

Abstract
Molecular inputs to chromatin via cellular metabolism are modifiers of the epigenome. These inputs — which include nutrient availability as a result of diet and growth factor signalling — are implicated in linking the environment to the maintenance of cellular homeostasis and cell identity. Recent studies have demonstrated that these inputs are much broader than previously known, encompassing the metabolism of a wide variety of sources, including alcohol and microbiotal metabolism. These factors modify DNA and histones and exert specific effects on cell biology, systemic physiology and pathology. In this Review, we discuss the nature of these molecular networks, highlight their role in mediating cellular responses, and explore their modifiability through dietary and pharmacological intervention.
Table of contents blurb
Various cellular metabolites provide the chemical moieties for DNA and histone modifications, resulting in complex interplay between metabolism and epigenetics. In this Review, Dai, Ramesh and Locasale discuss the metabolic regulation of diverse types of chromatin modifications, their functional consequences at molecular, cellular and organismal levels, as well as influences from diet and microbiota.
Introduction
Cells comprising a living organism contain near-identical genomic DNA that is processed and expressed differentially, owing to the presence of a molecular scaffold known as the nucleosome [G]. Within a single nucleosome, 147 bp of DNA wraps around an octamer of positively charged proteins called histones, present as two functional copies of histone type H2A, H2B, H3 and H4 proteins. Each nucleosome is then further condensed at increasing levels into a higher-order structure called chromatin, which can form a tightly packed barrier that restricts the access of molecular factors to the genome1. Cells contextually circumvent or reinforce this barrier by dynamically modifying DNA and histones at specific nucleotide or amino acid residues, establishing regions of the genome that are differentially exposed to cellular machinery. These modifications individually or synergistically influence various genome-associated processes — such as transcription, DNA replication and repair — by a variety of mechanisms including serving as recognition sites for proteins including transcription factors, histone chaperones, chromatin modifiers and chromatin remodellers [G], and changing the local and global structure of chromatin organization2–4.
Histones are modified on their free N-terminal tails, or their globular domains that physically interact with DNA, with chemical modifications including acetylation, methylation, phosphorylation, ubiquitinylation, acylation, hydroxylation, glycation, serotonylation, glycosylation, sumoylation and ADP ribosylation5,6. DNA is methylated at cytosine as well as adenine residues7,8. Chromatin is modified enzymatically, which we will discuss below, or non-enzymatically. Non-enzymatic chromatin modifications, which occur via the covalent adduction of histones and DNA to electrophilic moieties [G] derived from metabolism, are characteristic features of certain cellular abnormalities, but their functions are still poorly understood9. Finally, RNA can be methylated and acetylated10,11, referred to as epi-transcriptomic modifications that regulate RNA processing, mRNA half-life, translation, among other processes. These modifications, which for simplicity we refer to as epigenetic modifications, together form the epigenome and are linked to gene regulation and thus many physiological and pathological processes. Each are derived from intermediates in metabolism.
Metabolism is the result of networks of biochemical reactions that take in nutrients to process them to serve cellular demands including energy generation and biosynthesis12. Intermediates of these reactions are used as substrates and co-factors for a variety of epigenome-modifying enzymes13, allowing metabolism to directly communicate environmental changes to chromatin state14. The aberrant regulation of these molecular networks due to genomic mutations or environmental perturbations is associated with changes to embryonic development15, changes to cellular identity16, immune cell function17, tumorigenesis18, tumour progression19, and microbiome–host commensalism20. These networks can be manipulated both pharmacologically, and through diet and nutrition to have varying effects on altering physiology and disease.
Elucidating the ways in which chromatin can be modified by metabolism, and their functional importance, remains an active and growing area of research that is helping uncover fundamental cellular mechanisms underlying normal and disease states. In this Review, we discuss the principles underlying the regulation of metabolism and epigenetics, and the multifaceted ways in which chromatin can be modified by metabolic reactions. Beyond ‘canonical’ methylation and acetylation marks13,21–23 we describe our growing appreciation for diverse types of additional metabolite-derived chromatin modifications, such as histone acylation, homocysteinylation, monoaminylation, and many others. Furthermore, we explore the functional molecular consequences on chromatin biology, including chromatin structure, accessibility and transcription, as well as the resultant cellular and organismal effects, such as on cell fate, immune function and cancer. We also discuss emerging areas of metabolic influence on chromatin status and epigenetics such as diet, nutrition and microbiota.
Principles of the metabolic link to epigenetics
Despite the challenges in completely understanding the context-dependent roles of the metabolism–epigenetics axis and the complexity in metabolic pathways and chromatin modifications regulated by them, there are several universal principles underlying this cross-talk, that demonstrate the evolutionary emergence of specific molecular mechanisms that facilitate epigenomic dynamics under metabolic alterations.
The ability of epigenetic modifications to respond to fluctuations in metabolic activities is a consequence of the intrinsic thermodynamic and kinetic parameters of chromatin-modifying enzymes (Box 1). Addition and removal of most of these modifications are catalyzed by enzymes (i.e. ‘writers’ and ‘erasers’) that utilize metabolites as substrates or cofactors (i.e. chromatin-modifying metabolites (Figure 1)). Chromatin-modifying enzymes that use metabolite substrates whose physiological concentrations are close to or lower than the enzymes’ intrinsic Km and Kd values [G] are more susceptible to metabolic pathway alterations than those whose substrates are present in excess amounts13. This property thus enables metabolic fluctuations to influence the activities of certain chromatin-modifying enzymes and modulate the levels of specific epigenetic modifications, and the difference in substrate availabilities and Km values may determine relative sensitivities of epigenetic modifications to metabolic alterations24,25 (Box 1). On the other hand, chromatin modifications can also be added non-enzymatically, of which the detailed kinetic and thermodynamic properties are less well characterized but are influenced to some extent by the law of mass action.
Box 1: Kinetic and thermodynamic properties of chromatin-modifying enzymes.
Rates of enzyme reactions depend on many factors including intrinsic enzymatic parameters such as turnover number and Michaelis–Menten constant (Km) values, enzyme abundance, substrates, cofactors and allosteric activators or inhibitors, and other environmental factors such as pH, temperature, and local viscosity. These variables together determine the overall reaction rate through a quantitative relationship that depends on the molecular mechanism of the enzymatic reaction236. Studies describing the biochemistry and structural biology of chromatin-modifying enzymes have provided crucial insights about their catalytic mechanisms and regulators. Kinetic mechanisms of enzymes involved in DNA methylation, histone acetylation and histone methylation have been extensively investigated particularly when issues around drug development are concerned, showing that the exact catalytic mechanism for each chromatin-modifying enzyme is diverse and largely context-dependent (see the figure). For example, the histone acetyltransferases PCAF and GCN5 exhibit an ordered ternary catalytic mechanism in which acetyl-CoA binds to the enzyme before the histone peptide, which is followed by the release of acetylated histone and finally, the CoA molecule237–239. By contrast, p300 and HAT8 function through a ping-pong mechanism that starts with the binding of acetyl-CoA and ends with the release of acetylated histone240,241. Finally, random ternary kinetics was observed in the yeast histone acetyltransferase Rtt109242.
Given the diversity in catalytic mechanisms of chromatin-modifying enzymes and the variability in the specific enzymatic kinetic parameters, epigenetic modifications are likely to exhibit specificity due to these different mechanisms. The thermodynamic and kinetic properties of epigenetic modifications depend on the type of modification, the corresponding chromatin-modifying enzyme, the specific genomic locus, and the abundance of allosteric regulators and cofactors. This variability results in distinct dynamics of deposition and turnover in response to perturbations to metabolism. Directly targeting these epigenetic modifications has shown that the dynamics of histone acetylation is in general faster than changes in that of DNA and histone methylation243, and that acetylation and deacetylation occur at the timescale of minutes in vivo244,245. Histone and DNA methylation, on the other hand, are more stable compared to acetylation, which might constitute a type of epigenetic memory in response to stronger but transient perturbations243. An additional layer of complexity arises from the heterogenous affinity and activity of chromatin modifiers towards different chromatin modifications246 and different genomic loci247,248. Variation in the abundances of chromatin-modifying metabolites and apparent Km values in different cells and tissues might therefore lead to heterogeneity in the sensitivity of epigenetic marks to metabolic alterations in different contexts.
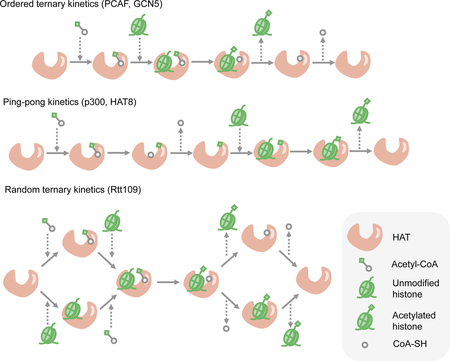
Figure 1. Overview of the mechanisms involved in the metabolic regulation of epigenetics.
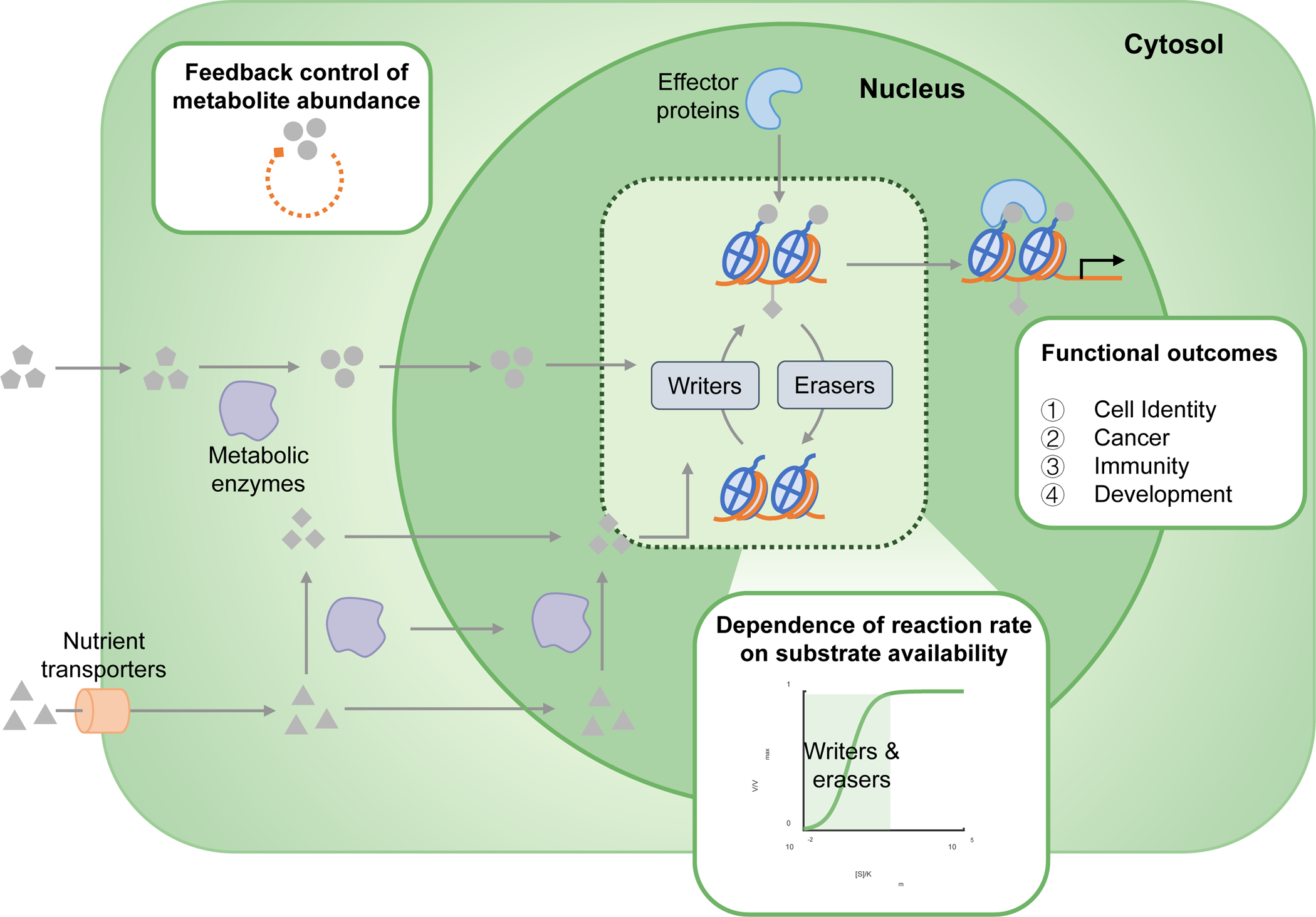
The abundance of chromatin-modifying metabolites is intracellularly regulated by several mechanisms. Metabolites that are taken up by cells can passively or actively diffuse through the plasma and nuclear membrane in order to modify chromatin. Alternatively, metabolites can be processed internally by the activity of metabolic enzymes that convert them into substrates or co-factors for chromatin-remodelling enzymes. These enzymes can also translocate to the nucleus where they can locally produce substrates for chromatin modification. The resultant consequence of metabolite abundance on the rate of chromatin modification is dependent upon the kinetic and thermodynamic parameters of the enzyme. Enzymes with initial [S]/Km ratios on the highlighted linear part of the displayed curve are more susceptible to perturbations to substrate concentrations — these include methyltransferases and acetyltransferases, among others. Finally, once the modifications have been deposited, effector proteins can recognize and bind them using specific protein-binding modules, upon which they determine a variety of intracellular fates including the regulation of homeostasis, development, immune regulation and tumorigenesis.
There are several additional mechanisms that enable the efficient and precise regulation of enzyme-catalyzed chromatin modifications by metabolic activity. Metabolic enzymes involved in the synthesis of chromatin-modifying metabolites, such as acetyl-CoA and S-adenosylmethionine (SAM), may be able to localize in the nucleus and interact with nucleosomes and chromatin-modifying enzymes to efficiently produce metabolites at specific genomic loci26–32. Levels of chromatin-modifying metabolites, such as SAM, are controlled by multiple mechanisms including both environmental inputs such as nutrient availability and intracellular methyl group sinks [G] that consume SAM33–35. Methyl group sinks are mediated by enzymes that metabolize SAM, allowing them to divert methyl groups away from enzymes such as histone methyltransferases, thus affecting their activity. These mechanisms provide avenues for control of metabolite levels and thus for chromatin to sense intracellular metabolic status.
Epigenetic modifications influence transcriptional programs through various mechanisms (Box 2). All of these outcomes can potentially be influenced by the metabolic regulation of the epigenome. Additionally, recent studies have found that chromatin compartments with differing transcriptional activity can segregate into membrane-less organelles through liquid–liquid phase separation [G] in response to chromatin modifications, establishing distinct chromatin domains with distinct patterns of regulation. Transcriptionally inactive heterochromatin can form phase-separated liquid droplets by interacting with heterochromatin protein 1 (HP1) which recognizes and binds to the histone modification H3K9me, allowing chromatin to stably condense inside these droplets36–38. On the other hand, active chromatin regions, such as those containing histone acetylation, enhancers [G] and super-enhancers [G], are also able to phase separate through interacting with binding proteins such as bromodomain [G] -containing proteins39–41. Similar effects promoting phase separation have also been found to be mediated by the interaction between N6-methyladenosine (m6A) in mRNA and the m6A-binding YTHDF proteins42. Although it is an open question as to whether chromatin phase separation is regulated by metabolism, these findings suggest that the ability of chromatin to phase-separate within cells may be regulated by epigenetic modifications derived from metabolites and might be sensitive to cellular metabolism. Furthermore, phase separation of other biomolecules has been shown to concentrate molecules in a certain phase to activate biochemical signalling processes43. Whether chromatin phase separation also results in localization of metabolites and activation or inhibition of chromatin-modifying reactions in a specific phase remains unknown, but it potentially serves as an additional mechanism for the precise control of local metabolite levels and chromatin modifications.
Box 2: Influences of epigenetic modifications on chromatin structure and gene expression.
The earliest clues suggesting the functional consequences of particular epigenetic modifications are their locus-specific enrichment and association with gene expression levels and gene regulation. Although not conclusively implying causality, these findings have been used to theorize on the existence of a ‘histone code’, which postulates that the presence and combination of specific DNA and histone modifications link to the regulation of transcriptional programs and gene expression events249. Studies over the past few decades have revealed two major ways for epigenetic modifications to regulate gene expression: by changing the local chromatin structure, or by influencing the recruitment of non-histone protein effectors to chromatin250. Chromatin can be roughly classified into two categories based on their structural and biochemical properties: condensed, transcriptionally silenced heterochromatin, and decondensed, actively transcribed euchromatin. Formation of heterochromatin and euchromatin is dependent on the existence of epigenetic modifications. Histone acetylation, typically enriched in euchromatin, is able to neutralize the positive charge of the modified lysine residue, thus disrupting the interaction between histone and DNA and resulting in an open chromatin structure that facilitates active transcription. Heterochromatin is typically enriched of trimethylation of histone 3 lysine 9 (H3K9me3), which is bound by heterochromatin protein 1 (HP1) that promotes compaction and spreading of heterochromatin251 and triggers liquid–liquid phase separation by forming oligomers38.
The chromatin-binding affinities of a plethora of proteins, including transcription factors, chromatin remodellers and components of the transcription machinery, can be modulated by these modifications. This is best demonstrated by the presence of chromatin-binding ‘reader’ modules in these proteins, such as the chromodomains and PHD fingers that recognize and bind to methylated histones, and the bromodomains and YEATS domains that bind to acetylated histones252,253. It has been hypothesized that the association between H3K4me3 and active transcription is largely mediated by the binding of H3K4me3 by the PHD finger of the TAF3 subunit of TFIID, a component of the RNA polymerase pre-initiation complex254. DNA methylation has recently been shown to reduce the binding affinity of most transcription factors while promoting the binding of PHD-containing proteins through hydrophobic interactions with the methylated cytosine255. Readers of some of the emerging, non-canonical histone modifications, such as succinylation116 and crotonylation256,257, have also been identified recently258.
Metabolism-derived chromatin modifications
DNA and histone methylation.
One of the most well studied chromatin modifications is the addition of a methyl (−CH3) group to the ε-amino group of lysine or arginine residues on histones and CpG islands of DNA44. This modification is derived from the metabolism of the essential amino acid methionine (Met) which in mammals is almost exclusively obtained from the diet45. Met uptake is followed by its conversion to the methyl-donor metabolite SAM46, which is then used as substrate for DNA and histone methyltransferases (Figure 2), producing S-adenosylhomocysteine (SAH), which competitively inhibits DNA and histone methyltransferases. Disruptions in Met metabolism and one-carbon metabolism [G], such as changes to threonine or methionine intake and activation or inhibition of metabolic enzymes in these pathways, have been shown in a variety of cellular systems to affect intracellular concentrations of SAM and SAH, therefore changing levels of DNA methylation47,48 and histone methylation. These metabolism-linked histone methylation alterations include: H3K4me3 in mouse and human embryonic stem cells (mESCs and hESCs)49,50, human colon cancer cells, mouse liver51,52 and Caenorhabditis elegans53; H3K9me1/2/3 in human colon cancer cells and mouse liver24; H3K27me3 in mESCs33; H3K36me3 in immune-activated macrophages54 and T cells55; and multiple tri-methylation marks in yeast34,35. These changes correlate with changes in gene expression to varying degrees.
Figure 2. Metabolic pathways producing chromatin-modifying metabolites.
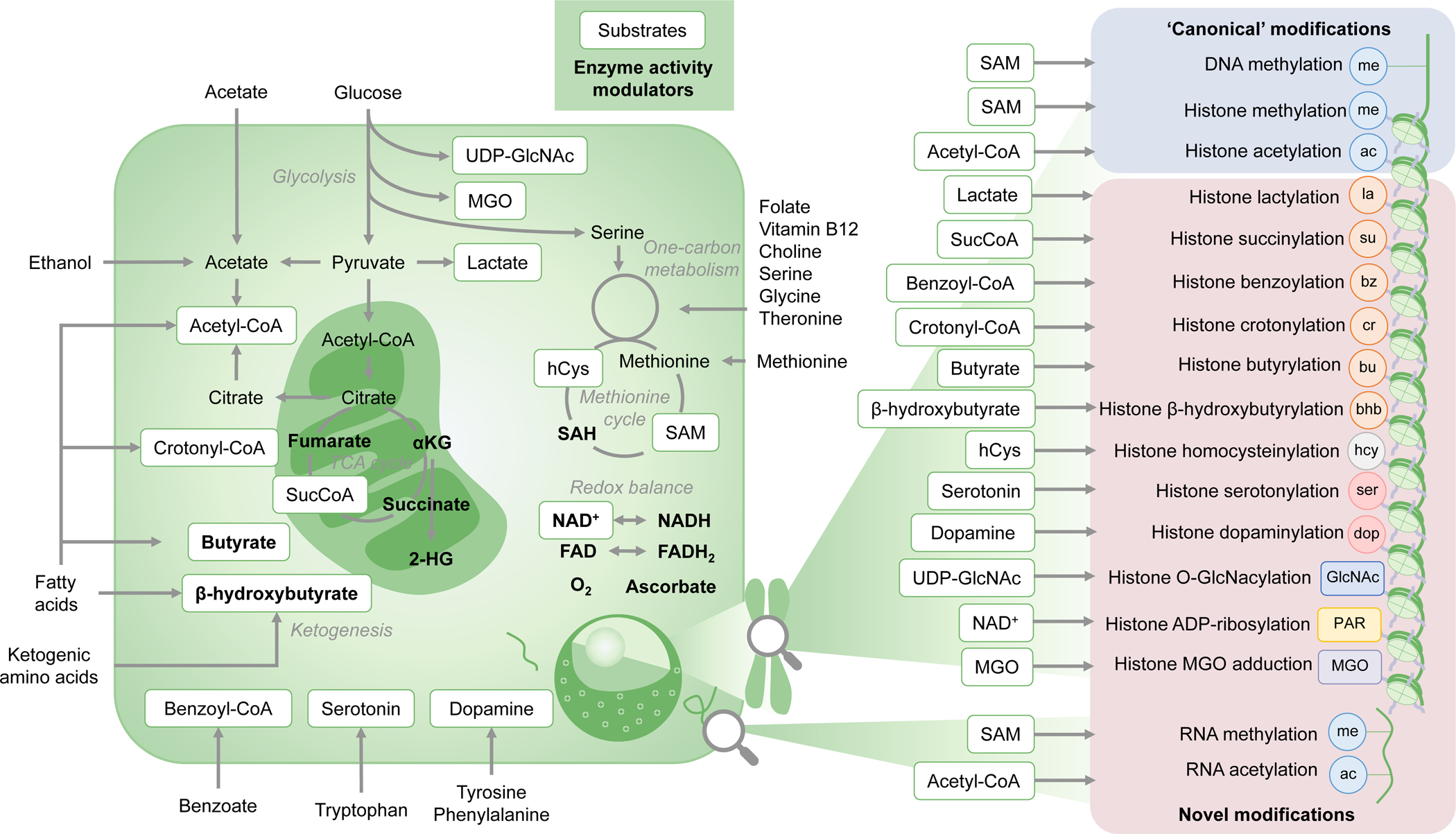
Nutrients such as glucose, fatty acids, amino acids and vitamins are utilized by cellular metabolic pathways to produce metabolites that are used as substrates or activity modulators of chromatin-modifying enzymes. These molecules are included in regulation of the abundance of a plethora of ‘canonical’ modifications, including histone acetylation, histone methylation and DNA methylation, and ‘emerging’ modifications including acylations, homocysteinylation, serotonylation etc. Central-carbon, one-carbon and methionine metabolism, acetate metabolism, ketogenesis and redox-related pathways feed the pools of several of these metabolites, and thus help regulate the epigenomic landscape in concert with chromatin modifiers, remodellers and transcription factors. 2-HG, 2-hydroxyglutarate; αKG, α-ketoglutarate; GlcNAc, β-N-acetylglucosamine; hCys, homocysteine (hcy when a histone modification); MGO, methylglyoxal; PAR, poly(ADP–ribose); SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SucCoA, succinyl-CoA; TCA, tricarboxylic acid.
Metabolism can also influence the activity of enzymes responsible for the turnover of chromatin methylation. Active removal of histone and DNA methylation is catalyzed by the TET family of DNA demethylases (TETs)56 and histone demethylases including JmjC-domain containing demethylases (JHDMs) and amine oxidases (LSDs)57. TETs and JHDMs belong to the class of α-ketoglutarate [G] (αKG)-dependent dioxygenases that use αKG and oxygen as substrates. These enzymes are inhibited by metabolism-derived structural analogues of αKG, including succinate, fumarate, and the oncometabolite 2-hydroxyglutarate (2-HG) produced by gain-of-function mutations of the metabolic enzymes isocitrate dehydrogenase 1 (IDH1) or IDH2. Catalysis by TETs and JHDMs is activated by ascorbate58 and also requires ferrous iron, Fe (II), as a cofactor, whereas LSD histone demethylases rely on flavin adenine dinucleotide (FAD) (Figure 2). Conditions involving alterations in metabolic pathways producing or consuming any of these molecules, such as glutamine deficiency59,60, iron deficiency61, and mutation of mitochondrial enzymes including IDH1/262, succinate dehydrogenase (SDH)63 and fumarate hydratase (FH)64, are thus able to shape the methylation landscape by modulating the activity of histone and DNA demethylases by affecting the activity of each of these aforementioned cofactors.
Another axis for the metabolic regulation of chromatin methylation is through the regulation of redox balance. Redox balance [G] ensures the maintenance of healthy levels of reactive oxygen species (ROS) which is crucial for cell survival. ROS generation during early development in C. elegans was recently shown to modulate the transcription of genes required for long-term survival by inhibiting the H3K4-methyltransferases MLL1–4 and reducing global levels of H3K4me365. Whether ROS generation as a result of metabolic reprogramming during this time involves the oxidation and depletion of SAM remains to be seen, but several potential mechanisms exist including the redox-responsiveness of the vitamin-B12 utilizing enzyme methionine synthase. Additionally, hypoxia can inhibit TETs and JHDMs such as KDM5A, KDM6A and KDM6B by limiting oxygen, resulting in global DNA66 and histone hypermethylation67,68.
Histone acetylation.
Another ‘canonical’ chromatin modification is the acetylation of histones, which involves the transfer of an acetyl-group derived from the high-energy metabolite acetyl-CoA to the ε-amino group of a histone lysine, catalyzed by acetyltransferases44. Acetyl-CoA in mammalian cells is primarily derived from carbon units provided by extracellular glucose which feeds into mitochondrial metabolism to generate citrate. Citrate is then exported to and lysed within the cytosol by the enzyme ATP–citrate lyase (ACLY) to generate acetyl-CoA69 (Figure 2). The availability of glucose and glycolytic activity subsequently influences global levels of histone acetylation through the generation of acetyl-CoA69,70. The short-chain fatty acid (SCFA) acetate is an additional source of acetyl-CoA for histone acetylation, through the function of the enzyme acetyl-CoA synthetase 2 (ACSS2). Acetate has been recently shown to be produced de novo from pyruvate, providing another pathway from glucose to histone acetylation71. Acetate and acetyl-CoA can also be generated from ethanol metabolism in the liver, which supports histone acetylation in the brain72 and other organs, or by the oxidative catabolism of lipids, which may contribute up to 90% of the acetylation of certain histone lysines73–75.
Like histone methylation, histone acetylation is actively turned over, in this case by a class of enzymes called histone deacetylases (HDACs) that are inhibited by metabolites including butyrate and β-hydroxybutyrate (β-OHB) that are produced during fatty acid oxidation, ketogenesis or derived from commensal microbiota76. Sirtuins, another class of HDACs, utilize NAD+ to deacetylate histones, and are sensitive to intracellular NAD+ levels77.
Histone acylation.
Perhaps less well understood but encompassing a broad landscape of emerging metabolic precursors are histone acylation modifications78, which are similar to their acetyl-counterparts in terms of their functionality and enzymatic regulation. These modifications are derived from short-chain acyl-group containing molecules, such as SCFAs, by enzymes that produce their respective acyl-CoA molecules, called acyl-CoA synthetases79,80 (Figure 2). If non-enzymatic and assuming the law of mass action, the relative abundance of acyl-histone modifications is proportional to the concentration of corresponding acyl-CoA metabolites81. The acyl-CoA metabolites may also be used by histone acetyltransferases such as p300 and KAT2A to catalyze histone-acylation reactions with a reduced binding affinity compared to the canonical substrate acetyl-CoA25,82, or they may react with histones in a non-enzymatic fashion83, especially for certain acyl-CoA metabolites such as succinyl-CoA and malonyl-CoA that are particularly reactive towards protein lysine residues84.
The concentration of acyl-CoA molecules can be dynamically modulated in diverse ways in response to perturbations in cellular environment. A recent study showed that lactate, the end product of glycolysis, can be used for histone lactylation, an emerging modification that correlates with transcriptional activation and is a functional output of metabolic reprogramming during macrophage polarization85. Histone succinylation has been shown to occur at least in part due to the nuclear localization of αKG dehydrogenase, which locally generates succinyl-CoA used by the acetyltransferase KAT2A to succinylate H3K79 around transcriptional start sites82. Histone benzoylation has been shown to dynamically respond to levels of sodium benzoate, a chemical food preservative, through the generation of benzoyl-CoA, and correlates with gene expression86. Histone crotonylation is derived from crotonyl-CoA produced from the SCFA crotonate, and used by the histone acetyltransferases p300/CBP to crotonylate histones and activate transcription20,25. The production of ketone bodies such as butyrate and β-OHB during metabolic fasting [G], can be used to butyrylate87 and β-hydroxybutyrylate histones88,89, which turns on genes involved in the starvation response. Since β-OHB is also an inhibitor of HDACs76, and histone butyrylation and crotonylation have additionally been shown to compete with acetylation87,90, the overall effect of β-OHB on gene expression and biological functions is determined by changes in both histone β-hydroxybutyrylation and acetylation, although acetylation could be more important given the multi-faceted roles of histone acetylation in regulating chromatin state, genome structure and gene expression. Further research is needed to uncover ways in which the levels of these acyl-CoA metabolites dynamically respond to metabolic reprogramming and compete with histone acetylation to modify chromatin.
Histone homocysteinylation.
Histone homocysteinylation is an emerging modification that has been observed in response to increased cellular homocysteine (Hcy) levels in human fetal brains during pregnancy (Figure 2). A study found 39 sites of histone lysine homocysteinylation in the four major histone variants, among which H3K79-Hcy negatively correlated with the expression of genes associated with neural tube closure, thus potentially contributing to the acquisition of neural tube defects91. Although it remains to be seen whether perturbations to methionine and one-carbon metabolism contribute to changes in histone homocysteinylation, this finding, together with the long-known association between maternal folate deficiency and neural tube defects in infants, suggests that the effects of folate deficiency on neural development may have some connection to dysregulated histone homocysteinylation.
Histone monoaminylation.
Another newly emerging histone modification is histone monoaminylation, which was recently shown to have roles in regulating neural functions and behaviours. Histones can be modified at glutamine residues by reacting with monoamine neural transmitters including serotonin and dopamine, which can sequentially influence chromatin biology, gene expression, neural function, and behaviours.
The uptake of the essential amino acid tryptophan is limiting for the generation of the neurotransmitter serotonin92,93 (Figure 2). Serotonin is involved in the maintenance of neuronal circuits. In tissues that produce the bulk of serotonin in humans, serotonin has been found to serotonylate H3K4me3-modified histones at glutamine residues, forming H3K4me3Q5ser. This modification is catalyzed by the enzyme transglutaminase 2 (TGM2), and is associated with transcriptional initiation and euchromatin, contributing to neuronal differentiation and signalling94.
Dopamine is a neurotransmitter with functions in the reward circuit of the brain. In neurons, dopamine is synthesized from tyrosine and phenylalanine95, and its release in the human brain is reduced by dietary depletion of the two amino acids96. In a brain region rich in dopaminergic neurons, dopamine has been associated with an emerging epigenetic modification, histone 3 glutamine 5 dopaminylation (H3Q5dop). H3Q5dop was decreased upon cocaine exposure but increased after drug withdrawal, and reduction in its level reversed changes in gene expression and drug-seeking behaviours upon drug withdrawal, indicating a causal relationship between H3Q5dop and addictive behaviours97.
Histone O-GlcNacylation.
Like cytosolic proteins, histones can be reversibly modified with β-N-acetylglucosamine (GlcNAc) at the hydroxyl group of serine or threonine residues, using UDP-GlcNAc as a substrate. UDP-GlcNAc is a byproduct of the hexosamine biosynthetic pathway [G]98, which requires metabolic precursors generated during central carbon metabolism [G], nitrogen metabolism, and fatty acid metabolism99 (Figure 2). O-GlcNAcylation is catalyzed by the canonical O-linked GlcNAc transferase (OGT) and turned-over by O-linked GlcNAc hydrolase (OGA)100. In mammalian cells, OGT appears to associate with TET proteins TET2 and TET3, which facilitate the transfer of the GlcNAc moiety to histones and help localize the modification toward transcriptional start sites. The modification helps regulate gene expression101, and might influence chromatin structure during DNA replication100.
Histone ADP–ribosylation.
Histones can be reversibly mono- or poly-ADP-ribosylated (PARylated), which involves the oligomeric addition of an ADP–ribose (PAR) moiety derived from NAD+ with the help of the poly(ADP–ribose) polymerase (PARP) family of enzymes102 (Figure 2). PAR modifications have been found to occur on histone lysines, where they can influence the placement of other modifications, and bind a variety of molecular effectors that regulate gene expression, chromatin structure, DNA replication and repair103. PARP enzymes can be activated in response to various physiological perturbations including the feeding of a high-fat diet, oxidative stress, ageing and DNA damage104.
Non-enzymatic chromatin modifications.
In certain circumstances, chromatin modifications are added without the participation of enzymes. These non-enzymatic chromatin modifications involve both canonical chromatin marks, such as acetylation and methylation, and modifications generated through adduction with electrophilic compounds105. Although histone acetylation and acylation occur through both enzyme-catalyzed and non-enzymatic mechanisms83, histone acylation has relatively lower rate constants [G]106. The reactivity towards non-enzymatic acetylation greatly varies across lysine residues and is largely determined by their biophysical properties such as surface exposure and local electrostatic interactions107. Non-enzymatic methylation of histones and DNA by SAM has also been reported108,109.
A poorly understood class of chromatin modifications is that enabled by electrophilic metabolites generated during glycolysis, lipid peroxidation [G]110 or exposure to environmental toxicants9,111,112, that form covalent adducts with nucleophilic functional groups [G] on histones and DNA. One example is methylglyoxal (MGO), a byproduct of glycolysis, amino acid metabolism and lipid metabolism113 (Figure 2). The formation of MGO adducts, termed advanced glycation end products [G] (AGEs), is closely related to ageing and chronic diseases including diabetes and cancer. MGO glycation of histones is upregulated upon enhanced glycolytic flux114, and is able to destabilize nucleosome structure and disrupt the landscape of chromatin modifications by competing with acetylation and methylation for the same residues115.
The functions of non-enzymatic chromatin modifications are still poorly understood. Paradoxically, several enzymes have been discovered that actively catalyze their removal from chromatin, offering clues about their potential functions. Non-enzymatically added histone acetyl and acyl groups are removed by sirtuins, which can lead to disease states when their activities are disrupted83, and recognized by their specific readers such as the YEATS domain116, suggesting that the maintenance of their genomic distribution can be enzymatically regulated and important in transcriptional control. Conversely, several intracellular detoxification systems including the activity of the enzymes DJ-1 and GLO-I/II115,117,118 and non-enzymatic buffering by the ketone body acetoacetate119 are required to detoxify MGO and suppress the formation of MGO adducts. These findings suggest that non-enzymatic histone adducts represent cellular stress-events that may require appropriate buffering in order to ensure proper cellular function.
RNA methylation and acetylation.
As with DNA and histones, RNA undergoes a variety of covalent modifications120, among which methylation and acetylation of mRNA are the best understood. Both modifications regulate mRNA degradation, splicing and translation through ‘readers’ that recognize and bind to the modified RNA molecules, and other functions10,121,122. The landscape of these chemical groups on mRNA molecules is sometimes termed the ‘epitranscriptome’, which can also be edited by enzymes that rely on metabolite substrates or cofactors. Formation of m6A, the most abundant form of mRNA methylation, is catalyzed by the METTL3–METTL14 complex and METTL16, which are SAM-dependent RNA methyltransferases. SAM depletion has been shown to reduce METTL16-dependent m6A of the MAT2A mRNA, thus promoting its stability and enhancing MAT2A expression. Since MAT2A encodes SAM synthetase, this mechanism appears to function as a negative feedback loop to increase SAM levels under SAM-deficient conditions123,124.
Removal of m6A is catalyzed by the RNA demethylases FTO and ALKBH5, both of which are αKG-dependent dioxygenases that are inhibited by succinate, fumarate, citrate and 2-HG125,126. Interestingly, the oncometabolite 2-HG and expression of mutant IDH1 was shown to exhibit tumour-suppressive activity by inhibiting FTO in leukaemia cells, resulting in enhanced global m6A and decreased stability of transcripts of the MYC oncogene and the gene CEBPA which positively regulates FTO expression127. Inhibition of FTO by 2-HG also increases methylation of small nuclear RNAs (snRNAs) and regulates mRNA splicing128. Perhaps in contrast to what we currently know about many of the non-canonical histone modifications, at this stage, there is an abundance of evidence illustrating the importance of RNA methylation on gene regulation.
Acetylation of mRNA is catalyzed by the acetyltransferase NAT10 whose activity depends on acetyl-CoA abundance and ATP129, generating N4-acetylcytidine (ac4C) that enhances translation efficiency10. Whether RNA deacetylation is enzyme-regulated and the molecular mechanism by which it occurs are currently unknown. Its role in gene regulation, although interesting to speculate, remains lesser known than for m6A.
Metabolism–epigenetics in cell-fate specification
The metabolic reprogramming of chromatin modifications is associated with several functional outcomes that include cell-fate specification, and by extension, processes such as development, ageing, immunology and the aetiology of diseases such as cancer (Figure 3). It is essential to understand whether or in what contexts metabolism per se drives cell fate transitions, or whether metabolic changes during cellular transitions occur independently of the driving events. Accumulating evidence has begun to demonstrate that metabolically driven chromatin dynamics directly affect the expression of genes related to cellular functions, and that their functional outcomes can be at least partially reversed by interfering with the metabolic changes or metabolically driven epigenomic changes, indicating that metabolism drives cell fate transition through regulation of the epigenome in many circumstances.
Figure 3. Physiological contexts of the metabolism–epigenetics axis.
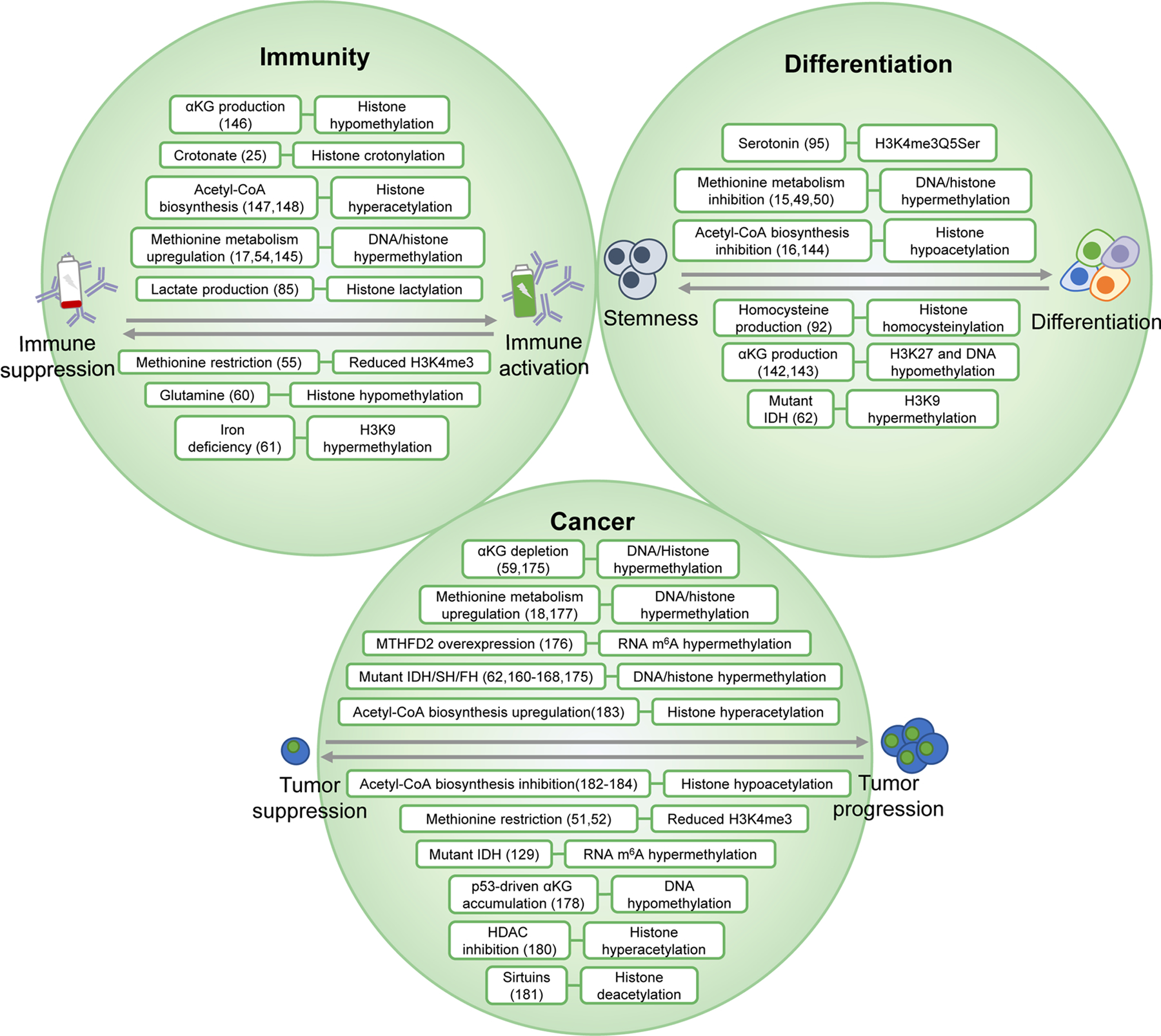
The intersection between metabolism and epigenetics is implicated in a variety of physiological contexts including lineage specification at the embryonic level, immune regulation and the oncogenic transformation of cells. The maintenance of stemness and pluripotency and the process of differentiation are characterized by changes to metabolism and subsequent dynamic changes to epigenetic modifications. This reprogramming is also implicated in the activation or retroactive suppression of a variety of immune cell types including T cells, B cells and macrophages, and the ability to mount an immune response in response to invading pathogens. Finally, the oncogenic transformation of cells can be driven by mutations in metabolic enzymes, or by genomic drivers that reprogram metabolism. These molecular networks offer therapeutic targets in the fields of developmental biology, immunotherapy and oncology. αKG, α-ketoglutarate; FH, fumarate hydratase; HDAC, histone deacetylase; IDH, isocitrate dehydrogenase; m6A, N6- methyladenosine; SDH, succinate dehydrogenase.
Differentiation.
The regulation of pluripotency and lineage-specification involves the participation of a variety of metabolic pathways in a context-dependent manner, which have been reported to fulfil bioenergetic demands during these transitions, and aid in cellular signalling pathways130. Metabolic pathway activity and nutrient availability have been associated with cell-fate-related outcomes, such as induced pluripotency131,132, maintenance of stemness133–136, and differentiation towards specific lineages137–140. Notably, these changes in fluxes through metabolic pathways can also modify the epigenome, in response to differentiation cues or nutrient availability.
Disruption to one-carbon metabolism has been shown to regulate embryonic stem cell (ESC) differentiation due to changes in SAM levels in culture49,50 and in mice15, along with changes to histone methylation and DNA methylation. Maintenance of the intracellular αKG/succinate ratio in mouse ESCs141, epiblast stem cells and primed human pluripotent stem cells142, can regulate differentiation by modulating TET- & JHDM-dependent DNA and histone methylation respectively. Modulating acetyl-CoA levels can affect the differentiation of ESCs and muscle stem cells, with concurrently occurring changes in histone acetylation16,143 and chromatin accessibility16. Emerging metabolically regulated modifications, including histone serotonylation (H3K4me3Q5ser) and histone homocysteinylation, have also been recently shown to play some role in cell-fate specification. As mentioned above, histone serotonylation at a particular site (H3K4me3Q5ser) can potentiate the differentiation of serotonergic neurons in cell culture and during mouse development94. Also as mentioned above, histone homocysteinylation has been shown to associate with increased Hcy levels in human fetal brains and the decreased expression of genes important for neural tube closure during development91.
Immunology.
The immune system is comprised of a diverse milieu of specialized cells that are activated or repressed in response to environmental inputs such as the presence of a pathogen and undergo dynamic changes in gene expression that regulate their function144. Metabolic reprogramming has been reported to drive the proliferation and differentiation of a myriad of immune cell populations, which also show concurrently occurring changes in chromatin state. Antigen receptor engagement in T-cells, for example, has been shown to increase metabolic flux through the methionine cycle, which upregulates DNA and histone methylation54. Methionine uptake has also been shown to maintain SAM synthesis and H3K4me3 levels in CD4+ T helper (Th) cells in culture, which regulates T-cell-mediated immune responses in vivo in a mouse model of multiple sclerosis55. The upregulation of SAM synthesis and levels of H3K36me3 are observed during lipopolysaccharide (LPS)-induced macrophage activation, which corresponds to increased IL-1β expression and production17. The metabolic regulation of TETs and JHDMs also regulates several aspects of immune cell biology. αKG production via glutaminolysis and other metabolic pathways, for example, is important for the activation of M2-macrophages and endotoxin clearance and involves the demethylation of repressive histone modifications at activating loci145. Glutamine availability in microenvironments of tumour-bearing mice, has conversely been shown to suppress T-cell activation, and promote tumorigenesis. Using a glutamine antagonist in these mice promoted T-cell activation by inducing a variety of changes including the reduction of αKG levels, and hypermethylation of activating histone modifications60. Iron availability in humans has recently been shown to correlate strongly with antibody production in response to vaccination. When these findings were further explored in cell culture and mice, iron (II) deficiency was found to induce defects in the humoral immune response [G] due to impaired activities of iron-dependent JHDMs and H3K9 hypermethylation at the promoter region of cyclin E, an important element for B-cell proliferation61.
The metabolic regulation of histone acetylation and acylation has also been reported during immune cell activation. The induction of glycolysis due to the upregulation of lactate dehydrogenase (LDHA) generates acetyl-CoA and histone acetylation in T-cells, which regulates production of the cytokine IFNγ146. Competition for nutrients such as glucose in tumour microenvironments can also restrict T-cell activation, which can be rescued by acetate supplementation. Acetate supplementation rescues histone acetylation, chromatin accessibility and subsequently cytokine and IFNγ production147. Lactate, the end product of glucose metabolism, can be utilized for histone lactylation and the activation of homeostatic genes during M1 macrophage polarization in response to bacterial infection85. Transcriptional responses in LPS-induced macrophage activation are regulated by the SCFA crotonate and its derivative crotonyl-CoA, which crotonylates histones at promoters of the activated genes and stimulates the production of chemokines and cytokines25. Each of these metabolic processes is likely to have some epigenetic/chromatin component to its function.
Cancer biology.
Metabolic reprogramming can underlie or support the transformation of non-malignant cells into tumour cells. This is driven either by upstream factors such as aberrations in oncogenes and tumour suppressors, or through direct mutations to metabolic genes, and has been hypothesized to support various cellular functions including the anabolic demands of uncontrolled proliferation148–151. Metabolically driven epigenomic conditioning is emerging as a key component of this reprogramming during tumorigenesis.
Oncogenic mutations exist in genes encoding all classes of the epigenetic machinery, including histones152, chromatin modifiers153,154, epigenetic ‘readers’155 and chromatin-remodellers156, indicating a selective pressure favouring epigenomic reprogramming for tumour progression14,157–160. Similarly, metabolic genes involved in producing chromatin-modifying metabolites are also frequently mutated in cancers, suggesting that the metabolically regulated epigenomic landscape has critical roles in cancer biology161,162. The most well-known example is the mutation of IDH1 or IDH2. According to an analysis of tumour exomes from The Cancer Genome Atlas project, mutant IDH1 serves as an oncogenic driver in at least seven cancer types, including those not typically known to harbour IDH mutations such as breast cancer162. As discussed previously, mutant IDH1 or IDH2 can lead to DNA and histone hypermethylation through the accumulation of 2-HG, resulting in the downregulation of genes associated with tumour suppression62,163–167. These findings have led to the development of inhibitors targeting mutant IDH that have been approved for use in acute myeloid leukaemia (AML), and are being studied in other malignancies168. However, in leukaemia cells, mutant IDH and accumulation of 2-HG have interestingly also been shown to suppress cancer cell proliferation by inhibiting the RNA demethylase FTO and destabilizing oncogenic MYC transcripts by increasing m6A, suggesting dual roles for 2-HG in cancer biology127.
Also frequently mutated in cancer are genes encoding the metabolic enzymes FH and SDH169,170, whose deficiency leads to the accumulation of fumarate and succinate respectively, both of which inhibit TETs and JHDMs171,172, resulting in genome-wide DNA and histone hypermethylation. This has been shown to enable oncogenic promoter–enhancer interactions63, and to induce epithelial-to-mesenchymal-transition (EMT)64. A recent study has further shown that histone hypermethylation caused by 2-HG, succinate and fumarate disrupts DNA repair, rendering cancer cells harbouring IDH, FH and SDH mutations vulnerable to PARP inhibition173. Similar to IDH, FH and SDH mutations, branched-chain amino acid [G] (BCAA) catabolism has recently been shown to induce oncogenic DNA hypermethylation in leukaemia cells by consuming αKG174. In renal cell carcinomas, the metabolic enzyme MTHFD2 [G] is overexpressed, contributing to increased global levels of m6A mRNA methylation by promoting the recycling of methionine through the folate cycle. This was shown in these cancers to increase HIF2α mRNA translation, and HIF2α-driven tumorigenesis175.
In addition to mutations in metabolic enzymes, cancer cells often exhibit altered metabolism in response to upstream drivers which can also reprogram the epigenome. Inactivation of the tumour suppressor LKB1 in a mouse model of KRAS-mutant pancreatic cancer resulted in the upregulation of one-carbon and methionine metabolism and DNA hypermethylation through the accumulation of SAM176, whereas expression of the p53 tumour suppressor increased levels of αKG and 5hmC, an intermediate of active DNA demethylation, resulting in premalignant differentiation and tumour suppression177.
Nearly all aspects of cancer cell metabolism, including the Warburg effect [G], hypoxia66,67, and dysregulated amino acid metabolism, result in epigenomic reprogramming to some extent, accompanied by changes in gene expression. Tumour initiating cells isolated from primary lung tumours have increased methionine cycle activity and histone methylation compared to their non-tumourigenic counterparts, rendering them sensitive to MAT2A inhibition18. Methionine uptake by cancer cells regulates global levels of H3K4me3 and the expression of cancer-associated genes, which can be modulated by restricting methionine in culture media51,52. This effect of methionine restriction on cancer-related gene expression could be associated with reduced tumour growth in mice178. Glutamine deficiency in the core region of melanoma tumours has been shown to result in histone hypermethylation compared to the periphery due to a decrease in αKG. Reducing glutamine levels was subsequently shown to impair cancer cell differentiation and lead to therapeutic resistance in these tumours59. In addition to histone methylation, histone acetylation can be modulated in cancers by regulating HDACs179, sirtuins180, and acetyl-CoA, which by titrating glucose or acetate in culture media has been shown to influence the expression of genes associated with cancer growth and metastasis181–183.
Diet and microbiota in metabolism–epigenetics
Lifestyle-related factors, such as exercise and nutrition, are important variables that influence health outcomes in humans. Although nutritional epidemiology is in general controversial, it is widely accepted that diet and nutrition have profound effects on physiology and disease outcomes, and that there is currently an urgent need to improve public health through encouraging adherence to healthy, if not as yet to be precisely defined, eating patterns184–186. Human foods and diets are extremely complex mixtures of countless chemical components that exist in varying abundance, but only a small fraction of these have been extensively investigated187. Many of these are macro- and micronutrients that feed into metabolism and chromatin biology.
Nutrients also interact with a highly complex and dynamic community of colonizing microorganisms, i.e. the microbiota, which further metabolize them and produce a variety of chromatin-modifying compounds. Although the effects of diet and microbiota on metabolism and epigenetics are less well understood compared to those of cell-autonomous factors such as metabolic enzyme activities, emerging research has started to demonstrate that metabolites derived from diet and microbiota have dynamic interplay with epigenetics and can play an important role in mediating the health-related effects of nutrition and other lifestyle variables (Figure 4), affecting all biological outcomes that we have discussed earlier, such as differentiation188–191, immunity192,193 and cancer51,52,178,179.
Figure 4. Influences of environmental factors on histone acetylation and methylation.

Environmental factors including nutrition, exercise and gut microbiome regulate histone methylation and acetylation by modulating the intracellular pools of metabolites, including S-adenosylmethionine (SAM) and acetyl-CoA that are used by histone methyltransferases (HMTs) and histone acetyltransferases (HATs), respectively. The activity of histone demethylases (HDMs) is supported by α-ketoglutarate (αKG) which can be derived from dietary glutamine, and inhibited by the limited oxygen availability in hypoxia. Ketone bodies and short-chain fatty acids (SCFAs) can provide acyl-CoA precursors for histone acylation, while also directly inhibiting the activity of histone deacetylases (HDACs).
Dietary profiles and associated epigenetic reprogramming.
As discussed earlier, dietary methionine restriction (MR) is able to reduce global histone methylation and influence gene expression by changing intracellular SAM levels51,52,178.The overall effect of methionine availability on global DNA methylation, however, has been shown to depend on the experimental protocol194 and tissue type47, as both global and site-specific hypo- and hypermethylation have been observed under MR. Nevertheless, these results together demonstrate that dietary methionine functions as an important regulator of the landscape of DNA and histone methylation by modulating methionine metabolism. As methionine levels differ greatly across human foods and diets (e.g. plant-based diets are generally lower in methionine)46, it is possible that each human diet is associated with a unique methylation signature, which contributes to the differential health outcomes associated with diets. In addition to methionine, other nutrients that feed into one-carbon and methionine metabolism, including folate, vitamin B-12 and choline, are also able to modulate the levels of SAM and its downstream metabolite SAH (which competes with SAM to inhibit the activities of DNA and histone methyltransferases) to induce epigenomic reprogramming195. In micropigs, dietary folate deficiency was shown to synergize with ethanol intake to decrease the SAM/SAH ratio in liver, resulting in global DNA hypomethylation and increased DNA damage196.
Calorie restriction (CR), which reduces total daily calorie intake by around 15–40% without causing malnutrition, is the most broadly recognized dietary intervention regimen with potential health benefits197. Prolonged CR has been previously shown to extend lifespan and generate positive health outcomes in model organisms such as yeast, fruit flies, mice and monkeys, through a variety of metabolic and physiological effects. In mice, the anti-ageing effects of CR have been associated with protection from the age-related reprogramming of DNA methylation, referred to as the ‘epigenetic clock’198,199, implying that epigenetic mechanisms could mediate the beneficial health outcomes of CR. It is unclear, however, what the precise mechanisms are that regulate these phenotypes, and whether metabolic regulation of the methylation landscape is involved. The generation of the ketone body β-OHB is another consequence of CR that mediates these phenotypes. β-OHB and other ketone bodies are produced through the breakdown of fatty acids and ketogenic amino acids, a process named ketogenesis, which is a metabolic adaptation to fasting200–202. β-OHB has multi-faceted roles in regulating chromatin modifications: it can inhibit class I HDACs, causing a global upregulation of histone acetylation76, and can serve as a substrate for histone β-hydroxybutyrylation, a mark enriched at active promoters and associated with upregulation of starvation-responsive metabolic pathways88. Ketogenesis and histone acetylation have also been observed in during exercise203,204, fasting197,205, and the intake of a ketogenic diet [G]206–208. The ketone body acetoacetate, which detoxifies MGO and potentially suppresses MGO adduction of histones, has also been shown to increase in response to a ketogenic diet119, implying an additional mechanism for the ketogenic diet to modulate chromatin state. Conversely, the generation of β-OHB is not observed during the intake of a non-ketogenic high-fat diet (HFD)209. HFD intake has additionally been shown to reduce levels of acetyl-CoA and histone acetylation by inhibiting the enzymes ACLY and ACSS2210, and to induce global reprogramming of circadian enhancer activities in mouse liver211. The distinct epigenetic responses to a spectrum of different diets suggest that multiple dietary factors may function together to shape the epigenomic landscape, and potentially regulate phenotypic outcomes. This remains an exciting area of future inquiry.
Interaction between diet and gut microbiota.
Dietary intake can influence nutrient availability in concert with the activity of numerous microorganisms that colonize the host body212,213. The interaction between diet and intestinal microbiome is bidirectional and complex: the composition of human gut microbiome can be substantially and rapidly changed by alteration in diet193,214,215, and nutrients from foods and diets are also metabolized by the bacterial species to produce a personalized microbiotal metabolome216 that influences host physiology including epigenetic programs. The digestion of dietary fibres by gut bacteria produces SCFAs, including acetate and butyrate217, which can be oxidized to feed the intracellular acetyl-CoA pool and histone acetylation. Butyrate, being also an inhibitor of HDACs, can influence levels of histone acetylation192 and crotonylation20. Therefore, a high-fibre diet is likely to enhance circulating levels of SCFAs and histone acetylation due to microbiotal activity. In mice, microbiotal colonization has been shown to result in a diet-dependent increase in H3 and H4 acetylation in different tissues, effects partially phenocopied by SCFA supplementation218. Colonization of butyrate-producing bacteria in colorectal tumour-bearing mice fed a high-fibre diet has tumour-suppressive effects through inhibition of HDACs by butyrate, which upregulates histone H3 acetylation, activates apoptotic genes and suppresses cancer cell proliferation179.
Another mechanism for microbiotal species to regulate the host epigenome is potentially by competing with host cells for nutrients. Colonization of choline-consuming strains of Escherichia coli in mouse gut was shown to reduce serum levels of methionine-cycle-related metabolites, to induce heritable changes to global DNA methylation, and to predispose these animals to HFD-induced metabolic disorder219.
Alcohol intake and the epigenome.
Alcohol consumption is a highly common practice around the world and is considered an important risk factor for a variety of pathologies. Alcohol is metabolized in the liver by alcohol dehydrogenase (ADH), forming acetaldehyde. Acetaldehyde is then further metabolized into acetate by aldehyde dehydrogenase (ALDH)220. A recent study showed that alcohol metabolism in mice feeds the acetate pool in circulation which in turn provides acetyl-CoA for histone acetylation in an ACSS2-dependent manner in the brain. This was shown to influence the activation of transcriptional programs related to learning and memory, influencing alcohol-related reward behaviour. In utero exposure to alcohol was additionally shown to influence histone acetylation in the developing fetal forebrain and midbrain, a potential mechanism for the incidence of fetal alcohol spectrum disorder [G]72. The contribution of alcohol to histone acetylation has also been demonstrated by in vivo 13C-tracing experiments221. These studies identify a molecular network connecting alcohol intake, epigenetics and health outcomes, which can potentially shape future therapeutic strategies to treat alcoholism or alcohol-related developmental disorders72.
Concluding remarks and future perspectives
In this Review, we have discussed how metabolism can shape the epigenomic landscape and potentially generate stable and even transgenerationally heritable functional consequences in different contexts. It is particularly exciting for both the epigenetics and metabolism fields to see that metabolically regulated epigenetic modifications include a broad spectrum of enzymatic and non-enzymatic modifications on histone, DNA and RNA molecules beyond the ‘canonical’ methylation and acetylation marks. Extensive work is needed to characterize the kinetic and thermodynamic behaviours of these ‘non-canonical’ marks and context-specific dynamics in response to metabolism.
All metabolically regulated chromatin-modifying enzymes mentioned so far are epigenetic ‘writers’ and ‘erasers’ for covalent chromatin modifications. Notably so far, we have not included ATP-dependent chromatin remodelling complexes222–224, owing to the high concentration of intracellular ATP, which far exceeds the Km values of ATPase domains of chromatin remodellers or any enzyme for that matter. Thus ATP-utilizing enzymes are substrate saturated and have minimal sensitivity towards changes in ATP concentrations. Nevertheless, the ability of chromatin remodellers to recognize and bind with various metabolically driven histone modifications, such as histone methylation and acetylation, is critical for their localization and function, hence metabolism may regulate the functions of these complexes through these modifications. Metabolites such as methionine, αKG, acetyl-CoA, ketone bodies, and redox agents all potentially regulate the function of chromatin remodellers in this way, yet this remains uncharacterized and requires further investigation.
Despite the exciting progress in discovering new metabolically regulated epigenetic marks, the understanding about functional outcomes of these epigenomic responses is still limited. Upcoming studies in the next few years should therefore focus on clarifying the causal role of the metabolically regulated epigenomic landscape in shaping phenotypic outcomes in physiology and disease. Recent advancements in state-of-the-art techniques offer promising toolkits for us to reach a comprehensive and quantitative understanding of the metabolism- epigenetics axis (Box 3).
Box 3: Technologies for dissecting the metabolic and epigenomic landscape.
Two major challenges in the characterization of the metabolically regulated epigenomic landscape are the lack of high-throughput techniques to collect multi-dimensional epigenomic and metabolomic data in a quantitative fashion with sufficient resolution, and the difficulty in demonstrating causality in the association between the two elements. Although chromatin immunoprecipitation followed by sequencing (ChIP-seq) is still the most widely applied technique for genome-scale profiling of histone modifications, alternatives have been developed to increase the coverage and resolution of epigenomic profiles in both bulk tissues and single cells. Global chromatin profiling based on targeted mass spectrometry techniques259 enables bulk-level, simultaneous quantification of 42 combinations of covalent modifications on histone H3, and has been applied to around 1,000 cancer cell lines in the Cancer Cell Line Encyclopedia (CCLE)260. Combined with DNA methylation, transcriptomic and metabolomic profiles261 in the same collection of cell lines, this multi-omic data set is a valuable resource for studying the quantitative relationship between metabolic activity and epigenomic landscape in cancer cells. A new chromatin-profiling technology termed Cleavage Under Targets and Release Using Nuclease (CUT&RUN) — in which DNA fragments bound to the modified histones are directly cleaved and released instead of undergoing crosslinking, sonication and immunoprecipitation as they do in standard ChIP-seq — has shown increased signal-to-noise ratio, efficiency and resolution and has enabled the profiling of chromatin modifications in very small number of cells262,263. Based on CUT&RUN, techniques for chromatin profiling at the single-cell level have also recently been developed264,265. Measurements of metabolomic profiles can achieve cellular or subcellular resolution through the application of mass spectrometry techniques266,267, potentially allowing the integration of metabolomic and epigenomic profiles at single-cell level.
Regarding the causality underlying any relationship between metabolism and epigenetics, it is of particular importance to understand whether changes in the abundance of a specific metabolite cause changes in the relevant epigenetic modifications, and whether these changes directly cause the observed functional and phenotypic outcomes. Isotope tracing, historically used for estimation of metabolic fluxes268, can be applied to quantify the flow of chemical groups from a metabolite to chromatin, thus offering a quantitative measurement of the direct contribution of metabolic pathway activity to chromatin modifications72,75,221,269. CRISPR–Cas9 based epigenome editing270,271 and synthetic biology approaches272, on the other hand, have enabled the targeted, locus-specific deposition or removal of specific epigenetic modifications and the programmable manipulation of components participating in chromatin regulation. These toolkits are providing a valuable opportunity towards us reaching a complete and mechanistic understanding of the metabolically regulated epigenomic landscape in a variety of physiological contexts.
Also unclear is if and how metabolism can dynamically influence the high-level architecture of chromosomes, such as chromatin accessibility, chromosomal looping, and physical properties such as liquid–liquid phase separation. Several theoretical studies have demonstrated that these structural properties of chromatin can be predicted by specific signatures of epigenetic modifications225–228, implying that changes in metabolism could probably alter the overall organization of genome. Furthermore, how metabolic heterogeneity in single cells can influence tissue or organ function through epigenetic regulation is still unknown229. This is especially interesting in the context of development where cells from different developmental states coexist. Single-cell multi-omics techniques that enable simultaneous profiling of gene expression, DNA methylation and chromatin accessibility in single cells could help understand this complexity230–233.
Finally, investigating the roles of metabolism and epigenetics in mediating health outcomes due to nutrition and microbiotal commensalism, are underexplored and promising fields of research. Given the genomic and metabolic heterogeneity among individuals, the wide global spectrum of diets and microbiotal composition, and the general complexity of chromatin, many functional links are still yet to be discovered that offer several promising avenues for future research. Integration of large-scale human datasets and machine learning methods along with rigorous biochemistry could be helpful in reconciling these disparate factors to predict health outcomes234,235 and shed light on future directions for incisive mechanistic studies.
Acknowledgements
J.W.L. thanks the Marc Lustgarten Foundation and the National Institutes of Health (R01CA193256) for their generous support.
Glossary terms
- Nucleosome
The basic structural unit of chromatin. Each nucleosome consists of 2 copies of the histones H2A, H2B, H3 and H4.
- Chromatin remodellers
Protein complexes that regulate gene expression by changing the organization of nucleosomes.
- Electrophilic moieties
Molecules that have the tendency to accept an electron pair by reacting with electron-rich nucleophiles.
- Km and Kd values
Quantities that describe the affinity of the substrate (Km) and inhibitor (Kd) to an enzyme. Smaller values for Km and Kd indicate higher binding affinity
- Methyl group sinks
Molecular pathways that regulate intracellular methyl group availability by consuming S-adenosylmethionine (SAM).
- Liquid–liquid phase separation
De-mixing of fluid into two or more distinct phases which can help compartmentalize molecules within a cell by forming membrane-less organelles.
- Enhancers
Gene-regulatory elements that can be coding or non-coding sequences that potentiate the transcription of genes proximal or distal to them.
- Super-enhancers
Large clusters of enhancers bound by multiple master transcription factors to activate transcription of cell-identity-related genes.
- Bromodomain
A protein domain that recognizes and binds to acetylated lysine residues.
- One-carbon metabolism
A metabolic network for transferring one-carbon units from nutrients to metabolites that support multiple physiological processes such as nucleotide synthesis.
- α-Ketoglutarate
(αKG). A metabolic intermediate of the tricarboxylic acid (TCA) cycle, which is derived from isocitrate and is further processed into succinyl-CoA. It can also be derived from glutamine metabolism.
- Redox balance
Reaction systems that help to maintain health levels of reactive oxygen species in intracellular compartments.
- Metabolic fasting
An intentional abstinence of food and drink during a period of time.
- Hexosamine biosynthetic pathway
A branch of glycolysis that utilizes substrates from amino acid, fatty acid, and nucleotide metabolism to generate substrates that participate in N-linked and O-linked protein glycosylation.
- Central carbon metabolism
Metabolic pathways involved in catabolism of carbohydrates, lipids, and amino acids for the production of ATP, biomass precursors, signaling, and redox status maintenance.
- Rate constants
A quantity that relates the speed of a chemical reaction to the concentrations of its substrates.
- Lipid peroxidation
A process by which lipids are endogenously rendered electrophilic by a degradative chemical reaction with free radicals.
- Nucleophilic functional groups
Chemical groups that have the tendency of donating an electron pair in reactions with electron-poor groups.
- Advanced glycation end products
(AGEs). Covalent adducts formed due to the chemical crosslinking of glycolytic byproducts to macromolecules such as DNA and protein.
- Humoral immune response
A branch of the immune system that involves the activation and differentiation of B-cells into plasma and memory cells, which mount antibody responses to invading pathogens.
- Branched-chain amino acid
The amino acids leucine, isoleucine and valine that each contain a branching aliphatic side chain.
- MTHFD2
The enzyme methylenetetrahydrofolate dehydrogenase 2, which couples the folate cycle to the methionine cycle, enabling the transfer of the one-carbon methyl-group to homocysteine to recycle methionine.
- Warburg effect
The phenomenon, first observed by German physiologist Otto Heinrich Warburg, that cancer cells hyperactivate glycolysis, even in the presence of sufficient oxygen.
- Ketogenic diet
A diet low in carbohydrate and high in fat, which precipitates the generation of ketone bodies by fatty acid catabolism.
- Fetal alcohol spectrum disorder
In utero exposure to alcohol that can give rise to the postnatal acquisition of developmental disorders.
Footnotes
Competing interests
J.W.L. advises Restoration Foodworks, Nanocare Technologies and Raphael Pharmaceuticals. Z.D. and V.R. declare no competing interests.
References
- 1.Alberts B Molecular biology of the cell. Sixth edition. edn, (Garland Science, Taylor and Francis Group, 2015). [Google Scholar]
- 2.Hyun K, Jeon J, Park K & Kim J Writing, erasing and reading histone lysine methylations. Exp Mol Med 49, e324–e324, doi: 10.1038/emm.2017.11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuettengruber B, Bourbon HM, Di Croce L & Cavalli G Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57, doi: 10.1016/j.cell.2017.08.002 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Stillman B Histone Modifications: Insights into Their Influence on Gene Expression. Cell 175, 6–9, doi: 10.1016/j.cell.2018.08.032 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Li X, Egervari G, Wang Y, Berger SL & Lu Z Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Bio 19, 563–578, doi: 10.1038/s41580-018-0029-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan JC & Maze I Nothing Is yet Set in (Hi)stone: Novel Post-Translational Modifications Regulating Chromatin Function. Trends Biochem Sci, doi: 10.1016/j.tibs.2020.05.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu TP et al. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 532, 329–333, doi: 10.1038/nature17640 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao C-L et al. N(6)-Methyladenine DNA Modification in the Human Genome. Molecular cell 71, 306–318. e307, doi: 10.1016/j.molcel.2018.06.015 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Galligan JJ & Marnett LJ Histone Adduction and Its Functional Impact on Epigenetics. Chemical Research in Toxicology 30, 376–387, doi: 10.1021/acs.chemrestox.6b00379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arango D et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 175, 1872–1886. e1824, doi: 10.1016/j.cell.2018.10.030 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yue Y, Liu J & He C RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Gene Dev 29, 1343–1355, doi: 10.1101/gad.262766.115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandel NS Navigating metabolism. (Cold Spring Harbor Laboratory Press, 2015). [Google Scholar]
- 13.Reid MA, Dai ZW & Locasale JW The impact of cellular metabolism on chromatin dynamics and epigenetics. Nature Cell Biology 19, 1298–1306, doi: 10.1038/ncb3629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavalli G & Heard E Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499, doi: 10.1038/s41586-019-1411-0 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Tang S et al. Methionine metabolism is essential for SIRT1-regulated mouse embryonic stem cell maintenance and embryonic development. The EMBO journal 36, 3175–3193, doi: 10.15252/embj.201796708 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yucel N et al. Glucose Metabolism Drives Histone Acetylation Landscape Transitions that Dictate Muscle Stem Cell Function. Cell reports 27, 3939–3955. e3936, doi: 10.1016/j.celrep.2019.05.092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu W et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Molecular Cell 75, 1147–1160. e1145, doi: 10.1016/j.molcel.2019.06.039 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Wang Z et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med 25, 825–837, doi: 10.1038/s41591-019-0423-5 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Reina-Campos M et al. Increased Serine and One-Carbon Pathway Metabolism by PKC λ/ι Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 35, 385–400. e389, doi: 10.1016/j.ccell.2019.01.018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fellows R et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nature Communications 9, 105, doi: 10.1038/s41467-017-02651-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaelin WG Jr. & McKnight SL Influence of metabolism on epigenetics and disease. Cell 153, 56–69, doi: 10.1016/j.cell.2013.03.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrer A & Wellen KE Metabolism and epigenetics: a link cancer cells exploit. Curr Opin Biotechnol 34, 23–29, doi: 10.1016/j.copbio.2014.11.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etchegaray JP & Mostoslavsky R Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol Cell 62, 695–711, doi: 10.1016/j.molcel.2016.05.029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haws SA et al. Methyl-Metabolite Depletion Elicits Adaptive Responses to Support Heterochromatin Stability and Epigenetic Persistence. Mol Cell 78, 210–223 e218, doi: 10.1016/j.molcel.2020.03.004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabari BR et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Molecular cell 58, 203–215, doi: 10.1016/j.molcel.2015.02.029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulusu V et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Reports 18, 647–658, doi: 10.1016/j.celrep.2016.12.055 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mews P et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–+, doi: 10.1038/nature22405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagaraj R et al. Nuclear Localization of Mitochondrial TCA Cycle Enzymes as a Critical Step in Mammalian Zygotic Genome Activation. Cell 168, 210–223 e211, doi: 10.1016/j.cell.2016.12.026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sivanand S et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Molecular Cell 67, 252–+, doi: 10.1016/j.molcel.2017.06.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masui K et al. mTORC2 links growth factor signaling with epigenetic regulation of iron metabolism in glioblastoma. Journal of Biological Chemistry 294, 19740–19751, doi: 10.1074/jbc.RA119.011519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sdelci S et al. MTHFD1 interaction with BRD4 links folate metabolism to transcriptional regulation. Nature Genetics 51, 990–+, doi: 10.1038/s41588-019-0413-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun RC et al. Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metabolism 30, 903–+, doi: 10.1016/j.cmet.2019.08.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sperber H et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nature Cell Biology 17, 1523–1535, doi: 10.1038/ncb3264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye CQ, Sutter BM, Wang Y, Kuang Z & Tu BP A Metabolic Function for Phospholipid and Histone Methylation. Molecular Cell 66, 180–+, doi: 10.1016/j.molcel.2017.02.026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye CQ et al. Demethylation of the Protein Phosphatase PP2A Promotes Demethylation of Histones to Enable Their Function as a Methyl Group Sink. Molecular Cell 73, 1115–+, doi: 10.1016/j.molcel.2019.01.012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larson AG et al. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547, 236–240, doi: 10.1038/nature22822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strom AR et al. Phase separation drives heterochromatin domain formation. Nature 547, 241–245, doi: 10.1038/nature22989 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanulli S et al. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575, 390–+, doi: 10.1038/s41586-019-1669-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibson BA et al. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484 e421, doi: 10.1016/j.cell.2019.08.037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nair SJ et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat Struct Mol Biol 26, 193–203, doi: 10.1038/s41594-019-0190-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, doi:ARTN eaar3958 10.1126/science.aar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ries RJ et al. m(6)A enhances the phase separation potential of mRNA. Nature 571, 424–+, doi: 10.1038/s41586-019-1374-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alberti S, Gladfelter A & Mittag T Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 176, 419–434, doi: 10.1016/j.cell.2018.12.035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell Research 21, 381–395, doi: 10.1038/cr.2011.22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ravanel S, Gakière B, Job D & Douce R The specific features of methionine biosynthesis and metabolism in plants. Proceedings of the National Academy of Sciences 95, 7805, doi: 10.1073/pnas.95.13.7805 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanderson SM, Gao X, Dai ZW & Locasale JW Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nature Reviews Cancer 19, 625–637, doi: 10.1038/s41568-019-0187-8 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Mattocks DAL et al. Short term methionine restriction increases hepatic global DNA methylation in adult but not young male C57BL/6J mice. Exp Gerontol 88, 1–8, doi: 10.1016/j.exger.2016.12.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang N Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim Nutr 4, 11–16, doi: 10.1016/j.aninu.2017.08.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shyh-Chang N et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226, doi: 10.1126/science.1226603 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shiraki N et al. Methionine Metabolism Regulates Maintenance and Differentiation of Human Pluripotent Stem Cells. Cell Metabolism 19, 780–794, doi: 10.1016/j.cmet.2014.03.017 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Dai Z, Mentch SJ, Gao X, Nichenametla SN & Locasale JW Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nature Communications 9, 1955, doi: 10.1038/s41467-018-04426-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mentch SJ et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell metabolism 22, 861–873, doi: 10.1016/j.cmet.2015.08.024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding W et al. Stress-responsive and metabolic gene regulation are altered in low S-adenosylmethionine. Plos Genet 14, doi:ARTN e1007812 10.1371/journal.pgen.1007812 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinclair LV et al. Antigen receptor control of methionine metabolism in T cells. eLife 8, e44210, doi: 10.7554/eLife.44210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roy DG et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab 31, 250–266. e259, doi: 10.1016/j.cmet.2020.01.006 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Kohli RM & Zhang Y TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479, doi: 10.1038/nature12750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi Y Histone lysine demethylases: emerging roles in development, physiology and disease. Nature Reviews Genetics 8, 829–833, doi:DOI 10.1038/nrg2218 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Young JI, Zuchner S & Wang GF Regulation of the Epigenome by Vitamin C. Annu Rev Nutr 35, 545–564, doi: 10.1146/annurev-nutr-071714-034228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan M et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nature cell biology 18, 1090–1101, doi: 10.1038/ncb3410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leone RD et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013, doi: 10.1126/science.aav2588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang Y et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nature Communications 10, 2935, doi: 10.1038/s41467-019-11002-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schvartzman J-M, Reuter VP, Koche RP & Thompson CB 2-hydroxyglutarate inhibits MyoD-mediated differentiation by preventing H3K9 demethylation. Proceedings of the National Academy of Sciences 116, 12851, doi: 10.1073/pnas.1817662116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flavahan WA et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 575, 229–+, doi: 10.1038/s41586-019-1668-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sciacovelli M et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547, doi: 10.1038/nature19353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bazopoulou D et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature, doi: 10.1038/s41586-019-1814-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thienpont B et al. Tumour hypoxia causes DNA hyper-methylation by reducing TET activity. Nature 537, 63–68, doi: 10.1038/nature19081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chakraborty AA et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science (New York, N.Y.) 363, 1217–1222, doi: 10.1126/science.aaw1026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Batie M et al. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363, 1222–+, doi: 10.1126/science.aau5870 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Wellen KE et al. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 324, 1076, doi: 10.1126/science.1164097 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cluntun AA et al. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer & Metabolism 3, 10, doi: 10.1186/s40170-015-0135-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu X et al. Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 175, 502–513. e513, doi: 10.1016/j.cell.2018.08.040 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mews P et al. Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–+, doi: 10.1038/s41586-019-1700-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sebastián C & Mostoslavsky R The Various Metabolic Sources of Histone Acetylation. Trends in Endocrinology & Metabolism 28, 85–87, doi: 10.1016/j.tem.2016.11.001 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Bose S, Ramesh V & Locasale JW Acetate Metabolism in Physiology, Cancer, and Beyond. Trends in Cell Biology 29, 695–703, doi: 10.1016/j.tcb.2019.05.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDonnell E et al. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Reports 17, 1463–1472, doi: 10.1016/j.celrep.2016.10.012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shimazu T et al. Suppression of Oxidative Stress by beta-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 339, 211–214, doi: 10.1126/science.1227166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chalkiadaki A & Guarente L Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol 8, 287–296, doi: 10.1038/nrendo.2011.225 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Sabari BR, Zhang D, Allis CD & Zhao Y Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 18, 90–101, doi: 10.1038/nrm.2016.140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dutta A, Abmayr SM & Workman JL Diverse Activities of Histone Acylations Connect Metabolism to Chromatin Function. Molecular cell 63, 547–552, doi: 10.1016/j.molcel.2016.06.038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hirschey MD & Zhao Y Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Molecular & cellular proteomics : MCP 14, 2308–2315, doi: 10.1074/mcp.R114.046664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simithy J et al. Characterization of histone acylations links chromatin modifications with metabolism. Nature Communications 8, 1141, doi: 10.1038/s41467-017-01384-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 552, 273–277, doi: 10.1038/nature25003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wagner GR & Hirschey MD Nonenzymatic Protein Acylation as a Carbon Stress Regulated by Sirtuin Deacylases. Molecular Cell 54, 5–16, doi: 10.1016/j.molcel.2014.03.027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Trefely S, Lovell CD, Snyder NW & Wellen KE Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol Metab, 100941, doi: 10.1016/j.molmet.2020.01.005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580, doi: 10.1038/s41586-019-1678-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang H et al. Lysine benzoylation is a histone mark regulated by SIRT2. Nature Communications 9, 3374, doi: 10.1038/s41467-018-05567-w (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goudarzi A et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Molecular cell 62, 169–180, doi: 10.1016/j.molcel.2016.03.014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xie Z et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Molecular cell 62, 194–206, doi: 10.1016/j.molcel.2016.03.036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang X et al. Molecular basis for hierarchical histone de-β-hydroxybutyrylation by SIRT3. Cell Discovery 5, 35, doi: 10.1038/s41421-019-0103-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gowans GJ et al. Recognition of Histone Crotonylation by Taf14 Links Metabolic State to Gene Expression. Molecular Cell 76, 909–921. e903, doi: 10.1016/j.molcel.2019.09.029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Q et al. Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nature Communications 9, 3436, doi: 10.1038/s41467-018-05451-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Biskup CS et al. Effects of Acute Tryptophan Depletion on Brain Serotonin Function and Concentrations of Dopamine and Norepinephrine in C57BL/6J and BALB/cJ Mice. PLOS ONE 7, e35916, doi: 10.1371/journal.pone.0035916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Solon-Biet SM et al. Branched-chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat Metab 1, 532–545, doi: 10.1038/s42255-019-0059-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Farrelly LA et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539, doi: 10.1038/s41586-019-1024-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meiser J, Weindl D & Hiller K Complexity of dopamine metabolism. Cell Commun Signal 11, 34, doi: 10.1186/1478-811X-11-34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Montgomery AJ, McTavish SF, Cowen PJ & Grasby PM Reduction of brain dopamine concentration with dietary tyrosine plus phenylalanine depletion: an [11C]raclopride PET study. Am J Psychiatry 160, 1887–1889, doi: 10.1176/appi.ajp.160.10.1887 (2003). [DOI] [PubMed] [Google Scholar]
- 97.Lepack AE et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 368, 197–201, doi: 10.1126/science.aaw8806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chiaradonna F, Ricciardiello F & Palorini R The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 7, 53, doi: 10.3390/cells7060053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ryczko MC et al. Metabolic Reprogramming by Hexosamine Biosynthetic and Golgi N-Glycan Branching Pathways. Sci Rep-Uk 6, 23043, doi: 10.1038/srep23043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang S, Roche K, Nasheuer H-P & Lowndes NF Modification of Histones by Sugar β-N-Acetylglucosamine (GlcNAc) Occurs on Multiple Residues, Including Histone H3 Serine 10, and Is Cell Cycle-regulated. Journal of Biological Chemistry 286, 37483–37495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen Q, Chen Y, Bian C, Fujiki R & Yu X TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564, doi: 10.1038/nature11742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Messner S & Hottiger MO Histone ADP-ribosylation in DNA repair, replication and transcription. Trends in cell biology 21, 534–542, doi: 10.1016/j.tcb.2011.06.001 (2011). [DOI] [PubMed] [Google Scholar]
- 103.Ciccarone F, Zampieri M & Caiafa P PARP1 orchestrates epigenetic events setting up chromatin domains. Seminars in Cell & Developmental Biology 63, 123–134, doi: 10.1016/j.semcdb.2016.11.010 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Bai P & Cantó C The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell metabolism 16, 290–295, doi: 10.1016/j.cmet.2012.06.016 (2012). [DOI] [PubMed] [Google Scholar]
- 105.Harmel R & Fiedler D Features and regulation of non-enzymatic post-translational modifications. Nature Chemical Biology 14, 244–252, doi: 10.1038/Nchembio.2575 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Kuo YM & Andrews AJ Quantitating the Specificity and Selectivity of Gcn5-Mediated Acetylation of Histone H3. Plos One 8, doi: 10.1371/journal.pone.0054896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baeza J, Smallegan MJ & Denu JM Site-Specific Reactivity of Nonenzymatic Lysine Acetylation. Acs Chem Biol 10, 122–128, doi: 10.1021/cb500848p (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Paik WK, Lee HW & Kim S Non-Enzymatic Methylation of Proteins with S-Adenosyl-L-Methionine. Febs Letters 58, 39–42, doi:Doi 10.1016/0014-5793(75)80220-1 (1975). [DOI] [PubMed] [Google Scholar]
- 109.Rydberg B & Lindahl T Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J 1, 211–216 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gaschler MM & Stockwell BR Lipid peroxidation in cell death. Biochemical and Biophysical Research Communications 482, 419–425, doi: 10.1016/j.bbrc.2016.10.086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen D, Fang L, Li H, Tang MS & Jin C Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J Biol Chem 288, 21678–21687, doi: 10.1074/jbc.M113.476630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Carrier EJ, Zagol-Ikapitte I, Amarnath V, Boutaud O & Oates JA Levuglandin Forms Adducts with Histone H4 in a Cyclooxygenase-2-Dependent Manner, Altering Its Interaction with DNA. Biochemistry 53, 2436–2441, doi: 10.1021/bi401673b (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kold-Christensen R & Johannsen M Methylglyoxal Metabolism and Aging-Related Disease: Moving from Correlation toward Causation. Trends Endocrinol Metab, doi: 10.1016/j.tem.2019.10.003 (2019). [DOI] [PubMed] [Google Scholar]
- 114.Luengo A et al. Reactive metabolite production is a targetable liability of glycolytic metabolism in lung cancer. Nature Communications 10, 5604, doi: 10.1038/s41467-019-13419-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Galligan JJ et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. P Natl Acad Sci USA 115, 9228–9233, doi: 10.1073/pnas.1802901115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Y et al. Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. P Natl Acad Sci USA 115, 2365–2370, doi: 10.1073/pnas.1717664115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Andreeva A et al. The apparent deglycase activity of DJ-1 results from the conversion of free methylglyoxal present in fast equilibrium with hemithioacetals and hemiaminals. J Biol Chem 294, 18863–18872, doi: 10.1074/jbc.RA119.011237 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Uribarri J et al. Dietary Advanced Glycation End Products and Their Role in Health and Disease. Advances in Nutrition 6, 461–473, doi: 10.3945/an.115.008433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Salomon T et al. Ketone Body Acetoacetate Buffers Methylglyoxal via a Non-enzymatic Conversion during Diabetic and Dietary Ketosis. Cell Chemical Biology 24, 935–+, doi: 10.1016/j.chembiol.2017.07.012 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Boccaletto P et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Research 46, D303–D307, doi: 10.1093/nar/gkx1030 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shi HL, Wei JB & He C Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Molecular Cell 74, 640–650, doi: 10.1016/j.molcel.2019.04.025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zaccara S, Ries RJ & Jaffrey SR Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Bio 20, 608–624, doi: 10.1038/s41580-019-0168-5 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Pendleton KE et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 169, 824–+, doi: 10.1016/j.cell.2017.05.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shima H et al. S-Adenosylmethionine Synthesis Is Regulated by Selective N-6-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Reports 21, 3354–3363, doi: 10.1016/j.celrep.2017.11.092 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Aik W et al. Structural Basis for Inhibition of the Fat Mass and Obesity Associated Protein (FTO). Journal of Medicinal Chemistry 56, 3680–3688, doi: 10.1021/jm400193d (2013). [DOI] [PubMed] [Google Scholar]
- 126.Gerken T et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318, 1469–1472, doi: 10.1126/science.1151710 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Su R et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–+, doi: 10.1016/j.cell.2017.11.031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mauer J et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nature Chemical Biology 15, 340–+, doi: 10.1038/s41589-019-0231-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ito S et al. Human NAT10 Is an ATP-dependent RNA Acetyltransferase Responsible for N-4-Acetylcytidine Formation in 18 S Ribosomal RNA (rRNA). Journal of Biological Chemistry 289, 35724–35730, doi: 10.1074/jbc.C114.602698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shyh-Chang N & Ng H-H The metabolic programming of stem cells. Genes Dev 31, 336–346, doi: 10.1101/gad.293167.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Folmes CD et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14, 264–271, doi: 10.1016/j.cmet.2011.06.011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Panopoulos AD et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res 22, 168–177, doi: 10.1038/cr.2011.177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tsogtbaatar E, Landin C, Minter-Dykhouse K & Folmes CDL Energy Metabolism Regulates Stem Cell Pluripotency. Front Cell Dev Biol 8, 87, doi: 10.3389/fcell.2020.00087 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ito K et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354, 1156–1160, doi: 10.1126/science.aaf5530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Takubo K et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61, doi: 10.1016/j.stem.2012.10.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ito K et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 18, 1350–1358, doi: 10.1038/nm.2882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tohyama S et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137, doi: 10.1016/j.stem.2012.09.013 (2013). [DOI] [PubMed] [Google Scholar]
- 138.Boon R et al. Amino acid levels determine metabolism and CYP450 function of hepatocytes and hepatoma cell lines. Nat Commun 11, 1393, doi: 10.1038/s41467-020-15058-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guijas C, Montenegro-Burke JR, Warth B, Spilker ME & Siuzdak G Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat Biotechnol 36, 316–320, doi: 10.1038/nbt.4101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Oburoglu L et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 15, 169–184, doi: 10.1016/j.stem.2014.06.002 (2014). [DOI] [PubMed] [Google Scholar]
- 141.Carey BW, Finley LWS, Cross JR, Allis CD & Thompson CB Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416, doi: 10.1038/nature13981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.TeSlaa T et al. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab 24, 485–493, doi: 10.1016/j.cmet.2016.07.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Moussaieff A et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab 21, 392–402, doi: 10.1016/j.cmet.2015.02.002 (2015). [DOI] [PubMed] [Google Scholar]
- 144.Buck MD, Sowell RT, Kaech SM & Pearce EL Metabolic Instruction of Immunity. Cell 169, 570–586, doi: 10.1016/j.cell.2017.04.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu P-S et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nature Immunology 18, 985–994, doi: 10.1038/ni.3796 (2017). [DOI] [PubMed] [Google Scholar]
- 146.Peng M et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484, doi: 10.1126/science.aaf6284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Qiu J et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Reports 27, 2063–2074. e2065, doi: 10.1016/j.celrep.2019.04.022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liberti MV & Locasale JW The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41, 211–218, doi: 10.1016/j.tibs.2015.12.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vander Heiden MG & DeBerardinis RJ Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, doi: 10.1016/j.cell.2016.12.039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pavlova NN & Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metabolism 23, 27–47, doi: 10.1016/j.cmet.2015.12.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Science Advances 2, doi:UNSP e1600200 10.1126/sciadv.1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nacev BA et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567, 473–+, doi: 10.1038/s41586-019-1038-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gozdecka M et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat Genet 50, 883–894, doi: 10.1038/s41588-018-0114-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Donaldson-Collier MC et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nature Genetics 51, 517–+, doi: 10.1038/s41588-018-0338-y (2019). [DOI] [PubMed] [Google Scholar]
- 155.Fujisawa T & Filippakopoulos P Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Bio 18, 246–262, doi: 10.1038/nrm.2016.143 (2017). [DOI] [PubMed] [Google Scholar]
- 156.Deribe YL et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med 24, 1047–+, doi: 10.1038/s41591-018-0019-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Suva ML, Riggi N & Bernstein BE Epigenetic Reprogramming in Cancer. Science 339, 1567–1570, doi: 10.1126/science.1230184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Flavahan WA, Gaskell E & Bernstein BE Epigenetic plasticity and the hallmarks of cancer. Science 357, doi:ARTN eaal2380 10.1126/science.aal2380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Feinberg AP The Key Role of Epigenetics in Human Disease Prevention and Mitigation. New Engl J Med 378, 1323–1334, doi: 10.1056/NEJMra1402513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huether R et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nature Communications 5, doi:ARTN 3630 10.1038/ncomms4630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schaefer IM, Hornick JL & Bovee JVMG The role of metabolic enzymes in mesenchymal tumors and tumor syndromes: genetics, pathology, and molecular mechanisms. Lab Invest 98, 414–426, doi: 10.1038/s41374-017-0003-6 (2018). [DOI] [PubMed] [Google Scholar]
- 162.Bailey MH et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–+, doi:ARTN 385.e1 10.1016/j.cell.2018.02.060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yan H et al. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine 360, 765–773, doi: 10.1056/NEJMoa0808710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Figueroa ME et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell 18, 553–567, doi: 10.1016/j.ccr.2010.11.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Losman J-A et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science (New York, N.Y.) 339, 1621–1625, doi: 10.1126/science.1231677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lu C et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478, doi: 10.1038/nature10860 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–+, doi: 10.1038/nature16490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Golub D et al. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front Oncol 9, 417–417, doi: 10.3389/fonc.2019.00417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.King A, Selak MA & Gottlieb E Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682, doi: 10.1038/sj.onc.1209594 (2006). [DOI] [PubMed] [Google Scholar]
- 170.Kaelin WG SDH5 Mutations and Familial Paraganglioma: Somewhere Warburg is Smiling. Cancer Cell 16, 180–182, doi: 10.1016/j.ccr.2009.08.013 (2009). [DOI] [PubMed] [Google Scholar]
- 171.Cervera AM, Bayley J-P, Devilee P & McCreath KJ Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Molecular cancer 8, 89–89, doi: 10.1186/1476-4598-8-89 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xiao M et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Gene Dev 26, 1326–1338, doi: 10.1101/gad.191056.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sulkowski PL et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature, doi: 10.1038/s41586-020-2363-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Raffel S et al. BCAT1 restricts alpha KG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–+, doi: 10.1038/nature24294 (2017). [DOI] [PubMed] [Google Scholar]
- 175.Green NH et al. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene 38, 6211–6225, doi: 10.1038/s41388-019-0869-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kottakis F et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395, doi: 10.1038/nature20132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Morris JP et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–+, doi: 10.1038/s41586-019-1577-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gao X et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–+, doi: 10.1038/s41586-019-1437-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Donohoe DR et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov 4, 1387–1397, doi: 10.1158/2159-8290.CD-14-0501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chalkiadaki A & Guarente L The multifaceted functions of sirtuins in cancer. Nature Reviews Cancer 15, doi: 10.1038/nrc3985 (2015). [DOI] [PubMed] [Google Scholar]
- 181.Lee JV et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell metabolism 20, 306–319, doi: 10.1016/j.cmet.2014.06.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lee JV et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca(2+)-NFAT signaling. Gene Dev 32, 497–511, doi: 10.1101/gad.311027.117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gruber JJ et al. HAT1 Coordinates Histone Production and Acetylation via H4 Promoter Binding. Molecular cell 75, 711–724. e715, doi: 10.1016/j.molcel.2019.05.034 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Afshin A et al. Health effects of dietary risks in 195 countries, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 393, 1958–1972, doi: 10.1016/S0140-6736(19)30041-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Steck SE & Murphy EA Dietary patterns and cancer risk. Nat Rev Cancer, doi: 10.1038/s41568-019-0227-4 (2019). [DOI] [PubMed] [Google Scholar]
- 186.Willett W et al. Food in the Anthropocene: the EAT-Lancet Commission on healthy diets from sustainable food systems. Lancet 393, 447–492, doi: 10.1016/S0140-6736(18)31788-4 (2019). [DOI] [PubMed] [Google Scholar]
- 187.Barabási A-L, Menichetti G & Loscalzo J The unmapped chemical complexity of our diet. Nature Food 1, 33–37, doi: 10.1038/s43016-019-0005-1 (2020). [DOI] [Google Scholar]
- 188.Taya Y et al. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 354, 1152–1155, doi: 10.1126/science.aag3145 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Ertl RP, Chen J, Astle CM, Duffy TM & Harrison DE Effects of dietary restriction on hematopoietic stem-cell aging are genetically regulated. Blood 111, 1709–1716, doi: 10.1182/blood-2007-01-069807 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cerletti M, Jang YC, Finley LW, Haigis MC & Wagers AJ Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell 10, 515–519, doi: 10.1016/j.stem.2012.04.002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Garcia-Caballero M et al. Role and therapeutic potential of dietary ketone bodies in lymph vessel growth. Nat Metab 1, 666–675, doi: 10.1038/s42255-019-0087-y (2019). [DOI] [PubMed] [Google Scholar]
- 192.Chang PV, Hao LM, Offermanns S & Medzhitov R The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. P Natl Acad Sci USA 111, 2247–2252, doi: 10.1073/pnas.1322269111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rangan P et al. Fasting-Mimicking Diet Modulates Microbiota and Promotes Intestinal Regeneration to Reduce Inflammatory Bowel Disease Pathology. Cell Reports 26, 2704–+, doi: 10.1016/j.celrep.2019.02.019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Waterland RA Assessing the effects of high methionine intake on DNA methylation. J Nutr 136, 1706s–1710s (2006). [DOI] [PubMed] [Google Scholar]
- 195.Mafra D et al. Methyl Donor Nutrients in Chronic Kidney Disease: Impact on the Epigenetic Landscape. J Nutr 149, 372–380, doi: 10.1093/jn/nxy289 (2019). [DOI] [PubMed] [Google Scholar]
- 196.Halsted CH et al. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. P Natl Acad Sci USA 99, 10072–10077, doi: 10.1073/pnas.112336399 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Di Francesco A, Di Germanio C, Bernier M & de Cabo R A time to fast. Science 362, 770–775, doi: 10.1126/science.aau2095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hahn O et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biology 18, doi:ARTN 56 10.1186/s13059-017-1187-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang TN et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biology 18, doi:ARTN 57 10.1186/s13059-017-1186-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Newman JC & Verdin E Ketone bodies as signaling metabolites. Trends Endocrin Met 25, 42–52, doi: 10.1016/j.tem.2013.09.002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Puchalska P & Crawford PA Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metabolism 25, 262–284, doi: 10.1016/j.cmet.2016.12.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rhoads TW et al. Caloric Restriction Engages Hepatic RNA Processing Mechanisms in Rhesus Monkeys. Cell Metabolism 27, 677–+, doi: 10.1016/j.cmet.2018.01.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sleiman SF et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. Elife 5, doi:ARTN e15092 10.7554/eLife.15092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sato S et al. Time of Exercise Specifies the Impact on Muscle Metabolic Pathways and Systemic Energy Homeostasis. Cell Metabolism 30, 92–+, doi: 10.1016/j.cmet.2019.03.013 (2019). [DOI] [PubMed] [Google Scholar]
- 205.Guo XL et al. Caloric restriction promotes cell survival in a mouse model of normal tension glaucoma. Sci Rep-Uk 6, doi:ARTN 33950 10.1038/srep33950 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Benjamin JS et al. A ketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. P Natl Acad Sci USA 114, 125–130, doi: 10.1073/pnas.1611431114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Roberts MN et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metabolism 26, 539–+, doi: 10.1016/j.cmet.2017.08.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tognini P et al. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metabolism 26, 523–+, doi: 10.1016/j.cmet.2017.08.015 (2017). [DOI] [PubMed] [Google Scholar]
- 209.Newman JC et al. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metabolism 26, 547–+, doi: 10.1016/j.cmet.2017.08.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Carrer A et al. Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels. Journal of Biological Chemistry 292, 3312–3322, doi: 10.1074/jbc.M116.750620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Guan DY et al. Diet-Induced Circadian Enhancer Remodeling Synchronizes Opposing Hepatic Lipid Metabolic Processes. Cell 174, 831–+, doi:ARTN 842.e12 10.1016/j.cell.2018.06.031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gilbert JA et al. Current understanding of the human microbiome. Nat Med 24, 392–400, doi: 10.1038/nm.4517 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schmidt TSB, Raes J & Bork P The Human Gut Microbiome: From Association to Modulation. Cell 172, 1198–1215, doi: 10.1016/j.cell.2018.02.044 (2018). [DOI] [PubMed] [Google Scholar]
- 214.David LA et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–+, doi: 10.1038/nature12820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Suez J et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514, 181–+, doi: 10.1038/nature13793 (2014). [DOI] [PubMed] [Google Scholar]
- 216.Blacher E et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474–+, doi: 10.1038/s41586-019-1443-5 (2019). [DOI] [PubMed] [Google Scholar]
- 217.den Besten G et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54, 2325–2340, doi: 10.1194/jlr.R036012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Krautkramer KA et al. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Molecular Cell 64, 982–992, doi: 10.1016/j.molcel.2016.10.025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Romano KA et al. Metabolic, Epigenetic, and Transgenerational Effects of Gut Bacterial Choline Consumption. Cell Host Microbe 22, 279–+, doi: 10.1016/j.chom.2017.07.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Schuckit MA Alcohol-use disorders. The Lancet 373, 492–501, doi: 10.1016/S0140-6736(09)60009-X (2009). [DOI] [PubMed] [Google Scholar]
- 221.Kriss CL et al. In Vivo Metabolic Tracing Demonstrates the Site-Specific Contribution of Hepatic Ethanol Metabolism to Histone Acetylation. Alcohol Clin Exp Res 42, 1909–1923, doi: 10.1111/acer.13843 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Narlikar GJ, Sundaramoorthy R & Owen-Hughes T Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490–503, doi: 10.1016/j.cell.2013.07.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bartholomew B Regulating the chromatin landscape: structural and mechanistic perspectives. Annu Rev Biochem 83, 671–696, doi: 10.1146/annurev-biochem-051810-093157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Clapier CR, Iwasa J, Cairns BR & Peterson CL Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol 18, 407–422, doi: 10.1038/nrm.2017.26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Qi YF & Zhang B Predicting three-dimensional genome organization with chromatin states. Plos Computational Biology 15, doi:ARTN e1007024 10.1371/journal.pcbi.1007024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Buckle A, Brackley CA, Boyle S, Marenduzzo D & Gilbert N Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci. Molecular Cell 72, 786–+, doi: 10.1016/j.molcel.2018.09.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.MacPherson Q, Beltran B & Spakowitz AJ Bottom-up modeling of chromatin segregation due to epigenetic modifications. P Natl Acad Sci USA 115, 12739–12744, doi: 10.1073/pnas.1812268115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Di Pierro M, Cheng RR, Aiden EL, Wolynes PG & Onuchic JN De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture. P Natl Acad Sci USA 114, 12126–12131, doi: 10.1073/pnas.1714980114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Xiao ZT, Dai ZW & Locasale JW Metabolic landscape of the tumor microenvironment at single cell resolution. Nature Communications 10, doi:ARTN 3763 10.1038/s41467-019-11738-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhou F et al. Reconstituting the transcriptome and DNA methylome landscapes of human implantation. Nature 572, 660–+, doi: 10.1038/s41586-019-1500-0 (2019). [DOI] [PubMed] [Google Scholar]
- 231.Bian SH et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362, 1060–+, doi: 10.1126/science.aao3791 (2018). [DOI] [PubMed] [Google Scholar]
- 232.Cao JY et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385, doi: 10.1126/science.aau0730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Clark SJ et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nature Communications 9, doi:ARTN 781 10.1038/s41467-018-03149-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Korem T et al. Bread Affects Clinical Parameters and Induces Gut Microbiome-Associated Personal Glycemic Responses. Cell Metabolism 25, 1243–+, doi: 10.1016/j.cmet.2017.05.002 (2017). [DOI] [PubMed] [Google Scholar]
- 235.Bashiardes S, Godneva A, Elinav E & Segal E Towards utilization of the human genome and microbiome for personalized nutrition. Curr Opin Biotech 51, 57–63, doi: 10.1016/j.copbio.2017.11.013 (2018). [DOI] [PubMed] [Google Scholar]
- 236.Cornish-Bowden A Fundamentals of enzyme kinetics. 4th, completely revised and greatly enlarged edition. edn, (Wiley-Blackwell, 2012). [Google Scholar]
- 237.Lau OD et al. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Journal of Biological Chemistry 275, 21953–21959, doi:DOI 10.1074/jbc.M003219200 (2000). [DOI] [PubMed] [Google Scholar]
- 238.Tanner KG, Langer MR & Denu JM Kinetic mechanism of human histone acetyltransferase P/CAF. Biochemistry 39, 11961–11969, doi: 10.1021/bi001272h (2000). [DOI] [PubMed] [Google Scholar]
- 239.Tanner KG, Langer MR, Kim YJ & Denu JM Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. Journal of Biological Chemistry 275, 22048–22055, doi:DOI 10.1074/jbc.M002893200 (2000). [DOI] [PubMed] [Google Scholar]
- 240.Thompson PR, Kurooka H, Nakatani Y & Cole PA Transcriptional coactivator protein p300 - Kinetic characterization of its histone acetyltransferase activity. Journal of Biological Chemistry 276, 33721–33729, doi:DOI 10.1074/jbc.M104736200 (2001). [DOI] [PubMed] [Google Scholar]
- 241.Wapenaar H et al. Enzyme kinetics and inhibition of histone acetyltransferase KAT8. Eur J Med Chem 105, 289–296, doi: 10.1016/j.ejmech.2015.10.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Albaugh BN, Kolonko EM & Denu JM Kinetic Mechanism of the Rtt109-Vps75 Histone Acetyltransferase-Chaperone Complex. Biochemistry 49, 6375–6385, doi: 10.1021/bi100381y (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bintu L et al. Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724, doi: 10.1126/science.aab2956 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Katan-Khaykovich Y & Struhl K Dynamics of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Gene Dev 16, 743–752, doi: 10.1101/gad.967302 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Evertts AG et al. Quantitative Dynamics of the Link between Cellular Metabolism and Histone Acetylation. Journal of Biological Chemistry 288, 12142–12151, doi: 10.1074/jbc.M112.428318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Horiuchi KY et al. Assay Development for Histone Methyltransferases. Assay Drug Dev Techn 11, 227–236, doi: 10.1089/adt.2012.480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Duncan EM, Chitsazan AD, Seidel CW & Alvarado AS Set1 and MLL1/2 Target Distinct Sets of Functionally Different Genomic Loci In Vivo. Cell Reports 13, 2741–2755, doi: 10.1016/j.celrep.2015.11.059 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kerimoglu C et al. KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Reports 20, 538–548, doi: 10.1016/j.celrep.2017.06.072 (2017). [DOI] [PubMed] [Google Scholar]
- 249.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45, doi:Doi 10.1038/47412 (2000). [DOI] [PubMed] [Google Scholar]
- 250.Kouzarides T Chromatin modifications and their function. Cell 128, 693–705, doi: 10.1016/j.cell.2007.02.005 (2007). [DOI] [PubMed] [Google Scholar]
- 251.Becker JS, Nicetto D & Zaret KS H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet 32, 29–41, doi: 10.1016/j.tig.2015.11.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Musselman CA, Lalonde ME, Cote J & Kutateladze TG Perceiving the epigenetic landscape through histone readers. Nature Structural & Molecular Biology 19, 1218–1227, doi: 10.1038/nsmb.2436 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Verdin E & Ott M 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 16, 258–264, doi: 10.1038/nrm3931 (2015). [DOI] [PubMed] [Google Scholar]
- 254.Lauberth SM et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152, 1021–1036, doi: 10.1016/j.cell.2013.01.052 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Yin Y et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, doi: 10.1126/science.aaj2239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Andrews FH et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nature Chemical Biology 12, 396–U333, doi: 10.1038/nchembio.2065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Xiong XZ et al. Selective recognition of Histone crotonylation by double PHD fingers of MOZ and DPF2. Nature Chemical Biology 12, 1111–+, doi: 10.1038/Nchembio.2218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhao S, Yue Y, Li YY & Li HT Identification and characterization of ‘readers’ for novel histone modifications. Curr Opin Chem Biol 51, 57–65, doi: 10.1016/j.cbpa.2019.04.001 (2019). [DOI] [PubMed] [Google Scholar]
- 259.Jaffe JD et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nature Genetics 45, 1386–+, doi: 10.1038/ng.2777 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–+, doi: 10.1038/s41586-019-1186-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Li HX et al. The landscape of cancer cell line metabolism. Nat Med 25, 850–+, doi: 10.1038/s41591-019-0404-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Skene PJ, Henikoff JG & Henikoff S Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nature Protocols 13, 1006–1019, doi: 10.1038/nprot.2018.015 (2018). [DOI] [PubMed] [Google Scholar]
- 263.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, doi:ARTN e21856 10.7554/eLife.21856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ku WL et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nature Methods 16, 323–+, doi: 10.1038/s41592-019-0361-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communications 10, doi:ARTN 1930 10.1038/s41467-019-09982-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Pareek V, Tian H, Winograd N & Benkovic SJ Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368, 283–290, doi: 10.1126/science.aaz6465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gilmore IS, Heiles S & Pieterse CL Metabolic Imaging at the Single-Cell Scale: Recent Advances in Mass Spectrometry Imaging. Annu Rev Anal Chem (Palo Alto Calif) 12, 201–224, doi: 10.1146/annurev-anchem-061318-115516 (2019). [DOI] [PubMed] [Google Scholar]
- 268.Dai ZW & Locasale JW Understanding metabolism with flux analysis: From theory to application. Metabolic Engineering 43, 94–102, doi: 10.1016/j.ymben.2016.09.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kopinski PK et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. P Natl Acad Sci USA 116, 16028–16035, doi: 10.1073/pnas.1906896116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kungulovski G & Jeltsch A Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet 32, 101–113, doi: 10.1016/j.tig.2015.12.001 (2016). [DOI] [PubMed] [Google Scholar]
- 271.Stricker SH, Koferle A & Beck S From profiles to function in epigenomics. Nature Reviews Genetics 18, 51–66, doi: 10.1038/nrg.2016.138 (2017). [DOI] [PubMed] [Google Scholar]
- 272.Park M, Patel N, Keung AJ & Khalil AS Engineering Epigenetic Regulation Using Synthetic Read-Write Modules. Cell 176, 227–+, doi: 10.1016/j.cell.2018.11.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]