Cellosaurus. These cells and their Ibrutinib-resistant derivatives (Zhao et al., 2017) were cultured in RPMI-1640 (GIBCO-Invitrogen) with penicillin (100 U/ml) and streptomycin (100 μg/ml) and maintained at 37°C in 5% CO2. Cell lines were routinely tested for mycoplasma using the Universal Mycoplasma Detection Kit from ATCC.
METHOD DETAILS
High-throughput small-molecule drug screens
Using a semi-automated platform, we tested the potency of a 60 small molecule annotated library in Jeko-1/SP49 parental and Ibrutinib-resistant derivative IR cells. Cell viability was estimated by using Resazurin (R&D Systems, AR002). In brief, cells were seeded in 384-well plates with 2,000 cells per well in 25 μL medium. Cells were cultured in the presence of different compounds at serial threefold diluted concentrations. After 3 (kinase inhibitors) or 6 days (epigenetic inhibitors) of treatment, 6 μL of Resazurin reagent was added into each well and incubated for 2 hr. Plates were read at 560/590 nm wavelength to estimate cell proliferation.
Cell-based imaging analysis of drug screening assay
Cells were seeded by a robotic pipettor in 384-well plates of a reconstructed lymphoma TME using the following components and concentrations: high physiological densities of primary patient cells (1–10 × 106 cells/ml) with lymphoma stromal cells (HK cells or autologous stromal cells, 2 × 105 cells/ml) suspended together in 600 μl RPMI 1640, 240 μl of 10x MEM, 240 μl of deionized H2O, 120 μl of 7.5% sodium bicarbonate solution, 600 μl of 1x RPMI 1640 and 1800 μl of 3.1 mg/ml Bovine collagen type I for 1 full 384-well plate, as detailed previously (Silva et al., 2015). After cell seeding, the 384-well plates were incubated at 37°C in 5% CO2 for 1–2 hr before an additional layer of media was added on top of the initial TME/tumor cell layer. A panel of drugs at five serial diluted concentrations was then added to the media, and plates were continuously imaged every 30 mins for 4 days (for cell line) or 6 days (for primary samples) by the Evos Auto FL microscope. All images were analyzed using a digital image analysis algorithm in ImageJ to detect cell viability based on membrane motion (pseudo-colored in green), and changes in viability were quantified by area under curve (AUC) and LD50 as described (Zhao et al., 2017; Silva et al., 2015, 2017). Maximum effect was determined by relative cell viability compared to media control at the highest drug dosage at 96 hours.
RNA-sequencing
All samples were prepared in biological triplicates. 1×106 cells were treated for 6 hr or 48 hr with either Ibrutinib, NVP-2, INCB054329 or DMSO at equal concentration as vehicle control. Total RNA was isolated using the RNeasy Plus Mini (QIAGEN Cat# 74134). Library prep was conducted using TruSeq Stranded mRNA Library Prep Kit (Illumina Cat #RS-122–2101/2) according to the manufacturer’s instructions. RNA sequencing was performed on HiSeq 2500v4 high output (50-bp, single-end reads). Tophat2 was used to align the Fastq files. TPM values were calculated and normalized using Cuffnorm. Genes that had a p < 0.05 and at least a two-fold change were considered to be significantly altered between treatments. Cutoff value for expressed genes was a TPM value equal to or higher than 1.
Chromatin immunoprecipitation followed by highly parallel sequencing (ChIP-Seq)
ChIP-seq was performed according to established protocols, with minor modifications. 50 million cells were used for ChIP-seq of H3K27ac. Crosslinking was performed in batches of 50 million cells in 50-mL tissue culture media by addition of one-tenth volume of 10X cross linking solution (11% formaldehyde, 50 mM HEPES pH 7.3, 100 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0). After 10 min of crosslinking at room temperature, formaldehyde was quenched with 125 mM glycine, cells were then washed three times in PBS pH 7.4, flash frozen in liquid nitrogen, and stored at −80°C. Frozen pellets were thawed on ice, resuspended in cold lysis buffer 1 (LB1; 5 mL per 50 million cells; 50 mM HEPES pH 7.3, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP- 40, and 0.25% Triton X-100, Roche protease inhibitor cocktail), and rotated for 10 min at 4°C. LB1 was removed and pellets were resuspended in cold lysis buffer 2 (LB2; 5 mL per 50 million cells; 10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA pH 8.0 and 0.5 mM EGTA pH 8.0, Roche protease inhibitor cocktail) and rotated for 10 min at 4°C. LB2 was removed and pellets were resuspended in cold sonication buffer (1.5 mL per 50 million cells; 50 mM HEPES pH 7.3, 140 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, Roche protease inhibitor cocktail). Samples were divided into 1.5 mL Bioruptor Plus TPX microtubes (Diagenode, #C30010010) at 250 μL per tube and sheared at 4°C using a water bath sonicator (Bioruptor, Diagenode; 22.5 minutes at high output; 30 s on, 30 s off). Sheared lysates were clarified by centrifuging at 20,000 × g at 4°C for 10 min and supernatants were collected together, setting aside 50 μL as an input sample. For all other ChIP-seq experiments, magnetic protein G beads (Dynabeads, ThermoFisher Scientific) were washed 3 times with, and resuspended in, 1-mL cold blocking buffer and then rotated with appropriate antibody overnight at 4°C using 100 μL of beads with 10 μg anti-H3K27ac (ABCAM ab4729) or 20 μL anti-RNAPII (Diagenode, #C15100055). Antibody:bead complexes were washed 3 times with cold blocking buffer, added to the diluted and clarified chromatin supernatant, and rotated overnight at 4°C. The bound chromatin was then washed twice with 1-mL cold sonication buffer, once with 1-mL cold sonication buffer supplemented with 500 mM NaCl, once with cold LiCl wash buffer (20 mM Tris pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate), and once with TE supplemented with 50 mM NaCl. Finally, beads were resuspended in 210 μL elution buffer (50 mM Tris-HCl pH 8, 10 mM EDTA, and 1% SDS) and chromatin was eluted by vortexing every 5 min while incubating at 65°C for 15 min. Beads were centrifuged at 20,000 × g for 1 min and the supernatant, together with input sample was placed at 65°C overnight to reverse crosslinks. RNA was digested with 0.2 mg/mL RNase A (Roche, 10109169001) at 37°C for 2 hr and protein was digested with 0.2 mg/mL proteinase K (Life Technologies, AM2546) at 55°C for 30 min. DNA was isolated with phenol chloroform extraction and ethanol precipitation.
Libraries for Illumina sequencing were prepared using ThruPLEX DNA-seq Kit (Rubicon) using 50 ng of DNA or less and amplifying according to manufacturer instructions. Amplified libraries were size-selected first using AMPure beads (Agencourt AMPure XP) and subsequently, using a 2% gel cassette in the Pippin Prep (SAGE Sciences) to capture fragments of 200–700 bp. Libraries were quantified by qPCR using the KAPA Biosystems library quantification kit, multiplexed with equimolar DNA content, and sequenced on an Illumina NextSeq 500 (single end 75 bp reads).
ChIP-Seq data processing
Sequence alignment
All datasets were aligned using Bowtie2 (version 2.2.1). All default parameters, except for –N 1 (reads that mapped uniquely to the genome with one or fewer mismatches) were used to align to human genome build NCBI37/HG19.
Identifying enriched regions
The MACS version 1.4.1 (Model based analysis of ChIP-Seq) 67 peak finding algorithm was used to identify regions of ChIP-Seq enrichment over background. A p value threshold of enrichment of 1e-9 was used for all datasets.
Calculating read density
We calculated the normalized read density of a ChIP-Seq dataset in any genomic region using the Bamliquidator (version 1.0) read density calculator. Briefly, ChIP-Seq reads aligning to the region were extended by 200 bp and the density of reads per base pair (bp) was calculated. For ChIP-seq, the density of reads in each region was normalized to the total number of million mapped reads producing read density in units of reads per million mapped reads per bp (rpm/bp).
Mapping typical enhancers and super-enhancers using H3K27ac enhancer definitions
H3K27ac super-enhancers (SEs) and typical enhancers (TEs) were mapped using the ROSE2 software package that has been previously described (Brown et al., 2014). MACS defined peaks were considered for rank-ordering by the ROSE2 algorithm. ROSE2 optimizes a stitching parameter on a per-sample basis for combining nearby peaks. Briefly, the algorithm optimizes for the enriched fraction of stitched regions. Read density within these regions was then quantified as noted above and stitched regions were ranked by this metric. Super-enhancers were called by re-scaling both the signal values and the ranks to fall between 0 and 1 and plotting a curve with scaled ranks on the x axis and scaled signal on the y axis. The x coordinate of the intersection point of the line of slope 1 that is tangent to the curve was used to define a cutoff for super-enhancers such that all stitched enhancers with a scaled rank greater than this cutoff were considered super-enhancers. Default ROSE2 parameters for stitching and region filtering, including exclusion of TSS-proximal signal (within 2.5 kb), were used. ROSE2 was also used to rank-order regions disproportionately enriched for POLII ChIP-seq signal as described for H3K27ac ChIP-seq above, except that no peak stitching was used and without excluding ± 2.5 kb from each TSS.
Activity-based protein profiling (ABPP)
Briefly, cell pellets were sonicated in IP/Lysis buffer, desalted and then depleted of endogenous ATP with Zeba spin column, and incubated with 10 μM desthiobiotin-ATP probes at room temperature for 10 min. The labeled proteins were reduced, alkylated and trypsin digested at 37°C for 2 hr. The labeled peptides were purified with high capacity streptavidin agarose resin, washed, eluted and subjected to LC-MS/MS for peptide sequencing. The peptide identification and relative quantification were performed using MaxQuant software (Version 1.2.2.5). The procedures of ABPP were as detailed previously (Zhao et al., 2017). Fold-change distribution of the ATP binding proteome was performed by GraphPad software. GO enrichment analysis was performed on the ABPP profile using Enrichr. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed on the increased protein kinases (comparing DTEP cells to parental cells) from 2 out of 3 cell lines. Raw Data are available via ProteomeXchange with identifier PXD010193.
Xenograft studies
Ten million Jeko-1 parental or IR were injected into the lower flank of NOD/SCID mice in a volume of 0.1 mL PBS. Mice were then randomized into control and treatment groups when tumor volume reached 0.2 cm3 (4 mice per group). Tumors were measured with calipers and when tumor volume reached 100–200 mm3 mice were randomized for treatment with Ibrutinib, NVP-2 or vehicle. Ibrutinib and NVP-2 was formulated in 30% (w/v) Captisol (pH 3.0). NVP-2 2.5 mg/kg was given i.p. twice a week and Ibrutinib was given 25 mg/kg oral lavage daily. Mice were humanely sacrificed when the control tumor reached ~4,000 mm3 or after the loss of more than 10% of body weight.
PDX model and imaging
Primary cells from patient specimen were transduced with the Vector: pCDH-EF-eFFLy-T2A-mCherry. Irradiation (200 cGy) was performed within 24 hours prior to xenografting. 1×106 of PDX cells were injected into the tail vain of NOD/SCID mice in a volume of 0.2 mL PBS. Mice were then randomized into control and treatment groups 4 days after tumor xenografting (4 mice per group). Drugs were given i.p. and the dose for NVP-2 was 2.5 mg/kg and Ibrutinib was 25 mg/kg. Mice were humanely sacrificed when the control tumor reached ~4,000 mm3 or after the loss of more than 10% of body weight. Images are taken 6 weeks after tumor xenofrafting. D-Luciferin (GoldBio Catalog # LUCK) was made as of 15 mg/mL in DPBS. 10 μL of Luciferin stock solution per gram of body weight (normally ~200 μL for a 20 g mouse for a standard 150 mg/kg injection) intraperitoneally was given about 10 mins before images were taken using in vivo imaging system IVIS200 (PerkinElmer).
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis (GSEA) was performed as described (Subramanian et al., 2005). The most differentially expressed genes ranked by log2 fold change for each comparison were used to generate a signature for GSEA analysis. The input activated or inactivated SE gene sets were extracted from H3K27ac ChIP-seq data. GSEA estimates whether the members of IR-specific gene set are found at the top or bottom of the NVP-2/INCB054329 treatment list, and if genes are specific to either parental or resistant cells, indicating they are associated with a specific phenotype, rather than being distributed uniformly or randomly across the list. An enrichment score (ES) is calculated to quantify the degree to which a gene set is over-represented at the top or bottom of the entire ranked list. After calculation of the scores for a collection of gene sets, an empirical phenotype-based permutation test procedure is used to estimate P values. GSEA normalizes the ES for each gene set to account for the variation in set sizes, yielding a normalized enrichment score (NES) and a false discovery rate (FDR). The FDR gives an estimate of the probability that a set with a given NES represents a false positive finding; it is computed by comparing the tails of the observed and permutation-computed null distributions for the NES.
ssGSEA (single sample GSEA)
To determine the relative activity of cancer pathways between Moffitt and MD Anderson cohorts, ssGSEA (version gsea2–2.2.1) was applied using patients’ gene expression profiles. To eliminate batch effects, we normalized gene expression by calculating the z-score within each cohort. Then, for each sample, we ranked all genes on the basis of their expression values to create a .rnk file as input for the software GSEAPreranked. The enrichment score was computed for HALLMARK as the assessment. HALLMARK signatures were downloaded from the Molecular Signatures MD Anderson cohort was downloaded from EGA. We also compared the increased HALLMARK in Ibrutinib resistant patient compared to Ibrutinib sensitive patients.
Enrichment map
Enriched gene-sets are graphically organized into a network, where each set is a node and edges represent gene overlap between sets; gene sets map to specific biological processes/pathways involved in Ibrutinib resistance. The Cytoscape network software (version 3.7.1) and the plugin “EnrichmentMap (version3.1.0)” and “AutoAnnotate (version 1.2)” were used to build the network. Plugin and source code are available at http://baderlab.org/Software/EnrichmentMap. Node color encodes the enrichment q-value. Node size is proportional to the total number of genes belonging to the corresponding gene-set (Merico et al., 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
P values of less than 0.05 were considered significant. Unless otherwise stated, comparison and statistical significance between two groups in this paper are based on two-sided t test. Analysis of variance (ANOVA) or the Kruskal-Wallis test was used for comparing data from multiple groups.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| cPARP | Cell Signaling Technologies | Cat# 5625; RRID: AB_10699459 |
| pAKT(Ser473) | Cell Signaling Technologies | Cat# 9271; RRID: AB_329825 |
| AKT | Cell Signaling Technologies | Cat# 4685; RRID: AB_2225340 |
| RNA pol II CTD phospho-Ser2 | Millipore | Cat# 04–1571-I; RRID: AB_11212363 |
| RNA pol II CTD phospho-Ser5 | Millipore | Cat# 04–1572-I; RRID: AB_11213421 |
| RNA pol II CTD phospho-Ser7 | Millipore | Cat# 04–1570-I; RRID: AB_2801298 |
| RNA pol II | Cell Signaling Technologies | Cat# 2629; RRID: AB_2167468 |
| MYC | Abcam | Cat# ab32072; RRID: AB_731658 |
| CDK7 | Cell Signaling Technologies | Cat# 2916; RRID: AB_2077142 |
| MCL-1 | Santa Cruz Biotechnology | Cat# sc-819; RRID: AB_2144105 |
| Actin | Santa Cruz Biotechnology | Cat# sc-47778 HP; RRID: AB_2714189 |
| RNAPII | Diagenode | Cat# C15100055; RRID: AB_2750842 |
| H3K27ac | Abcam | Cat# ab4729; RRID: AB_2118291 |
|
Bacterial and virus strains | ||
| pCDH-EF-eFFLy-T2A-mCherry Plasmid | Addgene | Cat# 104833; RRID: Addgene_104833 |
|
Biological samples | ||
| Primary Patient Specimens (MCL) | Moffitt Cancer Center | N/A |
|
Chemicals, peptides, and recombinant proteins | ||
| (+)-JQ1 | Cayman Chemical | Item# 11187 |
| (R)-PFI-2 | Cayman Chemical | Item# 14678 |
| A-1331852 | Selleckchem | Cat# S7801 |
| A-366 | Cayman Chemical | Item# 16081 |
| ABT-199 | Selleckchem | Cat# S8048 |
| ABT-263 | Selleckchem | Cat# S1001 |
| A-196 | Cayman Chemical | Item# 18317 |
| Alisertib | Selleckchem | Cat# S1133 |
| AZD7762 | Selleckchem | Cat# S1532 |
| AZD8055 | Selleckchem | Cat# S1555 |
| BAY-598 | Cayman Chemical | Item# 18238 |
| BEZ-235 | Selleckchem | Cat# S1009 |
| BI-9564 | Cayman Chemical | Item# 17897 |
| Bendamustine | TargetMol | Cat# T0095 |
| Bortezomib | Selleckchem | Cat# S1013 |
| Carfizomib | Selleckchem | Cat# S2853 |
| CPA7 | DC Chemicals | Cat# DC12009 |
| CPD23 | Dana-Farber Cancer Institute Jun Qi Lab | N/A |
| Dinaciclib | Selleckchem | Cat# S2768 |
| Doxorubicin | Selleckchem | Car# S1208 |
| GSK343 | Cayman Chemical | Item# 14094 |
| GSK484 | Cayman Chemical | Item# 17488 |
| GSK591 | Cayman Chemical | Item# 18354 |
| GSK864 | Cayman Chemical | Item# 18762 |
| GSK-J4 | Cayman Chemical | Item# 12073 |
| GSK-LSD1 | Cayman Chemical | Item# 16439 |
| Ibrutinib | Selleckchem | Cat# S2680 |
| I-CBP112 | Cayman Chemical | Item# 14468 |
| INCB052793 | Incyte Corporation (Wilmington, DE) | N/A |
| INCB054329 | Incyte Corporation (Wilmington, DE) | N/A |
| INCB059872 | Incyte Corporation (Wilmington, DE) | N/A |
| Lenalidomide | Selleckchem | Cat# 1029 |
| Lumpib | Selleckchem | Cat# S1069 |
| MK-1775 | Selleckchem | Cat# S1525 |
| MS049 | Cayman Chemical | Item# 18348 |
| NVP2 | MedChemExpress | HY-12214A |
| OF-1 | Cayman Chemical | Item# 17124 |
| OICR-9429 | Cayman Chemical | Item# 16095 |
| Olaparib | Selleckchem | Cat# 1060 |
| PFI-3 | Cayman Chemical | Cat# 15267 |
| PFI-4 | Cayman Chemical | Item# 17663 |
| PIK-75 | Selleckchem | Cat# S1205 |
| PRT1000220–005 | Incyte Corporation (Wilmington, DE) | N/A |
| R406 | Selleckchem | Cat# S2194 |
| Ruxolitinib | Selleckchem | Cat# S1378 |
| S63845 | ApexBio | Cat# A8737 |
| SCH-772984 | Selleckchem | Cat# S7101 |
| SGC0946 | Cayman Chemical | Item# 13967 |
| SGC707 | Cayman Chemical | Item# 17017 |
| Silvestrol | MedChemExpress | HY-13251 |
| THZ1 | Dana-Farber Cancer Institute Nathanael Gray Lab | N/A |
| THZ531 | Dana-Farber Cancer Institute Nathanael Gray Lab | N/A |
| Trametinib | Selleckchem | Cat# S2673 |
| UNC0642 | Cayman Chemical | Item# 14604 |
| UNC1215 | Cayman Chemical | Item# 13968 |
| UNC1999 | Cayman Chemical | Item# 14621 |
| VE-821 | Selleckchem | Cat# S8007 |
| Volasertib | Selleckchem | Cat# S2235 |
|
Critical commercial assays | ||
| Universal Mycoplasma Detection Kit | ATCC | Cat# 30–1012K |
| Resazurin | R&D Systems | Cat# AR002 |
| Bovine Type I Atelo-Collagen Solution | Advanced BioMatrix | Cat# 5005-B |
| Lymphoprep | STEMCELL Technologies | Cat# 07851 |
| RNeasy Plus Mini | QIAGEN | Cat# 74134 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# RS-122–2101/2 |
| 1.5 ml Bioruptor Plus TPX microtubes | Diagenode | Cat# C30010010 |
| Bioruptor | Diagenode | Cat# B01060010 |
| Dynabeads Protein G for Immunoprecipitation | ThermoFisher Scientific | Cat# 10007D |
| RNase A | Roche | Cat# 10109169001 |
| Proteinase K | Life Technologies | Cat# AM2546 |
| ThruPLEX DNA-seq Kit | TaKaRa | Cat# R400675 |
| AMPure Beads (Agencourt AMPure XP) | Beckman Coulter Life Science | Cat# A63882 |
| Pippin Prep | SAGE Sciences | Cat# PIP0001 |
| KAPA Biosystems Library Quantification Kit | KAPA Biosystems | Cat# KK4824 |
| Pierce Kinase Enrichment Kit with ATP Probe | ThermoFisher Scientific | Cat# 88310 |
| Zeba Spin Desalting Columns, 7K MWCO, 5 mL | ThermoFisher Scientific | Cat# 89892 |
| Captisol | Selleckchem | Cat# S4592 |
| D-Luciferin | GoldBio | Cat# LUCK |
|
Deposited data | ||
| RNaseq and CHIPseq | This Paper, Gene Expression Omnibus | GEO: GSE141336 |
| ABPP Proteomics | This Paper, Proteome Xchange | ProteomeXchange: PXD005734 |
|
Experimental models: cell lines | ||
| Jeko-1 | ATCC | Cat# CRL-3006; RRID: CVCL_1865 |
| Jeko-1 IR | Tao Lab | N/A |
| SP49 | University of Pennsylvannia Mariusz A. Wasik Laboratory | N/A |
| SP49 IR | Tao Lab | N/A |
| HK | Cellosaurus | Cat# CVCL_IY38; RRID: CVCL_IY38 |
| Autologous stromal cells | Tao Lab | N/A |
|
Experimental models: organisms/strains | ||
| Mouse: NOD-SCID (NOD-scid IL2Rgnull) | The Jackson Laboratory | Cat# 001303; RRID: IMSR_JAX:001303 |
|
Software and algorithms | ||
| Bamliquidator (version 1.0) | https://github.com/BradnerLab/pipeline/wiki/bamliquidator | N/A |
| ROSE2 | https://github.com/BradnerLab/pipeline/blob/master/ROSE2_main.py | Ref: Brown et al., 2014 |
| EMMA Platform Image Processing | Ref: Silva et al., 2015 | |
| ImageJ | https://imagej.nih.gov/ij/ | RRID:SCR_003070 |
| Eclipse (Mars) | https://www.eclipse.org/mars/ | N/A |
| MATLAB R2014b | https://www.mathworks.com/products/matlab.html | RRID: SCR_001622 |
| MATLAB R2016b | https://www.mathworks.com/products/matlab.html | RRID: SCR_001622 |
| GraphPad Prism 7 | https://www.graphpad.com/ | RRID: SCR_002798 |
| R (version 3.6.1) | https://cran.r-project.org/ | RRID: SCR_003005 |
| R Studio (version 1.1.456) | https://rstudio.com/ | N/A |
| Tophat2 | https://ccb.jhu.edu/software/tophat/index.shtml | RRID:SCR_013035 |
| Cuffnorm | http://cole-trapnell-lab.github.io/cufflinks/cuffnorm/index.html | N/A |
| Bowtie2 (version 2.2.1) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | RRID: SCR_016368, Ref: Langmead and Salzberg, 2012; Langmead et al., 2019 |
| MACS (version 1.4.1) | https://github.com/macs3-project/MACS | RRID:SCR_013291 |
| MaxQuant (version 1.2.2.5) | https://www.maxquant.org/ | RRID: SCR_014485, Ref: Cox et al.,2008 |
| KEGG | https://www.genome.jp/kegg/ | RRID: SCR_012773 |
| Enrichr | https://amp.pharm.mssm.edu/Enrichr | RRID: SCR_001575, Ref: Chen et al., 2013; Kuleshov et al., 2016 |
| GSEA | http://gsea-msigdb.org/gsea/index.jsp | RRID: SCR_003199, Ref: Subramanian et al., 2005 |
| ssGSEA (version gsea2–2.2.1) | https://github.com/broadinstitute/ssGSEA2.0 | N/A |
| Cytoscape (version 3.7.1) | https://cytoscape.org/ | RRID: SCR_003032 |
| EnrichmentMap Plugin (version 3.1.0) | http://baderlab.org/Software/EnrichmentMap | RRID: SCR_016052 |
| AutoAnnotate Plugin (version 1.2) | http://apps.cytoscape.org/apps/autoannotate | N/A |
Highlights.
Transcriptome reprogramming rewires kinome signaling in Ibrutinib-resistant MCL
Super-enhancer remodeling contributes to transcriptional changes in IR MCL
Targeting CDK9 or BRD4 compromises transcriptome and kinome rewiring to overcome IR
The EMMA platform can be a tool to predict clinical response in primary MCL samples
ACKNOWLEDGMENTS
We sincerely thank all members of the Tao, Qi, Cleveland, Sotomayor, and Shain laboratories for their technical support and comments and the Proteomics, Flow Cytometry, and Biostatistics Cores of the Moffitt Cancer Center and the Genomics Core of Dana Farber Cancer Institute for their outstanding service. This work was supported in part by grants from the National Cancer Institute (CA179062, CA134807, and CA137123 to J.T. and E.M.S.); a grant from the Lymphoma Research Foundation (to J.T.); research grants from Incyte Corporation (to J.T.); an LLS-SCOR grant (to J.L.C.); grants P01-CA066996–19 and P50-CA100707–15 (to J.Q.) and P30-CA076292 (to J.L.C); the Cortner-Couch Chair for Cancer Research from the University of South Florida School of Medicine (to J.L.C.); monies from Lesa France Kennedy (to J.L.C.); and funds from the State of Florida to the H. Lee Moffitt Cancer Center & Research Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108870.
REFERENCES
- Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, Thijssen R, Yeh P, Wong SQ, Ftouni S, et al. (2019). Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 25, 119–129. [DOI] [PubMed] [Google Scholar]
- Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, et al. (2013). MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 14, 249–256. [DOI] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, et al. (2014). NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 56, 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CY, Chihara D, Romaguera JE, Fowler NH, Seymour JF, Hagemeister FB, Champlin RE, and Wang ML (2015). Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann. Oncol. 26, 1175–1179. [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, et al. (2014). Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 4, 1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, Pop-Damkov P, Su N, Roamio Franklin VN, Sekhar Reddy Chilamakuri C, et al. (2020). AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 26, 922–934. [DOI] [PubMed] [Google Scholar]
- Colomer D, and Campo E (2014). Unlocking new therapeutic targets and resistance mechanisms in mantle cell lymphoma. Cancer Cell 25, 7–9. [DOI] [PubMed] [Google Scholar]
- Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. (2012). The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu Z, and Ribas A (2016). Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther. Adv. Med. Oncol. 8, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco J, Balaji U, Freinkman E, Witkiewicz AK, and Knudsen ES (2016). Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 14, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargano B, Amente S, Majello B, and Lania L (2007). P-TEFb is a crucial co-factor for Myc transactivation. Cell Cycle 6, 2031–2037. [DOI] [PubMed] [Google Scholar]
- Guo J, and Price DH (2013). RNA polymerase II transcription elongation control. Chem. Rev. 113, 8583–8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, and Zhou Q (2010). HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 38, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hing ZA, Mantel R, Beckwith KA, Guinn D, Williams E, Smith LL, Williams K, Johnson AJ, Lehman AM, Byrd JC, et al. (2015). Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood 125, 3128–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson C, Kopp N, Layer JV, Redd RA, Tschuri S, Haebe S, van Bodegom D, Bird L, Christie AL, Christodoulou A, et al. (2016). HSP90 inhibition overcomes ibrutinib resistance in mantle cell lymphoma. Blood 128, 2517–2526. [DOI] [PubMed] [Google Scholar]
- Jares P, Colomer D, and Campo E (2012). Molecular pathogenesis of mantle cell lymphoma. J. Clin. Invest. 122, 3416–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Lwin T, Zhao X, Ren Y, Li G, Moscinski L, Shah B, and Tao J (2019). Venetoclax as a single agent and in combination with PI3K-MTOR1/2 kinase inhibitors against ibrutinib sensitive and resistant mantle cell lymphoma. Br. J. Haematol. 184, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Jä nne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, and Halmos B (2005). EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9 (4), 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Wilks C, Antonescu V, and Charles R (2019). Scaling read aligners to hundreds of threads ongeneral-purpose processors. Bioinformatics 35 (3), 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, et al. (2014). Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 371, 1877–1888. [DOI] [PubMed] [Google Scholar]
- Martin P, Maddocks K, Leonard JP, Ruan J, Goy A, Wagner-Johnston N, Rule S, Advani R, Iberri D, Phillips T, et al. (2016). Postibrutinib outcomes in patients with mantle cell lymphoma. Blood 127, 1559–1563. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomie K, Jain P, Kotlov N, Segodin V, Cai Q, Liu Y, Navsaria L, Svekolkin V, Bagaev A, Frenkel F, et al. (2020). Multi-omics analysis of mantle cell lymphoma reveals an immune-cold tumor microenvironment associated with Ibrutinib resistance. J. Clin. Oncol. 38, 8055. [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, and Varmus H (2005). Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin BM, and Price DH (2006). Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305. [DOI] [PubMed] [Google Scholar]
- Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chan FC, Meissner B, Bhang HE, Ruddy D, Kauffmann A, et al. (2014). Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 20, 87–92. [DOI] [PubMed] [Google Scholar]
- Ren Y, Bi C, Zhao X, Lwin T, Wang C, Yuan J, Silva AS, Shah BD, Fang B, Li T, et al. (2018). PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. J. Clin. Invest. 128, 5517–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusan M, Li K, Li Y, Christensen CL, Abraham BJ, Kwiatkowski N, Buczkowski KA, Bockorny B, Chen T, Li S, et al. (2018). Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 8, 59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, Mussolin B, Kwak EL, Buscarino M, Lazzari L, et al. (2016). Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 6, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Jacobson T, Meads M, Distler A, and Shain K (2015). An Organotypic High Throughput System for Characterization of Drug Sensitivity of Primary Multiple Myeloma Cells. J. Vis. Exp, e53070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Silva MC, Sudalagunta P, Distler A, Jacobson T, Collins A, Nguyen T, Song J, Chen DT, Chen L, et al. (2017). An Ex Vivo Platform for the Prediction of Clinical Response in Multiple Myeloma. Cancer Res. 77, 3336–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Lin C, and Shilatifard A (2011). The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 25, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, et al. (2013). Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 369, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ML, Blum KA, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, et al. (2015). Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood 126, 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, and Zhou Q (2005). Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545. [DOI] [PubMed] [Google Scholar]
- Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares- Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, et al. (2017). Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Discov. 7, 302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, Bell T, Zhang H, Han G, Lorence E, et al. (2019a). Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 11, eaau1167. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wang HY, Liu X, Nunez-Cruz S, Jillab M, Melnikov O, Nath K, Glickson J, and Wasik MA (2019b). Cutting Edge: ROR1/CD19 Receptor Complex Promotes Growth of Mantle Cell Lymphoma Cells Independently of the B Cell Receptor-BTK Signaling Pathway. J. Immunol. 203, 2043–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gao Z, Rajthala S, Sapkota D, Dongre H, Parajuli H, Suliman S, Das R, Li L, Bindoff LA, et al. (2020). Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell. Mol. Life Sci. 77, 1115–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, Zhang L, Fu K, Bi C, Li J, et al. (2017). Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat. Commun. 8, 14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ren Y, Lawlor M, Shah BD, Park PMC, Lwin T, Wang X, Liu K, Wang M, Gao J, et al. (2019). BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models. Cancer Cell 35, 752–766.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucca E, and Bertoni F (2013). Toward new treatments for mantle-cell lymphoma? N. Engl. J. Med. 369, 571–572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNAseq and CHIPseq data reported in this paper is GEO: GSE141336.
The accession number for the ABPP proteomics data reported in this paper is ProteomeXchange: PXD005734.
Computational code used for ChIP-seq analyses can be obtained from the Jun Qi Laboratory github page. Namely, Bamliquidator was used to calculate read density which can be found at https://github.com/BradnerLab/pipeline/wiki/bamliquidator; and ROSE2, was used to identify enhancers and can be found at https://github.com/BradnerLab/pipeline/(ROSE2_main.py).
All algorithms and codes used to process images and perform downstream analyses for the EMMA platform were provided directly by the Silva Lab (Silva et al., 2015).