

Abstract
Laminopathies are rare diseases associated with mutations in LMNA, which encodes nuclear lamin A/C. LMNA variants lead to diverse tissue-specific phenotypes including cardiomyopathy, lipodystrophy, myopathy, neuropathy, progeria, bone/skin disorders, and overlap syndromes. The mechanisms underlying these heterogeneous phenotypes remain poorly understood, though post-translational modifications, including phosphorylation, are postulated as regulators of lamin function. We catalogued all known lamin A/C human mutations and their associated phenotypes, and systematically examined the putative role of phosphorylation in laminopathies. In silico prediction of specific LMNA mutant-driven changes to lamin A phosphorylation and protein structure was performed using machine-learning methods. Some of the predictions we generated were validated via assessment of ectopically expressed wild-type and mutant LMNA. Our findings indicate phenotype- and mutant-specific alterations in lamin phosphorylation, and that some changes in phosphorylation may occur independently of changes in lamin protein structure. Therefore, therapeutic targeting of phosphorylation in the context of laminopathies will likely require mutant- and kinase-specific approaches.
Keywords: laminopathy, intermediate filaments, mutation, post-translational modifications
Introduction
The lamin intermediate filament (IF) proteins are the primary structural proteins of the nuclear lamina. Their numerous functions include maintenance of nuclear shape and structure, transcription regulation, nuclear pore positioning, heterochromatin organization, and downstream pathways including differentiation, proliferation, and senescence (1-3). Lamins are type V intermediate filament proteins, subdivided into A- and B-types. Mutations in the genes encoding these proteins, the majority of which occur in LMNA (encoding lamin A/C), are associated with a group of rare heterogeneous diseases known as laminopathies. Laminopathies comprise a growing list of diseases with diverse tissue-specific phenotypes, including cardiomyopathies, lipodystrophies, muscular dystrophies, neuropathies, premature ageing syndromes (progeria), and bone/skin disorders.
The phenotypic diversity and tissue-specific nature of the laminopathies have driven extensive investigation into the mechanisms underlying these diseases. Two non-mutually exclusive putative mechanistic pathways have been postulated: 1) a “structural” pathway, whereby disrupted lamin protein structure leads to nuclear lamina fragility, nuclear deformation, and vulnerability to mechanical stresses; and 2) a “gene expression” pathway, whereby protein-protein interactions, heterochromatin organization, and genome-lamina contacts are altered, leading to changes in downstream gene expression (2, 4). The specific pathway invoked in each case is thought to depend on the location of the mutation within the structure of lamin A. Specifically, mutations in the helical rod domain tend to lead to cardiomyopathy and muscular dystrophy (“structural” pathway), while mutations in the tail or immunoglobulin (Ig)-like domain are associated with the lipodystrophy (“gene expression” pathway), though interplay between the pathways has also been surmised (4).
Still, the precise mediators of these potential pathways, particularly in light of potential therapeutic approaches, remain poorly understood. There is increasing evidence that post-translational modifications (PTMs) of IF proteins, including lamin A, have a major role in regulating their function, including structural aspects such as protein solubility and filament disassembly, as well as localization and partner protein interactions (5, 6). Phosphorylation in particular has been shown to have a significant role in regulating lamin A function, both in physiologic and pathologic contexts. Classically, lamin A phosphorylation occurs during mitosis, resulting in disassembly of the nuclear lamina (7, 8). More recent studies have also identified roles for interphase lamin A phosphorylation (9), including in regulation of lamin localization and distribution, protein-protein interactions, downstream signaling and transcription (10).
In the context of the laminopathies, available evidence primarily focuses on phosphorylation as part of a global stress response downstream of mutated lamin A. For example, in the LMNAH222P/H222P dilated cardiomyopathy mouse model, upregulation of the c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38α mitogen-activated protein kinase (MAPK) branches of the MAPK pathway has been demonstrated (11, 12). Furthermore, these stress-activated kinase pathways have shown promise as a therapeutic target: inhibitors of ERK1/2, JNK, and p38α have biochemical, phenotypic, and survival benefits in animal studies (13-17). In addition, the mammalian target of rapamycin (mTOR) kinase pathway, involved in such diverse cell functions as growth and proliferation, survival, and autophagy, has been implicated as a therapeutic target in lamin mutation-driven models of cardiomyopathy (18) and progeria (19).
In contrast, studies of direct lamin A phosphorylation and its potential role in laminopathies have been sparse. One study on phosphorylation in patients with Emery-Dreifuss muscular dystrophy (EDMD) versus healthy controls noted significantly decreased N-terminal phosphorylation of lamin A in the muscle cells of individuals with EDMD, though phosphorylation was similar between groups in fibroblasts (20). Meanwhile, the myopathy-associated mutations in the Ig-fold motif of lamin A have been associated with S458 phosphorylation, which is not present in other laminopathy types or in normal healthy controls (21).
To more comprehensively explore the precise roles of phosphorylation in the laminopathies, we compiled available data on all reported LMNA mutations with their associated phenotypes, as well as readily available clinical metadata such as sex and geographic origin. We subsequently performed in silico prediction of specific LMNA mutant-driven alterations in lamin A phosphorylation and protein structure. We then validated the in silico predictions using in vitro assessment of ectopically expressed wild-type and mutant LMNA phosphorylation. Our findings suggest disease- and mutant-specific alterations in lamin phosphorylation, and that some changes in phosphorylation may occur independent of changes in lamin protein structure. Therefore, therapeutic targeting of phosphorylation in the context of laminopathies may require a mutant- and kinase-specific approach.
Materials and Methods
Data Collection
LMNA mutation and phenotypic data were obtained from publicly available databases: the Human Intermediate Filament Database (22), the Universal Mutation Database (www.umd.be/LMNA), the UniProt Knowledgebase (23). Two additional reported mutations were provided by literature review of PubMed spanning 2017-2018 (24, 25). When available, additional clinical data were also collected, including patient sex and geographic origin. Additional nonsynonymous benign variant data were obtained from the Exome Aggregation Consortium (ExAC) (26). The workflow including data collection and computational analysis is summarized in Fig. 1. Additional data regarding domain assignment (start and end of lamin A domains) was obtained from UniProt (23). It should be noted that there is a lack of consensus in the literature regarding the residue position at which each domain should start and end, as several variations of domain position(s) have been reported (22, 23, 27-29). For our analyses, we have utilized UniProt’s domain designations consistently throughout. Importantly, there is disagreement among the field regarding the subdivision of coil 2 (into 2A, Linker 2, and 2B); based on recent literature (30), we have simply used coil 2 instead of subdivisions. Alignment of heptad repeats was performed using pooled references (23, 27, 28, 31).
Figure 1.
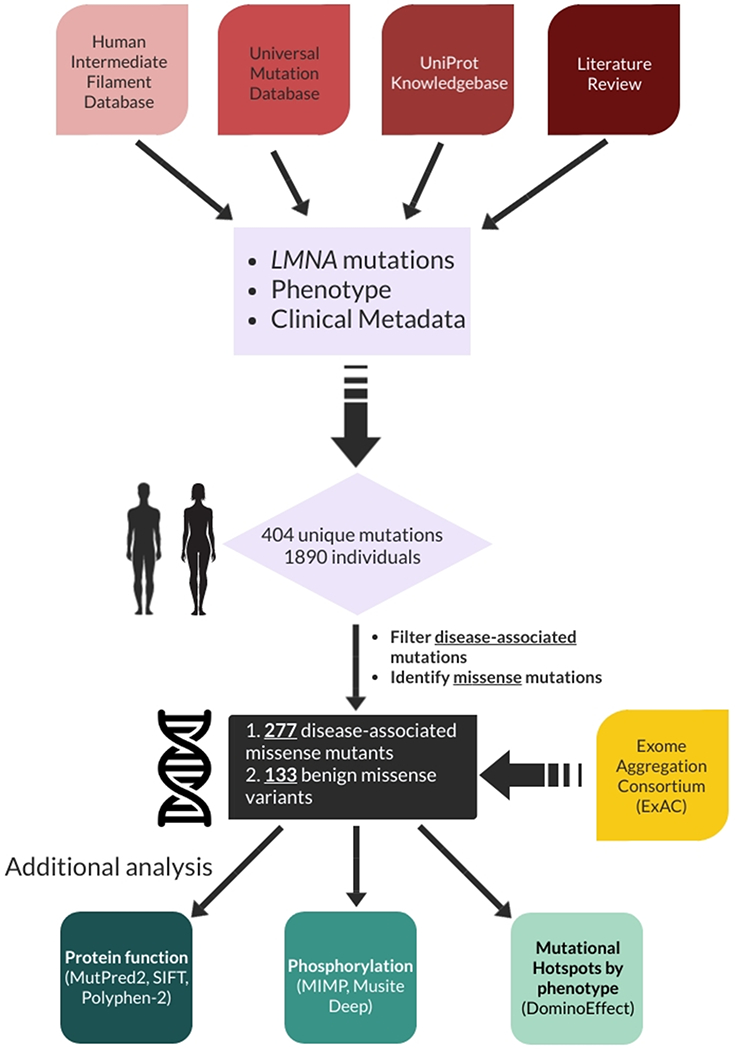
Schematic demonstrating overall workflow including source data acquisition, compilation, and analysis.
Protein structure and functional prediction
Mutational effects on protein structure and subsequent effects on function were performed using well-established, previously published software tools: MutPred2 (32), Sorting Intolerant from Tolerant (SIFT) (33), and Polymorphism Phenotyping v2 (PolyPhen-2) (34). MutPred2 is a machine-learning based method that integrates several protein parameters including metal-binding, structural stability, protein-binding, PTMs, and DNA-binding into its prediction algorithm. Meanwhile, SIFT and PolyPhen-2 rely on sequence homology and predicted alterations to protein structure in their algorithms. A “consensus prediction” between the 3 tools was obtained by identifying predicted protein disruptions that were consistent among all three tools with the highest confidence (see supplemental). The purpose of this was to eliminate potential effects on protein function due to predicted PTM alterations (only in MutPred2) and to minimize false positives, as computational tools have been shown to lack specificity in predicting significant protein disruption (35).
Phosphorylation prediction
Changes in lamin A phosphorylation due to mutational effects were predicted using two previously validated tools: 1) MIMP, a machine-learning algorithm developed originally for cancer networking analysis (36), and 2) MusiteDeep, a deep-learning algorithm that takes raw sequence data as input and uses convolutional neural networks to predict mutational impacts (37). MIMP works by constructing specificity models for kinases, which are then used to score phosphosites containing a mutation before and after the mutation to predict the impact it may have on phosphorylation (36). Training data for these algorithms were acquired from PhosphoELM (38), PhosphoSite Plus (39), HPRD (40, 41) and PhosphoNetwork (42). A consensus score, like the protein structural and functional prediction above, was created to minimize false positives.
In contrast to MIMP, MusiteDeep does not have a native algorithm to identify changes between native and mutant protein phosphorylation. Thus, native and mutant protein phosphorylation predictions were determined separately for each kinase (CDK, PKC, PKA, MAPK, CK2, Other—encompassing all other trained kinases), via input of an amino acid sequence. The resulting predicted phosphorylation scores (represented as a probability between 0 and 1) were then compared between the specific mutant sequence and the native sequence, and an overall likelihood of change (loss or gain) was determined via a z-score calculation (difference between mutant and native likelihood of phosphorylation divided by the standard deviation of phosphorylation scores in the native sequence). A ∣z-score∣ >= 1 was determined to be a probable change in phosphorylation status, as long as either the mutant or native absolute score was >=0.5 (threshold indicating that there is probable phosphorylation). As an example, if the standard deviation of native sequence MusiteDeep scores is 0.25, only a change of >=0.25 with at least one of the mutant or native values >=0.5 overall would yield a predicted change in phosphorylation.
Consensus scoring
A consensus score was calculated for both protein structural/functional and phosphorylation predictions. For protein structure/function, SIFT, PolyPhen-2, and MutPred2 were utilized for prediction. For each of these tools, there are different levels of confidence for each prediction. For SIFT, the native score assigns a "significance" between 0 and 1, and <0.1 was assigned a score of 1 (for "possible" damage) and <0.05 was assigned a score of 2 ("probable" damage). For PolyPhen-2, the HumDiv-trained model was used, with 10% false positive rate (fpr) given a score of 1 and 5% fpr given a score of 2. Finally, for MutPred2, a threshold of 0.68 (yields a fpr of 10%) was given a score of 1, and 0.80 (fpr 5%) was given a score of 2. These scores were summed (for a maximal score of 6), then converted to a consensus score between 0 and 1 (0-2 = 0; 3-4 = 1; 5-6 = 2). For phosphorylation prediction, a predicted change as indicated by either MIMP or MusiteDeep yielded a score of 1 (no change = 0). These 2 predictions were then converted to a consensus score between 0 and 1 by taking a mean (i.e. neither predicting a change = 0, one predicting change = 1, both predicting change = 2).
Hotspot analysis
To systematically and statistically define hotspots, the R package DominoEffect was utilized (43). DominoEffect uses a poisson distribution to systematically identify hotspots given a set of parameters such as window (length of peptide sequence considered) and threshold (the minimum proportion of total mutations represented by a single mutation). Hotspots were defined according to disease phenotype (e.g., hotspots for lipodystrophy were determined only using mutations associated with lipodystrophy).
Protein structural modeling
The protein structural predictions were supplemented with modeling and visualization of select mutations via DynaMut (44). There is no complete crystal structure of lamin A/C, so instead fragments including part of the globular tail domain and the coiled-coil domain were used. .PDB files were obtained from the RCSB Protein Data Bank (45). PDB 1IFR was used for the globular tail domain (46), and PDB 6JLB was used for the coil 2 of the rod domain (28).
Plasmids, transfection, and cell culture conditions
Human LMNA open reading frame was purchased from Origene (Rockville, MD) and subcloned into the pCMV6 vector with amino-terminal myc/DDK tag (Origene) as described (47). LMNA mutants were generated using the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Lipofectamine-2000 (Life Technologies, Carlsbad, CA) was used for transfection of human hepatoma Huh7 cells (American Type Culture Collection, Manassas, VA), which were maintained at 37 °C, 5% CO2, and plated at ~70% confluency one day prior to transfection. Where indicated, cells were treated with 500nM okadaic acid (Cayman Chemical, Ann Arbor, MI) for 1h prior to harvest.
Cell lysis and SDS-PAGE
Cells were solubilized in Triton lysis buffer (1% Triton X-100, 50mM Tris pH 8.0, 150mM NaCl, protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO)) or RIPA lysis buffer (50 mM Tris pH 8, 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail) as indicated. Where indicated, phosphatase inhibitors were added to lysis buffer: 1mM sodium orthovanadate, 1mM β-glycerophosphate, 1mM NaF. After centrifugation (12,000xg, 10min, 4°C), the supernatant protein concentration was determined (bicinchoninic acid assay, Thermo Fisher, Waltham, MA). The supernatant was then used for immunoprecipitation of tagged LMNA or diluted in Novex 2X Tris-glycine sample buffer with SDS (Invitrogen, Carlsbad, CA) with 2% β-mercaptoethanol for SDS-PAGE. For solubility experiments, the Triton-insoluble pellet was solubilized in SDS-containing sample buffer (95°C, 5min). Proteins were resolved using 4-20% Novex Tris-glycine gels (Invitrogen) and visualized by immunoblotting or Coomassie staining.
Immunoprecipitation and immunoblotting
Two days after transfection with myc/DDK-tagged LMNA, Huh7 cells were solubilized in RIPA lysis buffer (4°C, 30min), and tagged LMNA was immunoprecipitated using pooled anti-myc (Abcam, Cambridge, MA) and anti-FLAG (Sigma-Aldrich) antibodies and Dynabeads protein G (Invitrogen). Beads were washed with RIPA buffer followed by PBS, then stored at 4°C prior to in vitro kinase assay. For immunoblot analysis, lysates were resolved via SDS-PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA) for immunoblotting. Where indicated, data were quantified by densitometry using ImageJ version 1.52a.
In vitro kinase assays
The p38 kinase assay was performed per manufacturer guidelines (R & D Systems, Minneapolis, MN). Briefly, 100 ng recombinant p38α (R & D Systems; diluted to 10 ng/μl in 5 mM MOPS, pH 7.2, 5 mM MgCl2, 1 mM EGTA, 0.4 mM EDTA, 0.05 mM DTT, 2.5 mM β-glycerolphoshate, 40 ng/ml BSA) was added to beads with immunoprecipitated LMNA. Subsequently, 5 μl of water was added followed by 5 μl of [γ-32P] ATP assay cocktail (0.5μCi/μl [γ -32P]ATP (Perkin Elmer, Waltham, MA), 0.1mM ATP, 25mM MOPS pH 7.2, 25mM MgCl2, 5mM EGTA, 2mM EDTA, 0.25mM DTT, 12.5mM β-glycerophosphate), and the reaction was incubated at 30°C for 30 minutes. PKCα kinase assay was performed as described (48). Briefly, beads with immunoprecipitated LMNA were incubated with 50 ng PKCα (Abcam) and 5 μCi of [γ-32P]ATP (30°C, 30 min) in PKCα kinase buffer (20mM Tris pH 7.5, 5mM MgCl2, 0.2 mM CaCl2, 5 μg/mL phosphatidylserine, 0.5 μg/mL diolein, 5μM ATP). Kinase reactions were stopped by adding Laemmli buffer and heating (95°C< 4 min), then resolved via SDS-PAGE followed by autoradiography. Quantitation was performed by densitometry using ImageJ version 1.52a.
Statistical analysis
Statistical significance for comparisons of categorical distribution (e.g., domain) among phenotypes were performed using χ2 and Fisher’s exact tests (depending on the number of zero or low values). Hotspot vs. non-hotspot comparisons were analyzed with a two-proportions test. Hierarchical clustering and heatmap construction were performed by ComplexHeatmap (49). Numerical comparisons (e.g., likelihood score for phosphorylation change) were analyzed using non-parametric tests including Mann-Whitney and Kruskal-Wallis tests given lack of normality assumption (ordinal variables). Multiple comparisons were corrected for using the false discovery rate (FDR) method. For biochemical assays, two-tailed Student’s t-test or paired ANOVA (Friedman test) followed by multiple comparison adjustment using uncorrected Dunn’s test was used, as indicated. These tests were implemented using R and GraphPad Prism.
Results
Mutational patterns in the LMNA gene
To identify mutational patterns associated with the laminopathies, all available data on LMNA mutations were collected from publicly available databases and the medical literature (see Methods and Supplemental Table 1). In total, 399 disease-associated mutations encompassing 1619 individuals were identified (Fig. 2A), with an additional five mutations (not included in the 1619 total) recorded in available repositories that did not have a clear associated disease phenotype. Demographics of these 1619 patients are summarized in Table 1. The distribution of mutations and hotspots appeared to differ by phenotype (Fig. 2A). Of these disease-associated mutations, 369 (92.5%) were within coding regions of the LMNA gene, and 30 were intronic. There were 277 (75.1%) coding region missense mutations, with the remainder comprising other types of mutations such as frameshift, nonsense, and deletion/insertion (Fig. 2B). Notably, missense mutations occupied a larger proportion of disease-associated LMNA mutations compared to what had been previously observed in the Human Gene Mutation Database, a global analysis of all known disease-associated mutations in the human genome (Supplemental Fig. 1).
Figure 2. Observed disease-associated mutational patterns in LMNA.

A) Map depicting all known LMNA mutations distributed along the lamin A backbone (top: yellow bar depicts mature lamin A backbone, with the hashed -CaaX portion indicating pre-processed prelamin A). Black bars depict the positions of known mutations by phenotype, scaled in intensity by increasing number of individuals at that particular position. Taller red bars indicate identified hotspots via DominoEffect by phenotype. B) Summary of observed mutations in coding and non-coding (intronic) regions of LMNA (left), then subdivided by mutation type (right). Missense mutations are the most common LMNA mutation. C) Sex distribution differs significantly by phenotype (χ2 = 79.58, df = 5, p<0.0001). D) The distribution of mutation type differs significantly by phenotype (χ2 = 235.4, df = 10, p<0.0001). E) Mutations fall into different lamin A functional domains, depending on phenotype. There is a significant difference observed between groups (χ2 = 513.0, df = 10, p<0.0001). F) Distribution of mutation status (heterozygosity vs homozygosity) varies significantly by phenotype (Fisher’s exact test, simulated with B=1.0e+07: p<0.0001).
Table 1. Characteristics of laminopathy patients and the lamin mutations they harbor.
The table summarizes the disease phenotype, sex distribution, mutation status, and the type of mutation of the laminopathy patients we analyzed. “Other” mutations include any mutations that are not missense or intronic, such as frameshift, deletion, insertion, and nonsense mutations.
| Sex | Mutation status | Mutation Type | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phenotype | Male | Female | Unknown | Heterozygous | Compound Heterozygous |
Homozygous | Missense | Intronic | Other | |
| Striated | Myopathy | 21 (44%) | 27 (56%) | - | 46 (96%) | 2 (4%) | - | 43 (90%) | - | 5 (10%) |
| Cardiomyopathy | 250 (50%) | 203 (41%) | 46 (9%) | 494 (99%) | 4 (1%) | - | 289 (58%) | 38 (8%) | 172 (34%) | |
| Both | 233 (49%) | 200 (42%) | 44 (9%) | 465 (97%) | 6 (1%) | 6 (1%) | 329 (69%) | 38 (8%) | 110 (23%) | |
| Total | 504 (49%) | 430 (42%) | 90 (9%) | 1005 (98%) | 12 (1%) | 6 (1%) | 661 (65%) | 76 (7%) | 287 (28%) | |
| Lipodystrophy/Metabolic | 95 (28%) | 237 (69%) | 9 (3%) | 302 (89%) | 7 (2%) | 32 (9%) | 328 (96%) | 2 (1%) | 11 (3%) | |
| Neuropathy | 29 (63%) | 16 (35%) | 1 (2%) | 2 (4%) | - | 44 (96%) | 45 (98%) | - | 1 (2%) | |
| Progeria | 50 (41%) | 51 (41%) | 22 (18%) | 112 (91%) | 4 (3%) | 7 (6%) | 45 (37%) | 5 (4%) | 73 (59%) | |
| Overlap | 24 (31%) | 51 (65%) | 3 (4%) | 71 (91%) | 2 (3%) | 5 (6%) | 69 (88%) | - | 9 (12%) | |
| Other | 3 (43%) | 4 (57%) | - | 4 (57%) | - | 3 (43%) | 2 (29%) | 1 (14%) | 4 (57%) | |
| Total | 705 (43%) | 789 (49%) | 125 (8%) | 1496 (92%) | 25 (2%) | 97 (6%) | 1150 (71%) | 84 (5%) | 385 (24%) | |
Sex and geographic distribution differ by phenotype
Significant differences in the distribution by sex were observed among phenotypes (Fig. 2C). Specifically, lipodystrophy and metabolic disorders showed a female-skewed sex distribution, which was also observed in the overlap group. Males were relatively overrepresented in the neuropathy group. In contrast, relatively equal male-female distribution was observed in the other phenotypes. In contrast to the human situation, hepatocyte-specific null mice show a male predominance of developing spontaneous fatty liver disease as the mice age, and marked predisposition to steatohepatitis and fibrosis when challenged with a high fat diet (50).
Available data on geographic origin showed that most reported disease originated in Europe (Supplemental Table 2). The neuropathy phenotype, driven by those with Charcot-Marie-Tooth disease, mostly originated in Africa and was virtually absent from other geographic regions. The Americas showed a greater than expected number of patients with progeria. Meanwhile, the precise distribution of geographic origin among individuals with lipodystrophy is frequently not reported.
Phenotypic association with mutation type, protein domain and heterozygosity
In our comprehensive analysis of all available laminopathy genotype and phenotype data, striated muscle laminopathies (affecting either cardiac or skeletal muscle) and premature aging syndromes were associated with a greater proportion of non-missense (“other”) and intronic mutations than other phenotypes (Fig. 2D). In contrast, lipodystrophies, neuropathies, and bone/skin diseases were dominated by missense mutations.
Furthermore, striated muscle laminopathies and neuropathies were predominantly driven by mutations in the rod domain, while lipodystrophies, disorders affecting bone and skin [mandibuloacral dysplasia (MAD)], and premature aging syndromes were generally associated with mutations affecting the tail domain (Fig. 2E). Overlap syndromes (a single individual with two or more documented phenotypes, e.g. dilated cardiomyopathy and lipodystrophy simultaneously) had a relatively even distribution of mutations.
Laminopathy patients were most likely to have heterozygous LMNA mutations, with a small portion (25/1619 = 1.5%) demonstrating compound heterozygosity. Intriguingly, patients with Charcot-Marie-Tooth (CMT) neuropathy and mandibuloacral dysplasia were predominantly homozygous for their associated mutations (Fig. 2F)—driven mostly by their hotspots, R298C and R527H, respectively.
Disease-associated mutations frequently occur at specific amino acid residues and locations
Most commonly, disease-associated mutations resulted in a loss of a native arginine, followed by leucine, glutamic acid, and alanine (Fig. 3A). These most frequently led to a gain of proline, lysine, cysteine, histidine, and valine (Fig. 3A). When these changes were correlated with phenotype, the loss of a native leucine, glutamate, or alanine most frequently led to a striated muscle laminopathy phenotype, whereas loss of arginine uniquely led to a disproportionate number of patients with a lipodystrophy and neuropathy phenotype (Supplemental Fig. 2A). Meanwhile, loss of proline almost exclusively led to a striated muscle laminopathy phenotype, while gain of valine, histidine, or cysteine resulted in more heterogenous disease manifestations (Supplemental Fig. 2B).
Figure 3. Patterns of lost or gained amino acids in disease-associated missense mutations.

A) Shown are the absolute number of changes by amino acid residue (bars), with native (black) residues indicating those that are lost as a result of mutation, while mutant (gray) residues represent those that result from mutation. Specific residues are highlighted in red (loss) or blue (gain) if the absolute difference between native and mutant is at least 10. B) Schematic of coiled-coil dimerization based on heptad repeats, with example heptad from lamin A (residues 106-112), constructed with R package helixvis (71). Residues are lettered a-g to represent positions within the heptad, with colors representing charge/polarity (red: negative charge, blue: positive charge, yellow: neutral polar). C) Frequency of patients with mutations at a location within the rod domain corresponding to a heptad repeat, by position within the heptad. There is a significant difference among a-g (χ2 goodness of fit test: χ2= 222.0, df = 6, p<0.0001). D) Heatmap displaying phenotypes with at least 10 mutations corresponding to a heptad repeat. The color scale represents the number of patients with that phenotype that have a mutation corresponding to that position (a-g) within a heptad. DCM-CD, dilated cardiomyopathy with conduction disease; AR-CMT2, autosomal recessive Charcot-Marie-Tooth neuropathy type 2; LGMD1B, limb-girdle muscular dystrophy type 1B; L-CMD, LMNA-related congenital muscular dystrophy; FPLD, familial partial lipodystrophy; EDMD, Emery-Dreifuss muscular dystrophy.
We then considered how these mutations localized, particularly as it pertains to lamin dimer- and tetramerization, which may be adversely affected by mutations in the coiled-coil rod domain. Comprising this coiled-coil are a series of heptad repeats (Fig. 3B), which allow for stabilizing interactions between monomers. Intriguingly, disease-associated mutations more frequently occurred in the e and g positions of the heptad repeat, which are commonly form an oppositely charged ion pair (Fig. 3C). We then analyzed the phenotypes with the most mutations in the rod domain and noted distinct patterns in mutation frequency per position in the heptad repeat (Fig. 3D). In particular, dilated cardiomyopathy with conduction disease (DCM-CD) had relative parity among 6/7 heptad positions, whereas limb-girdle muscular dystrophy type 1b (LGMD1B), LMNA-related congenital muscular dystrophy (L-CMD), and Emery-Dreifuss muscular dystrophy (EDMD) showed a predominance of mutations at the e position. Curiously, the overlap syndrome DCM-CD and familial partial lipodystrophy (FPLD) showed a pattern more similar to the other skeletal muscular dystrophies and not DCM-CD alone.
Hotspot mutations account for the majority of patients with laminopathy
Hotspot mutations, residues with particularly high mutation rate, were found to be distributed throughout the entire protein, but were found to cluster by phenotype (Fig. 2A). There were 35 total hotspot residues encompassing 968/1619 (60%) patients with laminopathies (Fig. 4A). When associated with phenotype, hotspots formed 3 primary clusters, driven largely by the presence of either predominant adipose and cardiac involvement, predominant cardiac involvement, or relatively equal cardiac and skeletal muscle involvement (Fig. 4A). Hotspots primarily affected charged or non-polar native amino acid residues (31/35 = 94%), with arginine being the most commonly affected residue (16/35 = 46%). Relative to non-hotspot mutations, hotspots were most often driven by C to T transition mutations (Fig. 4B). Of missense mutations, analysis of amino acid transitions in hotspots versus non-hotspots revealed that hotspots were more commonly driven by native arginine residues mutated to a non-charged amino acid residue, while non-hotspots were more frequently leucine to proline mutations (Fig. 4C).
Figure 4. Hotspot mutations are associated with specific phenotypes and are driven by certain nucleotide and amino acid changes.

A) Heatmap showing hotspot mutations identified via DominoEffect and the relative number of individuals with involvement of each organ system/phenotype. B) Heatmap depicting differences in the proportion of total mutations in each group (hotspot vs. non-hotspot) represented by each specified nucleotide base change. There was a significant difference in C to T transition mutations favoring hotspot mutations (Two-proportions test, hotspots vs. non-hotspots: 0.3279 vs. 0.1347, adjusted p=0.0087). C) Heatmap depicting the proportion of total missense mutations per group (hotspot vs. non-hotspot) represented by each amino acid mutation. There were no significant individual amino acid changes after p-value correction for multiple testing. However, when grouped, mutations involving native arginine residues were found to be highly significant favoring hotspots (Two-proportions test, hotspots vs. non-hotspots: 0.592 vs. 0.202, adjusted p<0.0001), and mutations involving native leucine residues approached significance favoring non-hotspots (Two-proportions test, hotspots vs. non-hotspots: 0 vs. 0.145, adjusted p=0.095).
LMNA mutations can lead to overlapping phenotypes in certain individuals
Several mutations were found to affect multiple organ systems. There were 42 mutations leading to overlap syndromes (Fig. 5A), in which a single individual had multiple involved organ systems (e.g., both lipodystrophy and cardiomyopathy in a single patient). To identify phenotypic patterns among these mutations, hierarchical clustering was performed, revealing at least 6 distinct clusters. These clusters varied largely by the relative dominance (or lack thereof) of a single or pair of phenotypes. For example, cluster 1 was driven by the R482W familial partial lipodystrophy (FPLD) hotspot, which was found to only occasionally overlap with a limb girdle muscular dystrophy (LGMD) phenotype. Cluster 2 was driven by the CMT hotspot, R298C, with occasional striated muscle involvement. Cluster 3 demonstrated strong striated muscle involvement, with rare overlap of a lipodystrophy phenotype. Cluster 4 was characterized by a predominant lipodystrophy phenotype with relatively isolated bone/skin or cardiac disease. The remaining clusters demonstrated relatively equal overlap between organs—cluster 5 showed a consistent lipodystrophy phenotype with varying degrees of striated muscle involvement, while cluster 6 showed a persistent striated muscle phenotype combined with either neuropathy or progeroid syndromes.
Figure 5. LMNA mutations can be pleiotropic, sometimes causing overlapping phenotypes, disparate individual phenotypes, or both.

A) Heatmap displaying mutations leading to overlap syndromes (multiple organ system involvement in the same individual), with the relative involvement of each organ system/phenotype. Hierarchical clustering was performed on the mutations, demonstrating 6 distinct clusters, which are labeled. B) Heatmap depicting mutations leading to multiple, disparate syndromes (distinct organ involvement in different individuals), with the relative involvement of each organ system/phenotype. Hierarchical clustering was performed on the mutations, demonstrating 4 distinct clusters, which are labeled. C) Heatmap depicting mutations found in both (A) and (B), representing mutations that can lead to overlapping and multiple disparate phenotypes, with relative involvement of each organ system.
Single LMNA mutations can lead to multiple disparate phenotypes
In addition to the overlap syndromes, 16 mutations were found to lead to separate, disparate phenotypes, such that certain individuals had a phenotype affecting one organ system, while others with the same mutation had phenotypes affecting a different organ in a non-overlapping manner (e.g., some patients with isolated cardiac disease, others with isolated neuropathy). To identify phenotypic patterns among these mutations, hierarchical clustering was again performed, revealing 4 primary clusters (Fig. 5B). Clusters 1 and 2 were driven by hotspots. First, the G608G splicing Hutchinson-Gilford progeria syndrome (HGPS) hotspot mutation was noted to separately lead to a full progeroid syndrome versus relatively isolated skin involvement (restrictive dermopathy). Second, the R298C CMT hotspot led to either neuropathy or isolated cardiac disease. Cluster 3 included mutations associated with a range of phenotypic groups, without a dominant phenotype, while cluster 4 showed predominantly striated or cardiac muscle with lipodystrophy phenotypes.
Certain LMNA mutations are associated with both overlapping and disparate phenotypes
Seven mutations (6 missense, 1 nonsense) were associated with both overlapping and disparate phenotypes (Fig. 5C). Other than the R298C CMT hotspot, discussed above, and the R28W hotspot, which is associated with both lipodystrophy and striated muscle disease, these mutations were characterized by involvement of at least 3 organ systems. Interestingly, the R644C mutation was found to encompass all known major organ phenotypes of the laminopathies, other than isolated bone or skin disease.
Disease-associated mutations are predicted to alter lamin phosphorylation
To explore the mechanistic basis of the genotype-phenotype correlation in the laminopathies, we utilized a machine-learning based, computational approach to predict the direct impacts of specific missense mutations on lamin phosphorylation (see Methods for details on the tools used). As a control, “benign” variants lacking severe pediatric disease were obtained from the ExAC database. Relative to control, disease-associated mutations were predicted to be more likely to disrupt lamin phosphorylation (Fig. 6A). This was largely driven by a relative loss of phosphorylation (e.g., destruction of a kinase motif), while predicted gains in phosphorylation were rare and did not differ between pathogenic and benign groups (Supplemental Fig. 3A). Predicted changes in phosphorylation were mostly associated with mutations in the head or tail domains of the lamin A/C protein, with a relative paucity in the rod domain (Fig. 6B).
Figure 6. Predicted LMNA mutation-driven phosphorylation and protein structural changes are associated with disease.

A) Relative to variants restricted to asymptomatic individuals, disease-associated mutations have a significantly increased association with predicted phosphorylation changes (left, Mann-Whitney test: p=0.0004) and protein structural changes (right, Mann-Whitney test: p<0.0001), by likelihood score (see Materials and Methods). B) Left: Predicted phosphorylation changes are more likely in head and tail domains than the rod domain (Kruskal-Wallis test: p<0.0001; Dunn’s multiple comparisons test: head vs. rod, adjusted p=0.001; head vs. tail, adjusted p=0.002; tail vs. rod, adjusted p<0.0001). Right: Predicted changes in protein structure are associated with specific lamin domains (Kruskal-Wallis test: p<0.0001; Dunn’s multiple comparisons test: rod vs. tail, adjusted p<0.0001). C) Left: Predicted changes in phosphorylation differ by phenotype (Kruskal-Wallis test: p<0.0001). Striated laminopathies had significantly fewer predicted phosphorylation changes relative to the benign, asymptomatic ExAC group (Dunn’s multiple comparisons test: adjusted p=0.013). Lipodystrophy/metabolic disease, neuropathy and bone/skin disease had significantly greater predicted phosphorylation changes compared to the benign group (Dunn’s multiple comparisons test: lipodystrophy vs. benign, adjusted p<0.0001; neuropathy vs. benign, adjusted p<0.0001; bone/skin vs. benign, adjusted p<0.0001). Right: Predicted changes in protein structure differ by phenotype (Kruskal-Wallis test: p<0.0001). Striated laminopathies and neuropathies had significantly greater predicted protein structural changes relative to the benign ExAC group (Dunn’s multiple comparisons test: striated vs. benign, adjusted p<0.0001; neuropathy vs. benign, adjusted p<0.0001). D) Hotspots were associated with a greater degree of predicted phosphorylation changes compared to non-hotspot mutations (Mann-Whitney test: p<0.0001), but the same pattern was not observed in predicted protein structural changes (Mann-Whitney test: p=0.7897). E) Left: Penetrance (defined as the percentage of individuals with a specific variant that carry any disease) is associated with predicted phosphorylation status (e.g., loss or gain of phosphorylation) (Kruskal-Wallis test: p=0.009). This was specifically driven by a difference in the group predicted to lose phosphorylation, which was associated with decreased penetrance (Dunn’s multiple comparisons test: loss vs. no change, adjusted p=0.005). Right: Penetrance is associated with predicted likelihood of protein structural changes (Kruskal-Wallis test: p=0.0004). This was specifically driven by a difference between the unlikely group (score = 0) and the likely group (score = 1) (Dunn’s multiple comparisons test: unlikely vs. likely, adjusted p=0.0002).
Predicted lamin phosphorylation changes are associated with phenotype, hotspots, and penetrance
Certain phenotypes were associated with a greater likelihood of predicted phosphorylation changes. Lipodystrophy, neuropathy, and bone/skin groups were associated with a significantly increased likelihood of lamin phosphorylation change than the benign group (Fig. 6C). Striated muscle laminopathies, in contrast, were associated with fewer predicted phosphorylation changes than benign variants (Fig. 6C). These changes were driven largely by predicted loss of phosphorylation in the lipodystrophy, neuropathy, and bone/skin groups (Supplemental Fig. 3B).
Hotspot mutations were significantly more likely to be predicted to cause a change in lamin phosphorylation than other, non-hotspot mutations (Fig. 6D). Interestingly, phenotype penetrance appeared to correlate specifically with a predicted loss of phosphorylation (Fig. 6E).
In total, the phosphorylation prediction tools we utilized agreed on 207/277 (74.7%) mutations. They agreed on a phosphorylation change on 30 mutations, with complete agreement on the putative kinase family involved on 28 mutations (Supplemental Table 3). Notably, the most commonly lost kinase motif belonged to protein kinase C (PKC), with other lost motifs including protein kinase A (PKA), cyclin-dependent kinase (CDK), MAPK, and casein kinase 2 (CK2). Hotspots for CMT, FPLD, and MAD (R298C, R482W/Q/L, R527C/H) were all specifically associated with a predicted loss of PKC-mediated phosphorylation. The most commonly gained kinase families were CDK and MAPK—both invoked by the same mutations—with other gained kinases including PKA, Akt, and CK2.
Disease-associated mutations are predicted to alter protein structure
To investigate the possibility of alternative mechanisms of disease, we used several well-established computational tools (MutPred2, SIFT, PolyPhen-2) to predict the impact of missense mutations on protein structure, folding and function. Some select mutations were also modeled using DynaMut to further verify protein structural changes (Supplemental Fig. 4). When compared to benign variants from the Exome Aggregation Consortium (ExAC) database, disease-associated mutations were significantly more likely to be associated with predicted structural changes (Fig. 6A). In addition, prediction of altered protein structure was predicted to be significantly more likely in the rod domain relative to the tail (Fig. 6B), though there was no significant difference between the rod and head domains. Striated laminopathy and neuropathy were both associated with an increased likelihood of altered protein structure relative to the benign group (Fig. 6C), but notably this was not seen in other phenotypes. In contrast to phosphorylation predictions, predicted structural change was not more likely to segregate with hotspot mutations (Fig. 6D). Predicted protein structural changes were associated with disease penetrance, with higher likelihood of change associated with higher penetrance (Fig. 6E).
Predicted lamin phosphorylation and structural changes independently associate with phenotype
When predicted protein structural changes were compared to predicted phosphorylation changes, no significant correlation was observed (Fig. 7A). There was significant discordance (Fig. 7B), such that mutations tended to be predicted to induce either structural or phosphorylation changes, but not both. Intriguingly, different phenotypes had different patterns of predicted effects on phosphorylation and protein structure (Fig. 7C, D). While striated laminopathies primarily showed a relative dominance of protein structural changes, lipodystrophy and bone/skin diseases showed a large proportion of individuals with predicted changes in phosphorylation. Interestingly, patients with neuropathy (mostly hotspot R298C) were predicted to have both protein structural and phosphorylation changes as a result of their mutation.
Figure 7. Predicted LMNA mutation-driven phosphorylation and protein structural changes appear to represent separate processes.

A) Map depicting LMNA missense mutations, predicted phosphorylation changes, predicted protein structural changes along the lamin A protein backbone. Predicted phosphorylation changes are depicted in shades of orange, with the darker shade representing high confidence predictions (see Methods). Predicted protein structural changes/disruption are shown in shades of green, with darker shades indicated higher confidence. Red bars in the discordant row represent amino acid residues at which a missense mutation leads to a predicted change in one property, such as phosphorylation, but not the other (e.g., a specific mutation leads to a predicted change in phosphorylation without a corresponding change in protein structure). B) The distribution of missense mutations depending on discordance reveals greater than half of studied missense mutations lead to discordant predictions. A greater proportion of discordant mutations was represented by those with an isolated predicted protein structural change than with an isolated predicted phosphorylation change. C) Heatmap depicting relative contribution of changes in phosphorylation (yellow) and protein structure (blue), by individual patient, normalized to sample size per phenotype. Brighter colors represent a stronger contribution. Hotspots appear as larger, homogenous blocks of color since predictions will not vary among individuals with the same hotspot mutation. D) Summary of panel B results showing the average contributions of predicted phosphorylation or protein structural changes for each laminopathy category phenotype.
Selected lamin mutants demonstrate changes in phosphorylation in vitro
To further explore the effects LMNA mutation on phosphorylation as suggested by in silico prediction, we used site-directed mutagenesis to generate LMNA-variant constructs for selected hotspot mutations: T10I, R60G, and R482Q. We transiently transfected human hepatoma Huh7 cells with wild-type (WT) or mutant LMNA, followed by performing an in vitro kinase assay using immunoprecipitated WT or mutant lamin A as substrate. Notably, p38 MAPK, previously demonstrated to mediate downstream effects in cardiomyopathy (12), showed significantly decreased activity on the R60G mutant compared to WT LMNA, with a similar trend found for the T10I mutant (Fig. 8A). However, PKCα, which is known to directly interact with lamin A/C (51), did not show significant mutation-driven changes in activity. These data support kinase- and mutation-specific effects on lamin phosphorylation, consistent with our in silico prediction.
Figure 8. The disease-associated R60G mutation alters LMNA phosphorylation in vitro and reduces LMNA solubility.

(A) Huh7 cells were transfected with myc/DDK-tagged wild-type (WT) LMNA or the indicated LMNA mutant, then harvested in RIPA buffer followed by immunoprecipitation of LMNA using antibodies directed against the myc/DDK tag. Top left panel, WT and mutant LMNA were immunoprecipitated from cell lysates and analyzed by SDS-PAGE followed by immunoblot; tagged LMNA is indicated with an arrow. The immunoglobulin heavy chain (IgH) is labeled, and a non-specific band is indicated via arrowhead. Top right panel, in vitro phosphorylation of immunoprecipitated LMNA was carried out using recombinant p38 (top) or PKCα (bottom) followed by SDS-PAGE and autoradiography; 32P signal corresponding to myc/DDK-LMNA is indicated by an arrow, with an arrowhead indicating a non-specific band. Bottom panels, 32P-LMNA signal was quantitated relative to LMNA input using ImageJ software. Data shown were derived from n=3 independent experiments, and error bars represent standard error of the mean (SEM). Pairwise comparison using Student’s t test was used to determine statistical significance; *, P<0.05. (B) Top panels, Huh7 cells were transfected with myc/DDK-tagged WT LMNA or the indicated LMNA mutant, treated with 500 nM okadaic acid for 1 hour at 37 °C where indicated, and harvested in 1% Triton X-100 lysis buffer containing protease and phosphatase inhibitors. The Triton-insoluble pellet was then boiled in SDS buffer, and Triton-soluble and insoluble (pellet) fractions were analyzed by SDS-PAGE followed by immunoblot using the indicated primary antibodies. Coomassie-stained gels are shown to demonstrate equal protein loading. Myc/DDK-tagged LMNA is indicated via arrow; arrowhead indicates a non-specific band. Bottom panels, the soluble proportion of tagged LMNA was quantitated via ImageJ relative to WT LMNA under basal conditions, with WT LMNA under basal conditions arbitrarily set to 1.0. Data shown were derived from n=3 independent experiments, and error bars represent standard error of the mean (SEM). For each condition (basal and okadaic acid-treated), paired Friedman ANOVA was performed to compare WT to mutant LMNA, followed by uncorrected Dunn’s test for multiple comparison adjustment to determine statistical significance; *, P<0.05. M.W., apparent molecular weight; kDa, kilodaltons; IP, immunoprecipitate.
To explore whether the phosphorylation changes predicted in silico and observed in vitro might also occur in cells expressing mutant lamin A, WT and mutant lamin A were ectopically expressed in Huh7 cells, and cells were then treated with okadaic acid, a potent phosphatase inhibitor that regulates lamin phosphorylation (52). Immunoblotting of cell lysates with both phospho-specific antibodies and an antibody directed to the myc tag (detecting total lamin A) showed that after treatment with okadaic acid, there was an increase in phosphorylation of WT lamin A at S22 and S392 as well as an increase in WT lamin A solubility, as shown by an increase in the protein fraction found in supernatant (soluble fraction) compared to the pellet (insoluble fraction) (Fig. 8B). An increase in phosphorylated S22 and S392 was also observed after okadaic acid treatment with all of the tested lamin A mutants; it should be noted that S22 and S392 are thought to be primarily phosphorylated by CDK1 rather than p38 or PKC. However, the R60G and T10I mutants showed a relative reduction in solubility at baseline compared to WT. Importantly, although treatment with okadaic acid did increase their solubility somewhat, it did not rescue their solubility to WT levels, with a large portion of T10I and R60G remaining insoluble after okadaic acid treatment (though only the reduced solubility of R60G was statistically significant with respect to WT after okadaic acid). Notably, no change in R482Q solubility was observed relative to WT lamin A either at baseline or with okadaic acid treatment.
Discussion
The precise mechanisms underlying the laminopathies and their phenotypic diversity remain obscure. Studies seeking to elucidate these mechanisms frequently have been limited to single mutants, experimental models, or phenotypes. While the coexistence of multiple pathways of disease has been postulated, holistic approaches seeking to identify specific modulators of these pathways largely have not been explored. The importance of phenotypic clustering and genotype-phenotype correlation in the laminopathies is well established (11, 16, 53, 54). Here, we have assembled an updated and comprehensive repository of known LMNA variants with their associated phenotypes and additional available metadata, including mutation status and geographic. We then used high throughput, computational methods to predict mutation-specific effects on lamin A phosphorylation and protein structure with some experimental validation of our findings. To our knowledge, this is the most comprehensive analysis, classification and grouping of all known lamin mutations to date. Additionally, our novel application of machine-learning methods highlights the potential of computational approaches to systematically identify possible novel mechanisms of disease in genetic disorders. Altogether, our findings suggest a potential role for phosphorylation in the pathogenesis of laminopathies, and can serve as a foundation for further experimental exploration of these disease.
Laminopathy mutation profiles
Analysis of these data demonstrated patterns in mutation domain, patient sex, and heterozygosity and homozygosity. Laminopathy-associated mutations are mostly of the missense type, and affected individuals are typically heterozygous for mutant alleles—the notable exception being those with Charcot-Marie-Tooth disease, in which individuals are usually homozygous for the LMNA R298C mutation. Predominant heterozygosity in the setting of disease presumes dominant inheritance of most laminopathies. Rod domain mutations and non-missense mutations, particularly nonsense and frameshift mutations, are found most commonly in striated muscle laminopathies. In contrast, tail mutations are commonly associated with lipodystrophy-type disorders such as FPLD and MAD.
These key differences suggest separate or even dichotomous mechanisms underlying these different phenotypes. For instance, the higher prevalence of non-missense mutations in muscle laminopathies suggests that, relative to missense mutations, these mutation types may be overall more disruptive to protein structure and more significantly impair tissue tolerance to mechanical stress. Similarly, missense mutations that are more likely to disrupt protein structure (i.e. occurring within the rod domain) are also more prevalent in the striated muscle laminopathies. In contrast, diseases like lipodystrophies and mandibuloacral dysplasia are characterized by missense mutations in the tail domain. Importantly, the affected tissues in these diseases (adipose, bone, skin) are exposed to less mechanical stress compared to muscle, implying an alternative underlying mechanism.
Analysis of amino acid changes among laminopathy-associated missense mutations revealed patterns similar to those previously noted in keratins (55). For example, keratins have a relative scarcity of native cysteine and histidine residues, yet de novo cysteine or histidine introduction is seen in a significant proportion of disease-associated keratin mutations. This also appears to be the case in lamins, once again indicating cysteine or histidine may result in a “toxicity” that affects higher order structure formation, though previously under in vitro conditions, the keratin 14 mutant R125C was still able to form extensive filaments (56). In addition, proline was the most common mutant residue among laminopathy-associated missense mutations (Fig. 3). Interestingly, mutations resulting in a change to proline almost exclusively result in disease involving striated muscle, which invokes a common underlying mechanism. The addition of a proline can significantly alter protein structure given its conformationally rigid cyclic side chain; however, proline is also implicated in phosphorylation via proline-directed kinases, which target Ser/Thr that precede proline residues and encompass MAP kinases and CDKs. This could explain in part the efficacy of MAPK inhibitors in lamin-associated cardiomyopathies (12-14).
A deeper investigation of mutational patterns among the heptad repeats of the coiled-coil domain revealed that the e and g positions within the heptad were more likely to be mutated than other positions. A recent study illustrated that mutations causing L-CMD were more likely to be at the a d e g positions, which are critical for stabilizing lamin polymers (29). Here, we observed a relative predilection for skeletal muscle laminopathies to involve residues at the e position, whereas primary cardiac phenotypes showed a more even distribution. While this certainly requires further exploration, it could indicate a pathomechanical basis for skeletal muscle disorders that could even distinguish them from cardiac disorders.
Mutational patterns were largely driven by hotspot mutations, which frequently resulted in phenotypes involving multiple organ systems. Interestingly, comparing hotspot with non-hotspot mutations revealed a disproportionate number of cytosine to thymine transitions, which relates to a known phenomenon in which CpG dinucleotides are especially prone to mutation. This is due to vulnerability of cytosine to deamination, the frequent methylation of cytosine in CpG dinucleotides, with deamination of 5-methylcytosine resulting in thymidine (57). The vulnerability of these sites to mutation is responsible for the frequency of mutation in native arginine residues, given the high prevalence of cytosines within codons for arginine.
Several LMNA mutations lead to either overlapping syndromes or disparate phenotypes, in which a single mutation can lead to different organ system involvement in two different individuals. A curious example is the R644C mutation, which can lead to a wide array of disparate phenotypes including all major categories of laminopathies (progeria, neuropathy, striated muscle laminopathy, and lipodystrophy). This particular mutation also notably has an extremely low degree of penetrance, with only 31 out of 186 (16.7%) patients developing disease. The pleiotropy of such mutations suggests the presence of other modifiers, possibly spanning genetic, epigenetic, and post-translational realms, that modulate the development of a specific phenotype. Specifically, there has been evidence demonstrating that lamin A has a wide array of binding partners, including nucleoskeleton proteins such as Nespring2 and Nup88, transcription factors like Rb and SREBP1 a/c, kinases like PKCα, and chromatin directly (6). The presence of these binding partners is thought to vary based on tissue type, and similarly, different phosphorylation sites and states appear to depend on the specific cell or tissue type studied (6). Since many of these binding partners are thought to bind to the tail domain, it is possible that the overlapping and disparate phenotypes associated with a mutant like R644C (and several others in the tail region), may be due to variable binding of tissue-specific binding partners.
Crosstalk between lamin mutation-modulated phosphorylation and structural effects
PTMs likely impact disease mechanisms in a mutation-specific way. For example, previous studies have demonstrated a link between FPLD-associated mutations and disruption of lamin A SUMOylation (58-60). More recent studies have implicated loss of O-Glc-NAcylation to be associated with mutations resulting in HGPS (61). Moreover, lamin A/C classically undergoes several important physiology PTMs such as transient farnesylation, which allows lamin to initially associate with the inner nuclear membrane, and proteolytic cleavage into its mature form by ZMPSTE24. In HGPS, lamin A/C is unable to undergo this final cleavage step, resulting in permanent farnesylation. This has led to the basis for farnesyltransferase inhibitors in the treatment of HGPS (62). It is also thought that various PTMs may facilitate PTM crosstalk that may combine to regulate protein function (5).
Our findings demonstrated a significant correlation between disease and predicted mutation-induced changes to lamin phosphorylation and structure (Fig. 6, 7). Importantly, phosphorylation changes occurred predominantly in the head and tail domains of lamin A/C (Fig. 6B), which are the domains that typically harbor physiological phosphorylation for IFs in general (63). A limitation of our computational methods is that they are only able to make predictions on phosphorylation changes that are proximal to the given mutation (±16 or 25 amino acid residues, respectively) and cannot account for more distant effects that may occur due to protein folding. Therefore, it is possible that our approach will underestimate long-range mutational effects on phosphorylation.
Segregation of predicted phosphorylation and structural changes by phenotype demonstrated key differences, implicating possible phenotype-specific mechanisms of disease. Specifically, lipodystrophy and bone/skin phenotypes were associated with predicted changes in phosphorylation, while this was significantly less likely in striated muscle laminopathies, which were much more likely to be associated with structural change (Fig. 6C). Hotspot mutations were more likely to be associated with changes in phosphorylation, but the same trend was not observed among predicted structural changes (Fig. 6D, E). While it is possible that these findings may simply reflect a preferred localization of these disease mutations in the lamin protein without any direct physiological relevance, it could also suggest two alternative mechanisms by which mutations can lead to disease: 1) by directly disrupting protein structure, and 2) by inducing changes in phosphorylation, though these are not necessarily mutually exclusive.
In vitro assessment of mutation-associated changes in lamin phosphorylation revealed both kinase- and mutation-specific changes (Fig. 8). Utilizing an in vitro kinase assay with p38 MAP kinase indicated a decrease in phosphorylation in the T10I and R60G mutants (though only R60G was statistically significant), but not R482Q; in contrast, PKCα showed no significant changes across the mutants. Of note, lamin A/C contains more than 10 predicted PKCα sites, which limits the sensitivity of such an assay to detect a decrease in lamin phosphorylation. In addition, protein phosphorylation may impact protein conformation and solubility. We observed a significant reduction in baseline solubility of the T10I and R60G mutants relative to WT lamin A, and these mutants remained largely insoluble relative to WT after treatment with okadaic acid, a potent phosphatase inhibitor, even though phosphorylation of residues S22 and S392 could be detected for all mutants. These data suggest that the T10I and R60G mutants are relatively resistant to phosphorylation, though treatment with okadaic acid could also lead to global hyperphosphorylation, decreasing sensitivity for detecting site-specific phosphorylation changes.
Notably, T10I was predicted to be associated with loss of phosphorylation by our computational approach, while R60G was predicted to be associated with possible loss (only MusiteDeep predicted a loss). In contrast, neither T10I nor R60G were predicted to induce significant structural changes to lamin A/C, though our predictions do not consider effects beyond the monomeric form of the protein, and thus do not preclude downstream effects on aggregate formation that could also affect solubility (since relative insolubility could reflect either aggregation or an unaggregated insoluble pool). In this respect, it is quite possible that phosphorylation of specific amino acid residues may have an extensive impact on lamin polymer assembly.
Furthermore, while R482Q was predicted to be associated with a loss of phosphorylation, we did not observe a change in R482Q phosphorylation, and there was no noted change in solubility relative to WT lamin A/C. It is possible that solubility changes may be induced by phosphorylation in specific regions of the protein, such as the coiled-coil rod domain, while phosphorylation affecting tail regions may not have a similar effect. Interestingly, T10I is associated with progeroid syndromes, R60G with striated muscle laminopathies, and R482Q with lipodystrophy—again suggesting possible distinct mechanisms.
Further studies are needed to elucidate the precise phosphorylation sites that are altered in the setting of lamin mutations, as well as specific downstream effects that likely extend beyond protein conformation and solubility. To date, studies on lamin phosphorylation have focused on its role in cell cycle regulation, specifically phosphorylation of S22 and S392 during mitosis (9, 10). Previously, stress-induced kinase inhibition was shown to ameliorate the effects of lamin-associated cardiomyopathy and progeria (11, 12, 14, 17, 18). Our results suggest that the impact of kinase inhibitors likely reflects downstream phosphorylation-mediated effects that may be secondary to structural changes in the lamin A/C protein affecting polymerization, nuclear assembly, tolerance to mechanical stress, as shown in Figure 9. In addition, our findings demonstrate that an alternative phosphorylation-mediated pathway—direct lamin A/C phosphorylation—by itself directly associates with disease. Alterations to lamin A/C phosphorylation may specifically be involved in certain phenotypes, such as lipodystrophy, which have previously not been as amenable to stress-induced kinase inhibition. Mutations leading to changes in lamin A/C phosphorylation may have a myriad of downstream effects, including changes in localization, protein-protein interactions, lamin-chromatin interactions, and downstream signaling cascades (10). This ultimately leads to changes in gene expression, which likely represents a common downstream effect among the different phosphorylation-mediated pathways (Fig. 9).
Figure 9. Proposed phosphorylation-related laminopathy disease mechanisms.

Left: Global changes in phosphorylation occur in response to mutation-induced possible protein misfolding and aggregate formation, with subsequent alterations of dynamic filament assembly and nuclear morphology. This leads to decreased tolerance to cell-specific stress, such as mechanical and oxidative stress, which activates downstream stress-induced kinases such as ERK, mTOR, p38, and JNK. A phosphorylation-mediated signaling cascade ultimately results in altered gene expression, which leads to cell-specific phenotypes. Selective pharmacologic inhibitors against these stress-induced kinases are shown; previous studies have demonstrated attenuation of disease effects in cardiomyopathy and progeria models using such inhibitors. ARRY-371797 is currently in clinical trials for cardiomyopathy in humans. Chemical chaperones, which can stabilize native conformations of proteins (or reduce pathogenic misfolding), may have a role in laminopathies, but this has not been reported. Right: Lamin mutations lead to specific alteration of kinase-binding motifs on lamin itself, which leads to several downstream effects, including altered protein-protein interactions, altered lamin localization (e.g., an increased ratio of nucleoplasmic-to-nuclear envelope lamin), and direct lamin-chromatin/DNA interactions (e.g., lamin-associated domains). These could all lead to alterations in gene expression. Notably, protein and DNA interactions are likely cell-specific and may influence the specific phenotype observed. Other PTMs affected by mutations may also have an impact, with potential crosstalk between different PTMs. ERK = extracellular signal-related kinases, mTOR = mammalian target of rapamycin, p38 = p38 mitogen-activated protein kinases, JNK = c-Jun N-terminal kinases, LAP2α = lamina-associated polypeptide 2α, Rb = retinoblastoma protein.
Therapeutic implications
Distinct phosphorylation-mediated mechanisms of disease suggest the need for more than a one-size-fits-all approach. Previous studies demonstrating the benefits of stress-induced kinase inhibition have focused on the cardiomyopathy and progeria phenotypes, with a small number of mutations representing each phenotype studied (11, 15, 16, 18, 19, 64). Current clinical trials include only ARRY-371797, a p38 inhibitor, in the context of LMNA-related dilated cardiomyopathy (ClinicalTrials.gov). In some cases, kinase activation can also be beneficial. A recent study examining the interaction between prelamin A or mature lamin A/C with CK2 demonstrated that both prelamin A and mature lamin A/C inhibit CK2 via binding to its enzymatic core subunit, inducing early cellular senescence. Meanwhile, activation of CK2 with a small molecule spermidine conferred resistance to senescence and ameliorated progeroid features with extension of lifespan in mice (65). Our computational predictions also indicate mutation-dependent increases or decreases in phosphorylation, which further highlights the need for tailoring therapies depending on the precise mutation and phenotype in question.
Continued investigation elucidating the downstream pathways triggered by differential lamin phosphorylation may reveal additional therapeutic targets. For example, stress-induced kinase activation likely results from mechanical stress due to lamin mutation-induced structural alterations, which offers another opportunity for intervention at the protein folding step (Fig. 9). For example, in the skin fragility disorder epidermolysis bullosa simplex (EBS), treatment with 4-phenylbutyrate, a chemical chaperone, decreased keratin aggregation and reduced inflammation in a culture system (66, 67). Given evidence that multiple subtypes of laminopathy present with protein aggregation (1, 68-70), the use of chemical chaperones could provide a novel approach to laminopathy treatment.
Collectively, our findings suggest a role for direct lamin phosphorylation as a mutation-dependent and kinase-dependent modifier of tissue-specific defects and subsequent laminopathy phenotypes. Current therapeutic approaches may be limited to certain mutations or associated phenotypes, and thus may not fully encapsulate the myriad mechanisms of disease that are apparent in the laminopathies. This highlights the need to elucidate alternative disease mechanisms in order to construct specific, personalized treatment strategies. Here, we have re-purposed machine learning-based computational methods in a rare genetic disease, demonstrating the capability of these methodologies for generating hypotheses about mechanisms of disease.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01 DK47918 (M.B.O.), and R01 GM094231 (A.I.N.); a Liver Scholar Award from the American Liver Foundation (G.F.B.); and an institutional NIH award DK034933 to the University of Michigan. The authors declare no conflicts of interest.
Abbreviations
- CDK
cyclin-dependent kinase
- CK2
casein kinase 2
- CMT
Charcot-Marie-Tooth
- FPLD
familial partial lipodystrophy
- HGPS
Hutchinson-Gilford progeria syndrome
- IF
intermediate filaments
- Ig
immunoglobulin
- JNK
c-Jun N-terminal kinase
- LGMD
limb girdle muscular dystrophy
- MAD
mandibuloacral dysplasia
- MAPK
mitogen-activated protein kinase
- PKA
protein kinase A
- PKC
protein kinase C
- PTM
post-translational modifications
- SDS
Sodium dodecyl sulfate
- WT
wild-type
References
- 1.Brady GF, Kwan R, Bragazzi Cunha J, Elenbaas JS, and Omary MB (2018) Lamins and Lamin-Associated Proteins in Gastrointestinal Health and Disease. Gastroenterology 154, 1602–1619.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capell BC and Collins FS (2006) Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet 7, 940–952 [DOI] [PubMed] [Google Scholar]
- 3.Butin-Israeli V, Adam SA, Goldman AE, and Goldman RD (2012) Nuclear lamin functions and disease. Trends Genet. 28, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elzeneini E and Wickström SA (2017) Lipodystrophic laminopathy: Lamin A mutation relaxes chromatin architecture to impair adipogenesis. J. Cell Biol 216, 2607–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snider NT and Omary MB (2014) Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Mol. Cell Biol 15, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon DN and Wilson KL (2013) Partners and post-translational modifications of nuclear lamins. Chromosoma 122, 13–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peter M, Nakagawa J, Dorée M, Labbé JC, and Nigg EA (1990) In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 61, 591–602 [DOI] [PubMed] [Google Scholar]
- 8.Heald R and McKeon F (1990) Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61, 579–589 [DOI] [PubMed] [Google Scholar]
- 9.Kochin V, Shimi T, Torvaldson E, Adam SA, Goldman A, Pack C-G, Melo-Cardenas J, Imanishi SY, Goldman RD, and Eriksson JE (2014) Interphase phosphorylation of lamin A. J. Cell Sci 127, 2683–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torvaldson E, Kochin V, and Eriksson JE (2015) Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus 6, 166–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, and Worman HJ (2007) Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest 117, 1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, and Worman HJ (2012) Abnormal p38α mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet 21, 4325–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muchir A, Shan J, Bonne G, Lehnart SE, and Worman HJ (2009) Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet 18, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu W, Shan J, Bonne G, Worman HJ, and Muchir A (2010) Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim. Biophys. Acta - Mol. Basis Dis 1802, 632–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu W, Muchir A, Shan J, Bonne G, and Worman HJ (2011) Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in Lamin A/C gene. Circulation 123, 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muchir A, Reilly SA, Wu W, Iwata S, Homma S, Bonne G, and Worman HJ (2012) Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res 93, 311–319 [DOI] [PubMed] [Google Scholar]
- 17.Muchir A, Wu W, Sera F, Homma S, and Worman HJ (2014) Mitogen-activated protein kinase kinase 1/2 inhibition and angiotensin II converting inhibition in mice with cardiomyopathy caused by lamin A/C gene mutation. Biochem. Biophys. Res. Commun 452, 958–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, and Worman HJ (2012) Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med 4, 144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DuBose AJ, Lichtenstein ST, Petrash NM, Erdos MR, Gordon LB, and Collins FS (2018) Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc. Natl. Acad. Sci 115, 201802811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cenni V, Sabatelli P, Mattioli E, Marmiroli S, Capanni C, Ognibene A, Squarzoni S, Maraldi NM, Bonne G, Columbaro M, Merlini L, and Lattanzi G (2005) Lamin A N-terminal phosphorylation is associated with myoblast activation: Impairment in Emery-Dreifuss muscular dystrophy. J. Med. Genet 42, 214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitsuhashi H, Hayashi YK, Matsuda C, Noguchi S, Wakatsuki S, Araki T, and Nishino I (2010) Specific phosphorylation of Ser458 of A-type lamins in LMNA-associated myopathy patients. J. Cell Sci 123, 3893–3900 [DOI] [PubMed] [Google Scholar]
- 22.Szeverenyi I, Cassidy AJ, Cheuk WC, Lee BTK, Common JEA, Ogg SC, Chen H, Shu YS, Goh WLP, Kee WN, Simpson JA, Li LC, Goi HE, Li B, Lunny DP, Chuon D, Venkatesh A, Kian HK, McLean WHHI, Yun PL, and Lane EB (2008) The human intermediate filament database: Comprehensive information on a gene family involved in many human diseases. Hum. Mutat 29, 351–360 [DOI] [PubMed] [Google Scholar]
- 23.Bateman A (2019) UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Zhou Z-Y, Lu H-H, Xie Y, Li G, Huang J-F, and Zhao D-S (2018) Identification of a LMNA (c.646C>T) variant by whole-exome sequencing in combination with a dilated cardiomyopathy (DCM) related gene filter in a family with familiar DCM. J. Biomed. Res 32, 314–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishiyama A, Iida A, Hayashi S, Komaki H, Sasaki M, Nonaka I, Noguchi S, and Nishino I (2018) A novel LMNA mutation identified in a Japanese patient with LMNA-associated congenital muscular dystrophy. Hum. Genome Var 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Exome Aggregation Consortium, Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan L, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Cooper DN, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Elosua R, Florez JC, Gabriel SB, Getz G, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf J, Sklar P, Sullivan PF, Tuomilehto J, Watkins HC, Wilson JG, Daly MJ, and MacArthur DG (2015) Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herrmann H and Aebi U (2004) Intermediate Filaments: Molecular Structure, Assembly Mechanism, and Integration Into Functionally Distinct Intracellular Scaffolds. Annu. Rev. Biochem 73, 749–789 [DOI] [PubMed] [Google Scholar]
- 28.Ahn J, Jo I, Kang S. mi, Hong S, Kim S, Jeong S, Kim YH, Park BJ, and Ha NC (2019) Structural basis for lamin assembly at the molecular level. Nat. Commun 10, 3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertrand AT, Brull A, Azibani F, Benarroch L, Chikhaoui K, Stewart CL, Medalia O, Ben Yaou R, and Bonne G (2020) Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery–Dreifuss Muscular Dystrophy. Cells 9, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrmann H and Aebi U (2016) Intermediate filaments: Structure and assembly. Cold Spring Harb. Perspect. Biol 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bera M, Ainavarapu SRK, and Sengupta K (2016) Significance of 1B and 2B domains in modulating elastic properties of lamin A. Sci. Rep 6, 27879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD, and Radivojac P (2017) MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 134981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, and Ng PC (2012) SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hicks S, Wheeler DA, Plon SE, and Kimmel M (2011) Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat 32, 661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagih O, Reimand J, and Bader GD (2015) MIMP: Predicting the impact of mutations on kinase-substrate phosphorylation. Nat. Methods 12, 531–533 [DOI] [PubMed] [Google Scholar]
- 37.Wang D, Zeng S, Xu C, Qiu W, Liang Y, Joshi T, and Xu D (2017) MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 33, 3909–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, and Diella F (2011) Phospho.ELM: A database of phosphorylation sites-update 2011. Nucleic Acids Res. 39, D261–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015) PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TKB, Gronborg M, Ibarrola N, Deshpande N, Shanker K, Shivashankar HN, Rashmi BP, Ramya MA, Zhao Z, Chandrika KN, Padma N, Harsha HC, Yatish AJ, Kavitha MP, Menezes M, Choudhury DR, Suresh S, Ghosh N, Saravana R, Chandran S, Krishna S, Joy M, Anand SK, Madavan V, Joseph A, Wong GW, Schiemann WP, Constantinescu SN, Huang L, Khosravi-Far R, Steen H, Tewari M, Ghaffari S, Blobe GC, Dang CV, Garcia JGN, Pevsner J, Jensen ON, Roepstorff P, Deshpande KS, Chinnaiyan AM, Hamosh A, Chakravarti A, and Pandey A (2003) Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 13, 2363–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, Balakrishnan L, Marimuthu A, Banerjee S, Somanathan DS, Sebastian A, Rani S, Ray S, Harrys Kishore CJ, Kanth S, Ahmed M, Kashyap MK, Mohmood R, Ramachandra YI, Krishna V, Rahiman BA, Mohan S, Ranganathan P, Ramabadran S, Chaerkady R, and Pandey A (2009) Human Protein Reference Database - 2009 update. Nucleic Acids Res. 37, D767–D772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J, Rho HS, Newman RH, Zhang J, Zhu H, and Qian J (2014) Phospho networks: A database for human phosphorylation networks. Bioinformatics 30, 141–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buljan M, Blattmann P, Aebersold R, and Boutros M (2018) Systematic characterization of pan-cancer mutation clusters. Mol. Syst. Biol 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodrigues CH, Pires DE, and Ascher DB (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46, W350–W355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dhe-Paganon S, Werner ED, Chi Y-I, and Shoelson SE (2002) Structure of the globular tail of nuclear lamin. J. Biol. Chem 277, 17381–17384 [DOI] [PubMed] [Google Scholar]
- 47.Brady GF, Kwan R, Ulintz PJ, Nguyen P, Bassirian S, Basrur V, Nesvizhskii AI, Loomba R, and Omary MB (2018) Nuclear lamina genetic variants, including a truncated LAP2, in twins and siblings with nonalcoholic fatty liver disease. Hepatology 67, 1710–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delvecchio CJ and Capone JP (2008) Protein kinase C α modulates liver X receptor α transactivation. J. Endocrinol 197, 121–130 [DOI] [PubMed] [Google Scholar]
- 49.Gu Z, Eils R, and Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 [DOI] [PubMed] [Google Scholar]
- 50.Kwan R, Brady GF, Brzozowski M, Weerasinghe SV, Martin H, Park MJ, Brunt MJ, Menon RK, Tong X, Yin L, Stewart CL, and Omary MB (2017) Hepatocyte-Specific Deletion of Mouse Lamin A/C Leads to Male-Selective Steatohepatitis. CMGH 4, 365–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martelli AM, Bortul R, Tabellini G, Faenza I, Cappellini A, Bareggi R, Manzoli L, and Cocco L (2002) Molecular characterization of protein kinase C-α binding to lamin A. J. Cell. Biochem 86, 320–330 [DOI] [PubMed] [Google Scholar]
- 52.Thompson LJ, Bollen M, and Fields AP (1997) Identification of protein phosphatase 1 as a mitotic lamin phosphatase. J. Biol. Chem 272, 29693–29697 [DOI] [PubMed] [Google Scholar]
- 53.Scharner J, Gnocchi VF, Ellis JA, and Zammit PS (2010) Genotype–phenotype correlations in laminopathies: how does fate translate? Biochem. Soc. Trans 38, 257–262 [DOI] [PubMed] [Google Scholar]
- 54.Wu W, Iwata S, Homma S, Worman HJ, and Muchir A (2014) Depletion of extracellular signal-regulated kinase 1 in mice with cardiomyopathy caused by lamin A/C gene mutation partially prevents pathology before isoenzyme activation. Hum. Mol. Genet 23, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strnad P, Usachov V, Debes C, Gräter F, Parry DAD, and Omary MB (2011) Unique amino acid signatures that are evolutionarily conserved distinguish simple-type, epidermal and hair keratins. J. Cell Sci 124, 4221–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herrmann H, Wedig T, Porter RM, Lane EB, and Aebi U (2002) Characterization of early assembly intermediates of recombinant human keratins. J. Struct. Biol 137, 82–96 [DOI] [PubMed] [Google Scholar]
- 57.Fryxell KJ and Moon W-J (2005) CpG Mutation Rates in the Human Genome Are Highly Dependent on Local GC Content. Mol. Biol. Evol 22, 650–658 [DOI] [PubMed] [Google Scholar]
- 58.Simon DN, Domaradzki T, Hofmann WA, and Wilson KL (2013) Lamin A tail modification by SUMO1 is disrupted by familial partial lipodystrophy-causing mutations. Mol. Biol. Cell 24, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boudreau É, Labib S, Bertrand AT, Decostre V, Bolongo PM, Sylvius N, Bonne G, and Tesson F (2012) Lamin A/C Mutants Disturb Sumo1 Localization and Sumoylation in Vitro and in Vivo. PLoS One 7, e45918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, Yang C-S, Cubeñas-Potts C, Matunis MJ, and Paschal BM (2011) The defective nuclear lamina in Hutchinson-gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol. Cell. Biol 31, 3378–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simon D, Wriston A, Fan Q, Shabanowitz J, Florwick A, Dharmaraj T, Peterson S, Gruenbaum Y, Carlson C, Grønning-Wang L, Hunt D, and Wilson K (2018) OGT (O-GlcNAc Transferase) Selectively Modifies Multiple Residues Unique to Lamin A. Cells 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Young SG, Yang SH, Davies BSJ, Jung HJ, and Fong LG (2013) Targeting protein prenylation in progeria. Sci. Transl. Med 5, 171ps3–171ps3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Omary MB, Ku NO, Tao GZ, Toivola DM, and Liao J (2006) “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem. Sci 31, 383–394 [DOI] [PubMed] [Google Scholar]
- 64.Chatzifrangkeskou M, Yadin D, Marais T, Chardonnet S, Cohen-Tannoudji M, Mougenot N, Schmitt A, Crasto S, Di Pasquale E, MacQuart C, Tanguy Y, Jebeniani I, Pucéat M, Rodriguez BM, Goldmann WH, Ferro MD, Biferi MG, Knaus P, Bonne G, Worman HJ, and Muchir A (2018) Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet 27, 3060–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ao Y, Zhang J, Liu Z, Qian M, Li Y, Wu Z, Sun P, Wu J, Bei W, Wen J, Wu X, Li F, Zhou Z, Zhu W-G, Liu B, and Wang Z (2019) Lamin A buffers CK2 kinase activity to modulate aging in a progeria mouse model. Sci. Adv 5, eaav5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chamcheu JC, Siddiqui IA, and Mukhtar H (2016) Chemical chaperone therapy, a new strategy for genetic skin fragility disorders. Exp. Dermatol 25, 183–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spörrer M, Prochnicki A, Tölle RC, Nyström A, Esser PR, Homberg M, Athanasiou I, Zingkou E, Schilling A, Gerum R, Thievessen I, Winter L, Bruckner-Tuderman L, Fabry B, Magin TM, Dengjel J, Schröder R, and Kiritsi D (2019) Treatment of keratinocytes with 4-phenylbutyrate in epidermolysis bullosa: Lessons for therapies in keratin disorders. EBioMedicine 44, 502–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raharjo WH, Enarson P, Sullivan T, Stewart CL, and Burke B (2001) Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J. Cell Sci 114, 4447–4457 [DOI] [PubMed] [Google Scholar]
- 69.Hübner S, Eam JE, Wagstaff KM, and Jans DA (2006) Quantitative analysis of localization and nuclear aggregate formation induced by GFP-lamin A mutant proteins in living HeLa cells. J. Cell. Biochem 98, 810–826 [DOI] [PubMed] [Google Scholar]
- 70.Sylvius N, Hathaway A, Boudreau E, Gupta P, Labib S, Bolongo PM, Rippstein P, McBride H, Bilinska ZT, and Tesson F (2008) Specific contribution of lamin A and lamin C in the development of laminopathies. Exp. Cell Res 314, 2362–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wadhwa R, Subramanian V, and Stevens-Truss R (2018) Visualizing alpha-helical peptides in R with helixvis. J. Open Source Softw 3, 1008 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.