

Abstract
Corona Virus Disease 2019 (COVID-19) caused by Severe Acute Respiratory Syndrome coronavirus (SARS CoV-2) has been declared a worldwide pandemic by WHO recently. The complete understanding of the complex genomic structure of SARS CoV-2 has enabled the use of computational tools in search of SARS CoV-2 inhibitors against the multiple proteins responsible for its entry and multiplication in human cells. With this endeavor, 177 natural, anti-viral chemical entities and their derivatives, selected through the critical analysis of the literatures, were studied using pharmacophore screening followed by molecular docking against RNA dependent RNA polymerase and main protease. The identified hits have been subjected to molecular dynamic simulations to study the stability of ligand-protein complexes followed by ADMET analysis and Lipinski filters to confirm their drug likeliness. It has led to an important start point in the drug discovery and development of therapeutic agents against SARS CoV-2.
Keywords: RdRp, Mpro, SARS CoV-2, Natural products, Molecular docking, MD simulation
Abbreviations: ACE, Angiotensin converting enzyme; ADMET, Absorption, distribution, metabolism, excretion, and toxicity; COVID-19, Corona virus disease-2019; HB, Hydrogen bond; MD simulation, Molecular dynamic simulation; Mpro, Main protease; 3CLpro, 3-chymotrypsin-like protease; PLpro, Papain-like protease; RdRP, RNA-dependent RNA polymerase; EM, Electron microscopy; Dscore, Druggability score; nsp, Non-structural protein; RMSD, Root mean square deviation; RMSF, Root mean square fluctuation; RoG, Radius of gyration; ASL, Atom specification language; SARS CoV-2, Severe acute respiratory syndrome coronavirus 2; SASA, Solvent accessible surface area; SP, Standard precision; WHO, World health organization
Graphical abstract
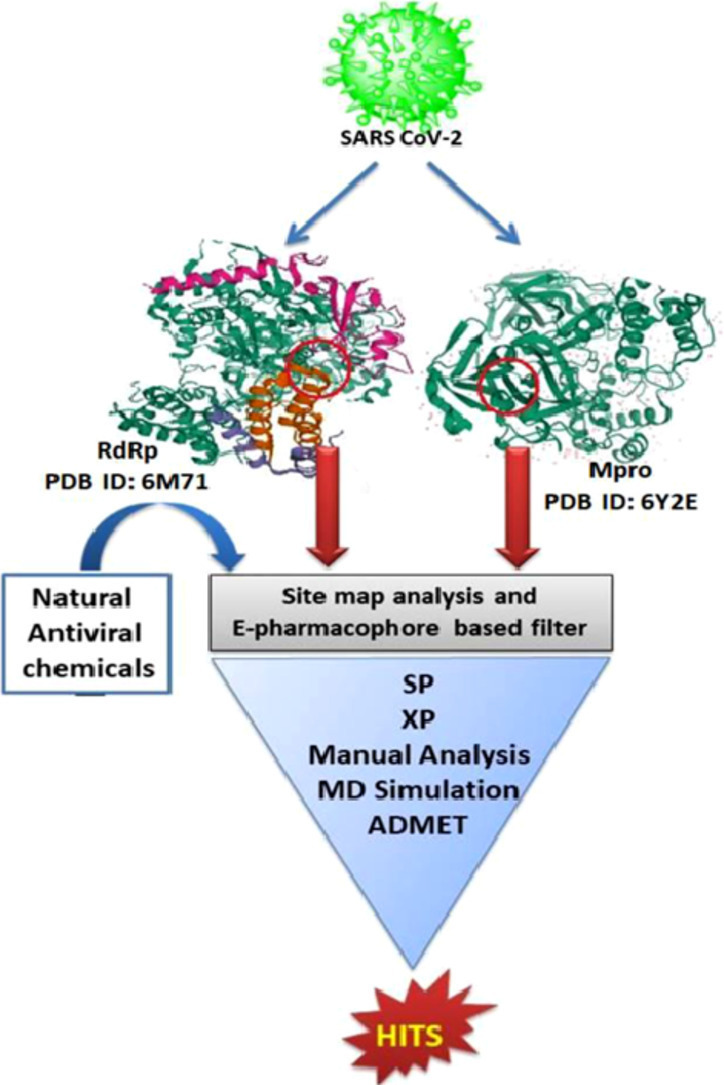
1. Introduction
Coronavirus disease (COVID-19) caused due to newly discovered and deadly coronavirus (family: Coronaviridae) is a highly contagious disease, named severe acute respiratory syndrome coronavirus disease-2 (SARS Cov-2). The name ‘coronavirus’ is coined due to the crown-like projections on its surface as in Latin, ‘Corona’ means ‘halo’ or ‘crown’[1]. The disease symptoms range from low-grade fever to severe breathing problems requiring artificial ventilation and in exceptional cases, fatal respiratory problems may be observed [2]. It can be transmitted through coughing or sneezing of infected patients without covering mouth and nose, physical contact with an infected human being through hand-shakes or infected surfaces, its exposure to mucous membranes of nasal, lacrimal or salivary fluids [1]. Since the identification of the first case of COVID-19 infection in December 2019 the disease has spread the world over [3]. World health organization (WHO) for the first time has declared the novel coronavirus as a high-level global risk on February 28, 2020 and COVID-19 as a global pandemic on March 11, 2020 [2,4]. More than 132,485,386 confirmed cases and more than 2875,672 deaths have been confirmed across the globe due to SARS CoV-2 till April 8, 2021. Massive vaccination drive world-over has begun but the disease spread is still rampant and till April 06, 2021, administration of about 650,382,819 vaccine doses have been achieved according to WHO [4]. Moreover, vaccines do not provide lifelong immunity.
All these facts necessitate the requirement of effective drugs for SARS CoV-2 which can be achieved through the understanding of its life cycle. The binding of SARS CoV-2 with the human angiotensin converting enzyme 2 (ACE2) receptors through its viral Spike (S) protein allowed its entry into the cytosol of the host cell, followed by its uncoating and biosynthesis of its polypeptides using host cell machinery [1,5]. Later, the synthesis of its RNA is achieved using the viral enzyme RNA dependent RNA polymerase (RdRP) [6]. Several structural and non-structural proteins required for its replication, catalyzed by Main protease (Mpro) also known as 3-chymotrypsin-like protease (3CLpro) and by papain-like protease (PLpro) shall be executed through mRNA to facilitate the multiplication of virus [7]. All these proteins can serve as druggable targets against COVID-19 in search of SARS CoV-2 inhibitors [8], [9], [10].
The use of in silico tools for drug discovery has been widely adopted nowadays [11], [12], [13] wherein molecular modeling might provide the key interactions between the ligands and the desired target of our interest to understand the mode of action of these agents [14], [15], [16]. Thus, many of the research groups have embarked on the repurposing of approved drugs and their in silico evaluation against SARS CoV-2 [17], [18], [19], [20]. The natural products [8,21,22] should not be underestimated in search of SARS CoV-2 as many of them have been reported to act against SARS CoV-2 through inhibition of RdRp [23] and Mpro [19,24,25]. This has urged the need for the drug design and drug discovery of new SARS CoV-2 inhibitors using in silico tools. From literature sources, we identified 177 phytochemicals and their derivatives having antiviral activity. These compounds were analyzed using pharmacophore-based screening followed by molecular docking against two main targets of SARS CoV-2 namely RdRp and Mpro. The successful candidates were subjected to molecular dynamic simulation for determining receptor-ligand complex stability. Further screening was carried out using ADMET assay and Lipinski filters to access their drug likeliness. The successful compounds are potential therapeutic agents for SARS CoV-2 and can be studied further using in vivo and in vitro efficacy and safety assays.
2. Results and discussion
2.1. Pharmacophore hypothesis
2.1.1. SiteMap analysis
Gao et al. reported the cryo-EM crystal structure of RdRp protein (resolution of 2.9 Å) comprising of nsp12 (non-structural protein) complexed with nsp7 and nsp8 [26]. Hilgenfeld and co-workers reported the unliganded dimeric structure of Mpro or 3CLPro having three domains for each monomer using X-ray diffraction with 1.75 Å of resolution [27]. These selected proteins for the key targets such as RdRp (PDB ID: 6M71) and Mpro (PDB ID: 6Y2E) have been deposited in protein data bank without any complexed or bound ligand. Hence, we felt the need to perform the SiteMap analysis for the selection of suitable binding or active sites for the proteins of interest [28,29]. Based on the volume of the pocket, enclosure, and the degree of hydrophobicity, the druggability assessment scores such as Dscore and SiteScore were calculated [30] to generate five binding sites (Table 1 ) and Dscore were taken into consideration for a further selection of the suitable binding site and generation of a pharmacophore model. The binding site with Dscore of 1.055 and Site score of 1.026 was selected for the RdRp which include residues Tyr122, Thr123, Tyr149, Asn150, Cys151, Phe165, Val166, Pro169, Leu172, Pro243, Thr246, Leu247, Arg249, Ala250, Leu251, Thr252, Ser255, Tyr265, Ile266, Lys267, Trp268, Asp269, Leu270, Val315, Leu316, Ser318, Thr319, Val320, Phe321, Pro322, Pro323, Phe326, Tyr346, Phe348, Arg349, Glu350, Thr394, Cys395, Phe396, Tyr456, Arg457, Tyr458, Asn459, Leu460, Pro461, Thr462, Met463, Ala625, Pro627, Asn628, Met629, Cys659, Ser664, Val675, Lys676, Pro677, Tyr788, Asn790, Asn791 and Val792 of chain A. In the same way, the active sites with Dscore of 0.934 and SiteScore 0.923, were selected for Mpro which include residues Thr24, Thr25, Thr26, Leu27, His41, Cys44, Thr45, Ser46, Met49, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, Arg188, Gln189, Thr190 and Gln192.
Table 1.
The obtained D scores and Site scores for the observed sites using site map analysis.
2.1.2. Pharmacophore development and ligand screening
The steric and electrostatic features of the ligands with binding sites were derived using the energy-based pharmacophore (E-pharmacophore) dependent hypothesis for the chosen active sites of these proteins using a Phase module [31]. The model has been generated with at least seven features (Fig. 1 ), mainly comprising of hydrogen bond acceptor (A), hydrogen bond donor (D), negatively charged ionizable atom (N), and aromatic ring (R) [such as ADNRRRR and AAADRRR for RdRp and MPro respectively].
Fig. 1.

(a) Pharmacophore hypothesis of the selected targets such as RdRp and (b) Mpro. The key features aromatic rings (R) and hydrogen bond acceptor (A), hydrogen bond donors (D), and negatively charged ionizable atoms (N) have been presented in brown colored rings, pink spheres with arrows, sky blue colored spheres and red-colored spheres, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The data set for 177 anti-viral compounds comprising of natural products and their structural congeners were constructed through an exhaustive literature survey using PubChem [32] and SciFinder [33] (Table S1, see supporting information). The previously generated hypotheses were validated using these ligands through the Phase module. Among the selected ligands, 55 (RdRp) and 117 (Mpro) ligands matched with a minimum of four identified features of the respective hypotheses.
2.2. Molecular docking
The receptor grids with size 10 Å for molecular docking were generated from the predicted binding sites with the highest d-score using the Grid-based Ligand Docking with Energetics (GLIDE) module of Schrodinger [34]. Molecular docking was performed to get detailed information about ligand-receptor binding affinity through standard (SP) and extra precision (XP) modules of GLIDE. The SP docking of ligands filtered through the Phase module against the active site of the respective proteins revealed 4 molecules (RdRp) and 18 molecules (Mpro) having docking score of ≤ −6.9 (Table S2, see supporting information). These molecules were further docked using the XP module to shortlist 4 molecules (RdRp) and 13 molecules (Mpro) with docking score of ≤ −7 (Table 2 ).
Table 2.
Docking scores of compounds having docking scores ≤ −7 in XP docking.
| Entry | Compounds | Docking score | PDB ID |
|---|---|---|---|
| 1 | Tannic acid | −16.5 | RdRp (6M71) |
| 2 | Dieckol | −10.0 | |
| 3 | Theaflavin-3,3′-digallate | −9.4 | |
| 4 | Phlorofucofuroeckol A | −8.3 | |
| 5 | Rutin | −11.8 | Mpro (6Y2E) |
| 6 | Dieckol | −11.3 | |
| 7 | 7-Phloroeckol | −10.9 | |
| 8 | Nictoflorin | −9.3 | |
| 9 | Procyanidin A2 | −9.2 | |
| 10 | Hyperoside | −9.1 | |
| 11 | Phlorofucofuroeckol A | −8.8 | |
| 12 | Epigallocatechin-3-gallate | −8.4 | |
| 13 | Juglanin | −8.2 | |
| 14 | Eckol | −7.9 | |
| 15 | Astragalin | −7.6 | |
| 16 | Procyanidin B1 | −7.4 |
The docked poses of selected ligands against RdRp with docking scores better than −7.0 are represented in Fig. 2 . Herein, tannic acid was found to fit well into the active site of RdRp with hydrogen bond (HB) forming interactions with catalytic residues including Ser318, Thr319, Phe321, Thr394, Lys391, Asp390, Asn140 and Asp454. Due to the presence of many hydroxyl groups in its structure and formed these HB interactions, the docked complex of tannic acid-RdRp could obtain the superior docking score of −16.5 (Table 2, Entry 1). Additionally, it could interact with the active site of protein via π–cation interactions with residues Arg249 and Arg349 (Fig. 2a). Dieckol fitted in the selected receptor binding site of the RdRp by the formation of HB with the Ile266, Val320 and Thr246, present in A chain of protein has been presented in Fig. 2b. Theaflavin-3,3′-digallate formed π-π interactions with Tyr265 and HB with Ile266, Thr252 and Asn459 with the ligand-binding site of the protein (Fig. 2c). Sadhukhan et al. also found the almost similar binding mode of theaflavin-3,3′-digallate against RdRp [35]. The tight binding mode has been also observed with Phlorofucofuroeckol A through π-cationic interactions with Arg349 and HB with Ile266, Ser255, Thr319, Asn459 and Thr394 residues of the RdRp (Fig. 2d).
Fig. 2.

Docked poses of tannic acid (a), dieckol (b), theaflavin-3,3′-digallate (c), and phlorofucofuroeckol A (d) in the catalytic triad of RdRp. The poses of docked compounds have been generated and represented using PyMol 2.4.0 [36]. Ligands and protein are represented as yellow-colored balls and stick models and colored cartoons, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Further, we analyzed and studied the docked poses of ligands having docking scores greater than −9.0 against Mpro (Fig. 3 ). With a docking score of −11.8 (Table 2, Entry 5), rutin could make HB with Leu141, Gly143, Glu166, Thr25 and Thr26 residues in S1 and S1’ subunits of target protein (Fig. 3a). These results have been found in consonance with the recently reported in silico analysis by Chhabria et al. highlighting rutin as a promising Mpro inhibitor [37]. Procyanidin got bound between the S2 and S4 subunits through π–π interactions with His41 and HB with Leu167, Glu166, His163, His164, Gly143 and Cys44 (Fig. 3b). Nictoflorin fitted snugly at the junction of catalytic sides of S1’, and S1 subunits along with the HB formation with Gly143, Glu166, Leu167 and Thr26 (Fig. 3c). The tight binding bode of hyperoside was observed due to HB interactions with Glu166, Leu141, Asn142, Gly143 and Thr26 residues of S1, S1’ and S4 subunits (Fig. 3d). Similar binding modes were observed with dieckol (Thr26, Gln189, His163, Glu166, Gly143 and Asn119, Fig. 3e) and 7-phloroeckol (Phe140, Asn142, Thr25, Gln189 and Glu166, Fig. 3f) through HB interactions.
Fig. 3.

Docking interactions of compounds with Mpro rutin (a), procyanidin (b), nictoflorin (c), hyperoside (d), dieckol (e), 7-phloroeckol (f). The poses of docked compounds were generated and represented using PyMol 2.4.0 [36] wherein ligands and protein are represented as a yellow-colored ball and stick models and colored cartoons, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Further, the docking poses of natural products including phlorofucofuroeckol A, epigallocatechin-3-gallate, juglanin, eckol, astragalin, procyanidin B1 against Mpro are represented in Fig. 4 using PyMol. The phlorofucofuroeckol A (Fig. 4a) formed HB with residues present in the active site of Mpro including Thr24, Thr26, Gly143, His163 and Glu166. Total three HB with residues His41, Phe140 and Glu166 (Fig. 4b) were formed by epigallocatechin-3-gallate with the main protease enzyme. The docked complex of juglanin with Mpro was stabilized by the formation of HB with residues including Thr25, Thr26, Gly143, His163 and Glu166 (Fig. 4c) at the cleft in between the S1 and S1’ domain of the enzyme. The eckol was found to bind at the junction of S1 and S4 subunits forming HB interactions with Gly143, His163, Glu166 and Gln189 residues of Mpro (Fig. 4d). A similar HB formation was recognized with astragalin (Thr26, Asn142, Gly143 and Glu166, Fig. 4e) docked in S1 along with deeply extending towards the S4 domain. Whereas, procyanidinB1 was bound at the centroid of the active site with a slight inclination towards the S4 domain by the formation of HB with His41, Cys44, Asn142, Glu166 and Gln189 (Fig. 4f).
Fig. 4.

Docking interactions of compounds with Mpro phlorofucofuroeckol A (a), epigallocatechin-3-gallate (b), juglanin (c), eckol (d), astragalin (e), procyanidin B1 (f). The poses of docked compounds have been generated and represented using PyMol 2.4.0 [36]. The ligands and protein are symbolized as yellow-colored ball and stick models and colored cartoons, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.3. MD simulation
Molecular dynamic (MD) simulations have been considered an important tool to claim the time-dependent stability of ligand into the active site of protein through several statistical parameters [38,39]. To address the stability of docked ligand into the active site of target protein, next, we carried out the molecular dynamic (MD) simulations for 10 ns using GROMACS 2020.1 [40,41] for the selected hits from results obtained through docking followed by manual analysis. However, MD run of ≤ 10 ns has been regarded as the more suitable and satisfactory for preliminary in silico studies as evident in scientific literatures [42], [43], [44], [45], [46], [47]. The time-dependent stability of the ligand in the active site was studied using statistical tools such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (RoG), solvent accessible surface area (SASA) and hydrogen bonds (HB).
The RMSD for the protein-ligand complex of Mpro-rutin has been found in the range of 0.04–0.58 nm with an average of 0.374 nm for ligand (Fig. 5a) and from 0.005 to 0.26 nm with an average of 0.19 nm for protein (Fig. 5b). This has suggested the stability of the complex without any major changes in orientation. The structural integrity and atomic mobility of the complex were evaluated using RMSF (< 0.39 nm, Fig. 5c). The plot of fluctuations in RoG (nm) against the time (ns) is presented in Fig. 5d to evaluate the compactness of the protein-ligand complex. Herein, RoG was found in the range from 2.183 to 2.248 nm with an average of 2.219 nm. The surface area (nm2) accessed by solvents or water molecules inside the active site was further studied using the plots of SASA versus run time (ns). We have observed the surface area in the range of 145.00–157.00 nm2 with an average of 150.549 nm2 (Fig. 5e) along with up to nine HB in the period of 10 ns (Fig. 5f) for the complex. The short-range electrostatic (Coul-SR) and van der Waals/hydrophobic (LJ-SR) energies were found to be −173.141 ± 6.1 kJ/mol and −110.926 ± 4.6 kJ/mol, respectively.
Fig. 5.

The schematic plots of RMSD-L (a), RMSD-P (b), RMSF (c), RoG (d), SASA (e) and HB (f) for the docked complex of Mpro-rutin.
Next, we performed the MD simulations of the Mpro-dieckol complex and studied these parameters to access the stability of dieckol into the active site of Mpro. The RMSD for ligand (0.04–0.7 nm with an average of 0.418 nm, Fig. 6a) and that for protein (0.005–0.27 nm with an average of 0.209 nm, Fig. 6b) unveiled its stability devoid of fluctuations in the orientation of dieckol against Mpro. The structural integrity and atomic mobility of the complex were found unique as evident from the plot of RMSF (not more than 0.9 nm, Fig. 6c). The least fluctuations in RoG (Fig. 6d) found in the range from 2.189 to 2.249 nm with an average of 2.204 nm supported our hypothesis of compactness of protein-ligand complex. The other plots for the obtained complex revealed the surface area in the range of 142.00–156.00 nm2 and with an average of 148.605 nm2 (Fig. 6e) along with ten hydrogen bonds during MD run of 10 ns (Fig. 6f). The short-range electrostatic potential (Coul-SR, −124.748 ± 4.8 kJ/mol) revealed the significant contribution of the electrostatic interactions as compared to hydrophobic interactions (LJ-SR, −189.294 ± 2.2 kJ/mol). All these results indicated the stability of dieckol at the active site of Mpro in agreement with the literature [37].
Fig. 6.

The RMSD-L (a), RMSD-P (b), RMSF (c), RoG (d), SASA (e) and H-bonds (f) plots for the complex of Mpro-dieckol.
In a similar fashion to rutin (Fig. 5) and dieckol (Fig. 6), we observed RMSD for the protein-ligand complex of RdRp-tannic acid in the range from 0.006 to 0.4 nm with an average of 0.305 nm for ligand (Fig. 7a) and from 0.009 to 0.36 nm with an average of 0.302 nm for protein (Fig. 7b) suggesting the stability of the complex. The RMSF value has been found with an average of 1.256 nm (Fig. 7c) supported the structural integrity and atomic mobility of the complex. The RoG of the complex has reached up to 5.01 nm after the continuous stability period up to 6.37 ns (Fig. 7d). The surface area accessed by solvents or water molecules inside the active site versus run time (ns) was found in the range of 398.00–432.00 nm2 along with an average of 417.741 nm2 (Fig. 7e). During the dynamic simulation, up to thirteen HB were formed by the ligand into the active site of protein (Fig. 7f). The short-range electrostatic (Coul-SR) and van der Waals/hydrophobic (LJ-SR) energies were found −326.319 ± 6.3 kJ/mol and −245.418 ± 6.3 kJ/mol respectively.
Fig. 7.

The RMSD-L (a), RMSD-P (b), RMSF (c), RoG (d), SASA (e) and H-bonds (f) plots for the complex of RdRp-Tannic acid.
Similarly, the RMSD for the protein-ligand complex of RdRp-dieckol was found in the range from 0 to 0.71 nm with an average of 0.573 nm for ligand (Fig. 8a) and from 0.09 to 0.35 nm with an average of 0.292 nm for protein (Fig. 8b). The RMSF of the present complex was found maximum up to 0.4 nm indicating the stability of the atoms in the complex (Fig. 8c). The RoG has ranged from 2.99 to 3.08 nm with an average of 3.041 nm (Fig. 8d) and SASA (Fig. 8e) were significantly ranging from 462 to 477 nm2 with an average of 469.97 nm2. Up to eight HB have been observed during the run time of 10 ns (Fig. 8f). The short-range electrostatic (Coul-SR) and van der Waals/hydrophobic (LJ-SR) energies have been found −100.311 ± 9 kJ/mol and −231.75 ± 1.3 kJ/mol respectively.
Fig. 8.

The RMSD-L (a), RMSD-P (b), RMSF (c), RoG (d), SASA (e) and H-bonds (f) plots for the complex of RdRp-Dieckol.
2.4. ADMET analysis
The ADMET analysis of selected hits using the QikProp module of Schrödinger [48] was performed to predict several physical properties such as molecular weight, octanol/water partition coefficient, aqueous solubility, QPPcaco for cell permeability, brain/blood partition coefficient, QPlogHERG for blockage of HERG K + channels, metab for the number of metabolic reactions, QPlogKhsa for prediction of binding affinity to human serum albumin and percentage human oral absorption and mutagenicity (Table 3 ).
Table 3.
ADMET parameters of the hits.a.
| Comp. | MWb | logPc | Log Sd | QPlog HERGe | QPPCacof | QPlog BBg | Metabh | QPlog Khsai | % Oral Absj |
|---|---|---|---|---|---|---|---|---|---|
| Dieckol | 742.55 | 0.994 | −4.183 | −7.011 | 0.40 | −5.426 | 11 | −0.279 | 0 |
| Theaflavin-3,3′-Digallate | 866.69 | −0.956 | −3.911 | −6.296 | 0.02 | −6.624 | 16 | −0.56 | 0 |
| Phlorofurofucoeckol A | 602.46 | 1.137 | −4.31 | −6.797 | 1.44 | −4.519 | 10 | −0.192 | 0 |
| Rutin | 610.52 | −2.595 | −2.134 | −5.19 | 0.74 | −4.593 | 10 | −1.263 | 0 |
| 7-Phloroeckol | 496.38 | 0.649 | −3.175 | −5.897 | 3.76 | −3.68 | 8 | −0.388 | 15.13 |
| Nictoflorin | 594.52 | −1.826 | −2.472 | −5.694 | 3.17 | −4.006 | 9 | −1.216 | 0 |
| Procyanidin A2 | 576.51 | 0.301 | −4.038 | −5.569 | 1.58 | −3.871 | 12 | −0.21 | 0 |
| Hyperoside | 464.38 | −1.397 | −2.656 | −5.379 | 2.79 | −3.808 | 8 | −0.901 | 0.839 |
| Epigallocatechin-3-gallate | 458.37 | −0.251 | −3.553 | −5.694 | 1.03 | −4.335 | 10 | −0.442 | 0 |
| Juglanin | 418.35 | −0.318 | −2.912 | −5.418 | 11.5 | −2.981 | 6 | −0.66 | 31.16 |
| Eckol | 372.28 | 0.476 | −2.38 | −4.825 | 20.2 | −2.397 | 6 | −0.434 | 40.16 |
| Astragalin | 448.38 | −0.748 | −2.452 | −4.933 | 9.63 | −2.937 | 7 | −0.751 | 14.25 |
| Procyanidin B1 | 578.52 | 0.461 | −4.723 | −6.564 | 0.81 | −4.671 | 14 | −0.22 | 0 |
The parameters were calculated using QikProp [48]. bMolecular weight in Dalton 130–725 Da
Partition coefficient (−2.0 to 6.5)
Solubility coefficient (−6.5 to 0.5).
Prediction of blockade of HERG K+ channels (>−5).
Permeability across gut in nm/s [<25 (poor) and >500 (excellent)].
Brain/blood partition coefficient (−3 to 1.2).
Number of metabolic reactions (1–8).
Extent of binding to human serum albumin (–1.5 to 1.5), and jExtent of human oral absorption [<25 (poor) and >80 (excellent)].
Being natural products, most of them have not fitted with the desired ranges for molecular weight and log P, the parameter for lipophilicity. From the value of logS, we can predict that most of the compounds have good aqueous solubility (−4.723 to −2.134). Several absorption enhancement methods during the formulation development may be adopted to tackle these limitations. Most of the hits have been observed with very poor gut cell permeability due to their hydrophilic nature. All the compounds except juglanin, eckol, and astragalin have been found with highly poor blood-brain barrier permeability due to its lipophobic nature indicating the least risk of CNS toxicity. Since, most of the compounds fit in the range of the number of metabolic reactions, blockage of HERG K + channels and binding to human serum albumin suggesting their good distribution and metabolic parameters. The poor oral absorption properties have indicated the mode of administration other than oral routes.
We also studied the Lipinski parameters for the selected drugs to test their vulnerability for drug likeliness [49]. Although Lipinski's rule of five has not been considered as the rigid standard to claim the drug likeliness as about half of USFDA approved drugs do not co-align with the rule of five along with the marketed natural products and their semisynthetic derivatives [50]. In our case, the selected natural products could not pass through the Lipinski filters sufficiently (Table S3, see supporting information) but should not limit the claim potential of these drugs as new SARS CoV-2 inhibitors and also, modification in the structure may alter the physicochemical features with the maintenance of their inhibitory effects.
3. Conclusions
In search of potent inhibitors of SARS CoV-2, the molecular modeling of selected 177 phytochemicals of medicinal importance against the predicted active site of RdRp and Mpro was performed using site map analysis and pharmacophore screening. Total four natural chemical entities with promising inhibition of RdRp along with twelve of these natural products inhibiting Mpro protein were identified through different modes of docking with significant docking score and binding interactions. The molecular dynamic simulation studies have further supported the time-dependent (10 ns) stability of selected ligands into the active site of the chosen targets along with their compactness, arrangement of secondary structure into 3-dimensional space without major alterations in binding poses of ligands. The dual targeting inhibitors including dieckol, theaflavin-3,3′-digallate and phlorofucofuroeckol A were found to inhibit both RdRp and Mpro significantly. Additionally, rutin derived from Ruta graveolens, Tephrosia purpurea and Eucalyptus sp. and 7-phloroeckol derived from Ecklonia bicyclis have immense potential to inhibit Mpro with sufficiently higher stability, suggesting their candidature as promising SARS CoV-2 inhibitors.
4. Experimental section
4.1. Preparation of ligands
Total 177 natural products and their structural congeners with known anti-viral activity were selected through literature survey and their 3-dimensional structures were downloaded from PubChem [32] and SciFinder [33]. The 2-dimensional structures of the selected ligands, their biological sources are presented in Table S1 (see supporting information). These ligands were prepared using LigPrep to process for generation of possible states of ionization at targeted pH 7.0+/−2.0 using Epik, the addition of metal binding states, de-saltation, and generation of tautomers and stereoisomers by employing OPLS3e (Optimized Potentials for Liquid Simulations) force field [51].
4.2. Preparation of proteins
The 3-dimensional structures of selected proteins RdRp (PDB ID: 6M71) and Mpro (PDB ID: 6Y2E) were downloaded from RCSB Protein Data Bank [52]. The structural correctness in terms of hydrogen consistency, bond orders, steric interactions, charges, optimization, energy minimization using OPLS3e forcefield were performed using protein preparation wizard (PrepWizard) of Schrodinger suite (Maestro 12.5) [53]. The minimized proteins were used for pharmacophore screening, sitemap analysis, receptor grid generation and as well as for molecular docking.
4.3. Site map analysis
The selected PDB IDs are deposited in the protein data bank without any specific or co-crystallized ligand or inhibitor. In search of the specific binding site for the protein, the site map analysis was performed for all three proteins [29]. To visualize and evaluate the top-most binding sites, the potential binding site regions were generated with a minimum of fifteen site points for each binding site under a more restrictive hydrophobic environment using the standard grid for the selected proteins. The receptor-binding site with the highest druggability score (D-score) from the generated top five binding sites were selected for further molecular modeling.
4.4. Pharmacophore development and screening
There are no ligands co-crystallized with selected proteins of interest including RdRp (PDB ID: 6M71) [26] and Mpro (PDB ID: 6Y2E) [27]. In search of potent inhibitors, we planned to generate the pharmacophore-based hypothesis using the Phase module of Schrödinger [31]. The receptor cavity-based E-pharmacophore method was adopted after specifying residues through atom specification language (ASL) of the best site map with the highest d-score. The model was generated for the selected binding site along with the generated seven features, mainly comprising of hydrogen bond acceptor (A), hydrogen bond donor (D), negative ionizable (N), and aromatic ring (R) [such as ADNRRRR and AAADRRR for RdRp and MPro respectively].
Next, with a view of screening of ligands against the developed hypothesis, the molecules prepared after the LigPrep were subjected to match a minimum four of the identified features of the pharmacophore model. For each ligand, a maximum of fifty conformers were generated which were processed for their energy minimization. The acceptor and donor were preset as negative and positive equivalent, respectively to calculate the Phase screen score for each conformer. Out of 177, 55 (RdRp) and 117 (Mpro) ligands matched with the set features of the hypothesis (Fig. 9 ).
Fig. 9.

Flow of the work adopted for the in silico studies of phytochemicals against RdRp and Mpro.
4.5. Molecular docking
4.5.1. Receptor grid generation
The receptor grids for all the proteins were generated on the predicted binding site from site map analysis with the highest d-score using the GLIDE module of Schrodinger [34]. The generated site map points were used to generate the grid box of size 10 Å.
4.5.2. Ligand docking
Next, molecular docking was carried out using GLIDE. The ligands matching through the developed hypothesis were docked on selected active sites of all three proteins using standard precision (SP) to generate 10 poses per ligand through flexible ligand sampling. The ligands with docking scores less than −6.9 obtained in SP docking were further docked on the same binding site using extra precision (XP) to generate 3 poses per ligand.
4.6. Molecular dynamic (MD) simulation
Further, selected compounds were assessed for performing the molecular dynamic simulation (Fig. 9) to check the ligand stability at the binding site in ligand-receptor complex using GROningen MAchine for Chemical Simulations (GROMACS) 2020.1 [40,41] software and CHARMM36 (Chemistry at Harvard Macromolecular Mechanics) as an all atom force field [54]. CHARMM General Force Field (CGenFF) server was used to generate topology for the ligand [55,56]. The complex was solvated using the TIP3P water model, neutralized using Na+ and Cl− ions, equilibrated using a canonical [number of particles (N), system volume (V) and temperature (T), NVT] and isobaric-isothermic [number of particles (N), system pressure (P) and temperature (T), NPT] ensemble for 100 picoseconds and finally, subjected to molecular dynamic run using NPT group at 300 K temperature and 1 bar pressure for 10 ns.
4.7. ADMET and lipinski filters
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis and Lipinski parameters of selected hits were carried out in the QuikProp module of Schrodinger [48] and SwissADME [57] to study the information about functional groups, physical and chemical properties, and ADME parameters for all the top ligands.
Credit author statement
Normi D. Gajjar: Methodology, Data generation, Docking, MD Simulations, ADMET Assay, Writing- Original draft and revised manuscript preparation; Tejas M. Dhameliya: Conceptualization, Supervision, Writing-Reviewing and Editing; and Gaurang B. Shah: Conceptualization, Supervision, Writing-Reviewing and Editing.
Declaration of Competing Interest
The authors declare no competing financial interest.
Acknowledgments
The work was supported under the Drug Discovery Hackathon 2020 (DDH-2020), a joint initiative of the All India Council of Technical Education (AICTE), Council of Scientific and Industrial Research (CSIR) and Government of India. The authors would like to acknowledge the support provided by Schrödinger Inc. for performing molecular modeling.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2021.130488.
Appendix. Supplementary materials
References
- 1.Ashour H.M., Elkhatib W.F., Rahman M.M., Elshabrawy H.A. Insights into the recent 2019 novel coronavirus (SARS-CoV-2) in light of past human coronavirus outbreaks. Pathogens. 2020;9:e186. doi: 10.3390/pathogens9030186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Coronavirus disease 2019 (COVID-19). Situation report –51, (2020). https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200311-sitrep-51-covid-19.pdf?sfvrsn=1ba62e57_10 (accessed February 2, 2021).
- 3.Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/(accessed April 8, 2021).
- 5.Zhou P., Lou Yang X., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Di Jiang R., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baric R.S., Yount B. Subgenomic Negative-Strand RNA Function during Mouse Hepatitis Virus Infection. J. Virol. 2000;74:4039–4046. doi: 10.1128/jvi.74.9.4039-4046.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 8.Huang F., Li Y., Leung E.L.H., Liu X., Liu K., Wang Q., Lan Y., Li X., Yu H., Cui L., Luo H., Luo L. A review of therapeutic agents and Chinese herbal medicines against SARS-CoV-2 (COVID-19) Pharmacol. Res. 2020;158 doi: 10.1016/j.phrs.2020.104929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva L.R., da Silva Santos-Júnior P.F., de Andrade Brandão J., Anderson L., Bassi Ê.J., Xavier de Araújo-Júnior J., Cardoso S.H., da Silva-Júnior E.F. Druggable targets from coronaviruses for designing new antiviral drugs. Bioorg. Med. Chem. 2020;28 doi: 10.1016/j.bmc.2020.115745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shyr Z.A., Gorshkov K., Chen C.Z., Zheng W. Drug discovery strategies for SARS-CoV-2. J. Pharmacol. Exp. Ther. 2020;375:127–138. doi: 10.1124/jpet.120.000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pancholia S., Dhameliya T.M., Shah P., Jadhavar P.S., Sridevi J.P., Yogeshwari P., Sriram D., Chakraborti A.K. Benzo[d]thiazol-2-yl(piperazin-1-yl)methanones as new anti-mycobacterial chemotypes: design, synthesis, biological evaluation and 3D-QSAR studies. Eur. J. Med. Chem. 2016;116:187–199. doi: 10.1016/j.ejmech.2016.03.060. [DOI] [PubMed] [Google Scholar]
- 12.Usha T., Shanmugarajan D., Goyal A.K., Kumar C.S., Middha S.K. Recent Updates on Computer-aided Drug Discovery: time for a Paradigm Shift. Curr. Top. Med. Chem. 2018;17:3296–3307. doi: 10.2174/1568026618666180101163651. [DOI] [PubMed] [Google Scholar]
- 13.Battisti V., Wieder O., Garon A., Seidel T., Urban E., Langer T. A Computational Approach to Identify Potential Novel Inhibitors against the Coronavirus SARS-CoV-2. Mol. Inform. 2020;39 doi: 10.1002/minf.202000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah P., Dhameliya T.M., Bansal R., Nautiyal M., Kommi D.N., Jadhavar P.S., Sridevi J.P., Yogeeswari P., Sriram D., Chakraborti A.K. N-Arylalkylbenzo[d]thiazole-2-carboxamides as Anti-mycobacterial Agents: design, New Methods of Synthesis and Biological Evaluation. Med. Chem. Commun. 2014;5:1489–1495. [Google Scholar]
- 15.Jadhavar P.S., Dhameliya T.M., Vaja M.D., Kumar D., Sridevi J.P., Yogeeswari P., Sriram D., Chakraborti A.K. Synthesis, Biological Evaluation and Structure-Activity Relationship of 2-Styrylquinazolones as Anti-tubercular Agents. Bioorg. Med. Chem. Lett. 2016;26:2663–2669. doi: 10.1016/j.bmcl.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 16.Dhameliya T.M., Tiwari R., Banerjee A., Pancholia S., Sriram D., Panda D., Chakraborti A.K. Benzo[d]thiazole-2-carbanilides as new anti-TB chemotypes: design, synthesis, biological evaluation, and structure-activity relationship. Eur. J. Med. Chem. 2018;155:364–380. doi: 10.1016/j.ejmech.2018.05.049. [DOI] [PubMed] [Google Scholar]
- 17.Meyer-Almes F.J. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: virtual screening and structure based drug design. Comput. Biol. Chem. 2020;88 doi: 10.1016/j.compbiolchem.2020.107351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferraz W.R., Gomes R.A., Novaes A.L.S., Goulart Trossini G.H. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Med. Chem. 2020;12:1815–1828. doi: 10.4155/fmc-2020-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavasotto C.N., Di Filippo J.I. In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Mol. Inform. 2021;40 doi: 10.1002/minf.202000115. [DOI] [PubMed] [Google Scholar]
- 20.Touret F., Gilles M., Barral K., Nougairède A., van Helden J., Decroly E., de Lamballerie X., Coutard B. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci. Reports. 2020;10:13093. doi: 10.1038/s41598-020-70143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Islam M.T., Sarkar C., El-Kersh D.M., Jamaddar S., Uddin S.J., Shilpi J.A., Mubarak M.S. Natural products and their derivatives against coronavirus: a review of the non-clinical and pre-clinical data. Phytother. Res. 2020;34:2471–2492. doi: 10.1002/ptr.6700. [DOI] [PubMed] [Google Scholar]
- 22.Antonio A.D.S., Wiedemann L.S.M., Veiga-Junior V.F. Natural products’ role against COVID-19. RSC Adv. 2020;10:23379–23393. doi: 10.1039/d0ra03774e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian L., Qiang T., Liang C., Ren X., Jia M., Zhang J., Li J., Wan M., YuWen X., Li H., Cao W., Liu H. RNA-dependent RNA polymerase (RdRp) inhibitors: the current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y., Liang C., Xin L., Ren X., Tian L., Ju X., Li H., Wang Y., Zhao Q., Liu H., Cao W., Xie X., Zhang D., Wang Y., Jian Y. The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanhed A.M., Patel D.V., Teli D.M., Patel N.R., Chhabria M.T., Yadav M.R. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol. Divers. 2021;25:383–401. doi: 10.1007/s11030-020-10130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., Guddat L.W., Wang Q., Lou Z., Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavasotto C.N., Lamas M.S., Maggini J. Functional and druggability analysis of the SARS-CoV-2 proteome. Eur. J. Pharmacol. 2021;890 doi: 10.1016/j.ejphar.2020.173705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrödinger Release 2020-3 . LLC, New York, NY; 2020. SiteMap, Schrödinger. [Google Scholar]
- 30.Vidler L.R., Brown N., Knapp S., Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012;55:7346–7359. doi: 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrödinger Release 2020-3 . LLC; New York, NY: 2020. Phase, Schrödinger. [Google Scholar]
- 32.PubChem. https://pubchem.ncbi.nlm.nih.gov/(accessed August 31, 2020).
- 33.CAS Sci-Finder. https://scifinder.cas.org/(accessed August 31, 2020).
- 34.Schrödinger Release 2020-3 . LLC; New York, NY: 2020. Glide, Schrödinger. [Google Scholar]
- 35.Singh S., Sk M.F., Sonawane A., Kar P., Sadhukhan S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: an in-silico analysis. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1796810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeLano W.L. DeLano Scientific LLC; San Carlos, CA: 2002. The PyMOL Molecular Genetics Graphics System. [Google Scholar]
- 37.Teli D.M., Shah M.B., Chhabria M.T. In silico Screening of Natural Compounds as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike RBD: targets for COVID-19. Front. Mol. Biosci. 2021;7 doi: 10.3389/fmolb.2020.599079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durrant J.D., McCammon J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hospital A., Goñi J.R., Orozco M., Gelpí J.L. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinforma. Chem. 2015;8:37–47. doi: 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.M.J. Abraham, Berk Hess, E. Lindahl, D. van der Spoel, GROMACS 2020.1 (Manual Version 2020.1) Zenodo, (2020). 10.5281/zenodo.4054996 (accessed September 10, 2020). [DOI]
- 41.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindah E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. [Google Scholar]
- 42.González Torres M., Villarreal-Ramírez E., de los A. Moyaho Bernal M., Álvarez M., González-Valdez J., Gutiérrez Uribe J.A., Leyva Gómez G., Cortez J.R.C. Insights into the application of polyhydroxyalkanoates derivatives from the combination of experimental and simulation approaches. J. Mol. Struct. 2019;1175:536–541. [Google Scholar]
- 43.Ramalho T.C., França T.C.C., Cortopassi W.A., Gonçalves A.S., Da Silva A.W.S., Da Cunha E.F.F. Topology and dynamics of the interaction between 5-nitroimidazole radiosensitizers and duplex DNA studied by a combination of docking, molecular dynamic simulations and NMR spectroscopy. J. Mol. Struct. 2011;992:65–71. [Google Scholar]
- 44.Abbas S., Nasir H.H., Zaib S., Ali S., Mahmood T., Ayub K., Tahir M.N., Iqbal J. Carbonic anhydrase inhibition of Schiff base derivative of imino-methyl-naphthalen-2-ol: synthesis, structure elucidation, molecular docking, dynamic simulation and density functional theory calculations. J. Mol. Struct. 2018;1156:193–200. [Google Scholar]
- 45.Batran R.Z., Khedr M.A., Abdel Latif N.A., Abd El Aty A.A., Shehata A.N. Synthesis, homology modeling, molecular docking, dynamics, and antifungal screening of new 4-hydroxycoumarin derivatives as potential chitinase inhibitors. J. Mol. Struct. 2019;1180:260–271. [Google Scholar]
- 46.Modi P., Patel S., Chhabria M. Structure-based design, synthesis and biological evaluation of a newer series of pyrazolo[1,5-a]pyrimidine analogues as potential anti-tubercular agents. Bioorg. Chem. 2019;87:240–251. doi: 10.1016/j.bioorg.2019.02.044. [DOI] [PubMed] [Google Scholar]
- 47.Hornak V., Simmerling C. Development of softcore potential functions for overcoming steric barriers in molecular dynamics simulations. J. Mol. Graph. Model. 2004;22:405–413. doi: 10.1016/j.jmgm.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 48.Schrödinger Release 2020-3 . LLC; New York, NY: 2020. QikProp, Schrödinger. [Google Scholar]
- 49.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and devlopment settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 50.Zhang M.Q., Wilkinson B. Drug discovery beyond the “rule-of-five. Curr. Opin. Biotech. 2007;18:478–488. doi: 10.1016/j.copbio.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Schrödinger Release 2020-3 . LLC; New York, NY: 2020. LigPrep, Schrödinger. [Google Scholar]
- 52.Protein Data Bank. https://www.rcsb.org/ (accessed August 30, 2020).
- 53.Protein Preparation Wizard . Schrödinger, LLC, New York, NY, 2020; Prime, Schrödinger, LLC, New York, NY; 2020. Epik, Schrödinger, LLC, New York, NY, 2020; Impact. 2020. [Google Scholar]
- 54.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., De Groot B.L., Grubmüller H., MacKerell A.D. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2016;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A.D. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu W., He X., Vanommeslaeghe K., MacKerell A.D. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012;33:2451–2468. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Reports. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.